

Abstract
Oxidative C–H/C–H coupling is a promising synthetic route for the streamlined construction of conjugated organic materials for optoelectronic applications. Broader adoption of these methods is nevertheless hindered by the need for catalysts that excel in forging core semiconductor motifs, such as ubiquitous oligothiophenes, with high efficiency in the absence of metal reagents. We report a (thioether)Pd-catalyzed oxidative coupling method for the rapid assembly of both privileged oligothiophenes and challenging hindered cases, even at low catalyst loading under Ag- and Cu-free conditions. A combined experimental and computational mechanistic study was undertaken to understand how a simple thioether ligand, MeS(CH2)3SO3Na, leads to such potent reactivity toward electron-rich substrates. The consensus from these data is that a concerted, base-assisted C–H cleavage transition state is operative, but thioether coordination to Pd is associated with decreased synchronicity (bond formation exceeding bond breaking) versus the “standard” concerted metalation-deprotonation (CMD) model that was formalized by Fagnou in direct arylation reaction. Enhanced positive charge build-up on the substrate results from this perturbation, which rationalizes experimental trends strongly favoring π-basic sites. The term electrophilic CMD (eCMD) is introduced to distinguish this mechanism from the standard model, even though both mechanisms locate in a broad concerted continuum. More O’Ferrall-Jencks analysis further suggests eCMD should be a general mechanism manifested by many metal complexes. A preliminary classification of complexes into those favoring eCMD or standard CMD is proposed, which should be informative for studies toward tunable catalyst-controlled reactivity.
Graphical Abstract

1. INTRODUCTION
Transition metal-catalyzed oxidative C–H/C–H coupling represents an appealing approach for the direct synthesis of biaryl motifs in conjugated organic materials because it bypasses the requisite synthesis of organometallic substrates for cross-coupling or metal reagents in conventional oxidative coupling methods.1 Oligothiophenes are particularly attractive synthetic targets for oxidative dehydrogenative coupling given their privileged2 status among conjugated materials with optical, electronic, and packing properties appropriate for organic field effect transistors (OFET),3 organic light-emitting diodes (OLED),4 organic photovoltaics (OPV),5 electrochromic devices (ECD),6 and liquid crystals.7 Compared to classic methods, such as the representative cases illustrated in Scheme 1 that require stoichiometric organolithium,8 hypervalent iodine,9 or mercury reagents,10 direct catalytic oxidative dehydrogenative coupling1,11 can construct the C−C linkage of these important conjugated materials in a potentially milder and more sustainable fashion. Existing methods for the latter nevertheless have practical drawbacks, and mechanistic uncertainties about what catalyst and ligand structures can accelerate the key C–H cleavage step hinders progress in this area.
Scheme 1.
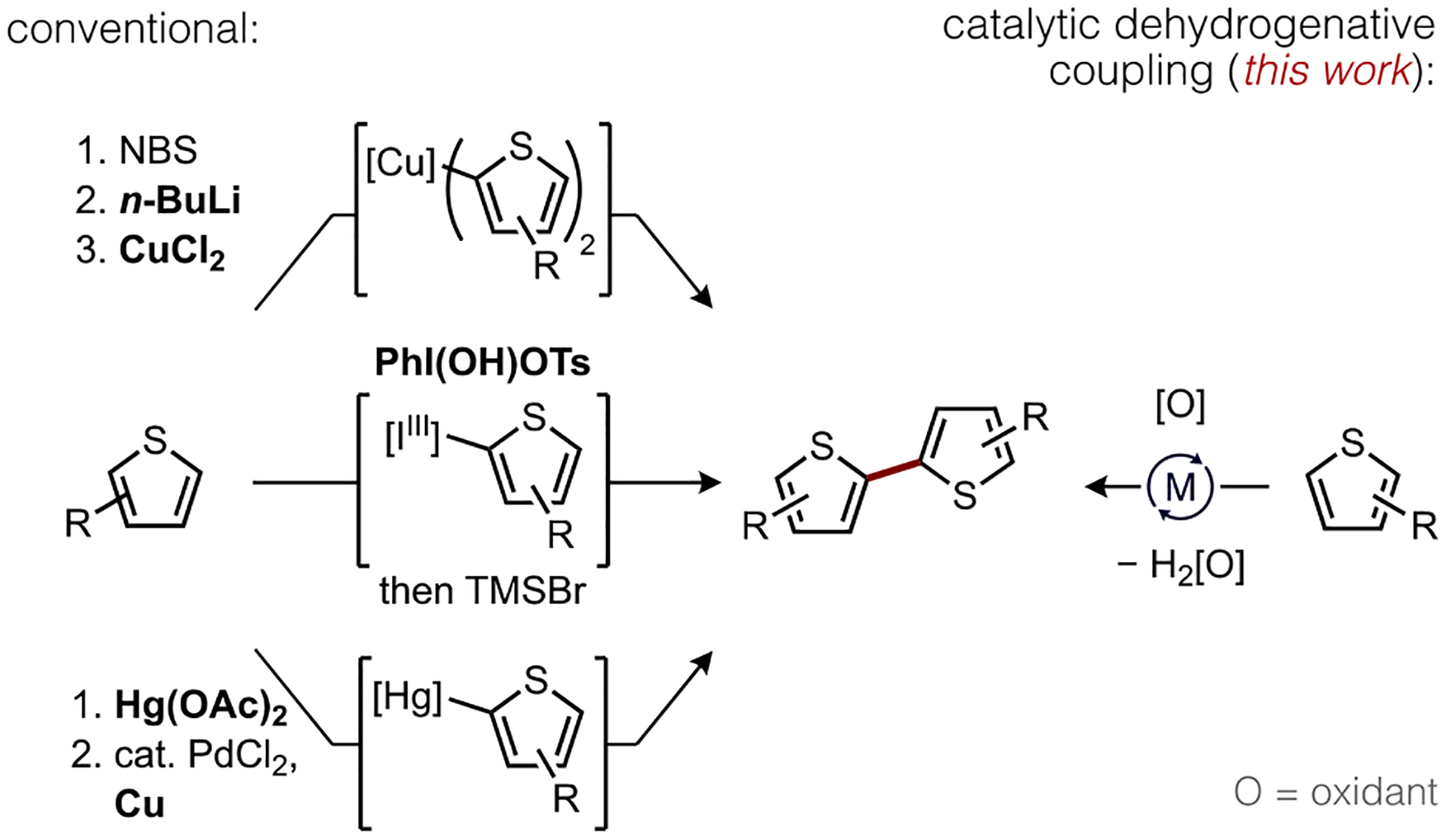
Illustrative Methods for Thiophene Coupling.
A leading palladium-catalyzed method for oxidative thiophene coupling requires AgF as a promoter and also generally favors reaction at more acidic, less hindered C–H bonds.12,13 Notwithstanding the need for silver as an essential additive in many dehydrogenative coupling reactions,14 which may correlate in some cases to a catalytic role for Ag(I) during the key (hetero)arene C–H activation step,15 the predominance of dative ligand-free (“ligandless”) Pd(II) catalysts in this area emphasizes the significant room for improvement that remains. In this regard, both catalytic efficiency and tunable site selectivity could benefit from ancillary ligand development.16,17,18 For instance, reactions that occur readily at more hindered C–H bonds are rare yet highly desirable for thiophene-based materials that are frequently decorated with fluorine, aryl, and/or alkyl substituents to augment processability and electronic properties.19
Because the “standard”20 concerted metalation-deprotonation (CMD) mechanism for C–H cleavage (Scheme 2) is believed to operate during many Pd(II)-catalyzed C–H functionalization reactions, which displays sensitivity to steric effects and tends to favor sites with lower C–H pKa (i.e., electron-poor substrates),21 it is fair to consider whether the standard CMD model is the preferable mechanistic manifold if the goal is to access new catalysts tailored for dehydrogenative coupling with electron-rich (hetero)arenes. It is thus instructive to consider (i) the limitations of the original CMD model in the context of dehydrogenative coupling reactions, (ii) what mechanisms are most effective for electron-rich (hetero)arene functionalizations, and (iii) what catalyst structures favor the latter.
Scheme 2.
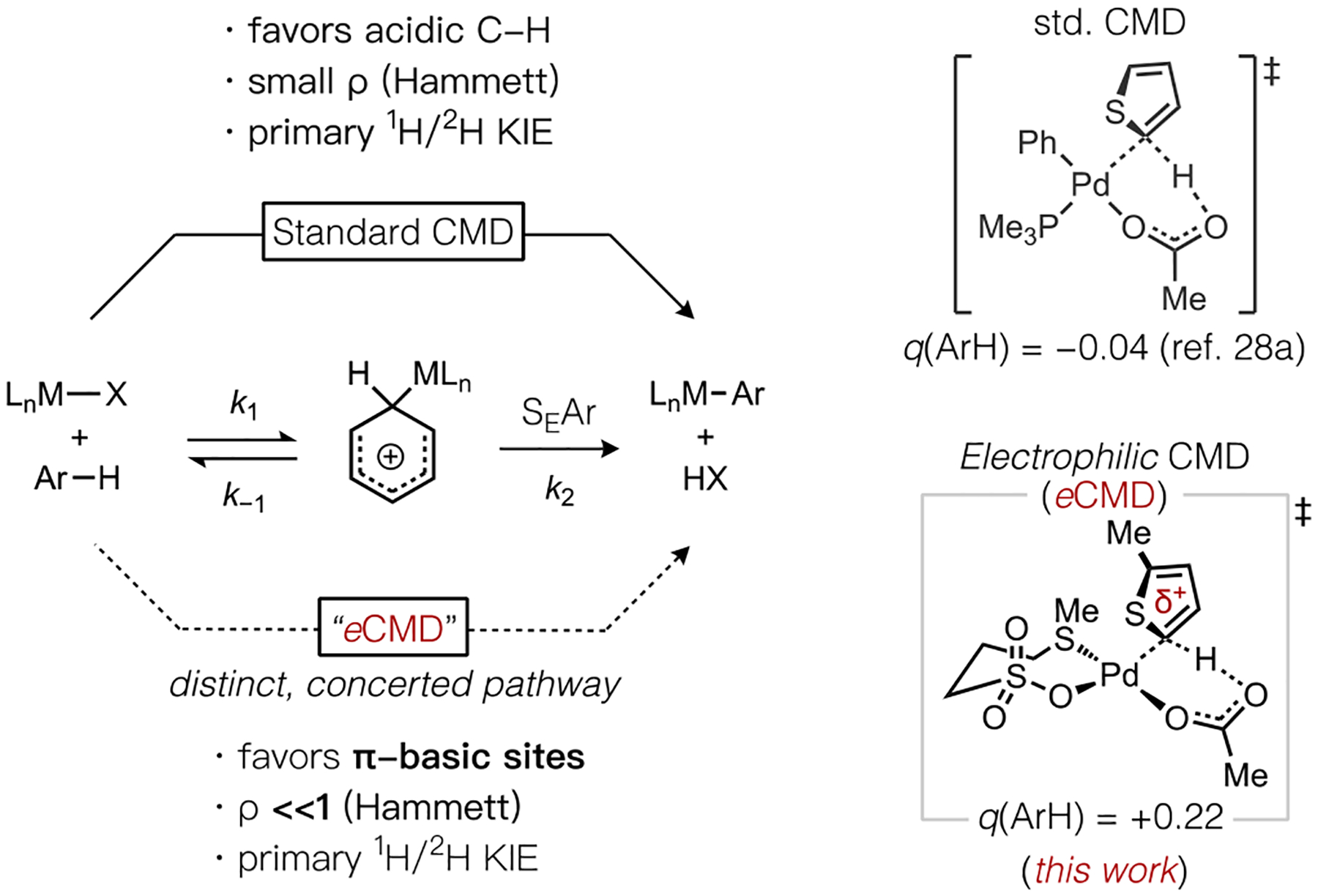
Representative Arene Activation Pathways and Transition States for Thiophene C–H Bond Cleavage.
Late transition metal complexes that catalyze a wide array of C–H functionalization reactions frequently feature an internal base (e.g., coordinated acetate) that can assist in the key bond cleavage step.22 Early experimental mechanistic studies of Pd(II)-mediated cyclometalation by Ryabov,23 and computational studies on the important role of carboxylate ligands during C–H bond cleavage of benzene or methane,24 supported the feasibility of a base-assisted mechanism of C–H cleavage. A general model was subsequently evolved by Davies and Macgregor, in studies of Pd(II), Ru(II), Rh(III), and Ir(III) promoted cyclometalations,25 and given the term “ambiphilic metal ligand activation” (AMLA) to highlight the important roles of both an electrophilic metal and nucleophilic base in facilitating C–H bond cleavage.26 Fagnou formalized an analogous concept as CMD from a series of detailed experimental and computational mechanistic studies of Pd-catalyzed direct arylation reactions.27 The standard CMD model has since become a widely adopted and effective construct for predicting reactivity and regioselectivity trends across a wide spectrum of C–H coupling reactions, which typically favor (hetero)arenes or sites with lower C–H bond pKa and less steric hindrance.28
Importantly, catalytic direct arylation reactions begin by oxidative addition of a haloarene to Pd(0). Consequently, the catalytic intermediates around which the standard CMD model was developed were organopalladium species possessing a strongly electron-releasing hydrocarbyl (aryl) ligand and frequently a strong σ-donor phosphine ligand as well. On that other hand, a number of exceptions have been noted where metal complexes appearing to cleave C–H bonds by a concerted, base-assisted mechanism do not adhere to the selectivity guidelines for CMD.23,29,30,31 These tend to be more electrophilic metal complexes, such as Pd(II) coordinated by weaker dative ligands (e.g., amine, sulfoxides) rather than strong σ-donor phosphines, and weaker X-type ligands (e.g., halides, sulfonates) rather than hydrocarbyl groups. For instance, classic cyclopalladation23,31 reactions by Pd(OAc)2 or arene mercurations with HgX2 (X = OAc, O2CCF3) exhibit reactivity patterns that parallel Friedel-Crafts chemistry but have been postulated to involve internal base assistance.30
Differing rationalizations have been put forward to account for such deviations from the standard CMD model, such as a stepwise SEAr-type mechanism. However, a primary kinetic isotope effect (KIE) is generally observed in these cases, which can only be explained in a stepwise pathway when deprotonation of a Wheland intermediate by external32 or internal33 base is rate-limiting (k2<<k−1, Scheme 2). A computational study of C–H activation by Pd(OAc)2 nevertheless found minimal charge build up on the catalyst-bound arene, which is inconsistent with the involvement of a σ-arenenium species.25a While the standard CMD model has been postulated to occur in a variety of reactions catalyzed or mediated by Pd(II), Rh(III), Ir(III), Ru(II), and Pt(II) complexes,22 the contradictory reactivity patterns noted above are not easily reconciled within a singular, concerted mechanistic manifold. Insights to explain how certain complexes simultaneously exbhibit mechanistic features of CMD together with reactivity patterns of stepwise SEAr pathways, and how ligand choice might dictate or amplify this behavior would significantly aid efforts to design new catalysts for dehydrogenative coupling with tunable reactivity.
Our group recently reported (thioether)Pd catalysts that accelerate electron-rich heteroarene C–H alkenylation and can enable C–H functionalization at hindered sites.34 The reactivity patterns observed with these catalysts also do not perfectly mirror the standard CMD model, raising questions about the potential generality of these catalysts and the details of their catalytic mechanisms for heteroarene functionalization. This motivated us to investigate whether thioether ligands might exert beneficial effects in oxidative heteroarene coupling because the putative organopalladium intermediate formed by C–H bond cleavage should be conserved in both C–H alkenylation and arene C–H/C–H coupling. We report here that thioether coordination to a Pd catalyst indeed exerts unique effects during oligothiophene synthesis through oxidative coupling, such as by enabling efficient catalytic turnover in the absence of Ag(I) or Cu(II) reagents and improved reactivity toward formation of hindered C−C bonds. Moreover, a combined experimental and computational mechanistic study suggests (thioether)Pd-promoted C–H bond cleavage remains concerted but features systematic differences versus the standard CMD model. It has previously been speculated that the standard CMD mechanism could be one of multiple possible mechanisms within a continuum spanning fully synchronous, concerted or stepwise pathways for base-assisted C–H bond cleavage, but an analysis by Gorelsky, Lapointe, and Fagnou previously failed to substantiate this hypothesis.28a We have revisited this idea using (thioether)Pd complexes as a model of catalysts that exhibit some characteristics contrasting the standard CMD model, and data are reported here suggesting certain combinations of ligands with Pd(II), and potentially many other metal complexes, may indeed perturb the nature of C–H bond cleavage mechanism into a distinct region of the mechanistic continuum in a predictable fashion.
