

Abstract
Although voltage-gated Ca2+ channels (VGCC) are a major Ca2+ entry pathway in vascular smooth muscle cells (VSMCs), several other Ca2+-influx mechanisms exist and play important roles in vasoreactivity. One of these is store-operated Ca2+ entry (SOCE), mediated by an interaction between STIM1 and Orai1. Although SOCE is an important mechanism of Ca2+ influx in non-excitable cells (cells that lack VGCC); there is debate regarding the contribution of SOCE to regulate VSMC contractility and the molecular components involved. Our previous data suggest acid-sensing ion channel 1a (ASIC1a) is a necessary component of SOCE and vasoconstriction in small pulmonary arteries. However, it is unclear if ASIC1a similarly contributes to SOCE and vascular reactivity in systemic arteries. Considering the established role of Orai1 in mediating SOCE in the systemic circulation, we hypothesize the involvement of ASIC1a in SOCE and resultant vasoconstriction is unique to the pulmonary circulation. To test this hypothesis, we examined the roles of Orai1 and ASIC1a in SOCE- and endothelin-1 (ET-1)-induced vasoconstriction in small pulmonary and mesenteric arteries. We found SOCE is coupled to vasoconstriction in pulmonary arteries but not mesenteric arteries. In pulmonary arteries, inhibition of ASIC1a but not Orai1 attenuated SOCE- and ET-1-induced vasoconstriction. However, neither inhibition of ASIC1a nor Orai1 altered ET-1-induced vasoconstriction in mesenteric arteries. We conclude that SOCE plays an important role in pulmonary, but not mesenteric, vascular reactivity. Furthermore, in contrast to the established role of Orai1 in SOCE in non-excitable cells, the SOCE response in pulmonary VSMCs is largely mediated by ASIC1a.
Introduction
Vascular smooth muscle cell (VSMC) contraction and relaxation play an important role in the regulation of vascular resistance and blood pressure control. It is well established that contraction is triggered by an increase in intracellular free calcium concentration ([Ca2+]i) mediated by a rapid Ca2+ release from intracellular stores and transmembrane Ca2+ influx through a variety of plasma membrane ion channels, exchangers, and transporters. Although Ca2+ influx in VSMC is thought to be mediated primarily by L-type voltage-gated Ca2+ channels (VGCC), it has become increasingly clear that Ca2+ influx through non-selective cation channels (NSCC) plays an important role in regulating vascular tone. These include 1) receptor-operated channels (ROCs) which are regulated by agonist-receptor interaction and downstream signal transduction [1]; 2) capacitative or store-operated channels (SOCs) which are activated by depletion of intracellular Ca2+ stores [2, 3]; 3) mechanosensitive or stretch-activated channels (SACs) which are activated by membrane stretch [4, 5]; and 4) constitutively active cation channels which are spontaneously active (reviewed in [6]). However, the molecular identity and the functional role of these channels to mediate vasoconstriction is still a matter of debate. Complicating matters is the fact that multiple Ca2+-permeable channels are expressed in a given vascular bed and the functional relevance of different Ca2+ channels within the vascular bed remains unclear. Therefore, it is necessary to gain a better understanding of the heterogeneity that exists in Ca2+ signaling among vascular beds.
Increases in VSMC [Ca2+]i in response to neurohumoral stimuli (norepinephrine/epinephrine) and vasoactive peptides (angiotensin II and endothelin-1) results from activation of phospholipase C (PLC) associated G-protein coupled receptors [7]. Activation of PLC leads to the hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2) generating the second messengers, inositol trisphosphate (IP3) and diacylglycerol (DAG). IP3 activates IP3 receptors (IP3R) on the sarcoplasmic reticulum (SR) and stimulates Ca2+ release. Plasma membrane SOCs are activated secondary to depletion of SR Ca2+ stores, a mechanism known as store-operated Ca2+ entry (SOCE) or capacitative Ca2+ entry. SOCE facilitates the refilling of Ca2+-depleted SR stores and is therefore critical to SR Ca2+ homeostasis [8]. In VSMCs, SOCE is known to participate in other physiological processes such as vascular tone regulation [9, 10], vasculogenesis, and cell proliferation [10, 11]. However, the coupling of SOCE to contraction has been shown to differ widely among vascular beds. Snetkov et al. demonstrated that even though induction of SOCE results in similar increases in [Ca2+]i in intrapulmonary, mesenteric, renal, femoral, and coronary arteries; the corresponding contraction was only observed in intrapulmonary arteries [12]. As the potential differential responses to SOCE between various vascular beds are likely due to regional differences in the molecular components of SOCE, the objective of this study is to identify the ion channels involved in coupling SOCE to vasoconstriction.
Stromal interaction molecule 1 (STIM1) is the key molecule involved in sensing levels of Ca2+ in the SR [13, 14]. Upon store depletion, STIM1 undergoes a conformational change, multimerizes, and translocate to regions of the SR adjacent to the plasma membrane where subsequent binding of STIM1 to Ca2+-permeable channels triggers the influx of Ca2+ across the plasma membrane [15]. Two different types of ionic current are evoked by store-depletion: 1) a high selectivity Ca2+ current mediated by a Ca2+ release-activated channel (CRAC) called Orai1 [16] and 2) NSCC current [17, 18]. Several members of the transient receptor potential canonical (TRPC) channel family have been proposed to act as SOCs (reviewed in [19, 20]), although this topic continues to be widely debated [21–23]. Previous work from our laboratory has shown that inhibition of acid-sensing ion channel 1a (ASIC1a) diminishes SOCE and associated vasoconstriction in pulmonary VSMC [24]. ASIC1a is a NSCC that belongs to the amiloride-sensitive degenerin/epithelial sodium channel (DEG/ENaC) superfamily. ASICs are known to be permeable to Na+, however homomeric ASIC1a channels can also conduct Ca2+ [25–27]. Although the ASICs are classically activated by extracellular acidosis; various non-proton ligands, effector proteins, and signaling molecules also regulate the function of ASICs [28, 29]. It is unclear if ASIC1a similarly contributes to SOCE and vascular reactivity in systemic arteries. Considering the established role of Orai1 in mediating SOCE in the systemic circulation, we hypothesize the involvement of ASIC1a in SOCE and resultant vasoconstriction is unique to the pulmonary circulation. To test this hypothesis, we have examined the roles of ASIC1a and Orai1 in SOCE- and endothelin-1-induced vasoconstriction in both small pulmonary and mesenteric arteries.
Materials and methods
All protocols employed were reviewed and approved by the Institutional Animal Care and Use Committee of the University of New Mexico School of Medicine (Albuquerque, NM) and abide by the National Institutes of Health guidelines for animal use. Fifty-two adult male Wistar rats (200–250 g body wt, Envigo) were used in this study. Animals were housed in polyacrylic cages (1–3 per cage) supplied with bedding (shredded paper) and polycarbonate rodent tunnels and other items for environmental enrichment. Animals were housed in a specific pathogen-free animal care facility and maintained on a 12:12 hour light-dark cycle. Water and standard chow (Teklad soy protein-free diet no. 2920; Envigo) were provided ad libitum. Rats were anesthetized with an overdose of pentobarbital sodium (200 mg/kg ip) and immediately euthanized by exsanguination after the loss of consciousness.
Assessment of SOCE and vasoreactivity in isolated, pressurized pulmonary and mesenteric resistance arteries
To determine simultaneous changes in vasoreactivity and [Ca2+]i, small resistance arteries were cannulated and pressurized for dimensional and fluorescence analysis as previously described [24, 30]. Following euthanasia, the lungs or mesentery were removed and immediately placed in PSS [pH adjusted to 7.4 with NaOH containing (in mM) 130 NaCl, 4 KC1, 1.2 MgSO4, 4 NaHCO3, 1.8 CaC12, 10 HEPES, 1.18 KH2PO4, 6 glucose]. Fourth- to fifth-order pulmonary (~ 125 μm inner diameter) and third- to fourth-order mesenteric arteries (~ 150 μm inner diameter; Table 2); of ∼1-mm length and without visible side branches were dissected free and transferred to a vessel chamber (CH-1, Living Systems). The proximal end of the artery was cannulated with a tapered glass pipette, secured in place with a single strand of silk ligature, and gently flushed to remove any blood from the lumen. The vessel was stretched longitudinally to approximate in situ length and pressurized with a servo-controlled peristaltic pump (Living Systems) to 12 mmHg (pulmonary) or 75 mmHg (mesenteric). Arteries were required to hold steady pressure on switching off the servo-control function to verify the absence of leaks; any vessel with apparent leaks was discarded. The vessel chamber was superfused with PSS; at 5 ml/min at 37°C. Images were obtained using an Eclipse TS100 microscope (Nikon) and IonOptix CCD100M camera to measure inner diameter, and dimensional analysis was performed by IonOptix Ion Wizard software (IonOptix). Arteries were incubated at room temperature with PSS containing the cell-permeable ratiometric Ca2+-sensitive fluorescent dye fura-2 acetoxymethyl ester (fura-2 AM, 2 μM; Life Technologies, F1201, lot #2021739) and 0.02% pluronic acid (Life Technologies, P3000MP, lot #1990297) for 45 min, as previously described [31]. Fura-2-loaded vessels were alternately excited at 340 and 380 nm at a frequency of 1 Hz with an IonOptix Hyperswitch dual-excitation light source, and the respective 510-nm emissions were collected with a photomultiplier tube. After subtracting background fluorescence, emission ratios (F340/F380) were calculated with Ion Wizard software (IonOptix) and recorded continuously throughout the experiment.
