

Abstract
Obesity is a significant health problem in westernized societies, particularly in the United States where it has reached epidemic proportions in both adults and children. The prevalence of childhood obesity has doubled in the past 30 years. The causation is complex with multiple sources, including an obesity promoting environment with plentiful highly dense food sources and overall decreased physical activity noted for much of the general population, but genetic factors clearly play a role. Advances in genetic technology using candidate gene approaches, genome-wide association studies, structural and expression microarrays, and next generation sequencing have led to the discovery of hundreds of genes recognized as contributing to obesity. Polygenic and monogenic causes of obesity are now recognized including dozens of examples of syndromic obesity with Prader–Willi syndrome, as a classical example and recognized as the most common known cause of life-threatening obesity. Genetic factors playing a role in the causation of obesity will be discussed along with the growing evidence of single genes and the continuum between monogenic and polygenic obesity. The clinical and genetic aspects of four classical but rare obesity-related syndromes (ie, Prader–Willi, Alström, fragile X, and Albright hereditary osteodystrophy) will be described and illustrated in this review of single gene and syndromic causes of obesity.
1. INTRODUCTION AND BACKGROUND
Obesity is an increasing serious health problem recognized worldwide and reaching epidemic status particularly in westernized societies. A role for genetic factors is now recognized as contributing to obesity and will be summarized in this review on single gene and syndromic causes of obesity. The United States currently leads as the most obese nation. The causation of obesity is clearly complex. The rising obesity prevalence is partially due to an obesity-promoting environment with highly dense, inexpensive, and plentiful food sources with a relatively sedentary lifestyle due to advances in modern technology leading to decreased energy expenditure for employees in the work place.1–3 A survival advantage over time has led to a thrifty phenotype due to a more efficient use of calories with fat deposition thereby contributing to the obesity epidemic in the presence of increased caloric intake.
Major health concerns related to long term obesity status include insulin resistance, type 2 diabetes, fatty liver, sleep apnea, cancer, hypertension, cardiovascular disease, stroke, and physical limitations with disabilities. Obesity, as a global public health problem, is on the rise as evidenced in 1997 at 19.4%; 24.5% in 2004, 33.8% in 2008, and 35.7% of adults in 2010.3 Childhood obesity is also present at 17% in the United States with 70% of obese adolescents becoming obese adults. The prevalence of childhood obesity has doubled in the past 30 years.4 The medical costs for treating complications from an overweight and obesity status are inordinate with estimates for treating people for overweight and obesity estimated at $72 billion and $198 billion, respectively impacted by the current obesogenic environment.3,5–10
Body mass index (BMI) is a commonly used measure of obesity in adults and children and defined as weight measured in kilograms divided by the square of height in meters (kg/m2), but this measure is not as accurate for determination of obesity in the pediatric age group. A BMI between 25 and 30 is considered overweight while BMIs greater than or equal to 30 are considered obese in adulthood. Since the 1980s, the mean BMI has increased by 0.4–0.5 kg/m2 per decade in adults. In childhood a BMI from the 85th to 95th percentile may be considered overweight and equal to or greater than the 95th percentile for obesity.3,11–13
Twin and family studies strongly implicate genetic factors as playing an important role in the development of obesity. Genetic predisposition for obesity can also be influenced by ethnicity. Monozygotic twin studies show concordance for obesity at a range from 70 to 90%, while in dizygotic twins range from 35 to 45%. Heritability estimates derived from twin studies report a representative number of 77% for BMI.14,15 Increased physical activity and exercise can lower the effects of genes impacting BMI and therefore obesity status. Hence, genes play an important central role in the determination of BMI and pathogenesis of obesity. In addition, strong genetic factors influence percentage body fat, waist circumference, energy expenditure, eating behavior, and level of physical activity. Specific gene mutations are found in 5–10% of obesity in childhood supported by a recent review of 370 recognized genes identified in the literature playing a role in obesity. 16 Several genes have been studied in relationship to obesity, particularly in mongenic causation; for example, MC4R, which is the single most commonly recognized gene causing childhood obesity and found in about 4% of cases.6,7 Structural chromosomal anomalies involving genomic regions containing causative genes for obesity have been reported such as 3p25 duplications and 16p11.2 deletions.17
With continually improved and varied complex genetic techniques requiring smaller quantities of DNA (and RNA) such as next generation sequencing further supported by prometaphase chromosome analysis, fluorescence in situ hybridization, linkage and genome-wide association studies (GWAS), copy number variant (CNV), and single nucleotide polymorphism (SNP) probes utilized in high resolution microarrays have been instrumental in identifying structural, chromosomal, and DNA abnormalities in genomic regions and candidate genes causing both rare and common forms of obesity. Specifically, chromosome abnormalities (deletions, duplications) have been recorded in humans with dysmorphic and syndromic obesity including chromosome 1p36 deletion; chromosome 2q37.3 deletion; chromosome 3p23 duplication; 3p25.3 duplication (contains the GHRL gene); chromosome 4q32.1 duplication; 4q35.1 duplication; chromosome 5p13.1 duplication; chromosome 6q16.2 deletion (SIM1 gene); 6q22.2 deletion; 6q24.3 duplication; 6q15-q21 deletion (SIM1 gene); 6q16-q21 deletion (SIM1 gene); 6q16.1-q16.3 deletion (SIM1 gene); chromosome 7q36 deletion; chromosome 9p23 deletion; 9q34 deletion; 9q34.3 deletion; 9q33.3q34 duplication; chromosome 10q22.3q23.2 duplication; chromosome 11p12-p14 deletion; 11p13-p14.2 deletion (BDNF gene); 11p11.2 deletion; chromosome 12p13.1 duplication; 12qter deletion; chromosome 14q32.2 hypomethylation status (maternal disomy 14); chromosome 16q13 duplication; 16p11.2 deletion (SH2B1 gene); 16q11.2-q13 duplication (FTO gene); chromosome 18q12.2-q21.1 deletion; chromosome 19q12-q13.2 duplication; 19q13.2 deletion; chromosome 20q13.13-q13.32 deletion; chromosome 22q11.2 deletion; chromosome Xq26.3-q27.3 deletion (FMR1 gene); Xq23q25 duplication; Xp11.3p21.1 duplication; Xp11.4q11.2 inversion; and Xq27.1-q28 deletion (FMR1 gene)17 (Table 1). Small deletions involving the chromosome 16p11.2 band have been reported in 0.5–0.7% of individuals with severe obesity and not found in nonobese healthy individuals. It includes the SH2B1 gene, an obesity-related gene that impacts leptin sensitivity and hence caloric intake.18 Chromosome microarray studies have shown rare CNVs which are greater than 2 Mb in size and found in 1.3% of individuals with obesity and evidence of disrupting multiple candidate genes for obesity (eg, POMC, UCP1, GHRL). Hence, the research of CNVs in the study of obesity has merits. Recently, Butler et al.16 summarized the literature and authoritative computer websites and found 370 clinically relevant and known genes reported for obesity and plotted the genes on chromosome ideograms to represent a visual display of gene distribution (Fig. 1A and B).
Table 1.
List of Chromosome Locations and Obesity Genes in Relationship to Obesity Phenotypes.
| Genetic Factors and Relationship to Obesity Phenotypes | Chromosome Location and Candidate Gene for Obesity |
|---|---|
| Obesity, early onset | 1p36.11 (NROB2); 2p23.3 (POMC); 18q21.32 (MC4R) |
| Obesity, severe | 3p25.2 (PPARG); 6q16.3 (SIM1); 11q13.4 (ULP3); 16p11.2 (SH2B1) |
| Obesity, generalized | 1p35.2 (SDC3); 3p25.3 (GHRL); 4q31.1 (UCP1); 5q13.2 (CART); 5q32 (ADRB2; PPARGC1B); 6q23.2 (ENPP1); 8p11.23 (ADRB3); 11p14.1 (BDNF); 13q14 (OLFM4); 14q32.2 (MEG-DMR); 16p11.2 (SH2B1); 16q12.2 (FTO); 16q22.1 (AGRP); 17q21 (HOXB5); 17q21.31 (PYY); 18q21.32 (MC4R) |
| Description | Gene of Interest |
| Genetic loci associated with child and/or adult BMI levels including extreme obesity identified by genome-wide studies (GWAS), linkage or with CNV/SNP microarrays | FTO; TMEM18; GNPDA2; INSIG2; MC4R; NEGR1; BDNF; KCTD15; PCSK1; CTNNBL1; MTCH2; NPC1; MAF; PTER; PRL; FAIM2; TFAP2B; SEC16B; ETV5; AIF1; GPRC5BB; MAP2K5; GIPR; FANCL; SDCCAG8; TNKS-MSRA; TNN13K; LRRN6C; NRXN3; FLJ35779; SLC39A8; TMEM160; CADM2; LRP1B; PRKD1; MTIF3; ZNF608; PTBP2; TUB; HMGA1; PPYR1 |
| Associated with childhood obesity—novel loci | SH2B1; EDIL3; S1PR5; FOXP2; TBCA; ABCB5; ZPLD1; KIF2B; ARL15; EPHA6-UNQ6114 |
| Associated with childhood and/or adult waist to hip ratio | LYPLAL1; C12orf51; LY86; HOXC13; RSPO3; VEGFA; TBX15; NFE2L3; GRB14; DNM3; PIGC; ITPR2; ADAMTS9; ZNRF3; NISCH; CPEB4 |
Figure 1.
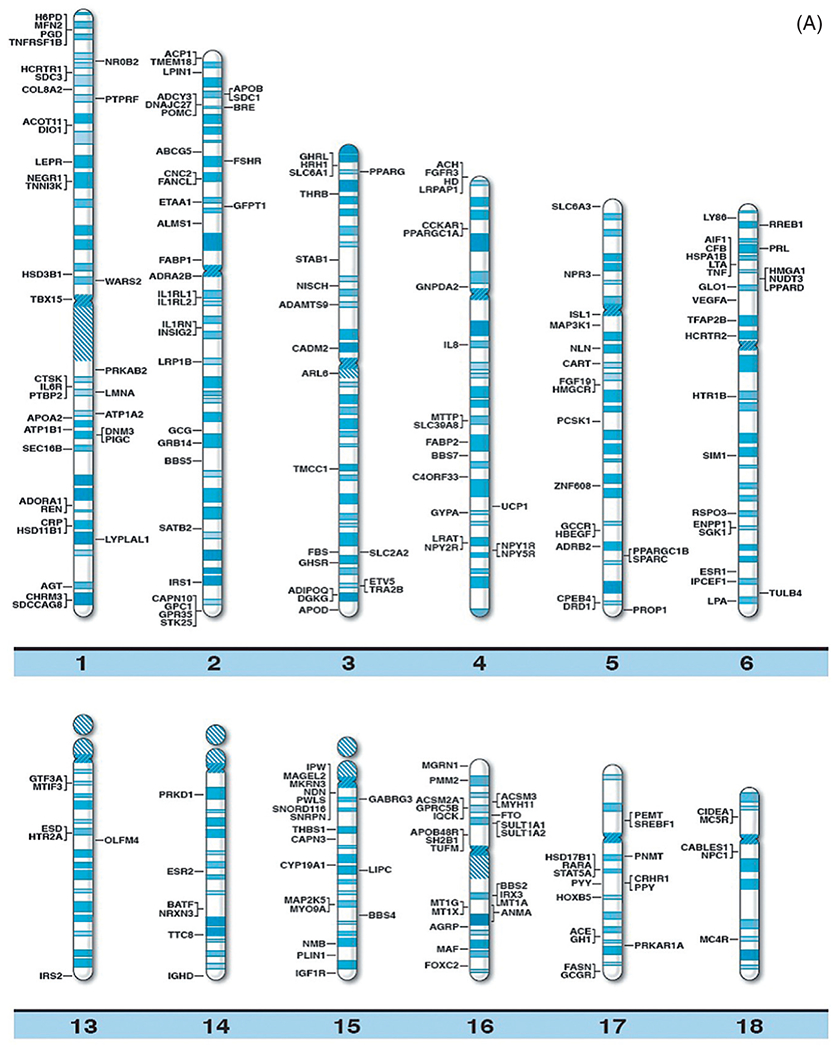
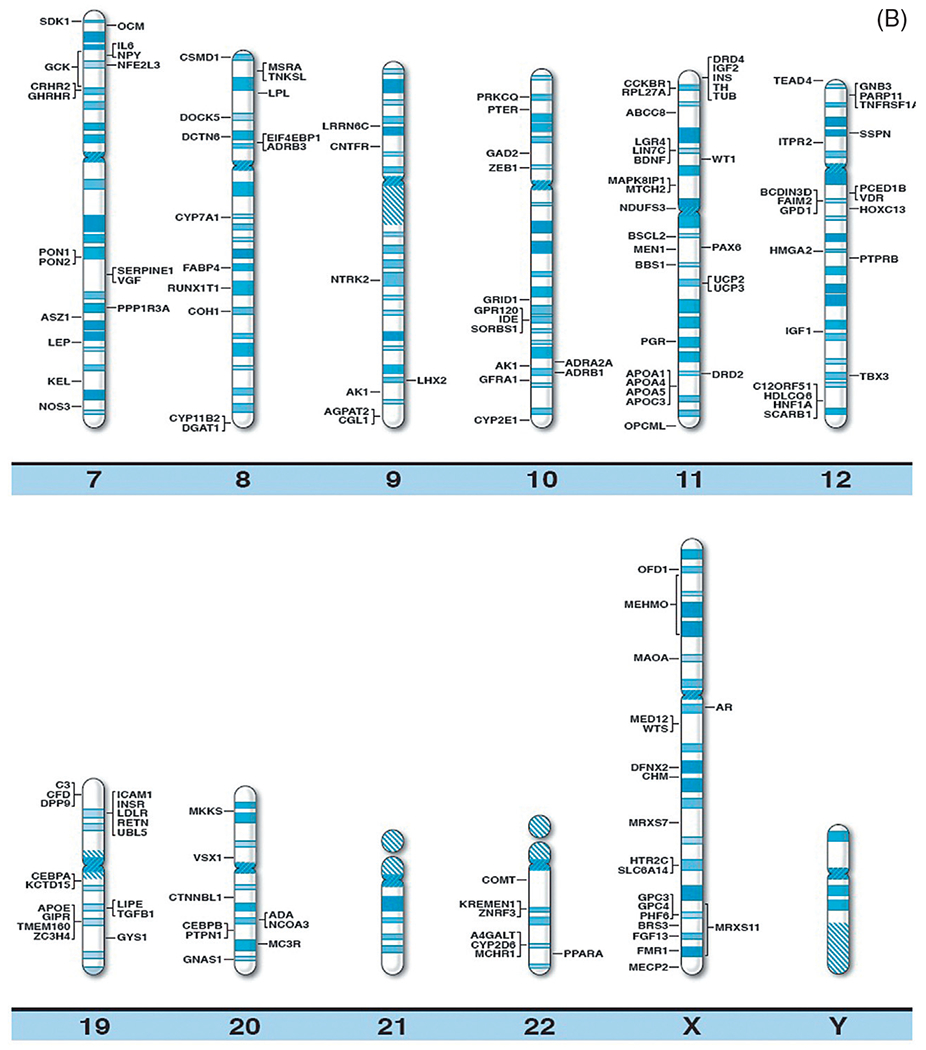
(A) Obesity gene ideogram part A. High resolution human chromosome ideograms (850 band level) with symbols representing recognized genes for obesity positioned at the chromosome band location. The upper “p” and lower “q” arms for each chromosome are separated by the centromere area.16 (B) Obesity gene ideogram part B. High resolution human chromosome ideograms (850 band level) with symbols representing recognized genes for obesity positioned at the chromosome band location. The upper “p” and lower “q” arms for each chromosome are separated by the centromere area.16
2. GENETIC VARIANT AND GENOME-WIDE ASSOCIATION STUDIES IN OBESITY
Identification of gene loci or markers for disease states became available in late 2005 and is used to discover a number of loci for obesity. The first study of its type to use DNA markers in childhood obesity was published in 2010 and two loci were found (ie, SDCCAG8 and TNKS/MSRA).19 Now larger, more advanced genetic loci studies have utilized 14 existing GWAS data sets to identify additional loci for obesity.3 The GWAS method is considered high-throughput with the advantage to assay millions of DNA markers as SNPs that span the entire human genome. When combined with powerful statistical approaches, the use of SNPs will identify additional loci for specific conditions including obesity.
