

Abstract
Cadherin EGF LAG seven-pass G-type receptor 3 (CELSR3) has been reported to exhibit a cancer-specific pattern. The present study aimed to investigate the clinical value and functional role of CELSR3 in hepatocellular carcinoma (HCC), and determine the underlying molecular mechanism in patients with HCC. CELSR3 expression in tumor and paracancerous HCC tissues was obtained from The Cancer Genome Atlas. Differential expression analysis was performed using the edgeR package. Pearson correlation analysis was used to analyze the correlation between methylation and mRNA levels of CELSR3. Pathways affected by aberrant CELSR3 expression were identified through Gene Set Enrichment Analysis. The results demonstrated that CELSR3 was highly expressed in the early stage of cancer and was present throughout the entire cancer process, which suggested that CELSR3 may serve a key role in the carcinogenesis of HCC. In addition, upregulation of CELSR3 was associated with its methylation level; high CELSR3 expression indicated a shorter overall survival time. Multiple candidate genes were screened by integrating differentially expressed (DE) mRNAs and target genes of DE microRNAs (miRs). Subsequent pathway enrichment analysis demonstrated that the upregulated genes were predominantly enriched in the ‘Neuroactive ligand-receptor interaction’ and ‘Cell cycle’ pathways, whereas the downregulated genes were primarily enriched in ‘Cytokine-cytokine receptor interaction’ and ‘Metabolic pathways’. CELSR3 and its connected nodes and edges were initially removed from the miRNA-mRNA regulatory network in order to prevent bias and compared with the network containing CELSR3 alone. The frequently dysregulated miRNAs were identified as miR-181 family members, and the results suggested that miR-181 and the Wnt/β-catenin signaling pathway influenced CELSR3 expression.
Keywords: CELSR3, hepatocellular carcinoma, prognosis
Introduction
Liver cancer is the third most common cause of cancer-associated mortality in humans worldwide, with a 5-year mortality rate <30% in 2018 (1), and hepatocellular carcinoma (HCC) is the most common type of primary liver carcinoma (2,3). Hepatitis B virus (HBV), HCV, alcoholic and non-alcoholic steatohepatitis are the most frequent causes of chronic liver disease, which remain high risk factors of HCC (4). The diagnosis, treatment and 5-year survival rate (5–9%) of HCC has improved over the past three decades (5). Particularly, the 5-year survival rate has been reported increase to 69% following early diagnosis and curative resection (6). However, the lack of diagnostic markers for early detection means the tumor is usually diagnosed at an advanced stage, which prevents curative therapies, including surgical resection and liver transplantation (7). In addition, the underlying molecular mechanism involved in the development and progression of HCC is not yet fully understood, which allows HCC to continue inducing a devastating effect on human health. Thus, further studies on the molecular pathological investigation and therapeutic intervention with high efficacy are required.
The Cadherin EGF LAG seven-pass G-type (CELSR) family derives from the cadherin EGF LAG seven-pass G-type receptor and is classified as a special subgroup of adhesion G-protein-coupled receptor due to cadherin repeating at the far N-terminal (8). CELSR receptor 3 (CELSR3) contains nine cadherin domains, which act as homophilic binding regions, and seven EGF-like domains involved in receptor-ligand interactions and cell adhesion, and is considered to serve a key role in cell-cell contacts (9,10). Dysregulation of CELSR3 DNA methylation is associated with several types of cancer, including pancreatic, hepatic, bladder and renal carcinomas (11–14). It has been reported that CELSR3 displays a cancer-specific pattern, and a 3.04-fold increase in its expression has been reported in tumor-associated stellate cells compared with inflammation-associated stellate cells (14).
These previous findings suggest that CELSR3 functions as an anticancer target in the aforementioned types of cancer. However, to the best of our knowledge, the clinicopathological significance and functional roles of upregulated CELSR3 in HCC have not yet been determined. Although CELSR3 has been demonstrated to act as an oncogene in several types of human cancer, previous studies have only focused on the dysregulation of its methylation and differential expression in cancer tissues (15–17), whereas limited focus has been placed on its function in tumorigenesis. Thus, further studies are required to understand the high expression molecular mechanism of CELSR3 in tumors.
microRNAs (miRNA) are a class of small, evolutionarily conserved, single-stranded non-coding RNA molecules, which are ~22 nucleotides long and regulate gene expression by binding to the 3′-untranslated regions of target mRNAs (18,19). This results in silencing and post-transcriptional regulation of gene expression (20,21). Dysregulation of miRNA has been associated with HCC progression, including evasion of cell apoptosis, irregular cell proliferation, angiogenesis, invasion and metastasis by targeting protein-coding genes (22,23).
The present study hypothesized that the upregulated CELSR3 expression may be partially due to upstream dysregulation of miRNAs, and therefore aimed to demonstrate the expression pattern of CELSR3 in HCC and determine its clinical significance, in order to improve the understanding of HCC and to provide a basis for future experimental and clinical research.
Materials and methods
Data collection
HCC-associated data from the liver hepatocellular carcinoma dataset (TCGA-LIHC) (24) were downloaded from The Cancer Genome Atlas (TCGA) database (https://cancergenome.nih.gov), which included 370 HCC tissues (survival time information was available) and 48 normal adjacent liver tissues. A total of 346 tumor samples were selected based on the availability of HCC progression from stages I–IV.
Identification of differentially expressed genes (DEGs)
Genes with an average count number <10 were excluded from the study. Patients were divided into low- and high-expression groups based on the median CELSR3 expression value of 7.32. ‘EdgeR’ v3.28.1 package (25) was used to identify DEGs between the two groups. DEGs were selected based on the following criteria: |log2[fold change (FC)]|>1, P<0.05 and false discovery rate (FDR)<0.05.
