

Abstract
Previously, we identified an arylstibonic acid, NSC13778 that specifically binds to the basic region of the C/EBPα BZIP domain and disrupts DNA binding. We now examine a panel of 14 additional arylstibonic acid derivatives of NSC13778 for their ability to inhibit the DNA binding of five B-ZIP dimers (c-Fos|JunD, VBP, C/EBPα, C/EBPβ, and CREB). They show various specificities at inhibiting the DNA binding of five B-ZIP domains. NSC13746 inhibits the DNA binding of C/EBPβ and CREB at 100 nM and promiscuously inhibiting the DNA binding of all five proteins in the 1 μM range. Dialysis experiments indicate that NSC 13746 binding to the B-ZIP domain is reversible. Thermal denaturation studies indicate that NSC13746 binds the B-ZIP domain. Some compounds specifically inhibit DNA binding, with VBP and c-Fos|JunD being most easily disrupted. These compounds inhibit, with similar specificities to the pure B-ZIP domains, the DNA binding of nuclear extract to the AP1 DNA sequence but no inhibition is observed to SP1 containing oligonucleotide. Transient transfection assays indicate that NSC13746 can inhibit the TPA induced activation of two B-ZIP dependent reporters. These experiments suggest that arylstibonic acids are promising leads for inhibiting the DNA binding of a group of B-ZIP proteins in cells.
Keywords: Arylstibonic acids, DNA binding, B-ZIP domain, leucine zipper, antimony, small-molecule inhibitors
1. Introduction
The B-ZIP domain dimerizes and binds to DNA in a sequence specific manner to regulate gene transcription. These transcription factors occur in approximately 55 genes in the human genome that have been grouped into 13 families with similar dimerization properties [1, 2]. Many of these B-ZIP domain containing genes are immediate early genes (IEGs) that are induced in a variety of pathologies including cancer [3]. For example, c-Jun, a member of the AP1 family of B-ZIP transcription factors, was the first nuclear transcription factor described as an oncogene [4, 5]. However, no inhibitors of the DNA binding of these proteins are in clinical trials, and only one compound has been described that can bind to the B-ZIP domain to inhibit DNA binding [6].
Targeting the DNA binding of the B-ZIP motif is a formidable task because of a) the absence of any enzymatic pocket that may be targeted specifically and b) the presence of a large and shallow interface (1000–3000 Å) that gets buried when B-ZIP proteins form homo- or heterodimers [7–9]. Potentially, binding of a B-ZIP protein to DNA can be disrupted in two ways: 1) targeting molecules that bind to the major or minor grooves of DNA causing changes in its topology and steric hindrances such that the transcription factor cannot bind to DNA [10, 11]; or 2) targeting the B-ZIP domain itself. The B-ZIP class of transcription factors binds to the major groove of DNA as homo- and/or heterodimers via their basic DNA binding domain. The dimerization domains of the B-HLH-ZIP domains, which are structurally similar to the B-ZIP domain, have been successfully targeted. A synthetic peptidomimetic that inhibits the protein-protein interaction between Myc-Max heterodimer has been described [12]. But due to poor cell permeability and high susceptibility to degradation, these peptides are poor pharmacological agents.
The potential clinical value of inhibiting the DNA binding of specific B-ZIP protein families has been demonstrated using transgenic mice [13, 14]. Previously, we designed potent dominant negatives for various B-ZIP and B-HLH-ZIP transcription factor families that dimerize with their wild type protein partners with high affinity and inhibit DNA binding [15–18]. These dominant negative proteins have been expressed in transgenic mice and shown to inhibit and/or regress papillomas in skin. For example, expression in the mouse epidermis of A-C/EBP, a dominant negative that inhibits the DNA binding of all C/EBP family members, prevents papilloma formation and can cause established papillomas to regress if expressed after papilloma formation [13]. Expression in the mouse epidermis of A-Fos, a dominant negative that inhibits AP1 DNA binding, converts papillomas into benign sebaceous adenomas that are not able to convert into carcinomas [14]. Additionally, inhibiting CREB by expressing A-CREB is able to prevent papilloma formation [19]. Together, these results suggest that inhibition of DNA binding activity of B-ZIP transcription factors offers a promising molecular target to develop an effective anticancer therapy.
In a previous study, we used a high-throughput fluorescence anisotropy screen and three secondary screens to identify small-molecule inhibitors that disrupt the DNA binding of the B-ZIP domains [6]. From a diverse set of 1,990 molecules, an arylstibonic acid, NSC13778, containing an antimony element, was identified that preferentially binds the C/EBPα basic region and inhibits DNA binding. Here we report the effect of 14 arylstibonic acid derivatives of the lead compound NSC13778 on the DNA binding activity of five B-ZIP dimers: c-Fos|JunD (AP1), VBP, C/EBPα, C/EBPβ, and CREB. In the present study, two in vitro and an in vivo assay were used to characterize these compounds. Electrophoretic Mobility Shift Assay (EMSA) was used to screen 14 derivatives of NSC13778 for their ability to inhibit B-ZIP-DNA binding in a dose-dependent manner. The two water-soluble compounds that were positive by EMSA were further characterized using isothermal circular dichroism (CD) spectroscopy and CD thermal denaturation studies. NSC13746 was also evaluated in vivo by a transient transfections luciferase reporter assay and shown to inhibit B-ZIP mediated transcriptional activity.
2. Materials and Methods
2.1. Compounds
NSC13778 and 14 arylstibonic acid derivatives were obtained from the Drug synthesis and Chemistry Branch, Developmental Therapeutics Program, National Cancer Institute, Bethesda, MD.
2.2. Protein expression and purification.
Amino acid sequences of B-ZIP domain proteins used in this study are given elsewhere: c-Fos [20], JunD [6], VBP [17], C/EBPα and C/EBPβ [15], CREB and its dominant negative A-CREB [16]. All recombinant proteins were expressed in E. coli BL21(LysE) strain and purified as described previously [18]. Final protein purification step involved HPLC using Vydac C14 reverse phase column. A linear gradient from 0–100% acetonitrile containing 0.01% trifluoroacetic acid over 45 minutes with a flow rate of 1ml/min was used to elute the proteins. All proteins have a 13 amino acid N-terminal ϕ10 epitope that was used for immuno detection of proteins in Western Blots.
2.3. Electrophoretic Mobility Shift Assay (EMSA)
28 mer oligonucleotides were purchased from Sigma-genosys and were HPLC purified. Single strand oligonucleotide was end labeled with [γ32P]ATP using T4 phage polynucleotide kinase. Labeled oligo was purified using a G-50 column (GE Healthcare, UK) according to manufacturer instructions and annealed to complementary unlabeled oligo resulting in radiolabeled double stranded DNA. Sequences of oligonucleotides used for EMSA experiments were:
AP1: 5’-GTCAGTCAGAATGACTCATATCGGTCAG-3’
VBP: 5’-GTCAGTCAGATTACGTAATATCGGTCAG-3’
C/EBP: 5’GTCAGTCAGATTGCGCAATATCGGTCAG-3’
CREB: 5’-GTCAGTCAGATGACGTCATATCGGTCAG-3’
Underlined nucleotides are the binding sequences for B-ZIP proteins. Protein-DNA interaction in presence and absence of a small-molecule was studied using EMSA. Six HPLC purified proteins, four homodimers (VBP, C/EBPα, C/EBPβ, CREB) and one heterodimer (c-Fos|JunD form AP1) were used in band shift experiments. Before EMSA protein stock solutions (10 mM) in CD buffer were heated to 50 °C for five minutes and cooled to room temperature. The heterodimer between c-Fos and JunD domains was generated by heating equimolar concentrations of c-Fos and JunD proteins at 65 °C in presence of 1 mM DTT for 15 min and cooling the mixture to room temperature. For EMSA, samples were prepared by mixing 10 nM dimer protein with the required concentration of small-molecule (0.1, 1.0 and 10 μM) in 15 μl gel shift reaction buffer (25 mM Tris; pH 8.0, 50 mM KCl, 0.5 mM EDTA, 2.5 mM DTT, 1% bovine serum albumin and 10% glycerol) and incubated for 5 min at 37 °C. 7 pM 32P-radiolabeled double stranded oligonuleotide was added and final volume was made up to 10 μl with gel shift reaction buffer and equilibrated at 37 °C for 15 min. Finally, the mixture was incubated for additional 5 min at room temperature and DNA|protein complexes were resolved on a 7.5% native PAGE using 25 mM Tris-Borate running buffer (pH 8.0) with 0.3 mM EDTA at 150 V potential (11 mA current) for 90 minutes. Gels were dried and autoradiographed after 18 hours exposure using a Kodak MR X-ray film. The shifted bands represent DNA|protein complexes.
2.4. Dialysis experiments
2 μM CREB B-ZIP dimer protein in absence and presence of 100 μM NSC13746 was dialyzed against CD buffer for 24 hours at 4 °C using a Slide-a-Layer dialysis tubing (Pierce) with a molecular weight cut off of 8,000. Dialyzed samples without heating and after heating to 50 °C for 5 min were mixed with radioactive probe and ran on 7.5% native PAGE as described above. Gel was dried and exposed to X-Ray film for 5 hours and developed subsequently.