Common variants in a number of genes encoding proteins involved with obesity include leptin and leptin receptors which regulate caloric intake and have been found to be associated with BMI and obesity status identified in several human populations. Another hormone that plays a role in glucose and fatty acid regulation and production is adiponectin which is coded by a specific gene and known to be lower in obese individuals and in those with type 2 diabetes. Other variants include the cannabinoid receptor 1 (CNR1), dopamine receptor 2 (DRD2), Fat mass and obesity associated (FTO), insulin induced gene 2 (INSIG2), serotonin receptor 2C (HTR2C), and SLC6A4. Another important obesity gene is peroxisome proliferator-activated receptor gamma (PPARγ) found to be associated with both obesity and type 2 diabetes along with the FTO gene which is considered the most robust common obesity–susceptibility locus found to date. The frequency for a minor allele (ie, rs9939609) of the FTO gene ranges from 38 to 44% in the Caucasian population and clearly correlates with weight gain by producing higher BMI levels and obesity.3,6,7,20–25
Using the GWAS approach, larger metaanalysis studies have been carried out by the Genetic Investigation of Anthropometric Traits (GIANT) consortium in Caucasians and continues to uncover obesity-related genes including transmembrane protein 18 (TMEM18), potassium channel tetramerization domain containing 15 (KCTD15), glucosamine-6-phosphate deaminase 2 (GNPDA2), SH2B adaptor protein 1 (SH2B1), mitochondrial carrier 2 (MTCH2), and neuronal growth regulator 1 (NEGR1). Furthermore, novel loci reported involve chromosome regions 1q25, 3q27, and 12q13. A more recent, expanded GIANT metaanalysis study involving 249,796 individuals has uncovered 32 BMI-associated genetic loci including 10 loci previously reported and associated with BMI and 4 associated with weight gain and/or increased waist-hip ratio (SEC16B, TFAP2B, FAIM2, NRXN3). Eighteen loci were reported to be novel (RBJ-ADCY3-POMC, GPRC5B-IQCK, MAP2K5-LBXCOR1, QPCTL-GIPR, TNNI3K, SLC39A8, FLJ35779-HMGCR, LRRN6C, TMEM160-ZC3H4, FANCL, CADM2, PRKD1, LRP1B, PTBP2, MTIF3-GTF3A, ZNF608, RPL27A-TUB, and NUDT3-HMGA1). Common CNVs were also found including a 21 kb deletion which was 50 kb upstream to the GPRC5B gene. Continuation of analytical studies by this consortium group have confirmed previously identified obesity loci such as FTO, SEC16B, MC4R, GIPR-QPCTL, ADCY3-DNAJC27, BDNF, and MAP2K5 and new additional gene loci (CDKAL1, PCSK1, GP2, GNPDA2, TFAP2B, PAX6, CDKAL1, and KLF9) associated with BMI.3 Hence, candidate gene, DNA linkage, and GWAS approaches have led to the identification of large sets of genes and involved genomic regions found to be associated with obesity and obesity-related endophenotypes. Evidence now exists for the presence of 370 genes playing a role and 153 genes associated with known reproduction and infertility with 21 of these genes in common in both obesity and infertility.16
3. RECOGNIZED SINGLE GENE CAUSES OF OBESITY
Genetic forms of obesity can be grouped into Mendelian or single gene and syndromic or multifactorial. These include recessive gene inheritance, partial gene deficiencies or duplications, genomic structural variations or CNVs, and polygenic causes. Monogenic forms or single gene conditions causing obesity have been reported for at least eight genes including leptin (LEP), leptin receptor (LEPR), proopiomelanocortin (POMC), prohormone convertase 1 (PCSK1), melanocortin 4 receptor (MC4R), single-minded homolog 1 (SIM1), brain-derived neurotrophic factor (BDNF), and the neurotrophic tyrosine kinase receptor type 2 gene (NTRK2). The hypothalamic leptin–melanocortin system is critical for regulating energy balance with disturbances leading to severe obesity disorders.3,6,7 These obesity genes are known to primarily affect common pathways involving lipid metabolism, deposition or transport, food seeking behavior and calorie selection types (ie, fat, protein, carbohydrates), level of physical activity, and forms of energy expenditure related to employment or recreation.
Several obesity-related syndromic genetic disorders are identified in humans, both common and rare and will be discussed later. Monogenic causes of morbid obesity are uncommon but marked obesity and extreme eating behavior (hyperphagia) are key features of several rare genetic syndromes including Prader–Willi, Alström, Bardet–Biedl, Albright hereditary osteodystrophy (AHO), Cohen, and fragile X syndromes. Recognized genes playing a role in these disorders are known [eg, SNRPN for Prader–Willi syndrome (PWS), GNAS for AHO, FMR1 for fragile X syndrome (FXS)],26 which will be discussed later. Understanding the molecular basis of these rare disorders and their genetic mechanisms involving the control of food intake and energy balance in the general population will be important to address the obesity epidemic. Coding and noncoding RNA expression patterns, specifically microRNAs and small nucleolar RNAs (snoRNAs) having important regulatory roles in biological processes, need to be better characterized including their impact on appetite regulation, gene–environment interaction, adipocyte differentiation, and biochemical pathways.16 Genetic variants have been reported near the MC4R and FTO genes which increase body weight in those carrying these variants, with mutations of the MC4R gene present in about 2% of all obese individuals. Male and female heterozygous carriers weigh 15–30 kg more, respectively compared with their relatives without the MC4R gene changes or mutations.3,6,7 More research is needed to examine genetic differences among obese and nonobese individuals, particularly rare CNVs to give novel insight into the genetic causation and architecture of obesity and associated infertility in the general population.
The consequences of increased weight and obesity can shorten life expectancy as well as affecting reproduction with dysfunction in ovulation, spontaneous abortions, and overall infertility. Adverse pregnancy outcomes are also noted including preeclampsia, fetal growth failure with premature delivery, and gestational diabetes. About 15% of all women in the United States are also infertile with advanced age playing a role. The prevalence of maternal obesity in the US population is increased with more women having obesity-related reproductive problems. Gradual and sustained maternal weight loss is needed to improve menstrual cycles and ovulation and thus reproductive rates and outcomes.27,28 Weight loss is considered the first line of treatment in those women with reproductive failure and obesity-related infertility. One of the most common causes of subfertility in women with obesity is polycystic ovarian syndrome which is associated with the androgen receptor (AR) gene.29,30 The AR gene produces a steroid hormone-activated transcription factor important in regulating androgen activity and sensitivity to sex hormones in both males and females involved in weight and body composition. This X-linked gene contains a polymorphic CAG trinucleotide repeat length that is inversely correlated with gene expression.31
Butler and Manzardo29 examined the polymorphic AR gene with CAG repeats and measures of weight and BMI in a cohort of nonsyndromic obese and lean controls, compared with PWS subjects. PWS is a rare obesity-related genetic disorder with growth hormone (GH) deficiency, hypogonadism, and natural sex hormone deficits. The effects of the AR gene CAG repeat length was examined in relationship to androgen-mediated response and obesity-related factors relevant to human infertility and reproduction. The average CAG repeat length in base pair size did not significantly differ by subject group but was significantly positively correlated with height among lean and obese males, but not present in PWS males. A negative correlation was also observed for weight among females when grouped together. The summary results support the role of sex hormones and the AR gene interaction in obesity and infertility, both cardinal features seen in PWS. The AR gene CAG repeat length is a marker for increased androgen sensitivity and shorter CAG repeat length in this study predicted smaller stature in non-PWS adult males. This process may accelerate fusion of bone growth plates and reduce the length of the growth phase. Hence, increased androgen effects from shorter CAG repeat lengths in non-PWS females could impact pregnancy-related weight gain and possibly pregnancy outcomes.
4. OBESITY GENES AND THEIR ENCODED PROTEINS
One of the key obesity-related proteins is leptin which is a 16-kDa secreted protein and encoded by the leptin (LEP) gene. This gene is expressed and secreted by adipocytes, but its receptor is primarily expressed in the hypothalamus. Leptin plays a major role in food intake regulation, energy balance, and body weight. Mutations and polymorphisms of the leptin and leptin receptor genes are associated with obesity in Caucasians as well as POMC with the gene located on chromosome 2. Its protein is produced by the hypothalamus and thus POMC plays a role in feeding behavior. The POMC gene expression level is positively regulated by leptin. Alterations in the POMC gene including a frame shift mutation have been reported to cause loss of function. This results in early-onset obesity, adrenal insufficiency, and red hair pigmentation. POMC also produces β-MSH and β-endorphin peptides via a proteolytic process involving the enzyme, prohormone convertase (PC), and when altered produces an aberrant fusion protein with lower binding affinity to the MC4R. This can have devastating effects because MC4R plays an important role in both food intake and body weight by controlling leptin effects and shown to cause severe obesity when disrupted in mice.6,7
Multiple nonsense and missense mutations in genes have been identified in humans and strongly associated with many obesity-related traits including the BDNF gene located on chromosome 11. Its receptor is a tyrosine receptor kinase B and encoded by the NTRK2 gene which regulates eating behavior and energy balance with growing evidence in humans as playing a role in obesity. In addition, the conditional knockout of BDNF gene in mice will develop obesity and hyperactivity. The SIM1 gene is located on chromosome 6 and also important in the differentiation and function of the central nervous system. When this gene is disturbed, early-onset obesity occurs along with increased linear growth, hyperinsulinemia, and hyperleptinemia in humans. The FTO gene in humans consists of nine exons and is located on chromosome 16. FTO gene expression is most abundant in the brain based on mouse studies, particularly in hypothalamic nuclei, an area involved in energy balance. The mRNA levels for the FTO gene are found specifically in the arcuate nucleus which regulates feeding patterns.3 Current research and supporting evidence directly identifies human disorders of energy balance and obesity such as Prader–Willi, Alström, and Albright hereditary osteodystrophy syndromes as targets for study in understanding the genetics of obesity. PWS and AHO are both due to errors in genomic imprinting or epigenetics. Epigenetics refers to changes in gene expression without altering the DNA code and influenced by the parent of origin. Epigenetics is an emerging field of study which impacts the development of multiple diseases and disorders including obesity and will require additional testing to gain a better understanding of pathogenesis.26
Genetic factors which predispose weight gain and development of obesity in humans can also impact the response to intervention in terms of weight loss. For example, those individuals with MC4R or POMC gene mutations appear to respond to a reduced caloric diet and exercise program but those with MC4R mutations fail to maintain weight lost after intervention. Hence, the gene–environment interaction needs to be considered when treating those affected with obesity. Access to a multidisciplinary team may be helpful in achieving weight loss including genetic evaluations and screening for obesity-related disorders or monogenic causes, dietary consultation, and behavior/pharmaceutical therapy. There is a growing list of pharmaceutical agents and drugs under clinical trial development to target known genetic causes of obesity, for example, PWS. Information learned through this process may have direct application on obesity in the general population. Bariatric surgery may also be considered. However, this is typically used as a final option for treating and managing exogenous obesity but with only limited experience in individuals with syndromic obesity.
Progress in clinical evaluations, awareness, and genetic testing will help to identify DNA factors contributing to obesity specific for each individual based on their family history, lifestyle, and obesogenic environment common in modern society. Other contributing factors may include environmental chemicals, heavy metals, and additive supplements. Next generation DNA sequencing including exome and whole-genome approaches will generate extensive genetic data and comprehensive genetic maps of potentially predisposing causative factors for obesity. This detailed picture of biological and molecular mechanisms and pathways will be key in understanding the development of childhood obesity and eating behavior.
Pediatric obesity in the United States is on the rise with about one-fifth of children being affected.4 Most or greater than 50% of cases of childhood obesity are exogenous in nature and result from excessive energy intake in relationship to energy expenditure for extended periods of time. Some cases of obesity are associated with hormonal or genetic factors, or syndromes. Hormone imbalances such as hypothyroidism, Cushing disease, GH deficiency, defective leptin signaling, and insulin resistance are influenced by genetic factors and account for a subset of cases including those with MC4R or POMC mutations. Examples of syndromic obesity include Prader–Willi, Alström, and Bardet–Biedl syndromes.
Critical features can be distinguished between the more rare endogenous obesity genetic disorders when compared with the more common exogenous forms of obesity. Those with endogenous obesity may have an earlier onset of obesity, lack of satiety, inadequate linear growth patterns, dysmorphic features, and cognitive/behavior problems. To identify differences between the two obesity forms a clinical genetics evaluation may be required to rule out dysmorphic/syndromic causes, obtain family history data, and request dietitian consultations for reviewing food records and calorie intake. In addition, appropriate hormonal and genetic laboratory testing would be ordered and results interpreted applicable for individual medical care.