2. RESULTS AND DISCUSSION
2.1. Identification of Active (Thioether)Pd Catalysts.
We initially focused on a model C–H/C–H coupling of 2-methylthiophene (1) using benzoquinone (BQ) as oxidant to identify ligand- accelerated reactions for this study (Table 1). The conditions for this screening lacked any stoichiometric metal reagent and were restricted to a short reaction time (2 h) and low [Pd] (0.5 mol%), which is ca. 6−20× lower than reported methods,12a,13 to more easily distinguish between highly active and poor catalysts. As a reference point, Pd(OAc)2 with no added dative ligand or with N-based ligands commonly used in oxidative Pd catalysis, such as pyridine (py), 2-fluoropyridine (2Fpy) and 4,5-diazafluorenone,17d,35 gave no bithiophene product within 2 h (entries 2, 3, and 5). Several monoprotected amino acid (MPAA) ligands pioneered by Yu were also considered (entries 6−10); examples lacking a thioether gave no product while Ac-Met-OH or Boc-Cys(Bzl)-OH gave low yield (4%−5%).18 A slight increase in product formation (3%−8%) occurred in the presence of a neutral thioether L1 (entries 11 and 12).34
Table 1.
Ligand Influence on C–H/C–H Coupling of 2-Methylthiophene.a
 | |||||
|---|---|---|---|---|---|
| entry | ligand | yield (%) | entry | ligand | yield (%) |
| 1b | — | 0 | 13 | L2 | 38 |
| 2b | py | 0 | 14 | L3 | 42 |
| 3b | 2Fpy | 0 | 15 | L4 | 43 |
| 4b | 2-py-(CH2)2SO3H | 0 | 16 | L5 | 10 |
| 5b | DAF | 0 | 17 | L6 | 43 |
| 6b | Ac-Val-OH | 0 | 18 | L7 | 60 |
| 7b | Boc-Val-OH Boc | 0 | 19c | L7 | 86 |
| 8b | Boc-Ile-OH | 0 | 20 | L8 | 43 |
| 9b | Ac-Met-OH | 5 | 21 | L9 | 10 |
| 10b | Boc-Cys(Bzl)-OH | 4 | 22b | L10 | 1 |
| 11b | L1 | 3 | 23b | L11 | 6 |
| 12 | L1 | 8 | 24b | L12 | 0 |
 | |||||
Conditions: thiophene (0.20 mmol), BQ (0.15 mmol), Pd(OAc)2, ligand, and HBF4∙Et2O stirred under air in AcOH (1.0 mL) at 60 °C for 2 h. Yield determined by 1H NMR versus 1,3,5-(CF3)3C6H3.
No HBF4.
CSA substituted for HBF4, AcOH/THF (1:1) solvent.
A more pronounced increase in bithiophene yield corresponded to the use of thioether ligands possessing a weakly-coordinating sulfonate anion (entries 13−21), and higher yields were generally associated with a more electron-rich thioether. Removal of an acetate ligand from Pd(OAc)2 by the action of acid cocatalyst appears to be important, which should generate a more electrophilic Pd species that could be more reactive toward C–H bond activation (vide infra).36 The highest yield (86%) was observed with substitution of HBF4 cocatalyst for camphorsulfonic acid (CSA) using L7 (entry 19). Modification of the anionic thioether to make it more strongly chelating (e.g., L6), or geometrically restricted from chelation to Pd (e.g., L9), gave inferior results (entries 17 and 21) suggesting hemi-lability is beneficial to catalysis. Substitution of the sulfonate moiety for a more basic carboxylate (entries 22 and 23) fully suppressed the reaction, which indicates the tethered anion does not function as the internal base.37 Lastly, the incorporation of a pendant sulfonate group into other dative ligand classes, such as pyridine (entry 4) or phosphine (entry 24), did not generate an active catalyst suggesting the thioether is a critical element for accessing a highly active Pd species. This catalyst survey suggested to us that (thioether)Pd complexes are exceptionally reactive toward catalytic processes featuring C–H cleavage of electron-rich heteroarenes. It was thus of interest to interrogate if, and how, anionic thioether (e.g., L7) coordination to Pd influences the mechanism of these reactions as compared to related catalytic systems.
2.2. Kinetic and Isotope Effect Experiments.
A kinetic analysis of 2-hexylthiophene (2-HT) coupling using L7/Pd(OAc)2 was conducted using the method of initial rates. The dependence of the rate on catalyst concentration (Figure 1a) was found to be first order but only for [Pd] >1.5 mM. At lower [Pd] (≤ 1.5 mM; ≤0.3 mol%), the kinetic behavior transitions to a second-order dependence of the rate on the (thioether)Pd catalyst.38 While complex, these kinetic data are clearly inconsistent with a simple monometallic mechanism for which a constant first-order dependence should be observed at all [Pd]. Instead, we interpret the kinetic trend as indicative of a switch in turnover-limiting step from one involving a single Pd center at high [Pd], such as C–H activation, to a scenario at low [Pd] in which reversible C–H activation precedes a later turnover-limiting step involving two Pd centers, such as Pd-to-Pd transmetalation (TM) or possibly reductive elimination (RE) from a dinuclear species (vide infra). A mechanistic study by Stahl on o-xylene homocoupling observed a similar concentration-dependent fluctuation in the kinetic order of a Pd catalyst.39
Figure 1.
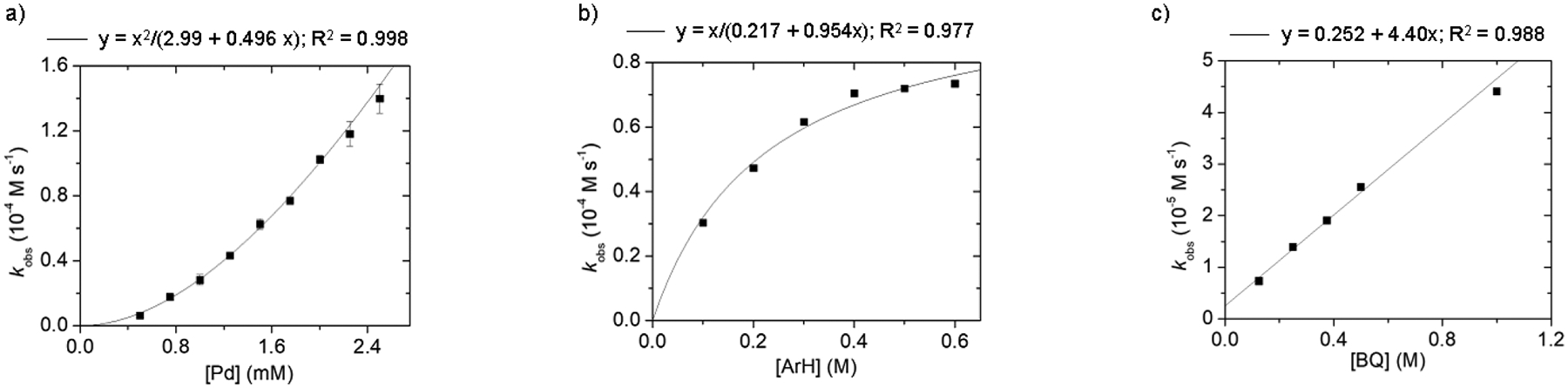
Dependence of the observed rate constant (kobs) on the concentration of (a) [Pd] (0.5−2.5 mM) using [arene] = 0.50 M and [BQ] = 0.38 M, (b) [arene] = 0.10−0.60 M using [Pd] = 2.5 mM and [BQ] = 0.25 M, or (c) [BQ] = 0.13−1.0 M using [Pd] = 1.0 mM and [arene] = 0.50 M during the coupling of 2-hexylthiophene in AcOH at 60 °C as determined by 1H NMR versus 1,3,5-(CF3)3C6H3 as standard. [Pd] = Pd(OAc)2/L7/CSA = 1:1:2 in all cases.
Saturation behavior was also observed when the dependence of the catalytic rate on [2-HT] was measured (Figure 1b),34 which is consistent with favorable substrate binding to Pd. Further support for strong substrate coordination was the observation of a shift in 1H and 19F NMR resonances corresponding to L4-coordinated Pd upon addition of 2-methylthiophene (Figure S18). Energy decomposition analysis further corroborates interaction energy as an important effect (see Section 2.4).
Another notable and unexpected kinetic observation was that the catalytic rate exhibits a positive order dependence on [BQ] during 2-HT coupling using 1.0 mM [Pd], a catalyst concentration for which a bimetallic turnover-limiting step is operative (Figure 1c). This kinetic behavior indicates BQ must be involved in a catalytic function beyond its typical role as a terminal oxidant to turnover Pd(0), potentially as a ligand for one Pd center of some bimetallic elementary step. An inner sphere role of BQ is further suggested by the alternative use of a more hindered quinone oxidant (e.g., p-xyloquinone or duroquinone) during coupling of 1, which suppressed catalytic activity (Table S6). The role of BQ in catalysis was explored in further detail computationally in Section 2.6.
The substrate 1H/2H kinetic isotope effect (KIE) was next investigated. For the reaction outlined in Table 2, the KIE was found to vary from 2.0−4.9 as determined from independent oxidative coupling reactions of 2-HT or 5-d-2-HT in acetic acid at varied [Pd] (2.5 to 0.5 mM, respectively). The trends of these isotope effects are consistent with a bimetallic turnover-limiting step following reversible C–H activation that would give rise to a squared dependence of the rate on [catalyst], which in turn should give rise to a squared dependence on the substrate isotope effect if both Pd complexes cleave a C–H bond. The mathematical basis for this relationship was determined by Stahl.39 Solvent isotope effects ranging from 1.2 to 0.57 were also determined for reactions with decreasing [Pd] (2.5 to 0.5 mM, respectively). This trend correlates to increased deuterium incorporation into 2-HT at lower [Pd], which again is consistent with a scenario where C–H activation occurs reversibly prior to another turnover-limiting step. In total, these KIE data are consistent with a scenario in which C–H activation and a bimetallic step, for instance Pd-to-Pd TM, occur at similar rates and their relative kinetic contributions are thus catalyst concentration dependent.
Table 2.
Substrate and Solvent 1H/2H Isotope Effects.
 | |||
|---|---|---|---|
| entry | [Pd] (mM) | substrate KIE | solvent KIE |
| 1 | 2.5 | 2.0 | 1.20 |
| 2 | 2.0 | 2.7 | 0.99 |
| 3 | 1.5 | 4.3 | 0.93 |
| 4 | 1.0 | 4.9 | 0.66 |
| 5 | 0.5 | 0.57 | |
2.3. Computational Analysis of C–H Activation.
The possibility of a stepwise electrophilic metalation (SEAr-type) mechanism is important to consider given the affinity of (thioether)Pd, and numerous other metal complexes,23,29b,30 for activation of more electron-rich (hetero)arenes. Using isotope effect data alone, it is difficult to unambiguously distinguish a stepwise mechanism involving rate-determining proton transfer (k2<<k−1, Scheme 2) from a concerted mechanism. Computational studies were thus conducted to provide important insights into the structures and energetics of relevant catalytic intermediates and reaction steps. Density functional theory (DFT) calculations were performed on reactions of (L7)Pd complexes with 2-methylthiophene. Geometry optimizations were performed using the selected spin-restricted M06-L functional with Stuttgart/Dresden ECP (SDD) basis set for Pd and 6–31+G(d) basis set for other atoms. Single point calculations were performed using the non-local functional M06 with IEFPCM (SCRF) solvent model (acetic acid), with SDD basis set for Pd and 6–311+G(d,p) for other atoms. Full computational details can be found in the Supporting Information, including assessment of other functionals and basis sets.
We previously reported experimental evidence of catalyst speciation in mixtures of Pd(OAc)2 with a neutral, monodentate thioether that occurs through equilibration of mono-, di-, and trinuclear Pd species possessing 1:2, 1:1, and 3:2 Pd/L ratios, respectively.34 To assess whether analogous speciation may occur with hemilabile anionic thioethers, we examined a 1:1 solution of Pd(OAc)2 and L4 in AcOH-d4 by 19F and 1H NMR spectroscopy. The obtained spectra suggested rich speciation also occurs in this case (Figure S18), but resonances were not sufficiently resolved for molecular weight determination by DOSY NMR and structure assignment. Computational evaluation of likely thioether-coordinated Pd species were instead pursued. Ground state energies of a variety of putative Pd species coordinated by L7 were calculated and compared (Scheme S19). In general, κ2-S,O coordination of L7 was favored after loss of one acetate ligand from Pd(OAc)2, which is presumed to occur under typical conditions that include catalytic amounts of a strong acid (e.g., CSA). Monomeric Pd(L7)2, dinuclear Pd2(OAc)2(L7)2, and trinuclear Pd3(OAc)4(L7)2 species are predicted to be close in free energy (ΔG = 2.8 kcal/mol) and likely equilibrate rapidly as indicated in eq 1, which is directly analogous to experimental observations reported previously for mixtures of Pd(OAc)2 and neutral, monodentate thioethers.34 The dinuclear complex 4 was selected as the starting point for subsequent calculations for simplicity, although it could be expected that complex kinetic orders in [Pd] ranging from 0.5 to 1 could arise from these off-cycle equilibria.
 |
(1) |
The L7-accelerated mechanism of C–H bond cleavage was next explored using DFT calculations (Figure 2). The reaction pathway was initiated by formation of a mononuclear species Pd(κ2S,O-L7)(κ2-OAc) (5) followed by reversible binding of substrate (e.g. 2-methylthiophene) to form a π-complex (6).24 Note that four possible diastereomers of the substrate-catalyst pair are possible due to cis/trans arrangements of the thioether and π-arene ligands bound to Pd and coordination of 2-methylthiophene through either the Re or Si face. The two more favorable isomers are illustrated in Figure 2 and the remaining possibilities shown in Figure S20. 2-Methylthiophene coordinates to Pd in an η2 fashion in cis-Si-6 with comparable Pd–C5 and Pd–C4 distances of 2.16 and 2.24 Å, respectively. The C5–H bond (1.09 Å) is distorted out of plane more so than the C4–H bond, as judged by the dihedral angle (ϕ) about C3–C4–C5–H (154.9°) or C2–C3–C4–H (165.9°). However, substantial double bond character remains as indicated by the C5–C4 bond length (1.42 Å) as compared to the σ-bond length in the solid-state structure of tetrahydrothiophene (1.520 Å)40 or calculated C5–C4 π-bond in free 2-methylthiophene (1.37 Å). These structural data are thus inconsistent with the formation of a σ-arenium (Wheland) intermediate in a stepwise electrophilic metalation pathway.
Figure 2.

(a) Computed energy profiles for reaction of the L7-Pd catalyst with 2-methythiophene and (b) transition states of the lowest energy pathways for concerted C–H bond cleavage. All energies given in kcal/mol and bond lengths in Å.
A concerted mechanism for cleavage of the substrate C–H bond was subsequently calculated to occur with the assistance of the adjacent acetate as an internal base via six-membered transition states cis-Si-TS7 and trans-Re-TS7, the latter of which was kinetically favored (ΔΔG‡ = 1.3 kcal/mol) but also endoergic by 13.8 kcal/mol. Isomerization of this less stable organo-Pd intermediate trans-Re-8 to cis-Si-8 should nevertheless be facile, so either mechanism for C–H cleavage may converge to the same organometallic product (e.g., cis-Si-8). Inspection of these two transition states at different levels of theory suggested that both cis and trans pathways occur with energy differences within the DFT computational error (Table S24), which lead us to conclude they are each kinetically relevant.