2.5. Nuclear extracts
Nuclear extracts were made from mouse liver [21] with some modifications. 10–15 grams of mouse liver was homogenized in 30 ml of homogenization buffer (HB) (10 mM HEPES (pH 7.6), 25 mM KCl, 0.15 mM spermine, 0.5 mM spermidine, 1 mM EDTA, 10 % glycerol, and 2 M sucrose) using a Teflon-glass homogenizer. 15 strokes of homogenizer were enough to get 90 % of cells lysed. 50 ml of HB buffer was added to make the final volume to 85 ml. 28 ml of homogenate was overlaid on 10 ml HB in three centrifuge tubes and spun at 24000 rpm for 30 min at −2 °C. Individual pellets containing nuclei from three tubes were pooled and re-suspended in 50 ml of 9:1 v/v HB and glycerol, divided into three parts and layered on 10 ml of HB buffer in three tubes and centrifugation step was repeated. Pellets were pooled again and suspended in nuclei lysis buffer (10 mM Hepes (pH 7.9), 100 mM KCl, 3 mM MgCl2, 0.1 mM EDTA, 1 mM DTT, 0.1 mM PMSF, and 10 % glycerol). Nuclei lysis was enhanced by 10 strokes in a glass Dounce homogenizer kept on ice and incubated for 40 min. Final centrifugation was performed for 30 min at 35,000 rpm and 4 °C. The pellet was discarded and supernatant was aliquoted and stored at −70 °C. Protein concentration of nuclear extract was measured by Bradford method (Bio-Rad, CA). For EMSA 5 μg of nuclear extracts were incubated with or without compounds (37 °C for 15 min) in gel shift buffer that contained 10 mM Hepes (pH 8.0), 6 % glycerol, 80 mM KCl, 0.05 mM EDTA, 1 mM MgCl2 and 1 mM DTT. In addition, 1 μg of polydI-dC was added to each reaction mixture to inhibit non-specific binding. 70 pM of radiolabeled DNA was added, mixture was incubated at room temperature and DNA|protein complexes were resolved using a 6 % native PAGE under the same condition as described for pure proteins. Gels were dried and autoradiographed. The following sequence was used for SP1 gel shifts: 5’-GTCAGTCAGGGGGCGGGGCATCGGTCAG-3’. Underlined nucleotides are the consensus binding site for SP1.
2.6. Circular Dichroism Spectroscopy.
Water-soluble NSC13746 and NSC13782 were used in circular dichroism (CD) studies. Samples for CD were prepared in phosphate buffer (12.5 mM phosphate buffer, pH 7.4, 150 mM KCl, 0.25 mM EDTA, 1 mM DTT). 2 μM dimer of each protein with required concentrations of compounds were incubated for two hours at 37 °C. CD spectra were recorded at 6 °C on JASCO J-720 spectropolarimeter in 5.0 mm quartz cuvette using a scanning rate of 100 nm/min at the spectral bandwidth of 2.0 nm. Each spectrum was an average of 10 individual spectra. Thermal denaturation studies of the B-ZIP domains in the presence and absence of compounds were carried out by heating the samples from 6–85/90 °C with a heating rate of 1 °C/min and ellipticity (θ) changes were recorded at 222 nm. The raw CD data were normalized to mean residue ellipticity (deg cm2/dmol) using the following relation
| (1) |
where θ is the observed ellipticity in millidegrees, C is the mean residue molar concentration of the protein, and l is the pathlength in centimeters.
Each thermal denaturation curve was analyzed as described earlier [22]. All data points ([θ]222, T) were fitted to the following equation
| (2) |
where T is temperature in °C, N and D are the values of θ222 of monomer and dimer, respectively, at temperature 0 °C, INTR is the temperature at which linear temperature-dependencies of dimer and monomer intercepts, Tm is the midpoint of thermal denaturation, ΔHm is the enthalpy change at Tm and R is the Gas constant. Values of Tm, ΔHm with predetermined ΔCp (heat capacity change on dimer formation) value of −1.2 kcal mol−1 dimer−1 K−1 [17] were used to obtain ΔGDi (Gibbs free energy change on dimer formation) according to the Gibbs-Helmholtz equation.
Binding constant for B-ZIP protein-small molecule interaction at Tm is calculated according to Waldron and Murphy (2003) using the following equation
| (3) |
where To and Tm are the midpoints of thermal denaturations of B-ZIP protein in absence and presence of small-molecule, respectively. ΔHm and ΔCp are the protein’s unfolding energetic parameters in absence of a small-molecule. R is the Gas constant and L is the free small-molecule concentration at Tm. (1)
2.7. Transient transfections
30,000 HepG2 cells were plated in each well in 24 well plates. Cells were co-transfected with Luc pGL3 basic vector (0.4 μg/well) with multiple binding sites for the AP1 and CREB B-ZIP proteins (AP1(7X), CRE(3X), and IL-2(5X) and β-galactosidase expressing plasmids (0.1 μg/well) using lipofectamine reagent according to manufacturer’s instructions (Invitrogen USA). Cells were allowed to attach (day 0). After 17 hours, NSC13746 that was dissolved in water was added to the wells at the final concentration of 10 μM (day 2). Cells were incubated for 24 hours in 37 °C incubator with 5% CO2 flow (day 3). The next day, half of the cells with or without compound, were induced by adding TPA (50 ng/well). Cells were further incubated for 4 hours at 37 °C, 5% CO2 environment. Finally, cells from each well were lysed in 100 μl of lysis buffer and analyzed for luciferase and β-galactosidase activities (Galacto-Light Plus™ system, AB Applied Biosystem, MA).
3. Results
3.1. Arylstibonic acid compounds
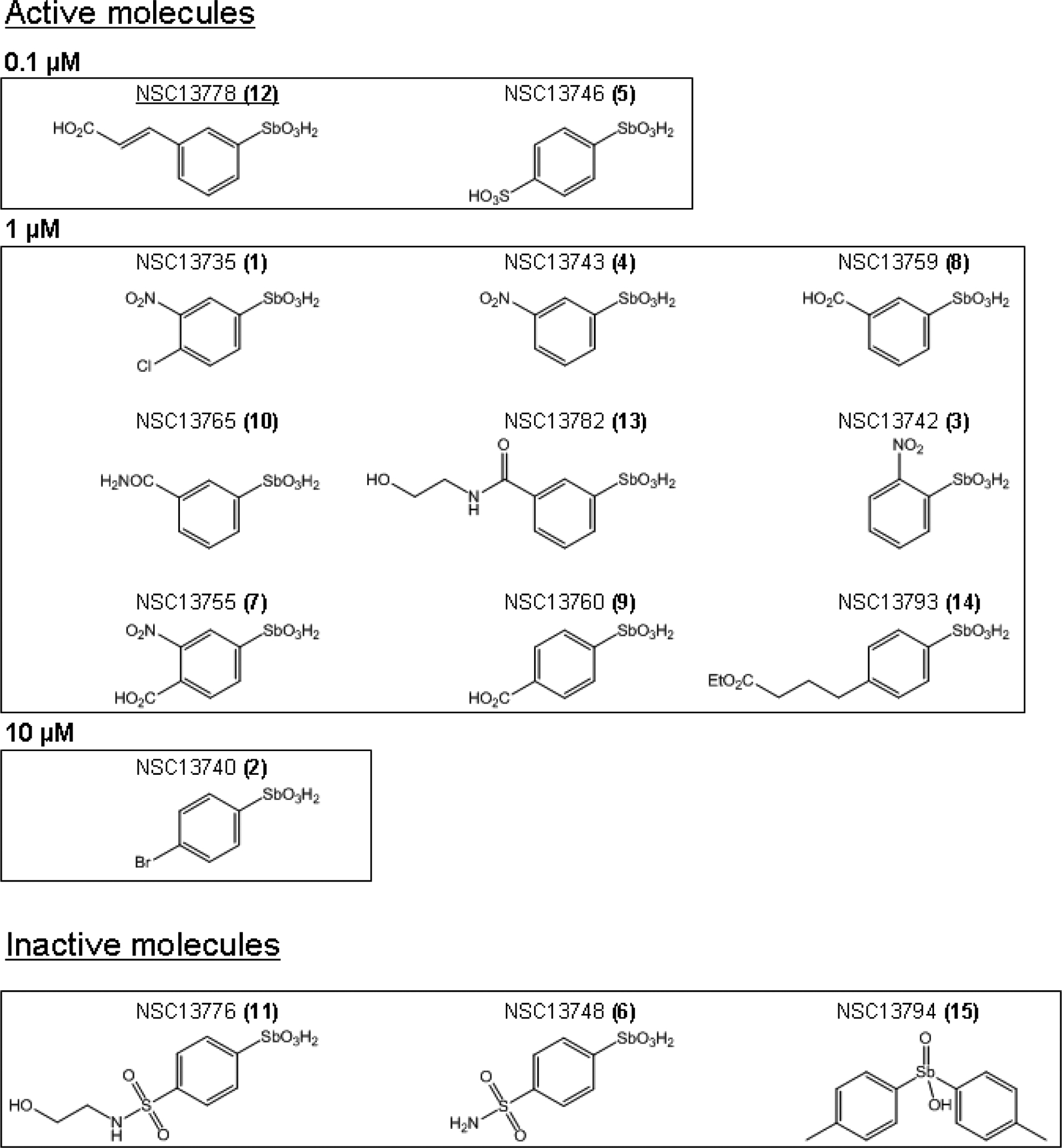
14 arylstibonic acid derivatives of NSC13778 with molecular weights ranging from 293 to 363 Daltons were examined for their ability to prevent the DNA binding of five B-ZIP dimers (AP1, VBP, C/EBPα, C/EBPβ, and CREB). The calculated logP values (log of the ratio of compound’s solubility in organic solvent to water) identified four compounds; NSC13746, NSC13748, NSC13776, and NSC13782 that could be dissolved in water and the 11 remaining compounds were dissolved in 100 % DMSO. Details of chemical and physical properties of the compounds can be found at http://129.43.27.140/ncidb2/. The chemical structures of the 12 active compounds and 3 inactive compounds are presented in Fig. 1.
