

ABSTRACT
Congenital erythropoietic porphyria (CEP or Gunther disease) is a very rare subtype of porphyria with a prevalence of <0.9 per 1 000 000. A 13-year-old female patient came to our hospital complaining of a severe cutaneous ulceration and scarring. The symptoms began in her first year of life as urine discoloration and skin blistering in sun-exposed areas. The family had been trapped in a high-risk conflict zone in Syria for many years, which precipitated the aggravation of symptoms. Based on clinical examination and laboratory tests, we diagnosed the patient with CEP and treated her with vitamin D supplementation alongside chronic blood transfusions, strict photoprotection and psychotherapy. After 7 months, there were no longer active ulcers or novel complications. Psychotherapy and patient education were important for her psychological development at this age. This treatment limited the deterioration of the symptoms and made the patient more committed to the periodic examinations.
Keywords: Gunther disease, congenital erythropoietic porphyria, ulceration, vitamin D, chronic blood transfusions
INTRODUCTION
Porphyria is a group of disorders that affect the heme biosynthesis pathway, which includes eight essential steps. Depending upon which step the deficiency occurs, particular porphyrins will accumulate and give rise to a specific spectrum of acute or non-acute symptoms [1]. Congenital erythropoietic porphyria (CEP or Gunther disease) is an extremely rare subtype of the non-acute group, estimating a prevalence of <0.9 in 1 000 000 [1, 2]. All organs are vulnerable to the porphyrin accumulation effects, but the most affected ones are the skin, hematopoietic system, teeth, bones and sclera. The clinical spectrum of CEP narrows by the age of onset, spanning from non-immune hydrops fetalis to isolated dermatological involvement in late-onset cases (Fig. 1) [2, 3]. Here we present the case of a 13-year-old Syrian female suffering from CEP; we also describe the management and follow-up for 7 months.
Figure 1.

An illustration of the heme biosynthesis pathway and CEP pathophysiology in a late normoblast. Each number represents the enzyme of the corresponding step: (1) aminolevulinic acid synthase, (2) aminolevulinic acid dehydratase, (3) porphobilinogen deaminase, (4) uroporphyrinogen III synthase, (5) uroporphyrinogen decarboxylase, (6) coproporphyrinogen-III oxidase, (7) protoporphyrinogen oxidase, (8) ferrochelatase. The gray–white rectangle represents the clinical spectrum that narrows by the increase in the age of onset, where the gray beginning expresses the concurrence of black-and-white-indexed involvements.
CASE PRESENTATION
A 13-year-old female patient came to our hospital with her parents complaining of severe cutaneous ulceration and scarring that span over her face and hands, accompanied with dyspigmentation. At first glance, we thought she was much older than just 13 years old (Fig. 2A). The symptoms began in her first year of life, where parents noticed severe skin redness and blistering in sun-exposed areas, mainly after outdoor activities. These lesions later progressed into ulcers with scarring and deformity on the nose, hands and fingers. Her parents also mentioned a history of urine discoloration in her early childhood. The family had been trapped in a high-risk conflict zone in northern Syria for many years. Unfortunately, the underprivileged security and healthcare conditions there prevented the patient from receiving adequate health services. There is no family history of consanguineous marriage between the parents or any of these symptoms in first-degree relatives. The patient, under the aforementioned circumstances, had received local antibiotics without any specific diagnosis.
Figure 2.
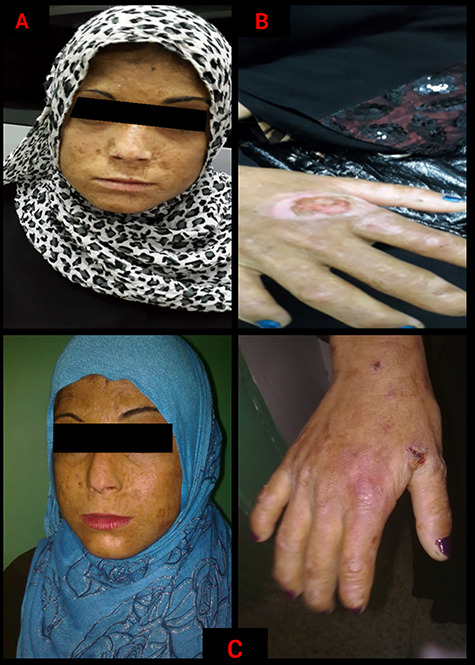
Congenital erythropoietic porphyria in a 13-year-old female: (A) a dyspigmented sclerodermatous appearance of the face accompanied with linear and spotted atrophic lesions. (B) Immense areas of ulceration over the dorsum of the hand, with acrolysis of the bones and soft tissues at the ends of the fingers. (C) Healing ulcers on the face and dorsum of the hand after 1 month of treatment; the skin improved in terms of pigmentation and elasticity, with no active ulcers.