Identification of differentially expressed miRNAs (DEMs)
The online GEO2R tool (https://www.ncbi.nlm.nih.gov/geo/geo2r) was used within the R software v3.4.3 package (https://www.r-project.org/) to identify DEMs between HCC tissues and normal adjacent tissues. miRNAs with an average count number <10 were excluded from the study. Samples were divided into two groups based on the median CELSR3 mRNA level of 7.32 as follows: High expression (>7.32) or low expression (<7.32). The ‘EdgeR’ package was used to identify DEMs between the two groups. DEMs were selected based on the following criteria: |log2(FC)|>1 and FDR<0.05. Heatmaps illustrating the expression levels of DEMs in samples were plotted using the ‘pheatmap’ R Bioconductor package (https://cran.r-project.org/web/packages/pheatmap). Volcano plots showing the differential expression status were generated using R v3.4.3 platform (https://www.r-project.org/).
Identifying target genes by inverse correlation and target prediction
Pearson's correlation analysis was applied to the DEMs and DEGs. Since miRNAs act as negative regulators (26), upregulated miRNAs resulted in downregulated target mRNAs, and vice versa. The prediction criterion was that the target gene must be identified in >2 of the following prediction databases: miRDB (27), miRTarBase (28) and TargetScan (29). Subsequently, target genes were combined with DEGs to select DEM-differentially expressed target gene pairs. Pearson's correlation analysis was used for DEM-differentially expressed target gene pairs, and pairs with r≤-0.4 were used to construct the miRNA-mRNA negative regulatory network using Cytoscape software v3.2.0 (https://cytoscape.org/) (30).
Copy number variation (CNV) and methylation analysis
The present study analyzed the association between CELSR3 expression and CNVs, as well as the methylation levels in HCC samples. Copy numbers ≤-1 or ≥1 confirmed the presence of CNV. Associations were estimated by Pearson correlation between CNV/methylation levels and CELSR3 mRNA level.
Gene set enrichment analysis
Gene set enrichment analysis of miRNA target genes was performed using KOBAS online tool v2.0 (http://kobas.cbi.pku.edu.cn/) (31), followed by Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis (https://www.genome.jp/kegg/). P<0.05 was considered to indicate significantly enriched pathways.
Cell culture
SK-Hep-1 cells were purchased from Nanjing KeyGen Biotech Co., Ltd. and incubated in MEM supplemented with 10% FBS (Gibco, Thermo Fisher Scientific, Inc.), at 37°C in 5% CO2. In addition, 0.25% EDTA-trypsin and PBS were purchased from Gibco (Thermo Fisher Scientific, Inc.) and used for cell dissociation and washes, respectively.
Cell transfection
Specific siRNA oligonucleotides of β-catenin and miR-181 inhibitor were designed by Qiagen GmbH. The oligonucleotide sequences were as follows: β-catenin, 5′-GGATGTGGATACCTCCCAAGT-3′; β-catenin siNC, 5′-UUCUCCGAACGUGUCACGUTT-3′; miR-181 inhibitor, 5′-ACUCACCGACAGCGUUGAAUGUU-3′; and NC inhibitor 5′-UUCUCCGAACGUGUCACGUTT-3′. SK-Hep-1 cells were seeded into 6-well plates at 70–90% confluence and transfected with Lipofectamine® 3000 (Thermo Fisher Scientific, Inc.) and siRNA (ratio, 1:1). Transfected cells were harvested after 72 h of transfection for RNA extraction. The siRNA concentration was 75 pmol/well.
Reverse transcription-quantitative (RT-q)PCR
Total RNA was extracted from SK-Hep-1 cells using the miRNeasy Mini kit (Qiagen GmbH) according to the manufacturer's protocol. Total RNA was measured using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Inc.) and reverse-transcribed into cDNA using the miScript II RT kit (Qiagen GmbH). The generated cDNA was used as a template for quantification of mRNA and miRNA, respectively. The following primer sequences were used for qPCR: miR-181 forward, 5′-AACATTCAACGCTGTCGGTGAGT-3′ and reverse, 5′-CAGTGCAGGGTCCGAGGT-3′; U6 forward, 5′-CTCGCTTCGGCAGCACA-3′ and reverse, 5′-AACGCTTCACGAATTTGCGT-3′; β-catenin forward, 5′-GCCACAAGATTACAAGAAACG-3′ and reverse, 5′-ATCAACTGGATAGTCAGCACC-3′; CELSR3 forward, 5′-CTACAGACAGCGAATCGGAGCA-3′ and reverse, 5′-CCACGCACATTGGACTTGAGG-3′; and β-actin forward, 5′-GACGTGGACATCCGCAAAGAC-3′ and reverse, 5′-CTGCTGGAAGGTGGACAGTGAG-3′. qPCR amplification was performed using the SYBR Premix Ex Taq kit (Roche Diagnostics), according to the manufacturer's protocol, using the ABI 7500 Sequence Detection system (Thermo Fisher Scientific, Inc.). The following thermocycling conditions were used for qPCR of mRNA: 95°C for 1 min, followed by 40 cycles of 95°C for 10 sec and 60°C for 30 sec. The following thermocycling conditions were used for qPCR of miRNA: 95°C for 10 min, followed by 40 cycles of 94°C for 10 sec, 55°C for 30 sec, and 70°C for 30 sec. Relative mRNAs and miRNAs expression levels were measured using the 2−ΔΔCq method (32) and normalized to the internal reference genes β-actin and RUN6, respectively. All experiments were performed in triplicate.
