

Abstract
Ionizing radiation (IR) is an important cancer treatment approach. However, radioresistance eventually occurs, resulting in poor outcomes in patients with cancer. Radioresistance is associated with multiple signaling pathways, particularly pro-survival signaling pathways. The extracellular signal-regulated kinase 1/2 (ERK1/2) cascade is an important signaling pathway that initiates several cellular processes and is regulated by various stimuli, including IR. Although numerous studies have demonstrated the pro-survival effects of active ERK, activation of ERK has also been associated with cell death, indicating that radiosensitization may occur by ERK stimulation. In this context, the present review describes the associations between ERK signaling, cancer and IR, and discusses the association between ERK and its pro-survival function in cancer cells, including stimuli, molecular mechanisms, clinical use of inhibitors and underlying limitations. Additionally, the present review introduces the view that active ERK may induce cell death, and describes the potential factors associated with this process. This review describes the various outcomes induced by active ERK to prompt future studies to aim to enhance radiosensitivity in the treatment of cancer.
Keywords: ionizing radiation, cancer, extracellular signal-regulated kinase, radiosensitivity, pro-survival, pro-death
1. Introduction
Ionizing radiation (IR) serves an essential role in modern cancer management due to its unique advantages, including non-invasiveness and a lack of intense systemic toxicity (1). As an integral component of adjuvant and palliative treatment strategies for primary and advanced/metastatic tumors, respectively, radiation therapy (RT) is administered for the management of nearly two-thirds of all types of cancer, including cancer of the prostate, cervix, bladder, head and neck, breast, lung, brain, pancreas and skin, anorectal cancer, and soft-tissue sarcomas (2). Although RT has demonstrated various degrees of success, recurrence and treatment failure may occur in patients due to intrinsic or external radioresistance (3). Therefore, strategies are urgently required for enhancing radiosensitivity in the treatment of cancer. Targeted molecular therapy has gained increasing attention for evaluating the effect of IR on targets of specific cancer-associated signaling pathways. Numerous preclinical and clinical studies have demonstrated that combination therapies using radiation and targeted molecular agents improve tumor response rates and clinical outcomes (4–6). Five potential therapy mechanisms have been described: i) Spatial cooperation; ii) temporal modulation; iii) biological cooperation; iv) cytotoxic enhancement; and v) normal tissue protection (7). Among the numerous signaling molecules, extracellular signal-regulated kinase (ERK) is one of the most important.
The ERK cascade functions as a crucial intermediary in intracellular signal transduction networks to transmit signals from extracellular stimuli, such as growth factors, hormones and neurotransmitters, among others (8). Increasing evidence indicates that activation of ERK induces cell proliferation and confers a survival advantage on cells, giving it a major role in human cancer (9). Therefore, pharmaceutical inhibitors targeting one of the common signals, the RAS/RAF/MEK/ERK signaling pathway, have been developed to improve the clinical outcomes of patients with cancer (6,10). However, even when tumors exhibit a positive primary response to these inhibitors, poor therapeutic effects may result from acquired resistance (11). Additionally, the mechanisms of resistance to ERK1/2 pathway inhibitors are unknown. Some studies have demonstrated that ERK activation leads to non-prosurvival effects in cancer cells (12–15). Particularly, active ERK may lead to cell death under different circumstances, such as location of ERK and time and extent of active ERK, which may result in radiosensitization in human cancer (4,5). Therefore, the true effect of ERK must be clarified before combining RT and ERK inhibitors for cancer treatment. In the current review, the dual effects of activated ERK on cancer cells and their respective potential mechanisms are summarized. The ways in which active ERK induces cell survival are described, including the molecular mechanisms, clinical use and limitations. Additionally, the association between ERK activation and cell death is described, as well as the influence of ERK on the response to IR in cancer cells. The present review provides a foundation for developing cancer therapies targeting the function of ERK.
2. ERK and cancer
ERKs belong to the family of mitogen-activated protein kinases (MAPKs), which also includes ERK5, c-JunNH2-terminal kinase and p38 MAPK (15). The present review focuses on ERKs, which include two isoforms, ERK1 and ERK2 (also known as p44 and p42 MAPK, respectively). Generally, ERK activation may occur in two ways; activated-ERK is associated with the autocrine/paracrine signaling of mitogenic growth factors through tyrosine kinase receptors, such as the epidermal growth factor receptor (EGFR), the insulin-like growth factor receptor or c-MET, induced by various stimuli (16). Additionally, ERK activation may occur as a result of abnormal activation or genetic alterations in its upstream signaling molecules, such as RAS, RAF and MEK1/2 (17). The RAS/RAF/MEK/ERK cascade is the typical signaling pathway following the three-stage enzymatic cascade of MAPKs (18). In this pathway, growth and survival factors activate RAS GTPases by promoting the release of GDP to allow GTP binding. Active RAS-GTP then binds to one of the RAF protein kinases, ARAF, BRAF or CRAF (also known as MAPK3K), resulting in their activation. Subsequently, RAF phosphorylates and activates MEK1/2 (also known as MAP2K1/2), which in turn phosphorylate and activate ERK1/2 (19). Once activated, ERK1/2 can regulate ~250 potential substrates, including transcription factors, protein kinases and phosphatases, cytoskeletal elements, regulators of apoptosis and a variety of other signaling-associated molecules (20,21). Activated-ERK can lead to various physiological responses, as shown in Fig. 1 (22).
Figure 1.
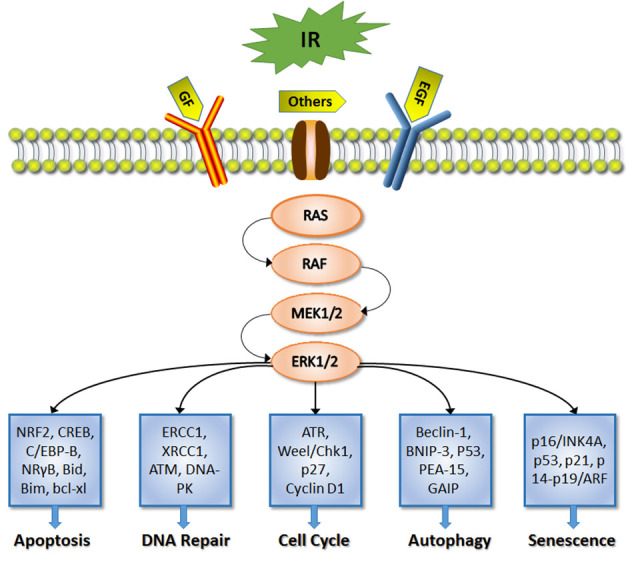
ERK signaling. The ERK signaling pathway can be stimulated by ionizing radiation via growth factor and EGFR. Active ERK participates in various cellular processes, such as apoptosis, DNA repair, cell cycle, autophagy and senescence. IR, ionizing radiation; GF, growth factor; EGF, epidermal GF; MEK1/2, mitogen-activated protein kinase 1/2; ERK, extracellular signal-regulated kinase 1/2; EGFR, epidermal growth factor receptor.
Under normal conditions, ERK signaling is regulated by feedback loops at multiple levels, which are essential for regulating cell growth and homeostasis. However, under abnormal circumstances, activated ERK may lead to various pathological changes (23), including tumorigenesis (24), diabetic nephropathy (25), viral infection (26), cardiovascular disease (27) and Alzheimer's disease (28). Nearly one-third of all types of cancer, including melanoma, uveal melanoma, and pancreatic, non-small cell lung, colorectal, basal-like breast and hepatic cancer, involve deregulated ERK (9). Hoshino et al (29) revealed that ERK was constitutively active in ~50 tumor cell lines (36.2%) in a tissue-specific manner; cell lines derived from the pancreas, colon, lung, ovary and kidney exhibited high frequencies of constitutive ERK activation, while those derived from the brain, esophagus, stomach and liver, and those of hematopoietic origin, exhibited low frequencies with a limited degree of ERK activation. Additionally, other stimuli, such as IR and chemotherapy drugs, can activate ERK and affect the efficacy of cancer therapy, with some exceptions (11,30). For example, Corn et al (31) observed that phosphorylated ERK-positive cancer cells became ERK-negative after RT in colorectal cancer. Overall, active ERK seems to be closely associated with the onset, development, invasion, metastasis and therapy-resistance of most types of tumor.