5. SYNDROMIC CAUSES OF OBESITY
Syndromic obesity may result from a single gene condition (eg, Cohen, Alström, Bardet–Biedl syndromes) or errors in imprinting (ie, epigenetics) as seen in PWS involving chromosome 15 or AHO involving chromosome 20. Other syndromes may have more than one cause, leading to the collection of findings such as obesity found in Down syndrome or the Prader–Willi phenotype (PWP) of FXS males. We will review the genetics of syndromic obesity and clinical presentation of classical and underreported rare causes of obesity. There are several syndromic causes of obesity in humans that are currently recognized; some are common such as Down syndrome while others are rare [eg, Alström syndrome (ALMS)] (Table 2).
Table 2.
Syndromic Causes of Obesity in Humans.
| Prader–Willi |
| Alstrom |
| Albright hereditary osteodystrophy |
| Bardet–Biedl |
| Cohen |
| Fragile X |
| Down |
| Carpenter |
| Kabuki |
| Turner |
| Borjeson-Forssman-Lehmann |
| Klinefelter |
| 1p36 deletion |
| Killian/Teschler-Nicola |
PWS is the first example of syndromic obesity to be discussed. It was first described in 1956 with nine cases and since that time well over 1000 cases have been reported.32 PWS is a neurodevelopmental disorder resulting from errors in genomic imprinting (epigenetics) with loss of only paternally expressed genes located on chromosome 15. It results usually from a de novo deletion of the 15q11-q13 region.33–36 A deletion of the 15q11-q13 region from the mother causes a different clinical disorder known as Angelman syndrome. About 70% of those with PWS will have the 15q11-q13 deletion while the remaining (25–30% of cases) will have both 15s from the mother or a defect of the imprinting center controlling the activity of the imprinted genes in the region found in only a few subjects. Those with an imprinting defect may have a high recurrence risk (up to 50%) for subsequently affected children with PWS in the family.33,35
PWS shows a range of mild learning deficits and characteristic behavior problems including self-injury (skin picking), outbursts, obsessive compulsions, temper tantrums, and food seeking with hyperphagia beginning in childhood. Additional clinical findings include growth and other hormone deficiencies leading to short stature, small hands and feet, hypogonadism, hypogenitalism, hypotonia, a poor suck, feeding difficulties during infancy, and a particular facies (Fig. 2). Infertility is present in both PWS males and females. Obesity can result if not controlled in early childhood and be life threatening.37 PWS is found in about 1 in 10,000–30,000 babies with a lower prevalence of 1 in 30,000–50,000 individuals; PWS affects between 350,000 and 400,000 people worldwide.38 Most cases are sporadic but PWS is considered one of the most common known genetic causes of life threatening obesity affecting all races and ethnic groups34 (Table 3).
Figure 2.
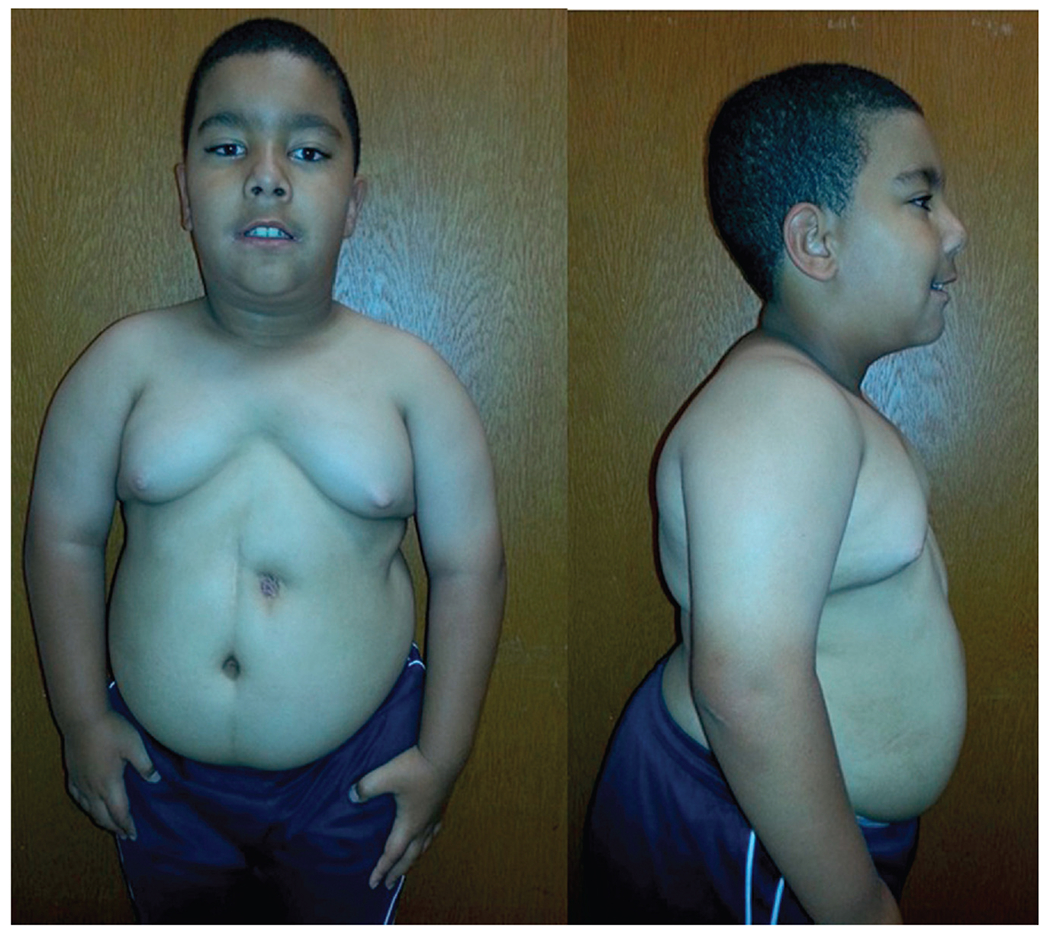
Frontal and profile view of an 8-year-old male with PWS showing the typical facial features, central obesity, and gastrostomy-tube site located on the abdomen.
Table 3.
Clinical, Cognitive, Behavior, and Genetic Findings Seen in PWS, PWP in FXS, ALMS, and AHO.
| Syndromes | Facial Features | Physical Features | Cognitive/Behavior Features | Genetics |
|---|---|---|---|---|
| Prader–Willi Syndrome (PWS) | Narrow forehead, almond-shaped eyes, strabismus, short nose with thin upper lip, downturned corners of mouth, dry sticky saliva, enamel hypoplasia | Severe hypotonia, short stature, obesity, osteoporosis, small hands and feet, scoliosis, hypopigmentation, head tilt forward, hypogenitalism | Mild learning impairment, hyperphagia, skin and rectal picking, difficulty with transitions, stubbornness, temper tantrums, perseverative speech, autism, obsessive compulsions, unusual skill with jigsaw puzzles, high pain tolerance | Paternally derived 15q11-q13 deletion (70% of cases), maternal disomy 15 (about 25% of cases), imprinting defects (1–3% of cases) |
| Prader–Willi Phenotype (PWP) in Fragile X Syndrome (FXS) | Round face, almond-shaped eyes, prominent ears | Obesity, delayed puberty, small penis, hypotonia | Developmental delay, food seeding behavior and hyperphagia, difficulty with transitions, perseverative speech, hand flapping, poor eye contact, autism, obsessive compulsions | FMR1 gene triplet repeat mutations (at chromosome Xq27.3) |
| Alström Syndrome (ALMS) | Round face, deep-set eyes, thickened skull, thick ears, frontal hair loss | Wide, flat feet with brachydactyly, scoliosis, dental anomalies, truncal obesity, short suture, hypogonadism, cardiomyopathy, vision (cone-rod dystrophy) and hearing loss, type 2 diabetes, progressive pulmonary, renal and hepatic problems with fibrosis | Developmental delay, balance disturbances and neurosensory deficits, depression, autism, obsessive compulsions | ALMS1 gene mutations (at chromosome 2p13) |
| Albright Hereditary Osteodystrophy (AHO) | Rounded face with short nose and short neck, delayed dental eruption or enamel hypoplasia | Moderate obesity, short fourth and fifth metacarpals and metatarsals, short distal phalanx of thumb, small stature, osteoporosis, subcutaneous mineral deposits and basal ganglia, thickened calvarium, variable hypocalcaemia and hyperphosphatemia, occasionally hypothyroidism, hypogonadism and infertility, lens opacity or cataracts, optic atrophy, scoliosis and vertebral anomalies | Mental deficiency (average IQ of 60) | Defects of the GNAS gene or imprinting center domain associated with different forms of pseudohypoparathyroidism (PHP) and pseudopseudohypoparathyroidism (PPHP) depending on parent of origin. Maternal inheritance leads to PHP-Ia (AHO plus hormone resistance) while paternal inheritance leads to PHPP or AHO without evidence of resistance to parathyroid hormone (PTH) |
FXS is the most common cause of intellectual disability that runs in families. It is due to a triplet repeat mutation of the FMR1 gene located at chromosome Xq27.3 while a carrier status or premutation form of this gene can be seen in females at risk of developing a full mutation in offspring in the next generation. FXS is associated with obesity but a subset has features in common with PWS referred to as the Prader–Willi phenotype (PWP). FXS generally affects males and was first reported in 1969.39 It is found in about 1 in 4000 males in the general population.40 Prominent clinical features include intellectual disability, prominent large ears, a narrow head, an elongated face with a flattened midface, and prognathism. Joint laxity, mitral valve prolapse, and macroorchidism are also common. Autism spectrum disorder can also be a common finding in those with classical FXS. As noted, a subset of males with this gene mutation have features similarly seen in PWS including hypotonia, developmental delay, short stature, a small penis, behavioral problems, excessive eating, and marked obesity41–43 (Table 3).
A third rare separate genetic obesity-related disorder is ALMS. This syndrome is due to mutations of a single gene (ALMS1) located on chromosome 2p13. ALMS occurs in about 1 in 1,000,000 individuals44 and is characterized by multiorgan involvement with fibrosis, progressive vision, and hearing loss, accompanied by obesity in childhood. Insulin resistance, type 2 diabetes mellitus, and high lipid levels are noted, along with hypogonadism; short stature; and cardiac, liver, lung, and kidney problems. The organ systems are often complicated by fibrotic changes noted throughout life. Obesity is observed in most children with ALMS and they are noted to have significant and rapid weight gain beginning with the first year of life.45,46 The obesity is predominantly distributed in both subcutaneous and visceral compartments. Several endocrine disturbances, including hypogonadism and hypothyroidism, are known to occur in this rare syndrome, as well as decreased growth velocity with advancing age. Most adolescents and adults are reported with short stature and low levels of insulin-like growth factors and GH. Chronic respiratory tract infections are common beginning in early childhood with some becoming severe and leading to chronic bronchitis, asthma, and obstructive pulmonary disease. Pulmonary hypertension is also common with severe interstitial obliterating fibrosis reported, consistent with the fibrotic changes occurring in other major organs.44,45
Most individuals with ALMS are reported to have normal intelligence, although mild to moderate delay in reaching major milestones may be noted, such as speech problems. Major depression, obsessive compulsions, and psychotic disturbances may occur particularly during adulthood in ALMS. The protein encoded by the ALMS1 gene is known to cause ALMS when disturbed and is related to ciliary function as similarly seen in an another genetic obesity disorder known as Bardet–Biedl syndrome which consists of more than one type.47 The ALMS1 gene contains 23 exons with several alternatively spliced transcripts (isoforms).48 ALMS occurs as a result of mutations of this protein coding gene and inherited as an autosomal recessive disorder (Table 3).45,46,49
A second obesity-related genetic disorder due to epigenetics is AHO. AHO was first reported in 194250 and is caused by errors in genomic imprinting found on chromosome 20.26 AHO results from an end-organ resistance to PTH and other associated hormones. Obesity is also a major manifestation along with small stature, mild mental deficiency, a particular facial appearance, short fourth and fifth metacarpals, skeletal anomalies, and delayed dental eruption. Osteoporosis, subcutaneous calcium deposits, hypothyroidism, hypogonadism, ocular problems, and cataracts are occasionally seen.42,51 Two major clinical variants are known for this complex disorder and referred to as PHP (PHP-Ia or PHP-Ib) and PPHP depending on the presence or absence of additional hormone resistance, along with the presence of the AHO phenotype and the specific pattern of inheritance (Table 3).
6. PRADER–WILLI SYNDROME
6.1. Genomic Imprinting Defects in Prader–Willi Syndrome
Most genes and transcripts on the chromosome 15q11-q13 region are subject to genomic imprinting with disturbances leading to PWS, a rare obesity-related disorder.35 Loss or deletion of gene alleles that are only active on the chromosome 15 received from the father and controlled by an imprinting center within the 15q11-q13 region leads to PWS. These same alleles when present on the maternal chromosome 15 are silenced by epigenetic factors usually through methylation. Loss of activity of the maternally expressed gene (ie, UBE3A) in the 15q11-q13 region, usually by the same deletion but from the mother leads to Angelman syndrome, an entirely different clinical disorder. PWS and Angelman syndrome were the first examples of errors in genomic imprinting causing disease in humans. It is now recognized that multiple syndromes affect growth, for example, Beckwith–Wiedemann syndrome with overgrowth as a manifestation and Silver–Russell syndrome with growth retardation as a feature are due to errors in genomic imprinting.26 About 1% of human genes are thought to be imprinted (estimated at about 150 in number); many code for growth factors, related proteins, and receptors and are expressed on only one chromosome depending on the parent of origin. Paternally expressed imprinted genes are thought to enhance the growth of the fetus while maternally expressed imprinted genes are more likely to inhibit growth. Genomic imprinting is now thought to play a role in other diseases such as malignancies, diabetes, and the aging process.26,52,53
Genomic imprinting is related to the methylation of cytosine bases in the CpG dinucleotides of the DNA molecule which are key regulatory elements of genes. Almost all imprinted genes have a CpG-rich differentially methylated region (DMR) which usually relates to allele repression. Many imprinted genes are arranged in clusters (imprinted domains) on different chromosomes under control of an imprinting center affecting animal growth, development, and viability. Imprinted genes may also contribute to behavior and language development, alcohol dependency, schizophrenia, and possibly bipolar affective disorders. In addition, the phenomena of genomic imprinting with abnormal imprinting and loss of heterozygosity (LOH) contributes to a wide range of malignancies.52–54
The expression of imprinted genes may be tissue- and stage specific with one of the parental alleles being differentially expressed only at a certain developmental stage or in certain cells. However, the monoallelic expression of an imprinted gene is not absolute. Thus, a potential role of genomic imprinting in the differentiation of tissue types may be to determine the transcription rate of genes that influence growth through a fine balance between the expression of the two parental alleles.55
Experimental evidence suggests that genomic imprinting evolved about 150 million years ago in a common live-born mammalian ancestor after divergence from egg-laying animals.56 Imprinting genes provide the paternal and maternal genomes the ability to exert counteracting growth effects during embryonic development.57 Approximately 1% of all mammalian genes are thought to be imprinted with the first gene (H19) reported to be imprinted in humans in 1992.58 Since then, many imprinted genes are now candidates for human disease including cancer, obesity, and diabetes.56
Imprinted genes are targets for environmental factors to influence expression through epigenetics whereby the expression level is altered without changing the DNA nucleotide coding structure. Imprinting disturbances have been reported in classical genetic disorders such as Beckwith–Wiedemann, Angelman, and Prader–Willi syndromes while the incidence of these disorders are increased in those individuals conceived with the use of assisted reproductive technology (ART). Hence, ART may increase imprinting defects by changing the regulation of imprinted genes.59
Epigenetics involve various processes altering gene activity without changing the primary nucleotide sequence of the DNA molecule. A common process for controlling gene activity is methylation. A gene that is methylated (inactivated) can be reactivated in male or female gametogenesis for the next generation. For example, a maternally imprinted gene (inactivated by methylation) may be unmethylated by male gametogenesis and transmitted as an active gene in the sperm.