On physical examination of the face, we observed areas of dyspigmentation and hypertrichosis, especially over the patient’s cheeks and forehead. Also, crusts were formed over the old ulcers. The central area of her face exhibited a sclerodermatous appearance with linear and spotted atrophic lesions. The nose was severely deformed and scarred (Fig. 2A). The teeth were reddish-brown discolored (erythrodontia) under the visible light. Eye examination revealed signs of conjunctivitis, scleromalacia and mild blepharitis; but there were no signs of corneal injury. The dorsum of the hands exhibited immense areas of ulceration, giving a similar sclerodermatous appearance to that in the face. Also, an acrolysis of the bones and soft tissues at the terminal ends of the fingers made the digits appear to be deformed and shorter than normal (Fig. 2B). The liver was palpable 2 cm under the costal margin, but the spleen was impalpable. Fluorescence spectroscopy using Wood’s lamp revealed a simultaneous bright red fluorescence of teeth and bright green fluorescence of interdigital spaces of the hands (Fig. 3).
Figure 3.

Fluorescence spectroscopy using Wood’s lamp revealed a simultaneous bright red fluorescence of teeth and bright green fluorescence of interdigital spaces.
The porphyrin profile in her urine revealed elevated uroporphyrins and coproporphyrin-I (Table 1). Based on the patient’s history, clinical examination and lab work, we diagnosed the patient with CEP. We informed the patient about her condition and the importance of strict photoprotection; also, we recommended using high sun protection factor sunscreens. Vitamin D supplementation was used to compensate for the lack of sun exposure alongside blood transfusions and deferasirox to chelate iron and modulate the symptoms. After 1 month of treatment, the patient showed noticeable improvement, there were no longer active ulcers, and the skin showed significant improvement in terms of elasticity and pigmentation (Fig. 2C). This improvement satisfied the patient and made her more committed to the periodic examinations. After 7 months of follow-up, there were no novel complications, and the patient’s symptoms improved significantly, but further psychotherapy is required. Also, we informed the patient that skin resurfacing treatment can provide a slight improvement to the old scars and dyspigmentation.
Table 1.
Porphyrins profile in the patient’s 24-h urine
| Test | Level | Reference range | Unit |
|---|---|---|---|
| Uroporphyrins | 9820 | Up to 30 | Nmol/24 h |
| Heptacarboxyporphyrin 1, 3 | 415 | Up to 9 | Nmol/24 h |
| Hexacarboxyporphyrin 1, 3 | 146 | Up to 8 | Nmol/24 h |
| Pentacarboxyporphyrin 1, 3 | 673 | Up to 10 | Nmol/24 h |
| Coproporphyrin 1 | 2514 | Up to 168 | Nmol/24 h |
DISCUSSION
The deficient activity of uroporphyrinogen III synthase (UROS), the fourth enzyme in the heme synthesis cycle, represents the cornerstone of the pathology in CEP [2]. This enzymatic deficiency was previously linked to a mutation in the UROS encoding gene, which explained the autosomal recessive inheritance pattern in the majority of CEP cases [2, 4]. Recently, it has been proven that several mutations in other genes can be involved in the pathogenesis, like GATA1 and aminolevulinate synthase genes, which also explained the X-linked inheritance pattern [2, 5].
These mutations result in a varied deficiency, but not an absence, of UROS. Hence, the downstream pathogenesis, clinical manifestations, diagnosis and management will depend on the accumulations and deficiencies of the heme cycle intermediates (Fig. 1) [1, 3]. Once hydroxymethylbilane is formed, it undergoes one of two pathways: (1) enzymatic conversion via UROS to uroporphyrinogen III (URO-III) and (2) non-enzymatic conversion to uroporphyrinogen-I and subsequently coproporphyrinogen-I [2, 3]. Normally, UROS dominates the process by converting the majority of hydroxymethylbilane to URO-III, which completes the cycle to form heme. But in CEP this enzyme is deficient, which shifts the process toward uroporphyrinogen-I and coproporphyrinogen-I formation, and further auto-oxidation to their corresponding porphyrins that accumulate in the erythroid precursors and subsequently in tissues due to hemolysis [1, 3]. The photoreactivity of these porphyrins induces free radical formation and cell death due to the photo-oxidation; this effect is amplified when the patient get exposed to wavelength 400–410 nm [2, 3]. A similar wavelength is used in phototherapy for neonatal jaundice but also predisposes to fatal sequelae in case of misdiagnosis before initiating phototherapy [6].
The porphyrin profiles, in addition to the clinical manifestations, represent the key diagnostic features of CEP [4]. In our case, the early onset of symptoms alongside the clinical examination and lab tests confirmed the diagnosis of CEP; we did not perform any genetic tests since they are unavailable in our hospital. According to a series of 29 CEP patients, approximately 76% of patients had an onset before the fifth year of life, with a greater likelihood for urine discoloration to be the initial symptom among other complications. Furthermore, the hematological complications was found to be the main predictors of poor prognosis, patients with an onset before the fifth year developed a more progressive form, involving neonatal jaundice and transfusion-dependent hemolytic anemia [7]. This is consistent with the patient’s history of urine discoloration in her early childhood preceding the onset of cutaneous and other complications, but neonatal jaundice was not reported.