3. ERK and irradiated cancer cells
IR is known to induce cell toxicity by damaging biological molecules directly and indirectly. High-linear energy transfer (LET) radiation, such as α particles and neutrons, directly ionizes cellular macromolecules, including DNA, RNA, lipids and proteins, while low-LET radiation, such as X-rays and γ-rays, indirectly damages biological macromolecules by generating reactive oxygen species, such as superoxide and hydroxide radicals (32). Indirect DNA damage from free radicals accounts for ~65% of radiation-induced DNA damage, which is characterized by both single- and double-stranded breaks (DSBs) in DNA, with the latter being more lethal than the former (30).
IR or DNA damage are important stimuli that can activate ERK, one of the most crucial signaling pathways (33,34). Notably, IR induces phosphorylation of ERK in a time-dependent manner; activation of ERK1/2 appears as early as 15 min after IR, with maximum activation observed after 24 h (35,36). The extent of ERK activation by radiation varies. For example, intense ERK activity is typically induced by low doses of radiation (1 Gy) rather than by high doses (6 Gy) (37). Additionally, IR can induce numerous other signaling pathways within cells, including EGFR, PI3K/AKT/mTOR and cell cycle checkpoint signaling pathways (38–40). These pathways in turn activate ERK and are upstream molecules of ERK (41). For example, Sambade et al (42) observed that activation of ERK1/2 by EGFR typically occurred at a later phase (60 and 90 min) rather than an early phase (15 and 30 min) after radiation.
Once activated, ERK participates in cell proliferation and survival through a variety of mechanisms that affect the radiosensitivity of tumor cells. In general, activated ERK protects tumor cells from radiation-induced death in numerous ways. Park et al (43) determined that ionizing radiation-induced MAPK can activate the progression elevated gene 3 promoter, thereby increasing the expression levels of vascular endothelial growth factor (VEGF) protein in glioblastoma. VEGF promotes tumor angiogenesis and is associated with endothelial radiosensitization (3). Similarly, activated ERK increases RAD51 expression in pancreatic cancer cells (44), which promotes homologous DNA repair, leading to RT resistance (45). In addition, a recent study determined that γ-ray irradiation alone can increase cell migration in vitro, which is mainly achieved by activating the EGFR/ERK/AKT signaling pathway and increasing the expression of nuclear factor γB (NFγB) (46). After pretreatment with olaparib, a poly(ADP-ribose) polymerase inhibitor, activation of the EGFR/ERK/AKT signaling pathway induced by γ-rays was inhibited, thereby reducing the metastatic capacity of tumor cells (46). However, some studies have found that activated ERK can promote RT-induced cell death, thereby increasing radiosensitivity (47). The mechanisms involved in this process mainly include abrogating radiation-induced G2/M arrest, apoptosis and autophagy (47–49). Therefore, activated ERK serves various roles in cancer cells, and the functions of activated ERK1/2 are influenced by numerous factors, including cell type, location of ERK, and time and extent of active ERK (5). The present review discusses two opposite outcomes of ERK activation: Pro-survival and pro-death.
Improvement in radiosensitivity by ERK inhibition
As aforementioned, radiation can induce rapid activation of ERK in various cancer cell types (50). A study has demonstrated that activated ERK protects cancer cells from the cytotoxic effects of radiation (51). ERKs promote cell survival in various ways, including via the inhibition of apoptosis, the induction of DNA damage repair and the arrest of the cell cycle.
Apoptosis, or programmed cell death type I, is an essential process leading to the removal of damaged cells without affecting normal cells, following DNA damage or during development (52). Apoptosis can be triggered by the caspase-mediated intrinsic signaling pathway, which is mainly regulated by the B-cell lymphoma 2 (Bcl-2) family of intracellular proteins, or by an extrinsic signaling pathway, which is closely regulated by the tumor necrosis factor (TNF) receptor family (53,54). Deregulation of apoptosis is associated with uncontrolled cell proliferation, cell growth, progression of cancer and cancer resistance to drug therapies (55,56). Therefore, apoptosis deregulation is considered a hallmark of cancer (55). Two types of proteins determine the cellular outcome: Pro-apoptotic proteins, such as Bcl-10, Bak, Bid, BAG, Bax, Blk, Bad and Bim, and anti-apoptotic proteins, such as Bcl-2, Bcl-x, Bcl-XS, Bcl-xl, Bcl-w, IAP and Mcl-1 (56). ERK activation induced by IR can activate various substrates, which transmit the signaling of ERK to apoptosis-associated proteins; common substrates include transcription factors, such as nuclear factor erythroid 2-related factor 2 (NRF2), cyclic AMP-responsive element binding protein (CREB) and CAAT/enhancer binding protein β (C/EBP-β) (57,58). Chen et al (57) observed that radiation-induced ERK1/2 phosphorylation increased NRF2 expression in osteosarcoma U-2 cells; activation of NRF2 served a radioprotective role by stimulating Bcl-2 and p65 expression, while inhibiting Bax and p53 expression. Similarly, ERK1/2 can induce Bcl-xl, Mcl-1 and c-FLIPs expression via CREB and C/EBP-β activated by ERK1/2 (38). Additionally, ERK is able to activate the NFγB1 dimer, a crucial regulator of anti-apoptotic genes, including genes encoding inhibitors of apoptotic proteins and members of the Bcl-2 family (59). Furthermore, ERK can directly stimulate Bid and Bim (38). Therefore, ERK1/2 is associated with apoptosis and can protect against cell death mainly by increasing or decreasing levels of anti- and pro-apoptotic proteins, respectively.
DNA damage repair is essential in the cancer cell response to IR and includes at least five main processes: Base excision repair, nucleotide excision repair, mismatch repair, homologous recombination (HR) repair and non-homologous end joining (NHEJ) (60). HR and NHEJ, the major repair pathways for DNA DSBs and closely associated with ERK (61), are mainly regulated by PI3K-like kinases, including ataxia telangiectasia mutated (ATM), RAD3-related protein (ATR) and DNA-dependent protein kinase (DNA-PK) (62). Additionally, DNA DSB repair is modulated directly or indirectly by other means, such as EGFR and the ERK axis (63). ERKs can directly activate ATM or DNA-PK, which mainly participate in NHEJ-mediated DSB repair (4). Furthermore, ERK can activate ATR followed by cell cycle arrest (22). Additionally, activation of ERK induced by IR has been associated with increased levels of transcriptional proteins (such as ERCC1 and XRCC1) involved in DNA repair in DU145 and LNCaP prostate carcinoma (64,65). However, in one study, radiation-induced ERK activation was affected by the extent or scope of DSB: Low-level DSBs (equivalent to 2 Gy) resulted in ERK activation, while high-level DSBs (>2 Gy) led to phosphatase-mediated ERK dephosphorylation and subsequent suppression of the ERK signaling pathway (4). Therefore, activation of ATM or ATR by activated ERK may depend on the radiation dose. Overall, ERK seems to be associated with DNA repair to protect against cell death.
Cell cycle arrest, which is governed by cell cycle checkpoint-associated proteins, such as cell cycle proteins (cyclins) and cyclin-dependent kinases, can maintain gene stability by blocking cell cycle progression and initiating processes to repair the detected damage (66). Some studies have demonstrated that ERK1/2 pathway activation following IR or DNA damage is critical for the activation of cell cycle checkpoints in response to radiation cytotoxicity; these studies confirmed that phosphorylation of ERK1/2 is a prerequisite for inducing ATR expression, which can activate Wee1 and checkpoint kinase 1 as key regulators of the G2/M checkpoint (67,68). Additionally, one study indicated that expression of basal breast cancer type 1 susceptibility protein (BRCA1) tumor suppressor is necessary for IR-induced activation of ERK, followed by G2/M arrest, in MCF-7 cells (69). Therefore, ERK is associated with key cell cycle checkpoint proteins to induce cell cycle arrest, and serves a protective role in response to radiation cytotoxicity.