A genome-wide search for imprinted genes in the human genome has identified over 150 candidate imprinted genes involving 115 chromosome bands.60 The number of human diseases or disorders due to genomic imprinting maybe greater than 100 conditions as a consequence of an inappropriate genetic alteration, such as a deletion or uniparental disomy involving a gene or chromosome region. Humans are predicted to have fewer imprinted genes than mice, but the types of human genes involved are markedly different from mice. Therefore, questions have been raised about the use of mice as models for human diseases, particularly those involved with imprinted genes, and assessing environmental factors that may impact on genes and their activity. Examples of classical human disorders related to alterations of genomic imprinting, besides Prader–Willi and Angelman syndromes, include Silver–Russell syndrome, Beckwith–Wiedemann syndrome, AHO, and more recently, uniparental disomy 14 (both paternal and maternal forms).26,61,62
Genes clustered together under the regulation of a single imprinting-controlling element suggest possible involvement of higher order regulatory elements showing allelic specific DNA replication. Genes contributed by the mother generally replicate or express at different rates than genes contributed by the father. However, inappropriate methylation may contribute to tumor formation by silencing tumor-suppressing genes or by activating growth-stimulating genes. In mammals, DNA methylation patterns are established and maintained during development by three distinct DNA cytosine methyltransferases (Dnmt1, Dnmt3a, and Dnmt3b). In mammalian somatic cells, cytosine methylation occurs in 60–80% of all CpG dinucleotides that are not randomly distributed in the genome. Heavily methylated heterochromatin and repetitive sequences contribute to gene silencing. Most CpG islands located at the promoter regions of many active genes are methylation free. Understanding the functions of DNA methylation and its regulation in mammalian development will help to elucidate how epigenetic mechanisms play a role in human diseases such as neurobehavioral problems and cancer.26,63
6.2. Genetic and Clinical Aspects of Prader–Willi Syndrome
The chromosome 15q11-q13 region contains several million DNA base pairs with dozens of imprinted genes and/or transcripts clustered and under the control of two imprinting-controlling centers (one for PWS and one for Angelman syndrome) in this cytogenetic region. There is also a nonimprinted domain or cluster of genes which are equally expressed from either of the parental chromosome 15s.33,35 Low copy DNA repeats called duplicons are located at the end of this chromosome region and are novel with designation at chromosome 15q11-q13 breakpoint sites.64,65 These repetitive DNA areas contain a functional HERC2 gene which is located distally at the chromosome 15q11-q13 region found at breakpoint BP3. Two HERC2 pseudogenes which are located at two proximal breakpoint sites (BP1 and BP2) are separated by about 500 kb size DNA segment66 (Fig. 3). Because these chromosome 15 breakpoint sites contain similar DNA sequences, they can contribute to nonhomologous or mispairing of the chromosome 15s during meiosis I leading to aberrant recombination or crossing-over events. This homologous chromosome 15 mispairing can therefore lead to deletions in the offspring and hence PWS when the deletion is from the father’s chromosome 15 or Angelman syndrome when the deletion is present on the chromosome 15 donated by the mother.
Figure 3.
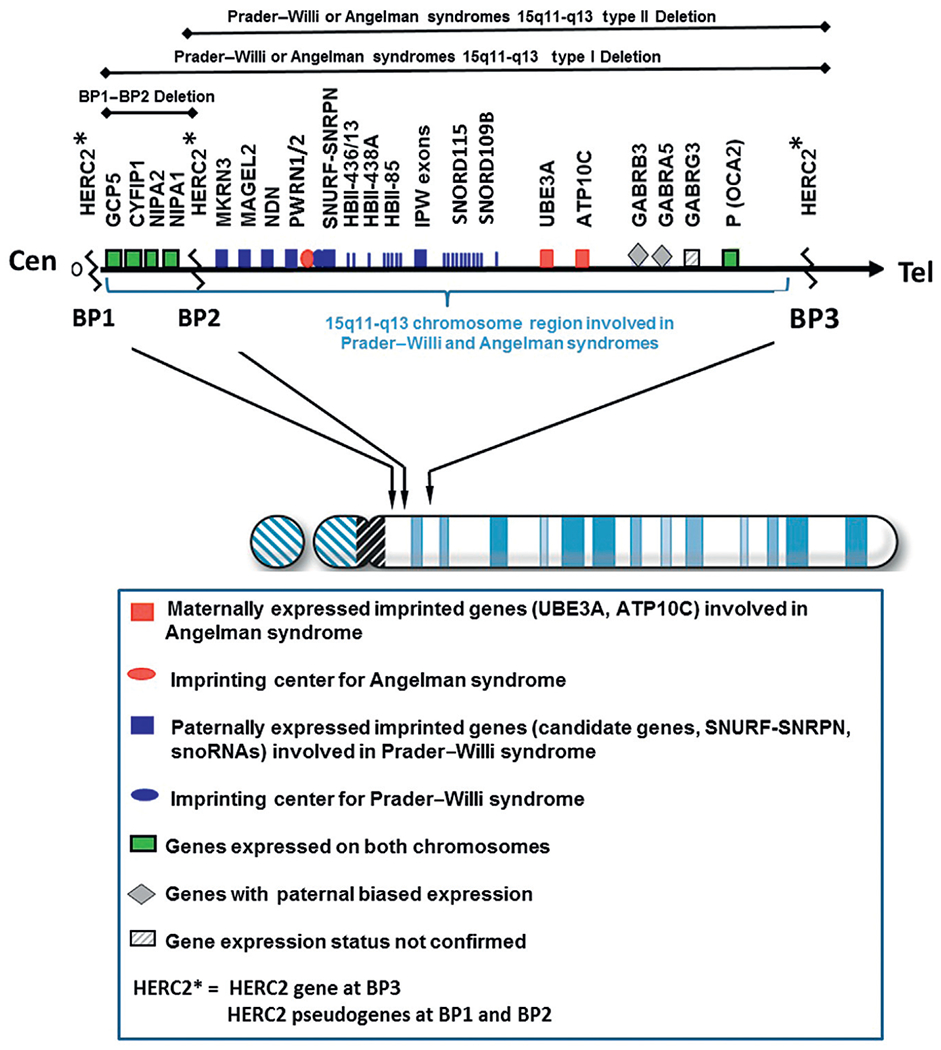
Chromosome 15 ideogram showing the location of genes in the 15q11-q13 region. BP1, BP2, and BP3 are the three common chromosome 15 breakpoints in the region at the site of breakage leading to the larger typical Type I deletion between BP1 and BP3 and the smaller Type II deletion between BP2 and BP3. The dark gray (blue in the web version) colored rectangle-shaped symbols represent paternally expressed genes (eg, MAGEL2) which when disturbed leads to PWS. The gray (red in the web version) colored square-shaped symbols represent maternally expressed genes and the UBE3A gene when disturbed causes Angelman syndrome. The light gray (green in the web version) colored rectangle-shaped symbols represent genes (eg, CYFIP1) that are expressed on both the maternal and paternal chromosome 15s.
About 70% of individuals with PWS will show the typical paternal deletion of the 15q11-q13 region and will be de novo in origin and not inherited. There are two types of the typical 15q11-q13 deletion, Type I and Type II with the Type I deletion being larger (about 6.5 Mb in size) and involving chromosome breakpoints BP1 andBP3.33,35 This is detectable by high resolution chromosomal microarray analysis (Fig. 4A). The smaller Type II deletion (about 5.3 Mb in size) involves breakpoints BP2 and BP3.67 Four genes (TUBGCP5, CYFIP1, NIPA1, and NIPA2) are located between BP1 and BP2 (Fig. 3) and are not imprinted. They show biallelic or normal expression from either the maternal or paternal chromosome 15. Individuals recently reported with speech delay and autistic characteristics are shown to have deletions and/or duplications of only the four genes between BP1 and BP2. In about 5% of individuals with PWS, an unusual or atypical deletion can be present that is either larger or smaller in size than the typical Type I or Type II deletion.68–70
Figure 4.
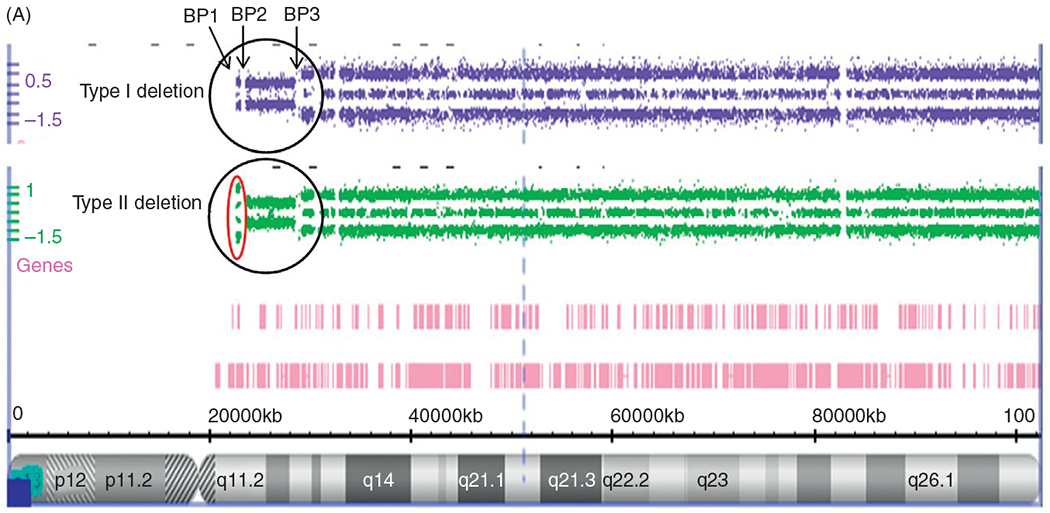
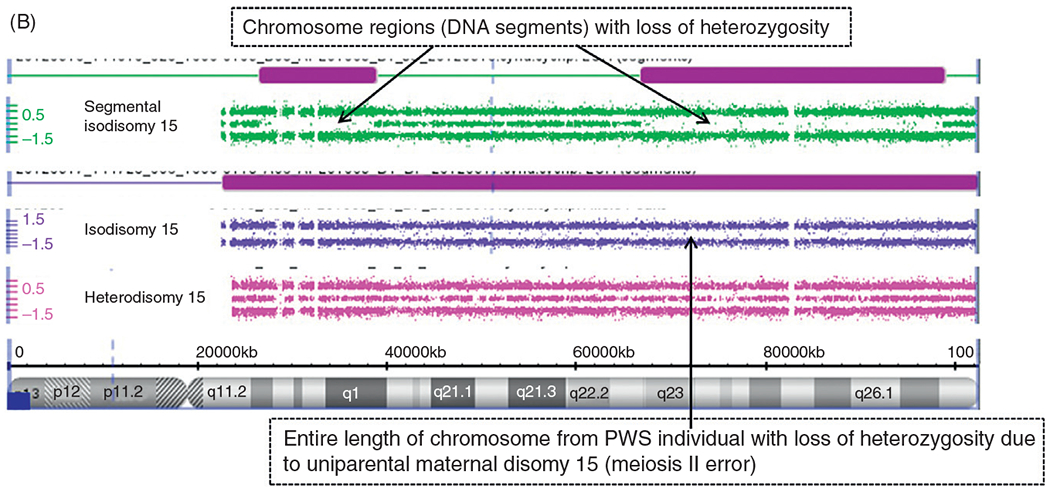
(A) Chromosome microarray results for PWS deletion subtypes. High resolution chromosome microarray using CNV and SNP probes to identify typical 15q11-q13 deletions in PWS classified as Type I involving breakpoints BP1 and BP3 and Type II involving breakpoints BP2 and BP3. (B) Chromosome Microarray Results for Uniparental Maternal Disomy 15. High resolution chromosome microarray using CNV and SNP probes to identify the maternal disomy 15 subtype (segmental isodisomy 15, isodisomy 15, and heterodisomy 15).
Several studies have shown that individuals with the larger typical 15q11–q13 Type I deletion have more severe neurodevelopmental symptoms, as compared to those with PWS or Angelman syndrome with the smaller typical Type II deletions.71,72 Bittel et al.73 later reported findings from mRNA isolated from lymphoblastoid cell lines established from males with PWS for four genes (ie, NIPA1, NIPA2, CYFIP1, and TUBGCP5) in the genomic area between BP1 andBP2 in the 15q11.2 chromosome band. They reported that 24–99% of the phenotypic variability in behavioral and academic measures obtained in their subjects could be explained by the individual gene expression levels. Dykens and Roof74 later examined and reported behavior findings in a mixed cohort of young and old subjects with PWS and showed a relationship between their genetic subtypes and age. They found negative associations between age and behavior in the 15q11–q13 Type I deletion subtype only which implicated nonimprinted genes between breakpoints BP1 and BP2, specifically the CYFIP1 gene. Disturbed expression of CYFIP1 is seen in other developmental disabilities including those with 15q disorders without PWS.
