

Abstract
The immune system is a vital determinant of cancer and shapes its trajectory. Notably, the immune reaction to cancer harbors dual potential for suppressing or promoting cancer development and progression. This polarity of the immune response is determined, in part, by the character of the interplay between innate and adaptive immunity. On the one hand, the innate immune compartment is a necessary proponent of cancer immunity by supporting an immunostimulatory state that enables T cell immunosurveillance. However, in the setting of cancer, innate immune cells are commonly polarized with immune-suppressive properties and as a result, orchestrate a tolerogenic niche that interferes with the cytotoxic potential of tumor antigen-specific T cells. Herein, we discuss the role of innate immunity as a positive and negative regulator of adaptive immunosurveillance; moreover, we highlight how tumor cells may skew leukocytes towards an immunosuppressive state and, as such, subvert the phenotypic plasticity of the immune compartment to advance disease progression. These observations establish the precedent for novel therapeutic strategies that aim to restore the tumor microenvironment to an immunoreactive state and, in doing so, condition and maintain the immunogenicity of tumors to yield deep and durable responses to immunotherapy.
Keywords: Cancer, immunotherapy, immune evasion, myeloid cells, T cells, maintenance therapy, conditioning therapy
1. Introduction
Cancer is a disease that finds its provenance in the accumulation of stochastic mutations which, over time, manifest in the uncontrolled growth of malignant cells.1 However, tumor initiation, progression, and metastasis do not unfold in a sequestered, cell-autonomous manner; rather, each are dynamic processes that arise from a confluence of tumor cell-intrinsic and -extrinsic factors.2,3 As such, cancers are influenced by a myriad of heterotypic interactions that occur between tumor cells and their local microenvironment.2
The conjecture that the immune system participates in the recognition and elimination of malignant neoplastic cells was initially put forth as the cancer immunosurveillance hypothesis by Paul Ehrlich and Thomas Burnet.4,5 The immunogenic capacity of cancer has since provided the impetus to fuel the development of therapies that seek to harness the immune system’s ability to survey and eliminate cancer cells.6,7 However, the lack of a universally efficacious response to immunotherapies, such as immune checkpoint blockade (i.e. anti-cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and anti-programmed death-ligand 1 (PDL1)/programmed cell death protein 1 (PD1)), suggests that multiple mechanisms of immune evasion will need to be addressed to broaden the therapeutic potential of immunotherapy.8
Tumor cell-driven mechanisms of immune evasion pose, arguably, the greatest barrier to the success of T cell-directed immunotherapies. Under conditions of immune pressure, tumor cells evolve in tandem to escape immune surveillance.4 This process, known as immunoediting (reviewed in detail elsewhere9,10) occurs in numerous ways, such as via the downregulation of class I human leukocyte antigen (HLA) expression on malignant cells, which allows tumors to circumvent antigen presentation and, by extension, immune detection and elimination.10,11 However, tumors may also exploit the immune system to act in its favor, most notably via the recruitment of tumor-associated myeloid cells and immunoregulatory leukocytes that orchestrate an immunosuppressive and inflammatory microenvironment.12–14 Conversely, tumors may instruct an immunological state that is deficient in key constituents (e.g. dendritic cells) that are necessary for generation of productive anti-tumor immunity. It is key, therefore, to recognize that the tumor-immune interaction is defined not only by the orientation (i.e. immunosuppressive or immunostimulatory) of the immune response but also its quality, which reflects the complexity of the immune reaction and its potential to invoke anti-tumor immunity (Figure 1). As such, effective immunotherapies must not only enhance the capacity of the immune system to eliminate tumors, but also address the regulatory mechanisms by which cancers capitalize upon the vulnerabilities of the immune system to enable disease progression.
Figure 1. The orientation and quality of the immune reaction defines the sensitivity of tumors to immunotherapy.
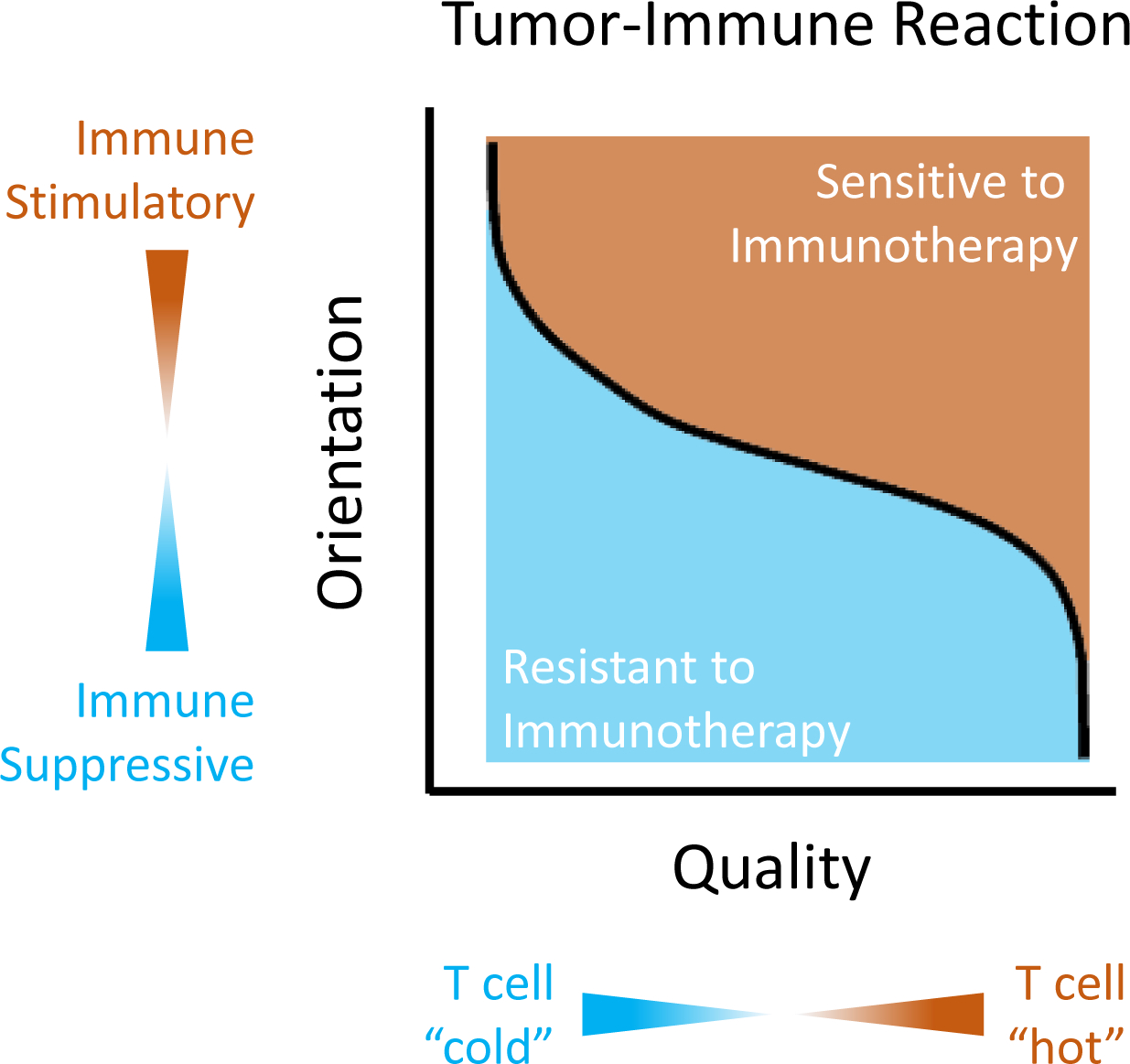
The immune reaction to cancer is determined by the orientation of the immune response, which refers to its inclination to support (i.e. immune stimulatory) or inhibit (i.e. immune suppressive) T cell activation, and the quality of the immune response, which refers to its ability to mediate T cell-dependent anti-tumor immunity. The quality of the immune response is dependent on multiple factors (e.g. dendritic cell infiltration and T cell chemoattractants) which determine the degree of T cell infiltration. T cell infiltration can range from “cold” (i.e. poor infiltration) to “hot” (i.e. brisk infiltration). Together, the orientation and quality of the immune response influence the likelihood of response to immunotherapy.
This review explores the bidirectionality of the tumor-immune interaction, and its implications in cancer immunotherapy. We begin with an overview of the mechanisms that comprise a favorable immunostimulatory state capable of supporting the elimination of malignant cells. We then discuss how these mechanisms are intricately regulated by innate immunity which is not only a pre-requisite for productive cancer immunosurveillance but also a vital determinant of the anti-tumor immune response. We emphasize that cancer immunoediting may jeopardize the maintenance of an immunostimulatory state and as a result, tumors may ultimately evolve mechanisms to escape immune elimination. In essence, immune pressure can trigger the evolution of a tolerogenic phenotype that compromises the immunogenicity of cancer. With this in mind, we highlight a need for therapies that condition tumors for immune elimination, while simultaneously maintaining an immunoreactive state within tumors by selectively derailing compensatory and cancer cell-driven mechanisms of immunosuppression.
2. The innate immune compartment is a critical determinant of T cell immunosurveillance in cancer
To unleash a potent T cell-mediated anti-tumor response, several sequential events that engage the innate and adaptive arms of the immune compartment must occur. This process, often referred to as the Cancer-Immunity Cycle15 consists, in brief, of the following steps: (i) uptake of tumor antigens, (ii) cross-presentation of tumor antigens to T cells by antigen presenting cells (APCs), (iii) priming and activation of naïve T cells, (iv) trafficking of activated T cells to the tumor microenvironment (TME), (v) T cell infiltration into tumors and (vi) elimination of malignant cells by activated cytotoxic T cells. In essence, the Cancer-Immunity Cycle is dependent on T cell priming (Steps i-iv) and effector phases (Steps v-vi) (Figure 2). These two phases of immunosurveillance are intricately regulated by innate immunity such that the generation of tumor antigen-specific T cells reflects a coordinated interaction between the innate and adaptive immune systems. This dynamic interplay is guided by the phenotype and function of innate immune cells, which influence and shape the biological state of tumor antigen specific T cells (e.g. tolerant or reactive). Herein, we discuss the role of innate immunity in promoting or inhibiting T cell priming and effector activity.
Figure 2. Adaptive immunity in cancer is defined by priming and effector phases.