Previous studies have demonstrated that ERK signaling serves a radioprotective role in cancer therapy and contributes to radioresistance (9,29,70). Therefore, inhibition of the ERK signaling pathway may provide a valuable approach to increase the radiosensitivity of cancer cells in response to IR. Various inhibitors for suppressing ERK signaling have been tested in preclinical and clinical investigations, including RAF inhibitors (vemurafenib, dabrafenib, LGX818, TAK-632, MLN2480 and PLX-4720), MEK inhibitors (trametinib, cobimetinib, MEK162, AZD6244, RO5126766, GDC-0623 and PD0325901) and ERK inhibitors (SCH772984, VTX11e and GDC-0994) (10,11,71,72). Radiation in combination with RAF and MEK inhibitors has been widely tested in vitro and in vivo for multiple types of cancer cells and various biological mechanisms (73). Estrada-Bernal et al (74) demonstrated that GSK212, a MEK1/2 inhibitor, downregulates several intermediates of DSB repair signaling, including BRCA1, RAD51, DNA-PK and PPM2, in irradiated pancreatic cancer cells, and suppresses DSB damage repair, particularly through HR repair pathways. Similar results were obtained by Marampon et al (75) using another MEK1/2 inhibitor, U0126, which reduced DNA-PK expression induced by IR. Furthermore, radiosensitization induced by a MEK inhibitor, AZD6244, was associated with a decreased cell cycle checkpoint response and increased mitotic catastrophe compared with no inhibitor present; in these in vitro and in vivo experiments, an apparent growth delay in xenografts of A549 cancer cells was observed after AZD6244 was combined with irradiation, compared with treatment by irradiation alone. Notably, the DNA repair pathway did not differ between the combined model and each treatment alone, which may be attributable to differences in molecular structure. A study observed that ERK inhibitors sensitized cancer cells to irradiation by downregulating specific molecules, such as transforming growth factor-α and TNF-α converting enzyme, which can stimulate a radioresistance mechanism by activating EGFR after inhibiting mutant RAS (76). Therefore, ERK inhibitors may increase cell death by suppressing phosphorylation of EGFR signaling pathways. Other potential mechanisms, such as ERK inhibition, can disrupt the production of c-Myc induced by IR, which can promote the onset, progression and resistance to targeted therapy in numerous types of cancer, such as prostate cancer (6,77). Active ERK-induced apoptotic effects have been confirmed by promoting pro-apoptotic and inhibiting anti-apoptotic proteins (59). The aforementioned data suggest that inhibition of ERK signaling may be an effective treatment for some types of cancer cells.
The effects of inhibitors of ERK signaling on tumor suppression are being evaluated in various clinical trial phases; however, the observed poor outcomes are the result of acquired resistance, clinical side effects, varying functional times, intensity or other unclear reasons, which are limiting their value for clinical application (78–80). The main mechanisms of acquired resistance include: i) NRAS or KRAS mutations, amplification of BRAF V600E, mutations in MEK1/2 and loss of CDKN2A, which lead to BRAF inhibitor resistance; ii) MEK mutations or BRAF amplification, which result in MEK inhibitor resistance; iii) ERK mutations, which contribute to ERK inhibitor resistance; iv) other abnormally activated or elevated levels of molecules, such as PI3K/AKT, CCND1, receptor tyrosine kinase and CRAF128; v) a decreased dependency of tumor cells on the ERK signaling pathway for growth; and vi) rebound of MEK/ERK after inhibition treatment or compensatory mechanisms (11,18,81).
Numerous studies have revealed less satisfactory outcomes regarding ERK inhibition (11,12). Therefore, the opposing functions of active ERK must be considered, particularly when evaluating whether radiosensitivity may be improved by inducing ERK activation.
Activation of ERK: A potential strategy for radiosensitization
Some studies have suggested that activation of ERK by IR or other compounds may lead to cell death; active ERK can have a pro-death or growth-arrest role in cancer cells (15,82). Dual effects are the result of the numerous mechanisms upstream and downstream of ERK. However, the association between the pro-death effect of active ERK and radiosensitivity has not been systematically evaluated in cancer cells.
Studies have identified various stimuli that can further induce cancer cell death by activating ERK, including antitumor agents, such as taxol (83), carboplatin (48), etoposide (84), doxorubicin and cisplatin (85), elements, such as cadmium (86,87) and benzo(a)pyrene (88), irradiation (89,90), naturally derived products, such as chelerythrine (91) and piperlongumine (92), and others (82,93). Additionally, constitutively activated ERK mediates cell death. In two diffuse large B-cell lymphoma (DLBCL) cell lines, CD40-sensitive DLBCL cells were induced to undergo apoptosis by CD40 ligand (CD40L) only when ERK was constitutively activated, and this effect disappeared when the MEK inhibitor U0126 inhibited ERK phosphorylation; by contrast, CD40-resistant DLBCL cells exhibited no response to CD40L due to a lack of constitutively activated ERK (82). Three main pro-death or anti-proliferative mechanisms were involved in this process, including apoptosis-induced cell death, autophagic programmed cell death and senescence (15,94,95).
As aforementioned, both constitutive and stimuli-induced ERK signaling contribute to apoptosis and are influenced by numerous factors, some of which are closely associated with IR or radiosensitivity. Lee et al (47) demonstrated that overexpression of protein kinase C δ (PKCδ), a PKC isoform, contributed to the expression of phosphorylated ERK; the PKCδ-ERK signaling pathway further enhanced radiation-induced apoptosis and radiosensitivity by abrogating radiation-induced G2/M arrest. Another isoform of PKC, PKCε, also led to radiation-induced cell death by mediating ERK activation; this pro-death effect was inhibited when NIH3T3 cells were pretreated with the MEK inhibitor PD98059 (69,89). Watanabe et al (90) demonstrated that PD98059 significantly inhibited radiation-induced apoptosis, further leading to radioresistance, indicating that active ERK may serve a radiosensitizing role in rat cells. In terms of ERK substrates, several studies have demonstrated that activated ERK in numerous cancer cells can phosphorylate p53, an essential tumor suppressor that serves a pivotal role in protecting genome integrity and mediating cell death (96–98), and is key to the radiation response in tumor cells. Functional p53 regulates irradiated cancer cell death by inducing apoptosis or senescence, and by inhibiting autophagy-associated cell survival (30,49,99). Pseudo-ginsenoside-Rh2, which is a derivative of ginsenoside Rh2, has been shown to suppress cell growth and induce intrinsic apoptotic pathways through activation of the Ras/Raf/ERK/p53 signaling pathway, subsequently upregulating Bax expression and downregulating Bcl-2 and Bcl-xl expression (100). Furthermore, excessive activation of the Ras/Raf/ERK/p53-p21 signaling pathway was shown to induce apoptosis and G2/M arrest in human lung carcinoma cells (101). A study demonstrated that activation of ERK/p53/Beclin-1 could mediated autophagic cell death in A549 cells (102). Therefore, p53 upregulation seems to be an essential mechanism of ERK-induced cell death (Fig. 2). However, other studies have suggested that ERK induces cell death independently of p53 such as cPLA2-FasL and DAPK-TNF pathway (83,103).
Figure 2.