Chai et al.75 showed that these four genes are highly conserved and biallelically expressed. NIPA1 or nonimprinted in Prader–Willi/Angelman syndrome 1 gene is the best studied gene and associated with autosomal dominant hereditary spastic paraplegia.76 NIPA1 also mediates Mg2+ transport and is highly expressed in neuronal tissue. Jiang et al.77 later reported childhood absence epilepsy when mutations were found in the NIPA2 gene. The TUBGCP5 gene or tubulin gamma complex associated protein 5 gene is involved in neurobehavioral disorders including ADHD and OCD [24]. CYFIP1 or cytoplasmic fragile X mental retardation 1 (FMR1) interacting protein 1 gene interacts with FMRP in a ribonucleoprotein complex. FMRP is the product of the FMR1 gene which is associated with FXS, the most common cause of familial intellectual disability that primarily affects males.40 Hence, the importance of these four genes in causation of neurological development and functions but not all individuals with defects within the 15q11.2 band (ie, microdeletions or microduplications) share a clinical phenotype or are clinically affected. Therefore, this region contains genetic material that shows incomplete or low penetrance along with variable expressivity. Population surveys show that about 0.25% of controls will have the 15q11.2 BP1–BP2 microdeletion with penetrance estimated at 10.4% representing a twofold increase over the general population.78,79
In summary, studies of the 15q11.2 BP1–BP2 microdeletion or the Burnside Butler syndrome found that affected individuals will show developmental and language delay, neurobehavioral disturbances, and psychiatric problems that can vary from person to person. However, this emerging syndrome is now recognized with a prevalence ranging from 0.57–1.27% of patients presenting for high resolution microarray analysis accounting for a two- to fourfold increase compared with controls.79 Autism is reported in this disorder and this chromosome anomaly is considered one of the more common findings in those present for microarray analysis. Seizures, schizophrenia, and mild dysmorphic features are less commonly seen in this condition but are at risk.
The second most common genetic cause of PWS is maternal disomy 15 where both chromosome 15s come from the mother and is found in about 25% of affected individuals.35 There are three recognized forms of maternal disomy 15. These include maternal heterodisomy 15 with two different chromosome 15s from the mother due to errors in the first stage of meiosis (meiosis I) from nondisjunction and without cross-over events or shuffling of genes from the two maternal chromosome 15s. A second form is maternal isodisomy 15 with two identical chromosome 15s from the mother due to errors in the second division of meiosis (meiosis II) or the equational phase due to nondisjunction. The third form is segmental maternal isodisomy 15 with two partially different chromosome 15s received in the offspring from the mother due to errors in meiosis I from nondisjunction with cross-over events leading to segments of isodisomy or DNA sequence regions with identical gene alleles. High resolution CNV/SNP microarray studies have shown that if DNA segments of 10 Mb size area or larger are seen with LOH on a specific chromosome with a normal copy number, then uniparental disomy is present. For example, if this LOH occurs on chromosome 15 in the presence of DNA methylation testing showing a PWS genetic pattern, then this represents maternal disomy 15 (Fig. 4B). An individual with PWS and maternal disomy 15 can be at risk for a second genetic condition, if the mother is a carrier of an autosomal recessive gene mutation on chromosome 15 for a disorder if the gene is located in the LOH or isodisomic region with identical DNA sequences representing two copies of the same gene allele.33,35
Maternal disomy 15 is thought to arise from an error in gametogenesis in the female with the egg containing two chromosome 15s from the mother and if fertilized by a normal sperm with a single chromosome 15 then a trisomy 15 zygote results.26,80 Trisomy 15 is lethal and a relatively common cause of spontaneous abortions. In an event of trisomy 15 rescue, the extra chromosome 15 is not passed in the next cell division in the developing embryo. Thus, a normal 46 chromosome number is now established in the embryo from an abnormal 47 chromosome count and leads to viability of the fetus. If the father’s chromosome 15 is lost then the two remaining chromosome 15s from the mother will lead to maternal disomy 15 and the fetus is born with PWS due to his genetic subtype.
Another genetic phenomenon can occur in females with PWS due to maternal disomy 15 which involves the X chromosome. Females have two X chromosomes (one from the father and one from the mother) while males have only one X chromosome; however, the number of active X-linked genes remains constant in both sexes due to gene dosage compensation in females with or without PWS. Females generally inactivate one of their X chromosomes at random which then equals the number of X-linked genes found in the male. This process of X chromosome inactivation occurs very early in pregnancy. Occasionally, this process is not random and skewness occurs which can allow for expression of X-linked conditions in females. Hence, the trisomy 15 rescue event in the early pregnancy of a developing female with PWS and maternal disomy 15 may allow for a small number of cells to survive and to populate embryo development. These small number of cells rescued by the trisomy event may have the same X chromosome active leading to X chromosome inactivation skewness. This allows for the presence of an X-linked condition if the mother is a carrier of an X-linked gene and PWS due to maternal disomy 15.35
The third major category of genetic subtypes in PWS is an imprinting defect. These defects may be due to microdeletions of the imprinting center or due to epimutations through DNA methylation errors in gametogenesis. If the father carries an imprinting defect (microdeletion) that he inherits from his mother’s chromosome 15, he is unaffected due to the presence of his father’s normal chromosome 15. However, when he passes the imprinting defect in his chromosome 15 onto his offspring, that offspring will then have PWS. The risk for him to pass the defect to his offspring would be 50%.37
6.3. Molecular Genetics and Prader-Willi Syndrome
There are dozens of genes and transcripts located in the 15q11-q13 region with most under an imprinting center control. About 10 genes are imprinted and all but 2 (UBE3A and ATP10C) are paternally expressed and regulated by DNA methylation (methylation equates to gene inactivity and unmethylation equates to gene activity). The maternally expressed UBE3A gene causes Angelman syndrome.35,36
The SNRPN (small nuclear ribonucleoprotein N) and a second protein coding sequence (SNURF, or SNRPN upstream reading frame) are located in the 15q11-q13 region. Exons 4–10 of the complex bicistronic SNURF–SNRPN gene encode a core spliceosomal protein (SmN) involved in mRNA splicing in the brain, whereas exons 1–3 encode a 71-amino-acid protein enriched in arginine residues. A disruption of this complex locus will cause loss of function of paternally expressed genes in this region, leading to PWS.35,66 Multiple copies of noncoding C/D box snoRNAs or SNORDs involved in RNA processing are embedded within the long SNURF-SNRPN transcript. These include SNORD64, SNORD107, SNORD108, SNORD109A, and SNORD109B (previously referred to as HBII-13), HBII-436, HBII-437, HBII-438A, and HBII-438B, respectively. Deletions of other snoRNAs also located in the same region have been implicated in causing a PWS phenotype,81 specifically SNORD115 (HBII-52) and SNORD116 (HBII-85). Other imprinted genes that are not components of the SNURF-SNRPN gene complex locus and located proximally are MKRN3, MAGEL2, NDN, and C15orf2. They are involved in brain development and function.35,36
Necdin (NDN) is a paternally expressed gene from the melanoma-associated protein (MAGE) family and required for cell cycle proliferation and differentiation. It is expressed in the hypothalamus, thalamus, and pons suggesting a role in brain development and axon growth. Mice lacking this gene show delayed migration of the sympathetic neurons, neonatal lethality, and respiratory problems. A second gene in this region is the MAGEL2 gene which is paternally expressed in various brain regions including the hypothalamus. It appears to play a role in circadian rhythm, brain structure, behavior, and maintenance of fertility recently reported to be associated with autism.82 The MKRN3 gene is a member of the makorin (MKRN) RING finger protein gene family that encodes a specific group of proteins (makorins) and present in a wide variety of eukaryotes. The MKRN3 is abundantly expressed (paternal only) in the developing brain and nervous system. Mutations of this gene have recently been found in individuals with precocious puberty.83
Another class of genes located in the distal area of the 15q11-q13 region are not imprinted and include three gamma aminobutyric acid (GABA) receptor subunits (GABRB3, GABRA5, and GABRG3) with evidence of unequal expression (paternal bias).66 The disturbances of receptor subunit genes for GABA, a major inhibitory neurotransmitter, have been implicated in a number of symptoms associated with PWS including hunger, obsessive–compulsive disorder, and altered visual perception and memory. The P gene encodes a protein required for pigment production and is located in the distal end of the chromosome 15q11-q13 region and noted to have equal expression from each parental allele. Mutations of this gene are known to cause oculocutaneous albinism II.33,35,36
The UBE3A and ATP10C genes are located distal to the paternally expressed SNURF–SNRPN complex gene locus and imprinted with maternal expression only. A maternal 15q11-q13 deletion including the UBE3A gene or mutations of this gene causes Angelman syndrome. Several transcripts in the 15q11-q13 region are thought to read in an antisense direction, which is complementary to DNA sequences of other genes in the region, but in a reverse direction thereby impacting on the regulation or control of gene activity including the UBE3A antisense transcript.
Butler et al.68 reported a submicroscopic deletion of the 15q11-q13 region approximately 100–200 kb in size in a 5-year-old female with PWS in 1996 as one of the first PWS subjects with an atypical small deletion detectable only with molecular genetic or cytogenetic techniques and included the imprinting center controlling element with adjoining genetic regions now recognized as the snoRNAs (SNORDs). Later, Sahoo et al.81 described a male child with features of PWS and a paternal deletion involving the SNORDs, particularly SNORD109A, the SNORD116 gene cluster, and part of the snoRNA SNORD115 cluster. De Smith et al.84 also reported a 19-year-old male with a PWS phenotype and a 187 kb microdeletion of chromosome 15q11-q13 encompassing SNORD116. Therefore, growing evidence including genetic data in mice support that paternal loss of SNORD116 may contribute to energy homeostasis, growth pattern, and reproduction; all disturbed in PWS. Larger deletions have also been reported which include the 15q14 band and in individuals with features of PWS but with additional findings. These include hearing deficits, cutaneous ear tags, and congenital heart defects. The expanded 15q11-q14 deletion contains about 60 protein-coding genes and making it larger (about twice the size) than the typical 15q11-q13 deletion seen in PWS.69
6.4. Clinical Features in Prader–Willi Syndrome
PWS is a complex genomic imprinting disorder characterized by mental, behavior, and physical findings with obesity as the most significant health problem. Key neuroendocrine peptides produced by the gastrointestinal system are known to play a role in feeding and eating behaviors that are disturbed in PWS.85,86 For example, ghrelin stimulates eating and peptide YY inhibits eating, but when disturbed can lead to abnormal eating patterns and obesity. Plasma ghrelin levels are elevated in PWS as noted during infancy and early childhood.71 This elevation potentially contributes significantly to hyperphagia seen as a cardinal feature in PWS and usually begins in early childhood, if not controlled. In addition, the noncoding snoRNA SNORD115 is thought to regulate alternative splicing of the human serotonin 5-HT2C receptor gene, an important receptor that contributes to normal eating behavior in humans.87 When this snoRNA expression pattern is disturbed through loss of function (eg, deleted in PWS) then an altered receptor results leading to excessive eating behavior and obesity.
Bittel et al.88 also reported on gene expression patterns in males with PWS and found several disturbed genes when compared with control males. Several of these genes were involved in eating behavior and obesity (ADIPOR2, MC2R, SAG, HCRT, STAR, OXTR) and serotonin receptors (eg, HTR2B). Hence, evidence to date supports both disturbed neuropeptide and gene/transcript products playing a role in the PWS phenotype requiring further studies.
PWS is generally divided into two major stages of clinical course development. The first stage is characterized by infantile hypotonia, temperature instability, a weak cry and poor suck, feeding difficulties, developmental delay, and hypogonadism/hypogenitalism. The second stage occurs in early childhood (2–5 years of age) and is characterized by developmental delay, behavioral problems (skin picking, tantrums, obsessive compulsions), speech delay, an insatiable appetite, food seeking with rapid weight gain, and subsequent obesity, if diet is not controlled. There are now several recognized nutritional phases and subphases referred to as 0, 1a, 1b, 2a, 3, and 4. These occur from the time of pregnancy with decreased fetal movements (ie, phase 0) then followed by severe hypotonia and feeding difficulties in infancy. Transitions then occur to an increased weight phase with or without increased caloric intake and followed by food seeking with hyperphagia in early childhood and progressed into adulthood (ie, phases 3 and 4).89
Short stature and small hands/feet also occur in PWS due to GH deficiency, along with rumination, sleep problems, physical inactivity, and decreased pain sensitivity. Hypopigmentation, hypogenitalism/hypogonadism, scoliosis, sleep apnea, enamel hypoplasia, and decreased saliva are more apparent later in life and accompanied by almond-shaped palpebral fissures, down-turned corners of the mouth, a narrow bifrontal diameter, a short nose, and a small chin.34 Several of these features may be present in infancy but become more evident in childhood into adolescence and adulthood. Hypothalamic dysfunction is implicated in many manifestations of this genetic obesity-related syndrome with decreased growth velocity, endocrine disturbances accompanied by behavioral and intellectual disabilities (an average IQ of 65).
Due to developmental delay, PWS infants sit independently by 1 year of age, crawl at 16 months, walk at about 2 years, and talk (10 words) at 39 months but generally have a weak or absent cry with little spontaneous activity.34,90 This is due to decreased muscle tone and strength. Excessive sleepiness with diminished swallowing and sucking reflexes are common and often necessitate gavage feedings with use of special nipples or gastrostomy tube placement to address the feeding problems. Growth parameters should be assessed regularly using recently established standardized growth charts developed for PWS infants.91 Calories are adjusted accordingly, but fats required for brain growth should not be restricted as PWS infants usually require fewer calories than recommended to avoid rapid weight gain. Vitamin and mineral intake such as calcium should be added, as well as caloric intake monitored closely by a dietitian. Developmental assessments, early stimulation programs, and occupational and physical therapy services are recommended.37 Endocrine disturbances including thyroid, growth, and sex hormones and cortisol levels should be monitored from infancy. Myopia and impaired vision may be recognized in early childhood.
Many children with PWS will be mainstreamed in the school setting, but special education and support services are often required. PWS children will generally have strengths related to reading, and with visual and long-term memory skills, but may have weaker math, sequential processing, and short-term memory skills. Verbal skills may be relative strengths particularly in those with PWS having maternal disomy 15. Unusual skill of working with jigsaw puzzles are also noted and more common in those with the 15q11-q13 deletion.37,92
During adolescence, hypogonadism and hypogenitalism becomes more pronounced in the vast majority of individuals with PWS due to hypothalamic hypogonadism leading to low testosterone and estrogen levels. About 90% of males will have cryptorchidism along with a micropenis and an underdeveloped scrotum.34 A hypoplastic labia majora and minora with a small clitoris are often seen in PWS females and puberty is generally absent or delayed in both males and females. Menarche in females may occur but delayed until about 30 years of age but in rare cases, pregnancies in PWS females have been reported.37 PWS males are infertile.