Statistical analysis
Data are presented as the mean ± standard deviation (n=3). All statistical analyses were performed using R v3.4.3 (https://www.r-project.org/). Univariate analysis was performed using the χ2 test to assess the association between CELSR3 expression and clinicopathological characteristics in patients with HCC. The difference in CELSR3 expression between cancer tissues and adjacent normal tissues was analyzed using a paired two-sided Student's t-test. ANOVA followed by LSD post hoc test were also used to compare CELSR3 expression levels among multiple groups with distinct gene copy numbers. The prognostic value of CELSR3 in HCC was assessed using the Kaplan-Meier online database (http://kmplot.com/analysis). Pearson's correlation coefficient analysis was performed using expression values of DEGs and the methylation levels in HCC data obtained from TCGA in order to construct the miRNA-mRNA negative regulatory network. Significant differences among RT-qPCR data were assessed using one-way ANOVA with Tukey's post hoc analysis. P<0.05 was considered to indicate a statistically significant difference.
Results
CELSR3 is associated with clinicopathological characteristics in patients with HCC
HCC samples were divided into two groups, with the median CELSR3 mRNA levels used as the threshold. The χ2 test and two-sided Student's t-test were used to determine whether age, sex, pathological stage and OS status were associated with CELSR3 expression, the results of which are presented in Table I. The results demonstrated that age was significantly associated with CELSR3 expression, as high CELSR3 expression was observed in elderly patients (P=0.020). In addition, CELSR3 was identified as a clinical stage-associated gene (P=0.011). However, univariate analysis revealed that CELSR3 expression was not associated with sex (P=0.406) or OS status (P=0.159) (data not shown).
Table I.
Association between CELSR3 expression and the demographic and clinicopathological characteristics of patients with HCC.
| CELSR3 expression | ||||
|---|---|---|---|---|
| Characteristic | High, n=209 | Low, n=209 | χ2 | P-value |
| Age, mean ± SD | 63.22±12.72 | 61.09±14.64 | 0.020a | |
| Sex | 0.6898 | 0.406 | ||
| Female | 65 | 74 | ||
| Male | 144 | 135 | ||
| Pathologic stage | 6.4034 | 0.011a | ||
| I–II | 137 | 121 | ||
| III–V | 61 | 27 | ||
| NA | 11 | 61 | ||
| Overall survival status | 1.9800 | 0.159 | ||
| Dead | 88 | 73 | ||
| Alive | 121 | 136 | ||
P<0.05. NA, not available.
CELSR3 expression is high in HCC
Information obtained from TCGA data portal on age, sex and clinical stage demonstrated higher CELSR3 expression in HCC tissues compared with in normal liver tissues (P<0.001; Fig. 1). Additionally, CELSR3 expression was significantly elevated in HBV- (P=0.038) or HCV-infected (P=0.007) HCC samples compared with those without any infections (Fig. S1). Overall, the present results suggested that CELSR3 may serve a key role in the carcinogenesis of HCC.
Figure 1.

CELSR3 expression is increased in hepatocarcinogenesis. CELSR3 expression level was significantly elevated in liver tumor tissues compared with normal adjacent tissues. CELSR3, cadherin EGF LAG seven-pass G-type receptor 3.
Association between CNV/methylation and CELSR3 mRNA level
HCC samples exhibited decreased copy numbers through homozygous deletions, whereas CELSR3 expression did not decrease with copy number loss (P=0.089; Fig. 2B). In addition, CELSR3 expression was poorly negatively correlated with the methylation level of CELSR3 based on Pearson's correlation coefficient (P=0.059; r=−0.095; Fig. 2A).
Figure 2.
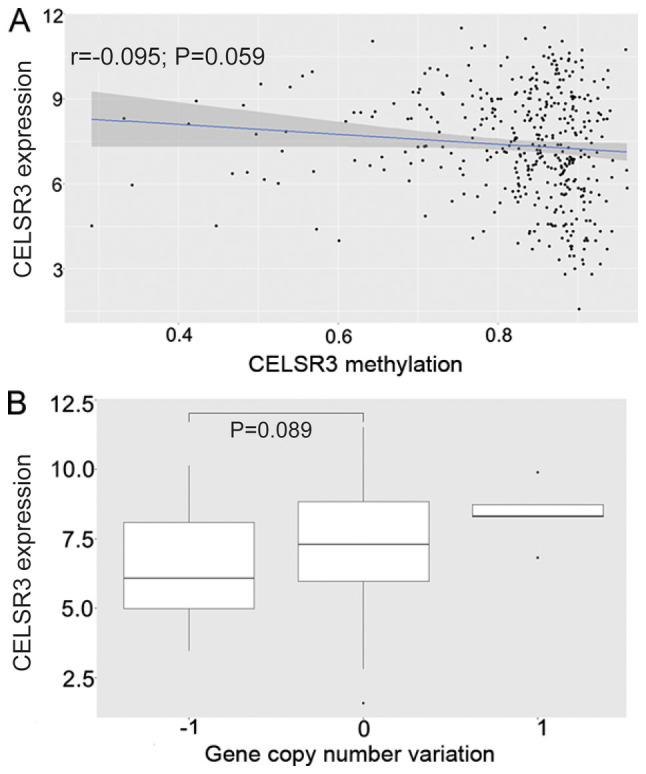
Univariate analysis of CELSR3 expression, with its methylation and CNVs. (A) CELSR3 expression was negatively associated with its methylation (P=0.059). (B) CELSR3 expression did not decrease with copy number loss (P=0.089). CELSR3, cadherin EGF LAG seven-pass G-type receptor 3; CVN, copy number variation.