Table 2. Baseline inner diameter and vessel wall [Ca2+]i in pulmonary and mesenteric arteries treated with vehicle, diltiazem (50 μM), PcTX1 (20 nM), or AnCoA4 (20 μM).
| Pulmonary | Mesenteric | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Diameter | P-Value | Fura-2 ratio (F340/F380) | P-Value | N | Diameter | P-Value | Fura-2 ratio (F340/F380) | P-Value | N | |
| (μm) | (μm) | |||||||||
| Vehicle | 124 ± 7 | 0.79 ± 0.06 | 7 | 149 ± 15 | 0.66 ± 0.06 | 6 | ||||
| Diltiazem | 125 ± 19 | >0.9999 | 0.61 ± 0.11 | 0.9665 | 4 | 165 ± 12 | 0.8575 | 0.64 ± 0.02 | 0.9823 | 6 |
| PcTX1 | 119 ± 9 | 0.9763 | 0.82 ± 0.06 | 0.9699 | 7 | 166 ± 11 | 0.8648 | 0.75 ± 0.03 | 0.5156 | 5 |
| AnCoA4 | 122 ± 11 | 0.9982 | 0.78 ± 0.06 | 0.9999 | 7 | 139 ± 17 | 0.9535 | 0.59 ± 0.05 | 0.6305 | 7 |
Experiments were conducted in the absence or presence of the L-type VGCC inhibitor, diltiazem (50 μM; Sigma, D2521, lot #MKCD7486); the specific ASIC1a inhibitor, psalmotoxin 1 (PcTX1; 20 nM; Phoenix Peptides, 063–22, lot #433599); or the specific Orai1 inhibitor, AnCoA4 (20 μM; Millipore Sigma, 532999, lot #3030527). We have previously demonstrated that this concentration of diltiazem (50 μM) prevents increases in the vessel [Ca2+]i and vasoconstriction-induced by the depolarizing stimulus, KCl (50 mM) [32]. PcTX1 has been reported to selectively inhibit ASIC1a isoform over other ASICs [33]. Furthermore, we have previously shown that this concentration of PcTX1 has similar effects to those seen in pulmonary VSMCs of ASIC1 null mice [34, 35]. AnCoA4 reduces the Orai1 association to STIM1 and consequently blocks SOCE [36]. We have previously determined this concentration of AnCoA4 inhibits SOCE in pulmonary arterial endothelial cells and pulmonary microvascular endothelial cells [37].
Store-operated Ca2+ entry
Fura-2-loaded arteries were superfused with Ca2+-free, PSS (in mM: 130 NaCl, 4 KC1, 1.2 MgSO4, 4 NaHCO3, 10 HEPES, 1.18 KH2PO4, 6 glucose, 3 EGTA; pH adjusted to 7.4 with NaOH) containing 50 μM diltiazem to prevent Ca2+ entry through L-type VGCC, and 10 μM cyclopiazonic acid (CPA; Calbiochem, 23905) to deplete intracellular Ca2+ stores and prevent Ca2+ reuptake through the sarcoplasmic reticulum Ca2+-ATPase for 15 minutes before replenishing the perfusate with Ca2+. The changes in [Ca2+]i were determined upon the repletion of HEPES-based PSS containing 1.8 mM CaCl2 in the continued presence of diltiazem and CPA. SOCE was calculated as the change (Δ) in fura-2 ratio between Ca2+-depleted state and Ca2+-depleted state.
Endothelin-1 responses
Endothelin-1 (ET-1; Sigma-Aldrich, E7764, lot #088M4849V) induced vasoconstrictor reactivity and changes in the vessel wall [Ca2+]i were assessed by superfusion (5 ml/min at 37°C) of cumulative concentrations of ET-1 in isolated pulmonary (10−11 to 10−7 M) and mesenteric (10−11 to 10−8 M) arteries.
Generation of primary pulmonary and mesenteric smooth muscle cell cultures
Intrapulmonary and mesenteric arteries (~2nd-5th order) were dissected from surrounding tissue and enzymatically digested. Pulmonary arteries were digested in reduced-Ca2+ Hank’s balanced salt solution (HBSS) containing papain (26 U/ml), type-I collagenase (1,750 U/ml), dithiothreitol (1 mg/ml), and BSA (2 mg/ml) at 37°C for 30 min. Mesenteric arteries were digested in reduced-Ca2+ HBSS containing HEPES (15 mM), elastase (11.25 U/ml), soybean trypsin inhibitor type 1-S (1 mg/ml), type-I collagenase (180 U/ml), and BSA (2 mg/ml) at 37°C for 45 min. Single smooth muscle cells were dispersed by gentle trituration with a fire-polished pipette in Ca2+-free HBSS. Pulmonary VSMC were plated in Ham’s F-12 media supplemented with 5% fetal bovine serum and 1% penicillin/streptomycin. Mesenteric VSMC were plated in DMEM media supplemented with 10% fetal bovine serum, L-glutamine (2 mM), HEPES buffer (25 mM), and 1% penicillin/streptomycin. All cells were grown in a humidified atmosphere of 5% CO2-95% air at 37°C. Pulmonary and mesenteric VSMC purity was verified by morphological appearance and the presence of smooth muscle 22α (transgelin; S1 Fig). Furthermore, PCR showed the appearance of smooth muscle α-actin and lacked expression of the neuronal marker, calcitonin gene-related peptide (S1 Fig).
Determination of ASIC1a and Orai1 expression
RT-PCR
Total RNA was extracted using TRIzol from pulmonary and mesenteric arteries. Brain tissue was used as a positive control. One μg of total RNA was reversed transcribed into cDNA using the Transcription First-Strand cDNA Synthesis kit (Roche). PCR was performed on cDNA with the iCycler PCR system (Bio-Rad) using REDExtract-N_Amp PCR ReadyMix (Sigma) and specific primers (Table 1) to detect transcripts for ASIC1a, Orai1, and β-actin. PCR products were separated using gel electrophoresis on a 3% agarose gel and stained with ethidium bromide for visualization under UV light.
Table 1. Primers and base pair (bp) product size used for RT-PCR for ASIC1a, Orai1, and β-actin.
| Primer Pair Sequence | Product Size, bp | |
|---|---|---|
| Orai1 | 357 bp | |
| Forward | 5’-ACGTCCACAACCTCAACTCC-3’ | |
| Reverse | 5’-ACTGTCGGTCCGTCTTATGG-3’ | |
| ASIC1a | 305 bp | |
| Forward | 5’-GCCTATGAGATCGCAGGG-3’ | |
| Reverse | 5’-AAAGTCCTCAAACGTGCCTC-3’ | |
| β-actin | 244 bp | |
| Forward | 5’-AGTGTGACGTTGACATCCGT-3’ | |
| Reverse | 5’-GACTCATCGTACTCCTGCTT-3’ |
Western blotting analysis
Orai1 and ASIC1a protein expression were determined by Western blot analysis. Pulmonary and mesenteric arteries were homogenized in 10 mM Tris·HCl homogenization buffer (containing 255 mM sucrose, 2 mM EDTA, 12 μM leupeptin, 1 μM pepstatin A, and 0.3μM aprotinin) with a glass homogenizer. The lysate (60 μg) was separated by SDS-PAGE (7.5% or 12% Tris/glycine) and transferred to a polyvinylidene difluoride membrane. The blot was blocked for 1 h with 5% milk and then incubated at 4°C with rabbit anti-Orai1 (1.5 hrs @ 1:300; Proteintech: 14443-1-AP; expected MW 35–44 kDa) or rabbit anti-ASIC1a (48 hrs @ 1:500; Millipore: AB5674P; reported MW ~60 and ~100 kDa). For immunochemical labeling, blots were incubated with anti-rabbit IgG-horseradish peroxidase (1 hr @ 1:3,000; Bio-Rad). Following chemiluminescence labeling (ECL; Pierce), Proteins were detected by exposing the blot to chemiluminescence-sensitive film (GeneMate).
Determination of ASIC1a-STIM1 and Orai1-STIM1 colocalization
Protein-protein interactions were determined in smooth muscle cells using Duolink in situ proximity ligation assay (PLA) as previously described [34, 38]. Cells were plated on 18-well slides (Ibidi) and grown until ~75% confluent. In some experiments, cells were pretreated with Ca2+ free PSS plus CPA to induce SOCE before the cells were fixed with 2% paraformaldehyde. Following fixation, samples were incubated with Duolink blocking buffer for 30 min at 37°C then incubated overnight with mouse anti-STIM1 (1:50; BD Biosciences 610954) and goat anti-ASIC1a (1:50; Santa Cruz Biotechnology: sc-13903) or rabbit anti-Orai1 (1:100; Alomone ACC-062). We have previously determined the specificity of goat anti-ASIC1a using wild-type and knockout mice [39], and the specificity of rabbit anti-Orai1 using control and siRNA-treated cells [37]. Cells were then incubated with anti-mouse PLUS and anti-goat MINUS or anti-rabbit MINUS PLA probes (1:5) for 1 h at 37°C. Negative controls were completed by incubation of each primary antibody individually. Samples were amplified with Duolink In Situ Detection Reagent Orange (excitation/emission: 554/579 nm; Sigma- Aldrich) for 100 min at 37°C. SYTOX Green (1:10,000; Invitrogen) was used as a nuclear stain and actin was stained with Alexa Fluor 647 Phalloidin (1:100; Invitrogen). Samples were mounted with Duolink mounting media and Z-stack images of the PLA interaction were acquired using a confocal microscope (TCS SP5; Leica). Each puncta was considered a positive protein-protein interaction. The number and size (pixel2) of puncta per cell were determined using ImageJ (National Institutes of Health).
Calculations and statistics
All data are expressed as means ± SE. Values of n refer to the number of animals in each group unless otherwise stated. Statistical significance was tested at the 95% (P < 0.05) confidence level using an unpaired t-test, one-way analysis of variance (ANOVA), or two-way ANOVA as appropriate (GraphPad Prism). If differences were detected by ANOVA, individual groups were compared with the recommended post-hoc test which is specified in the figure legends. Normal distribution was tested using the Shapiro-Wilks Normality Test (P > 0.05), any data sets that were not normally distributed were analyzed by non-parametric analysis. Data that were represented as percent were normalized by arcsine transformation before statistical analysis.