About 90% of individuals with PWS without GH treatment will develop short stature by adulthood. The average adult male will have a height of 155 cm and the adult female will be 147 cm tall.34,37 Small hands and feet are common during adolescence and adulthood and often accompanied by scoliosis and kyphosis. Bracing or surgery may be required. PWS adolescents may weigh more than 300 pounds if caloric intake is not controlled and if GH was not administered at a younger age. Eating related deaths do occur including choking on gorged food and gastric necrosis with rupture.93 Therefore, to avoid these complications, locking the refrigerator and food cabinets to prevent access to food and excessive eating are often required and accompanied by close supervision by all care givers inside and outside of the home setting.94 Although psychotropic drugs are often prescribed to help control behavior problems, no specific medication has been consistently effective. Rigorous control of the food environment which should be secured is recommended to avoid hyperphagia and include regular exercise programs (about 30 min/day) to manage weight and the excessive eating behavior and associated health complications.37,95 Mild mental deficiency is common for the family background and obsessive compulsions, outbursts, and temper tantrums may be triggered by withholding food. Self-injury (eg, skin picking) is also common beginning in childhood which correlates with the PWS genetic subtype, particularly those with the 15q11-q13 deletion.71 Behavioral problems may begin by 3 years of age and manifest as poor peer relationships, immaturity, and inappropriate social behavior, which are reminiscent of autism.
As short stature is a main feature of PWS individuals due to GH deficiency, recombinant human GH which is FDA approved is now prescribed for PWS beginning in infancy and throughout childhood to achieve maximum adult height ultimately within the normal range.33,37 Beneficial effects are noted on stature, weight, and body composition (increased muscle and decreased fat mass) with GH treatment in PWS.96–98 Positive effects on motor development, strength, and cognitive effects have also been reported in both children and adults during GH treatment.99 The quality of life is also improved in adults.100 The prevalence of scoliosis in PWS is high (30–80%),33,37 and thus a concern in those treated with GH and needs to be closely monitored. Recommended starting GH dose in children is 0.18–0.3 mg/kg per week given as daily subcutaneous injections with careful evaluation of clinical status, bone age measures, surveillance for scoliosis, and serum IGF-1 levels at regular intervals should be performed as well as monitoring regular caloric intake records regularly.101 Plotting the individual growth parameters (height, weight, head circumference) on syndrome-specific standardized PWS growth charts is recommended for standard of care.91,102
Weight control requires a strict dietary plan and coordination with a dietitian. Increased physical activity is also encouraged and recorded. Caloric intake is restricted to 6–8 cal/cm of height for weight loss beginning in early childhood and to 10–12 cal/cm of height to maintain weight for nongrowth hormone treated individuals with PWS particularly during childhood. A general recommendation for dietary intake for PWS adolescents or adults may include 800 cal/day to lose weight or 1000–1200 cal/day to maintain weight if food seeking is controlled and exercise program in place.95 A diet plan should be adjusted to include calories from protein at 30–35%; 45% for carbohydrates; and about 30% for fat.37 Calcium, vitamin supplements, and fish oil are recommended throughout life.
6.5. Clinical Findings Associated with Typical 15q11-q13 Deletions (Type I and Type II) in Prader–Willi Syndrome
Differences in cognitive, psychological, and behavioral findings in young adults with PWS was first reported by Butler et al.71 in 2004 with the longer 15q11-q13 Type I deletion involving breakpoints BP1 andBP3 versus those with the shorter Type II deletion involving breakpoints BP2 and BP3. PWS individuals with the longer Type I deletion score significantly worse in self-injurious (skin picking) assessments and in maladaptive behavior findings when compared to those with the smaller Type II deletion. Obsessive-compulsive behavior was also more frequently seen in PWS subjects with the Type I deletion. Academic achievement scores did differ between PWS subjects with the shorter Type II versus longer Type I typical 15q11-q13 deletion. This study supported that the loss of genes between BP1 and BP2 when deleted would increase the severity of the behavioral and psychological problems seen in PWS. For example, the adaptive behavior scores were generally worse in PWS individuals having the Type I deletion, such as obsessive-compulsive behaviors. Those with Type I deletion generally had poorer math and reading skills. Additional problems included poorer visual-motor integration, worse adaptive behavior, and more compulsions in those subjects with Type II deletion, particularly relating to grooming and bathing skills and compulsions that disrupted daily living. Intellectual ability and academic achievement were also poorer in those PWS subjects with Type I deletions as well as visual processing, compared with those with Type II deletions.71
7. FRAGILE X SYNDROME AND THE PRADER–WILLI PHENOTYPE
As obesity is a cardinal feature of PWS, it is also common in a subset of individuals with the FXS.103 A recent survey of BMI data collected from 718 children with FXS showed a prevalence rate of obesity (31%) which was higher than that found in age matched control children (18%).43,104 FXS is considered the most common cause of familial intellectual disability and due to a CGG triplet repeat expansion greater than 200 in size in the 5’ untranslated region of the fragile X mental retardation 1 (FMR1) gene.40,43,104 Those with a smaller repeat size (50–200) would be considered as carriers or having a premutation form while the normal number of CGG repeats is less than 50 and usually less than 30. The expanded full mutation usually leads to methylation and little to no mRNA is transcribed; thus, resulting in a lack of production of the fragile X mental retardation protein (FMRP) encoded by the FMR1 gene.40,104 Lack of this protein is correlated with an increased childhood growth rate in FXS patients and supported by evidence from the FXS knockout mouse model showing enhanced growth rate for the mice and obesity. Interestingly, about 10% of individuals with FXS will have severe obesity, hyperphagia, hypogonadism, or delayed puberty as seen in PWS.43 This subset of FXS patients, termed the Prader-Willi phenotype (PWP), do not have chromosome 15q11-q13 gene deletions or abnormalities.103
Mild to profound intellectual disability is seen in affected males with an IQ in the 30–55 range but can extend into the borderline normal range. Hand flapping or biting (self-injury) is frequently seen along with poor eye contact. The speech is often repetitive (echolalia) and clustered. Complete lack of speech is seen in those with more severe and profound range of intellectual disability. Attention problems, hyperactivity, and overstimulation are common leading to abnormal behavioral outcomes, hyperkinetic activity, and emotional instability. Autism is seen in most FXS patients and may be recorded as high as 60%.42 Females may also have the full mutation resulting in the same learning and behavioral problems seen in males. Females who carry a premutation may have social or generalized anxiety, and math skill deficits. They are at risk of having premature ovarian failure and twin pregnancies. Other occasional clinical findings seen in individuals with FXS include eye problems such as strabismus, nystagmus, and myopia. Seizures may also be present along with torticollis, pectus excavatum, kyphoscoliosis, and a submucous cleft palate. Some individuals also present cerebral gigantism. Expansion of the carrier gene status (premutation) to a full mutation occurs only in females and correlates with the size of the premutation. Males with the premutation are prone to anxiety and deficits in executive function and tremors (Parkinson-like) with advancing age.42,104,105
The FMR1 gene is located at the chromosome Xq27.3 band consisting of 17 coding exons that span 38 kb in size.40,104 FMRP is transported back and forth between the nucleus and the cytoplasm with binding and transportation of mRNAs in the neurons at the synapse level in the brain. FMRP stabilizes mRNA or can enhance degradation of mRNA. It appears to shape the pattern of mRNA regulation throughout human fetal development. Interestingly, a lower expression of a gene located in the 15q11-q13 region involved with PWS that encodes the cytoplasmic FMR1 interacting protein 1 (CYFIP1) also works in concert with FMRP and is associated with PWP.106
Hence, the classic features seen in FXS patients include an elongated face with a prominent forehead and ears, flat feet, mitral valve prolapse, soft fleshy hands, and hyperextensible joints. These indicate the presence of a connective tissue disorder. For example, individuals with FXS can have macroorchidism (two to four times larger testicular volume than seen in normal adult males). Hand callouses from self-injury and a plantar crease between the first and second toes are helpful indicators for the FXS. Characteristic behavioral findings include anxiety, attention deficit hyperactivity disorder, autistic tendencies such as hyperarousal to sensory stimuli for sound and food texture, self-injury, and often obsessive–compulsive disorder.42,104,105 The family history often supports an X-linked inheritance pattern with intellectual disability and/or autism in affected males (uncles and nephews on the maternal side). The diagnosis of FXS and molecular genetic testing to identify the number of CGG repeats on the FMR1 gene would be indicated. The obsessions seen in males with FXS when focused on food can lead to overeating, food seeking at night, and obesity. This can become severe and, thus, lead to the PWP status. PWP individuals are described as having a lack of satiation leading to hyperphagia by 5 years of age and truncal obesity involving the torso and abdomen as seen in PWS by 10 years of age. Other PWP features in common with PWS include a round face, hypotonia, short stature, delayed puberty, and hypogenitalism with a ravenous appetite including the consumption of inedible food such as raw meat and lack of satiety requiring locking refrigerators.43
8. ALSTRöM SYNDROME: A RARE OBESITY-RELATED SINGLE GENE DISORDER
ALMS is another obesity-related disorder due to a single gene defect with an autosomal recessive inheritance pattern. It has a prevalence of about 1 per 1,000,000 individuals and considered very rare.44 Due to multiorgan involvement and fibrosis, individuals with ALMS have a reduced life expectancy of less than 50 years. This obesity syndrome is caused by mutations in the ALMS1 gene located on chromosome 2p13. The symptoms typically begin during infancy. Affected individuals have visual impairment; cone-rod dystrophy; hearing loss; childhood truncal obesity; developmental delay; insulin resistance and type 2 diabetes mellitus; hypertriglyceridemia; short stature; scoliosis and kyphosis; dilated cardiomyopathy; and progressive pulmonary, hepatic, and renal dysfunction. Fibrosis can also develop in multiple organs.45,46 Photoreceptor dystrophy which is present in 100% of subjects, begins at birth to 15 months of age and is accompanied by nystagmus leading to early blindness. Progressive sensorineural hearing loss is severe and occurs in the first 10 years of life in about three-fourths of affected individuals.
During the first 5 years of life, truncal obesity develops along with insulin resistance, acanthosis nigricans, hyperinsulinemia, and hyperlipidemia.44 Other endocrine problems include hypothyroidism, GH alterations, polycystic ovarian syndrome, acute pancreatitis, and type 2 diabetes mellitus. About 45% of individuals with ALMS will have developmental delay and impaired learning skills along with sleep disturbances.
The protein coded by the ALMS1 gene when disturbed involves organs throughout the body. Its function is unclear but a role in ciliary function, intracellular trafficking, and adipocyte differentiation is reported.46,47 This is similar to what is found in another obesity-related genetic disorder, Bardet–Biedl syndrome. The diagnosis of ALMS is usually made based on clinical features and confirmed by molecular genetic testing of disease causing mutations in the ALMS1 gene.
Cardiomyopathy and congestive heart failure (CHF) are seen in about 70% of individuals with this syndrome and account for major causes of morbidity and mortality. The onset of CHF can occur as early as infancy or develop later leading to fibrosis and myocardial hypertrophy with dilation and restrictive impairment of both ventricles.46 Hepatic changes are seen including cirrhosis, steatosis, and hepatosplenomegaly followed by gastrointestinal bleeding, inflammation, and fibrosis. End-stage liver disease is the cause of death in about 10% of individuals with ALMS. Renal disease is a major cause of morbidity in this syndrome. Progressive renal impairment and varying degrees of glomerular disease, albuminuria, and interstitial fibrosis may be found with end-stage renal disease occurring as early as the teenage years. Hypogonadism is seen in both sexes with ALMS. Low–normal levels of testosterone and elevated gonadotropins are seen in males along with a small penis and testicular atrophy. Secondary sex characteristics may be normal in males. Hypogonadism in females becomes evident by puberty with delays in secondary sex characteristics and in menarche. As seen in other obesity-related disorders, infertility is a consistent finding in ALMS.45,47,49
Heterozygous carriers of mutations in the ALMS1 gene are generally asymptomatic. Exon 8 contains a large tandem repeat domain which encodes 34 imperfect repeats of about 50 amino acids constituting 40% of the protein.48,49 Over 100 different mutations have been reported in the ALMS1 gene with the majority being of the nonsense or frameshift type. The coding regions of exons 8, 10, and 16 are most commonly affected. Population studies suggest founder effects in certain ethnic backgrounds with lack of disease causing mutations in the 5’ half of the coding region of ALMS1 which include exons 1 through 7, seen as lethal to the developing embryo.45,47,49
Hyperphagia is seen in several rare but classical genetic obesity-related syndromes such as PWS, ALMS, and FXS, specifically the PWP which is seen in a subset of males (about 10%) with FXS. Using advanced genetic microarray methods to compare coding and noncoding gene expression patterns in adult males with PWS, ALMS, and nonsyndromic obesity relative to nonobese males using readily available lymphoblastoid cells, we attempted to identify disease-specific molecular signature patterns and disturbed mechanisms in obesity.107 Through this study, we found 231 genes that were upregulated in ALMS males compared with nonobese males using stringent classification criteria, but no genes were upregulated in either obese or PWS adult males. We found 124 genes downregulated in ALMS using this high resolution expression microarray approach. Interestingly, the metallothionein gene (MT1X) which is involved in metal toxicity was significantly downregulated in ALMS and also in obese males but not in PWS males. Only the complex SNRPN gene locus was disturbed or downregulated in PWS along with several downregulated snoRNAs in the 15q11-q13 region including SNORD116, SNORD109B, SNORD109A, and SNORD107. Several snoRNAs that were disturbed in ALMS males and found outside chromosome 15 targeted multiple genes that impacted rRNA processing, developmental pathways, and associated diseases. Fifty-two miRNAs were upregulated in ALMS, and four were shared with obese males but not PWS males. Our data supported that the cell cycle (eg, PPP3CA), transcription (eg, POLE2), and developmental processing may be impacted by the upregulated genes found in ALMS. The downregulated genes found in ALMS were involved with metabolic processes (eg, FABP3), immune responses (eg, IL32), and cell signaling (eg, IL1B) when examined using computer generated pathway analyses. The high number of gene and noncoding RNA disturbances found in ALMS did contrast the observations seen in PWS and in males with nonsyndromic obesity. This contrast may reflect the progressing multiorgan pathology seen in ALMS and the ongoing disease process related to a shortened life expectancy.