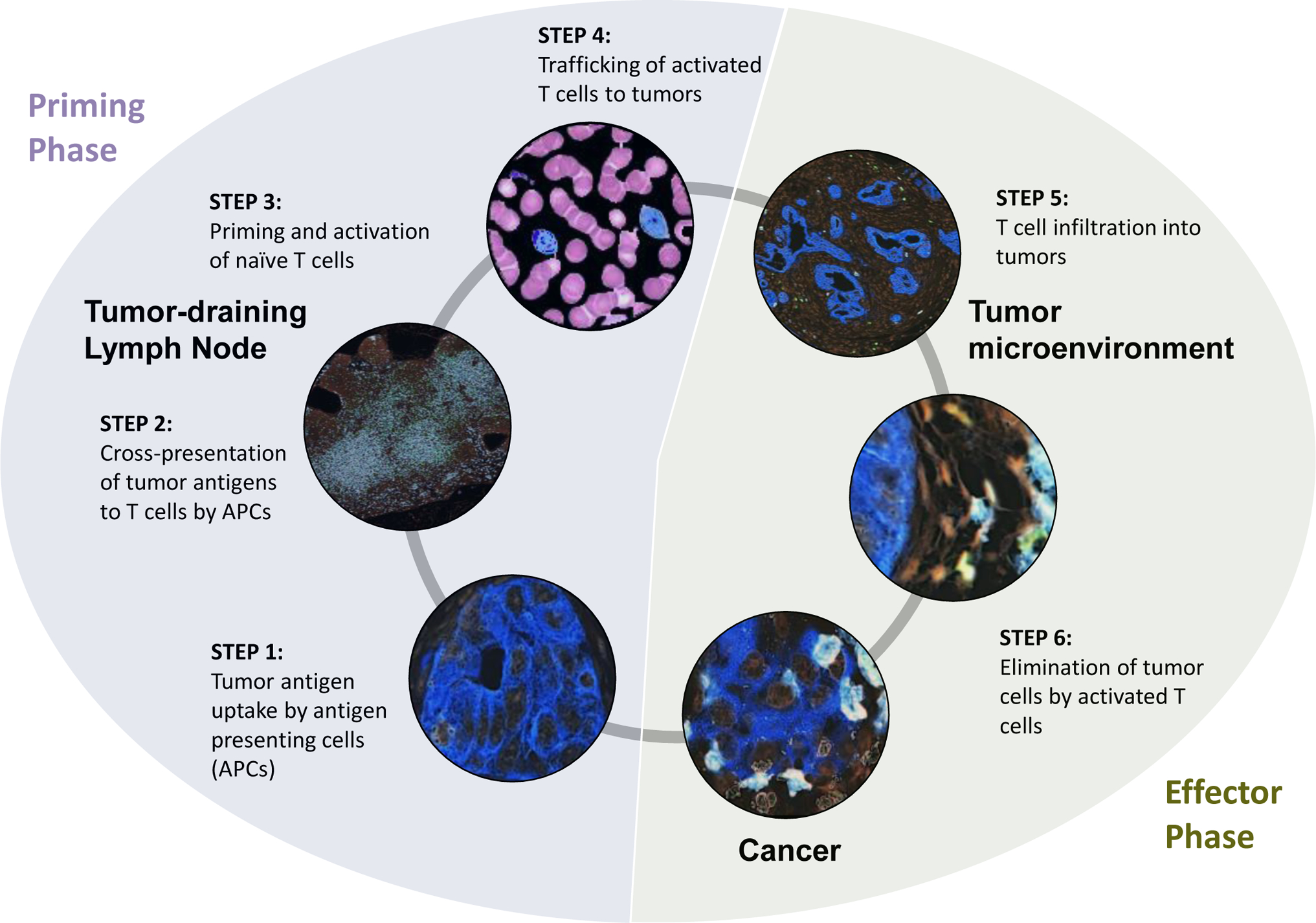
T cell immunosurveillance in cancer is dependent on steps (1–4) that support T cell priming and activation (Priming Phase) and steps (5–6) which involve T cell effector activity (Effector Phase). Steps associated with the Priming Phase include (1) the release of tumor antigens which are captured by antigen presenting cells (APCs), (2) cross-presentation of tumor antigens to T cells by APCs in tumor-draining lymph nodes, (3) priming and activation of naïve T cells by antigen-loaded APCs, and (4) trafficking of T cells from lymph node into the blood stream and to tumors. Steps associated with the Effector Phase include (5) T cell infiltration into tumors and (6) T cell recognition and elimination of tumor cells.
2.1. Dendritic cells are the classical drivers of antigen presentation and T cell priming
Dendritic cells (DCs) are a subset of innate immune cells that serve as key mediators of anti-tumor immunity.16 As APCs in the TME, DCs initiate the Cancer-Immunity Cycle by crosspresenting tumor-associated antigens to naive T cells. In mice, conventional dendritic cells (cDCs) are critical regulators of T cell priming. cDCs have been classified into two functionally distinct lineages: CD103+ cDC1 lineage which are responsible for CD8+ cytotoxic T lymphocyte (CTL) priming and the CD11b+ cDC2 lineage which have been implicated in priming of helper CD4+ T cells.17 It is noteworthy that despite their relatively sparse abundance in tumors, cDC1s are critical to coordinating T cell infiltration into tumors. Specifically, in a mouse model of melanoma, CTL recruitment to the TME was shown to require Batf3+ CD103+ cDC1s which facilitate T cell recruitment by releasing chemoattractants, specifically C-X-C motif chemokine ligand 9/10 (CXCL9/10).18 To this end, strategies to expand and activate cDC1s at tumor sites can improve T cell immunosurveillance in cancer.19,20 For example, cDC1s are enriched in tumors in response to FMS-like tyrosine kinase 3 ligand (FLT3L) which when combined with a TLR3 agonist sensitizes tumors to PDL1 immune checkpoint blockade.19,20 Within the tumor bed, this small but vital population of CD103+ DCs acquires antigens derived from apoptotic and necroptotic tumor cells.19,21 CD103+ DCs possess migratory capacity and traffic from the tumor core to secondary lymphoid organs to cross-present antigens to naive T cells.22 In contrast, CD8ɑ+ cDC1s reside in lymph nodes (LNs), where they capture and present soluble antigens that drain from tumor lymphatics.23
Recently, the importance of cDC2s in the regulation of immunosurveillance in cancer has also been appreciated. cDC2s are a heterogeneous cell population comprising multiple subsets and coordinate CD4+ T cell priming.24 Single cell analyses have revealed that cDC2 subsets are generated by distinct developmental pathways and transcriptional regulators.25 While defined subsets of cDC2s display similar capacity to support regulatory T cell (Treg) differentiation, they demonstrate distinct capacities to promote differentiation of conventional CD4+ T cell subsets, suggesting that they may have unique roles in regulating anti- and pro-inflammatory immune responses.25 cDC2s are enriched in tumors in response to granulocyte-macrophage colony-stimulating factor (GM-CSF).19 In a mouse model of melanoma, cDC2 frequency in tumors positively correlated with Treg frequency.24 Notably, genetic depletion of Tregs was found to enhance cDC2 migration to tumor-draining lymph nodes; promote antigen-specific CD4+ T cell priming; and improve CD4+ T cell differentiation in vivo. Further, in patients with newly diagnosed head and neck cancer, increased cDC2 frequency along with a decrease in Treg frequency was found to associate with improved progression-free survival.24 In melanoma patients, increased cDC2s in tumors is seen in responders to anti-PD1 therapy.24 Thus, cross-talk between cDC2s and Tregs may shape the immune response to cancer and in doing so, influence outcomes to immunotherapy.
Antigen-loaded DCs are, under the right conditions, incredibly potent stimulators of T cell activation. However, before they may invoke the expansion of antigen-specific CTLs, DCs must undergo a process known as ‘licensing,’ which imbues them with the capacity to prime naive T cells.26, 27 The proper licensing of DCs is contingent upon the activation of the CD40-CD40L pathway. CD40 is a member of the tumor necrosis factor (TNF) receptor superfamily, and is expressed abundantly on APCs, including DCs, macrophages, and B cells.28 Upon recognition of their cognate antigen, CD40L-expressing CD4+ T helper cells bind to their complementary CD40 receptor on the surface of DCs.29,30 Activation of the CD40 pathway increases expression of Major Histocompatibility Complex (MHC) II, CD80 (B7–1), and CD86 (B7–2) on the surface of DCs, which together support T cell priming.31–34 Recent literature shows that CD4+ T cell-mediated licensing of DCs also stimulates the release of C-C motif chemokine ligand 5 (CCL5), which recruits C-C chemokine receptor 5 (CCR5) naïve CD8+ T cells to the site of the licensed DCs.35 As such, DCs cooperate with cells of the adaptive immune system to coordinate a series of events that culminate in the generation of antigen-specific T cells.
2.2. The phenotypic plasticity of macrophages confers them with regulatory properties that affect T cell priming
Similar in many ways to dendritic cells, macrophages are a subset of innate immune cells with the capacity to promote or hamper the priming of effector T cells. However, macrophages comprise the most abundant myeloid-derived immune infiltrate in the majority of solid cancers.36 Their functional heterogeneity arises by virtue of their incredible phenotypic plasticity, and endows them with the ability to assume a range of distinct activation states. Thus, macrophages are dynamic cells whose functional properties are directed by environmental cues that emanate from their surrounding microenvironment.
The unique ability of macrophages to modulate their physiological properties in a context-dependent manner is a corollary of the opposing roles that macrophages play in the maintenance of tissue homeostasis. Macrophage polarity has been studied extensively in vitro where distinct stimuli can skew macrophages with either pro- or anti-tumor activity.37,38 For example, interferon (IFN)-γ and Toll-like receptor (TLR) agonists induce macrophages with an M1 phenotype associated with anti-tumor activity. In contrast, interleukin (IL)-4 and IL-13 engender macrophages with an M2 phenotype associated with pro-tumor activity. This binary M1-M2 schema can be a useful conceptual model, though oversimplified, for defining extremes of macrophage polarization.39 For instance, ‘classically’ activated M1 macrophages primarily coordinate an immune response against foreign pathogens.40 As such, M1 macrophages secrete a robust profile of proinflammatory cytokines (e.g. IL-6, IL-12, and IL-23) and differentially upregulate the expression of MHC II molecules.41,42 In contrast, ‘alternatively’ activated M2 macrophages assist in wound healing and secrete anti-inflammatory cytokines, such as IL-4 and IL-10, to regulate the immune response and resolve residual tissue damage induced by the M1 response.43 In the context of cancer and tumorigenesis, the induction of an M1-like phenotype is typically seen to enable adaptive immunosurveillance; whereas polarization toward an M2-like state tends to instill macrophages with functional properties that suppress T cell anti-tumor immunity.
Although macrophages are more commonly associated with suppression of cancer immunity, tumor-infiltrating macrophages can promote the induction of a T cell-mediated immune response in cancer. Certainly, migratory and lymphoid-resident DCs comprise the major population of APCs, but M1-like macrophages in the TME can also phagocytose malignant cells and cross-present tumor antigens to prime CD8+ T cells.44 In addition, recent studies show that extra-tumoral tissue-resident macrophages may enhance the ability of DCs to prime and activate CTLs. For example, a subset of splenic marginal zone macrophages are known to transfer phagocytosed antigens to DCs, which then go on to enable an adaptive immune response by inducing the cross-priming of CTLs.35,45,46 Therefore, under the appropriate conditions macrophages can support anti-tumor activity by promoting the proliferation of tumor antigen-specific T cells.
Despite their potential to stimulate cancer immunity, macrophages are usually exploited by tumors to suppress adaptive immunosurveillance. For instance, the secretion of IL-10 by M2-like macrophages upregulates N-glycan branching on the surface of effector T cells, a post-translational modification which impedes T cell activation by disrupting the ability of the T cell receptor (TCR) to bind to its CD8 coreceptor.47 Similarly, macrophage-derived reactive nitrogen species can nitrate tyrosine residues on the TCR of CD8+ T cells and, in turn, disrupt the ability of the TCR to bind peptide-loaded MHC.48 Because the TCR must bind its cognate antigen in order to achieve full T cell activation, macrophages mediate biochemical alterations that induce T cell tolerance to tumor-associated antigens. In addition, tumor-infiltrating macrophages can express multiple immune checkpoint proteins, such as PDL1 and B7-H4, which propagate the transduction of inhibitory signals that inhibit T cell activation.49,50 As such, macrophages are potent regulators of T cell biology, and can restrain the elaboration of an adaptive immune response against cancer.