The early transition state cis-Si-TS7 features Pd–Cipso and Cipso–H bond distances of 2.09 Å and 1.30 Å, respectively. In contrast, the late transition state trans-Re-TS7 is characterized by a slightly shortened Pd–Cipso bond (2.06 Å) and elongated Cipso −H bond (1.43 Å). In both cases the indicated bond lengths suggest the transition state is considerably asynchronous with a greater extent of Pd–C bond formation than C–H bond cleavage. We speculate the result is a CMD-type mechanism featuring characteristics of SEAr-type reactions (i.e., complete Pd–C bond formation prior to C–H bond cleavage), which is apparently facilitated by coordination of an anionic thioether ligand to Pd. This type of transition state lowers the kinetic barrier for activation of electron-rich heteroarenes and also helps to rationalize the observed site selectivity and reactivity patterns for our catalysts, favoring more π-basic sites, that do not align well with trends established for direct arylation.
Other plausible transition states, such as those involving alternative internal proton acceptors other than coordinated acetate (e.g., CF3CO2−, CH3SO3−) or external base, were also evaluated (Figures S21−S23) and occurred in all cases with higher kinetic barriers than the pathways shown in Figure 2. Alternative mechanisms involving intact Pd aggregates during C–H bond cleavage also cannot be conclusively ruled out,34,41 but the lack of terminal acetate ligands in the di- and tripalladium species formed by coordination of L7 to Pd (eq 1) suggest a mononuclear active species is favored, which agrees with our previous study of a related (thioether)Pd catalyst and the majority of mechanisms proposed in other Pd-catalyzed dehydrogenative coupling reactions.42 The resulting thienylpalladium species formed after C–H bond cleavage (e.g., cis-Si-8) was the starting point for evaluations of subsequent catalytic steps for biaryl formation, which are discussed in Section 2.6.
2.4. Distinctions Between the Standard CMD Model and “eCMD”.
As mentioned above, the standard CMD model correlates reactivity/selectivity with C–H bond pKa and less sterically hindered sites in most cases.28a Ess has proposed this general preference may originate from a kinetic-thermodynamic connection between site selectivity during C–H bond cleavage and the strength of the forming Pd–C bond.43 However, exceptions to the “pKa rule” have been widely noted even when evidence suggests a concerted, base-assisted mechanism remains operative. Accounting for how differences in the metal complex structure give rise to these “non-classic” CMD mechanisms and extraction of guidelines to predict what catalysts should display a particular reactivity pattern would be informative. In this regard, the concept of a mechanistic continuum for C–H cleavage mechanisms has been postulated both for C(sp2)–H and C(sp3)–H bond cleavage and could potentially provide a basis to rationalize multiple types of concerted pathways.25c,28a,44 Such a continuum can be analyzed by More O’Ferrall-Jencks analysis, and the plot shown in Figure 3a does so by mapping the simultaneous change of metal-carbon and carbon-hydrogen bond order during transition states for C–H bond cleavage. The reaction coordinate begins at the upper-left of the plot and proceeds to the lower-right, with the diagonal dashed line representing a perfectly concerted and synchronous trajectory. Transition states falling above or below the diagonal occur with decreasing synchronicity and lie either toward canonical stepwise SEAr (upper-right corner) or C–H deprotonation (lower-left corner) pathways, respectively.
Figure 3.
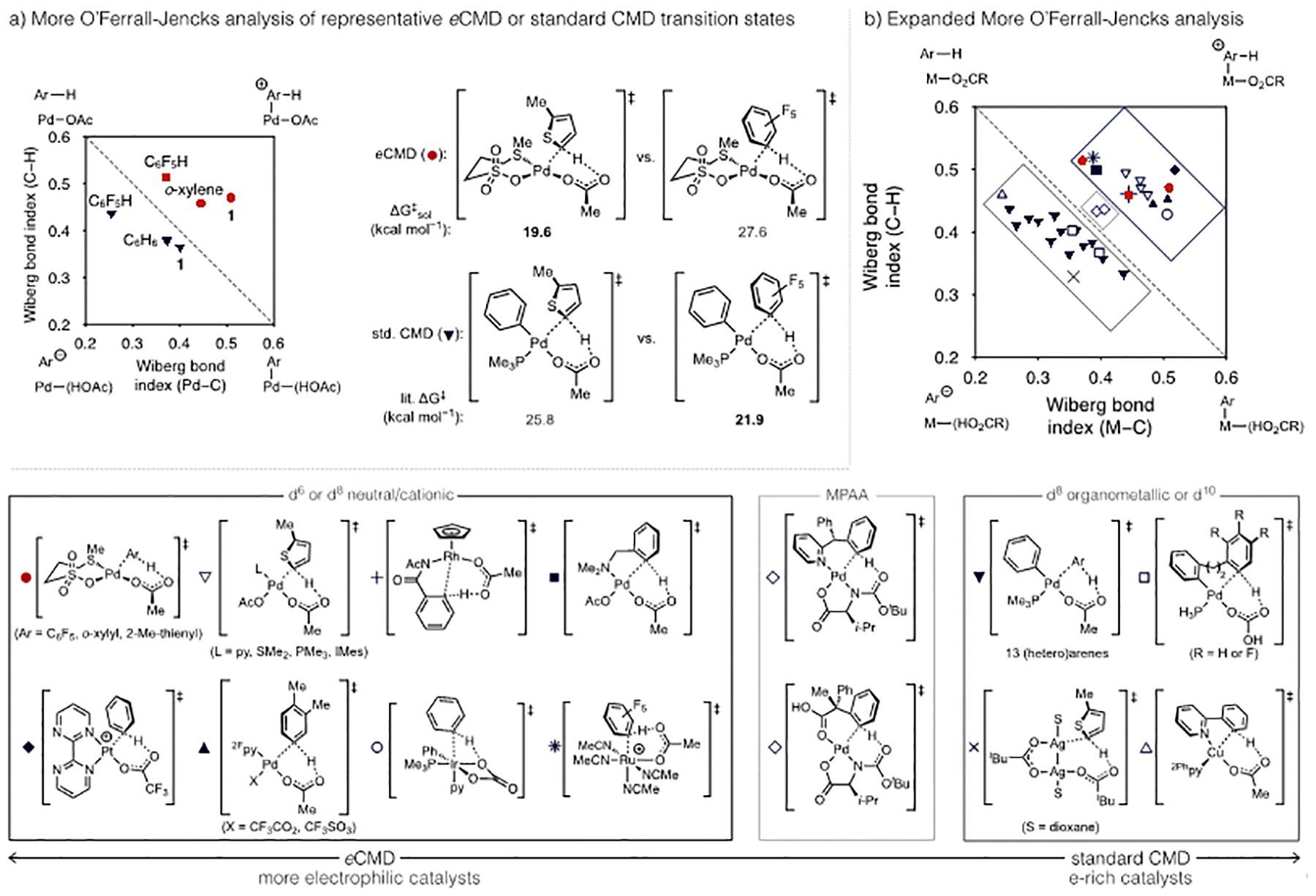
(a) More O’Ferrall-Jencks analysis of the effect of representative electron-poor, -neutral, or -rich substrates on the transition state location and free energy of activation, relative to free arene and Pd(PMe3)(Ph)(κ2-OAc) for standard CMD or Pd(κ2S,O-L7)2 for eCMD and (b) expanded analysis of other concerted, base-assisted C–H cleavage transition states. Bond indices were calculated using literature DFT data for transition states represented by plus (Fagnou),45 square (Macgregor),25a diamond (Periana),46 up triangles (Stahl),36 open circle (Gorelsky and Woo),47 star (Larrosa)48 open diamonds (Yu),49 down triangles (Fagnou),28a open squares (Echavarren),21b,21c cross (Sanford),15a or open/up triangle (Roithova)50 symbols.
Two distinct clusters are apparent in this More O’Ferrall-Jencks plot (Figure 3a) when representative transition states for activation of an electron poor (C6F5H), neutral (benzene/xylene), or electron-rich (thiophene) substrate are plotted. Importantly, the substrate identity does not appear to dictate whether a transition state locates above or below the diagonal. Instead, substrate variation leads to displacement parallel to the diagonal and results in an earlier or later transition state for more electron poor (e.g., Ar = C6F5) or more electron-rich (e.g., Ar = 2-Me-thienyl) cases, respectively. This substrate effect does not substantially alter the polarization because such parallel movement in the plot correlates to roughly equal and opposite fluctuations in bond breaking and formation.
The group of transition states falling below the synchronous trajectory in Figure 3a correspond to C–H cleavage by organopalladium complexes associated with direct arylation reactions, which manifest standard CMD. This cluster should thus display some characteristics of the limiting mechanism in this part of the continuum (e.g., C–H deprotonation), which does align with typical experimental site selectivity in direct arylation that favors more acidic C–H bonds. The other cluster of transition states residing above the diagonal are relatively less synchronous (i.e., larger average displacement perpendicular to the diagonal) and reside in the region of the continuum toward stepwise SEAr. As such, reactivity favoring more nucleophilic sites should be expected for reactions involving these metal complexes. Because this latter cluster of transition states populates a region of the concerted continuum that is characterized by metal-carbon bond formation that is more advanced than carbon-hydrogen bond cleavage, we propose that they constitute another type of CMD mechanism that may be described as electrophilic CMD or “eCMD”.
If eCMD is indeed a concerted mechanism with electrophilic characteristics, it should be expected reactions featuring this pathway could give rise to different reactivity patterns versus reactions proceeding through standard CMD. To test this, we calculated several C–H cleavage barriers (Figure 3a) for a complex proposed to react by eCMD, Pd(L7)(OAc), which is found to be much more reactive toward an electron-rich (e.g., 2-methylthiophene) rather than electron-poor (e.g., C6F5H) substrate (ΔΔG‡ ca. 8 kcal/mol). This reactivity trend is fully reversed for standard CMD, as judged from reported energies for activation of C6F5H or 2-methylthiophene by Pd(PMe3)(Ph)(OAc) that reflect strong preference for the electron-poor arene (ΔΔG‡ ca. 4 kcal/mol). These counterpoised energy differences are corroborated through intermolecular competition experiments (Scheme 3). Catalytic C–H/C–H coupling of an equimolar mixture of 2-methylthiophene and C6F5H using L7/Pd(OAc)2 formed exclusively 3 in 66% yield as determined by NMR versus internal standard; no cross-product or octafluorobiphenyl were detectable and >99% C6F5H remained after 2 h. On the other hand, direct arylation under Fagnou’s conditions27 using p-bromofluorobenzene, PtBu2Me/Pd(OAc)2 as a catalyst, and an equimolar mixture of 2-hexylthiophene and C6F5H, generated 2,3,4,4’,5,6-hexafluorobiphenyl in high NMR yield (86%) with no detectable products corresponding from activation of the electron-rich substrate. These results emphasize that standard CMD and eCMD type transition states can correlate to potentially significant experimental reactivity differences. Several additional analyses, using the representative models shown in Scheme 4, were subsequently conducted to test this conclusion further.
Scheme 3.
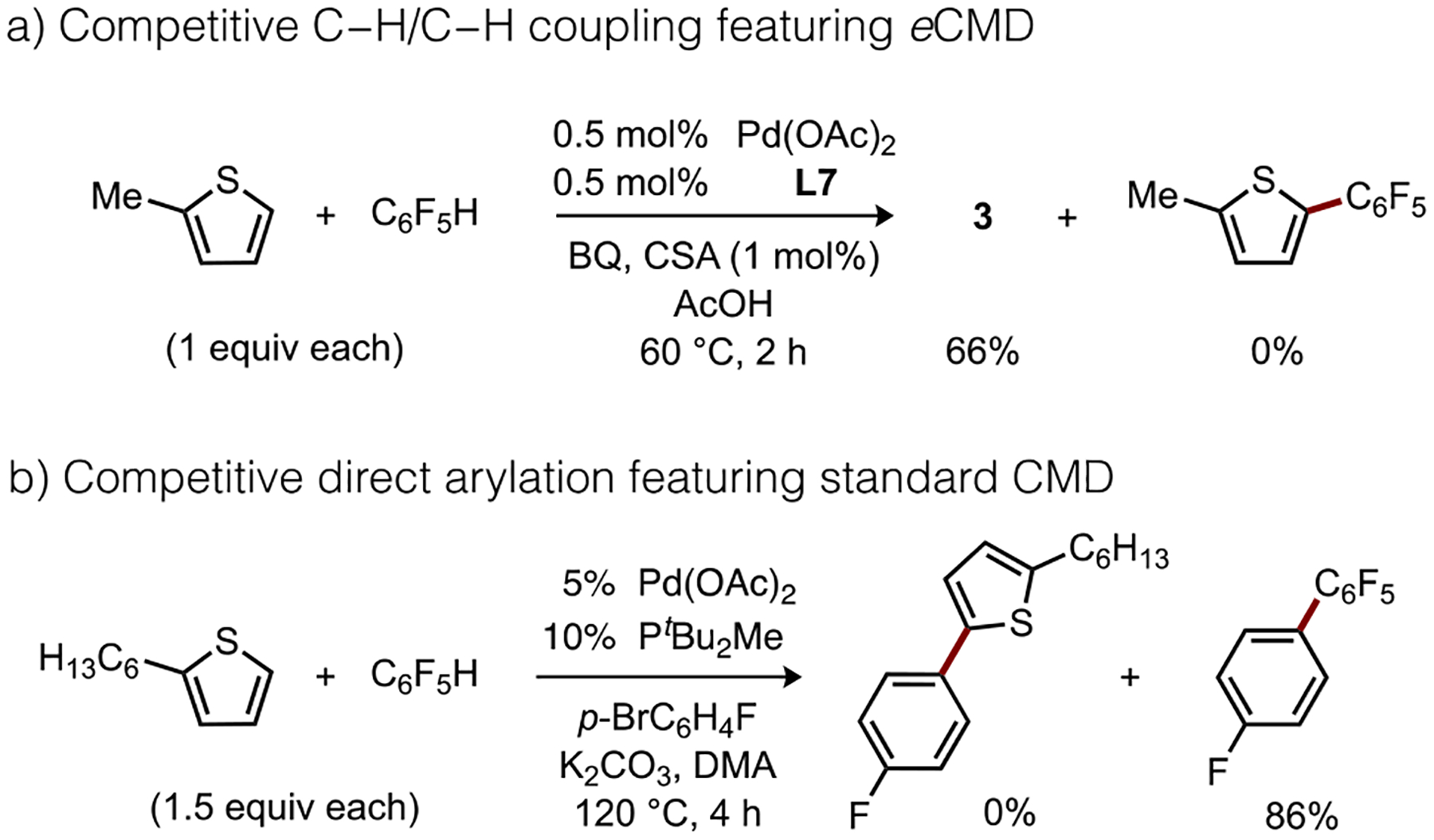
Catalytic competition reactions featuring (a) eCMD or (b) standard CMD mechanisms.
Scheme 4.