Results
L-type Ca2+ channels contribute to ET-1-induced constriction of mesenteric but not pulmonary arteries
Since L-type Ca2+ channels are considered to be a major contributor to VSMC Ca2+ influx, we first determined the role of L-type VGCC to ET-1 induced vasoconstriction in small pulmonary and mesenteric arteries. In pulmonary arteries, inhibition of L-type VGCC with diltiazem did not significantly alter baseline diameter or vessel wall [Ca2+]i (Table 2). Furthermore, diltiazem did not affect vasoconstriction (Fig 1A, top panel) or changes in vessel wall Ca2+ (Fig 1A, bottom panel) in response to increasing doses of ET-1. In mesenteric arteries, inhibition of L-type VGCC with diltiazem did not significantly alter baseline diameter or vessel wall [Ca2+]I (Table 2). However, ET-1-induced vasoconstriction in mesenteric arteries was significantly attenuated by diltiazem (Fig 1B, top panel) and tended to decrease changes in vessel wall Ca2+ but was not statistically significant (Fig 1B, bottom panel).
Fig 1. L-type Ca2+ channels contribute to ET-1 induced vasoconstriction in mesenteric but not pulmonary arteries.

Representative traces (top panels) and summary data showing vasoconstriction (percent baseline inner diameter; middle panels) and changes in fura-2 ratio (F340/F380; bottom panels) in response to endothelin-1 (ET-1; 10−11–10−7 or 10−11–10−8) in the presence or absence of diltiazem (50 μM) in small pulmonary (A; left panels) or mesenteric (B; right panels) arteries. n = 4–6 per group, values are means ± SEM. *p ≤ 0.05 vs. vehicle; ** p < 0.01; analyzed by two-way ANOVA followed by Sidak’s multiple comparisons test.
Orai1 and ASIC1a mRNA and protein expression in pulmonary and mesenteric arteries
We next evaluated mRNA and protein expression of Orai1 and ASIC1 in small pulmonary and mesenteric arteries. We found Orai1 and ASIC1 transcripts (Fig 2A) and protein (Fig 2B) were expressed in both pulmonary and mesenteric arteries. Brain tissue was used as a positive control. Orai1 was detected around the expected MW of 35 kDa and a larger band around 75 kDa (a potential dimer). Similar to previous reports [40], we detect two bands for ASIC1 protein, ~100 kDa and 60 kDa. Interestingly, the 60 kDa band is more intense in mesenteric arteries compared to pulmonary arteries or brain samples.
Fig 2. Orai1 and ASIC1a are expressed in pulmonary and mesenteric arteries and VSMC.
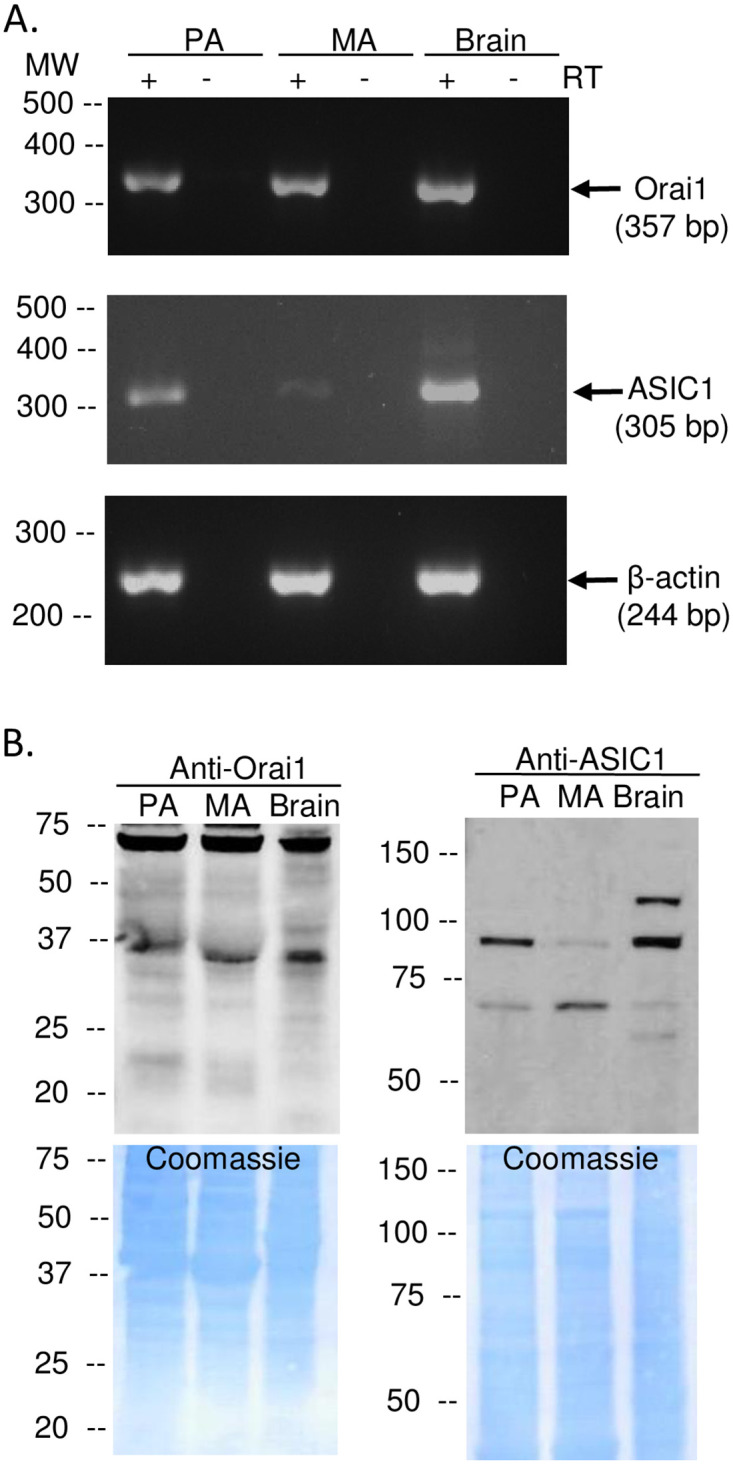
Representative PCR gels (A) showing expression of Orai1 (357 bp) and ASIC1a (305 bp) in intact pulmonary arteries (PA), mesenteric arteries (MA), and brain tissue (positive control). All lanes were loaded with 5 μL cDNA. β-actin (244 bp) was used as a loading control for intact arteries. Representative western blots (B left) showing protein expression of Orai1 and ASIC1 (~100 kDa and 60 kDa) in intact PA, MA, and brain tissue (positive control). Coomassie blue staining shows equal protein loading between samples (B; right). Each experiment was replicated 3 times.
Store-operated Ca2+ entry is coupled to vasoconstriction in pulmonary but not mesenteric arteries
Induction of SOCE in isolated pressurized small pulmonary arteries resulted in a substantial and sustained increase in [Ca2+]i (Fig 3A and 3B) that was associated with an ~25% vasoconstriction (Fig 3C and 3D). In mesenteric arteries, induction of SOCE also resulted in a sustained increase in [Ca2+]i. However, SOCE was significantly less compared to pulmonary arteries (Fig 3A and 3B). Furthermore, SOCE was not associated with a significant change in vessel inner diameter in mesenteric arteries (Fig 3C and 3D).
Fig 3. Store-operated Ca2+ entry is coupled to vasoconstriction in pulmonary but not mesenteric arteries.

Representative traces and summary data of SOCE (A & B) and SOCE-induced vasoconstriction (C & D) in pulmonary and mesenteric resistance arteries. All studies were done in the presence of diltiazem (50μM) and cyclopiazonic acid (10μM). n = 7–10 animals/group. ***p ≤ 0.001 or **** p ≤ 0.0001 vs. pulmonary; analyzed by unpaired t-test.
Store-operated Ca2+ entry is dependent on ASIC1a in pulmonary but not mesenteric arteries
To determine the contribution of Orai1 and ASIC1a to SOCE in small pulmonary and mesenteric arteries, we repeated experiments in the presence of the ASIC1a inhibitor, PcTX1; or the Orai1 inhibitor, AnCoA4. Inhibition of ASIC1a largely attenuated the SOCE and associated vasoconstrictor responses in pulmonary arteries; however, Orai1 inhibition did not alter responses to store depletion compared to the vehicle (Fig 4A). In contrast, we observed a minimal SOCE response, in mesenteric arteries, which did not induce vasoconstriction. SOCE was not altered by PcTX1 in mesenteric arteries. Although AnCoA4 tended to blunt SOCE in mesenteric arteries, this effect was not significant (P = 0.0717). Neither PcTX1 nor AnCoA4 affected the lack of vasoconstriction in mesenteric arteries. However, the power of statistical comparison is low (0.435) in mesenteric arteries and may be due to the small effect size that we see in the mesenteric SOCE response. Together, these data demonstrate SOCE in mesenteric arteries is a negligible Ca2+ influx pathway compared to pulmonary arteries.
Fig 4. Store-operated Ca2+ entry is dependent on ASIC1a in pulmonary but not mesenteric arteries.

SOCE-induced changes in fura-2 ratio (Δ F340/F380; top panels) and associated vasoconstriction (% baseline; bottom panels) in small pulmonary and mesenteric arteries, respectively. Studies were conducted in the presence or absence of PcTX1 (20 nM) or AnCoA4 (20 μM). Vehicle data are the same as Fig 3. n = 5–10 animals/group. **p ≤ 0.01 vs. vehicle; *** p ≤ 0.001 vs. vehicle; analyzed by one-way ANOVA and individual groups compared with the Tukey’s multiple comparison test.