Macrophages may also engage in mechanisms of indirect interference that culminate in a state of T cell dysfunction. An emerging hypothesis is that macrophages phagocytose and withhold tumor antigens from APCs.50 This is of particular consequence in the context of the tumor-draining lymph nodes, which house a subset of CD169+ subcapsular sinus (SCS) macrophages. SCS macrophages line the marginal zone of the LN, a region which occupies the interstitial space between the lymphatic fluid and the lymphoid tissue.51,52 As such, SCS macrophages are among the first subsets of immune cells to encounter tumor-associated antigens and apoptotic cellular debris contained in the afferent lymphatic fluid.53 In murine models, CD169+ splenic marginal zone macrophages (MZMs) promote anti-tumor immunity by phagocytosing tumor-associated antigens to cross-present to CD8+ T cells.54 However, emerging evidence suggests that CD169+ macrophages may also play a dual role in suppressing the adaptive immune response. For example, splenic MZMs can confer immunosuppressive effects and protect the host from leveraging a T cell-directed response against auto- or self-antigens.55,56 One potential explanation for this finding is that CD169+ macrophages capture antigens to prevent them from inducing an immune response. Therefore, in the context of cancer, it is possible that SCS macrophages in the LN may ‘hoard’ tumor-associated antigens and in doing so, prevent their cross-presentation to T cells.
2.3. Innate immune cells coordinate formation of T cell inflamed (“hot”) and non-inflamed (“cold”) tumors
To eliminate malignant cells, activated tumor antigen-specific T cells must first traverse the TME and infiltrate the tumor bed. Therefore, sufficient intratumoral T cell infiltration is a prerequisite for T cell effector activity. The immune contexture of the TME can exhibit an incredible degree of spatial heterogeneity, and the composition of the immune infiltrate may differ markedly both within and across tumors. T cell-infiltrated (or ‘inflamed’) tumors are characterized by a robust infiltrate of CD8+ T cells which serve as strong predictors of improved prognostic and survival outcomes across several cancer types.57–60 On the molecular level, the transcriptomic profiling of T cell-rich tumors indicates that immunoreactive tumor beds upregulate the expression of IFN signaling genes.60–62 On the contrary, T cell ‘cold’ tumors, which correlate with poor patient survival, are marked by an abundance of Tregs and immunosuppressive myeloid cells.63,64 These observations imply that the innate immune system regulates the phenotypic plasticity of the TME and may dictate the formation of T cell ‘hot’ and ‘cold’ tumors.
Innate immune cells facilitate anti-tumor immunity by promoting the migration of activated T cells into the TME. Recent data suggest that cytokines and soluble factors derived from innate immune cells are critical to supporting the formation of T cell-inflamed tumors.65 As such, the innate immune compartment is an enabler of T cell cytolytic activity by facilitating the migration of tumor antigen-specific T cells from secondary lymphoid organs (i.e. the site of T cell priming and activation) to the tumor bed. IFNγ stimulation, for instance, induces the secretion of CXCL9 by APCs, which in turn promotes the recruitment of CXCR3-expressing T cells into tumors.66 Batf3-dependent CD8α+ DCs, aside from their roles in antigen presentation and T cell priming, have also been shown to function as a major source of CXCL10, the release of which promotes the infiltration of CXCR3+ activated T cells into the tumor nest.18,67 As such, myeloid-derived cytokines prime the tumor bed for productive anti-tumor immunity by mediating the infiltration of tumor antigen-specific T cells.
However, there is concomitant evidence to suggest that the innate immune compartment may also foster the formation of T cell-cold tumors. Clinical studies of patient-derived tissues have shown that tumors that are scarce in infiltrating T cells generally possess an enriched myeloid infiltrate.68,69 The immune-based phenotyping of tumor tissues collected from patients with pancreatic ductal adenocarcinoma (PDAC) shows that an increased density of tumor-infiltrating myeloid cells, including CD163+ macrophages and CD66b+ granulocytes, is predictive of reduced overall survival and poor response to neoadjuvant immunotherapy.70 Similarly, myeloid cell infiltration inversely correlates with CD8+ T cell infiltrates in breast cancer.71 As such, these correlative observations indicate that the myeloid compartment can have a key role in orchestrating T cell exclusion in cancer.
Experimental studies confirm that innate immune cells can preclude the entry of T cells into the tumor nest. In a mouse model of breast cancer, the therapeutic depletion of tumor-associated macrophages (TAMs) via colony-stimulating factor-1 receptor (CSF-1R) blockade significantly increases the proportion of tumor-infiltrating CD8+ T cells.71 This suggests that macrophages can promote tumor progression by excluding CD8+ T cells from entering the microenvironment and engaging malignant cells. Similarly, the elimination of Ly6CloF4/80+ extratumoral macrophages in a murine model of spontaneous PDAC increases the infiltration of CD8+ T cells into tumors.72 On a mechanistic level, one means by which myeloid cells facilitate T cell exclusion is via the secretion of reactive nitrogen species (RNS), which can induce post-translational modifications on T cell chemoattractants.73 For instance, the nitration of CCL2 by myeloid-derived RNS restricts T cells to the peritumoral stroma and impedes their direct entry into the tumor nest.74 In addition, stromal cells adjacent to tumors may release cytokines that exclude T cells from the tumor nest. Recent studies have shown that stromal leukocytes, acting in concert with cancer-associated fibroblasts, may inhibit T cell entry by initiating the activation of transforming growth factor beta (TGF-β) signaling. Notably, pharmacologic inhibition of TGF-β signaling was found to provoke T cell entry into tumors in murine models when combined with PDL1 blockade75,76 Collectively, these findings demonstrate that the innate immune reaction to cancer is a critical determinant of the productivity of T cell immunosurveillance. Thus, factors which are extrinsic to tumors cells, such as macrophages, granulocytes and dendritic cells, can directly shape the immunogenicity of cancer.
3. Tumors evolve mechanisms of immune escape that threaten to abrogate productive cancer immunosurveillance
The utility of the Cancer-Immunity Cycle rests upon the premise that tumors possess a degree of immunogenicity that is sufficient to yield productive immune surveillance. However, an emerging corpus of literature suggests that malignant cells can acquire mechanisms to escape anti-tumor immunity. Two dominant modes have been defined by which cancers may evade immune destruction. First, malignant cells can evolve phenotypic changes that result in the Darwinian selection of less immunogenic tumor variants.77 Second, in addition to cell-intrinsic determinants, tumors can resist immune elimination via the modulation of the host immune system. Notably, tumor cells can: (i) suppress the development and productivity of adaptive immune surveillance, and/or (ii) redirect the immune compartment to drive disease progression. Therefore, cancer evolves to selectively inhibit the anti-tumor properties of the immune system, while amplifying its tumorigenic potential.2 To this end, both neoplastic cells and the host immune compartment are culpable in enabling the process of immunoediting.
3.1. Cancers evolve alterations that culminate in less immunogenic tumor variants
To escape immune elimination, tumors cells may evolve properties that render them less susceptible to immune recognition.12,13 Phenotypically, this often manifests via deficiencies in antigen-presenting machinery (APM), or in the downregulation of MHC class I molecules on malignant cells. Indeed, transcriptomic characterization of patient tumors has shown that a subset of genes that regulate the assembly of the APM are commonly downregulated in several malignancies.78–81 This suggests that neoplastic cells may escape immune recognition by functionally disrupting the translocation of intracellular fragments for presentation on the MHC complex. In addition, compelling evidence exists to suggest that tumors may evade detection due to genetic changes that result in loss of heterozygosity (LOH) at the MHC locus.82 The advent of novel bioinformatic tools has made it possible to infer these LOH events by quantifying the differences in MHC haplotype-specific copy number between germline and tumor genomes captured from a single patient.83,84 Strikingly, the application of this technology to malignant tissues isolated from 90 treatment-naïve non-small-cell lung cancer patients has shown that approximately 40% of tumor samples exhibit loss of one parental MHC haplotype.83 Because haplotype diversity is essential to drive the expression of distinct MHC molecules with differential antigen binding affinities, LOH at the MHC locus allows malignant cells to selectively present only the neoepitopes that bind most strongly to the conserved haplotype. Consequently, the loss of an MHC haplotype may encourage the selection of less immunogenic tumors variants with the capacity to evade host immune elimination by downregulating the presentation of tumor-associated antigens.
3.2. Cancer cell-intrinsic mechanisms suppress the elaboration of T cell immunity
Tumors employ several mechanisms to inhibit adaptive immune surveillance. Because T cell priming is a multi-step process that involves a number of cell types, there are several stages at which malignant cells can intervene to disrupt the generation of tumor antigen-specific T cells. On the level of antigen-presentation, malignant cells can hinder the migration of CD103+ DCs into the TME, where they recognize and acquire tumor antigens for cross-presentation. Consistent with this, in a Pten-negative BRAFV600E model of melanoma, the induction of Wnt/β-catenin signaling was found to silence the expression of CCL4 which, in turn, impeded the migration of CD103+ DCs into the tumor.85,86 Similarly, the release of tumor-derived cholesterol metabolites, such as liver X receptor-ɑ, can downregulate the expression of CCR7 on DCs and impede tumor-antigen loaded DCs from migrating into tumor-draining lymph nodes.87 Additional tumor-derived soluble factors have also been shown to negatively regulate DC biology. For example, the release of IL-10 and IL-6 by tumors promotes the formation of tolerogenic DCs that fail to elicit an adaptive immune response against tumor-specific antigens.69 Similarly, the secretion of vascular endothelial growth factor (VEGF) inhibits the differentiation of hematopoietic progenitor cells into mature DCs.88 Thus, cancer cell-intrinsic mechanisms may drive the functional dysregulation of key events within the Cancer-Immunity Cycle.
Tumors may also induce immune tolerance by inhibiting effector T cells that infiltrate the TME. A major mode by which this occurs is via the activation of canonical immune checkpoint pathways. For instance, PDL1 on tumor and myeloid cells directly inhibits T cell effector activity by engaging its complementary receptor, PD1, on the surface of activated T cells.89 Interestingly, recent work reveals that tumor-derived exosomes captured from patients with metastatic melanoma contain PDL1, which suggests that tumor-derived cellular cargo may further compound this immunoregulatory response.90 Tumors can also alter the biochemical milieu of the TME to stimulate T cell dysfunction. To this end, expression of indoleamine 2,3-dioxygenase (IDO) by tumor cells and myeloid cells catalyze the breakdown of the amino acid, tryptophan, to its metabolite, kynurenine.91 This disruption of the nutritional homeostasis at the level of the TME suppresses T cell function, which requires tryptophan to retain proliferative capacity.92 In addition to disrupting the metabolic state of a tumor, tumor cells and myeloid cells collectively secrete a range of cytokines that impinge upon T cell cytolytic effector activity. For instance, tumor-derived TGF-β transcriptionally silences genes that regulate the expression of perforin and granzyme, two proteins which cooperate to induce T cell-mediated cytotoxicity.93 Both tumor cells and myeloid cells can also produce chemokines that engage the recruitment of CCR2+ and CSF1R+ macrophages as well as CXCR2+ granulocytes which together instruct a vicious cycle of immune suppression with tumors. As such, innate immunity along with tumor cells share roles in instructing the TME to wield an incredibly diverse arsenal of mechanisms with potent capacity to suppress T cell immunity.