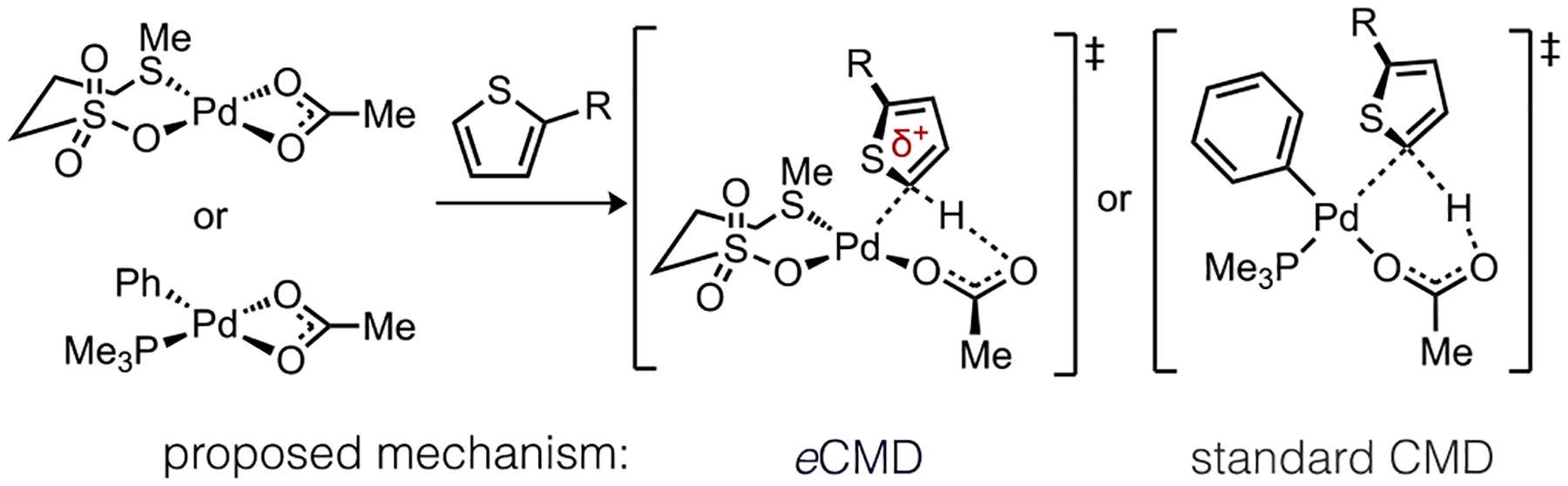
Representative Transition State Structures for C–H Bond Cleavage of a 2-Substituted Thiophene by Pd(II).
A Hammett analysis was performed using L7/Pd(OAc)2 to catalyze C–H/C–H coupling of a series of six 2-substituted thiophenes (Figure 4a). Kinetic data were derived from individual reactions using the method of initial rates under conditions described in Table 1, entry 19. Calculation of relative transition state energies (e.g., cis-Si-TS7) were also used in this analysis; the slope of these data (ρ = −7.1) signifies accumulation of substantial positive charge in the eCMD transition state. While thioether L7 appears important for enhancing this electrophilic behavior, other reactions that locate in the eCMD region, such as the cyclometalation of substituted N,N-dimethylbenzylamines studied by Ryabov,23 also show electrophilic characteristics (ρ = −1.6). The trend reverses for organometallic species associated with standard CMD, with Pd(PMe3)(Ph)(OAc) considered here as a representative case (Figure 4b). Literature calculated values for these reactions trend closely with experiment,28a and the slope of these data reflect modest accumulation of negative charge on the substrate (ρ = 1.3) in the concerted transition state.
Figure 4.
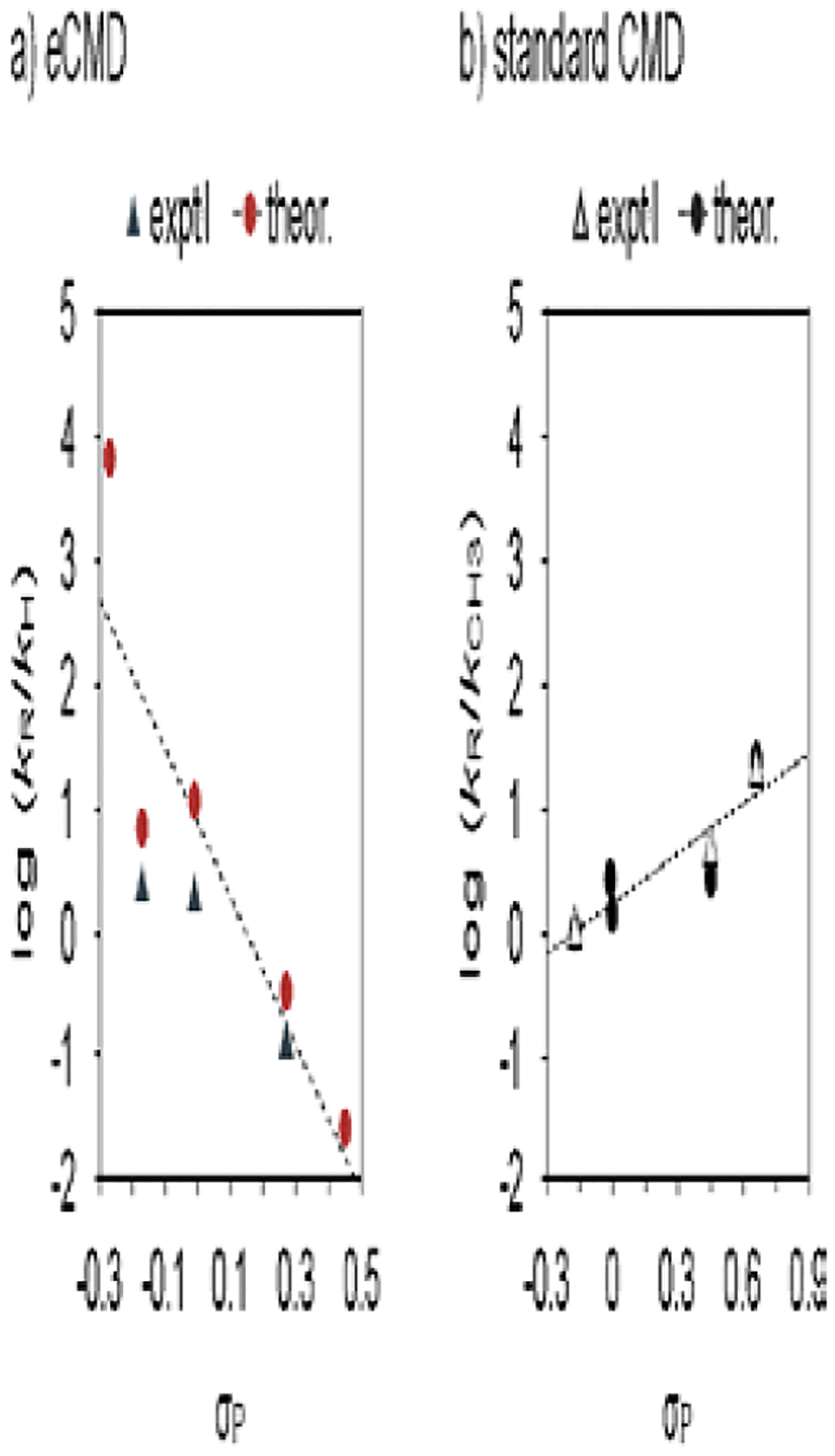
Hammett analysis of 2-substituted thiophenes during C–H activation by (a) eCMD during C–H/C–H coupling reactions catalyzed by L7/Pd(OAc)2 or (b) literature data for standard CMD during direct arylation.28a Calculated values correspond to Scheme 4 transition states.
Charge accumulation in the concerted transition states was also assessed by natural bond order (NBO) analysis for the representative cases in Scheme 4. The sum of atomic charges (q) on the thiophene substrate (Table 3) agrees with the trends observed by Hammett analysis suggesting eCMD exhibits a larger degree of charge build-up on the substrate, which ranges from 0.16 to 0.25, depending on the thiophene identity. On the other hand, charge accumulation during standard CMD is considerably less (0.03 to 0.11) suggesting a minimally polarized transition state. Together these Hammett and NBO data reinforce the notion that eCMD is a more polarized, concerted C–H cleavage mechanism.
Table 3.
Calculated atomic charges (NBO) on 2-substituted thiophenes for transition states depicted in Scheme 4.
| eCMD | standard CMD | |||
|---|---|---|---|---|
| R | ΔG‡sol (kcal/mol)a | q(ArH)c | ΔG‡ (kcal/mol)b | q(ArH)c |
| OMe | 6.7 | 0.25 | ||
| Me | 10.8 | 0.22 | 25.8 | 0.11 |
| Ph | 10.5 | 0.21 | 25.2 | 0.09 |
| H | 11.7 | 0.20 | 25.6 | 0.09 |
| C6F5 | 13.3 | 0.18 | ||
| CO2Me | 14.9 | 0.16 | 25.2 | 0.06 |
| CN | 23.9 | 0.03 | ||
Energies were calculated at the M06/6–311+G(d,p)-SDD/PCM(AcOH)//M06L/6–31+G(d)-SDD level of theory.
Literature gas phase values calculated at the B3LYP/DZVP(Pd)/TZYP level of theory.28a
Determined by natural bond order (NBO) analysis.
A distortion-interaction analysis was also performed on the same representative Pd complexes. Data from this analysis shown in Figure 5a indicate that interaction energy is more significant in magnitude for eCMD and is associated with a considerable sensitivity of ΔE‡ to the functional group on the thiophene substrate. On the other hand, the interaction energies are smaller in magnitude and overall activation barriers are relatively flat across the series for CMD (Figure 5b; tabular data in Table S28). These data signify that substrate-catalyst interactions are an important influence on reactions involving eCMD and could enhance the catalyst preference for activation of π-basic sites. These data are also consistent with kinetic observations of saturation behavior in [arene] during reactions with (thioether)Pd catalysts, which are not often observed in dehydrogenative coupling reactions using other catalysts.
Figure 5.

Distortion-interaction analysis of representative (a) eCMD or (b) classic CMD transition states for reactions depicted in Scheme 4.
The four lines of evidence discussed above (More O’Ferrall-Jencks, Hammett, NBO charge, and energy decomposition analyses) help to solidify how eCMD differs from the standard CMD pathway. A logical next question to consider is, what structure features of the transition metal complex are most important with regard to whether eCMD or standard CMD is favored? To probe this, we calculated transition states for an expanded selection of Pd(OAc)2 complexes coordinated by a dative ligand other than L7, such as pyridines, a neutral thioether, phosphine, or N-heterocyclic carbene that span a range of σ-donicity. As can be seen in Figure 3b (open triangles), activation of 2-methylthiophene by each of these complexes give rise to a transition state that locates in the eCMD region suggesting the identity of the L-type ligand may be not the determinant factor. In fact, a commonality of Pd complexes manifesting this mechanism is the absence of a strong X-type ligand (e.g., aryl, alkyl), which is substituted for a weak donor, such as carboxylate or sulfonate. In other words, the reaction mechanism differs by whether the complex is organometallic (std. CMD) or a more electrophilic, inorganic Pd(II) complex (eCMD).
Moving this analysis beyond Pd(II), a range of other metal complexes proposed to cleave C–H bonds by a concerted mechanism were considered to probe the potential generality of the eCMD model. Representative Ir(III),47 Rh(III),45 and Ru(II)48 complexes reported in the literature were analyzed and each locates in the eCMD region of the More O’Ferrall-Jencks plot consistent with their established electrophilic properties. If electrophilic d8 and d6 complexes enforce eCMD, it might then be predicted that complexes with higher d-electron count would favor standard CMD by rendering the metal relatively more nucleophilic. In this regard, a dinuclear d10, Ag(I) complex recently studied by Sanford was plotted and indeed locates in the standard CMD region,15a which is consistent with its observed reactivity pattern. Another d10 metal complex, Cu(2Phpy)2(OAc) (2Phpy = 2-phenylpyridine),50 also appears to promote standard CMD.
Because the eCMD and CMD mechanisms are partitioned by a perfectly synchronous transition state within a putative mechanistic continuum, it would be anticipated that their reactivity differences are most amplified when a catalyst structure shifts either class of mechanism farther from this crossing point (i.e., perpendicular displacement from the synchronous diagonal in the More O’Ferrall-Jencks plot). For instance, an electrophilic Pt(II) cation developed by Periana for Shilov-type reactions is not surprisingly the most polarized eCMD-type transition state considered here.46 Likewise, a d10 metal (e.g., Sanford’s dinuclear Ag(I) complex) is one of the most polarized in the standard CMD region. On the other hand, more synchronous and less polarized transition states should blur the boundaries between eCMD and CMD and might exhibit subtler ligand effects on reactivity and selectivity. Experimental observations by Yu of ligand-switchable selectivity using different MPAA-Pd complexes (Figure 3b, open squares) is consistent with this notion, given the central location of these transition states in the plot.37,49
It is striking that such a wide variety of base-assisted, concerted C–H cleavage transition states spanning a diverse combination of metal, ancillary ligands, and (hetero)arene substrate can display eCMD characteristics distinct from the electron-rich complexes established to promote standard CMD. Based on these distinctions, we propose eCMD is a general class of C–H cleavage mechanism, and it may be the case that many complexes operating via eCMD have previously been conflated with standard CMD simply for lack of a more fitting model that can rationalize SEAr-type behavior together with mechanistic features of concerted C–H cleavage. The addition of this new model also substantiates Fagnou’s postulation that multiple classes of concerted mechanism could exist within a broader continuum.28a Importantly, this analysis also suggests the extent of nucleophilic or electrophilic character in the catalyst, as dictated by the ancillary ligands and metal electronic configuration, overrides the influence of the substrate as to what type of C–H cleavage mechanism occurs. These observations should be broadly informative to ongoing efforts to realize predictable and tunable site selectivity in catalytic, nondirected C–H functionalizations.
2.5. Scope of Thiophene C–H/C–H Coupling Using a Thioether-Pd Catalyst.
The above mechanistic analysis highlights that Pd(L7)(OAc) is one of the most electrophilic Pd catalysts in the eCMD region, as depicted by its location away from the synchronous diagonal. This helps to rationalize why this catalyst is particularly reactive toward activation of electron-rich heteroarene C–H bonds.34 To further demonstrate the utility of this (thioether)Pd catalyst, we applied it to the synthesis of a range of oligothiophenes (Table 4). Bithiophenes 2, 3, and 9 along with known p- and n-type semiconductors 13 and 14 were smoothly formed (Table 4, a) within 2 h using only 0.5 mol% [Pd] giving good isolated yields (77%−85%).51,52 Opportunities for streamlining the syntheses of oligothiophene motifs are exemplified by 12, a liquid crystal precursor, which was prepared previously from 2-(para-methoxyphenyl)-3-hexylthiophene by a conventional three-step sequence of bromination, Grignard formation, then Kumada coupling that occurred in 45% yield.7 On the other hand, direct C–H/C–H coupling using our (thioether)Pd catalyst generated 12 in a single step in 93% isolated yield from the same precursor. A privileged n-type material (15) was also formed in good isolated yield (70%) using 2 mol% [Pd].53
Table 4.