Fig. 1.
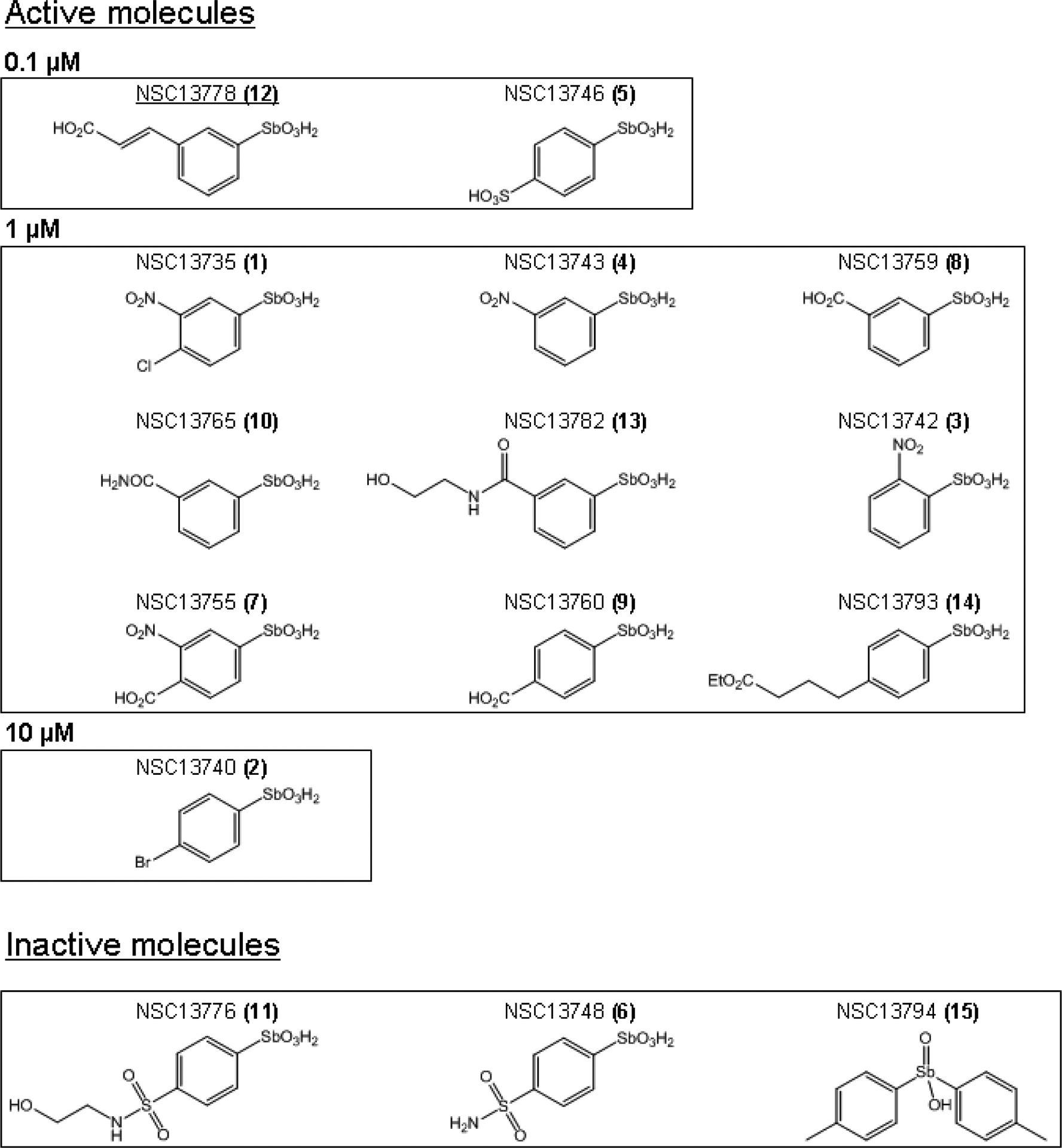
Structure of 15 arylstibonic acids used in this study. Structures of 12 compounds that inhibit B-ZIP DNA binding at 0.1–10 μM. NSC13778 identified in our previous study [6] is underlined. Structures of 3 inactive molecules are also included. The numbers (1–15) in the parenthesis after NSC numbers are used in subsequent Fig.s to identify compounds.
3.2. Interactions of 15 arylstibonic acids with five B-ZIP domains assayed by EMSA
Fig. 2 presents EMSA used to evaluate which of the 15 antimony compounds at three concentrations (0.1 μM, 1 μM and 10 μM) could inhibit the formation of five DNA|B-ZIP complexes (AP1, VBP, C/EBPα, C/EBPβ and CREB). All five B-ZIP proteins used here bind with KD of pM-nM (KD, the dissociation constant is the B-ZIP protein concentration at which bound and free probe are of equal intensities) [23]. Each sample contained 10 nM dimer of pure protein mixed with 7 pM of radioactive DNA in the absence and presence of compound. The absence or decrease in the intensity of the DNA|B-ZIP complex indicates that the compound is preventing formation of the DNA|B-ZIP complex and therefore is active.
Fig. 2.
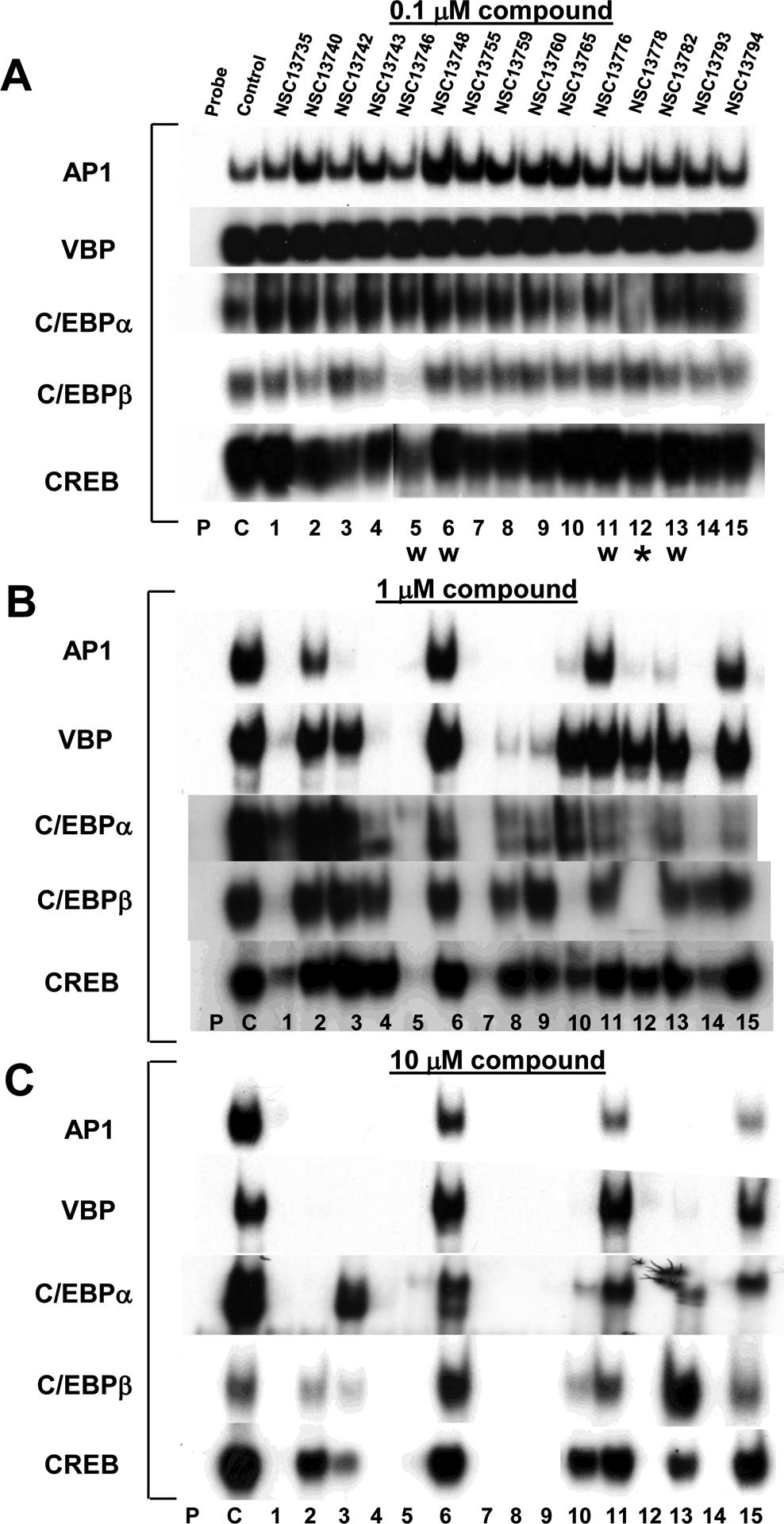
Inhibition of DNA binding of five B-ZIP transcription factors by 15 arylstibonic acids at increasing concentrations. A) EMSA showing the inhibition of DNA binding of AP1 (c-Fos|JunD heterodimer), VBP, C/EBPα, C/EBPβ and CREB by 15 arylstibonic acids at 0.1 μM. Samples contain 10 nM dimer of each protein, 0.1 μM compound, and 7 pM of 28 bp radiolabeled double stranded DNA containing unique binding site for each transcription factor. Probe (P) is radioactive DNA only, control (C) is DNA plus B-ZIP domain and 1–15 contain DNA, protein, and arylstibonic acids identified in Fig. 1. W indicate water-soluble compounds, all other molecules were dissolved in 100 % DMSO. B) Inhibition of B-ZIP DNA binding activity in 1 μM compound. C) Inhibition of B-ZIP DNA binding in 10 μM compound. * identifies NSC13778 described previously [6]. For ease of comparison, only the DNA|B-ZIP complex is shown.