3.3. Cancer cells enable immune-mediated mechanisms of tumorigenesis
In leveraging its tumorigenic capacity, cancer cells commandeer the immune system to advance tumor growth. This occurs primarily via the recruitment of selective populations of myeloid cells which, in the context of the TME, potently suppress adaptive immunosurveillance and intensify the proliferative capacity of cancer. Tumors, therefore, exploit the plasticity of the TME to establish a permissive niche, or site of ‘immune privilege,’ that shelter cells from immune elimination.12,13,94,95
Tumors establish an immunotolerant environment via the recruitment of suppressive immune cells. This process is highly dependent on paracrine signaling mechanisms wherein malignant cells secrete a repertoire of cytokines (e.g. CXCL1, granulocyte-macrophage colony-stimulating factor (GM-CSF), IL-1β, IL-4, and IL-13) to lure selective immune cells into the tumor core.96, 97 In a genetic model of pancreatic cancer, tumor-derived GM-CSF promotes the infiltration of a heterogeneous population of CD11b+Gr1+ myeloid cells.98 CD11b+Gr1+ cells, which may include neutrophils and monocytes, among other innate immune cells, encourage tumorigenesis in several ways. For example, the release of arginase-1 by CD11b+Gr1+ neutrophils catalyzes the catabolic degradation of arginine and in doing so, depletes the microenvironment of essential nutrients.98 In turn, this scarcity of arginine impairs the proliferative capacity of T cells and downregulates the expression of CD3ζ, a critical component of the TCR that is necessary to induce T cell activation.99–101 Moreover, CD11b+Gr1+ myeloid cells can sculpt an inflammatory milieu that supports tumor initiation and growth. For instance, preclinical work has shown that IL-1β, a pro-inflammatory cytokine, not only recruits myeloid cells, but also promotes the initiation of gastric neoplasia via the activation of NF-κB signaling in CD11b+Gr1+ cells.102 Recently, it was also shown that TLR4 activation in epithelial cells drives activation of the NLRP3 inflammasome leading to release of IL-1β and the formation of an immunosuppressive microenvironment in mouse models of pancreatic cancer.103 Immune cells within tumors can also react to signals, such as ATP, that are released by tumor cells undergoing cell death. Extracellular ATP (eATP) is a damage-associated molecular pattern (DAMP) that incites inflammation by acting as a chemoattractant for circulating neutrophils.104–108 eATP is also processed by ectonucleotidases, including CD39 and CD73, to generate adenosine which promotes immunosuppression.109 The immunosuppressive functions of adenosine are mediated by adenosine A2 receptors which when triggered by adenosine, impair T cell biology109 and support the immunosuppressive properties of myeloid cells.110 Thus, cancer cells coordinate a TME that empowers innate immunity to suppress adaptive immunosurveillance and to give rise to a host of pleiotropic effects that collectively stimulate tumor growth.
In addition to recruiting immunosuppressive cells, tumor-induced alterations in the cytokine profile of the TME may impart immune cells with pro-tumorigenic properties. This applies particularly to macrophages which comprise, arguably, the most pliable and phenotypically heterogeneous subset of innate immune cells. Tumor-derived cytokines such as IL-4, IL-13, CCL2, and colony-stimulating factor (CSF), can polarize macrophages with functional attributes that tumor cells co-opt to support disease progression.111,112 TAMs have been extensively shown to suppress anti-tumor immunity. For example, in a murine model of mammary carcinoma macrophage-derived IL-10 abrogates the elaboration of an adaptive T cell response by inhibiting the production of IL-12 in DCs.113 TAMs can also potentiate the metastatic outgrowth of tumors by stimulating neovascularization.114 In response to TGFβ and hypoxic conditions in the TME, tumor-infiltrating macrophages upregulate their expression of VEGF protein, a key mediator of angiogenesis.115,116 Thus, in addition to suppressing T cell immunity, TAMs may also supply tumors with the essential growth factors needed to further nourish their growth.
4. The coevolution of tumor immunoediting necessitates a novel approach to cancer immunotherapy
To escape immune recognition and subsequent elimination, tumor cells can decrease their antigenicity (i.e. expression and presentation of tumor antigens), reduce their immunogenicity (i.e. potential to alert tumor-specific immunity), form an immunosuppressive microenvironment, and impair the functional capacity of the immune system (i.e. the ability of the immune system to produce anti-tumor activity). These hallmarks of immune escape (reviewed in detail elsewhere12) undermine T cell immunosurveillance in cancer. One cancer that exemplifies each of these hallmarks is pancreatic cancer, which is characterized by few mutated neo-proteins (i.e. limited antigenicity)117,118, a poor T cell response (i.e. weak immunogenicity)70,119–121, an immunosuppressive microenvironment directed by myeloid cells, fibroblasts, and Tregs122,123, and a systemic inflammatory reaction that associates with T cell dysfunction.124 Based on this biology, it is perhaps unsurprising that T cell-directed immunotherapies, such as single-agent and even dual ICB, have failed to trigger significant regressions in pancreatic cancer.119,125,126 Similar findings have been seen in other classically T cell ‘cold’ tumors such as glioblastoma where immunotherapy has yet to produce durable and significant clinical outcomes.127,128 The failure of immunotherapy in these T cell “cold” cancers, though, implies that distinct mechanisms of immune resistance, such as the immunological state of the TME, are critical to defining the therapeutic potential of immunotherapy.
Most solid cancers do not respond to immunotherapy. As such, it may be necessary to first “condition” tumors with an immunostimulatory state prior to implementing immunotherapy that is designed to trigger the generation of tumor-specific T cells and to enhance their functional capacity. Given the propensity of tumors to adapt to immune pressure, strategies that seek to “maintain” tumor immunogenicity may also be necessary to achieve durable responses. In this regard, we propose a biphasic approach to the application of immunotherapy in cancer, which we discuss in detail in subsequent sections (Figure 3). The first phase, which we call the ‘conditioning’ phase, consists of converting the TME from a tolerogenic to immunostimulatory state. The priority of this act of conditioning is to sensitize tumors to immunotherapy. Conditioning may be achieved through multiple mechanisms such as (i) the depletion or inhibition of tumor-associated myeloid cell populations and (ii) re-polarization of immune cells with anti-tumor properties.129 The second phase, or ‘maintenance’ phase, involves the inhibition of potential mechanisms of acquired resistance to prevent tumors from shifting their TME to an immunosuppressive state. The productiveness of this approach may be sequence-dependent and, as such, the maintenance phase may need to be initiated after implementing T cell-directed immunotherapy. This framework of ‘conditioning’ and ‘maintenance’ phases may potentiate novel combinatorial strategies by which to increase the clinical utility of immunotherapy. However, it should be noted that not all tumors may require this approach of applying ‘conditioning’ and ‘maintenance’ regimens. For example, for many patients with melanoma, incorporation of ICB is sufficient to trigger a productive and durable anti-tumor immune response.7,130,131 This observation implies that in melanoma, sufficient antigenicity is often present and that T cells have ample capacity to be invigorated with anti-tumor activity. In addition, it suggests that the immunological state of the TME that surrounds cancer cells in melanoma is receptive to immunotherapy. Nonetheless, this scenario is atypical and most patients with solid cancers do not respond to T cell-directed immunotherapies (e.g. anti-PD1/PDL1 and anti-CTLA-4) with deep and durable tumor regressions. Thus, for most cancers, strategies to sensitize tumors to immunotherapy will be needed. Here, we discuss potential strategies for conditioning and maintaining tumors in an immunostimulatory state as an approach for broadening the potential of immunotherapy.
Figure 3. A biphasic approach to cancer immunotherapy.
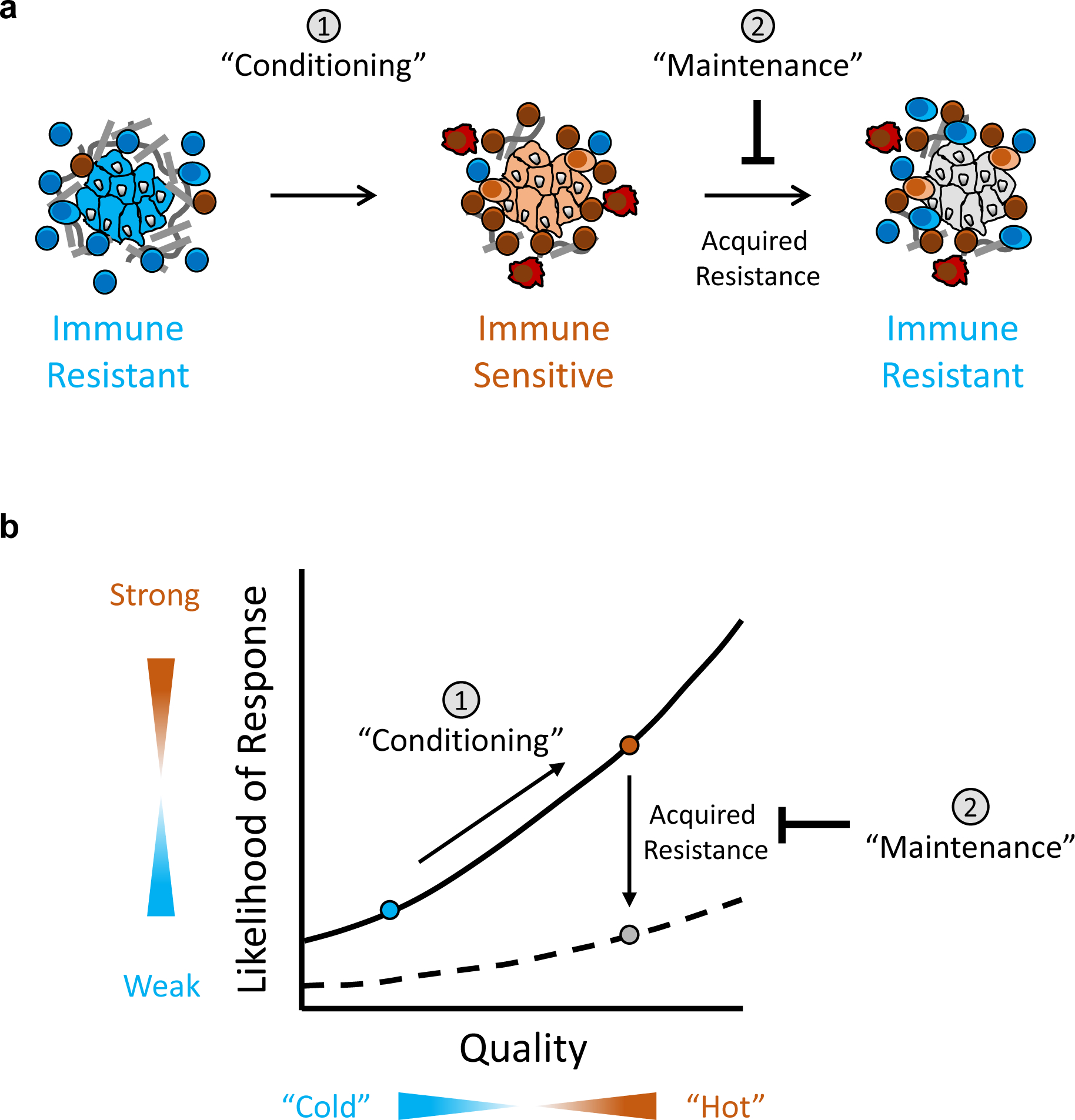
(a) Conceptual model displaying two phases of immunotherapy. In the first phase, tumors are “conditioned” with sensitivity to immunotherapy. In the setting of immunotherapy, tumor cells may then adapt to immune pressure (i.e. acquired resistance) which undermines the success of immunotherapy. As such, the second phase incorporates strategies to “maintain” tumors as immune sensitive and to prevent their reversion to immune resistant. (b) Correlation plot showing the relationship between the likelihood of responding to immunotherapy and the quality of the immune reaction to cancer. The “conditioning” phase of therapy aims to shift an immune resistant tumor (blue circle) to immune sensitive (orange circle). However, tumors may also acquire therapeutic resistance (e.g. recruitment of immune suppressive cells) which abate a productive immune response. The “maintenance” phase of therapy aims to prevent this shift to a state (grey circle) of decreased likelihood of response to immunotherapy.