Scope of Oxidative C–H/C–H Coupling Using a (Thioether)Pd Catalyst.a
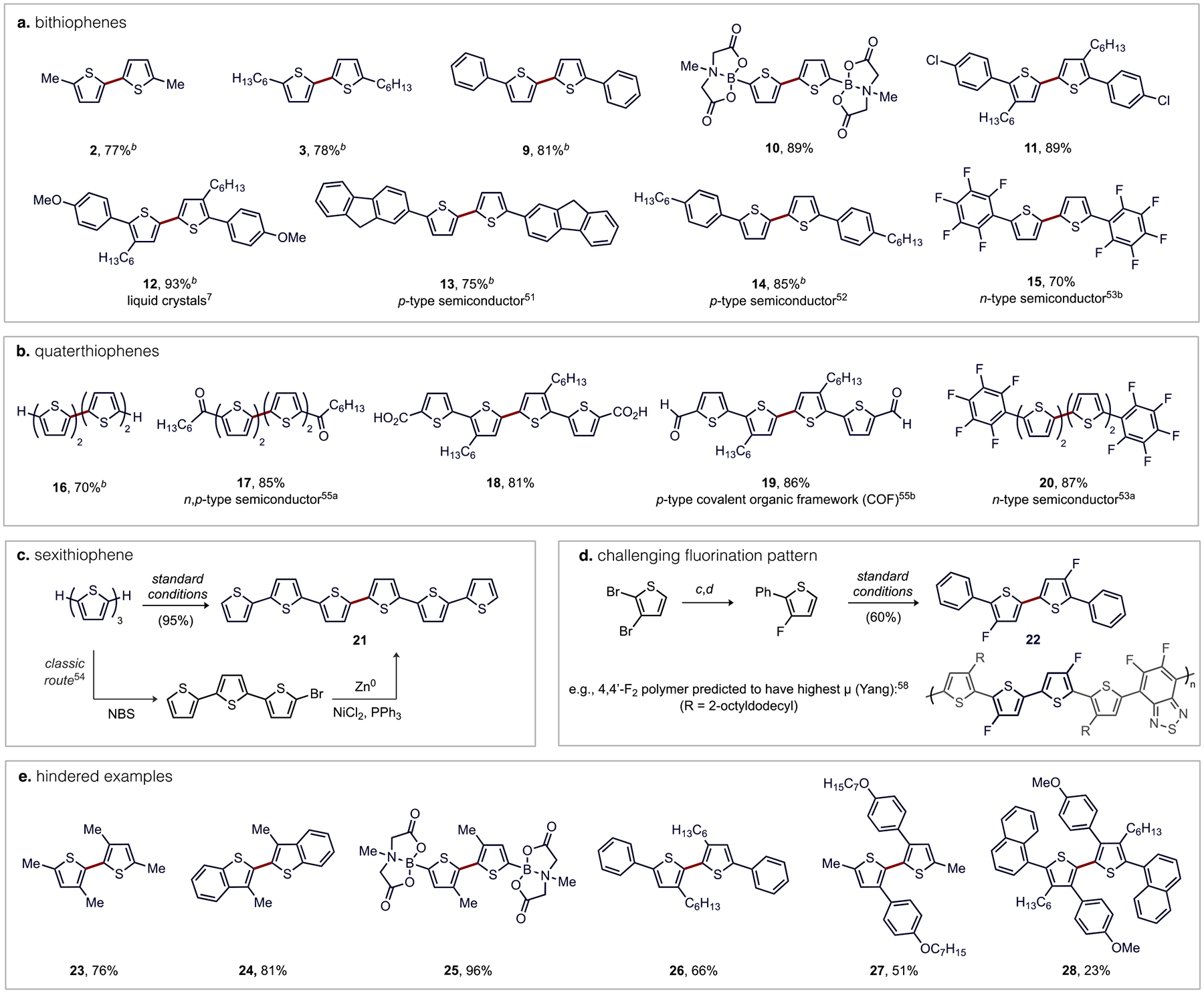 |
Isolated yields. Standard conditions: thiophene (0.5 mmol), BQ (0.38 mmol), Pd(OAc)2 (2 mol%), L7 (2 mol%), and CSA (4 mol%) stirred under air in AcOH/THF (1:1) at 60 °C.
0.5 mol% Pd(OAc)2, 0.5 mol% L7, and 1 mol% CSA; 2 h.
Pd(PPh3)4 (2.0 mol%), Na2CO3 (2 equiv) in toluene/H2O at 120 °C for 20 h (81%).
n-BuLi, Et2O, −78 °C, then NFSI (53%).
Parent α-quaterthiophene (16) was prepared in a single step from bithiophene with high selectivity and good isolated yield (70%), aided by precipitation of this oligomer at the mild reaction temperature (60 °C). α-Sexithiophene (21) was prepared with similar selectivity and high isolated yield (95%) using terthiophene as the substrate, which is a greener alternative to the conventional route involving bromination followed by reductive coupling using stoichiometric Zn metal (Table 4, c).54 The use of 2 mol% [Pd] was generally effective for other more challenging oligomers, such as those possessing an organometallic group (e.g., 10) or quaterthiophenes featuring deactivating electron-withdrawing substituents, such as polyfluoroaryl, aldehyde, ketone, or carboxylic acid (e.g., 17−20) groups giving good isolated yields (81%−89%) of several privileged p- and n-type semiconductor materials within 12 h.53,55 Another example relates to the fluorinated bithiophene (FBT) motif. A flurry of recent studies on device performance have noted a sensitivity to incorporation of fluorine into organic materials but also to the relative positioning of fluorines within the macromolecular structure.19b,56,57 C–H/C–H coupling should be useful to form uncommon FBT motifs from simple fluorothiophene building blocks. In particular, the 4,4’-FBT isomer remains rare in known conjugated oligomers or polymers yet has been predicted to enable improved hole mobility versus analogues.58 A model of this motif (22) was assembled in good isolated yield (60%) as shown in Table 4d, which highlights that (thioether)Pd catalysts can indeed forge new fluorinated 5,5’-biaryl-2,2’-bithiophene59 materials in a convergent fashion.
Lastly, we tested the potential for coupling at hindered C–H bonds (Table 4, e), which has generally not been practical using other Pd-catalyzed dehydrogenative coupling methods. Construction of bithiophenes 23−25 from moderately hindered 3-methyl substituted thiophenes occurred in high isolated yields (76%−96%) using the standard protocol. Increasing the substituent size to 3-hexyl12b or 3-aryl groups was still tolerated by the L7-Pd(OAc)2 catalyst to form 26 and 27 in reasonable yields (51%−66%). Even the use of the particularly hindered 2-(1-naphthyl-4-(para-methoxyphenyl)thiophene still gave a modest isolated yield (23%) of a fully substituted, tetraaryl bithiophene 28. These cases highlight a considerable improvement in steric tolerance by this L7-Pd(OAc)2 catalyst that allows assembly of highly substituted structures commonly encountered in modern organic materials.
Feasibility for cross selectivity was also tested (Scheme 5, a). Reaction of an equimolar mixture of 2-phenyl-4-hexylthiophene and 2-phenylthiophene under the standard conditions generated an 87% combined NMR yield strongly favoring the cross-product 29 over the two homo-coupling products (29/(9+26) = 20:1); 29 was isolated in 60% yield. The high cross selectivity is curious because at least one C–H activation by this catalyst must occur preferentially at the most sterically hindered yet π-basic site (C5 in 2-phenyl-4-hexylthiophene). While the typical rationalization for cross selectivity in dehydrogenative coupling is sequential C–H activation at a single metal center by two different mechanisms, our kinetic data suggesting a bimetallic step that occurs after C–H activation, such as Pd-to-Pd TM or RE, is rate-determining (see Section 2.2). This opens the possibility that one of these downstream catalytic steps could also be selectivity-determining.60 Either TM and RE steps could be slower for the hindered homocoupling pathway leading to 26 (Scheme 5, b). Additionally, the absence of the homocoupling product 9 can only be rationalized by a kinetic selectivity by L7-Pd(OAc)2 favoring C–H cleavage at the more hindered substrate. This highlights a key feature of Pd coordinated by an anionic thioether ligand – selectivity that is able to override steric control in favor of electronic control, which remains rare in nondirected C–H functionalization.
Scheme 5.
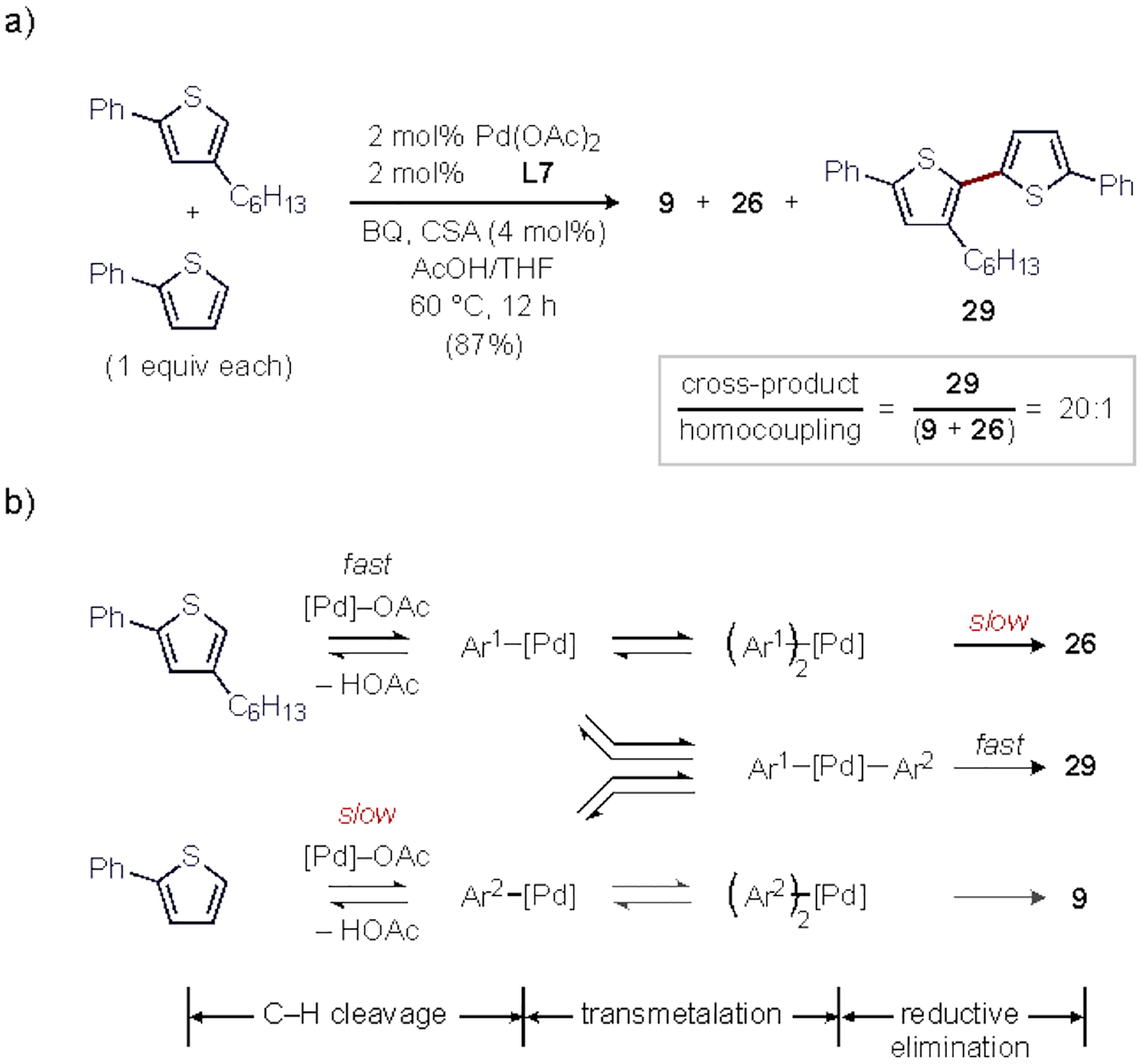
(a) Cross C–H/C–H coupling, and (b) proposed origin of selectivity.
2.6. Pd-to-Pd Transmetalation and Ligand-Accelerated Reductive Elimination.
The kinetic data in Figure 2 are consistent with a bimetallic turnover-limiting step involving an unusual 2:1 molecularity of Pd and BQ in the catalytic intermediate. The potential structure(s) of such a species is not intuitive. Furthermore, the role of anionic thioether in facilitating a bimetallic TM/RE step remained unclear from experimental data alone. These lingering questions about the elementary mechanisms and intermediates involved in thioether-Pd catalysis were thus evaluated computationally. A putative pathway to the penultimate diaryl-Pd intermediate leading to biaryl formation could occur by sequential activation of two substrates at a single metal center, as depicted in Figure 6. We found that the calculated barrier for a second C–H cleavage step by such as pathway is large. Thiophene coordination to form intermediate 33 then C–H cleavage by standard CMD through TS34 (ΔG‡ = 15.1 kcal/mol) renders the overall mononuclear pathway to a diorgano-Pd intermediate (e.g., 35) quite unfavorable.
Figure 6.
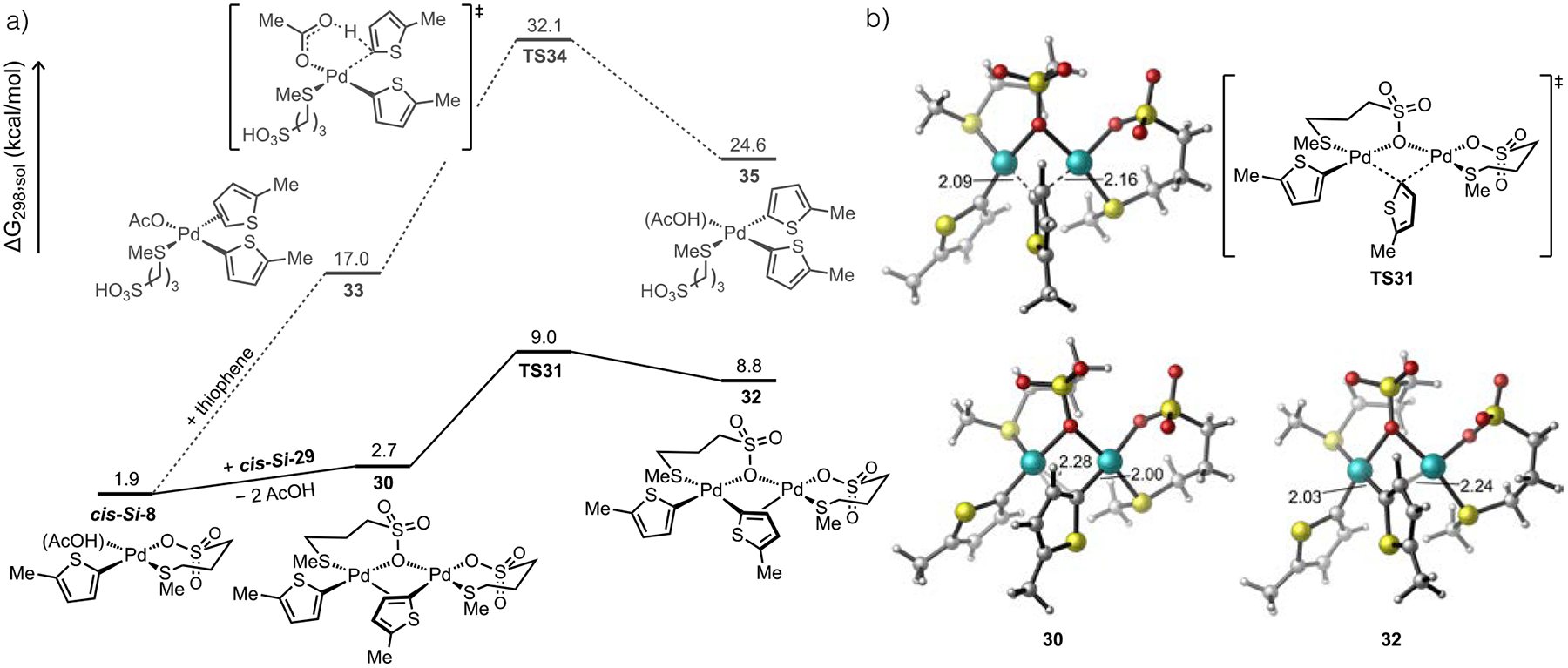
(a) Computed energy profiles for formation of diorgano-Pd intermediate by sequential C–H activation at a single Pd center versus Pd-to-Pd TM and (b) key intermediates and transition state for the TM pathway. All energies given in kcal/mol and bond lengths in Å.
Alternatively, we were able to locate another pathway involving disproportionation through Pd-to-Pd TM (Figure 6). Adduct formation between two equivalents of cis-Si-8 initially forms a dinuclear complex 30 possessing one μO-sulfonate bridging ligand from L7 and a κ1,η2-bridging mode from one thienyl ligand. From this species, Pd-to-Pd TM can occur through a low energy transition state TS31 (9.0 kcal/mol) to form a new dinuclear complex 32 possessing one diaryl-Pd fragment and one inorganic Pd fragment. The facile transmetalation between two aryl-Pd(II) species, which has been observed previously,61 is far lower in energy than the monometallic pathway (ΔΔG‡ = 23.1 kcal/mol). Importantly, the low barrier for Pd-to-Pd TM pathway also means it is unlikely to be the turnover- limiting step during catalytic C–H/C–H coupling. These results also reveal an additional beneficial role of the anionic thioether ligands we developed (e.g., L7), which is the ability to assemble reactive multimetallic structures with short metal-metal distances (e.g., Pd−Pd = 2.94 Å in 30) thereby facilitating cooperativity. We were also unable to find any computational support for an accelerating effect of BQ in this TM pathway; any intermediates with one Pd atom coordinated by BQ could not be located. The low energy barrier calculated for Pd-to-Pd TM and lack of evidence for BQ acceleration of TM forced us to consider the kinetic relevance of the product forming step, RE, to fully reconcile the kinetic data.