ASIC1a and Orai1 interact with STIM1 in VSMC
As Ca2+ levels in the SR decrease, STIM1 undergoes a conformational change, multimerizes, and translocates to regions of the SR adjacent to the plasma membrane where subsequent binding of STIM1 to Ca2+-permeable channels triggers an influx of Ca2+ across the plasma membrane [15]. Thus we first examined the mRNA and protein levels of STIM1 in pulmonary and mesenteric arteries and found that STIM1 was expressed in both vascular beds (S2 Fig). Furthermore, an examination of STIM1, Orai1, and ASIC1 mRNA showed expression in both primary cultured pulmonary and mesenteric VSMCs (S3 Fig). Therefore, we examined the potential interaction and clustering of STIM1 with both Orai1 and ASIC1a in pulmonary and mesenteric VSMCs using Duolink proximity ligation assay. Under basal conditions, we found that Orai1 interacts with STIM1 in both pulmonary (Fig 5A and 5C) and mesenteric (Fig 5D and 5F) VSMCs. Upon store depletion with CPA in pulmonary VSMC, there was neither a difference in the number of puncta (interactions) per cell (Fig 5B) nor puncta size (Fig 5C). In mesenteric VSMC there tended to be both an increase in the number of puncta per cell (p = 0.0593) and a significant increase in puncta size, suggesting an increase in Orai1-STIM1 clustering. However, the size of the puncta is substantially smaller than the puncta measured in pulmonary arteries. The changes observed in mesenteric VSMC may be due to possible phenotypic changes that can occur in VSMs in cell culture from a quiescent to the proliferative state. Furthermore, the power of statistical comparison was lower than expected when analyzing the number of puncta per cell in mesenteric VSMCs (0.623).
Fig 5. Store depletion increases the interaction between Orai1 and STIM1 in VSMCs.
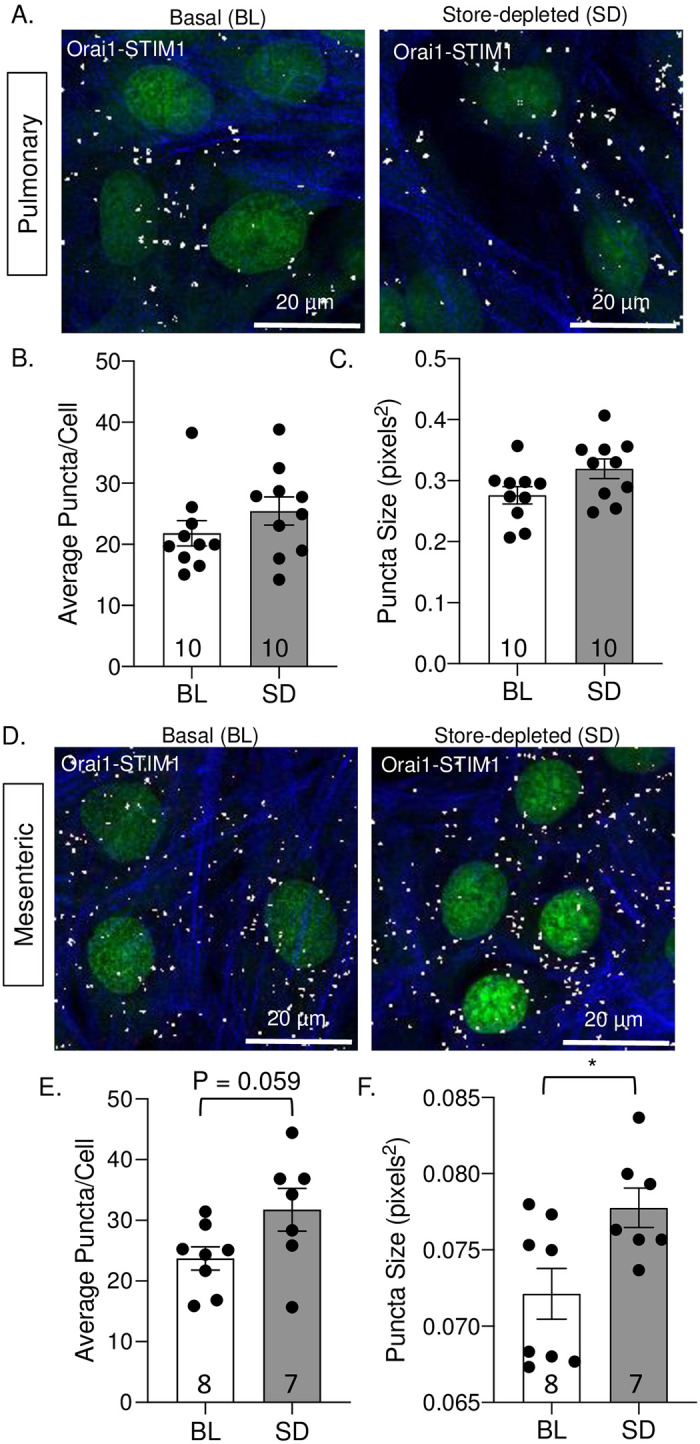
Representative images of Orai1/STIM1 interaction in pulmonary (A) and mesenteric (D) VSMCs under basal conditions (BL; left) or following store-depletion (SD; right). Summary data showing the average number of puncta per cell (B and E) and puncta size (C and F) in pulmonary (B-C) and mesenteric (E-F) VSMCs. The nuclei are labeled with SYTOX (green), actin is labeled with Alexa Fluor 647 Phalloidin (blue), and puncta formation (white). N = 7–10; each data point represents an individual well taken from 1–3 experiments (cell cultures generated from different animals and ran on different days). *P ≤ 0.05 vs. baseline.
ASIC1a also interacts with STIM1 under basal conditions in both pulmonary (Fig 6A) and mesenteric (Fig 6D) VSMCs. Upon store depletion in pulmonary VSMC there was a significant increase in the number of puncta (interactions) per cell (Fig 6B) and a significant increase in puncta size (Fig 6C), indicating clustering of ASIC1a and STIM1 proteins. Compared to pulmonary VSMC, there were significantly fewer puncta/cell in mesenteric VSMC. In addition, store depletion in mesenteric VSMC resulted in a significant decrease in the number of puncta (interactions), per cell (Fig 6E) but no change in puncta size (Fig 6F).
Fig 6. Store depletion increases the interaction between ASIC1a and STIM1 in pulmonary, but not in mesenteric VSMCs.
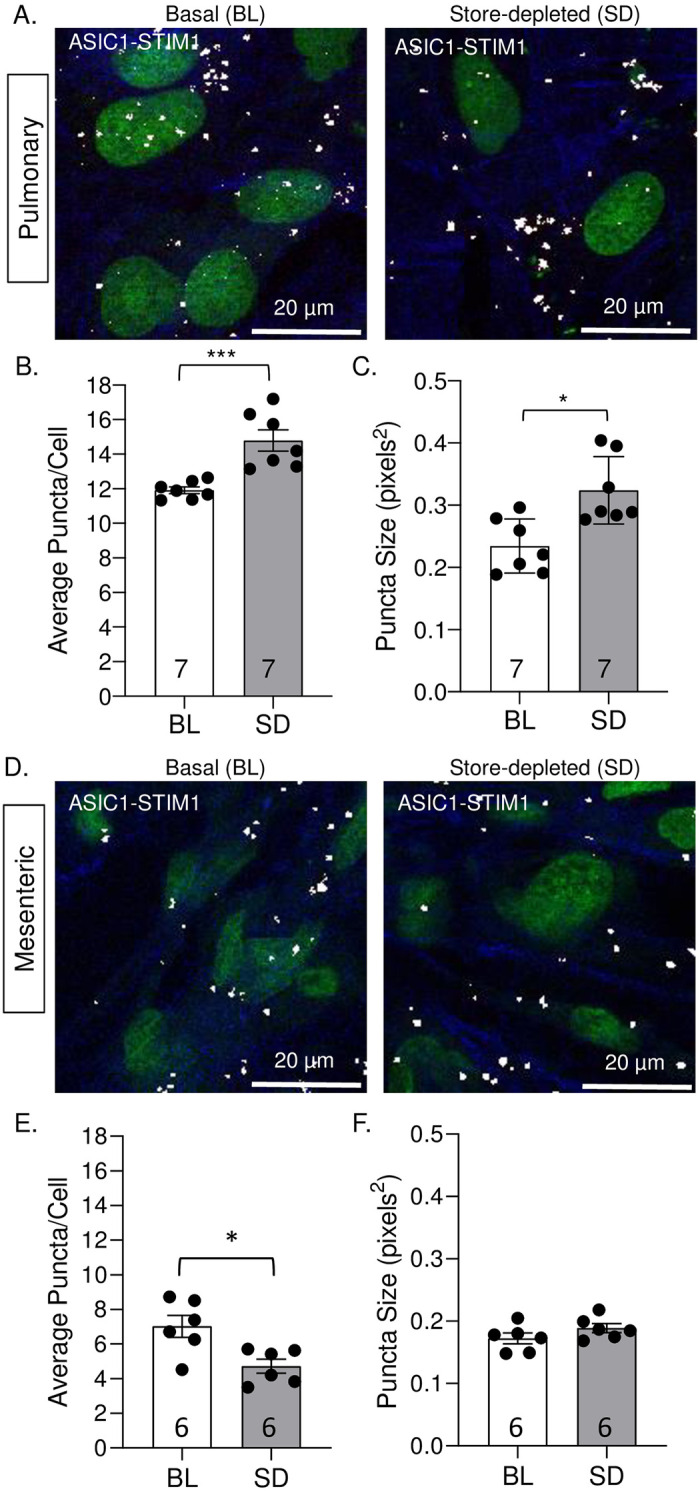
Representative images of ASIC1a/STIM1 interaction in pulmonary (A) and mesenteric (D) VSMCs under basal conditions (BL; left) and following store-depletion (SD; right). Summary data showing the average number of puncta per cell (B and E) and puncta size (C and F) in pulmonary (B-C) and mesenteric (E-F) VSMCs. The nuclei are labeled with SYTOX (green), actin is labeled with Alexa Fluor 647 Phalloidin (blue), and puncta formation (white). n = 6–7 wells; each data point represents an individual well taken from 1–3 experiments (cell cultures generated from different animals and ran on different days). *P < 0.05 vs. baseline; **P < 0.01 vs. baseline; *** p < 0.001 vs. baseline; analyzed by unpaired t-test, except for C which was analyzed using Mann-Whitney test.