High CELSR3 expression is associated with poor HCC prognosis
HCC samples were divided into low and high expression groups, with the median CELSR3 mRNA levels set as the threshold. A significantly shorter overall survival time was observed in patients with high CELSR3 expression compared with those in the low CELSR3 expression group (P=0.024; Fig. 3A). No significant effect on OS was demonstrated between CELSR3 expression and sex (P=0.090; Fig. 3B), indicating that CELSR3 affected HCC prognosis independently of sex.
Figure 3.
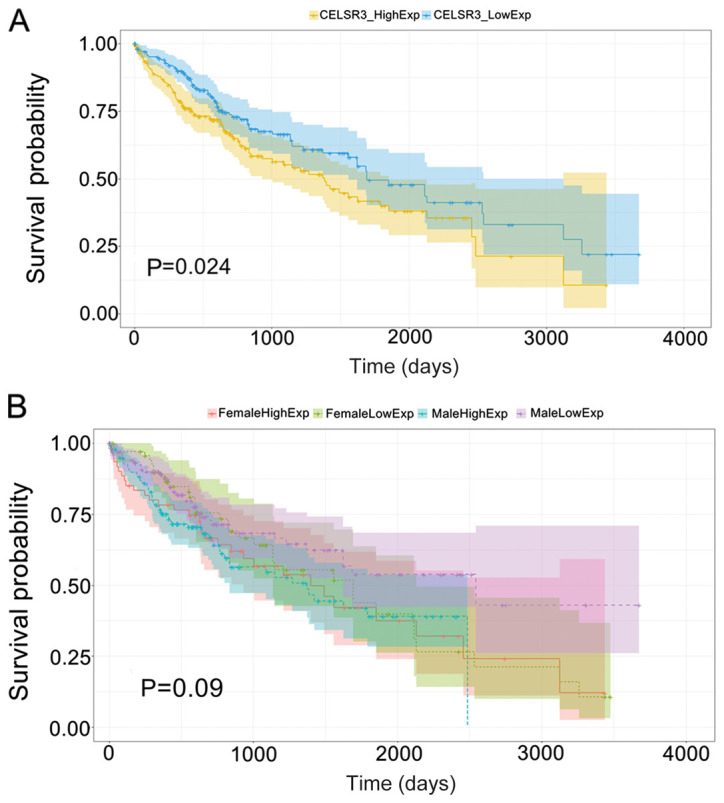
High CELSR3 expression is associated with poor prognosis of hepatocellular carcinoma. (A) Patients with high CELSR3 expression exhibited a shorter OS time compared with patients with low CELSR3 expression. (B) Regarding sex, no significant difference in OS was observed between the high and low CELSR3 expression groups. CELSR3, cadherin EGF LAG seven-pass G-type receptor 3; OS, overall survival.
Analysis of DEGs and DEMs
miRNA expression in HCC samples with low CELSR3 expression was compared with that in samples with high CELSR3 expression, which identified 5 downregulated and 74 upregulated miRNAs. A heatmap and volcano plot of the DEMs were generated using the R v3.4.3 platform (Fig. 4A). The aforementioned downregulated and upregulated miRNAs corresponded to 3,602 and 17,514 target genes, respectively. The same analysis procedure was applied for the DEGs, which identified 625 downregulated and 2,077 upregulated genes. A heatmap and volcano plot of the DEGs were generated (Fig. 4B). KEGG pathway analysis was subsequently performed on 175 upregulated and 411 downregulated common genes between the DEGs and the target genes of DEMs, and the top 20 pathways with the highest enrichment were selected for plotting (Fig. 5A and B).
Figure 4.

Identification of miRNAs and core genes among screened DEMs and DEGs. Volcano plot and heatmap of (A) DEMs and (B) DEGs. DEMs and DEGs with log2(FC)>1 are highlighted in red, and DEMs with log2(FC)≤1 are highlighted in green. miRNA, microRNA; DEM, differentially expressed miRNA; DEG, differentially expressed gene; FDR, false discovery rate; up, upregulated; down, downregulated; no, no change.
Figure 5.
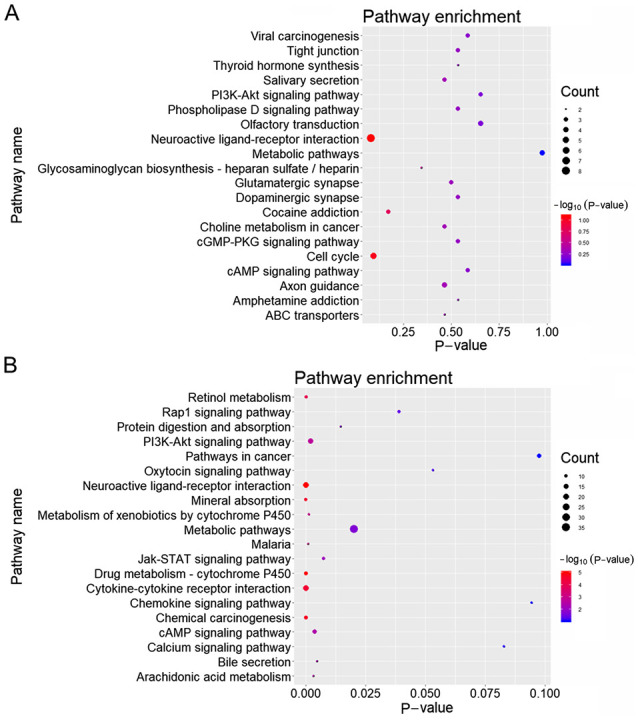
KEGG enrichment analysis of differentially expressed microRNA target genes. KEGG enrichment analysis of (A) upregulated and (B) downregulated target genes. KEGG, Kyoto Encyclopedia of Genes and Genomes.