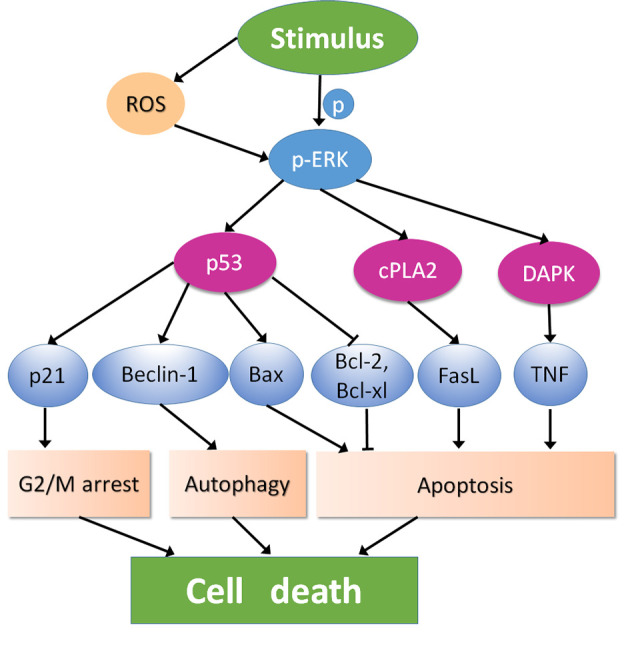
Mechanisms of ERK-induced cell death. Stimulus activate ERK directly or indirectly. p-ERK could induce cell death dependent of p53 or independent of p53. ROS, reactive oxygen species; p-ERK, phosphorylated extracellular signal-regulated kinase; DAPK, death-associated protein kinase; cPLA2, cytosolic phospholipase A2; TNF, tumor necrosis factor; FasL, Fas ligand; Bcl, B-cell lymphoma.
In addition, there are a number of other factors for which the intensity and localization of ERK activation may be key in the choice between cell survival or cell death (104). Tang et al (84) demonstrated that etoposide and IR resulted in DNA damage that activated ERK with the same intensity; however, low-intensity DNA damage resulted in cell cycle arrest, while high-intensity DNA damage caused apoptosis. Inactive ERK localizes in the cytoplasm, while activated ERK1/2 typically exerts its role by entering the nucleus and phosphorylating transcription factors (105). However, some studies have suggested that active ERK1/2 can access specific substrates and affect cell conditions by translocating to other organelles, such as the mitochondria (106), the endoplasmic reticulum (107) and various membranes (108). Studies have demonstrated that nuclear ERK mainly improves cell proliferation or oncogenic transformation and migration in various types of cancer either by inducing oncogenic signals, such as c-Myc or c-Fos, or by inhibiting tumor suppressors such as Tob or Foxo3a (109–112). However, stimulation of mitochondrial ERK is involved in both cell survival and apoptosis (105). For example, Cook et al (106) demonstrated that activation of the mitochondrial ERK1/2 signaling pathway promoted mitochondrial fission or fragmentation, with the latter being involved in the onset of cell apoptosis. A previous review stated that the specific mechanisms of the aforementioned translocation are mediated by a number of anchoring and scaffold proteins (105).
Overall, the aforementioned data suggest that activating, rather than inhibiting, ERK may increase cancer cell death. Therefore, an activator of ERK, honokiol, has been tested in vitro and in vivo. Honokiol exhibits the potential to treat cancer by inhibiting cell growth and migration by inducing ERK-dependent apoptosis and autophagy (113,114). Therefore, ERK-induced cell death may be a potential therapeutic strategy that requires further evaluation.
4. Conclusions and perspectives
RT is an indispensable tumor treatment; however, its effectiveness is limited by radioresistance. Radiobiology has revealed that increasing the radiation dose improves local tumor control, but also causes unavoidable damage to normal organs at higher doses. Therefore, the identification of novel RT strategies, such as changing the segmentation method and increasing radiosensitivity, is required to improve RT efficiency. At present, ERK is a promising target for enhancing the radiosensitivity of tumors, which are dependent on the survival-promoting role of ERK activation; however, there are numerous challenges and limitations that require further evaluation to improve clinical treatment. Since ERK activation has dual roles, the exact function of ERK should be clarified and the potential mechanisms elucidated, such as the intracellular localization of ERK and the corresponding microenvironment, before combining RT with ERK inhibitors. A number of studies have indicated that ERK activation serves a role in promoting cell death associated with the degree and duration of activation. The main role served by ERK at different doses and treatment times for different current RT regimens, such as stereotactic body and conventional RT, should be further investigated. Combining RT with targeted molecular therapy and chemotherapy may markedly enhance the therapeutic window for RT use. Clarification of the individual and comprehensive effects of treatment measures on the direction of ERK function is required before implementation of combined treatment. Therefore, in order to develop an effective treatment plan for patients with tumors by targeting the ERK signaling pathway, it is crucial to understand the mechanism of action and characteristics of combined therapy on ERK function.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the National Natural Science Foundation of China (grant no. 81703034), the Project of Science and Technology Department of Jilin Province (grant nos. 20190103089JH and 20190303151SF), the Project of Bethune Plan Research of Jilin University (grant no. 20181336), the Project of Health and Family Planning Commission of Jilin Province (grant no. 2014ZC054) and the Bethune Special Research of Science and Technology Department of Jilin Province (grant no. 20160101079JC).
Availability of data and materials
Not applicable.
Authors' contributions
GC and BL conceived the review. YLu and BL wrote the review. YLu and XY revised the review. YLi proofread the manuscript and revised the manuscript for important intellectual content. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Alicikus ZA, Yamada Y, Zhang Z, Pei X, Hunt M, Kollmeier M, Cox B, Zelefsky MJ. Ten-year outcomes of high-dose, intensity-modulated radiotherapy for localized prostate cancer. Cancer. 2011;117:1429–1437. doi: 10.1002/cncr.25467. [DOI] [PubMed] [Google Scholar]
- 2.Miller KD, Siegel RL, Lin CC, Mariotto AB, Kramer JL, Rowland JH, Stein KD, Alteri R, Jemal A. Cancer treatment and survivorship statistics, 2016. CA Cancer J Clin. 2016;66:271–289. doi: 10.3322/caac.21349. [DOI] [PubMed] [Google Scholar]
- 3.Barker HE, Paget JT, Khan AA, Harrington KJ. The tumour microenvironment after radiotherapy: Mechanisms of resistance and recurrence. Nat Rev Cancer. 2015;15:409–425. doi: 10.1038/nrc3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hawkins AJ, Golding SE, Khalil A, Valerie K. DNA double-strand break-induced pro-survival signaling. Radiother Oncol. 2011;101:13–17. doi: 10.1016/j.radonc.2011.05.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ding M, Zhang E, He R, Wang X. Newly developed strategies for improving sensitivity to radiation by targeting signal pathways in cancer therapy. Cancer Sci. 2013;104:1401–1410. doi: 10.1111/cas.12252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ciccarelli C, Di Rocco A, Gravina GL, Mauro A, Festuccia C, Del Fattore A, Berardinelli P, De Felice F, Musio D, Bouché M, et al. Disruption of MEK/ERK/c-Myc signaling radiosensitizes prostate cancer cells in vitro and in vivo. J Cancer Res Clin Oncol. 2018;144:1685–1699. doi: 10.1007/s00432-018-2696-3. [DOI] [PubMed] [Google Scholar]
- 7.Morris ZS, Harari PM. Interaction of radiation therapy with molecular targeted agents. J Clin Oncol. 2014;32:2886–2893. doi: 10.1200/JCO.2014.55.1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26:3291–3310. doi: 10.1038/sj.onc.1210422. [DOI] [PubMed] [Google Scholar]