In negative control experiments, we incubated each primary antibody individually and observed no positive interactions (S4 Fig). Taken together, these data demonstrate a physical interaction between ASIC1a and STIM1 in pulmonary VSMCs and support our findings that ASIC1a functions as a SOC in the pulmonary circulation.
Vasoconstrictor and arterial wall [Ca2+]i responses to ET-1 are attenuated in response to ASIC1a inhibition and augmented in response to Orai1 inhibition in pulmonary arteries
Orai1 channels have also been shown to play a vasoactive role that is independent of SOCE [41]. It is possible that both ASIC1a and Orai1 participate in ligand-activated Ca2+ entry in VSMCs and vasoconstriction through a store-independent mechanism; therefore, we further assessed the role of ASIC1a and Orai1 in ET-1-induced vasoconstriction. In both pulmonary and mesenteric arteries, inhibition of ASIC1a or Orai1 did not significantly alter baseline diameter or vessel wall [Ca2+]i (Table 2). Consistent with previous studies showing ASIC1a contributes to agonist-induced vasoreactivity in small pulmonary arteries [31, 39, 42], PcTX1 reduced ET-1-mediated vasoconstriction and arterial wall [Ca2+]i responses in small pulmonary arteries (Fig 7A). In contrast, Orai1 inhibition with AnCoA4 enhanced ET-1-induced vasoconstriction but did not alter [Ca2+]i responses in pulmonary arteries (Fig 7A). Interestingly, in mesenteric arteries we found that PcTX1 did not affect ET-1-induced vasoconstriction but significantly attenuated arterial wall [Ca2+]i responses in small mesenteric arteries at the highest concentration of ET-1 (Fig 7B). Further examination with AnCoA4 indicated no change in vasoconstriction or arterial wall [Ca2+]i in response to ET-1 (Fig 7B). Together, these studies suggest that ASIC1a and Orai1 play unique roles in pulmonary but not mesenteric artery constriction.
Fig 7. Vasoconstrictor and arterial wall [Ca2+]i responses to ET-1 are attenuated in response to ASIC1a inhibition and augmented in response to Orai1 inhibition in pulmonary arteries.

Vasoconstriction (percent baseline inner diameter; top panels) and changes in fura-2 ratio (F340/F380; bottom panels) in response to endothelin-1 (ET-1; 10−11–10−7 or 10−11–10−8) in the presence or absence of PcTX1 (20 nM) or AnCoA4 (20 μM) in small pulmonary (A) and mesenteric (B) arteries. N = 4–7 per group, values are means ± SEM. *p ≤ 0.05 vs. vehicle; ** p < 0.01, *** p ≤ 0.001, **** p ≤ 0.0001; analyzed by two-way anova followed by Dunnett’s multiple comparisons test.
Discussion
The goal of this study was to compare the contribution of SOCE to vasoconstriction and potential mediators of this response between small pulmonary and mesenteric arteries. The major findings of this study are that 1) the contribution of L-type VGCC to agonist-induced vasoconstriction in pulmonary and mesenteric arteries is minimal; 2) SOCE is functionally linked to vasoconstriction in pulmonary but not mesenteric arteries; and 3) ASIC1a is the major contributor of SOCE and SOCE-induced vasoconstriction in pulmonary arteries; while Orai1 plays a minimal role in vasoconstriction in either bed. Together, these data demonstrate a unique role for ASIC1a in the pulmonary circulation where it contributes to both SOCE-induced and agonist-induced vasoconstriction; yet ASIC1a does not contribute to vasoconstriction in the mesenteric circulation.
A variety of Ca2+ influx pathways have been identified in VSMC, including (but not limited to) VGCC, SOC, and ROC. The contribution of these various Ca2+ influx pathways utilized by VSMC to elicit contraction varies widely across different vascular beds. Therefore, we first investigated the role of L-type VGCC in both pulmonary and mesenteric agonist-induced vasoconstriction, using the antagonist diltiazem. Similar to other reports [31], there was little effect of diltiazem on agonist-induced vasoconstriction of pulmonary arteries, suggesting little role for Ca2+-influx through L-type VGCC in ET-1-induced vasoconstriction. Although diltiazem significantly reduced ET-1 induced vasoconstriction in mesenteric arteries, a large proportion of the vasoconstrictor response remained along with the increased [Ca2+]i. These findings are consistent with studies by Kawanabe et al. [43, 44] in rabbit basilar and internal carotid arteries showing a large proportion of ET-1-induced contraction is L-type VGCC independent. Although there is likely a component of T-type VGCC to ET-1 increases in Ca2+ influx, general inhibition of all VGCC does not abolish the inward calcium currents caused by ET-1 [45]. Together, these data suggest the increases in [Ca2+]i and vasoconstriction in response to ET-1 are mediated by a variety of sources.
Although SOCE has been demonstrated in several VSMC preparations, the relationship between Ca2+ entry via this pathway and contraction remains unclear. Contraction due to SOCE has been demonstrated in the aorta, intrapulmonary, cerebral, mesenteric and femoral arteries [12, 46–48]; while others report that elevated [Ca2+]i due to SOCE appears to be dissociated from contraction in the aorta, coronary, renal, and mesenteric arteries [12, 49]. Although Snetkov et al., reported that SOCE resulted in a similar increase in Ca2+ between pulmonary and mesenteric arteries, regardless of coupling to contraction [12], we found the SOCE response to be significantly less in mesenteric compared to pulmonary arteries. Consequently, it is possible that the smaller SOCE response in mesenteric arteries is not sufficient to induce vasoconstriction. Interestingly, this smaller SOCE response in mesenteric arteries seems to be associated with lower ASIC1 mRNA and protein expression in mesenteric compared to pulmonary arteries, although these were not statistically compared. The failure of SOCE to elicit mesenteric vasoconstriction is supported by the observations that ASIC1a and Orai1 inhibition did not affect ET-1-induced vasoconstriction. These data suggest the component of ET-1-induced vasoconstriction that is VGCC-independent is not mediated by SOCE but rather other Ca2+ signaling pathways. ROCs such as TRP channels or other non-selective cation channels may play a role in ET-1-induced Ca2+ in mesenteric arteries. Furthermore, cytosolic Ca2+ levels may be altered due to the production and signaling of IP3 to activate IP3 receptors and release Ca2+ from Ca2+ stores. It is also possible that Ca2+ sensitization is playing a role in ET-1-induced vasoconstriction. Specifically, Ca2+ sensitization occurs when vasoconstriction is regulated by increasing phosphorylation of the myosin regulatory light chain independent of changes in [Ca2+]i [50]. Therefore, it is possible that the ET-1-induced vasoconstrictor responses we see are due to ET-1-stimulated Ca2+ sensitization.
Expression of Orai1 was similar between pulmonary and mesenteric arteries; and although inhibition of Orai1 tended to diminish SOCE in mesenteric arteries, it did not affect pulmonary SOCE. Furthermore, inhibition of Orai1 did not affect vasoconstrictor responses in mesenteric vessels. This is in contrast to other studies showing that silencing of Orai1 or its functional inhibition with monoclonal antibodies diminishes SOCE and agonists-induced contraction in coronary and aortic rings [10, 47]. These studies suggest that in some vascular beds, Orai1 is present and functions as a SOC. Although the role of Orai1 in VSMC has been studied in several cultured VSMC preparations, it is important to note that the above studies ([10, 47]; as well as Figs 5 & 6) were conducted in freshly isolated tissue as opposed to cultured cells. Indeed, it has been shown that expression of Orai isoforms is relatively low in contractile VSMCs; however, expression of Orai1 and STIM1 dramatically increase along with SOCE in cultured VSMCs [51, 52]. VSMCs in culture are known to undergo a phenotypic switch to a synthetic or proliferative state, which often correlates with a more prominent SOCE response [53, 54]. This may explain why we observed increased clustering (indicated by increased puncta size) between Orai1 and STIM1 in response to store depletion in transiently cultured mesenteric VSMC even though Orai1 did not significantly contribute to SOCE responses in freshly isolated tissue. Together these data suggest there may be a minimal contribution of vascular Orai1-dependent SOCE to normal vascular homeostasis; however, SOCE may become a more important mechanism of Ca2+ influx in cardiovascular diseases involving VSMC dysfunction leading to a more proliferative, synthetic state of the VSMC [46, 55].