4.1. Depletion of immunosuppressive myeloid cells can sensitize tumors to immunotherapy
The immunosuppressive state of cancer is governed by a variety of mechanisms of which myeloid cells play a significant role. Inhibiting the recruitment of pro-tumorigenic myeloid cells offers a means by which to shift the immunological state of the TME from immunosuppressive to immunostimulatory.132 Immunosuppressive myeloid cells may physically exclude T cells from the tumor bed, or directly compromise their cytolytic capacity in the TME.133 Thus, myeloid cells are key orchestrators of a tolerogenic niche. To this end, disrupting the trafficking of these cells into the TME should, in theory, increase not only T cell infiltration but also enhance the quality (i.e., effector function) of infiltrating T cells. For example, in a murine model of PDAC, the administration of a monoclonal antibody (mAb) against CSF-1R has been shown to trigger depletion of TAMs and subsequent, accumulation of tumor-infiltrating T cells.134 Remarkably, in this study CSF-1R mAb treatment sensitized pancreatic tumors to CTLA-4 blockade in a CD8+ T cell-dependent manner. Similarly, in a mouse model of melanoma, CSF-1R inhibition was shown to sensitize tumors to combined anti-CTLA-4 and anti-PD1 treatment.135 These findings show that anti-CSF-1R-mediated TAM depletion can shift the TME to an immunoreactive state which supports the infiltration of tumor-reactive T cells and sensitizes tumors to immune checkpoint blockade.
Disrupting the activity of myeloid chemoattractants is a common approach being explored to convert the immunological state of a tumor from immune-suppressive to -stimulatory. For example, CCL2 derived from tumor and stromal cells is a potent regulator of CCR2+ monocyte recruitment into the TME. In a model of hepatocellular carcinoma (HCC), CCL2 blockade depletes TAM recruitment, increases T cell infiltration, and induces tumor regression in a T cell-dependent manner.136 In addition to CCL2, tumors are often rich in CXCR2-ligands, including CXCL1, CXCL2, and CXCL5, which are chemoattractants for neutrophils.137 In a mouse model of PDAC, the concomitant delivery of a CXCR2 inhibitor and anti-PD1 was shown to significantly prolong survival and augments the therapeutic potential of immunotherapy.138 Similarly, in a mouse model of rhabdomyosarcoma, anti-CXCR2 mAb therapy inhibited the recruitment of suppressive myeloid cells to tumors and in turn, conditioned tumors for increased responsiveness to anti-PD1 therapy.139 Together, these findings indicate that conditioning the TME with myeloid-depleting agents can convert tumors from treatment-resistant to -sensitive.
4.2. Educating myeloid cells with anti-tumor properties can augment cancer immunosurveillance
Despite its ability to fuel tumorigenesis, the myeloid compartment is also an indispensable mediator of cancer immunity. As such, myeloid cells can be polarized with anti-tumor properties and harnessed to sensitize the TME to immunotherapy. Recent work has shown that activation of select innate immune signaling pathways can impart myeloid cells with attributes that enable cancer immunosurveillance. For example, in a mouse model of pancreatic cancer, CD40 stimulation in combination with chemotherapy produces tumor regression by augmenting the cross-presentation of tumor antigens by Batf3+ DCs.140 In this case, the addition of chemotherapy is thought to enhance the ability of CD40-activated DCs to prime T cells by inducing an immunogenic form of tumor cell death and, by extension, the release of tumor-associated antigens that serve to fuel DCs for the priming of T cells.141 CD40 agonists have also shown to combine synergistically with immune checkpoint blockade to promote T cell dependent anti-tumor immunity in mouse models of colon and pancreatic cancer.142,143 The therapeutic potential of CD40 agonists, though, extends beyond promoting tumor-specific adaptive immunity. For instance, CD40 agonists can also invoke a systemic immune reaction that triggers anti-tumor responses that are independent of T cells and dependent on innate immunity.122,144 To this end, treatment with a CD40 agonist was found to imbue macrophages with the capacity to eliminate tumor cells and degrade the fibrotic stroma that surrounds tumor cells in a model of pancreatic cancer.122,144 In addition, systemic CD40 activation was shown to condition tumors for enhanced sensitivity to chemotherapy144, thereby demonstrating the potential to shift the biological state of tumors from treatment-resistant to treatment-sensitive.
Other myeloid agonists have also been studied for their potential to leverage the myeloid reaction to cancer for therapeutic benefit. For instance, Toll-like receptor (TLR) agonists constitute an approach by which to engender the myeloid compartment with anti-tumor potential. Bacterial or fungal-derived moieties, which express shared motifs called pathogen-associated molecular patterns, induce TLR signaling to initiate an innate immune response to foreign infection.145 Notably, the activation of TLR signaling polarizes macrophages to an M1-like state.146 In this regard, the induction of TLR signaling with synthetic ligands has been shown to educate macrophages with immunostimulatory properties that are conducive to the elimination of tumor cells.147 For example, in ICB-naïve patients with metastatic melanoma, intratumoral injection of a TLR9 agonist combined with PD1 blockade was shown to induce tumor regression and increase the number of tumor-infiltrating T cells relative to baseline.148 Similarly, in a mouse model of pancreatic cancer, partial activation of CD11b using a small molecule agonist was found to polarize tumor-associated macrophages with immunostimulatory capacity and in turn, render tumors sensitive to immune checkpoint inhibitors.149
However, not all tumors appear to respond to myeloid agonism implying that in some settings additional mechanisms may exist that lock tumors into an immunosuppressive state. For example, tumor-secreted factors including CXCL1, IL-1β and GM-CSF that are released by tumors may endow their surrounding microenvironment with an immune privileged status that must be undone before the benefit of immunotherapy can be realized.97,98,103,150 For example, CXCL1 released by tumor cells abrogates the efficacy of a CD40 agonist in combination with immune checkpoint blockade in a mouse model of pancreatic cancer.97 Similarly, IL-1β produced by tumor cells in response to TLR activation of the NLRP3 inflammasome can establish a microenvironment that resists immunotherapy.103 EPHA2 signaling in tumor cells and subsequent expression of cyclooxygenase-2 has also been shown to restrict the activity of immunotherapy in a mouse model of pancreatic cancer.151 Thus, tumor-intrinsic factors can shape the immunoreactive state of the TME and in doing so, impede the efficacy of immunotherapy.
4.3. Overcoming acquired immune resistance by maintaining tumors in an immunoreactive state
Although immunotherapy has produced complete tumor remissions for some patients with cancer, achieving a universally durable response remains an elusive goal. One plausible explanation for this sobering epidemiological observation involves the activation of treatment-induced mechanisms of acquired resistance. The immune system is an extraordinarily complex network, and one that exhibits an exceptional degree of functional redundancy. To that end, selectively targeting one feature of immunity can inadvertently trigger the elaboration of compensatory mechanisms to re-establish a state of biological equilibrium.
For many solid cancers especially gastrointestinal cancers, the treatment approach is grounded in conventional cytotoxic chemotherapy delivered until disease progression. However, this paradigm is now being challenged with the advent of less toxic strategies that can maintain and even consolidate responses produced after a short induction phase of chemotherapy. For example, PARP inhibitors are now approved as a form of maintenance therapy for patients with BRCA1/2-mutant PDAC who have not progressed after 4 months of platinum-based chemotherapy.152 This concept of sequencing therapies is particularly amenable to immunotherapy which can provoke compensatory mechanisms of immune resistance which limit therapeutic efficacy. For example, depletion of immunosuppressive CXCR2+ neutrophils sensitizes pancreatic tumors to chemotherapy.68 Interestingly, though, this synergistic effect subsides as the inhibition of CXCR2+ neutrophils triggers an increase in the recruitment of CCL2+ monocytes. To circumvent this resistance, dual inhibition of CXCR2+ neutrophils and CCL2+ monocytes has been shown to maintain the immunoreactive state of the tumor and further enhance the efficacy of chemotherapy.68 A similar effect is seen with depletion of CCL2+ macrophages in a model of breast cancer. For instance, whereas anti-CCL2 therapy shows capacity to restrain mammary tumor growth, cessation of therapy triggers a surge in the mobilization of bone marrow-derived myeloid cells which, in turn, drives pulmonary metastasis in an IL-6 and VEGF-dependent manner.153 In this case, it is possible that cessation of CCL2 inhibition causes the immune compartment to overshoot and recruit an immense number of TAMs to compensate for the prolonged period of monocyte depletion. Nonetheless, these unprecedented consequences of CCL2 blockade illustrate that it is profoundly difficult to modulate the immune system in an insular manner.
For many cancers, achieving deep and durable remissions may require sequential administration of immune therapeutics. This approach may allow for coaxing tumors into a state of immune vulnerability that is permissive for complete remission.154 For example, in a mouse model of lung cancer, treatment with PD-1 blockade induces initial disease control but ultimately, tumors adapt with upregulation of alternative immune checkpoint molecules including TIM-3.155 Notably, sequential treatment with anti-PD-1 with subsequent addition of anti-TIM-3 was found to prolong survival thereby illustrating the potential to sequence immune checkpoint blockade for improved outcomes. Additionally, in a subset of patients with metastatic melanoma, PD-1 blockade has been shown to induce the expansion of CD8+ effector memory T cells which associates with response to therapy.156 This finding suggests that development of long-term immunological memory may be fundamental to achieving complete and durable remissions.157
The appropriate sequencing of myeloid directed therapies may also improve treatment responses. For example, in a mouse model of pancreatic cancer, a CD40 agonist delivered prior to cytotoxic chemotherapy produces transient anti-tumor activity. However, introducing a maintenance phase of myeloid inhibition using an inhibitor of focal adhesion kinase after the CD40 agonist produces prolonged disease control.158 Importantly, the sequence of combining myeloid agonists with myeloid inhibitors may be particularly critical. In this regard, delivery of a CSF1R inhibitor prior to treatment with a TLR9 agonist has been shown to abrogate TLR9-dependent anti-tumor activity.159 Taken together, the immune compartment is a highly dynamic and inter-dependent network. As such, the induction of even a single alteration may unintentionally trigger the dysregulation of an array of seemingly disparate pathways. Thus, future therapies must identify ways in which to proactively inhibit these treatment-induced mechanisms of immune-suppression. To this end, incorporating rationally designed treatment phases (e.g. conditioning and maintenance phases) offers an approach for enhancing and sustaining a productive anti-tumor immune response.