- 9.Neuzillet C, Tijeras-Raballand A, De Mestier L, Cros J, Faivre S, Raymond E. MEK in cancer and cancer therapy. Pharmacol Ther. 2014;141:160–171. doi: 10.1016/j.pharmthera.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 10.Kidger AM, Sipthorp J, Cook SJ. ERK1/2 inhibitors: New weapons to inhibit the RAS-regulated RAF-MEK1/2-ERK1/2 pathway. Pharmacol Ther. 2018;187:45–60. doi: 10.1016/j.pharmthera.2018.02.007. [DOI] [PubMed] [Google Scholar]
- 11.Samatar AA, Poulikakos PI. Targeting RAS-ERK signalling in cancer: Promises and challenges. Nat Rev Drug Discov. 2014;13:928–942. doi: 10.1038/nrd4281. [DOI] [PubMed] [Google Scholar]
- 12.Bonner JA, Vroman BT, Christianson TJ, Karnitz LM. Ionizing radiation-induced MEK and Erk activation does not enhance survival of irradiated human squamous carcinoma cells. Int J Radiat Oncol Biol Phys. 1998;42:921–925. doi: 10.1016/S0360-3016(98)00325-3. [DOI] [PubMed] [Google Scholar]
- 13.Mebratu Y, Tesfaigzi Y. How ERK1/2 activation controls cell proliferation and cell death: Is subcellular localization the answer? Cell Cycle. 2009;8:1168–1175. doi: 10.4161/cc.8.8.8147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Teixeiro E, Daniels MA. ERK and cell death: ERK location and T cell selection. FEBS J. 2010;277:30–38. doi: 10.1111/j.1742-4658.2009.07368.x. [DOI] [PubMed] [Google Scholar]
- 15.Cagnol S, Chambard JC. ERK and cell death: Mechanisms of ERK-induced cell death-apoptosis, autophagy and senescence. FEBS J. 2010;277:2–21. doi: 10.1111/j.1742-4658.2009.07366.x. [DOI] [PubMed] [Google Scholar]
- 16.Keshet Y, Seger R. The MAP kinase signaling cascades: A system of hundreds of components regulates a diverse array of physiological functions. Methods Mol Biol. 2010;661:3–38. doi: 10.1007/978-1-60761-795-2_1. [DOI] [PubMed] [Google Scholar]
- 17.Guo YJ, Pan WW, Liu SB, Shen ZF, Xu Y, Hu LL. ERK/MAPK signalling pathway and tumorigenesis. Exp Ther Med. 2020;19:1997–2007. doi: 10.3892/etm.2020.8454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Luca A, Maiello MR, D'alessio A, Pergameno M, Normanno N. The RAS/RAF/MEK/ERK and the PI3K/AKT signalling pathways: Role in cancer pathogenesis and implications for therapeutic approaches. Expert Opin Ther Targets. 2012;16(Suppl 2):S17–S27. doi: 10.1517/14728222.2011.639361. [DOI] [PubMed] [Google Scholar]
- 19.Ramos JW. The regulation of extracellular signal-regulated kinase (ERK) in mammalian cells. Int J Biochem Cell Biol. 2008;40:2707–2719. doi: 10.1016/j.biocel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 20.Yoon S, Seger R. The extracellular signal-regulated kinase: Multiple substrates regulate diverse cellular functions. Growth Factors. 2006;24:21–44. doi: 10.1080/02699050500284218. [DOI] [PubMed] [Google Scholar]
- 21.Carlson SM, Chouinard CR, Labadorf A, Lam CJ, Schmelzle K, Fraenkel E, White FM. Large-scale discovery of ERK2 substrates identifies ERK-mediated transcriptional regulation by ETV3. Sci Signal. 2011;4:rs11. doi: 10.1126/scisignal.2002010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun Y, Liu WZ, Liu T, Feng X, Yang N, Zhou HF. Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J Recept Signal Transduct Res. 2015;35:600–604. doi: 10.3109/10799893.2015.1030412. [DOI] [PubMed] [Google Scholar]
- 23.Kim EK, Choi EJ. Compromised MAPK signaling in human diseases: An update. Arch Toxicol. 2015;89:867–882. doi: 10.1007/s00204-015-1472-2. [DOI] [PubMed] [Google Scholar]
- 24.Balmanno K, Cook SJ. Tumour cell survival signalling by the ERK1/2 pathway. Cell Death Differ. 2009;16:368–377. doi: 10.1038/cdd.2008.148. [DOI] [PubMed] [Google Scholar]
- 25.Sakai N, Wada T, Furuichi K, Iwata Y, Yoshimoto K, Kitagawa K, Kokubo S, Kobayashi M, Hara A, Yamahana J, et al. Involvement of extracellular signal-regulated kinase and p38 in human diabetic nephropathy. Am J Kidney Dis. 2005;45:54–65. doi: 10.1053/j.ajkd.2004.08.039. [DOI] [PubMed] [Google Scholar]
- 26.DuShane JK, Wilczek MP, Mayberry CL, Maginnis MS. ERK is a critical regulator of JC polyomavirus infection. J Virol. 2018;92:e01529–17. doi: 10.1128/JVI.01529-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muslin AJ. MAPK signalling in cardiovascular health and disease: Molecular mechanisms and therapeutic targets. Clin Sci (Lond) 2008;115:203–218. doi: 10.1042/CS20070430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rai SN, Dilnashin H, Birla H, Singh SS, Zahra W, Rathore AS, Singh BK, Singh SP. The role of PI3K/Akt and ERK in neurodegenerative disorders. Neurotox Res. 2019;35:775–795. doi: 10.1007/s12640-019-0003-y. [DOI] [PubMed] [Google Scholar]
- 29.Hoshino R, Chatani Y, Yamori T, Tsuruo T, Oka H, Yoshida O, Shimada Y, Ari-I S, Wada H, Fujimoto J, Kohno M. Constitutive activation of the 41-/43-kDa mitogen-activated protein kinase signaling pathway in human tumors. Oncogene. 1999;18:813–822. doi: 10.1038/sj.onc.1202367. [DOI] [PubMed] [Google Scholar]
- 30.Park HS, You GE, Yang KH, Kim JY, An S, Song JY, Lee SJ, Lim YK, Nam SY. Role of AKT and ERK pathways in controlling sensitivity to ionizing radiation and adaptive response induced by low-dose radiation in human immune cells. Eur J Cell Biol. 2015;94:653–660. doi: 10.1016/j.ejcb.2015.08.003. [DOI] [PubMed] [Google Scholar]
- 31.Corn BW, Kovner F, Bek S, Wexler I, Lifschits B, Seger R. ERK signaling in colorectal cancer: A preliminary report on the expression of phosphorylated ERK and the effects of radiation therapy. Am J Clin Oncol. 2008;31:255–258. doi: 10.1097/COC.0b013e31815a5647. [DOI] [PubMed] [Google Scholar]
- 32.Panganiban RA, Snow AL, Day RM. Mechanisms of radiation toxicity in transformed and non-transformed cells. Int J Mol Sci. 2013;14:15931–15958. doi: 10.3390/ijms140815931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dent P, Yacoub A, Fisher PB, Hagan MP, Grant S. MAPK pathways in radiation responses. Oncogene. 2003;22:5885–5896. doi: 10.1038/sj.onc.1206701. [DOI] [PubMed] [Google Scholar]
- 34.Yan Y, Black CP, Cowan KH. Irradiation-induced G2/M checkpoint response requires ERK1/2 activation. Oncogene. 2007;26:4689–4698. doi: 10.1038/sj.onc.1210268. [DOI] [PubMed] [Google Scholar]
- 35.Williams TM, Flecha AR, Keller P, Ram A, Karnak D, Galbán S, Galbán CJ, Ross BD, Lawrence TS, Rehemtulla A, Sebolt-Leopold J. Cotargeting MAPK and PI3K signaling with concurrent radiotherapy as a strategy for the treatment of pancreatic cancer. Mol Cancer Ther. 2012;11:1193–1202. doi: 10.1158/1535-7163.MCT-12-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cuneo KC, Geng L, Fu A, Orton D, Hallahan DE, Chakravarthy AB. SU11248 (sunitinib) sensitizes pancreatic cancer to the cytotoxic effects of ionizing radiation. Int J Radiat Oncol Biol Phys. 2008;71:873–879. doi: 10.1016/j.ijrobp.2008.02.062. [DOI] [PubMed] [Google Scholar]
- 37.Carter S, Auer KL, Reardon DB, Birrer M, Fisher PB, Valerie K, Schmidt-Ullrich R, Mikkelsen R, Dent P. Inhibition of the mitogen activated protein (MAP) kinase cascade potentiates cell killing by low dose ionizing radiation in A431 human squamous carcinoma cells. Oncogene. 1998;16:2787–2796. doi: 10.1038/sj.onc.1201802. [DOI] [PubMed] [Google Scholar]