At 0.1 μM, most arylstibonic acid derivatives were inactive (Fig. 2A). However, as demonstrated in our previous study, NSC13778 inhibited the DNA binding activity of C/EBPα [6] while NSC13746, a sulfonic acid derivative of NSC13778, disrupted the DNA binding of C/EBPβ and CREB. Increasing the concentration of the compounds to 1 μM generated a complex pattern of DNA binding inhibition (Fig. 2B). Two compounds, NSC13742 and NSC13782, showed the highest specificity as they completely disrupted the DNA binding of the AP1 B-ZIP transcription factor only. Four compounds (NSC13743, NSC13759, NSC13760, and NSC13793) displayed similar binding patterns being most active against AP1 and VBP, only weakly active against C/EBPα, and had no effect on the DNA binding of C/EBPβ and CREB. Compound NSC13765 was active for AP1 and C/EBPβ only. Consistent with previous results, NSC13778 disrupted the DNA |AP1 and DNA|C/EBPβ complex at 1 μM [6]. Only three compounds were inactive against all B-ZIP proteins (NSC13748, NSC13776, NSC13794), while two compounds disrupted the DNA binding of all the B-ZIP transcription factors (NSC13746 and NSC13755).
Raising compound concentrations to 10 μM produced more inhibition of DNA binding, again with a complex pattern. Eight compounds inhibited the DNA binding of all B-ZIPs (NSC13735, NSC13743, NSC13746, NSC13755, NSC13759, NSC13760, NSC13778, NSC13793), while the remaining compounds showed limited activity mainly against AP1 and VBP. Any specific inhibition displayed by the compounds at 1μM was lost at 10 μM.
Antimony is known to interact with nitrogen and thiol containing ligands raising the possibility that the interaction of the arylstibonic acids and the B-ZIP domains may be non-specific [24]. EMSA assay in presence of glutathione was used to find if these antimony compounds interact non-specifically with protein’s functional groups. CREB binding to a consensus CRE DNA sequence in the presence of 10 μM compounds is not affected by the presence of 1 or 5 mM glutathione suggesting that these antimony containing molecules are binding specifically to CREB protein (Supplementary Fig. 1).
3.3. Activity of arylstibonic acids in nuclear extracts
Fig. 3 examines the effect of the 15 arylstibonic acids on the DNA binding of nuclear extracts to oligonucleotides containing consensus AP1 and SP1 binding sites. Fig. 3A presents competition assays that demonstrate the DNA binding specificity of the nuclear extracts. We use two labeled (SP1 and AP1) and three unlabeled double stranded oligonucleotides (SP1, AP1 and C/EBP, see Materials and Methods) mixed with mouse liver nuclear extract in EMSA to examine the binding specificity of the nuclear extracts. Lanes 1–4 represent binding reactions using labeled SP1 probe. SP1 binding activity shows two distinct bands (lanes 1, 3–4) as has been reported earlier [25]. The competition assay using 100 fold excess of unlabeled SP1 oligonucleotide effectively inhibit the SP1 binding activity (lane 2) whereas binding remains same when similar amount of unlabeled AP1 or C/EBP oligonucleotides were used (lane 3–4) suggesting the bound activity is specific for SP1. Lanes 5–8 show nuclear extract binding activity when labeled AP1 probe was used. 100 fold excess of only unlabeled AP1 oligonucleotide effectively competes out the binding (lane 7). In contrast, 100 fold excess of unlabeled SP1 or C/EBP oligonuleotides (lanes 6 and 8) do not affect the binding.
Fig. 3.
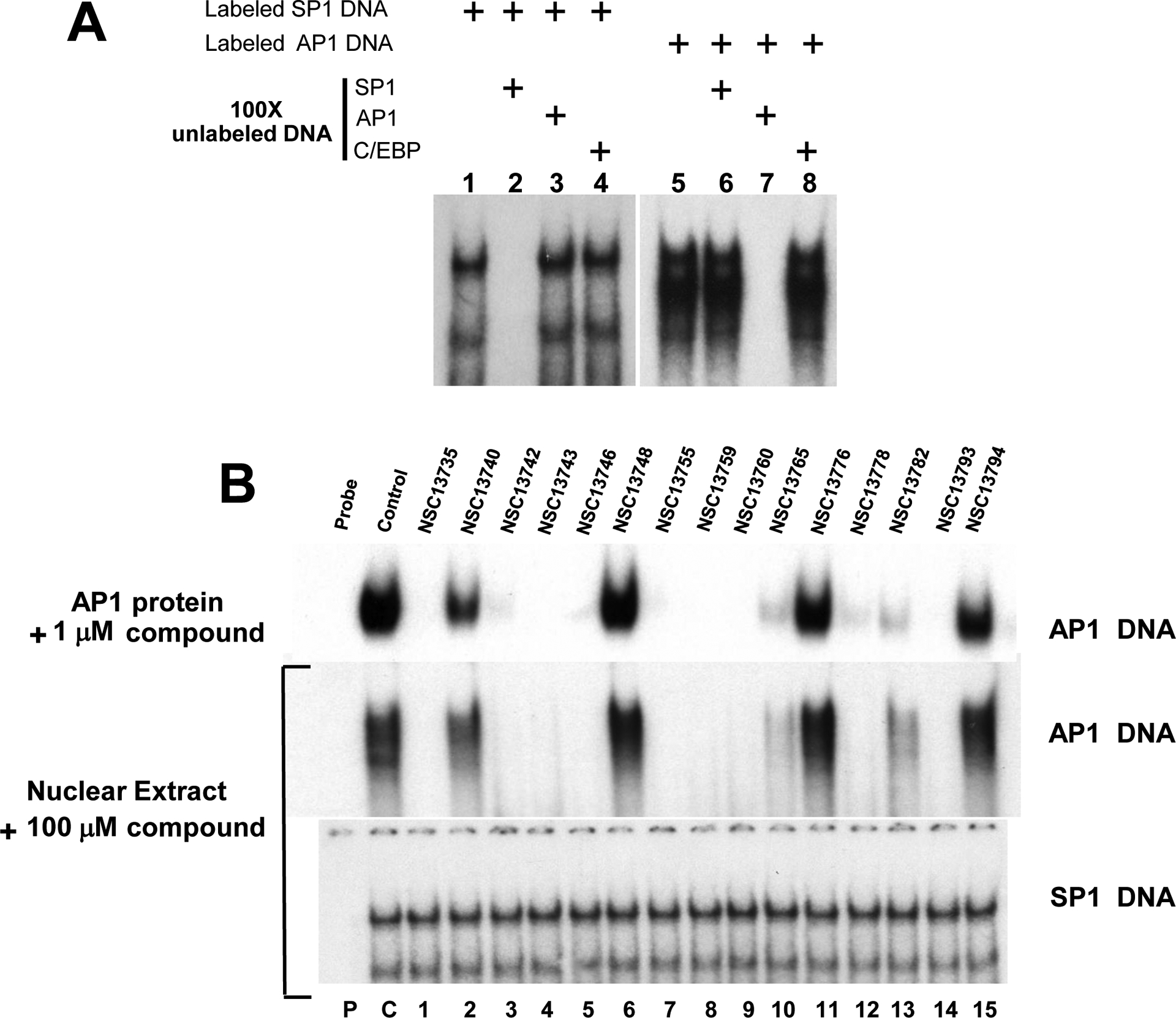
EMSA using nuclear extracts and CD studies show the inhibiting activities and specificity of compound(s). A) Competition assay using AP1 and SP1 probe. Lanes 1–4 and 5–8 represent binding results using mouse liver nuclear extracts mixed with AP1 and SP1 probe respectively. SP1 binding is totally abrogated by 100 fold excess of unlabeled SP1 probe only whereas excess amount of unlabeled AP1 and C/EBP probes have no effect. Similarly AP1 activity (lanes 5–8) can be inhibited only by excess unlabeled AP1 probe (lane 7). These results show that SP1 and AP1 proteins bind specifically to their corresponding probe in nuclear extracts. B) EMSA using mouse nuclear extracts and AP1 probe produces similar results as pure B-ZIP protein. Upper panel shows the DNA binding of pure c-Fos|JunD (AP1) B-ZIP domains to an AP1 containing oligonucleotide in the presence of 1 μM compounds (Fig. 2B). Bottom two panels show EMSA using mouse liver nuclear extracts showing the DNA binding activity to either a consensus AP1 or SP1 DNA binding site in the presence of antimony compounds. P, C and numbers 1–15 have the same meaning as in Fig. 2.
Fig. 3B initially presents EMSA binding patterns generated by incubating 1 μM compound with AP1 pure B-ZIP domains (c-Fos|JunD) (Fig. 2B). This pattern is reproduced when 100 μM compound is incubated with nuclear extracts. The similarity in compound activity suggests that these compounds retain their specificity of action in the complex nuclear extract environment. In contrast, the arylstibonic acids did not affect the ability of nuclear extracts to bind to an oligonucleotide containing a consensus SP1 binding site. SP1 belongs to the zinc finger family of transcription factors and does not share structural homology with B-ZIP transcription factors.