The possibility of turnover-limiting RE through a bimetallic mechanism initially seemed unlikely. First, it is generally assumed that C(sp2)−C(sp2) RE of simple hydrocarbyl groups is facile from Pd(II) complexes.62 Furthermore, Stahl concluded from a related study of o-xylene C–H/C–H coupling that Pd-to-Pd TM was the rate-determining catalytic step, though it is not clear if RE was explicitly ruled out as a kinetically relevant step in this study.39 We nevertheless interrogated several plausible pathways to bithiophene formation starting from the dinuclear complex 32 formed after TM. Dissociation of the η2-coordination mode of the thienyl ligand is first needed so the reacting ligand can adopt the appropriate orientation for the C−C bond-forming RE transition state. Because BQ is well documented as a promoter of RE reactions,63 we modeled coordination of BQ to break the bridging thienyl coordination mode to generate 36 (Figure 7). Alternatively, solvent could occupy this site after loss of η2-thienyl coordination to generate an alternative intermediate 39. In either of these cases, we found that spontaneous redistribution of a thioether ligand (L7) occurs concomitantly with formation of a new dinuclear complex. Both 36 and 39 exist as a μ-O adduct between a diorgano-Pd fragment and an inorganic Pd(L7)2 fragment.
Figure 7.
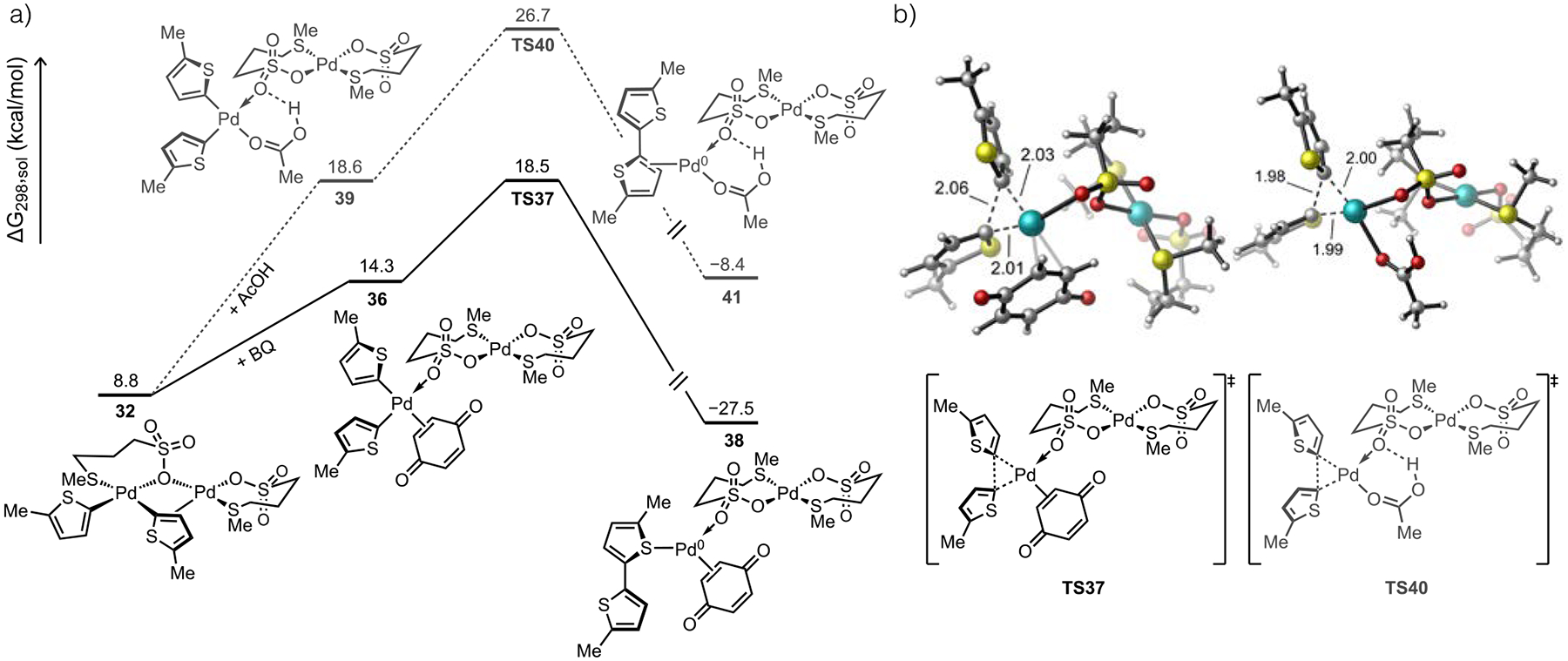
(a) Computed energy profiles for RE from diorgano-Pd intermediates and (b) transition states for RE with or without assistance from BQ coordination. All energies given in kcal/mol and bond lengths in Å.
Reductive elimination from the BQ-coordinated intermediate 36 is predicted to occur with a substantially lower barrier compared to the competing reaction through intermediate 39 lacking coordinated BQ (ΔΔG‡ = 8.2 kcal/mol). Monometallic RE pathways were also considered (Figure S25−S26) by initial fragmentation of 32, both with and without coordinated BQ, and these pathways were predicted to occur with higher energy barriers. The bimetallic, BQ-accelerated pathway to biaryl formation thus rationalizes the final outstanding experimental observations. First, the second order dependence of the catalytic rate on [Pd] and the 2:1 molecularity of Pd and BQ at low catalyst loading agrees with the low energy pathway proceeding through intermediate 36 that possesses two Pd atoms and a single coordinated BQ on the Pd atom poised to undergo RE. More importantly, the relative energy barriers for C–H activation and RE are predicted to be near isoergic (ΔΔG‡ = 0.2 kcal/mol). This result aligns well with the experimental observation that C–H/C–H coupling occurs with a first order dependence on catalyst concentration at high [Pd] because this reaction step is limited by C–H cleavage whereas a second-order dependence of the rate on [Pd] is observed at low catalyst concentrations when two molecules of catalyst are required to undergo reversible TM followed by irreversible and turnover-limiting RE.
A catalytic cycle is proposed in Scheme 6 for (thioether)Pd-catalyzed C–H/C–H coupling based on the above interpretations of kinetic, isotope effect, and computational data. An aryl-Pd intermediate is initially generated by C–H activation after reversible substrate coordination, but a second C–H activation step does not subsequently occur at this species. Instead, Pd-to-Pd TM occurs to generate a diaryl-Pd complex that remains associated to an inorganic Pd(L7)2 fragment. Coordination of BQ to this dinuclear complex accelerates the RE step to form the biaryl product. Finally, catalyst regeneration by oxidation of Pd(0) completes the cycle and regenerates the acetate ligands need for subsequent eCMD steps. This catalytic mechanism thus occurs by dual ligand catalysis in which both the thioether ancillary ligand and BQ, acting as a π-acid, operate in concert to achieve high catalytic rates under mild conditions.
Scheme 6.
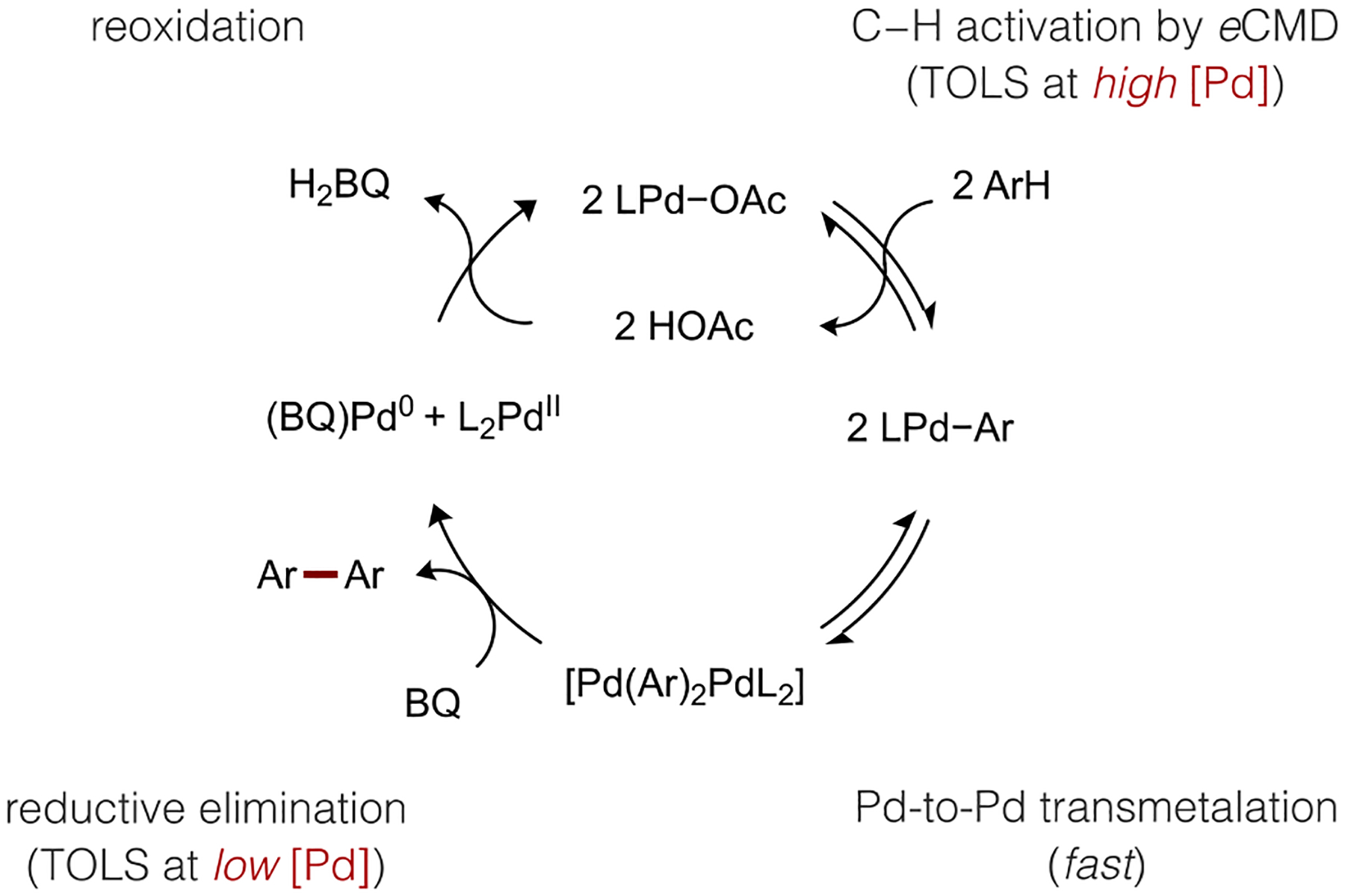
Proposed Catalytic Cycle for C–H/C–H Coupling.
L = L7; Ar = thienyl; TOLS = turnover-limiting step.
3. CONCLUSION
In summary, a mild (thioether)Pd-catalyzed oxidative coupling method was developed that constructs oligothiophene motifs in a modular fashion, with low catalyst loading, and in absence of stoichiometric metal promoters. A combined experimental and computational study was undertaken to reveal several unexpected mechanistic features of this system. First, a bimetallic pathway was revealed wherein the penultimate diorgano-Pd intermediate is generated by one C–H activation event at two Pd centers followed by reversible disproportionation through Pd-to-Pd transmetalation. Second, a mechanistic picture emerged that involves dual ligand acceleration where the thioether promotes C–H activation and BQ promotes bimetallic reductive elimination. Each of these steps occur with a similar energy barrier but different kinetic order dependence on [Pd], thus the turnover-limiting step is catalyst concentration dependent. These findings provide new insights into how ligands affect reactivity in Pd-catalyzed C–H/C–H coupling and emphasize that steps after C–H activation should be considered as potential selectivity determining steps during (cross)dehydrogenative coupling reactions. In the present reactions, reductive elimination is actually the rate- and selectivity-determining step under standard conditions.
Evaluation of acetate-assisted C–H bond cleavage at Pd(II) coordinated by an anionic thioether (e.g., L7) by DFT, Hammett, NBO charge, and distortion-interaction analyses suggest a concerted mechanism is operative that we describe as electrophilic CMD, or eCMD. The eCMD mechanism is characterized by more advanced metal-carbon bonding and less C–H bond cleavage in the transition state as compared to standard concerted metalation-deprotonation (CMD) that was formulated from reactions involving electron-rich catalysts. These features of eCMD give rise to higher polarization and positive charge build-up on the substrate during C–H activation, which strongly favors reactions at more π-basic sites in contrast to established trends for standard CMD that favors more acidic sites.
The generality of eCMD was supported through a broader analysis of other metal complexes in a More O’Ferrall-Jencks plot, from which we conclude that (i) many complexes other than Pd(L7)OAc can promote eCMD and (ii) a particular complex locates within the eCMD or standard CMD region as a function of its structure rather than by the identity of the reacting substrate. More electron poor complexes, such as inorganic d8 (e.g., inorganic Pd(II)) and d6 complexes (e.g., Rh(III), Ir(III), Ru(II)) routinely reside in the eCMD region of the mechanistic continuum whereas more electron-rich complexes, such as organometallic d8 (e.g., arylpalladium) and d10 complexes, generally locate in the standard CMD region. These correlations agree very well with established reactivity patterns for each cluster of complexes with CMD favoring reactions at more acidic substrates and sites and eCMD favoring reactions with more electron-rich substrates and sites. Furthermore, the highly active catalyst Pd(L7)OAc identified in this study is one of the most electrophilic species in the eCMD region, as visualized by the perpendicular displacement of its transition state from the synchronous trajectory in the More O’Ferrall-Jencks plot. This accounts for its exceptional reactivity toward C–H functionalization of electron-rich (hetero)arenes. The catalyst structure-mechanism correlations that were established help to rectify electrophilic patterns of reactivity in base-assisted (hetero)arene C–H cleavage that are captured by the standard CMD model. Instead, eCMD represents another mechanism within a broader continuum that also includes standard CMD. Preliminary guidelines to predict what catalyst structures enable eCMD or standard CMD should be informative for future efforts focused on switchable, catalyst-controlled reactivity.
Supplementary Material
ACKNOWLEDGMENT
Financial support was provided by Princeton University and NIH R35GM128902. We thank Xinyi Chen for helping set up Gaussian computations.
Footnotes
Supporting Information
Experimental procedures and spectral data for new compounds.
Spectra and tabular data for kinetic experiments.
The Supporting Information is available free of charge on the ACS Publications website.