- 38.Hein AL, Ouellette MM, Yan Y. Radiation-induced signaling pathways that promote cancer cell survival (review) Int J Oncol. 2014;45:1813–1819. doi: 10.3892/ijo.2014.2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, Egia A, Sasaki AT, Thomas G, Kozma SC, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008;118:3065–3074. doi: 10.1172/JCI34739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cho JH, Hong WG, Jung YJ, Lee J, Lee E, Hwang SG, Um HD, Park JK. Γ-Ionizing radiation-induced activation of the EGFR-p38/ERK-STAT3/CREB-1-EMT pathway promotes the migration/invasion of non-small cell lung cancer cells and is inhibited by podophyllotoxin acetate. Tumour Biol. 2016;37:7315–7325. doi: 10.1007/s13277-015-4548-y. [DOI] [PubMed] [Google Scholar]
- 41.Lee HC, An S, Lee H, Woo SH, Jin HO, Seo SK, Choe TB, Yoo DH, Lee SJ, Hong YJ, et al. Activation of epidermal growth factor receptor and its downstream signaling pathway by nitric oxide in response to ionizing radiation. Mol Cancer Res. 2008;6:996–1002. doi: 10.1158/1541-7786.MCR-08-0113. [DOI] [PubMed] [Google Scholar]
- 42.Sambade MJ, Camp JT, Kimple RJ, Sartor CI, Shields JM. Mechanism of lapatinib-mediated radiosensitization of breast cancer cells is primarily by inhibition of the Raf>MEK>ERK mitogen-activated protein kinase cascade and radiosensitization of lapatinib-resistant cells restored by direct inhibition of MEK. Radiother Oncol. 2009;93:639–644. doi: 10.1016/j.radonc.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Park JS, Qiao L, Su ZZ, Hinman D, Willoughby K, McKinstry R, Yacoub A, Duigou GJ, Young CS, Grant S, et al. Ionizing radiation modulates vascular endothelial growth factor (VEGF) expression through multiple mitogen activated protein kinase dependent pathways. Oncogene. 2001;20:3266–3280. doi: 10.1038/sj.onc.1204258. [DOI] [PubMed] [Google Scholar]
- 44.Zhang X, Ma N, Yao W, Li S, Ren Z. RAD51 is a potential marker for prognosis and regulates cell proliferation in pancreatic cancer. Cancer Cell Int. 2019;19:356. doi: 10.1186/s12935-019-1077-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.King HO, Brend T, Payne HL, Wright A, Ward TA, Patel K, Egnuni T, Stead LF, Patel A, Wurdak H, Short SC. RAD51 is a selective DNA repair target to radiosensitize glioma stem cells. Stem Cell Reports. 2017;8:125–139. doi: 10.1016/j.stemcr.2016.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chowdhury P, Dey P, De D, Ghosh U. Gamma ray-induced in vitro cell migration via EGFR/ERK/Akt/p38 activation is prevented by olaparib pretreatment. Int J Radiat Biol. 2020;96:651–660. doi: 10.1080/09553002.2020.1711461. [DOI] [PubMed] [Google Scholar]
- 47.Lee YJ, Soh JW, Dean NM, Cho CK, Kim TH, Lee SJ, Lee YS. Protein kinase Cdelta overexpression enhances radiation sensitivity via extracellular regulated protein kinase 1/2 activation, abolishing the radiation-induced G(2)-M arrest. Cell Growth Differ. 2002;13:237–246. [PubMed] [Google Scholar]
- 48.Singh S, Upadhyay AK, Ajay AK, Bhat MK. p53 regulates ERK activation in carboplatin induced apoptosis in cervical carcinoma: A novel target of p53 in apoptosis. FEBS Lett. 2007;581:289–295. doi: 10.1016/j.febslet.2006.12.035. [DOI] [PubMed] [Google Scholar]
- 49.Cheng G, Kong D, Hou X, Liang B, He M, Liang N, Ma S, Liu X. The tumor suppressor, p53, contributes to radiosensitivity of lung cancer cells by regulating autophagy and apoptosis. Cancer Biother Radiopharm. 2013;28:153–159. doi: 10.1089/cbr.2012.1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Premkumar K, Shankar BS. Involvement of MAPK signalling in radioadaptive response in BALB/c mice exposed to low dose ionizing radiation. Int J Radiat Biol. 2016;92:249–262. doi: 10.3109/09553002.2016.1146829. [DOI] [PubMed] [Google Scholar]
- 51.Lee SY, Jeong EK, Ju MK, Jeon HM, Kim MY, Kim CH, Park HG, Han SI, Kang HS. Induction of metastasis, cancer stem cell phenotype, and oncogenic metabolism in cancer cells by ionizing radiation. Mol Cancer. 2017;16:10. doi: 10.1186/s12943-016-0577-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kaczanowski S. Apoptosis: Its origin, history, maintenance and the medical implications for cancer and aging. Phys Biol. 2016;13:031001. doi: 10.1088/1478-3975/13/3/031001. [DOI] [PubMed] [Google Scholar]
- 53.Gerl R, Vaux DL. Apoptosis in the development and treatment of cancer. Carcinogenesis. 2005;26:263–270. doi: 10.1093/carcin/bgh283. [DOI] [PubMed] [Google Scholar]
- 54.Wong RS. Apoptosis in cancer: From pathogenesis to treatment. J Exp Clin Cancer Res. 2011;30:87. doi: 10.1186/1756-9966-30-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pistritto G, Trisciuoglio D, Ceci C, Garufi A, D'Orazi G. Apoptosis as anticancer mechanism: Function and dysfunction of its modulators and targeted therapeutic strategies. Aging (Albany NY) 2016;8:603–619. doi: 10.18632/aging.100934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Elmore S. Apoptosis: A review of programmed cell death. Toxicol Pathol. 2007;35:495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen N, Zhang R, Konishi T, Wang J. Upregulation of NRF2 through autophagy/ERK 1/2 ameliorates ionizing radiation induced cell death of human osteosarcoma U-2 OS. Mutat Res. 2017;813:10–17. doi: 10.1016/j.mrgentox.2016.11.006. [DOI] [PubMed] [Google Scholar]
- 58.Boucher MJ, Morisset J, Vachon PH, Reed JC, Lainé J, Rivard N. MEK/ERK signaling pathway regulates the expression of Bcl-2, Bcl-X(L), and Mcl-1 and promotes survival of human pancreatic cancer cells. J Cell Biochem. 2000;79:355–369. doi: 10.1002/1097-4644(20001201)79:3<355::AID-JCB20>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 59.Kurland JF, Voehringer DW, Meyn RE. The MEK/ERK pathway acts upstream of NF kappa B1 (p50) homodimer activity and Bcl-2 expression in a murine B-cell lymphoma cell line. MEK inhibition restores radiation-induced apoptosis. J Biol Chem. 2003;278:32465–32470. doi: 10.1074/jbc.M212919200. [DOI] [PubMed] [Google Scholar]
- 60.Chatterjee N, Walker GC. Mechanisms of DNA damage, repair, and mutagenesis. Environ Mol Mutagen. 2017;58:235–263. doi: 10.1002/em.22087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Golding SE, Morgan RN, Adams BR, Hawkins AJ, Povirk LF, Valerie K. Pro-survival AKT and ERK signaling from EGFR and mutant EGFRvIII enhances DNA double-strand break repair in human glioma cells. Cancer Biol Ther. 2009;8:730–738. doi: 10.4161/cbt.8.8.7927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Valerie K, Povirk LF. Regulation and mechanisms of mammalian double-strand break repair. Oncogene. 2003;22:5792–5812. doi: 10.1038/sj.onc.1206679. [DOI] [PubMed] [Google Scholar]