3.4. NSC13746 binds reversibly to the CREB B-ZIP protein domain
To determine whether the binding of the arylstibonic acids to the B-ZIP protein is non-covalent and reversible, the most active compound (NSC13746) was examined. CREB protein samples in the absence and presence of NSC13746 were extensively dialyzed against CD buffer for 24 hours at 4 °C. In the EMSA assay, 10 μM of NSC13746 inhibited the DNA binding of CREB B-ZIP protein domain but subsequent dialysis at 4 °C and heating the sample to 50 °C for 5 minutes restored DNA binding indicating that the compound’s interaction with the B-ZIP domain is non-covalent and reversible (Fig. 4, compare lane 2 to lanes 6 and 7). If the sample is not heated after dialysis, DNA binding is not fully restored (Fig. 4, lane 4 and 5), a phenomenon repeatedly observed with B-ZIP proteins that have been stored at 4 °C and it is independent of the presence of compound. We interpret this to suggest that the basic region forms a ‘metastable’ structure that is not competent to bind to DNA that can be reversed upon heating.
Fig. 4.
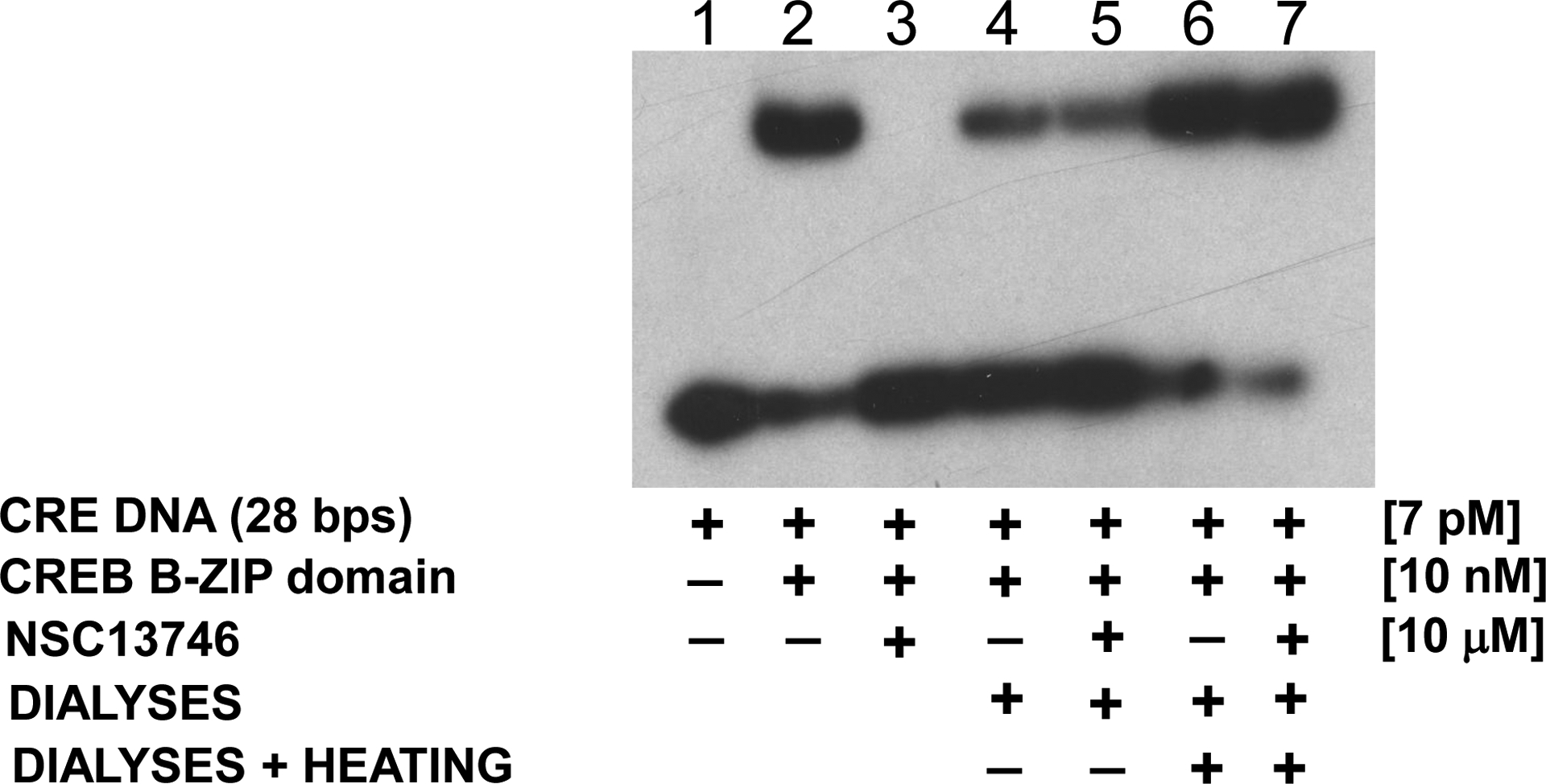
EMSA showing the non-covalent reversible interaction of NSC13746 with the CREB B-ZIP domain. Lanes 2–3 represent protein samples that were not dialyzed whereas samples in lanes 4–7 were dialysed against CD buffer for 24 hour at 4 °C. 1) Radiolabelded double stranded 28 base pair DNA probe containing the consensus CREB binding site. Lane 2) DNA plus 10 nM dimer of the CREB B-ZIP domain produces a DNA|protein complex. Lane 3) DNA, CREB B-ZIP domain, and 10 μM NSC13746. Lane 4) CREB protein sample without NSC13746 was dialyzed overnight against CD buffer (12.5 mM phosphate buffer, pH 7.4, 0.25 mM EDTA, 1 mM DTT with 150 mM KCl), mixed with radiolabeled probe and ran on the gel without heating. Lane 5) Sample containing CREB protein with NSC13746 was treated similarly to lane 4. Lane 6) Sample from lane 4 was heated to 50 °C for 5 minutes. Lane 7) Sample from lane 5 was heated to 50 °C for 5 minutes. DNA binding of CREB protein incubated with NSC13746 was completely recovered after dialyses and heat treatment (see text for details) indicating that NSC13746 interact non-covalently and reversibly with CREB protein.
3.5. Circular dichroism spectroscopy shows NSC13746 disrupts DNA|CREB complex and binds to B-ZIP protein domains
The interaction between NSC13746 and the CREB B-ZIP domain was further tested by both circular dichroism (CD) spectra and thermal denaturation spectroscopy. B-ZIP transcription factors have an N-terminal basic region responsible for sequence specific DNA binding and a four-heptad or longer C-terminal region termed the leucine zipper that dictates homo-and/or heterodimerization [1]. The basic region is unstructured in the absence of DNA, but becomes helical upon DNA binding. Fig. 5A shows four CD wavelength scans (200–240 nm) of 2 μM 28 bp double stranded CRE DNA, 2 μM CREB B-ZIP domain dimer, and their equimolar mixture, and the CREB|DNA mixture with NSC13746. The spectrum of 2 μM CREB B-ZIP homodimer shows well-defined minima at 208 nm and 222 nm characteristic of dimeric α-helices. In the presence of DNA, there is an increase in the negative ellipticity at 208 and 222 nms that we interpret as the unstructured basic region forming α-helices upon DNA binding [23]. When 100 μM NSC13746 was added to the DNA|CREB complex, incubated at 37 °C for 30 min and rescanned, the spectrum reverts to the CREB only spectrum. This suggests that NSC13746 disrupted the DNA|CREB complex, and subsequently caused the basic region to become un-helical.
Fig. 5.