Although inhibition of Orai1 did not significantly alter SOCE in pulmonary arteries, it unexpectedly augmented the vasoconstrictor response in these endothelium-intact vessels. Receptors for ET-1 are expressed on both VSMC (ETA and ETB), where activation results in vasoconstriction, and endothelial cells (ETB), which mediate vasodilation. Since we have previously shown an important role of Orai1 in SOCE response in pulmonary arterial endothelial cells and pulmonary microvascular endothelial cells [37], it is likely AnCoA4 inhibited endothelial Orai1 and thus ET-1 vasodilatory responsiveness leading to augmented ET-1 vasoconstriction. We did not observe a corresponding increase in the vessel wall [Ca2+]i, however, this is likely a result of Ca2+ sensitization of the contractile apparatus that occurs in pulmonary arteries [56]. It is possible that within the pulmonary circulation, ASIC1a plays a larger role in VSMC SOCE while Orai1 mediates endothelial SOCE. This is consistent with the idea that in non-excitable cells whole-cell currents activated by Ca2+ store depletion are predominantly mediated by Ca2+ release-activated Ca2+ current (ICRAC) through Orai1. Whereas, in excitable muscle cells there is a greater contribution of Ca2+-permeable NSCC with a linear current-voltage relationship that contributes more to the SOC current (ISOC) (reviewed in [57]). Although it remains controversial, transient receptor potential (TRP) channels have long been proposed as SOC candidates [22, 58].
Numerous TRP channels contribute to the regulation of membrane potential and vascular tone. Of the various subtypes, it is the canonical (TRPC) subfamily, particularly TRPC1 that has been proposed as a SOC. Several studies using siRNA knockdown or neutralizing antibodies towards TRPC1 show incomplete inhibition of SOCE in various VSMCs including the aorta, pulmonary, cerebral, and portal vein [59–62]. In addition to minimal inhibition of SOCE by targeting TRPC1, these studies were also completed in cultured cells which may alter the SOCE response. Interestingly, Dietrich et al. [63] demonstrated VSMCs of TRPC1-/- mice freshly isolated from aortas and cerebral arteries showed no difference in SOCE induced by thapsigargin, IP3, or cyclopiazonic acid compared to cells from wild-type mice. Further investigation into the role of TRPC1 in SOCE found that TRPC1 only contributed to SOCE when both STIM1 and Orai1 were present, suggesting that TRPC1 is not be directly activated by store depletion, but rather is activated secondarily to STIM1 and Orai1 [64–66]. Therefore, although VSMC TRPC channels play an important role in vascular homeostasis, it is unlikely through SOCE.
In contrast to TRPC1-/- mice where there was no observable change in VSMC SOCE response [63], ASIC1a-/- mice have significantly reduced SOCE in pulmonary VSMCs [34, 38, 39]. Furthermore, transfection of the ASIC1a gene into pulmonary VSMCs from ASIC1a−/− mice restores SOCE to a level similar to that in wildtypes, further demonstrating ASIC1a, per se, is an essential component of SOCE [39]. ASIC1a conducts both Na+ and Ca2+ [25–27], and it is possible ASIC1a-mediated Na+ influx leads to VSMC depolarization and secondary activation of VGCC. However, this effect is unlikely in the present study since all SOCE responses were performed in the presence of diltiazem. Further, diltiazem and PcTX1 had very different effects on ET-1-induced vasoconstriction, suggesting different mechanisms are involved. Consistent with the possibility that ASIC1a functions as a SOC in pulmonary VSMC, we found an increased interaction between ASIC1a and STIM1 in pulmonary VSMC that was not present in mesenteric VSMCs in response to store depletion. Furthermore, we observed an increase in the ASIC1a-STIM1 association upon store depletion in pulmonary VSMCs. Although we observed an association between ASIC1a and STIM1 in mesenteric VSMC, we did not measure any increase after store depletion. Therefore, suggesting that ASIC1a is not involved in SOCE in mesenteric VSMCs. It is possible that the basal interaction that we observe between ASIC1a and STIM1 in mesenteric VSMC is due to the effects of cell culture where the cells can undergo phenotypic switches possibly leading to enhanced expression of ASIC1a and STIM1 in the cells. Although we did not investigate the role of ASIC1a to SOCE in other systemic vascular beds beyond the mesenteric circulation, these studies suggest ASIC1a may have a pulmonary-specific role. Indeed, we found ASIC1a-/- mice are protected from the development of chronic hypoxia-induced pulmonary hypertension [39]. More specifically, in response to chronic hypoxia, ASIC1a-/- mice showed no increase in the SOCE response, vasoconstrictor reactivity, vascular remodeling, right ventricular systolic pressure or right ventricular hypertrophy observed in wildtype mice [39], suggesting a unique role for ASIC1a in development of pulmonary hypertension.
In addition to pulmonary hypertension, SOCE dysregulation is associated with several vascular disorders including atherosclerosis, systemic hypertension, and restenosis. In STIM1 smooth muscle and endothelial cell-specific knockout animals, basal systolic blood pressures were similar to those of wild-type animals [67]. However, angiotensin-II-induced hypertension was significantly attenuated in the smooth muscle-specific STIM1-/- compared to wildtype animals [68], suggesting that STIM1 within the smooth muscle is a critical player in the role of angiotensin-II-induced hypertension. Similarly, the mRNA and protein levels of STIM1 and Orai1 were significantly higher and there was a significant increase in SOCE and force generation in aortic rings in stoke-prone spontaneously hypertensive rats (SHRSP) compared to Wistar-Kyoto (WKY) controls [47]. In addition, SOCE-induced contraction was dramatically increased in mesenteric arteries from aged (22 months) compared to young (3 months) rats [46]. These data suggest SOCE plays a larger role in vascular function in diseased/aged states. Therefore, although we do not see a role for ASIC1a and Orai1 in basal SOCE responses in mesenteric arteries, these channels may exhibit a larger role in the enhanced SOCE response associated with cardiovascular diseases.
In summary, this study demonstrates the vast heterogeneity that exists between vascular beds. Whereas SOCE is very prominent in the pulmonary circulation and contributes to pulmonary vasoconstriction; in the mesenteric circulation the role of SOCE to vascular reactivity is negligible. Moreover, we have found that VSMC expression of ASIC1a plays a unique role in the pulmonary circulation to mediate vasoconstriction. As this study only investigated pulmonary versus mesenteric arteries, further investigation is warranted to determine the importance of SOCE and ASIC1a in other systemic vascular beds. In addition, it is important to determine whether SOCE is enhanced and becomes a fundamental signaling pathway that mediates the vascular dysfunction that occurs with many cardiovascular diseases. This knowledge, and the specific ion channels involved, will provide potential molecular targets to treat cardiovascular diseases.
Supporting information
A: representative immunofluorescence images of pulmonary and mesenteric VSMC stained for smooth muscle 22α. B: representative PCR gels showing expression of smooth muscle α actin (F: 5’-ACTGCTGCTTCCTCTTCTTC-3’; R: 5’-GGCCAGCTTCGTCATACTCC-3’), calcitonin gene-related peptide (F: 5’-GTTCTCCCCTTTCCTGGTTG-3’; R: 5-’CTGGGGCTGTTATCTGTTCA-3’) and β-actin.
(PDF)
Representative A: PCR gel showing mRNA expression of STIM1 in pulmonary (PA) and mesenteric (MA) arteries and brain (positive control; F: 5’-ATGCCAATGGTGATGTGGAT-3’; R: 5’-CCATGGAAGGTGCTGTGTTT-3’). B: Representative western blot showing protein expression in PA, MA, and brain (top; Abcam: ab108994), Coomassie blue was used to measure even loading (B-bottom).
(PDF)
Representative PCR gel showing mRNA expression of STIM1, Orai1, and ASIC1 in both pulmonary and mesenteric primary cultured VSMC.
(PDF)
Representative images of negative control experiments where each primary antibody was incubated individually with subsequent incubation with probes.
(PDF)
(PDF)
Data Availability
The authors have uploaded the data in a repository at https://nih.figshare.com/ with DOI: 10.35092/yhjc.12576890.