- 63.Meyn RE, Munshi A, Haymach JV, Milas L, Ang KK. Receptor signaling as a regulatory mechanism of DNA repair. Radiother Oncol. 2009;92:316–322. doi: 10.1016/j.radonc.2009.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yacoub A, McKinstry R, Hinman D, Chung T, Dent P, Hagan MP. Epidermal growth factor and ionizing radiation up-regulate the DNA repair genes XRCC1 and ERCC1 in DU145 and LNCaP prostate carcinoma through MAPK signaling. Radiat Res. 2003;159:439–452. doi: 10.1667/0033-7587(2003)159[0439:EGFAIR]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 65.Yacoub A, Park JS, Qiao L, Dent P, Hagan MP. MAPK dependence of DNA damage repair: Ionizing radiation and the induction of expression of the DNA repair genes XRCC1 and ERCC1 in DU145 human prostate carcinoma cells in a MEK1/2 dependent fashion. Int J Radiat Biol. 2001;77:1067–1078. doi: 10.1080/09553000110069317. [DOI] [PubMed] [Google Scholar]
- 66.Engeland K. Cell cycle arrest through indirect transcriptional repression by p53: I have a DREAM. Cell Death Differ. 2018;25:114–132. doi: 10.1038/cdd.2017.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhao H, Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol. 2001;21:4129–4139. doi: 10.1128/MCB.21.13.4129-4139.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Abbott DW, Holt JT. Mitogen-activated protein kinase kinase 2 activation is essential for progression through the G2/M checkpoint arrest in cells exposed to ionizing radiation. J Biol Chem. 1999;274:2732–2742. doi: 10.1074/jbc.274.5.2732. [DOI] [PubMed] [Google Scholar]
- 69.Yan Y, Black CP, Cao PT, Haferbier JL, Kolb RH, Spieker RS, Ristow AM, Cowan KH. Gamma-irradiation-induced DNA damage checkpoint activation involves feedback regulation between extracellular signal-regulated kinase 1/2 and BRCA1. Cancer Res. 2008;68:5113–5121. doi: 10.1158/0008-5472.CAN-07-5818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dossett LA, Kudchadkar RR, Zager JS. BRAF and MEK inhibition in melanoma. Expert Opin Drug Saf. 2015;14:559–570. doi: 10.1517/14740338.2015.1011618. [DOI] [PubMed] [Google Scholar]
- 71.Caunt CJ, Sale MJ, Smith PD, Cook SJ. MEK1 and MEK2 inhibitors and cancer therapy: The long and winding road. Nat Rev Cancer. 2015;15:577–592. doi: 10.1038/nrc4000. [DOI] [PubMed] [Google Scholar]
- 72.Affolter A, Muller MF, Sommer K, Stenzinger A, Zaoui K, Lorenz K, Wolf T, Sharma S, Wolf J, Perner S, et al. Targeting irradiation-induced mitogen-activated protein kinase activation in vitro and in an ex vivo model for human head and neck cancer. Head Neck. 2016;38(Suppl 1):E2049–E2061. doi: 10.1002/hed.24376. [DOI] [PubMed] [Google Scholar]
- 73.Frémin C, Meloche S. From basic research to clinical development of MEK1/2 inhibitors for cancer therapy. J Hematol Oncol. 2010;3:8. doi: 10.1186/1756-8722-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Estrada-Bernal A, Chatterjee M, Haque SJ, Yang L, Morgan MA, Kotian S, Morrell D, Chakravarti A, Williams TM. MEK inhibitor GSK1120212-mediated radiosensitization of pancreatic cancer cells involves inhibition of DNA double-strand break repair pathways. Cell Cycle. 2015;14:3713–3724. doi: 10.1080/15384101.2015.1104437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Marampon F, Gravina GL, Di Rocco A, Bonfili P, Di Staso M, Fardella C, Polidoro L, Ciccarelli C, Festuccia C, Popov VM, et al. MEK/ERK inhibitor U0126 increases the radiosensitivity of rhabdomyosarcoma cells in vitro and in vivo by downregulating growth and DNA repair signals. Mol Cancer Ther. 2011;10:159–168. doi: 10.1158/1535-7163.MCT-10-0631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chung EJ, Urick ME, Kurshan N, Shield W, III, Asano H, Smith PD, Scroggins BS, Burkeen J, Citrin DE. MEK1/2 inhibition enhances the radiosensitivity of cancer cells by downregulating survival and growth signals mediated by EGFR ligands. Int J Oncol. 2013;42:2028–2036. doi: 10.3892/ijo.2013.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hayes TK, Neel NF, Hu C, Gautam P, Chenard M, Long B, Aziz M, Kassner M, Bryant KL, Pierobon M, et al. Long-Term ERK inhibition in KRAS-mutant pancreatic cancer is associated with MYC degradation and senescence-like growth suppression. Cancer Cell. 2016;29:75–89. doi: 10.1016/j.ccell.2015.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kroeze SG, Fritz C, Hoyer M, Lo SS, Ricardi U, Sahgal A, Stahel R, Stupp R, Guckenberger M. Toxicity of concurrent stereotactic radiotherapy and targeted therapy or immunotherapy: A systematic review. Cancer Treat Rev. 2017;53:25–37. doi: 10.1016/j.ctrv.2016.11.013. [DOI] [PubMed] [Google Scholar]
- 79.Luebker SA, Koepsell SA. Diverse mechanisms of BRAF inhibitor resistance in melanoma identified in clinical and preclinical studies. Front Oncol. 2019;9:268. doi: 10.3389/fonc.2019.00268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lito P, Rosen N, Solit DB. Tumor adaptation and resistance to RAF inhibitors. Nat Med. 2013;19:1401–1409. doi: 10.1038/nm.3392. [DOI] [PubMed] [Google Scholar]
- 81.Jaiswal BS, Durinck S, Stawiski EW, Yin J, Wang W, Lin E, Moffat J, Martin SE, Modrusan Z, Seshagiri S. ERK mutations and amplification confer resistance to ERK-Inhibitor therapy. Clin Cancer Res. 2018;24:4044–4055. doi: 10.1158/1078-0432.CCR-17-3674. [DOI] [PubMed] [Google Scholar]
- 82.Hollmann CA, Owens T, Nalbantoglu J, Hudson TJ, Sladek R. Constitutive activation of extracellular signal-regulated kinase predisposes diffuse large B-cell lymphoma cell lines to CD40-mediated cell death. Cancer Res. 2006;66:3550–3557. doi: 10.1158/0008-5472.CAN-05-2498. [DOI] [PubMed] [Google Scholar]
- 83.Bacus SS, Gudkov AV, Lowe M, Lyass L, Yung Y, Komarov AP, Keyomarsi K, Yarden Y, Seger R. Taxol-induced apoptosis depends on MAP kinase pathways (ERK and p38) and is independent of p53. Oncogene. 2001;20:147–155. doi: 10.1038/sj.onc.1204062. [DOI] [PubMed] [Google Scholar]
- 84.Tang D, Wu D, Hirao A, Lahti JM, Liu L, Mazza B, Kidd VJ, Mak TW, Ingram AJ. ERK activation mediates cell cycle arrest and apoptosis after DNA damage independently of p53. J Biol Chem. 2002;277:12710–12717. doi: 10.1074/jbc.M111598200. [DOI] [PubMed] [Google Scholar]
- 85.Mukherjee S, Dash S, Lohitesh K, Chowdhury R. The dynamic role of autophagy and MAPK signaling in determining cell fate under cisplatin stress in osteosarcoma cells. PLoS One. 2017;12:e0179203. doi: 10.1371/journal.pone.0179203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Martin P, Poggi MC, Chambard JC, Boulukos KE, Pognonec P. Low dose cadmium poisoning results in sustained ERK phosphorylation and caspase activation. Biochem Biophys Res Commun. 2006;350:803–807. doi: 10.1016/j.bbrc.2006.09.126. [DOI] [PubMed] [Google Scholar]
- 87.Martin P, Pognonec P. ERK and cell death: Cadmium toxicity, sustained ERK activation and cell death. FEBS J. 2010;277:39–46. doi: 10.1111/j.1742-4658.2009.07369.x. [DOI] [PubMed] [Google Scholar]
- 88.Lin T, Mak NK, Yang MS. MAPK regulate p53-dependent cell death induced by benzo[a]pyrene: Involvement of p53 phosphorylation and acetylation. Toxicology. 2008;247:145–153. doi: 10.1016/j.tox.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 89.Lee YJ, Soh JW, Jeoung DI, Cho CK, Jhon GJ, Lee SJ, Lee YS. PKC epsilon-mediated ERK1/2 activation involved in radiation-induced cell death in NIH3T3 cells. Biochim Biophys Acta. 2003;1593:219–229. doi: 10.1016/S0167-4889(02)00392-0. [DOI] [PubMed] [Google Scholar]