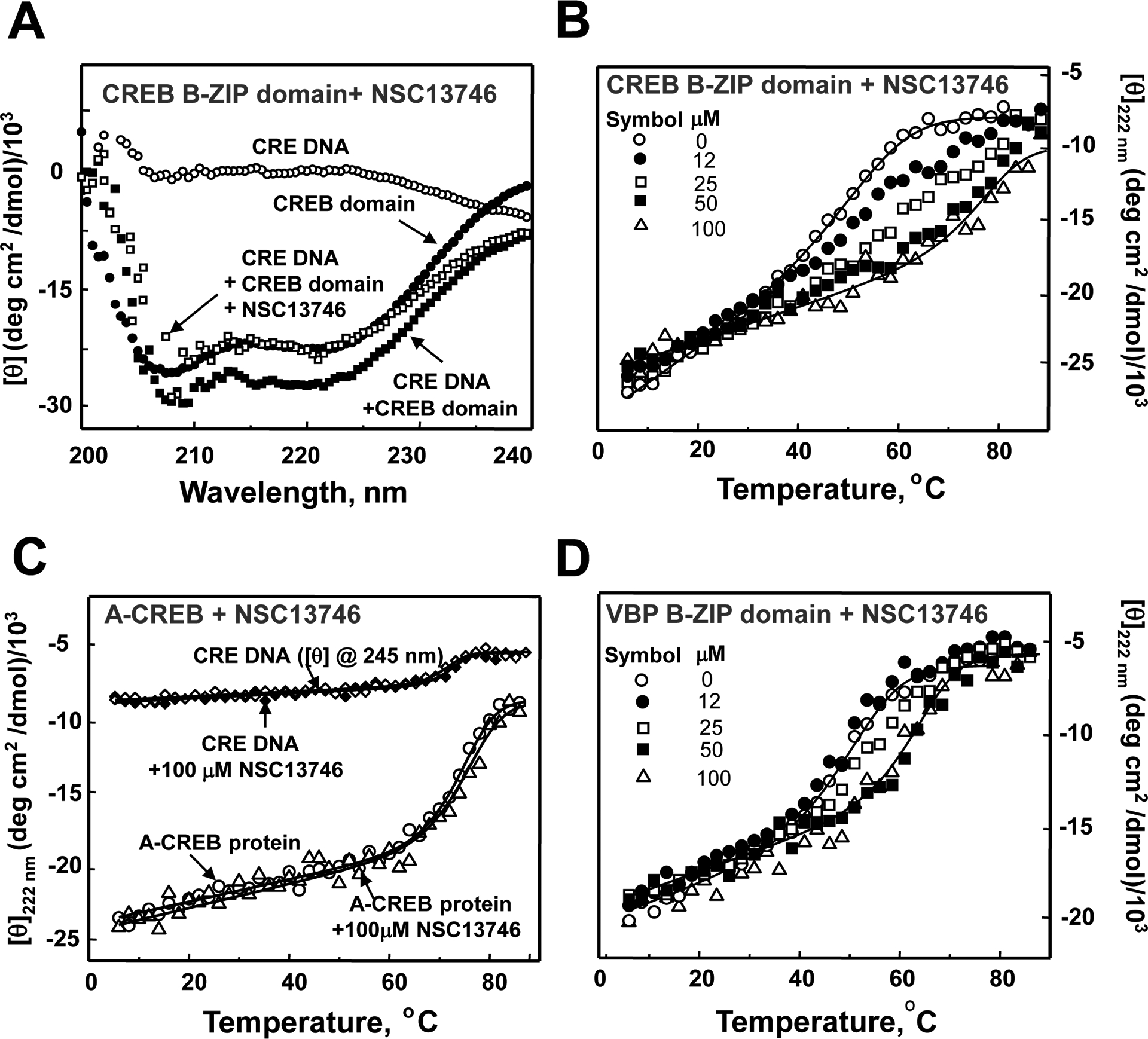
NSC13746 binds to the basic DNA binding domain of CREB. A) CD characterization of NSC13746 interaction with CREB B-ZIP domain. CD spectrum of 2 μM 28 bp double stranded DNA containing a consensus CRE sequence (○), 2 μM of the CREB B-ZIP domain dimer (●), mixture of 2 μM CRE DNA and 2 μM CREB B-ZIP domain dimer (■), mixture of 2 μM CRE DNA, 2 μM CREB B-ZIP domain dimer and 100 μM of NSC13746 heated to 50 °C and cooled to 6 °C (□). B) Thermal stability monitored by CD profiles at 222 nm of CREB B-ZIP domains (2 μM dimer) in the presence of four concentrations of NSC13746 absence (○) and presence of 12 μM (●), 25 μM (□), 50 μM (■), and 100 μM (Δ) of NSC13746. C) CD thermal denaturation studies at 245 nm of 2 μM 28 bp DNA containing unique CRE site and A-CREB protein (222 nm) in absence and presence of NSC13746. A-CREB is a dominant negative form of CREB in which basic DNA binding domain of CREB is replaced by a designed acidic extension such that its homodimer or heterodimer can not bind DNA. Thermal stability of CRE DNA (in absence (◊) and presence (♦)) and A-CREB protein (absence (○) and presence (Δ)) of NSC13746 remains unchanged. D) Thermal denaturation studies of VBP B-ZIP domain in presence of 0–100 μM NSC13746.
The disruption of CREB|DNA binding by NSC13746 could be caused by compound binding to either DNA or CREB. In order to determine if NSC13746 binds to CREB protein, CD thermal denaturation studies were carried out in absence and presence of compound. Fig. 5B shows thermal denaturations of CREB protein with increasing concentrations of NSC13746 (12–100 μM). If a small-molecule binds to the native state (dimer), the stability of the protein is increased as seen by an increase in melting temperature (Tm) [26, 27]. This property was exploited previously in quantifying the interaction between the lead compound NSC13778 and B-ZIP proteins [6]. With increasing concentrations of NSC13746, Tm of CREB increases. At a saturating concentration of 100 μM NSC13746 the denaturation curves are symmetric. These curves were analyzed for thermodynamic parameters and values are presented in Table 1.
TABLE 1.
Thermal stability parameters of B-ZIP domains with arylstibonic acids
| Protein | Compound conc., μM | Tma,°C | ΔHm, kcal mol−1 | ΔGDib, kcal mol−1
|
Kaapp(Tm)c |
|---|---|---|---|---|---|
| c-Fos|JunD | 0 | 42.5 | −45 ± 5 | −8.5 ± 0.1 | |
| (AP1) | 100 | 66.1 | −58 ± 6 | −12.6 ± 0.4 | 6.0 ×106 |
| VBP | 0 | 50.7 | −51 ± 6 | −9.8 ± 0.2 | |
| 100 | 63.5 | −59 ± 7 | −12.4 ± 0.3 | 3.3 ×105 | |
| C/EBPα | 0 | 45.8 | −56 ± 2 | −9.2 ± 0.1 | |
| 100 | 62.7 | −65 ± 7 | −12.6 ± 0.5 | 1.7 ×106 | |
| C/EBPβ | 0 | 64.4 | −71 ± 4 | −13.4 ± 0.1 | |
| 100 | 82.4 | −77 ± 6 | −17.5 ± 0.5 | 4.9 ×106 | |
| CREB | 0 | 49.3 | −53 ± 5 | −9.7 ± 0.1 | |
| 100 | 77.6 | −59 ± 5 | −14.5 ± 0.3 | 6.2 ×108 | |
| c-Fos|JunD | 0 | 42.5 | −45 ± 5 | −8.5 ± 0.1 | |
| (AP1) | 100 | 44.7 | −47 ± 4 | −8.7 ± 0.1 | 5.9 ×103 |
| VBP | 0 | 50.7 | −51 ± 6 | −9.8 ± 0.2 | |
| 100 | 56.2 | −54 ± 5 | −10.8 ± 0.2 | 2.6 ×104 | |
| C/EBPβ | 0 | 64.4 | −71 ± 4 | −13.4± 0.1 | |
| 100 | 65.2 | −69 ± 5 | −13.5 ± 0.5 | - |
The values represent mean of three independent experiments. Error in Tm values was ≤1°C.
The error in the ΔGDi value is due to error in ΔHm and ΔCp (1.2 ± 0.2 kcal mol−1) values.
Binding constants of compounds for each protein were calculated using equation 3.
Superscript ‘app’ represents the apparent values.
CD thermal denaturation of CRE DNA at 245 nm in the absence and presence of 100 μM compound shows no interaction with DNA (Fig. 6C). Next NSC13746 binding to a designed dominant negative of CREB i.e., A-CREB [16] was tested. A-CREB contains the CREB leucine zipper dimerization domain fused to a designed acidic extension that replaces the DNA-binding region. The Tm of the A-CREB protein does not change in presence of 100 μM NSC13746.
Fig. 6.
Thermal stability monitored by CD profiles at 222 nm showing binding of NSC13782 to AP1 and VBP but not C/EBPβ. A) Heat-induced denaturation of AP1 in absence (○) and presence of 100 μM NSC13782 (Δ). B) Thermal denaturation of VBP in absence (○) and presence of 100 μM NSC13782 (Δ). C) Thermal denaturation of C/EBPβ in absence(○) and presence (Δ) of 100 μM NSC13782.
The ability of NSC13746 to stabilize the VBP B-ZIP protein domain was also evaluated by CD thermal denaturation experiments (Fig. 5D). Tm of VBP B-ZIP protein domain increases in presence of NSC13746 compound suggesting that the compound is binding to the VBP protein dimer.
3.6. NSC13746 binds to five B-ZIP motifs with different affinities
The stability of the interaction between NSC13746 and different B-ZIP domains was evaluated using CD thermal denaturation monitored at 222 nm. EMSA showed that NSC13746 inhibited the DNA binding activity of C/EBPβ and CREB at 0.1 μM, and AP1, VBP and C/EBPα at 1 μM (Fig. 2A–B). To quantify the interaction of NSC13746 with B-ZIP proteins, thermal stability of heterodimeric AP1, and homodimeric C/EBPα, and C/EBPβ was also measured (Fig. 5B and D and Supplementary Fig. 2A–C). Thermal denaturation curves in the presence of 100 μM NSC13746 were fitted according to equation 2 and analyzed for thermodynamic parameters as described in the preceding paragraph. Values obtained from curve fitting are given in Table 1. Binding constants of NSC13746 for each of the five B-ZIP dimers were calculated according to equation 3 and are included in Table 1. Since the denaturation of B-ZIP proteins may not be two-state type, binding constants in presence of NSC13746 are given as ‘apparent’ values. NSC13746 binds each of the five B-ZIP proteins with different affinities. For example, the compound has a binding constant of 6.2×108 for CREB B-ZIP protein domain that is three magnitudes more than for VBP. This partly recapitulates the EMSA results where 100 nM NSC13746 inhibits the DNA binding of C/EBPβ and CREB protein.
CD studies to examine the thermal stability of B-ZIP domains mixed with different antimony compounds could not be performed for those that are only soluble in DMSO because of high interference due to DMSO in the 200–240 nm wavelength range. NSC13782, the only antimony compound besides NSC13746 that is water-soluble and shows some specificity of action in the EMSA, was used in CD thermal denaturation studies. Fig. 6A–B show that 100 μM compound increased the Tm of AP1 and VBP B-ZIP domain from 42.5 °C to 44.7 °C and 50.7 °C to 56.2 °C respectively, whereas C/EBPβ stability was not changed (Fig. 6C). This correlates to the EMSA data in which NSC13782 specifically inhibits the DNA bindings of AP1 and VBP B-ZIP dimer but not C/EBPβ (Fig. 2C). Stability parameters and binding constants of NSC13782 for AP1 and VBP are given in Table 1. Thus, CD thermal denaturation data demonstrate that the antimony containing compounds interact with B-ZIP domains with different specificities.