5. Concluding remarks
The TME functions, in many ways, as the nexus between the immune system and tumor cells. As such, the remarkably complex bidirectional interactions that transpire between the immune compartment and malignant cells in the space of the TME can regulate the course of disease. In this review, we discussed the interplay between innate and adaptive immunity and the impact of this interaction on the immunoreactive state of cancer which can either support or inhibit cancer growth. We highlight that tumors leverage the tumorigenic potential of the immune system and, in doing so, alter the phenotypic landscape of the TME in a manner that suppresses productive adaptive immunosurveillance. We also propose that leveraging the inherent plasticity of the TME is a means by which to overcome cancer cell-driven mechanisms of resistance. As such, future treatment paradigms may need to focus not only on identifying modes by which to condition the landscape of the TME for improved immunotherapy efficacy but also on strategies to maintain the initial immune response to overcome mechanisms of acquired resistance that emerge in the face of cancer immunotherapy.
Acknowledgements:
This work was supported by grants from the National Institutes of Health grants R01 CA197916, R01 CA245323, U01 CA224193-01, the Stand Up to Cancer (SU2C) Innovative Research Grant SU2C-AACR-IRG 13-17, and grant support from the Robert L. Fine Cancer Research Foundation. The content is solely the responsibility of the authors and does not necessarily represent the official views of the Robert L. Fine Cancer Research Foundation.
Abbreviations
- APC
antigen presenting cell
- APM
antigen-presenting machinery
- cDC
conventional dendritic cell
- CCL
C-C motif chemokine ligand
- CCR
C-C chemokine receptor
- CSF
colony-stimulating factor
- CSF-1R
colony-stimulating factor-1 receptor
- CTL
cytotoxic T lymphocyte
- CTLA-4
cytotoxic T-lymphocyte-associated protein 4
- CXCL
C-X-C motif chemokine ligand
- CXCR
C-X-C motif chemokine receptor
- DAMP
damage-associated molecular pattern
- DC
dendritic cell
- eATP
extracellular ATP
- FLT3L
FMS-like tyrosine kinase 3 ligand
- GM-CSF
granulocyte-macrophage colony-stimulating factor
- HCC
hepatocellular carcinoma
- HLA
human leukocyte antigenIDO, indoleamine 2,3-dioxygenase
- IFN
interferon
- IL
interleukin
- LN
lymph node
- LOH
loss of heterozygosity
- MHC
major histocompatibility complex
- MZM
marginal zone macrophage
- LN
lymph node
- mAb
monoclonal antibody
- PD1
programmed cell death protein 1
- PDL1
programmed death-ligand 1
- PDAC
pancreatic ductal adenocarcinoma
- RNS
reactive nitrogen species
- SCS
subcapsular sinus
- TAM
tumor-associated macrophage
- TCR
T cell receptor
- TGF-β
transforming growth factor beta
- TLR
toll-like receptor
- TME
tumor microenvironment
- TNF
tumor necrosis factor
- Treg
regulatory T cell
- VEGF
vascular endothelial growth factor
Footnotes
Disclosure of Potential Conflicts of Interest: G.L.B. is a consultant/advisory board member for Seattle Genetics, Aduro Biotech, AstraZeneca, Bristol-Myers Squibb, Incyte, Genmab, Takeda, Merck, and BiolineRx; reports receiving commercial research grants from Incyte, Bristol-Myers Squibb, Verastem, Halozyme, Biothera, Newlink, Novartis, Arcus, and Janssen; and is an inventor of intellectual property and recipient of royalties from Novartis and Advaxis, Inc. No potential conflicts of interest were disclosed by the other authors.
References
- 1.Loeb LA, Loeb KR, Anderson JP. Multiple mutations and cancer. Proc Natl Acad Sci.2003;100:776–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell. 2011;144:646–674. [DOI] [PubMed] [Google Scholar]
- 3.Pitt JM, Vétizou M, Daillère R, et al. Resistance Mechanisms to Immune-Checkpoint Blockade in Cancer: Tumor-Intrinsic and -Extrinsic Factors. Immunity. 2016;44:1255–1269. [DOI] [PubMed] [Google Scholar]
- 4.Dunn GP, Old LJ, Schreiber RD. The Immunobiology of Cancer Immunosurveillance and Immunoediting. Immunity. 2004;21:137–148. [DOI] [PubMed] [Google Scholar]
- 5.Swann JB, Smyth MJ. Immune surveillance of tumors. J Clin Invest. 2007;117:1137–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lynch TJ, Bondarenko I, Luft A, et al. Ipilimumab in Combination With Paclitaxel and Carboplatin As First-Line Treatment in Stage IIIB/IV Non–Small-Cell Lung Cancer: Results From a Randomized, Double-Blind, Multicenter Phase II Study. J Clin Oncol. 2012;30:2046–2054. [DOI] [PubMed] [Google Scholar]
- 7.Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N Engl J Med. 2019;381:1535–1546. [DOI] [PubMed] [Google Scholar]
- 8.Vonderheide RH. The Immune Revolution: A Case for Priming, Not Checkpoint. Cancer Cell. 2018;33:563–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smyth MJ, Dunn GP, Schreiber RD. Cancer immunosurveillance and immunoediting: the roles of immunity in suppressing tumor development and shaping tumor immunogenicity. Adv Immunol. 2006;90:1–50. [DOI] [PubMed] [Google Scholar]
- 10.Schreiber RD, Old LJ, Smyth MJ. Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion. Science. 2011;331:1565–1570. [DOI] [PubMed] [Google Scholar]
- 11.Campoli M, Ferrone S. HLA antigen changes in malignant cells: epigenetic mechanisms and biologic significance. Oncogene. 2008;27:5869–5885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beatty GL, Gladney WL. Immune Escape Mechanisms as a Guide for Cancer Immunotherapy. Clin Cancer Res. 2015;21:687–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stone ML, Beatty GL. Cellular determinants and therapeutic implications of inflammation in pancreatic cancer. Pharmacol Ther. 2019;201:202–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wellenstein MD, de Visser KE. Cancer-Cell-Intrinsic Mechanisms Shaping the Tumor Immune Landscape. Immunity. 2018;48:399–416. [DOI] [PubMed] [Google Scholar]
- 15.Chen DS, Mellman I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity. 2013;39:1–10. [DOI] [PubMed] [Google Scholar]
- 16.Mildner A, Jung S. Development and function of dendritic cell subsets. Immunity.2014;40:642–656. [DOI] [PubMed] [Google Scholar]
- 17.Gardner A, Ruffell B. Dendritic Cells and Cancer Immunity. Trends Immunol. 2016;37:855–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spranger S, Dai D, Horton B, et al. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell. 2017;31:711–723.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Broz ML, Binnewies M, Boldajipour B, et al. Dissecting the Tumor Myeloid Compartment Reveals Rare Activating Antigen-Presenting Cells Critical for T Cell Immunity. Cancer Cell. 2014;26:638–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Salmon H, Idoyaga J, Rahman A, et al. Expansion and Activation of CD103 + Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity. 2016;44:924–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kotera Y, Shimizu K, Mulé JJ. Comparative analysis of necrotic and apoptotic tumor cells as a source of antigen(s) in dendritic cell-based immunization. Cancer Res. 2001;61:8105–8109. [PubMed] [Google Scholar]
- 22.Roberts EW, Broz ML, Binnewies M, et al. Critical Role for CD103 + /CD141 + Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer Cell. 2016;30:324–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fu C, Jiang A. Dendritic Cells and CD8 T Cell Immunity in Tumor Microenvironment. Front Immunol. 2018;9:3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Binnewies M, Mujal AM, Pollack JL, et al. Unleashing Type-2 Dendritic Cells to Drive Protective Antitumor CD4+ T Cell Immunity. Cell. 2019;177:556–571.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brown CC, Gudjonson H, Pritykin Y, et al. Transcriptional Basis of Mouse and Human Dendritic Cell Heterogeneity. Cell. 2019;179:846–863.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reis e Sousa C Dendritic cells in a mature age. Nat Rev Immunol. 2006;6:476–483. [DOI] [PubMed] [Google Scholar]
- 27.Immunology Lanzavecchia A.. Licence to kill. Nature. 1998;393:413–414. [DOI] [PubMed] [Google Scholar]
- 28.Elgueta R, Benson MJ, de Vries VC, et al. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol Rev. 2009;229:152–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hernandez MGH, Shen L, Rock KL. CD40-CD40 Ligand Interaction between Dendritic Cells and CD8 + T Cells Is Needed to Stimulate Maximal T Cell Responses in the Absence of CD4 + T Cell Help. J Immunol. 2007;178:2844–2852. [DOI] [PubMed] [Google Scholar]
- 30.Flinsenberg TWH, Spel L, Jansen M, et al. Cognate CD4 T-Cell Licensing of Dendritic Cells Heralds Anti-Cytomegalovirus CD8 T-Cell Immunity after Human Allogeneic Umbilical Cord Blood Transplantation. J Virol. 2015;89:1058–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vonderheide RH, Bajor DL, Winograd R, et al. CD40 immunotherapy for pancreatic cancer. Cancer Immunol Immunother. 2013;62:949–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clatza A, Bonifaz LC, Vignali DAA, et al. CD40-Induced Aggregation of MHC Class II and CD80 on the Cell Surface Leads to an Early Enhancement in Antigen Presentation. J Immunol. 2003;171:6478–6487. [DOI] [PubMed] [Google Scholar]
- 33.Beyranvand Nejad E, van der Sluis TC, van Duikeren S, et al. Tumor Eradication by Cisplatin Is Sustained by CD80/86-Mediated Costimulation of CD8+ T Cells. Cancer Res. 2016;76:6017–6029. [DOI] [PubMed] [Google Scholar]
- 34.Haile ST, Bosch JJ, Agu NI, et al. Tumor Cell Programmed Death Ligand 1-Mediated T Cell Suppression Is Overcome by Coexpression of CD80. J Immunol. 2011;186:6822–6829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kurts C, Robinson BWS, Knolle PA. Cross-priming in health and disease. Nat Rev Immunol. 2010;10:403–414. [DOI] [PubMed] [Google Scholar]
- 36.Cassetta L, Pollard JW. Targeting macrophages: therapeutic approaches in cancer. Nat Rev Drug Discov. 2018;17:887–904. [DOI] [PubMed] [Google Scholar]
- 37.Murray PJ, Allen JE, Biswas SK, et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity. 2014;41:14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mantovani A, Sica A, Locati M Macrophage Polarization Comes of Age. Immunity. 2005;23:344–346. [DOI] [PubMed] [Google Scholar]
- 39.Aras S, Zaidi MR. TAMeless traitors: macrophages in cancer progression and metastasis. Br J Cancer. 2017;117:1583–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leopold Wager CM, Wormley FL. Classical versus alternative macrophage activation: the Ying and the Yang in host defense against pulmonary fungal infections. Mucosal Immunol. 2014;7:1023–1035. [DOI] [PubMed] [Google Scholar]
- 41.Long KB, Beatty GL. Harnessing the antitumor potential of macrophages for cancer immunotherapy. OncoImmunology. 2013;2:e26860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11:889–896. [DOI] [PubMed] [Google Scholar]