In order to further investigate the molecular mechanisms, five target genes of the downregulated miRNAs were paired with the upregulated genes, and 74 target genes of the upregulated miRNAs were paired with the downregulated genes to identify overlapping genes. Subsequently, a miRNA-mRNA regulatory network was constructed based on the regulatory association between miRNAs and mRNAs. In order to prevent bias, CELSR3 and its connected nodes and edges were initially removed from the miRNA-mRNA regulatory network (Fig. 6A). This was subsequently compared with the miRNA-mRNA regulatory network containing CELSR3 alone (Fig. 6B), which identified miR-181 family members, suggesting that miR-181 downregulated CELSR3 gene expression in HCC.
Figure 6.
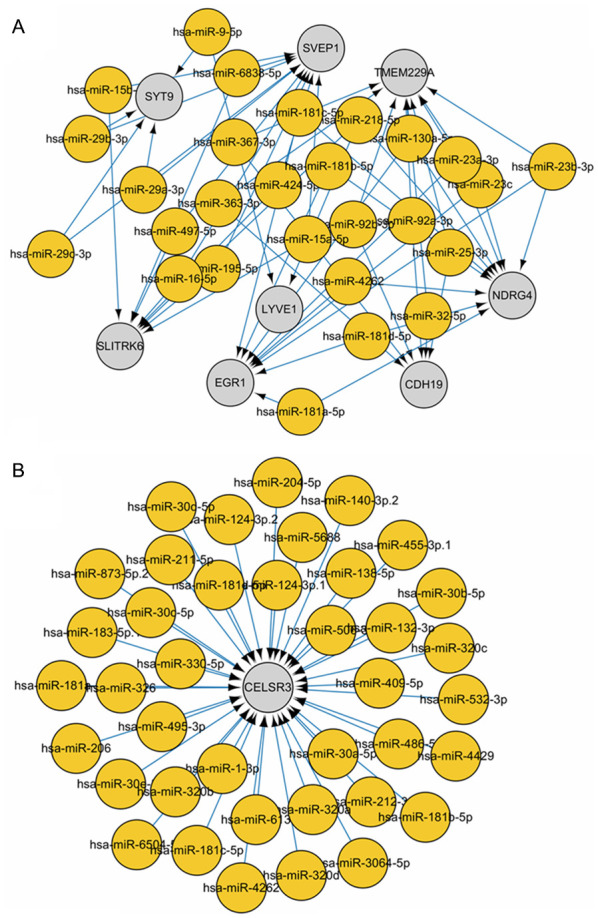
Integrated miRNA-mRNA correlation network analysis. (A) Following elimination of CELSR3, its connected nodes and edges, and core DEGs and DEMs, a correlation network was identified based on the following filtering criteria: r<-0.3 and P<0.05. (B) DEM-DEG regulatory network diagram containing CELSR3 alone. miRNA, microRNA; CELSR3, cadherin EGF LAG seven-pass G-type receptor 3; DEGs, differentially expressed genes; DEMs, differentially expressed miRNAs.
Wnt/β-catenin signaling pathway upregulates CELSR3 expression by downregulating miRNA-181 expression
The present study performed RT-qPCR in SK-Hep-1 cells transfected with β-catenin siRNA or miR-181 inhibitor to determine the associations between the Wnt/β-catenin signaling pathway, miR-181 and CELSR3 expression in HCC progression. β-catenin expression was significantly decreased following transfection with β-catenin siRNA (Fig. 7A). In addition, β-catenin knockdown significantly decreased miR-181 expression (Fig. 7B), whereas CELSR3 expression was significantly upregulated following transfection with β-catenin siRNA compared with the control siRNA group (Fig. 7C). miR-181 expression significantly decreased following transfection with miR-181 inhibitor (Fig. 7D). Notably, miR-181 inhibition did not affect β-catenin expression (Fig. 7E), whereas CELSR3 expression was significantly upregulated following transfection with miR-181 inhibitor compared with the control group (Fig. 7F). Overall, these results demonstrated that inhibiting the Wnt/β-catenin signaling pathway upregulated CELSR3 expression by downregulating miR-181 expression.
Figure 7.

Reverse transcription-quantitative PCR analysis. Expression levels of (A) β-catenin, (B) miR-181 and (C) CELSR3 in SK-Hep-1 cells transferred with β-catenin siRNA and control siRNA. Expression levels of (D) miR-181, (E) β-catenin and (F) CELSR3 in SK-Hep-1 cells transferred with the miR-181 inhibitor and control inhibitor. Error bars indicate mean ± standard deviation (n=3). The significance was assessed via one-way ANOVA (with Tukey's post hoc analysis). **P<0.01 vs. respective control. miR, microRNA; CELSR3, cadherin EGF LAG seven-pass G-type receptor 3; si, small interfering.