3.7. NSC13746 specifically inhibits in vivo transcription of TPA induced AP1 and CREB dependent reporters
NSC13746 that was active in EMSA, and CD was also used to test the inhibition of transiently transfected AP1 and CREB-dependent luciferase reporters in HepG2 cells (Fig. 7). Cells were transfected with reporter plasmids containing multiple binding sites for AP1 and CREB (see Materials and Methods) in the absence and presence of 10 μM NSC13746 compound. Plasmid expressing β-galactosidase under the control of CMV promoter was co-transfected and was used for data normalization. A control experiment was performed in absence of NSC13746 and its value was arbitrarily set as 1. HepG2 cells were induced by TPA treatment (50 ng/well) and transcriptional activities were measured in absence and presence of compound. Compound NSC13746 reduced the AP1 and CREB-mediated transcription. The inhibition was stronger for CREB, which is in agreement with EMSA and CD results. IL-2, which is not a B-ZIP transcription factor was not affected by NSC13746, demonstrating the specificity of NSC13746 in inhibiting the transcriptional activities mediated by B-ZIP proteins only.
Fig. 7.

A) Activation of transiently transfected luciferase reporters in the presence of NSC13746. HepG2 hepatoma cells were treated with 10 μM NSC13746 and transfected with luciferase reporter plasmids each containing multiple DNA binding sites in their promoters for AP1(7X), C/EBP (3X), CREB(4X), and IL-2(5X). Cells were co-transfected with β-galactosidase plasmid as an internal control. TPA induction of the TRE (AP1) and CRE reporter but not IL2 reporter are inhibited by 10 μM NSC13746.
4. Discussion
A hallmark of cancer is misregulated transcription due to overactive oncogenes and/or down regulation of tumor-suppressor genes [3]. The involvement of B-ZIP transcription factors, like AP1, in malignancies is well documented [28, 29]. Unfortunately, identifying small molecules that disrupt these non-enzymatic DNA-protein interactions has been difficult. Previously, an arylstibonic acid NSC13778 was identified that bound the basic region of a B-ZIP domain and prevented its binding to DNA [6]. Here we have extended that study to include 14 derivatives of NSC13778 to ascertain if any of these compounds binds to five B-ZIP protein domains. All 15 small molecule compounds used in this study share a common stibono-benzoic acid structure that contains an antimony element with a benzene ring (Fig. 1), and they follow Lipinski’s “Rule-of-Five” indicating that they are drug-like molecules [30].
Antimony containing compounds are used in various pharmacological formulations [31]. For example, the antimony containing compound, hydrated potassium antimonytartate is used as an anti-schistosomol drug, a cough suppressant, as well as an emetic [32]. Antimony has also been used in cancer therapy, for example, studies using lung cancer cell lines showed that the cytotoxic effect of antimony tartrate was comparable to other anti-cancer drugs like cisplatin and doxorubicin [33]. Likewise, sodium stibogluconate acts synergistically with interferon-α (INF-α) to eliminate INF-α resistant human cancer cells in a mouse model system [34]. NSC13778 and two of its antimony containing analogs (NSC13755 and NSC13759) were used to block the HIV-1 entry into cells by disrupting the viral gp120 and CD4 receptor interaction [35]. NSC13778 was also effective in inhibiting the poxvirus type I topoisomerase [36]. Stivers and coworkers [37] showed that NSC13778 and analogs inhibit the activity of Ape1 by binding to both the enzyme and enzyme-substrate complex and suggested that the arylstibonic acids mimic the phosphates of DNA backbone and can bind to multiple sites in the enzyme. Using human topoisomerase IB as molecular target Kim and colleagues, screened the library of arylstibonic acids and found a few analogs to be competitive inhibitors and others non essential activators of the enzyme [38]. Furthermore, NSC13778 is non-toxic at concentrations as high as 400 μM in tissue culture [35].
A structure–activity relationship (SAR) of these compounds is most revealing by the examination of the structures of the three compounds that have no activity in our assay. The arylstibonic acid is important for activity because replacing a hydroxyl group on the stibonic acid with a methyl phenyl group (NSC13794) destroyed activity (Fig. 1). Two compounds (NSC13776 and NSC13748) contain an amidation of the sulfonic acid in NSC13746 that renders them completely inactive (Fig. 1) showing that the negative charge on the molecule is important for its activity. These results suggest that interaction between NSC13746 and the B-ZIP proteins contain an electrostatic component.
A couple of structural features are common for the 12 active arylstibonic acids. They all contain an additional polar or charged group that are typically in the meta, para or both positions except NSC13742 where nitro group is at ortho position.
Recently, Stivers’ group has found that arystibonic acids inhibit endonuclease Ape1 [37] and topoisomerase IB [38] enzymatic activities. Our results with B-ZIP DNA binding show a different activity profile compared to the enzyme inhibitors identified by Stivers’ group. For example, NSC13755 is the most potent arylstibonic acid in both enzyme inhibition assays but has intermediate activity at inhibiting B-ZIP binding with no specificity for different B-ZIP domains. Additionally, NSC13742 that contains an ortho group is not active in enzyme assays but is active in our DNA inhibition assay.
The binding site of the arylstibonic acids on the B-ZIP domain can be studied using B-ZIP mutants. Previously, we observed that NSC13778 bound the C/EBPα B-ZIP domain. Mutants in the basic region near the N-terminus of the leucine zipper structure inhibited binding suggesting this is the site of binding. Additionally, mutants where the basic region was either removed or replaced also could not bind NSC13778 supporting the suggestion that binding was in the basic region just N-terminal of the leucine zipper [6]. The results with NSC13746 are similar; it can bind the CREB, C/EBPα, C/EBPβ, AP1 and VBP B-ZIP domains. Thermal stability of A-CREB protein, that lacks a basic DNA binding domain, instead has a rationally designed acidic peptide, remains unchanged in presence and absence of NSC13746 suggesting that the compound binds to the N-terminal of the coiled-coil. A structural feature common to all the proteins is the presence of basic DNA binding domain and N-terminal coiled coil structure containing many amines from the helical backbone that we speculate may interact with the arylstibonic acid. This suggestion helps explain the promiscuous nature of the arystibonic acids to all the B-ZIP domains examined. Structural studies will be needed to unambiguously determine the nature of the interaction between these arystibonic acids and the B-ZIP domain.
Small-molecules that inhibit the dimerization of the Myc|Max transcription factor have been described. These compounds destabilize transcription factor by disrupting protein-protein interactions critical for dimerization and subsequent DNA binding [12, 39]. In contrast, NSC13778 and its active analogs (NSC13746 and NSC13782) act differently. They bind to the B-ZIP domain and stabilize them as indicated by the increase in the melting temperature of the proteins (Fig. 5–6). The B-ZIP domain bound to the antimony compound is not able to bind the DNA. These arylstibonic acids show a range of specificities against different B-ZIP protein domains but presumably more complex derivatives will have more specificity. This promiscuous property, however, may actually be helpful because now a single drug can inhibit the function of several transcription factors. It is observed that compounds that promiscuously inhibit different kinases are more effective against certain cancers [40]. In fact, poly-pharmacology may be a more prevalent aspect of clinical medicine than previously appreciated [41, 42].
This study shows that the arylstibonic acid NSC13746 bind specifically to B-ZIP proteins at micro molar concentrations and can inhibit their DNA binding activity both in vitro and in vivo. The inhibition of the DNA binding of B-ZIP domains by these compounds suggests this molecular scaffold is a promising avenue to explore additional more diverse compounds.
Supplementary Material
Supplementary Fig. 1. EMSA showing the inhibition of DNA binding of CREB B-ZIP transcription factors by 10 μM each of the 15 arylstibonic acids in absence and presence of A) 0 (panel from Fig. 2C), B) 1 mM and C) 5 mM glutathione. Experimental conditions used for EMSA were same as in Fig. 2C except that the samples contained 1 mM (B) and 5 mM (C) of glutathione.
Supplementary Fig. 2. CD characterization of NSC13746 interaction with different B-ZIP domains. A-C) Thermal stability monitored by CD profiles at 222 nm of three B-ZIP domains (2 μM dimer) in the presence of four concentrations of NSC13746, absence (○) and presence of 12 μM (●), 25 μM (□), 50 μM (■), and 100 μM (Δ) of NSC13746. A) AP1, B) C/EBPα and C) C/EBPβ.