- 43.Mantovani A, Marchesi F, Malesci A, et al. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. 2017;14:399–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tseng D, Volkmer J-P, Willingham SB, et al. Anti-CD47 antibody-mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. Proc Natl Acad Sci. 2013;110:11103–11108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Backer R, Schwandt T, Greuter M, et al. Effective collaboration between marginal metallophilic macrophages and CD8+ dendritic cells in the generation of cytotoxic T cells. Proc Natl Acad Sci. 2010;107:216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van Dinther D, Veninga H, Iborra S, et al. Functional CD169 on Macrophages Mediates Interaction with Dendritic Cells for CD8+ T Cell Cross-Priming. Cell Rep. 2018;22:1484–1495. [DOI] [PubMed] [Google Scholar]
- 47.Smith LK, Boukhaled GM, Condotta SA, et al. Interleukin-10 Directly Inhibits CD8+ T Cell Function by Enhancing N-Glycan Branching to Decrease Antigen Sensitivity. Immunity. 2018;48:299–312.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nagaraj S, Gupta K, Pisarev V, et al. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med. 2007;13:828–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kuangy D-M, Zhao Q, Peng C, et al. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J Exp Med. 2009;206:1327–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.DeNardo DG, Ruffell B. Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol. 2019;19:369–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Asano K, Kikuchi K, Tanaka M. CD169 macrophages regulate immune responses toward particulate materials in the circulating fluid. J Biochem (Tokyo). 2018;164:77–85. [DOI] [PubMed] [Google Scholar]
- 52.Moran I, Grootveld AK, Nguyen A, et al. Subcapsular Sinus Macrophages: The Seat of Innate and Adaptive Memory in Murine Lymph Nodes. Trends Immunol. 2019;40:35–48. [DOI] [PubMed] [Google Scholar]
- 53.Louie DAP, Liao S. Lymph Node Subcapsular Sinus Macrophages as the Frontline of Lymphatic Immune Defense. Front Immunol. 2019;10:347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Asano K, Nabeyama A, Miyake Y, et al. CD169-Positive Macrophages Dominate Antitumor Immunity by Crosspresenting Dead Cell-Associated Antigens. Immunity. 2011;34:85–95. [DOI] [PubMed] [Google Scholar]
- 55.McGaha TL, Chen Y, Ravishankar B, et al. Marginal zone macrophages suppress innate and adaptive immunity to apoptotic cells in the spleen. Blood. 2011;117:5403–5412. [DOI] [PubMed] [Google Scholar]
- 56.Qiu C-H, Miyake Y, Kaise H, et al. Novel Subset of CD8α + Dendritic Cells Localized in the Marginal Zone Is Responsible for Tolerance to Cell-Associated Antigens. J Immunol. 2009;182:4127–4136. [DOI] [PubMed] [Google Scholar]
- 57.Dahlin AM, Henriksson ML, Van Guelpen B, et al. Colorectal cancer prognosis depends on T-cell infiltration and molecular characteristics of the tumor. Mod Pathol. 2011;24:671–682. [DOI] [PubMed] [Google Scholar]
- 58.Hiraoka K, Miyamoto M, Cho Y, et al. Concurrent infiltration by CD8+ T cells and CD4+ T cells is a favourable prognostic factor in non-small-cell lung carcinoma. Br J Cancer. 2006;94:275–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Curiel TJ, Coukos G, Zou L, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–949. [DOI] [PubMed] [Google Scholar]
- 60.Gajewski TF, Schreiber H, Fu Y-X. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14:1014–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hervas-Stubbs S, Perez-Gracia JL, Rouzaut A, et al. Direct Effects of Type I Interferons on Cells of the Immune System. Clin Cancer Res. 2011;17:2619–2627. [DOI] [PubMed] [Google Scholar]
- 62.Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480:480–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tu J-F, Ding Y-H, Ying X-H, et al. Regulatory T cells, especially ICOS+ FOXP3+ regulatory T cells, are increased in the hepatocellular carcinoma microenvironment and predict reduced survival. Sci Rep. 2016;6:35056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kumar V, Patel S, Tcyganov E, et al. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016;37:208–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nagarsheth N, Wicha MS, Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol. 2017;17:559–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dangaj D, Bruand M, Grimm AJ, et al. Cooperation between Constitutive and Inducible Chemokines Enables T Cell Engraftment and Immune Attack in Solid Tumors. Cancer Cell. 2019;35:885–900.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Spranger S, Luke JJ, Bao R, et al. Density of immunogenic antigens does not explain the presence or absence of the T-cell–inflamed tumor microenvironment in melanoma. Proc Natl Acad Sci. 2016;113:E7759–E7768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nywening TM, Belt BA, Cullinan DR, et al. Targeting both tumour-associated CXCR2 + neutrophils and CCR2 + macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut. 2018;67:1112–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Binnewies M, Roberts EW, Kersten K, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24:541–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tsujikawa T, Kumar S, Borkar RN, et al. Quantitative Multiplex Immunohistochemistry Reveals Myeloid-Inflamed Tumor-Immune Complexity Associated with Poor Prognosis. Cell Rep. 2017;19:203–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.DeNardo DG, Brennan DJ, Rexhepaj E, et al. Leukocyte Complexity Predicts Breast Cancer Survival and Functionally Regulates Response to Chemotherapy. Cancer Discov. 2011;1:54–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Beatty GL, Winograd R, Evans RA, et al. Exclusion of T Cells From Pancreatic Carcinomas in Mice Is Regulated by Ly6Clow F4/80+ Extratumoral Macrophages. Gastroenterology. 2015;149:201–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science. 2015;348:74–80. [DOI] [PubMed] [Google Scholar]
- 74.Molon B, Ugel S, Del Pozzo F, et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J Exp Med. 2011;208:1949–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mariathasan S, Turley SJ, Nickles D, et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554:544–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tauriello DVF, Palomo-Ponce S, Stork D, et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. 2018;554:538–543. [DOI] [PubMed] [Google Scholar]
- 77.Rooney MS, Shukla SA, Wu CJ, et al. Molecular and Genetic Properties of Tumors Associated with Local Immune Cytolytic Activity. Cell. 2015;160:48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Leone P, Shin E-C, Perosa F, et al. MHC Class I Antigen Processing and Presenting Machinery: Organization, Function, and Defects in Tumor Cells. JNCI J Natl Cancer Inst. 2013;105:1172–1187. [DOI] [PubMed] [Google Scholar]
- 79.O’Donnell JS, Teng MWL, Smyth MJ. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol. 2019;16:151–167. [DOI] [PubMed] [Google Scholar]
- 80.Kloor M, Becker C, Benner A, et al. Immunoselective pressure and human leukocyte antigen class I antigen machinery defects in microsatellite unstable colorectal cancers. Cancer Res. 2005;65:6418–6424. [DOI] [PubMed] [Google Scholar]
- 81.Mehta AM, Jordanova ES, Kenter GG, et al. Association of antigen processing machinery and HLA class I defects with clinicopathological outcome in cervical carcinoma. Cancer Immunol Immunother. 2007;57:197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Torres MJ, Ruiz-Cabello F, Skoudy A, et al. Loss of an HLA haplotype in pancreas cancer tissue and its corresponding tumor derived cell line. Tissue Antigens. 1996;47:372–381. [DOI] [PubMed] [Google Scholar]
- 83.McGranahan N, Rosenthal R, Hiley CT, et al. Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution. Cell. 2017;171:1259–1271.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.The TRACERx consortium, Rosenthal R, Cadieux EL, et al. Neoantigen-directed immune escape in lung cancer evolution. Nature. 2019;567:479–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature. 2015;523:231–235. [DOI] [PubMed] [Google Scholar]
- 86.Spranger S, Gajewski TF. A new paradigm for tumor immune escape: β-catenin-driven immune exclusion. J Immunother Cancer. 2015;3:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Villablanca EJ, Raccosta L, Zhou D, et al. Tumor-mediated liver X receptor-α activation inhibits CC chemokine receptor-7 expression on dendritic cells and dampens antitumor responses. Nat Med. 2010;16:98–105. [DOI] [PubMed] [Google Scholar]
- 88.Oyama T, Ran S, Ishida T, et al. Vascular endothelial growth factor affects dendritic cell maturation through the inhibition of nuclear factor-kappa B activation in hemopoietic progenitor cells. J Immunol Baltim Md 1950. 1998;160:1224–1232. [PubMed] [Google Scholar]
- 89.Dong H, Strome SE, Salomao DR, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat Med. 2002;8:793–800. [DOI] [PubMed] [Google Scholar]
- 90.Chen G, Huang AC, Zhang W, et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature. 2018;560:382–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Munn DH, Mellor AL. IDO in the Tumor Microenvironment: Inflammation, Counter-Regulation, and Tolerance. Trends Immunol. 2016;37:193–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Uyttenhove C, Pilotte L, Théate I, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. 2003;9:1269–1274. [DOI] [PubMed] [Google Scholar]
- 93.Yang L, Pang Y, Moses HL. TGF-β and immune cells: an important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010;31:220–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Munn DH, Mellor AL. The tumor-draining lymph node as an immune-privileged site. Immunol Rev. 2006;213:146–158. [DOI] [PubMed] [Google Scholar]
- 95.Mellor AL, Munn DH. Creating immune privilege: active local suppression that benefits friends, but protects foes. Nat Rev Immunol. 2008;8:74–80. [DOI] [PubMed] [Google Scholar]
- 96.Vesely MD, Kershaw MH, Schreiber RD, et al. Natural Innate and Adaptive Immunity to Cancer. Annu Rev Immunol. 2011;29:235–271. [DOI] [PubMed] [Google Scholar]
- 97.Li J, Byrne KT, Yan F, et al. Tumor Cell-Intrinsic Factors Underlie Heterogeneity of Immune Cell Infiltration and Response to Immunotherapy. Immunity. 2018;49:178–193.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bayne LJ, Beatty GL, Jhala N, et al. Tumor-Derived Granulocyte-Macrophage Colony-Stimulating Factor Regulates Myeloid Inflammation and T Cell Immunity in Pancreatic Cancer. Cancer Cell. 2012;21:822–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mollinedo F Neutrophil Degranulation, Plasticity, and Cancer Metastasis. Trends Immunol. 2019;40:228–242. [DOI] [PubMed] [Google Scholar]
- 100.Geiger R, Rieckmann JC, Wolf T, et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell. 2016;167:829–842.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Munder M, Schneider H, Luckner C, et al. Suppression of T-cell functions by human granulocyte arginase. Blood. 2006;108:1627–1634. [DOI] [PubMed] [Google Scholar]
- 102.Tu S, Bhagat G, Cui G, et al. Overexpression of Interleukin-1β Induces Gastric Inflammation and Cancer and Mobilizes Myeloid-Derived Suppressor Cells in Mice. Cancer Cell. 2008;14:408–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Das S, Shapiro B, Vucic EA, et al. Tumor Cell-Derived IL-1β Promotes Desmoplasia and Immune Suppression in Pancreatic Cancer. Cancer Res. 2020;canres.2080.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Di Virgilio F, Dal Ben D, Sarti AC, et al. The P2X7 Receptor in Infection and Inflammation. Immunity. 2017;47:15–31. [DOI] [PubMed] [Google Scholar]