9. ALBRIGHT HEREDITARY OSTEODYSTROPHY
AHO was first reported in 1942 and is due to an end-organ resistance to PTH and other hormones.50 Obesity is a major manifestation. In addition to obesity, other clinical features of AHO consist of small stature (adult height of 54–60 in.), mild mental deficiency with an average IQ of 60 (range of 20–99), a round face with a low nasal bridge, a short nose and neck, delayed dental eruption with enamel hypoplasia, short metacarpals and metatarsals especially of the fourth and fifth digits, and short distal phalanges of the thumb.42 These skeletal changes represent early epiphyseal fusion and may not be evident until several years of age. In addition, arm span is decreased for height. Reproductive dysfunction is common due to partial resistance to gonadotropins. Other features seen in this disorder include osteoporosis, areas of subcutaneous mineralization with calcium deposits including the basal ganglia, a thickened calvarium, variable low plasma calcium and/or high phosphate levels, and seizures secondary to low calcium levels. Hypogonadism and hypothyroidism are seen along with abnormal eye findings including lens opacity or cataracts, optic atrophy, diplopia, microphthalmia, ocular, and macular degeneration. Pancreatic dysfunction, vertebral anomalies, and advanced bone age are reported in affected individuals.42,51,108,109
There are two major clinical variants of this disorder referred to as PHP (PHP-Ia or PHP-Ib) and PPHP. The clinical presentation depends on the presence or absence of hormonal resistance and the AHO phenotype with the pattern of inheritance. Individuals with PHP-Ia have features of AHO, along with hypocalcemia, hyperphosphatemia, mild hypothyroidism, and hypogonadism with an abnormal response to the GH releasing hormone at the brain level. Affected individuals also have high serum PTH levels. Individuals with PPHP have the characteristic features of AHO, but without identified resistance to parathyroid and thyroid stimulating hormones and gonadotropins. PHP-Ia and PPHP have been reported in the same families, but depend on the parent of origin thereby indicating the role of the genomic imprinting status in causation of this disorder. Both disease variant types result from decreased activity of the alpha subunit of the membrane bound trimeric G subunit-regulatory signaling protein (GNAS).42,51
GNAS is involved in several complex cellular pathways and mechanisms by stimulating cellular adenyl cyclase activity. PHP individuals present PTH-resistance but lack features of AHO including obesity and are defined as having the PHP-Ib subtype. Most PHP-Ib cases are sporadic in origin and typically lack GNAS gene mutations. PHP-la and PPHP are caused by heterozygous mutations in exons of the GNAS gene which leads to gene inactivity. GNAS encodes the alpha subunit of the stimulatory guanine nucleotide-binding protein (G-protein) subunit alpha (Gs-alpha) and the autosomal dominant form of PHP-lb. This condition is caused by heterozygous mutations that disrupt an imprinting controlling element of the GNAS gene and not within the exons of the gene.110,111
Gs-alpha is a trimeric regulatory protein which functions by coupling to membrane receptors for adenyl cyclase and thus stimulates cyclic adenosine monophosphate. GNAS is expressed biallelically on both maternal and paternal alleles of chromosome 20 in most tissues but expressed monoallelically from only the maternal GNAS allele in a selected number of tissues (eg, the pituitary and thyroid glands, proximal tubules of the kidneys and gonads). Other gene transcripts of the GNAS are known and produced or expressed only from the maternal or paternal GNAS imprinted gene allele. The promoter of the gene lacks methylation. Individuals with PHP-Ia and features of AHO also have mutations of the GNAS gene or cytogenetic deletions of chromosome 20q13 including the complex imprinted GNAS gene locus. Those with PPHP (or those AHO patients without evidence of hormone resistance) also carry heterozygous inactivating GNAS gene mutations. Maternal inheritance of a mutation can lead to PHP-Ia (AHO with hormone resistance) while paternal inheritance of the same mutation leads to PPHP or AHO alone. When the altered gene is inherited from the affected father with either PHP-Ia or PPHP, then PPHP occurs in the offspring. If the same GNAS mutation is present in the mother affected with either PHP-Ia or PPHP, then the offspring will be affected with PHP-Ia.42,110 Hence, the parent of origin of GNAS gene disturbances lead to different clinical disorders and inheritance patterns.
GNAS is a complex imprinted locus on chromosome 20q13.11. Several transcripts are produced using alternative promoters and splice sites. As illustrated when altered, these transcripts can lead to several clinical disorders or presentations. The best characterized GNAS transcript encodes the G protein Gs-alpha subunit. This transcript is biallelically expressed in most tissues, but expressed on one allele (maternal) in the gonads, pituitary, and thyroid glands and in the proximal renal tubules. Other GNAS transcripts are expressed exclusively from either the paternal or the maternal GNAS allele. Hence, the GNAS locus encodes four main transcripts: Gs-alpha, XLAS, NESP55, and the A/B transcript, and an antisense transcript (GNASAS). Gs-alpha is found ubiquitously and stimulates adenyl cyclase when coupled with a membrane receptor that generates the second messenger cyclic AMP (cAMP). Many hormones and neurotransmitters exert their actions through receptors coupled to Gs-alpha. Gs-alpha is involved in AHO, XLAS is paternally expressed, and NESP55 is exclusively maternally expressed and encodes a chromogranin-like neuroendocrine secretory protein. NESP55 does not share any amino acid sequences with Gs-alpha because of a stop codon in its unique first exon sequence. The A/B transcript uses an alternative first exon A/B or exon 1A and the antisense GNAS transcript is expressed from the paternal GNAS allele; all involved in cellular processes and when disturbed can lead to disease.111,112 The XLAS transcript contains a previously undescribed large variant of the Gs-alpha subunit and expressed exclusively from the paternal GNAS allele that is located upstream of exon 1. It occurs most often in neuroendocrine and neurological tissue.
Reproductive- and health-related problems are common in individuals with AHO having GNAS gene disturbances. Clinical and genetic evaluations with testing are required to identify the specific genetic subtype and to supply appropriate and accurate genetic counseling leading to better medical care for affected or at-risk individuals.
In summary, as common or exogenous obesity represents an interaction of complex multifactorial agents including susceptibility genes with an obesogenic environment characterized by increased consumption of high caloric foods and a sedentary lifestyle, genetic mutations and variants are becoming more recognized as playing a role in response to our emerging environment. Research with known dietary components (high-fat food or high saturated-fat food) and population-based studies are needed to identify and develop targeted treatment protocols for personalized nutritional or pharmaceutical therapy for individuals at an increased risk for genetic factors and developing childhood obesity. In addition, the growing evidence learned about the role of genes from single gene obesity-related disorders such as Alström, Prader–Willi, and fragile X syndromes (with or without the PWP) and mutations or gene variants present in the general population need to be better characterized and information applied in the clinical setting beginning in early childhood to avoid obesity and its manifestations.
REFERENCES
- 1.Friedman JM. Modern science versus the stigma of obesity. Nat Med. 2004;10(6): 563–569. [DOI] [PubMed] [Google Scholar]
- 2.Lyon HN, Hirschhorn JH. Genetics of common forms of obesity: a brief overview. AmJClin Nutr. 2005;82(1):215S–217S. [DOI] [PubMed] [Google Scholar]
- 3.Xia Q, Grant S. The genetics of human obesity. Ann NY Acad Sci. 2013;1281(1): 178–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mason K, Page L, Balikcioglu PG. Screening for hormonal, monogenic, and syndromic disorders in obese infants and children. PediatrAnn. 2014;43(9):e218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Behan DF, Cox SH, Lin Y, et al. Obesity and its Relation to Mortality and Morbidity Costs. Schaumburg, Illinois: Society of Actuaries: Committee on Life Insurance Research; 2010: 1–78 [Google Scholar]
- 6.Choquet H, Meyre D. Genetics of obesity: what have we learned? Curr Genomics. 2011;12(3):169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choquet H, Meyre D. Molecular basis of obesity: current status and future prospects. CurrGenomics. 2011;12(3):154–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Must A Does overweight in childhood have an impact on adult health? Nutr Rev. 2003;61:139–142. [DOI] [PubMed] [Google Scholar]
- 9.Nicklas TA, Baranowski T, Cullen KW, et al. Eating patterns, dietary quality and obesity. JAmColl Nutr. 2001;20(6):599–608. [DOI] [PubMed] [Google Scholar]
- 10.Parsons TJ, Power C, Logan S, et al. Childhood predictors of adult obesity: a systematic review. IntJ ObesRelat Metab Disord. 1999;23(suppl 8):S1–S107. [PubMed] [Google Scholar]
- 11.Daniels SR, Arnett DK, Eckel RH, et al. Overweight in children and adolescents: pathophysiology, consequences, prevention, and treatment. Circulation. 2005;111: 1999–2012. [DOI] [PubMed] [Google Scholar]
- 12.Flegal KM, Wei R, Ogden C. Weight-for-stature compared with body mass index-forage growth charts for the United States from the centers for disease control and prevention. AmJClin Nutr. 2002;75:761–766. [DOI] [PubMed] [Google Scholar]
- 13.Himes JH, Dietz WH. Guidelines for overweight in adolescent preventive services: recommendations from an expert committee. The Expert Committee on Clinical Guidelines for Overweight in Adolescent Preventive Services. Am J Clin Nutr. 1994;59:307–316. [DOI] [PubMed] [Google Scholar]
- 14.Farooqi IS, O’Rahilly S. New advances in the genetics of early onset obesity. IntJObes. 2005;29:1149–1152. [DOI] [PubMed] [Google Scholar]
- 15.Stunkard AJ, Foch TT, Hrubec Z. A twin study of human obesity. JAMA. 1986;256: 51–54. [PubMed] [Google Scholar]
- 16.Butler MG, McGuire AM, Manzardo AM. Clinically relevant known and candidate genes for obesity and their overlap with human infertility and reproduction. J Assist ReprodGenet. 2015;32(4):495–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dasouki MJ, Youngs EL, Hovanes K. Structural chromosome abnormalities associated with obesity: report of four new subjects and review of literature. Curr Genomics. 2011;12(3):190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walters RG,Jacquemont S, Valsesia A, et al. A new highly penetrant form of obesity due to deletions on chromosome 16p11. Nature. 2010;463:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scherag A, Dina C, Hinney A, et al. Two new Loci for body-weight regulation identified in a joint analysis of genome-wide association studies for early-onset extreme obesity in French and German study groups. PLoS Genet. 2010;6:e1000916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boissel S, Reish O, Proulx K, et al. Loss-of-function mutation in the dioxygenase-encoding FTO gene causes severe growth retardation and multiple malformations. AmJHumGenet. 2009;85:106–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deliard S, Panossian S, Mentch FD, et al. The missense variation landscape of FTO, MC4R, and TMEM18 in obese children of African Ancestry. Obesity. 2013;21(1): 159–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loos RJ, Lindgren CM, Li S, et al. Common variants near MC4R are associated with fat mass, weight and risk of obesity. Nat Genet. 2008;40:768–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meyre D, Proulx K, Kawagoe-Takaki H, et al. Prevalence of loss-of-function FTO mutations in lean and obese individuals. Diabetes. 2009;59:311–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thorleifsson G, Walters GB, Gudbjartsson DF, et al. Genome-wide association yields new sequence variants at seven loci that associate with measures of obesity. Nat Genet. 2009;41:18–24. [DOI] [PubMed] [Google Scholar]
- 25.Willer CJ, Speliotes EK, Loos RJF, et al. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat Genet. 2009;41:25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Butler MG. Genomic imprinting disorders in humans: a mini-review. J Assist Reprod Genet. 2009;26(9–10):477–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jungheim ES, Travieso JL, Carson KR, et al. Obesity and reproductive function. Obstet Gynecol Clin North Am. 2012;39:479–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Venkatesh T, Suresh PS, Tsutsumi R. New insights into the genetic basis of infertility. Appl Clin Genet. 2014;7:235–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Butler MG, Manzardo AM. Androgen receptor (AR) gene CAG trinucleotide repeat length associated with body composition measures in non-syndromic obese, non-obese and Prader-Willi syndrome individuals. J Assist Reprod Genet. 2015;32:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saute JA, Silva AC, Souza GN, et al. Body mass index is inversely correlated with the expanded CAG repeat length in SCA3/MJD patients. Cerebellum. 2012;11:771–774. [DOI] [PubMed] [Google Scholar]
- 31.Barber TM, Franks S. Genetics of polycystic ovary syndrome. Front Horm Res. 2013;40:28–39. [DOI] [PubMed] [Google Scholar]
- 32.Prader A, Labhart A, Willi H. Ein sydnrom von adipositas, kleinwuchs, kryptorchismus und oligophrenie nach myatonieartigem zustand im neugeborenenalter. Schweizerische MedizinishceWochenschrift. 1956;6(3):1260–1261. [Google Scholar]
- 33.Angulo MA, Butler MG, Cataletto ME. Prader–Willi syndrome: a review of clinical, genetic, and endocrine findings. JEndocrinol Invest. 2015;38:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Butler MG. Prader-Willi syndrome: current understanding of cause and diagnosis. Am J Med Genet. 1990;35(3):319–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Butler MG. Prader–Willi syndrome: obesity due to genomic imprinting. CurrGenomics. 2011;12:204–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cassidy SB, Schwartz S, Miller JL, et al. Prader–Willi syndrome. Genet Med. 2012;14 (1):10–26. [DOI] [PubMed] [Google Scholar]