Acknowledgments
This research was supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. government.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Vinson C, Myakishev M, Acharya A, Mir AA, Moll JR and Bonovich M, Classification of human B-ZIP proteins based on dimerization properties, Mol Cell Biol 22 (2002) 6321–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Newman JR and Keating AE, Comprehensive identification of human bZIP interactions with coiled-coil arrays, Science 300 (2003) 2097–101. [DOI] [PubMed] [Google Scholar]
- [3].Darnell JE Jr., Transcription factors as targets for cancer therapy, Nat Rev Cancer 2 (2002) 740–9. [DOI] [PubMed] [Google Scholar]
- [4].Vogt PK, Bos TJ and Doolittle RF, Homology between the DNA-binding domain of the GCN4 regulatory protein of yeast and the carboxyl-terminal region of a protein coded for by the oncogene jun, Proc Natl Acad Sci U S A 84 (1987) 3316–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Vogt PK, Jun, the oncoprotein, Oncogene 20 (2001) 2365–77. [DOI] [PubMed] [Google Scholar]
- [6].Rishi V, Potter T, Laudeman J, Reinhart R, Silvers T, Selby M, Stevenson T, Krosky P, Stephen AG, Acharya A, Moll J, Oh WJ, Scudiero D, Shoemaker RH and Vinson C, A high-throughput fluorescence-anisotropy screen that identifies small molecule inhibitors of the DNA binding of B-ZIP transcription factors, Anal Biochem 340 (2005) 259–71. [DOI] [PubMed] [Google Scholar]
- [7].Lo Conte L, Chothia C and Janin J, The atomic structure of protein-protein recognition sites, J Mol Biol 285 (1999) 2177–98. [DOI] [PubMed] [Google Scholar]
- [8].Jones S and Thornton JM, Principles of protein-protein interactions, Proc Natl Acad Sci U S A 93 (1996) 13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hu Z, Ma B, Wolfson H and Nussinov R, Conservation of polar residues as hot spots at protein interfaces, Proteins 39 (2000) 331–42. [PubMed] [Google Scholar]
- [10].Olenyuk BZ, Zhang GJ, Klco JM, Nickols NG, Kaelin WG Jr. and Dervan PB, Inhibition of vascular endothelial growth factor with a sequence-specific hypoxia response element antagonist, Proc Natl Acad Sci U S A 101 (2004) 16768–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Dervan PB, Molecular recognition of DNA by small molecules, Bioorg Med Chem 9 (2001) 2215–35. [DOI] [PubMed] [Google Scholar]
- [12].Berg T, Cohen SB, Desharnais J, Sonderegger C, Maslyar DJ, Goldberg J, Boger DL and Vogt PK, Small-molecule antagonists of Myc/Max dimerization inhibit Myc-induced transformation of chicken embryo fibroblasts, Proc Natl Acad Sci U S A 99 (2002) 3830–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Oh WJ, Rishi V, Orosz A, Gerdes MJ and Vinson C, Inhibition of CCAAT/enhancer binding protein family DNA binding in mouse epidermis prevents and regresses papillomas, Cancer Res 67 (2007) 1867–76. [DOI] [PubMed] [Google Scholar]
- [14].Gerdes MJ, Myakishev M, Frost NA, Rishi V, Moitra J, Acharya A, Levy MR, Park SW, Glick A, Yuspa SH and Vinson C, Activator protein-1 activity regulates epithelial tumor cell identity, Cancer Res 66 (2006) 7578–88. [DOI] [PubMed] [Google Scholar]
- [15].Krylov D, Olive M and Vinson C, Extending dimerization interfaces: the bZIP basic region can form a coiled coil, Embo J 14 (1995) 5329–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ahn S, Olive M, Aggarwal S, Krylov D, Ginty DD and Vinson C, A dominant-negative inhibitor of CREB reveals that it is a general mediator of stimulus-dependent transcription of c-fos, Mol Cell Biol 18 (1998) 967–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Moll JR, Olive M and Vinson C, Attractive interhelical electrostatic interactions in the proline- and acidic-rich region (PAR) leucine zipper subfamily preclude heterodimerization with other basic leucine zipper subfamilies, J Biol Chem 275 (2000) 34826–32. [DOI] [PubMed] [Google Scholar]
- [18].Rishi V, Gal J, Krylov D, Fridriksson J, Boysen MS, Mandrup S and Vinson C, SREBP-1 dimerization specificity maps to both the helix-loop-helix and leucine zipper domains: use of a dominant negative, J Biol Chem 279 (2004) 11863–74. [DOI] [PubMed] [Google Scholar]
- [19].Rozenberg J, Rishi V, Orosz A, Moitra J, Glick A and Vinson C, Inhibition of CREB function in mouse epidermis reduces papilloma formation, Mol Cancer Res 7 (2009) 654–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Olive M, Krylov D, Echlin DR, Gardner K, Taparowsky E and Vinson C, A dominant negative to activation protein-1 (AP1) that abolishes DNA binding and inhibits oncogenesis, J Biol Chem 272 (1997) 18586–94. [DOI] [PubMed] [Google Scholar]
- [21].Gorski K, Carneiro M and Schibler U, Tissue-specific in vitro transcription from the mouse albumin promoter, Cell 47 (1986) 767–76. [DOI] [PubMed] [Google Scholar]
- [22].Krylov D, Mikhailenko I and Vinson C, A thermodynamic scale for leucine zipper stability and dimerization specificity: e and g interhelical interactions, Embo J 13 (1994) 2849–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Moll JR, Acharya A, Gal J, Mir AA and Vinson C, Magnesium is required for specific DNA binding of the CREB B-ZIP domain, Nucleic Acids Res 30 (2002) 1240–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Basinger MA and Jones MM, Structural requirements for chelate antidotal efficacy in acute antimony(III) intoxication, Res Commun Chem Pathol Pharmacol 32 (1981) 355–63. [PubMed] [Google Scholar]
- [25].Zhu WG, Srinivasan K, Dai Z, Duan W, Druhan LJ, Ding H, Yee L, Villalona-Calero MA, Plass C and Otterson GA, Methylation of adjacent CpG sites affects Sp1/Sp3 binding and activity in the p21(Cip1) promoter, Mol Cell Biol 23 (2003) 4056–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Brandts JF and Lin LN, Study of strong to ultratight protein interactions using differential scanning calorimetry, Biochemistry 29 (1990) 6927–40. [DOI] [PubMed] [Google Scholar]
- [27].Waldron TT and Murphy KP, Stabilization of proteins by ligand binding: application to drug screening and determination of unfolding energetics, Biochemistry 42 (2003) 5058–64. [DOI] [PubMed] [Google Scholar]
- [28].Weiss C and Bohmann D, Deregulated repression of c-Jun provides a potential link to its role in tumorigenesis, Cell Cycle 3 (2004) 111–3. [PubMed] [Google Scholar]
- [29].Milde-Langosch K, The Fos family of transcription factors and their role in tumourigenesis, Eur J Cancer 41 (2005) 2449–61. [DOI] [PubMed] [Google Scholar]
- [30].Lipinski CA, Lombardo F, Dominy BW and Feeney PJ, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings, Adv Drug Deliv Rev 46 (2001) 3–26. [DOI] [PubMed] [Google Scholar]
- [31].McCallum RI, Antimony in medicine, Rep Proc Scott Soc Hist Med 93-94 (1992) 1–16. [PubMed] [Google Scholar]
- [32].McCallum RI, President’s address. Observations upon antimony, Proc R Soc Med 70 (1977) 756–63. [PMC free article] [PubMed] [Google Scholar]
- [33].Duffin J and Campling BG, Therapy and disease concepts: the history (and future?) of antimony in cancer, J Hist Med Allied Sci 57 (2002) 61–78. [DOI] [PubMed] [Google Scholar]
- [34].Yi T, Pathak MK, Lindner DJ, Ketterer ME, Farver C and Borden EC, Anticancer activity of sodium stibogluconate in synergy with IFNs, J Immunol 169 (2002) 5978–85. [DOI] [PubMed] [Google Scholar]
- [35].Yang QE, Stephen AG, Adelsberger JW, Roberts PE, Zhu W, Currens MJ, Feng Y, Crise BJ, Gorelick RJ, Rein AR, Fisher RJ, Shoemaker RH and Sei S, Discovery of small-molecule human immunodeficiency virus type 1 entry inhibitors that target the gp120-binding domain of CD4, J Virol 79 (2005) 6122–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Bond A, Reichert Z and Stivers JT, Novel and specific inhibitors of a poxvirus type I topoisomerase, Mol Pharmacol 69 (2006) 547–57. [DOI] [PubMed] [Google Scholar]
- 37.] Seiple LA, Cardellina JH 2nd, Akee R and Stivers JT, Potent inhibition of human apurinic/apyrimidinic endonuclease 1 by arylstibonic acids, Mol Pharmacol 73 (2008) 669–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kim H, Cardellina JH 2nd, Akee R, Champoux JJ and Stivers JT, Arylstibonic acids: novel inhibitors and activators of human topoisomerase IB, Bioorg Chem 36 (2008) 190–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Xu Y, Shi J, Yamamoto N, Moss JA, Vogt PK and Janda KD, A credit-card library approach for disrupting protein-protein interactions, Bioorg Med Chem 14 (2006) 2660–73. [DOI] [PubMed] [Google Scholar]
- [40].Wilhelm S, Carter C, Lynch M, Lowinger T, Dumas J, Smith RA, Schwartz B, Simantov R and Kelley S, Discovery and development of sorafenib: a multikinase inhibitor for treating cancer, Nat Rev Drug Discov 5 (2006) 835–44. [DOI] [PubMed] [Google Scholar]
- [41].Overington JP, Al-Lazikani B and Hopkins AL, How many drug targets are there? Nat Rev Drug Discov 5 (2006) 993–6. [DOI] [PubMed] [Google Scholar]
- [42].Hopkins AL, Mason JS and Overington JP, Can we rationally design promiscuous drugs? Curr Opin Struct Biol 16 (2006) 127–36. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. 1. EMSA showing the inhibition of DNA binding of CREB B-ZIP transcription factors by 10 μM each of the 15 arylstibonic acids in absence and presence of A) 0 (panel from Fig. 2C), B) 1 mM and C) 5 mM glutathione. Experimental conditions used for EMSA were same as in Fig. 2C except that the samples contained 1 mM (B) and 5 mM (C) of glutathione.
Supplementary Fig. 2. CD characterization of NSC13746 interaction with different B-ZIP domains. A-C) Thermal stability monitored by CD profiles at 222 nm of three B-ZIP domains (2 μM dimer) in the presence of four concentrations of NSC13746, absence (○) and presence of 12 μM (●), 25 μM (□), 50 μM (■), and 100 μM (Δ) of NSC13746. A) AP1, B) C/EBPα and C) C/EBPβ.