- 90.Watanabe H, Kurabayashi T, Miura M. Inhibition of the extracellular signal-regulated kinase (ERK) pathway and the induction of radioresistance in rat 3Y1 cells. Int J Radiat Biol. 2004;80:451–457. doi: 10.1080/09553000410001702355. [DOI] [PubMed] [Google Scholar]
- 91.Yang R, Piperdi S, Gorlick R. Activation of the RAF/mitogen-activated protein/extracellular signal-regulated kinase kinase/extracellular signal-regulated kinase pathway mediates apoptosis induced by chelerythrine in osteosarcoma. Clin Cancer Res. 2008;14:6396–6404. doi: 10.1158/1078-0432.CCR-07-5113. [DOI] [PubMed] [Google Scholar]
- 92.Randhawa H, Kibble K, Zeng H, Moyer MP, Reindl KM. Activation of ERK signaling and induction of colon cancer cell death by piperlongumine. Toxicol In Vitro. 2013;27:1626–1633. doi: 10.1016/j.tiv.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhao L, Li J, Hao YH, Gao YB, Wang SM, Zhang J, Dong J, Zhou HM, Liu SC, Peng RY. Microwave-induced apoptosis and cytotoxicity of NK cells through ERK1/2 signaling. Biomed Environ Sci. 2017;30:323–332. doi: 10.3967/bes2017.043. [DOI] [PubMed] [Google Scholar]
- 94.Han HY, Kim H, Jeong SH, Lim DS, Ryu MH. Sulfasalazine induces autophagic cell death in oral cancer cells via Akt and ERK pathways. Asian Pac J Cancer Prev. 2014;15:6939–6944. doi: 10.7314/APJCP.2014.15.16.6939. [DOI] [PubMed] [Google Scholar]
- 95.Bastola T, An RB, Kim YC, Kim J, Seo J. Cearoin induces autophagy, ERK activation and apoptosis via ROS generation in SH-SY5Y neuroblastoma cells. Molecules. 2017;22(pii):E242. doi: 10.3390/molecules22020242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tian X, Geng J, Zheng Q, Wang L, Huang P, Tong J, Zheng S. Single high dose irradiation induces cell cycle arrest and apoptosis in human hepatocellular carcinoma cells through the Ras/Raf/MEK/ERK pathways. Int J Radiat Biol. 2020:1–8. doi: 10.1080/09553002.2020.1694188. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 97.Wang Z, Ma L, Su M, Zhou Y, Mao K, Li C, Peng G, Zhou C, Shen B, Dou J. Baicalin induces cellular senescence in human colon cancer cells via upregulation of DEPP and the activation of Ras/Raf/MEK/ERK signaling. Cell Death Dis. 2018;9:217. doi: 10.1038/s41419-017-0223-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Brown L, Benchimol S. The involvement of MAPK signaling pathways in determining the cellular response to p53 activation: Cell cycle arrest or apoptosis. J Biol Chem. 2006;281:3832–3840. doi: 10.1074/jbc.M507951200. [DOI] [PubMed] [Google Scholar]
- 99.Xu J, Patel NH, Saleh T, Cudjoe EK, Jr, Alotaibi M, Wu Y, Lima S, Hawkridge AM, Gewirtz DA. Differential radiation sensitivity in p53 Wild-Type and p53-Deficient tumor cells associated with senescence but not apoptosis or (Nonprotective) Autophagy. Radiat Res. 2018;190:538–557. doi: 10.1667/RR15099.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wang Y, Xu H, Lu Z, Yu X, Lv C, Tian Y, Sui D. Pseudo-Ginsenoside Rh2 induces A549 cells apoptosis via the Ras/Raf/ERK/p53 pathway. Exp Ther Med. 2018;15:4916–4924. doi: 10.3892/etm.2018.6067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lv C, Hong Y, Miao L, Li C, Xu G, Wei S, Wang B, Huang C, Jiao B. Wentilactone A as a novel potential antitumor agent induces apoptosis and G2/M arrest of human lung carcinoma cells, and is mediated by HRas-GTP accumulation to excessively activate the Ras/Raf/ERK/p53-p21 pathway. Cell Death Dis. 2013;4:e952. doi: 10.1038/cddis.2013.484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Liu Y, Yang Y, Ye YC, Shi QF, Chai K, Tashiro S, Onodera S, Ikejima T. Activation of ERK-p53 and ERK-mediated phosphorylation of Bcl-2 are involved in autophagic cell death induced by the c-Met inhibitor SU11274 in human lung cancer A549 cells. J Pharmacol Sci. 2012;118:423–432. doi: 10.1254/jphs.11181FP. [DOI] [PubMed] [Google Scholar]
- 103.Woessmann W, Chen X, Borkhardt A. Ras-mediated activation of ERK by cisplatin induces cell death independently of p53 in osteosarcoma and neuroblastoma cell lines. Cancer Chemother Pharmacol. 2002;50:397–404. doi: 10.1007/s00280-002-0502-y. [DOI] [PubMed] [Google Scholar]
- 104.Pognonec P. ERK and cell death: Overview. FEBS J. 2010;277:1. doi: 10.1111/j.1742-4658.2009.07365.x. [DOI] [PubMed] [Google Scholar]
- 105.Wainstein E, Seger R. The dynamic subcellular localization of ERK: Mechanisms of translocation and role in various organelles. Curr Opin Cell Biol. 2016;39:15–20. doi: 10.1016/j.ceb.2016.01.007. [DOI] [PubMed] [Google Scholar]
- 106.Cook SJ, Stuart K, Gilley R, Sale MJ. Control of cell death and mitochondrial fission by ERK1/2 MAP kinase signalling. FEBS J. 2017;284:4177–4195. doi: 10.1111/febs.14122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Denoyelle C, Abou-Rjaily G, Bezrookove V, Verhaegen M, Johnson TM, Fullen DR, Pointer JN, Gruber SB, Su LD, Nikiforov MA, et al. Anti-oncogenic role of the endoplasmic reticulum differentially activated by mutations in the MAPK pathway. Nat Cell Biol. 2006;8:1053–1063. doi: 10.1038/ncb1471. [DOI] [PubMed] [Google Scholar]
- 108.Fehrenbacher N, Bar-Sagi D, Philips M. Ras/MAPK signaling from endomembranes. Mol Oncol. 2009;3:297–307. doi: 10.1016/j.molonc.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Shaul YD, Seger R. The MEK/ERK cascade: From signaling specificity to diverse functions. Biochim Biophys Acta. 2007;1773:1213–1226. doi: 10.1016/j.bbamcr.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 110.Klemke RL, Cai S, Giannini AL, Gallagher PJ, de Lanerolle P, Cheresh DA. Regulation of cell motility by mitogen-activated protein kinase. J Cell Biol. 1997;137:481–492. doi: 10.1083/jcb.137.2.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ajenjo N, Canon E, Sanchez-Perez I, Matallanas D, León J, Perona R, Crespo P. Subcellular localization determines the protective effects of activated ERK2 against distinct apoptogenic stimuli in myeloid leukemia cells. J Biol Chem. 2004;279:32813–32823. doi: 10.1074/jbc.M313656200. [DOI] [PubMed] [Google Scholar]
- 112.Maik-Rachline G, Hacohen-Lev-Ran A, Seger R. Nuclear ERK: Mechanism of translocation, substrates, and role in cancer. Int J Mol Sci. 2019;20(pii):E1194. doi: 10.3390/ijms20051194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Huang K, Chen Y, Zhang R, Wu Y, Ma Y, Fang X, Shen S. Honokiol induces apoptosis and autophagy via the ROS/ERK1/2 signaling pathway in human osteosarcoma cells in vitro and in vivo. Cell Death Dis. 2018;9:157. doi: 10.1038/s41419-017-0166-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Yeh PS, Wang W, Chang YA, Lin CJ, Wang JJ, Chen RM. Honokiol induces autophagy of neuroblastoma cells through activating the PI3K/Akt/mTOR and endoplasmic reticular stress/ERK1/2 signaling pathways and suppressing cell migration. Cancer Lett. 2016;370:66–77. doi: 10.1016/j.canlet.2015.08.030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.