REFERENCES
- (1).(a) Yeung CS; Dong VM; Catalytic Dehydrogenative Cross-Coupling: Forming Carbon−Carbon Bonds by Oxidizing Two Carbon−Hydrogen Bonds. Chem. Rev 2011, 111, 1215–1292; [DOI] [PubMed] [Google Scholar]; (b) Cho SH; Kim JY; Kwak J; Chang S; Recent advances in the transition metal-catalyzed twofold oxidative C-H bond activation strategy for C-C and C-N bond formation. Chem. Soc. Rev 2011, 40, 5068–5083; [DOI] [PubMed] [Google Scholar]; (c) Yang Y; Lan J; You J; Oxidative C–H/C–H Coupling Reactions between Two (Hetero)arenes. Chem. Rev 2017, 117, 8787–8863. [DOI] [PubMed] [Google Scholar]
- (2).Perepichka I; Perepichka F, Handbook D of Thiophene-Based Materials: Applications in Organic Electronics and Photonics; John Wiley & Sons, Ltd: New York, 2009. [Google Scholar]
- (3).Wang C; Dong H; Hu W; Liu Y; Zhu D; Semiconducting π-Conjugated Systems in Field-Effect Transistors: A Material Odyssey of Organic Electronics. Chem. Rev 2012, 112, 2208–2267. [DOI] [PubMed] [Google Scholar]
- (4).(a) Geiger F; Stoldt M; Schweizer H; Bäuerle P; Umbach E; Electroluminescence from oligothiophene-based light-emitting devices. Adv. Mater 1993, 5, 922–925; [Google Scholar]; (b) Yoshida Y; Tanigaki N; Yase K; Hotta S; Color-Tunable Highly Polarized Emissions from Uniaxially Aligned Thin Films of Thiophene/Phenylene Co-oligomers. Adv. Mater 2000, 12, 1587–1591. [Google Scholar]
- (5).(a) Segura JL; Martin N; Guldi DM; Materials for organic solar cells: the C60/π-conjugated oligomer approach. Chem. Soc. Rev 2005, 34, 31–47; [DOI] [PubMed] [Google Scholar]; (b) Zhang Q; Kan B; Liu F; Long G; Wan X; Chen X; Zuo Y; Ni W; Zhang H; Li M; Hu Z; Huang F; Cao Y; Liang Z; Zhang M; Russell TP; Chen Y; Small-molecule solar cells with efficiency over 9%. Nat. Photonics 2014, 9, 35. [Google Scholar]
- (6).Nielsen CB; Angerhofer A; Abboud KA; Reynolds JR; Discrete Photopatternable π-Conjugated Oligomers for Electrochromic Devices. J. Am. Chem. Soc 2008, 130, 9734–9746. [DOI] [PubMed] [Google Scholar]
- (7).Cheng X; Dong X; Wei G; Prehm M; Tschierske C; Liquid-Crystalline Triangle Honeycomb Formed by a Dithiophene-Based X-Shaped Bolaamphiphile. Angew. Chem. Int. Ed 2009, 48, 8014–8017. [DOI] [PubMed] [Google Scholar]
- (8).(a) Kagan J; Arora SK; The Synthesis of Alpha-thiophene Oligomers by Oxidative Coupling of 2-Lithiothiophenes. Bull. Chem. Soc. Jpn 1970, 20, 1937–1940; [Google Scholar]; (b) Hiroyuki H; Taketoshi N; Haruki K; Juro O; Tatsuo W; Hiroyuki S; Synthesis and Properties of α,ω-Disubstituted Oligo(3-hexylthiophene)s and Oligothienoquinonoids in Head-to-head Orientation. Bull. Chem. Soc. Jpn 1995, 68, 2363–2377. [Google Scholar]
- (9).(a) Dohi T; Morimoto K; Kiyono Y; Maruyama A; Tohma H; Kita Y; The synthesis of head-to-tail (H-T) dimers of 3-substituted thiophenes by the hypervalent iodine(iii)-induced oxidative biaryl coupling reaction. Chem. Commun 2005, 2930–2932; [DOI] [PubMed] [Google Scholar]; (b) Kita Y; Morimoto K; Ito M; Ogawa C; Goto A; Dohi T; Metal-Free Oxidative Cross-Coupling of Unfunctionalized Aromatic Compounds. J. Am. Chem. Soc 2009, 131, 1668–1669; [DOI] [PubMed] [Google Scholar]; (c) Morimoto K; Yamaoka N; Ogawa C; Nakae T; Fujioka H; Dohi T; Kita Y; Metal-Free Regioselective Oxidative Biaryl Coupling Leading to Head-to-Tail Bithiophenes: Reactivity Switching, a Concept Based on the Iodonium(III) Intermediate. Org. Lett 2010, 12, 3804–3807. [DOI] [PubMed] [Google Scholar]
- (10).(a) Kretchmer RA; Glowinski R; Organomercury compounds as synthetic intermediates. Coupling of arylmercuric salts. J. Org. Chem 1976, 41, 2661–2662; [Google Scholar]; (b) McClain MD; Whittington DA; Mitchell DJ; Curtis MD; Novel Poly(3-alkylthiophene) and Poly(3- alkylthienyl ketone) Syntheses via Organomercurials. J. Am. Chem. Soc 1995, 117, 3887–3888; [Google Scholar]; (c) Curtis MD; McClain MD; A New Poly(3-alkylthiophene) Synthesis via Pd-Catalyzed Coupling of Thienyl Mercuric Chlorides. Chem. Mater 1996, 8, 936–944. [Google Scholar]
- (11).(a) Lyons TW; Sanford MS; Palladium-Catalyzed Ligand-Directed C–H Functionalization Reactions. Chem. Rev 2010, 110, 1147–1169; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wencel-Delord J; Droge T; Liu F; Glorius F; Towards mild metal-catalyzed C-H bond activation. Chem. Soc. Rev 2011, 40, 4740–4761. [DOI] [PubMed] [Google Scholar]
- (12).(a) Masui K; Ikegami H; Mori A; Palladium-Catalyzed C–H Homocoupling of Thiophenes: Facile Construction of Bithiophene Structure. J. Am. Chem. Soc 2004, 126, 5074–5075; [DOI] [PubMed] [Google Scholar]; (b) Takahashi M; Masui K; Sekiguchi H; Kobayashi N; Mori A; Funahashi M; Tamaoki N; Palladium-Catalyzed C–H Homocoupling of Bromothiophene Derivatives and Synthetic Application to Well-Defined Oligothiophenes. J. Am. Chem. Soc 2006, 128, 10930–10933; [DOI] [PubMed] [Google Scholar]; (c) He C-Y; Wang Z; Wu C-Z; Qing F-L; Zhang X; Pd-catalyzed oxidative cross-coupling between two electron rich heteroarenes. Chem. Sci 2013, 4, 3508–3513. [Google Scholar]
- (13).Aerobic, Ag-free conditions have also been developed for thiophene C–H/C–H coupling. See:; Li N-N; Zhang Y-L; Mao S; Gao Y-R; Guo D-D; Wang Y-Q; Palladium-Catalyzed C–H Homocoupling of Furans and Thiophenes Using Oxygen as the Oxidant. Org. Lett 2014, 16, 2732–2735. [DOI] [PubMed] [Google Scholar]
- (14).Bay KL; Yang Y-F; Houk KN; Multiple roles of silver salts in palladium-catalyzed C–H activations. J. Organomet. Chem 2018, 864, 19–25. [Google Scholar]
- (15).(a) Lotz MD; Camasso NM; Canty AJ; Sanford MS; Role of Silver Salts in Palladium-Catalyzed Arene and Heteroarene C–H Functionalization Reactions. Organometallics 2017, 36, 165–171; [Google Scholar]; (b) Whitaker D; Burés J; Larrosa I; Ag(I)-Catalyzed C–H Activation: The Role of the Ag(I) Salt in Pd/Ag-Mediated C–H Arylation of Electron-Deficient Arenes. J. Am. Chem. Soc 2016, 138, 8384–8387. [DOI] [PubMed] [Google Scholar]
- (16).For early precedents of ligand-acceleration of C–H bond cleavage by late metal catalysts, see:; (a) Periana RA; Taube DJ; Gamble S; Taube H; Satoh T; Fujii H; Platinum Catalysts for the High-Yield Oxidation of Methane to a Methanol Derivative. Science 1998, 280, 560–564; [DOI] [PubMed] [Google Scholar]; (b) Zhong HA; Labinger JA; Bercaw JE; C–H Bond Activation by Cationic Platinum(II) Complexes: Ligand Electronic and Steric Effects. J. Am. Chem. Soc 2002, 124, 1378–1399; [DOI] [PubMed] [Google Scholar]; (c) Davies HML; Manning JR; Catalytic C–H functionalization by metal carbenoid and nitrenoid insertion. Nature 2008, 451, 417; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Yanagisawa S; Sudo T; Noyori R; Itami K; Direct C–H Arylation of (Hetero)arenes with Aryl Iodides via Rhodium Catalysis. J. Am. Chem. Soc 2006, 128, 11748–11749. [DOI] [PubMed] [Google Scholar]
- (17).For leading examples of pyridine ligand-acceleration of palladium catalysis featuring C–H bond cleavage, see:; (a) Ferreira EM; Stoltz BM; Catalytic C–H Bond Functionalization with Palladium(II): Aerobic Oxidative Annulations of Indoles. J. Am. Chem. Soc 2003, 125, 9578–9579; [DOI] [PubMed] [Google Scholar]; (b) Cho SH; Hwang SJ; Chang S; Palladium-Catalyzed C–H Functionalization of Pyridine N-Oxides: Highly Selective Alkenylation and Direct Arylation with Unactivated Arenes. J. Am. Chem. Soc 2008, 130, 9254–9256; [DOI] [PubMed] [Google Scholar]; (c) Zhang Y-H; Shi B-F; Yu J-Q; Pd(II)-Catalyzed Olefination of Electron-Deficient Arenes Using 2,6-Dialkylpyridine Ligands. J. Am. Chem. Soc 2009, 131, 5072–5074; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Emmert MH; Cook AK; Xie YJ; Sanford MS; Remarkably High Reactivity of Pd(OAc)2/Pyridine Catalysts: Nondirected C–H Oxygenation of Arenes. Angew. Chem. Int. Ed 2011, 50, 9409–9412; [DOI] [PubMed] [Google Scholar]; (e) Kubota A; Emmert MH; Sanford MS; Pyridine Ligands as Promoters in PdII/0-Catalyzed C–H Olefination Reactions. Org. Lett 2012, 14, 1760–1763; [DOI] [PubMed] [Google Scholar]; (f) Wang P; Verma P; Xia G; Shi J; Qiao JX; Tao S; Cheng PTW; Poss MA; Farmer ME; Yeung K-S; Yu J-Q; Ligand-accelerated non-directed C–H functionalization of arenes. Nature 2017, 551, 489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).For leading examples of monoprotected amino acid (MPAA)-accelerated palladium catalysis featuring C–H bond cleavage, see:; (a) Shi B-F; Maugel N; Zhang Y-H; Yu J-Q; PdII-Catalyzed Enantioselective Activation of C(sp2)–H and C(sp3)–H Bonds Using Monoprotected Amino Acids as Chiral Ligands. Angew. Chem. Int. Ed 2008, 47, 4882–4886; [DOI] [PubMed] [Google Scholar]; (b) Engle KM; Wang D-H; Yu J-Q; Ligand-Accelerated C–H Activation Reactions: Evidence for a Switch of Mechanism. J. Am. Chem. Soc 2010, 132, 14137–14151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).(a) Hu H; Chow PCY; Zhang G; Ma T; Liu J; Yang G; Yan H; Design of Donor Polymers with Strong Temperature-Dependent Aggregation Property for Efficient Organic Photovoltaics. Acc. Chem. Res 2017, 50, 2519–2528; [DOI] [PubMed] [Google Scholar]; (b) Zhang Q; Kelly MA; Bauer N; You W; The Curious Case of Fluorination of Conjugated Polymers for Solar Cells. Acc. Chem. Res 2017, 50, 2401–2409. [DOI] [PubMed] [Google Scholar]
- (20).For clarity, the term “standard CMD” is used to disambiguate this classic model developed by Fagnou from another type of concerted mechanism (eCMD) proposed in the present work.
- (21).(a) Kalyani D; Sanford MS; Regioselectivity in Palladium-Catalyzed C–H Activation/Oxygenation Reactions. Org. Lett 2005, 7, 4149–4152; [DOI] [PubMed] [Google Scholar]; (b) García-Cuadrado D; Braga AAC; Maseras F; Echavarren AM; Proton Abstraction Mechanism for the Palladium-Catalyzed Intramolecular Arylation. J. Am. Chem. Soc 2006, 128, 1066–1067; [DOI] [PubMed] [Google Scholar]; (c) García-Cuadrado D; de Mendoza P; Braga AAC; Maseras F; Echavarren AM; Proton-Abstraction Mechanism in the Palladium-Catalyzed Intramolecular Arylation: Substituent Effects. J. Am. Chem. Soc 2007, 129, 6880–6886; [DOI] [PubMed] [Google Scholar]; (d) Gorelsky SI; Lapointe D; Fagnou K; Analysis of the Concerted Metalation-Deprotonation Mechanism in Palladium-Catalyzed Direct Arylation Across a Broad Range of Aromatic Substrates. J. Am. Chem. Soc 2008, 130, 10848–10849. [DOI] [PubMed] [Google Scholar]
- (22).Lapointe D; Fagnou K; Overview of the Mechanistic Work on the Concerted Metallation–Deprotonation Pathway. Chem. Lett 2010, 39, 1118–1126. [Google Scholar]
- (23).Ryabov AD; Sakodinskaya IK; Yatsimirsky AK; Kinetics and mechanism of ortho-palladation of ring-substituted NN-dimethylbenzylamines. J. Chem. Soc., Dalton Trans 1985, 2629–2638. [Google Scholar]
- (24).Biswas B; Sugimoto M; Sakaki S; C–H Bond Activation of Benzene and Methane by M(η2-O2CH)2 (M = Pd or Pt). A Theoretical Study. Organometallics 2000, 19, 3895–3908. [Google Scholar]
- (25).(a) Davies DL; Donald SMA; Macgregor SA; Computational Study of the Mechanism of Cyclometalation by Palladium Acetate. J. Am. Chem. Soc 2005, 127, 13754–13755; [DOI] [PubMed] [Google Scholar]; (b) Davies DL; Al-Duaij O; Fawcett J; Giardiello M; Hilton ST; Russell DR; Room-temperature cyclometallation of amines, imines and oxazolines with [MCl2Cp*]2 (M = Rh, Ir) and [RuCl2(p-cymene)]2. Dalton Trans. 2003, 4132–4138; [Google Scholar]; (c) Davies DL; Donald SMA; Al-Duaij O; Macgregor SA; Pölleth M; Electrophilic C–H Activation at {Cp*Ir}: Ancillary-Ligand Control of the Mechanism of C–H Activation. J. Am. Chem. Soc 2006, 128, 4210–4211. [DOI] [PubMed] [Google Scholar]
- (26).Boutadla Y; Davies DL; Macgregor SA; Poblador-Bahamonde AI; Mechanisms of C–H bond activation: rich synergy between computation and experiment. Dalton Trans. 2009, 5820–5831. [DOI] [PubMed] [Google Scholar]
- (27).Lafrance M; Rowley CN; Woo TK; Fagnou K; Catalytic Intermolecular Direct Arylation of Perfluorobenzenes. J. Am. Chem. Soc 2006, 128, 8754–8756. [DOI] [PubMed] [Google Scholar]
- (28).(a) Gorelsky SI; Lapointe D; Fagnou K; Analysis of the Palladium-Catalyzed (Aromatic)C–H Bond Metalation–Deprotonation Mechanism Spanning the Entire Spectrum of Arenes. J. Org. Chem 2012, 77, 658–668; [DOI] [PubMed] [Google Scholar]; (b) Gorelsky SI; Origins of regioselectivity of the palladium-catalyzed (aromatic)CH bond metalation–deprotonation. Coord. Chem. Rev 2013, 257, 153–164. [Google Scholar]
- (29).(a) Park C-H; Ryabova V; Seregin IV; Sromek AW; Gevorgyan V; Palladium-Catalyzed Arylation and Heteroarylation of Indolizines. Org. Lett 2004, 6, 1159–1162; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Li L; Brennessel WW; Jones WD; C–H Activation of Phenyl Imines and 2-Phenylpyridines with [Cp*MCl2]2 (M = Ir, Rh): Regioselectivity, Kinetics, and Mechanism. Organometallics 2009, 28, 3492–3500; [Google Scholar]; (c) Sommai P-A; Tetsuya S; Yoshiki K; Masahiro M; Masakatsu N; Palladium-Catalyzed Arylation of Azole Compounds with Aryl Halides in the Presence of Alkali Metal Carbonates and the Use of Copper Iodide in the Reaction. Bull. Chem. Soc. Jpn 1998, 71, 467–473. [Google Scholar]
- (30).(a) Winstein S; Traylor TG; Mechanisms of Reaction of Organomercurials. II. Electrophilic Substitution on Saturated Carbon. Acetolysis of Dialkylmercury Compounds. J. Am. Chem. Soc 1955, 77, 3747–3752; [Google Scholar]; (b) Fung CW; Khorramdel-Vahed M; Ranson RJ; Roberts RMG; Kinetics and mechanism of mercuriation of aromatic compounds by mercury trifluoroacetate in trifluoroacetic acid. Journal of the Chemical Society, Perkin Transactions 2 1980, 267–272; [Google Scholar]; (c) Lau W; Kochi JK; Arene activation with mercury(II) and thallium(III) electrophiles. Mechanistic relevance of charge-transfer transitions in .pi.-complexes as intermediates. J. Am. Chem. Soc 1986, 108, 6720–6732. [Google Scholar]
- (31).Davidson JM; Triggs C; Reaction of metal ion complexes with hydrocarbons. Part I. ‘Palladation’ and some other new electrophilic substitution reactions. The preparation of palladium(I). J. Chem. Soc. A 1968, 1324–1330. [Google Scholar]
- (32).(a) Sweet JR; Graham WAG; Cationic η2-arene complexes of rhenium in carbon-hydrogen bond activation. J. Am. Chem. Soc 1983, 105, 305–306; [Google Scholar]; (b) Vigalok A; Uzan O; Shimon LJW; Ben-David Y; Martin JML; Milstein D; Formation of η2 C–H Agostic Rhodium Arene Complexes and Their Relevance to Electrophilic Bond Activation. J. Am. Chem. Soc 1998, 120, 12539–12544. [Google Scholar]
- (33).Joo JM; Touré BB; Sames D; C–H Bonds as Ubiquitous Functionality: A General Approach to Complex Arylated Imidazoles via Regioselective Sequential Arylation of All Three C–H Bonds and Regioselective N-Alkylation Enabled by SEM-Group Transposition. J. Org. Chem 2010, 75, 4911–4920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Gorsline BJ; Wang L; Ren P; Carrow BP; C–H Alkenylation of Heteroarenes: Mechanism, Rate, and Selectivity Changes Enabled by Thioether Ligands. J. Am. Chem. Soc 2017, 139, 9605–9614. [DOI] [PubMed] [Google Scholar]
- (35).(a) Izawa Y; Stahl SS; Aerobic Oxidative Coupling of o-Xylene: Discovery of 2-Fluoropyridine as a Ligand to Support Selective Pd-Catalyzed C–H Functionalization. Adv. Synth. Catal 2010, 352, 3223–3229; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Campbell AN; Meyer EB; Stahl SS; Regiocontrolled aerobic oxidative coupling of indoles and benzene using Pd catalysts with 4,5-diazafluorene ligands. Chem. Commun 2011, 47, 10257–10259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Wang D; Stahl SS; Pd-Catalyzed Aerobic Oxidative Biaryl Coupling: Non-Redox Cocatalysis by Cu(OTf)2 and Discovery of Fe(OTf)3 as a Highly Effective Cocatalyst. J. Am. Chem. Soc 2017, 139, 5704–5707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Cheng G-J; Yang Y-F; Liu P; Chen P; Sun T-Y; Li G; Zhang X; Houk KN; Yu J-Q; Wu Y-D; Role of N-Acyl Amino Acid Ligands in Pd(II)-Catalyzed Remote C–H Activation of Tethered Arenes. J. Am. Chem. Soc 2014, 136, 894–897. [DOI] [PubMed] [Google Scholar]
- (38).The linear regression (dashed line) in Figure 1a, inset was fit to data points between [Pd] = 0.5–1.5 mM.