- 105.Idzko M, Ferrari D, Eltzschig HK. Nucleotide signalling during inflammation. Nature. 2014;509:310–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chen Y, Yao Y, Sumi Y, et al. Purinergic Signaling: A Fundamental Mechanism in Neutrophil Activation. Sci Signal. 2010;3:ra45–ra45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chen Y, Corriden R, Inoue Y, et al. ATP Release Guides Neutrophil Chemotaxis via P2Y2 and A3 Receptors. Science. 2006;314:1792–1795. [DOI] [PubMed] [Google Scholar]
- 108.Cauwels A, Rogge E, Vandendriessche B, et al. Extracellular ATP drives systemic inflammation, tissue damage and mortality. Cell Death Dis. 2014;5:e1102–e1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Vijayan D, Young A, Teng MWL, et al. Targeting immunosuppressive adenosine in cancer. Nat Rev Cancer. 2017;17:709–724. [DOI] [PubMed] [Google Scholar]
- 110.Cekic C, Day Y-J, Sag D, et al. Myeloid Expression of Adenosine A 2A Receptor Suppresses T and NK Cell Responses in the Solid Tumor Microenvironment. Cancer Res. 2014;74:7250–7259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mantovani A, Sozzani S, Locati M, et al. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23:549–555. [DOI] [PubMed] [Google Scholar]
- 112.Ribatti D, Nico B, Crivellato E, et al. Macrophages and tumor angiogenesis. Leukemia. 2007;21:2085–2089. [DOI] [PubMed] [Google Scholar]
- 113.Ruffell B, Chang-Strachan D, Chan V, et al. Macrophage IL-10 Blocks CD8+ T Cell-Dependent Responses to Chemotherapy by Suppressing IL-12 Expression in Intratumoral Dendritic Cells. Cancer Cell. 2014;26:623–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. 2004;4:71–78. [DOI] [PubMed] [Google Scholar]
- 115.Harmey JH, Dimitriadis E, Kay E, et al. Regulation of macrophage production of vascular endothelial growth factor (VEGF) by hypoxia and transforming growth factor β−1. Ann Surg Oncol. 1998;5:271–278. [DOI] [PubMed] [Google Scholar]
- 116.Jeon S-H, Chae B-C, Kim H-A, et al. Mechanisms underlying TGF-β1-induced expression of VEGF and Flk-1 in mouse macrophages and their implications for angiogenesis. J Leukoc Biol. 2007;81:557–566. [DOI] [PubMed] [Google Scholar]
- 117.Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Balachandran VP, Łuksza M, Zhao JN, et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature. 2017;551:512–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Beatty GL, Eghbali S, Kim R. Deploying Immunotherapy in Pancreatic Cancer: Defining Mechanisms of Response and Resistance. Am Soc Clin Oncol Educ Book. 2017;267–278. [DOI] [PubMed] [Google Scholar]
- 120.Stromnes IM, Hulbert A, Pierce RH, et al. T-cell Localization, Activation, and Clonal Expansion in Human Pancreatic Ductal Adenocarcinoma. Cancer Immunol Res. 2017;5:978–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Carstens JL, Correa de Sampaio P, Yang D, et al. Spatial computation of intratumoral T cells correlates with survival of patients with pancreatic cancer. Nat Commun. 2017;8:15095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Beatty GL, Chiorean EG, Fishman MP, et al. CD40 Agonists Alter Tumor Stroma and Show Efficacy Against Pancreatic Carcinoma in Mice and Humans. Science. 2011;331:1612–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lutz ER, Wu AA, Bigelow E, et al. Immunotherapy Converts Nonimmunogenic Pancreatic Tumors into Immunogenic Foci of Immune Regulation. Cancer Immunol Res. 2014;2:616–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Xu J, Sai H, Li Y, et al. Peripheral Blood T-Cell Fitness Is Diminished in Patients With Pancreatic Carcinoma but Can Be Improved With Homeostatic Cytokines. Cell Mol Gastroenterol Hepatol. 2019;8:656–658.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.O’Reilly EM, Oh D-Y, Dhani N, et al. Durvalumab With or Without Tremelimumab for Patients With Metastatic Pancreatic Ductal Adenocarcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019;5:1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Royal RE, Levy C, Turner K, et al. Phase 2 Trial of Single Agent Ipilimumab (Anti-CTLA-4) for Locally Advanced or Metastatic Pancreatic Adenocarcinoma: J Immunother. 2010;33:828–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Jackson CM, Choi J, Lim M. Mechanisms of immunotherapy resistance: lessons from glioblastoma. Nat Immunol. 2019;20:1100–1109. [DOI] [PubMed] [Google Scholar]
- 128.Ribas A Tumor Immunotherapy Directed at PD-1. N Engl J Med. 2012;366:2517–2519. [DOI] [PubMed] [Google Scholar]
- 129.Olive KP. Fanning the Flames of Cancer Chemoresistance: Inflammation and Anticancer Therapy. J Oncol Pract. 2017;13:181–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Callahan MK, Kluger H, Postow MA, et al. Nivolumab Plus Ipilimumab in Patients With Advanced Melanoma: Updated Survival, Response, and Safety Data in a Phase I Dose-Escalation Study. J Clin Oncol. 2018;36:391–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Postow MA, Chesney J, Pavlick AC, et al. Nivolumab and Ipilimumab versus Ipilimumab in Untreated Melanoma. N Engl J Med. 2015;372:2006–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Engblom C, Pfirschke C, Pittet MJ. The role of myeloid cells in cancer therapies. Nat Rev Cancer. 2016;16:447–462. [DOI] [PubMed] [Google Scholar]
- 133.Peranzoni E, Lemoine J, Vimeux L, et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti–PD-1 treatment. Proc Natl Acad Sci. 2018;115:E4041–E4050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zhu Y, Knolhoff BL, Meyer MA, et al. CSF1/CSF1R Blockade Reprograms Tumor-Infiltrating Macrophages and Improves Response to T-cell Checkpoint Immunotherapy in Pancreatic Cancer Models. Cancer Res. 2014;74:5057–5069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Holmgaard RB, Zamarin D, Lesokhin A, et al. Targeting myeloid-derived suppressor cells with colony stimulating factor-1 receptor blockade can reverse immune resistance to immunotherapy in indoleamine 2,3-dioxygenase-expressing tumors. EBioMedicine. 2016;6:50–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Li X, Yao W, Yuan Y, et al. Targeting of tumour-infiltrating macrophages via CCL2/CCR2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut. 2017;66:157–167. [DOI] [PubMed] [Google Scholar]
- 137.Jablonska J, Wu C-F, Andzinski L, et al. CXCR2-mediated tumor-associated neutrophil recruitment is regulated by IFN-β: IFN-β regulated neutrophil recruitment. Int J Cancer. 2014;134:1346–1358. [DOI] [PubMed] [Google Scholar]
- 138.Steele CW, Karim SA, Leach JDG, et al. CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell. 2016;29:832–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Highfill SL, Cui Y, Giles AJ, et al. Disruption of CXCR2-Mediated MDSC Tumor Trafficking Enhances Anti-PD1 Efficacy. Sci Transl Med. 2014;6:237ra67–237ra67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Byrne KT, Vonderheide RH. CD40 Stimulation Obviates Innate Sensors and Drives T Cell Immunity in Cancer. Cell Rep. 2016;15:2719–2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Vonderheide RH. CD40 Agonist Antibodies in Cancer Immunotherapy. Annu Rev Med. 2020;71:annurev-med-062518–045435. [DOI] [PubMed] [Google Scholar]
- 142.Zippelius A, Schreiner J, Herzig P, et al. Induced PD-L1 Expression Mediates Acquired Resistance to Agonistic Anti-CD40 Treatment. Cancer Immunol Res. 2015;3:236–244. [DOI] [PubMed] [Google Scholar]
- 143.Winograd R, Byrne KT, Evans RA, et al. Induction of T-cell Immunity Overcomes Complete Resistance to PD-1 and CTLA-4 Blockade and Improves Survival in Pancreatic Carcinoma. Cancer Immunol Res. 2015;3:399–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Long KB, Gladney WL, Tooker GM, et al. IFN and CCL2 Cooperate to Redirect Tumor-Infiltrating Monocytes to Degrade Fibrosis and Enhance Chemotherapy Efficacy in Pancreatic Carcinoma. Cancer Discov. 2016;6:400–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Mogensen TH. Pathogen Recognition and Inflammatory Signaling in Innate Immune Defenses. Clin Microbiol Rev. 2009;22:240–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Vidyarthi A, Khan N, Agnihotri T, et al. TLR-3 Stimulation Skews M2 Macrophages to M1 Through IFN-αβ Signaling and Restricts Tumor Progression. Front Immunol. 2018;9:1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Feng Y, Mu R, Wang Z, et al. A toll-like receptor agonist mimicking microbial signal to generate tumor-suppressive macrophages. Nat Commun. 2019;10:2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ribas A, Medina T, Kummar S, et al. SD-101 in Combination with Pembrolizumab in Advanced Melanoma: Results of a Phase Ib, Multicenter Study. Cancer Discov. 2018;8:1250–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Panni RZ, Herndon JM, Zuo C, et al. Agonism of CD11b reprograms innate immunity to sensitize pancreatic cancer to immunotherapies. Sci Transl Med. 2019;11:eaau9240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Pylayeva-Gupta Y, Lee KE, Hajdu CH, et al. Oncogenic Kras-Induced GM-CSF Production Promotes the Development of Pancreatic Neoplasia. Cancer Cell. 2012;21:836–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Markosyan N, Li J, Sun YH, et al. Tumor cell–intrinsic EPHA2 suppresses antitumor immunity by regulating PTGS2 (COX-2). J Clin Invest. 2019;129:3594–3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Golan T, Hammel P, Reni M, et al. Maintenance Olaparib for Germline BRCA -Mutated Metastatic Pancreatic Cancer. N Engl J Med. 2019;381:317–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Bonapace L, Coissieux M-M, Wyckoff J, et al. Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature. 2014;515:130–133. [DOI] [PubMed] [Google Scholar]
- 154.Balachandran VP, Beatty GL, Dougan SK. Broadening the Impact of Immunotherapy to Pancreatic Cancer: Challenges and Opportunities. Gastroenterology. 2019;156:2056–2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Koyama S, Akbay EA, Li YY, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. 2016;7:10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Ribas A, Shin DS, Zaretsky J, et al. PD-1 Blockade Expands Intratumoral Memory T Cells. Cancer Immunol Res. 2016;4:194–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Anandappa AJ, Wu CJ, Ott PA. Directing Traffic: How to Effectively Drive T Cells into Tumors. Cancer Discov. 2020;10:185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Stone ML, Graham K, Aldridge D, et al. Abstract 115: A CD40 agonist potentiates the efficacy and immune-stimulatory capacity of chemotherapy in combination with a focal adhesion kinase inhibitor in a mouse model of pancreatic ductal adenocarcinoma. In: Tumor Biology. American Association for Cancer Research:115–115. [Google Scholar]
- 159.Liu M, O’Connor RS, Trefely S, et al. Metabolic rewiring of macrophages by CpG potentiates clearance of cancer cells and overcomes tumor-expressed CD47−mediated ‘don’t-eat-me’ signal. Nat Immunol. 2019;20:265–275. [DOI] [PMC free article] [PubMed] [Google Scholar]