- 37.Butler MG, Lee PDK, Whitman BY, eds. In: ManagementofPrader–WilliSyndrome 3rded. NewYork, NY: Springer Science+BusinessMedia;2006:. [Google Scholar]
- 38.Butler MG, Thompson T. Prader–Willi syndrome: clinical and genetic finding. Endocrinology. 2000;10:3S–16S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lubs HA. A marker X chromosome. Am J Hum Genet. 1969;21(3):231. [PMC free article] [PubMed] [Google Scholar]
- 40.Hagerman RJ, Hagerman PJ. Fragile X syndrome: model of gene-brain-behavior relationships. Mol Genet Metab. 2001;74(1):89–97. [DOI] [PubMed] [Google Scholar]
- 41.Chudley AE, Hagerman RJ. Fragile X syndrome. JPediatr. 1987;110(6):821–831. [DOI] [PubMed] [Google Scholar]
- 42.Jones KL, ed. In: Smiths Recognizable Patterns of Human Malformation 6th ed. Philadelphia, PA:WB Saunders Company;2006:. [Google Scholar]
- 43.Lozano R, Rosero CA, Hagerman RJ. Fragile X spectrum disorders. Intractable Rare Dis Res. 2014;3(4):134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Álvarez-Satta M, Castro-Sánchez S, Valverde D. Alström syndrome: current perspectives. Appl Clin Genet. 2015;8:171–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marshall JD, Bronson RT, Collin GB, et al. New Alström syndrome phenotypes based on the evaluation of 182 cases. Arch InternMed. 2005;165(6):675–683. [DOI] [PubMed] [Google Scholar]
- 46.Marshall JD, Maffei P, Collin GB, Naggert JK. Alström syndrome: genetics and clinical overview. Curr Genomics. 2011;12(3):225–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Waters AM, Beales PL. Ciliopathies: an expanding disease spectrum. Pediatr Nephrol. 2011;26(7):1039–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Collin GB, Marshall JD, Ikeda A, et al. Mutations in ALMS1 cause obesity, type 2 diabetes and neurosensory degeneration in Alström syndrome. Nat Genet. 2002;31(1): 74–78. [DOI] [PubMed] [Google Scholar]
- 49.Marshall JD, Beck S, Maffei P, et al. Alström syndrome. EurJHum Genet. 2007;15(12): 1193–1202. [DOI] [PubMed] [Google Scholar]
- 50.Albright F, Burnett CH, Smith PH, et al. Pseudohypoparathyroidism—an example of “Seabright-Bantam syndrome”: report of three cases. Endocrinology. 1942;30:922–932. [Google Scholar]
- 51.Levine MA. Clinical spectrum and pathogenesis of pseudohypoparathyroidism. Rev Endocr Metab Disord. 2000;1(4):265–274. [DOI] [PubMed] [Google Scholar]
- 52.Bartolomei MS, Tilghman SM. Genomic imprinting in mammals. Annu Rev Genet. 1997;31:493–525. [DOI] [PubMed] [Google Scholar]
- 53.Walter J, Paulsen M. Imprinting and disease. Semin Cell Dev Biol. 2003;14:101–110. [DOI] [PubMed] [Google Scholar]
- 54.Delaval K, Wagschal A, Feil R. Epigenetic deregulation of imprinting in congenital diseases of aberrant growth. Bioessays. 2006;28(5):453–459. [DOI] [PubMed] [Google Scholar]
- 55.Platonov ES, Isaev DA. Genomic imprinting in the epigenetics of mammals. Genetika. 2006;42(9):1235–1249. [PubMed] [Google Scholar]
- 56.Murphy SK, Jirtle RL. Imprinting evolution and the price of silence. Bioessays. 2003;25 (6):577–588. [DOI] [PubMed] [Google Scholar]
- 57.Haig D, Graham C. Genomic imprinting and the strange case of the insulin-like growth factor II receptor. Cell. 1991;64(6):1045–1046. [DOI] [PubMed] [Google Scholar]
- 58.Zhang Y, Tycko B. Monoallelic expression of the human H19 gene. NatGenet. 1992;1 (1):40–44. [DOI] [PubMed] [Google Scholar]
- 59.Niemitz EL, Feinberg AP. Epigenetics and assisted reproductive technology: a call for investigation. Am J Hum Genet. 2004;74(4):599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Luedi PP, Dietrich FS, Weidman JR, et al. Computational and experimental identification of novel human imprinted genes. GenomeRes. 2007;17(12):1723–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Falk MJ, Curtis CA, Bass NE, et al. Maternal uniparental disomy chromosome 14: case report and literature review. PediatrNeurol. 2005;32(2):116–120. [DOI] [PubMed] [Google Scholar]
- 62.Temple K, Shrubb V, Lever M, et al. Isolated imprinting mutation of the DLK1/GTL2 locus associated with a clinical presentation of maternal uniparental disomy of chromosome 14. J Med Genet. 2007;44:637–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zakharova IS, Shevchenko AI, Zakian SM. Monoallelic gene expression in mammals. Chromosoma. 2009;118(3):279–290. [DOI] [PubMed] [Google Scholar]
- 64.Nicholls RD, Knepper JL. Genome organization, function, and imprinting in Prader–Willi and Angelman syndromes. AnnuRevGenomicsHumGenet. 2001;2:153–175. [DOI] [PubMed] [Google Scholar]
- 65.Christian SL, Robinson WP, Huang B, et al. Molecular characterization of two proximal deletion breakpoint regions in both Prader–Willi and Angelman syndrome patients. Am J Hum Genet. 1995;57(1):40–48. [PMC free article] [PubMed] [Google Scholar]
- 66.Bittel DC, Butler MG. Prader–Willi syndrome: clinical genetics, cytogenetics and molecular biology. ExpertRev Mol Med. 2005;7(14):1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Butler MG, Fischer W, Kibiryeva N, et al. Array comparative genomic hybridization (aCGH) analysis in Prader–Willi syndrome. AmJMed Genet A. 2008;146(7):854–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Butler MG, Christian SL, Kubota T, et al. A 5-year-old white girl with Prader–Willi syndrome and a submicroscopic deletion of chromosome 15q11q13. AmJ Med Genet. 1996;65(2):137–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Butler MG, Bittel DC, Kibiryeva N, et al. An interstitial 15q11-q14 deletion: expanded Prader–Willi syndrome phenotype. AmJMed Genet A. 2010;152A(2):404–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kim SJ, Miller JL, Kuipers PJ, et al. Unique and atypical deletions in Prader–Willi syndrome reveal distinct phenotypes. Hum Mol Genet. 2012;20(3):283–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Butler MG, Bittel DC, Talebizadeh Z. Plasma peptide YY and ghrelin levels in infants and children with Prader–Willi syndrome. J Pediatr Endocrinol Metab. 2004;17: 1177–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zarcone J, Napolitano D, Peterson C, et al. The relationship between compulsive behaviour and academic achievement across the three genetic subtypes of Prader–Willi syndrome. J Intellect Disabil Res. 2007;51(pt 6):478–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bittel DC, Kibiryeva N, Butler MG. Expression of4 genes between chromosome 15 breakpoints 1 and 2 and behavioral outcomes in Prader–Willi syndrome. Pediatrics. 2006;118(4):e1276–e1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dykens EM, Roof E. Behavior in Prader–Willi syndrome: relationship to genetic subtypes and age. J Child Psychol Psychiatry. 2008;49(9):1001–1008. [DOI] [PubMed] [Google Scholar]
- 75.Chai JH, Locke DP, Greally JM, et al. Identification of four highly conserved genes between breakpoint hotspots BP1and BP2 of the Prader–Willi/Angelman syndromes deletion region that have undergone evolutionary transposition mediated by flanking duplicons. AmJHum Genet. 2003;73(4):898–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Svenstrup K, Moller RS, Christensen J, et al. NIPA1 mutation in complex hereditary spastic paraplegia with epilepsy. EurJ Neurol. 2011;18(9):1197–1199. [DOI] [PubMed] [Google Scholar]
- 77.Jiang Y, Zhang Y, Zhang P, et al. NIPA2 located in 15q11. 2 is mutated in patients with childhood absence epilepsy. Hum Genet. 2012;131(7):1217–1224. [DOI] [PubMed] [Google Scholar]
- 78.Burnside RD, Pasion R, Mikhail FM, et al. Microdeletion/microduplication of proximal 15q11.2 between BP1 and BP2: a susceptibility region for neurological dysfunction including developmental and language delay. Hum Genet. 2011;130(4): 517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cox DM, Butler MG. The 15q11.2 BP1-BP2 microdeletion syndrome: a review. Int J Mol Sci. 2015;16(2):4068–4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cassidy SB, Lai LW, Erickson RP, et al. Trisomy 15 with loss of the paternal 15asa cause of Prader–Willi syndrome due to maternal disomy. AmJHumGenet. 1992;51:701–708. [PMC free article] [PubMed] [Google Scholar]
- 81.Sahoo T, del Gaudio D, German JR, et al. Prader–Willi phenotype caused by paternal deficiency for the HBII-85 C/D box small nucleolar RNA cluster. Nat Genet. 2008;40 (6):719–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Schaaf CP, Gonzalez-Garay ML, Xia F, et al. Truncating mutations of MAGEL2 cause Prader–Willi phenotypes and autism. Nat Genet. 2013;45(11):1405–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Macedo DB, Abreu AP, Reis AC, et al. Central precocious puberty that appears to be sporadic caused by paternally inherited mutations in the imprinted gene makorin ring finger 3. J Clin Endocrinol Metab. 2014;99(6):E1097–E1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.de Smith AJ, Purmann C, Walters RG, et al. A deletion of the HBII-85 class of small nucleolar RNAs (snoRNAs) is associated with hyperphagia, obesity and hypogonadism. Hum Mol Genet. 2009;18(17):3257–3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cummings DE, Clement K, Purnell JQ, et al. Elevated plasma ghrelin levels in Prader–Willi syndrome. Nat Med. 2002;8(7):643–644. [DOI] [PubMed] [Google Scholar]
- 86.Irizarry K, Bain J, Butler MG, et al. Metabolic profiling in Prader–Willi syndrome and non-syndromic obesity: sex differences and the role of growth hormone treatment. Clin Endocrinol (Oxf). 2015;83(6):797–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kishore S, Stamm S. The snoRNA HBII-52 regulates alternative splicing of the serotonin receptor 2C. Science. 2006;311(5758):230–232. [DOI] [PubMed] [Google Scholar]
- 88.Bittel DC, Kibiryeva N, Sell SM, et al. Whole genome microarray analysis of gene expression in Prader–Willi syndrome. Am J Med Genet. 2007;143A(5):430–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Miller JL, Lynn CH, Driscoll DC, et al. Nutritional phases in Prader–Willi syndrome. AmJMed Genet A. 2011;155A(5):1040–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Butler MG, Meaney FJ, Palmer CG. Clinical and cytogenetic survey of 39 individuals with Prader–Labhart–Willi syndrome. Am J Med Genet. 1986;23(3):793–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Butler MG, Sturich J, Lee J, et al. Growth standards of infants with Prader–Willi syndrome. Pediatrics. 2011;127(4):687–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dykens EM. Are jigsaw puzzle skills “spared” in persons with Prader–Willi syndrome? J Child Psychol Psychiatry. 2002;43(3):343–352. [DOI] [PubMed] [Google Scholar]
- 93.Stevenson DA, Heinemann J, Angulo M, et al. Gastric rupture and necrosis in Prader–Willi syndrome. JPediatrGastroenterolNutr. 2007;45(2):272–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.McCandless SE. Clinical report-health supervision for children with Prader–Willi syndrome. Pediatrics. 2011;127(1):195–204. [DOI] [PubMed] [Google Scholar]
- 95.Butler MG. Management of obesity in Prader–Willi syndrome. Nat ClinPractEndocrinol Metab. 2006;2(11):592–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Angulo M, Castro-Magana M, Mazur B, et al. Growth hormone secretion and effects of growth hormone therapy on growth velocity and weight gain in children with Prader–Willi syndrome. J Pediatr Endocrinol Metab. 1996;9(3):393–400. [DOI] [PubMed] [Google Scholar]
- 97.Carrel AL, Myers SE, Whitman BY, et al. Long-term growth hormone therapy changes the natural history of body composition and motor function in children with Prader–Willi syndrome. JClin Endocrinol Metab. 2010;95:1131–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Eiholzer U, l’Allemand D. Growth hormone normalizes height, prediction of final height and hand length in children with Prader–Willi syndrome after 4 years of therapy. HormRes. 2000;53(4):185–192. [DOI] [PubMed] [Google Scholar]
- 99.Höybye C, Thomn M, Bohm B. Cognitive, emotional, physical and social effects of growth hormone treatment in adults with Prader–Willi syndrome. JIntellectDisabilRes. 2005;49:245–252. [DOI] [PubMed] [Google Scholar]
- 100.Butler MG, Smith BK, Lee J, et al. Effects of growth hormone treatment in adults with Prader–Willi syndrome. Growth Horm IGF Res. 2013;23(3):81–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Deal CL, Tony M, Hoybye C, et al. Growth hormone research society workshop summary: consensus guidelines for recombinant human growth hormone therapy in Prader–Willi syndrome. JClin Endocrinol Metab. 2013;98:E1072–E1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Butler MG, Lee J, Manzardo AM, et al. Growth charts for non-growth hormone treated Prader–Willi syndrome. Pediatrics. 2015;135(1):e126–e135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.De Vries BB, Fryns JP, Butler MG, et al. Clinical and molecular studies in fragile X patients with a Prader–Willi-like phenotype. J Med Genet. 1993;30(9):761–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hagerman PJ, Hagerman RJ. Fragile X-associated tremor/ataxia syndrome. Ann NY Acad Sci. 2015;1338(1):58–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Niu YQ, Yang JC, Hall DA, et al. Parkinsonism in fragile X-associated tremor/ataxia syndrome (FXTAS): revisited. Parkinsonism Relat Disord. 2014;20(4):456–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nowicki ST, Tassone F, Ono MY, et al. The Prader–Willi phenotype of fragile X syndrome. J Dev Behav Pediatr. 2007;28(2):133–138. [DOI] [PubMed] [Google Scholar]
- 107.Butler MG, Wang K, Marshall JD, et al. Coding and noncoding expression patterns associated with rare obesity-related disorders: Prader–Willi and Alström syndromes. Adv Genomics Genet. 2015;5:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bastepe M The GNAS locus and pseudohypoparathyroidism. Adv Exp Med Biol. 2008;626:27–40. [DOI] [PubMed] [Google Scholar]
- 109.Fitch N Albright’s hereditary osteodystrophy: a review. Am J Med Genet. 1982;11 (1):11–29. [DOI] [PubMed] [Google Scholar]
- 110.Jüppner H. Genetic and epigenetic defects at the GNAS locus cause different forms of pseudohypoparathyroidism. Annales d’endocrinologie. 2015;76(2):92–97. [DOI] [PubMed] [Google Scholar]
- 111.Mantovani G, Elli FM, Spada A. GNAS epigenetic defects and pseudohypoparathyroidism: time for a new classification? Horm Metab Res. 2012;44(10):716–723. [DOI] [PubMed] [Google Scholar]
- 112.Bastepe M, Juppner H. GNAS locus and pseudohypoparathyroidism. Horm Res Paediatr. 2005;63(2):65–74. [DOI] [PubMed] [Google Scholar]