- (39).Wang D; Izawa Y; Stahl SS; Pd-Catalyzed Aerobic Oxidative Coupling of Arenes: Evidence for Transmetalation between Two Pd(II)-Aryl Intermediates. J. Am. Chem. Soc 2014, 136, 9914–9917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).CCDC 1055553.
- (41).(a) Haines BE; Berry JF; Yu J-Q; Musaev DG; Factors Controlling Stability and Reactivity of Dimeric Pd(II) Complexes in C–H Functionalization Catalysis. ACS Catal. 2016, 6, 829–839; [Google Scholar]; (b) Canty AJ; Ariafard A; Sanford MS; Yates BF; Mechanism of Pd-Catalyzed Ar–Ar Bond Formation Involving Ligand-Directed C–H Arylation and Diaryliodonium Oxidants: Computational Studies of Orthopalladation at Binuclear Pd(II) Centers, Oxidation To Form Binuclear Palladium(III) Species, and Ar···Ar Reductive Coupling. Organometallics 2013, 32, 544–555; [Google Scholar]; (c) Yang Y-F; Cheng G-J; Liu P; Leow D; Sun T-Y; Chen P; Zhang X; Yu J-Q; Wu Y-D; Houk KN; Palladium-Catalyzed Meta-Selective C–H Bond Activation with a Nitrile-Containing Template: Computational Study on Mechanism and Origins of Selectivity. J. Am. Chem. Soc 2014, 136, 344–355. [DOI] [PubMed] [Google Scholar]
- (42).(a) Davies DL; Macgregor SA; McMullin CL; Computational Studies of Carboxylate-Assisted C–H Activation and Functionalization at Group 8–10 Transition Metal Centers. Chem. Rev 2017, 117, 8649–8709; [DOI] [PubMed] [Google Scholar]; (b) Ackermann L; Carboxylate-Assisted Transition-Metal-Catalyzed C–H Bond Functionalizations: Mechanism and Scope. Chem. Rev 2011, 111, 1315–1345. [DOI] [PubMed] [Google Scholar]
- (43).Petit A; Flygare J; Miller AT; Winkel G; Ess DH; Transition-State Metal Aryl Bond Stability Determines Regioselectivity in Palladium Acetate Mediated C–H Bond Activation of Heteroarenes. Org. Lett 2012, 14, 3680–3683. [DOI] [PubMed] [Google Scholar]
- (44).(a) Ess DH; Nielsen RJ; Goddard Iii WA; Periana RA; Transition-State Charge Transfer Reveals Electrophilic, Ambiphilic, and Nucleophilic Carbon−Hydrogen Bond Activation. J. Am. Chem. Soc 2009, 131, 11686–11688; [DOI] [PubMed] [Google Scholar]; (b) Ess DH; Goddard WA; Periana RA; Electrophilic, Ambiphilic, and Nucleophilic C–H Bond Activation: Understanding the Electronic Continuum of C–H Bond Activation Through Transition-State and Reaction Pathway Interaction Energy Decompositions. Organometallics 2010, 29, 6459–6472; [Google Scholar]; (c) Vastine BA; Hall MB; Carbon−Hydrogen Bond Activation: Two, Three, or More Mechanisms? J. Am. Chem. Soc 2007, 129, 12068–12069. [DOI] [PubMed] [Google Scholar]
- (45).Guimond N; Gorelsky SI; Fagnou K; Rhodium(III)-Catalyzed Heterocycle Synthesis Using an Internal Oxidant: Improved Reactivity and Mechanistic Studies. J. Am. Chem. Soc 2011, 133, 6449–6457. [DOI] [PubMed] [Google Scholar]
- (46).Ziatdinov VR; Oxgaard J; Mironov OA; Young KJH; Goddard WA; Periana RA; Carboxylic Solvents and O-Donor Ligand Effects on CH Activation by Pt(II). J. Am. Chem. Soc 2006, 128, 7404–7405. [DOI] [PubMed] [Google Scholar]
- (47).García-Melchor M; Gorelsky SI; Woo TK; Mechanistic Analysis of Iridium(III) Catalyzed Direct C-H Arylations: A DFT Study. Chemistry – A European Journal 2011, 17, 13847–13853. [DOI] [PubMed] [Google Scholar]
- (48).Simonetti M; Perry GJP; Cambeiro XC; Juliá-Hernández F; Arokianathar JN; Larrosa I; Ru-Catalyzed C–H Arylation of Fluoroarenes with Aryl Halides. J. Am. Chem. Soc 2016, 138, 3596–3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Cheng G-J; Chen P; Sun T-Y; Zhang X; Yu J-Q; Wu Y-D; A Combined IM-MS/DFT Study on [Pd(MPAA)]-Catalyzed Enantioselective C–H Activation: Relay of Chirality through a Rigid Framework. Chemistry – A European Journal 2015, 21, 11180–11188. [DOI] [PubMed] [Google Scholar]
- (50).Gray A; Tsybizova A; Roithova J; Carboxylate-assisted C–H activation of phenylpyridines with copper, palladium and ruthenium: a mass spectrometry and DFT study. Chem. Sci 2015, 6, 5544–5553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Meng H; Bao Z; Lovinger AJ; Wang B-C; Mujsce AM; High Field-Effect Mobility Oligofluorene Derivatives with High Environmental Stability. J. Am. Chem. Soc 2001, 123, 9214–9215. [DOI] [PubMed] [Google Scholar]
- (52).Mushrush M; Facchetti A; Lefenfeld M; Katz HE; Marks TJ; Easily Processable Phenylene−Thiophene-Based Organic Field-Effect Transistors and Solution-Fabricated Nonvolatile Transistor Memory Elements. J. Am. Chem. Soc 2003, 125, 9414–9423. [DOI] [PubMed] [Google Scholar]
- (53).(a) Facchetti A; Yoon M-H; Stern CL; Katz HE; Marks TJ; Building Blocks for n-Type Organic Electronics: Regiochemically Modulated Inversion of Majority Carrier Sign in Perfluoroarene-Modified Polythiophene Semiconductors. Angew. Chem. Int. Ed 2003, 42, 3900–3903; [DOI] [PubMed] [Google Scholar]; (b) Yoon M-H; Facchetti A; Stern CE; Marks TJ; Fluorocarbon-Modified Organic Semiconductors: Molecular Architecture, Electronic, and Crystal Structure Tuning of Arene- versus Fluoroarene-Thiophene Oligomer Thin-Film Properties. J. Am. Chem. Soc 2006, 128, 5792–5801. [DOI] [PubMed] [Google Scholar]
- (54).(a) Nakayama J; Konishi T; Murabayashi S; Hoshino M; Preparation of α-Quater-, α-Sexi-, and α-Octithiophenes. Heterocycles 1987, 26, 1793–1796; [Google Scholar]; (b) Martinez F; Voelkel R; Naegele D; Naarmann H; Thiophene Oligomers: Synthesis and Characterization. Mol. Cryst. Liq. Cryst 1989, 167, 227–232. [Google Scholar]
- (55).(a) Yoon M-H; DiBenedetto SA; Facchetti A; Marks TJ; Organic Thin-Film Transistors Based on Carbonyl-Functionalized Quaterthiophenes: High Mobility N-Channel Semiconductors and Ambipolar Transport. J. Am. Chem. Soc 2005, 127, 1348–1349; [DOI] [PubMed] [Google Scholar]; (b) Keller N; Bessinger D; Reuter S; Calik M; Ascherl L; Hanusch FC; Auras F; Bein T; Oligothiophene-Bridged Conjugated Covalent Organic Frameworks. J. Am. Chem. Soc 2017, 139, 8194–8199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).(a) Zhao W; Li S; Yao H; Zhang S; Zhang Y; Yang B; Hou J; Molecular Optimization Enables over 13% Efficiency in Organic Solar Cells. J. Am. Chem. Soc 2017, 139, 7148–7151; [DOI] [PubMed] [Google Scholar]; (b) Zhang Q; Yan L; Jiao X; Peng Z; Liu S; Rech JJ; Klump E; Ade H; So F; You W; Fluorinated Thiophene Units Improve Photovoltaic Device Performance of Donor–Acceptor Copolymers. Chem. Mater 2017, 29, 5990–6002. [Google Scholar]
- (57).(a) Sakamoto Y; Komatsu S; Suzuki T; Tetradecafluorosexithiophene: The First Perfluorinated Oligothiophene. J. Am. Chem. Soc 2001, 123, 4643–4644; [DOI] [PubMed] [Google Scholar]; (b) Wang Z; Li Z; Liu J; Mei J; Li K; Li Y; Peng Q; Solution-Processable Small Molecules for High-Performance Organic Solar Cells with Rigidly Fluorinated 2,2′-Bithiophene Central Cores. ACS Appl. Mater. Interfaces 2016, 8, 11639–11648. [DOI] [PubMed] [Google Scholar]
- (58).Qiu M; Zhu D; Yan L; Wang N; Han L; Bao X; Du Z; Niu Y; Yang R; Strategy to Manipulate Molecular Orientation and Charge Mobility in D–A Type Conjugated Polymer through Rational Fluorination for Improvements of Photovoltaic Performances. J. Phys. Chem. C 2016, 120, 22757–22765. [Google Scholar]
- (59).Salzner U; Effects of Perfluorination on Thiophene and Pyrrole Oligomers. J. Phys. Chem. A 2010, 114, 5397–5405. [DOI] [PubMed] [Google Scholar]
- (60).Kuhl N; Hopkinson MN; Glorius F; Selective Rhodium(III)-Catalyzed Cross-Dehydrogenative Coupling of Furan and Thiophene Derivatives. Angew. Chem. Int. Ed 2012, 51, 8230–8234. [DOI] [PubMed] [Google Scholar]
- (61).(a) Tan Y; Barrios-Landeros F; Hartwig JF; Mechanistic Studies on Direct Arylation of Pyridine N-Oxide: Evidence for Cooperative Catalysis between Two Distinct Palladium Centers. J. Am. Chem. Soc 2012, 134, 3683–3686; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Casado AL; Casares JA; Espinet P; An Aryl Exchange Reaction with Full Retention of Configuration of the Complexes: Mechanism of the Aryl Exchange between [PdR2L2] Complexes in Chloroform (R = Pentahalophenyl, L = Thioether). Organometallics 1997, 16, 5730–5736; [Google Scholar]; (c) Ozawa F; Fujimori M; Yamamoto T; Yamamoto A; Mechanism of the reaction of trans-bis(diethylphenylphosphine)di-m-tolylpalladium(II) with methyl iodide affording m-xylene. Evidence for a reductive elimination process involving the intermolecular exchange of organic groups. Organometallics 1986, 5, 2144–2149. [Google Scholar]
- (62).(a) Ananikov VP; Musaev DG; Morokuma K; Theoretical Insight into the C−C Coupling Reactions of the Vinyl, Phenyl, Ethynyl, and Methyl Complexes of Palladium and Platinum. Organometallics 2005, 24, 715–723; [Google Scholar]; (b) Pérez-Rodríguez M; Braga AAC; Garcia-Melchor M; Pérez-Temprano MH; Casares JA; Ujaque G; de Lera AR; Álvarez R; Maseras F; Espinet P; C−C Reductive Elimination in Palladium Complexes, and the Role of Coupling Additives. A DFT Study Supported by Experiment. J. Am. Chem. Soc 2009, 131, 3650–3657; [DOI] [PubMed] [Google Scholar]; (c) Xue L; Lin Z; Theoretical aspects of palladium-catalysed carbon–carbon cross-coupling reactions. Chem. Soc. Rev 2010, 39, 1692–1705. [DOI] [PubMed] [Google Scholar]
- (63).(a) Hull KL; Sanford MS; Mechanism of Benzoquinone-Promoted Palladium-Catalyzed Oxidative Cross-Coupling Reactions. J. Am. Chem. Soc 2009, 131, 9651–9653; [DOI] [PubMed] [Google Scholar]; (b) Ishikawa A; Nakao Y; Sato H; Sakaki S; Pd(ii)-promoted direct cross-coupling reaction of arenes via highly regioselective aromatic C–H activation: a theoretical study. Dalton Trans. 2010, 39, 3279–3289; [DOI] [PubMed] [Google Scholar]; (c) Alexandre V; Jacques M; Jean LB; Ubiquitous Benzoquinones, Multitalented Compounds for Palladium-Catalyzed Oxidative Reactions. Eur. J. Org. Chem 2015, 2015, 4053–4069. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.