Discussion
CELSR3 has been reported to display a cancer-specific pattern (15,16). A 3.04-fold increase in CALSR3 expression has been reported in tumor-associated stellate cells compared with inflammation-associated stellate cells (14). However, the clinicopathological significance and functional role of CELSR3 upregulation in HCC has not yet been fully determined. The present study aimed to investigate the biological function of CELSR3 and determine its underlying molecular mechanism in HCC based on information obtained from TCGA database. The results demonstrated that CELSR3 was highly expressed in the early stages of cancer and throughout the cancer process. CNVs have been used to determine prognosis and subsequent treatment strategies for different types of cancer (33). Methylation of DNA cytosine residues at the carbon 5 position in cytosine-guanine dinucleotides is a common epigenetic mechanism in eukaryotic DNA, which serves a key role in regulating gene activities (34,35). Furthermore, CELSR3 expression was not correlated with the DNA methylation level of HCC. High CELSR3 expression was predictive of poor overall survival time in patients with liver cancer. Taken together, these results suggested that CELSR3 may serve a key role in HCC tumorigenesis and prognosis, which, to the best of our knowledge, has not been reported in previous studies.
In the present study, multiple candidate genes were screened following integration of DEGs and target genes of DEMs, which identified 175 upregulated and 411 downregulated genes. Subsequent pathway enrichment analysis demonstrated that the upregulated genes were predominantly enriched in ‘Neuroactive ligand-receptor interaction’ and ‘Cell cycle’, whereas the downregulated genes were primarily enriched in pathways of ‘Cytokine-cytokine receptor interaction’ and ‘Metabolic pathways’. Neuroactive ligand-receptor interaction may be associated with HCC, since genes expressed in human liver were previously reported to be involved in neuroactive ligand receptor interaction pathways (36). In addition, a previous study has demonstrated that neuroactive ligand-receptor interaction is observed in each stage of HCC (37). Thus, this pathway appears to serve a key role in HCC progression. C-kit is a receptor of the stem cell factor, the activation of which is considered crucial for cell proliferation and migration (38). Of note, the activation of c-kit has been reported to attribute to the cell proliferation and cirrhosis of HCC (39).
miRNA and mRNA expression profiling analyses were performed in the present study using the data obtained from TCGA. A total of five target genes of the downregulated miRNAs were paired with the upregulated genes, and 74 target genes of the upregulated miRNA were paired with the downregulated genes to identify overlapping genes. This was combined with the integration analysis between DEMs and DEGs to construct a miRNA-mRNA regulatory network. In order to prevent bias, CELSR3, along with its connected nodes and edges were initially removed from the miRNA-mRNA regulatory network. This was subsequently compared with the miRNA-mRNA regulatory network containing CELSR3 alone, which identified miR-181 family members as the most common dysregulated miRNAs. The results of the present study identified a novel association between miR-181 and CELSR3 gene expression in HCC. The Wnt/β-catenin signaling pathway has been demonstrated to directly regulate miR-181 expression in HCC (40).
Although an integrated in silico analysis was performed and a potential miRNA-mRNA regulatory network was constructed, a number of limitations exist in the present study. For example, implementing the median CELSR3 mRNA expression levels as the cut-off values to divide high- and low-risk patients is an arbitrary method, which makes it difficult to set a threshold for prognostic marker detection (41–43). Furthermore, the sample size of TCGA dataset included in the present study was too small to demonstrate effective outcomes; thus, future studies will aim to increase the patient sample size to validate the respective findings.
In conclusion, the results of the present study demonstrated that aberrant CELSR3 expression served an important role in the pathogenesis and prognosis of HCC. In addition, CELSR3 expression was not correlated with the DNA methylation level of HCC. Notably, a novel association was identified between miR-181 and CELSR3 mRNA expression in HCC, suggesting that the miR-181-CELSR3 pair may regulate HCC progression. Upregulation of CELSR3 may provide a potential therapeutic target for HCC, since the protein encoded by this gene is located at the plasma membrane and has intriguing signaling capabilities (44). Based on their known biological functions, it is worth further investigating the association between miR-181 and CELSR3 expression, their molecular mechanism and therapeutic value.
Supplementary Material
Acknowledgements
Not applicable.
Funding
The present study was funded by the Science Foundation of the Hunan Province, Key Development Program (grant no. 2017SK2054).
Availability of data and materials
The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request. The TCGA-LIHC dataset is available from TCGA database (https://cancergenome.nih.gov).