Funding Statement
This work was supported by the National Heart, Lung and Blood Institute grants R01 HL-111084 (to N.L. Jernigan), F31 HL145836 (to S.M. Garcia) and T32 HL007736 (to T.C. Resta)
References
- 1.Bolton TB. Mechanisms of action of transmitters and other substances on smooth muscle. Physiol Rev. 1979;59(3):606–718. 10.1152/physrev.1979.59.3.606 [DOI] [PubMed] [Google Scholar]
- 2.Parekh AB, Penner R. Store depletion and calcium influx. Physiol Rev. 1997;77(4):901–30. 10.1152/physrev.1997.77.4.901 [DOI] [PubMed] [Google Scholar]
- 3.Putney JW Jr. Capacitative calcium entry revisited. Cell Calcium. 1990;11(10):611–24. 10.1016/0143-4160(90)90016-n [DOI] [PubMed] [Google Scholar]
- 4.Davis MJ, Meininger GA, Zawieja DC. Stretch-induced increases in intracellular calcium of isolated vascular smooth muscle cells. Am J Physiol. 1992;263(4 Pt 2):H1292–9. 10.1152/ajpheart.1992.263.4.H1292 [DOI] [PubMed] [Google Scholar]
- 5.Kirber MT, Walsh JV Jr., Singer JJ. Stretch-activated ion channels in smooth muscle: a mechanism for the initiation of stretch-induced contraction. Pflugers Arch. 1988;412(4):339–45. 10.1007/BF01907549 [DOI] [PubMed] [Google Scholar]
- 6.Albert AP, Large WA. Signal transduction pathways and gating mechanisms of native TRP-like cation channels in vascular myocytes. J Physiol. 2006;570(Pt 1):45–51. Epub 2005/09/29. 10.1113/jphysiol.2005.096875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wynne BM, Chiao CW, Webb RC. Vascular Smooth Muscle Cell Signaling Mechanisms for Contraction to Angiotensin II and Endothelin-1. J Am Soc Hypertens. 2009;3(2):84–95. 10.1016/j.jash.2008.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Manjarres IM, Rodriguez-Garcia A, Alonso MT, Garcia-Sancho J. The sarco/endoplasmic reticulum Ca(2+) ATPase (SERCA) is the third element in capacitative calcium entry. Cell Calcium. 2010;47(5):412–8. 10.1016/j.ceca.2010.03.001 [DOI] [PubMed] [Google Scholar]
- 9.Park KM, Trucillo M, Serban N, Cohen RA, Bolotina VM. Role of iPLA2 and store-operated channels in agonist-induced Ca2+ influx and constriction in cerebral, mesenteric, and carotid arteries. Am J Physiol Heart Circ Physiol. 2008;294(3):H1183–7. 10.1152/ajpheart.01148.2007 [DOI] [PubMed] [Google Scholar]
- 10.Dominguez-Rodriguez A, Diaz I, Rodriguez-Moyano M, Calderon-Sanchez E, Rosado JA, Ordonez A, et al. Urotensin-II signaling mechanism in rat coronary artery: role of STIM1 and Orai1-dependent store operated calcium influx in vasoconstriction. Arterioscler Thromb Vasc Biol. 2012;32(5):1325–32. 10.1161/ATVBAHA.111.243014 [DOI] [PubMed] [Google Scholar]
- 11.Barlow CA, Rose P, Pulver-Kaste RA, Lounsbury KM. Excitation-transcription coupling in smooth muscle. J Physiol. 2006;570(Pt 1):59–64. 10.1113/jphysiol.2005.098426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Snetkov VA, Aaronson PI, Ward JP, Knock GA, Robertson TP. Capacitative calcium entry as a pulmonary specific vasoconstrictor mechanism in small muscular arteries of the rat. Br J Pharmacol. 2003;140(1):97–106. 10.1038/sj.bjp.0705408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE, et al. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol. 2005;15(13):1235–41. 10.1016/j.cub.2005.05.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol. 2005;169(3):435–45. 10.1083/jcb.200502019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Prakriya M, Lewis RS. Store-Operated Calcium Channels. Physiol Rev. 2015;95(4):1383–436. 10.1152/physrev.00020.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lewis RS. The molecular choreography of a store-operated calcium channel. Nature. 2007;446(7133):284–7. 10.1038/nature05637 [DOI] [PubMed] [Google Scholar]
- 17.Xu SZ, Beech DJ. TrpC1 is a membrane-spanning subunit of store-operated Ca2+ channels in native vascular smooth muscle cells. Circ Res. 2001;88(1):84–7. 10.1161/01.res.88.1.84 [DOI] [PubMed] [Google Scholar]
- 18.Albert AP, Large WA. Store-operated Ca2+-permeable non-selective cation channels in smooth muscle cells. Cell Calcium. 2003;33(5–6):345–56. 10.1016/S0143-4160(03)00048-4 [DOI] [PubMed] [Google Scholar]
- 19.Ambudkar IS, Ong HL, Liu X, Bandyopadhyay BC, Cheng KT. TRPC1: the link between functionally distinct store-operated calcium channels. Cell Calcium. 2007;42(2):213–23. 10.1016/j.ceca.2007.01.013 [DOI] [PubMed] [Google Scholar]
- 20.Worley PF, Zeng W, Huang GN, Yuan JP, Kim JY, Lee MG, et al. TRPC channels as STIM1-regulated store-operated channels. Cell Calcium. 2007;42(2):205–11. 10.1016/j.ceca.2007.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Choi S, Maleth J, Jha A, Lee KP, Kim MS, So I, et al. The TRPCs-STIM1-Orai interaction. Handb Exp Pharmacol. 2014;223:1035–54. 10.1007/978-3-319-05161-1_13 [DOI] [PubMed] [Google Scholar]
- 22.DeHaven WI, Jones BF, Petranka JG, Smyth JT, Tomita T, Bird GS, et al. TRPC channels function independently of STIM1 and Orai1. J Physiol. 2009;587(Pt 10):2275–98. 10.1113/jphysiol.2009.170431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ong HL, de Souza LB, Ambudkar IS. Role of TRPC Channels in Store-Operated Calcium Entry. Adv Exp Med Biol. 2016;898:87–109. 10.1007/978-3-319-26974-0_5 [DOI] [PubMed] [Google Scholar]
- 24.Jernigan NL, Paffett ML, Walker BR, Resta TC. ASIC1 contributes to pulmonary vascular smooth muscle store-operated Ca(2+) entry. Am J Physiol Lung Cell Mol Physiol. 2009;297(2):L271–85. Epub 2009/06/02. 10.1152/ajplung.00020.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Waldmann R, Champigny G, Bassilana F, Heurteaux C, Lazdunski M. A proton-gated cation channel involved in acid-sensing. Nature. 1997;386(6621):173–7. 10.1038/386173a0 [DOI] [PubMed] [Google Scholar]
- 26.Xiong ZG, Zhu XM, Chu XP, Minami M, Hey J, Wei WL, et al. Neuroprotection in ischemia: blocking calcium-permeable acid-sensing ion channels. Cell. 2004;118(6):687–98. 10.1016/j.cell.2004.08.026 [DOI] [PubMed] [Google Scholar]
- 27.Yermolaieva O, Leonard AS, Schnizler MK, Abboud FM, Welsh MJ. Extracellular acidosis increases neuronal cell calcium by activating acid-sensing ion channel 1a. Proc Natl Acad Sci U S A. 2004;101(17):6752–7. Epub 2004/04/13. 10.1073/pnas.0308636100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu Y, Chen Z, Li WG, Cao H, Feng EG, Yu F, et al. A nonproton ligand sensor in the acid-sensing ion channel. Neuron. 2010;68(1):61–72. 10.1016/j.neuron.2010.09.001 [DOI] [PubMed] [Google Scholar]
- 29.Zha XM. Acid-sensing ion channels: trafficking and synaptic function. Mol Brain. 2013;6:1 Epub 2013/01/02. 10.1186/1756-6606-6-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Naik JS, Osmond JM, Walker BR, Kanagy NL. Hydrogen sulfide-induced vasodilation mediated by endothelial TRPV4 channels. Am J Physiol Heart Circ Physiol. 2016;311(6):H1437–H44. Epub 2016/10/07. 10.1152/ajpheart.00465.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jernigan NL, Herbert LM, Walker BR, Resta TC. Chronic hypoxia upregulates pulmonary arterial ASIC1: a novel mechanism of enhanced store-operated Ca2+ entry and receptor-dependent vasoconstriction. Am J Physiol Cell Physiol. 2012;302(6):C931–40. Epub 2011/12/30. 10.1152/ajpcell.00332.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jernigan NL, Broughton BR, Walker BR, Resta TC. Impaired NO-dependent inhibition of store- and receptor-operated calcium entry in pulmonary vascular smooth muscle after chronic hypoxia. Am J Physiol Lung Cell Mol Physiol. 2006;290(3):L517–25. 10.1152/ajplung.00308.2004 [DOI] [PubMed] [Google Scholar]
- 33.Escoubas P, De Weille JR, Lecoq A, Diochot S, Waldmann R, Champigny G, et al. Isolation of a tarantula toxin specific for a class of proton-gated Na+ channels. J Biol Chem. 2000;275(33):25116–21. 10.1074/jbc.M003643200 [DOI] [PubMed] [Google Scholar]
- 34.Gonzalez Bosc LV, Plomaritas DR, Herbert LM, Giermakowska W, Browning C, Jernigan NL. ASIC1-mediated calcium entry stimulates NFATc3 nuclear translocation via PICK1 coupling in pulmonary arterial smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2016;311(1):L48–58. 10.1152/ajplung.00040.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Herbert LM, Resta TC, Jernigan NL. RhoA increases ASIC1a plasma membrane localization and calcium influx in pulmonary arterial smooth muscle cells following chronic hypoxia. Am J Physiol Cell Physiol. 2018;314(2):C166–C76. Epub 2017/10/25. 10.1152/ajpcell.00159.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sadaghiani AM, Lee SM, Odegaard JI, Leveson-Gower DB, McPherson OM, Novick P, et al. Identification of Orai1 channel inhibitors by using minimal functional domains to screen small molecule microarrays. Chem Biol. 2014;21(10):1278–92. 10.1016/j.chembiol.2014.08.016 [DOI] [PubMed] [Google Scholar]