Our differential diagnosis also involved porphyria cutanea tarda (PCT), hepatoerythropoietic porphyria (HEP), epidermolysis bullosa and xeroderma pigmentosum. We ruled out PCT and HEP biochemically; epidermolysis bullosa was excluded clinically since it involves areas with physical injury despite light exposure. Xeroderma pigmentosum was excluded under the absence of tumors and neurological complications [1, 8].
As for the management, it involves vitamin D supplementation with photoprotection, chronic blood transfusion and allogeneic bone marrow transplantation, which represents the only effective therapy for severe CEP cases [1, 4]. Also, promising alternatives have emerged recently: afamelanotide, a melanocortin-stimulating hormone analog, was used for improving light tolerance by stimulating melanosynthesis; proteasome inhibitors were used to inhibit URO-III degradation in cases of certain mutations; and gene therapy [2, 9]. For our patient, vitamin D supplementation alongside strict photoprotection and blood transfusion have succeeded in reducing the deterioration of the symptoms to the date of this article.
This case presents a clinical picture of CEP in a patient who was impeded from receiving healthcare services for many years due to the war. Also, we discuss the efficiency of preventive therapy along with blood transfusion. More studies are required to replace this symptomatic management with effective pharmacological therapies.
DECLARATIONS
ETHICAL APPROVAL
Not applicable.
CONSENT FOR PUBLICATION
Written informed consent was obtained from the patient for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
AVAILABILITY OF DATA AND MATERIAL
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
COMPETING INTERESTS
The authors declare no conflicts of interest.
FUNDING
There are no sources of funding.
GUARANTOR
Ali Kahila.
ACKNOWLEDGEMENTS
None.
ABBREVIATIONS AND ACRONYMS
- CEP
congenital erythropoietic porphyria
- UROS
uroporphyrinogen III synthase
- URO-III
uroporphyrinogen III
- PCT
porphyria cutanea tarda
- HEP
hepatoerythropoietic porphyria
REFERENCES
- 1. Stolzel U, Doss MO, Schuppan D. Clinical guide and update on Porphyrias. Gastroenterology 2019;157:365–81 e4. doi: 10.1053/j.gastro.2019.04.050. [DOI] [PubMed] [Google Scholar]
- 2. Di Pierro E, Brancaleoni V, Granata F. Advances in understanding the pathogenesis of congenital erythropoietic porphyria. Br J Haematol 2016;173:365–79. doi: 10.1111/bjh.13978. [DOI] [PubMed] [Google Scholar]
- 3. Desnick RJ, Astrin KH. Congenital erythropoietic porphyria: advances in pathogenesis and treatment. Br J Haematol 2002;117:779–95. doi: 10.1046/j.1365-2141.2002.03557.x. [DOI] [PubMed] [Google Scholar]
- 4. Karim Z, Lyoumi S, Nicolas G, Deybach JC, Gouya L, Puy H. Porphyrias: a 2015 update. Clin Res Hepatol Gastroenterol 2015;39:412–25. doi: 10.1016/j.clinre.2015.05.009. [DOI] [PubMed] [Google Scholar]
- 5. Phillips JD, Steensma DP, Pulsipher MA, Spangrude GJ, Kushner JP. Congenital erythropoietic porphyria due to a mutation in GATA1: the first trans-acting mutation causative for a human porphyria. Blood 2007;109:2618–21. doi: 10.1182/blood-2006-06-022848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Baran M, Eliacik K, Kurt I, Kanik A, Zengin N, Bakiler AR. Bullous skin lesions in a jaundiced infant after phototherapy: a case of congenital erythropoietic porphyria. Turk J Pediatr 2013;55:218–21. [PubMed] [Google Scholar]
- 7. Katugampola RP, Badminton MN, Finlay AY, Whatley S, Woolf J, Mason N et al. Congenital erythropoietic porphyria: a single-observer clinical study of 29 cases. Br J Dermatol 2012;167:901–13. doi: 10.1111/j.1365-2133.2012.11160.x. [DOI] [PubMed] [Google Scholar]
- 8. Erwin A, Balwani M, Desnick RJ. Congenital erythropoietic porphyria In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews((R)). Seattle (WA): University of Washington, Seattle, 1993. Available from: https://www.ncbi.nlm.nih.gov/books/NBK154652/ [PubMed] [Google Scholar]
- 9. Howard M, Hall A, Ramsay D. Congenital erythropoietic porphyria (Gunther disease) - long-term follow up of a case and review. Dermatol Online J 2017;23:13030/qt10n7k90g. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.