Authors' contributions
ZW and XO contributed to the design of the study, wrote the manuscript and analysed the data. ZW revised the manuscript. GZ acquired, analysed and interpreted the data. LY made substantial contributions to the conception and design of the present study and revised the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Allemani C, Matsuda T, Di Carlo V, Harewood R, Matz M, Nikšić M, Bonaventure A, Valkov M, Johnson CJ, Estève J, et al. Global surveillance of trends in cancer survival 2000–14 (CONCORD-3): Analysis of individual records for 37 513 025 patients diagnosed with one of 18 cancers from 322 population-based registries in 71 countries. Lancet. 2018;391:1023–1075. doi: 10.1016/S0140-6736(17)33326-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Venook AP, Papandreou C, Furuse J, de Guevara LL. The incidence and epidemiology of hepatocellular carcinoma: A global and regional perspective. Oncologist. 2010;15(Suppl 4):S5–S13. doi: 10.1634/theoncologist.2010-S4-05. [DOI] [PubMed] [Google Scholar]
- 3.Stepien M, Fedirko V, Duarte-Salles T, Ferrari P, Freisling H, Trepo E, Trichopoulou A, Bamia C, Weiderpass E, Olsen A, et al. Prospective association of liver function biomarkers with development of hepatobiliary cancers. Cancer Epidemiol. 2016;40:179–187. doi: 10.1016/j.canep.2016.01.002. [DOI] [PubMed] [Google Scholar]
- 4.Bhangui P, Adam R, Vibert E, Azoulay D, Samuel D, Castaing D. 41 resection or transplantation for early hepatocellular carcinoma in a cirrhotic liver-size does matter. J Clin Exp Hepatol. 2011;1:151. doi: 10.1016/S0973-6883(11)60179-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Simoneau E, Hassanain M, Madkhali A, Salman A, Nudo CG, Chaudhury P, Metrakos P. (18)F-Fluorodeoxyglucose positron-emission tomography could have a prognostic role in patients with advanced hepatocellular carcinoma. Curr Oncol. 2014;21:e551–e556. doi: 10.3747/co.21.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Trevisani F, Cantarini MC, Wands JR, Bernardi M. Recent advances in the natural history of hepatocellular carcinoma. Carcinogenesis. 2008;29:1299–1305. doi: 10.1093/carcin/bgn113. [DOI] [PubMed] [Google Scholar]
- 7.Zheng H, Zou AE, Saad MA, Wang XQ, Kwok JG, Korrapati A, Li P, Kisseleva T, Wang-Rodriguez J, Ongkeko WM. Alcohol-dysregulated microRNAs in hepatitis B virus-related hepatocellular carcinoma. PLoS One. 2017;12:e0178547. doi: 10.1371/journal.pone.0178547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang XJ, Zhang DL, Xu ZG, Ma ML, Wang WB, Li LL, Han XL, Huo Y, Yu X, Sun JP. Understanding cadherin EGF LAG seven-pass G-type receptors. J Neurochem. 2014;131:699–711. doi: 10.1111/jnc.12955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Usui T, Shima Y, Shimada Y, Hirano S, Burgess RW, Schwarz TL, Takeichi M, Uemura T. Flamingo, a seven-pass transmembrane cadherin, regulates planar cell polarity under the control of Frizzled. Cell. 1999;98:585–595. doi: 10.1016/S0092-8674(00)80046-X. [DOI] [PubMed] [Google Scholar]
- 10.Langenhan T, Aust G, Hamann J. Sticky signaling-adhesion class G protein-coupled receptors take the stage. Sci Signal. 2013;6:re3. doi: 10.1126/scisignal.2003825. [DOI] [PubMed] [Google Scholar]
- 11.Jeong P, Ha YS, Cho IC, Yun SJ, Yoo ES, Kim IY, Choi YH, Moon SK, Kim WJ. Three-gene signature predicts disease progression of non-muscle invasive bladder cancer. Oncol Lett. 2011;2:679–684. doi: 10.3892/ol.2011.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anwar SL, Lehmann U. DNA methylation, microRNAs, and their crosstalk as potential biomarkers in hepatocellular carcinoma. World J Gastroenterol. 2014;20:7894–7913. doi: 10.3748/wjg.v20.i24.7894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ricketts CJ, Morris MR, Gentle D, Brown M, Wake N, Woodward ER, Clarke N, Latif F, Maher ER. Genome-wide CpG island methylation analysis implicates novel genes in the pathogenesis of renal cell carcinoma. Epigenetics. 2012;7:278–290. doi: 10.4161/epi.7.3.19103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Erkan M, Weis N, Pan Z, Schwager C, Samkharadze T, Jiang X, Wirkner U, Giese NA, Ansorge W, Debus J, et al. Organ-, inflammation- and cancer specific transcriptional fingerprints of pancreatic and hepatic stellate cells. Mol Cancer. 2010;9:88. doi: 10.1186/1476-4598-9-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gu X, Li H, Sha L, Mao Y, Shi C, Zhao W. CELSR3 mRNA expression is increased in hepatocellular carcinoma and indicates poor prognosis. PeerJ. 2019;7:e7816. doi: 10.7717/peerj.7816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khor GH, Froemming GR, Zain RB, Abraham TM, Lin TK. Involvement of CELSR3 hypermethylation in primary oral squamous cell carcinoma. Asian Pac J Cancer Prev. 2016;17:219–223. doi: 10.7314/APJCP.2016.17.1.219. [DOI] [PubMed] [Google Scholar]
- 17.Xie Z, Dang Y, Wu H, He R, Ma J, Peng Z, Rong M, Li Z, Yang J, Jiang Y, et al. Effect of CELSR3 on the cell cycle and apoptosis of hepatocellular carcinoma cells. J Cancer. 2020;11:2830–2844. doi: 10.7150/jca.39328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hayes CN, Chayama K. MicroRNAs as biomarkers for liver disease and hepatocellular carcinoma. Int J Mol Sci. 2016;17:280. doi: 10.3390/ijms17030280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tat TT, Maroney PA, Chamnongpol S, Coller J, Nilsen TW. Cotranslational microRNA mediated messenger RNA destabilization. Elife. 