- 37.Zhang B, Naik JS, Jernigan NL, Walker BR, Resta TC. Reduced membrane cholesterol after chronic hypoxia limits Orai1-mediated pulmonary endothelial Ca2+ entry. Am J Physiol Heart Circ Physiol. 2018;314(2):H359–H69. 10.1152/ajpheart.00540.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Herbert LM, Nitta CH, Yellowhair TR, Browning C, Gonzalez Bosc LV, Resta TC, et al. PICK1/calcineurin suppress ASIC1-mediated Ca2+ entry in rat pulmonary arterial smooth muscle cells. Am J Physiol Cell Physiol. 2016;310(5):C390–400. 10.1152/ajpcell.00091.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nitta CH, Osmond DA, Herbert LM, Beasley BF, Resta TC, Walker BR, et al. Role of ASIC1 in the development of chronic hypoxia-induced pulmonary hypertension. Am J Physiol Heart Circ Physiol. 2014;306(1):H41–52. Epub 2013/11/05. 10.1152/ajpheart.00269.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lin LH, Jin J, Nashelsky MB, Talman WT. Acid-sensing ion channel 1 and nitric oxide synthase are in adjacent layers in the wall of rat and human cerebral arteries. J Chem Neuroanat. 2014;61–62:161–8. Epub 2014/10/23. 10.1016/j.jchemneu.2014.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang X, Zhang W, Gonzalez-Cobos JC, Jardin I, Romanin C, Matrougui K, et al. Complex role of STIM1 in the activation of store-independent Orai1/3 channels. J Gen Physiol. 2014;143(3):345–59. 10.1085/jgp.201311084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jernigan NL. Smooth muscle acid-sensing ion channel 1: pathophysiological implication in hypoxic pulmonary hypertension. Exp Physiol. 2015;100(2):111–20. 10.1113/expphysiol.2014.081612 [DOI] [PubMed] [Google Scholar]
- 43.Kawanabe Y, Hashimoto N, Masaki T. Characterization of Ca2+ channels involved in endothelin-1-induced contraction of rabbit basilar artery. J Cardiovasc Pharmacol. 2002;40(3):438–47. 10.1097/00005344-200209000-00013 [DOI] [PubMed] [Google Scholar]
- 44.Kawanabe Y, Hashimoto N, Masaki T. Involvements of voltage-independent Ca2+ channels and phosphoinositide 3-kinase in endothelin-1-induced PYK2 tyrosine phosphorylation. Mol Pharmacol. 2003;63(4):808–13. 10.1124/mol.63.4.808 [DOI] [PubMed] [Google Scholar]
- 45.Tykocki NR, Watts SW. The interdependence of endothelin-1 and calcium: a review. Clin Sci (Lond). 2010;119(9):361–72. 10.1042/CS20100145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang Y, Zhu J, Wang X, Xue N, Du J, Meng X, et al. Contrasting Patterns of Agonist-induced Store-operated Ca2+ Entry and Vasoconstriction in Mesenteric Arteries and Aorta With Aging. J Cardiovasc Pharmacol. 2015;65(6):571–8. 10.1097/FJC.0000000000000225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Giachini FR, Chiao CW, Carneiro FS, Lima VV, Carneiro ZN, Dorrance AM, et al. Increased activation of stromal interaction molecule-1/Orai-1 in aorta from hypertensive rats: a novel insight into vascular dysfunction. Hypertension. 2009;53(2):409–16. 10.1161/HYPERTENSIONAHA.108.124404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bergdahl A, Gomez MF, Wihlborg AK, Erlinge D, Eyjolfson A, Xu SZ, et al. Plasticity of TRPC expression in arterial smooth muscle: correlation with store-operated Ca2+ entry. Am J Physiol Cell Physiol. 2005;288(4):C872–80. 10.1152/ajpcell.00334.2004 [DOI] [PubMed] [Google Scholar]
- 49.Tosun M, Paul RJ, Rapoport RM. Coupling of store-operated Ca2+ entry to contraction in rat aorta. J Pharmacol Exp Ther. 1998;285(2):759–66. [PubMed] [Google Scholar]
- 50.Somlyo AP, Somlyo AV. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev. 2003;83(4):1325–58. 10.1152/physrev.00023.2003 [DOI] [PubMed] [Google Scholar]
- 51.Berra-Romani R, Mazzocco-Spezzia A, Pulina MV, Golovina VA. Ca2+ handling is altered when arterial myocytes progress from a contractile to a proliferative phenotype in culture. Am J Physiol Cell Physiol. 2008;295(3):C779–90. 10.1152/ajpcell.00173.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Potier M, Gonzalez JC, Motiani RK, Abdullaev IF, Bisaillon JM, Singer HA, et al. Evidence for STIM1- and Orai1-dependent store-operated calcium influx through ICRAC in vascular smooth muscle cells: role in proliferation and migration. FASEB J. 2009;23(8):2425–37. 10.1096/fj.09-131128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bisaillon JM, Motiani RK, Gonzalez-Cobos JC, Potier M, Halligan KE, Alzawahra WF, et al. Essential role for STIM1/Orai1-mediated calcium influx in PDGF-induced smooth muscle migration. Am J Physiol Cell Physiol. 2010;298(5):C993–1005. 10.1152/ajpcell.00325.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gonzalez-Cobos JC, Zhang X, Zhang W, Ruhle B, Motiani RK, Schindl R, et al. Store-independent Orai1/3 channels activated by intracrine leukotriene C4: role in neointimal hyperplasia. Circ Res. 2013;112(7):1013–25. 10.1161/CIRCRESAHA.111.300220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guo RW, Yang LX, Li MQ, Pan XH, Liu B, Deng YL. Stim1- and Orai1-mediated store-operated calcium entry is critical for angiotensin II-induced vascular smooth muscle cell proliferation. Cardiovasc Res. 2012;93(2):360–70. 10.1093/cvr/cvr307 [DOI] [PubMed] [Google Scholar]
- 56.Jernigan NL, Walker BR, Resta TC. Chronic hypoxia augments protein kinase G-mediated Ca2+ desensitization in pulmonary vascular smooth muscle through inhibition of RhoA/Rho kinase signaling. Am J Physiol Lung Cell Mol Physiol. 2004;287(6):L1220–9. 10.1152/ajplung.00196.2004 [DOI] [PubMed] [Google Scholar]
- 57.Avila-Medina J, Mayoral-Gonzalez I, Dominguez-Rodriguez A, Gallardo-Castillo I, Ribas J, Ordonez A, et al. The Complex Role of Store Operated Calcium Entry Pathways and Related Proteins in the Function of Cardiac, Skeletal and Vascular Smooth Muscle Cells. Front Physiol. 2018;9:257 10.3389/fphys.2018.00257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Alonso-Carbajo L, Kecskes M, Jacobs G, Pironet A, Syam N, Talavera K, et al. Muscling in on TRP channels in vascular smooth muscle cells and cardiomyocytes. Cell Calcium. 2017;66:48–61. 10.1016/j.ceca.2017.06.004 [DOI] [PubMed] [Google Scholar]
- 59.Brueggemann LI, Markun DR, Henderson KK, Cribbs LL, Byron KL. Pharmacological and electrophysiological characterization of store-operated currents and capacitative Ca(2+) entry in vascular smooth muscle cells. J Pharmacol Exp Ther. 2006;317(2):488–99. 10.1124/jpet.105.095067 [DOI] [PubMed] [Google Scholar]
- 60.Li J, Sukumar P, Milligan CJ, Kumar B, Ma ZY, Munsch CM, et al. Interactions, functions, and independence of plasma membrane STIM1 and TRPC1 in vascular smooth muscle cells. Circ Res. 2008;103(8):e97–104. 10.1161/CIRCRESAHA.108.182931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ng LC, O'Neill KG, French D, Airey JA, Singer CA, Tian H, et al. TRPC1 and Orai1 interact with STIM1 and mediate capacitative Ca(2+) entry caused by acute hypoxia in mouse pulmonary arterial smooth muscle cells. Am J Physiol Cell Physiol. 2012;303(11):C1156–72. 10.1152/ajpcell.00065.2012 [DOI] [PubMed] [Google Scholar]
- 62.Rodriguez-Moyano M, Diaz I, Dionisio N, Zhang X, Avila-Medina J, Calderon-Sanchez E, et al. Urotensin-II promotes vascular smooth muscle cell proliferation through store-operated calcium entry and EGFR transactivation. Cardiovasc Res. 2013;100(2):297–306. 10.1093/cvr/cvt196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dietrich A, Kalwa H, Storch U, Mederos y Schnitzler M, Salanova B, Pinkenburg O, et al. Pressure-induced and store-operated cation influx in vascular smooth muscle cells is independent of TRPC1. Pflugers Arch. 2007;455(3):465–77. 10.1007/s00424-007-0314-3 [DOI] [PubMed] [Google Scholar]
- 64.Cheng KT, Liu X, Ong HL, Ambudkar IS. Functional requirement for Orai1 in store-operated TRPC1-STIM1 channels. J Biol Chem. 2008;283(19):12935–40. 10.1074/jbc.C800008200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cheng KT, Liu X, Ong HL, Swaim W, Ambudkar IS. Local Ca2+ entry via Orai1 regulates plasma membrane recruitment of TRPC1 and controls cytosolic Ca2+ signals required for specific cell functions. PLoS Biol. 2011;9(3):e1001025 10.1371/journal.pbio.1001025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xu X, Star RA, Tortorici G, Muallem S. Depletion of intracellular Ca2+ stores activates nitric-oxide synthase to generate cGMP and regulate Ca2+ influx. J Biol Chem. 1994;269(17):12645–53. [PubMed] [Google Scholar]
- 67.Kassan M, Zhang W, Aissa KA, Stolwijk J, Trebak M, Matrougui K. Differential role for stromal interacting molecule 1 in the regulation of vascular function. Pflugers Arch. 2015;467(6):1195–202. 10.1007/s00424-014-1556-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kassan M, Ait-Aissa K, Radwan E, Mali V, Haddox S, Gabani M, et al. Essential Role of Smooth Muscle STIM1 in Hypertension and Cardiovascular Dysfunction. Arterioscler Thromb Vasc Biol. 2016;36(9):1900–9. 10.1161/ATVBAHA.116.307869 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A: representative immunofluorescence images of pulmonary and mesenteric VSMC stained for smooth muscle 22α. B: representative PCR gels showing expression of smooth muscle α actin (F: 5’-ACTGCTGCTTCCTCTTCTTC-3’; R: 5’-GGCCAGCTTCGTCATACTCC-3’), calcitonin gene-related peptide (F: 5’-GTTCTCCCCTTTCCTGGTTG-3’; R: 5-’CTGGGGCTGTTATCTGTTCA-3’) and β-actin.
(PDF)
Representative A: PCR gel showing mRNA expression of STIM1 in pulmonary (PA) and mesenteric (MA) arteries and brain (positive control; F: 5’-ATGCCAATGGTGATGTGGAT-3’; R: 5’-CCATGGAAGGTGCTGTGTTT-3’). B: Representative western blot showing protein expression in PA, MA, and brain (top; Abcam: ab108994), Coomassie blue was used to measure even loading (B-bottom).
(PDF)
Representative PCR gel showing mRNA expression of STIM1, Orai1, and ASIC1 in both pulmonary and mesenteric primary cultured VSMC.
(PDF)
Representative images of negative control experiments where each primary antibody was incubated individually with subsequent incubation with probes.
(PDF)
(PDF)
Data Availability Statement
The authors have uploaded the data in a repository at https://nih.figshare.com/ with DOI: 10.35092/yhjc.12576890.