2016;5:e12880. doi: 10.7554/eLife.12880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang X, Xu X, Ge G, Zang X, Shao M, Zou S, Zhang Y, Mao Z, Zhang J, Mao F, et al. miR498 inhibits the growth and metastasis of liver cancer by targeting ZEB2. Oncol Rep. 2019;41:1638–1648. doi: 10.3892/or.2018.6948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu H. MicroRNAs in breast cancer initiation and progression. Cell Mol Life Sci. 2012;69:3587–3599. doi: 10.1007/s00018-012-1128-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chu R, Mo G, Duan Z, Huang M, Chang J, Li X, Liu P. miRNAs affect the development of hepatocellular carcinoma via dysregulation of their biogenesis and expression. Cell Commun Signal. 2014;12:45. doi: 10.1186/s12964-014-0045-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fang T, Lv H, Lv G, Li T, Wang C, Han Q, Yu L, Su B, Guo L, Huang S, et al. Tumor-derived exosomal miR-1247-3p induces cancer-associated fibroblast activation to foster lung metastasis of liver cancer. Nat Commun. 2018;9:191. doi: 10.1038/s41467-017-02583-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, Kovatich AJ, Benz CC, Levine DA, Lee AV, et al. An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell. 2018;173:400–416.e11. doi: 10.1016/j.cell.2018.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Robinson MD, McCarthy DJ, Smyth GK. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu TX, Rothenberg ME. MicroRNA. J Allergy Clin Immunol. 2018;141:1202–1207. doi: 10.1016/j.jaci.2017.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kertesz M, Iovino N, Unnerstall U, Gaul U, Segal E. The role of site accessibility in microRNA target recognition. Nat Genet. 2007;39:1278–1284. doi: 10.1038/ng2135. [DOI] [PubMed] [Google Scholar]
- 28.Rehmsmeier M, Steffen P, Hochsmann M, Giegerich R. Fast and effective prediction of microRNA/target duplexes. RNA. 2004;10:1507–1517. doi: 10.1261/rna.5248604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chung TK, Lau TS, Cheung TH, Yim SF, Lo KW, Siu NS, Chan LK, Yu MY, Kwong J, Doran G, et al. Dysregulation of microRNA-204 mediates migration and invasion of endometrial cancer by regulating FOXC1. Int J Cancer. 2012;130:1036–1045. doi: 10.1002/ijc.26060. [DOI] [PubMed] [Google Scholar]
- 30.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xie C, Mao X, Huang J, Ding Y, Wu J, Dong S, Kong L, Gao G, Li CY, Wei L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011;39:W316–W322. doi: 10.1093/nar/gkr483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 33.Lee JW, Son MH, Cho HW, Ma YE, Yoo KH, Sung KW, Koo HH. Clinical significance of MYCN amplification in patients with high-risk neuroblastoma. Pediatr Blood Cancer. 2018;65:e27257. doi: 10.1002/pbc.27257. [DOI] [PubMed] [Google Scholar]
- 34.Laisné M, Gupta N, Kirsh O, Pradhan S, Defossez PA. Mechanisms of DNA methyltransferase recruitment in mammals. Genes (Basel) 2018;9:617. doi: 10.3390/genes9120617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roos L, van Dongen J, Bell CG, Burri A, Deloukas P, Boomsma DI, Spector TD, Bell J. Integrative DNA methylome analysis of pan-cancer biomarkers in cancer discordant monozygotic twin-pairs. Clin Epigenetics. 2016;8:7. doi: 10.1186/s13148-016-0172-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li H, Zhao X, Li C, Sheng C, Bai Z. Integrated analysis of lncRNA-associated ceRNA network reveals potential biomarkers for the prognosis of hepatitis B virus-related hepatocellular carcinoma. Cancer Manag Res. 2019;11:877–897. doi: 10.2147/CMAR.S186561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao Y, Xue F, Sun J, Guo S, Zhang H, Qiu B, Geng J, Gu J, Zhou X, Wang W, et al. Genome-wide methylation profiling of the different stages of hepatitis B virus-related hepatocellular carcinoma development in plasma cell-free DNA reveals potential biomarkers for early detection and high-risk monitoring of hepatocellular carcinoma. Clin Epigenetics. 2014;6:30. doi: 10.1186/1868-7083-6-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kazi JU, Rönnstrand L. FMS-like Tyrosine kinase 3/FLT3: From basic science to clinical implications. Physiol Rev. 2019;99:1433–1466. doi: 10.1152/physrev.00029.2018. [DOI] [PubMed] [Google Scholar]
- 39.Lebron MB, Brennan L, Damoci CB, Prewett MC, O'Mahony M, Duignan IJ, Credille KM, DeLigio JT, Starodubtseva M, Amatulli M, et al. A human monoclonal antibody targeting the stem cell factor receptor (c-Kit) blocks tumor cell signaling and inhibits tumor growth. Cancer Biol Ther. 2014;15:1208–1218. doi: 10.4161/cbt.29523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ji J, Yamashita T, Wang XW. Wnt/beta-catenin signaling activates microRNA-181 expression in hepatocellular carcinoma. Cell Biosci. 2011;1:4. doi: 10.1186/2045-3701-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lu Y, Wang L, Liu P, Yang P, You M. Gene-expression signature predicts postoperative recurrence in stage I non-small cell lung cancer patients. PLoS One. 2012;7:e30880. doi: 10.1371/journal.pone.0030880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhu CQ, Ding K, Strumpf D, Weir BA, Meyerson M, Pennell N, Thomas RK, Naoki K, Ladd-Acosta C, Liu N, et al. Prognostic and predictive gene signature for adjuvant chemotherapy in resected non-small-cell lung cancer. J Clin Oncol. 2010;28:4417–4424. doi: 10.1200/JCO.2009.26.4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Subramanian J, Simon R. Gene expression-based prognostic signatures in lung cancer: Ready for clinical use? J Natl Cancer Inst. 2010;102:464–474. doi: 10.1093/jnci/djq025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Feng J, Xu Y, Wang M, Ruan Y, So KF, Tissir F, Goffinet A, Zhou L. A role for atypical cadherin Celsr3 in hippocampal maturation and connectivity. J Neurosci. 2012;32:13729–13743. doi: 10.1523/JNEUROSCI.1965-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request. The TCGA-LIHC dataset is available from TCGA database (https://cancergenome.nih.gov).