

Abstract
Purpose of Review
To highlight recent findings on how picornavirus infections of the airways and cardiac tissues impact cellular inflammation and remodeling events.
Recent Findings
Recent published work has revealed that although many picornavirus infections appear to be initially asymptomatic, there are significant disease sequelae that result from chronic or persistent infections and the long-term, pathogenic effects on host tissues.
Summary
Because many acute picornavirus infections are asymptomatic, it is difficult to diagnose these pathologies at the early stages of disease. As a result, we must rely on preventative measures (i.e., vaccination) or discover novel treatments to reverse tissue damage and remodeling in affected individuals. Both of these strategies will require a comprehensive knowledge of virus-and cell-specific replication determinants and how these processes induce pathogenic effects in infected cells and tissues.
Keywords: Picornavirus, Rhinovirus, Coxsackievirus, Inflammation, Remodeling, Asthma, Cystic fibrosis, Myocarditis
Introduction
Picornaviruses are small non-enveloped viruses whose sizes vary from 30 to 32 nm. The capsid is composed of 60 identical protomers assembled in an icosahedral structure protecting the viral genome, which consists of a non-segmented, positive-strand RNA ranging from 6.7 to 10.1 kb. The family Picornaviridae belongs to the order Picornavirales. To date, more than 110 species have been described, grouped in more than 47 genera [1, 2]. These viruses are primarily transmitted by the fecal-oral route or via saliva and respiratory droplets. They can infect a number of different organs, including the central nervous system, heart, liver, skin, gastrointestinal tract, and upper respiratory tract. Each species uses a dedicated receptor(s) to enter the host cell, and viral replication steps take place in the cytoplasm.
Following infection of host cells, the innate immune system detects viral components and triggers a response. It is known that melanoma differentiation–associated gene 5 (MDA5) recognizes dsRNA of picornaviruses [3, 4]. Moreover, retinoic acid–inducible gene I (RIG-I) is cleaved during picornavirus infection, suggesting a role in the innate response to these viruses [5]. MDA5 and RIG-I both stimulate the production of cytokines, which allows the recruitment of immune cells that will trigger a response to eliminate the pathogenic agent. These inflammatory mechanisms are dependent on host cells (and their tissue of origin) as well as the virus [6]. In most cases, such inflammatory mechanisms are good for the host; however, on some occasions an excessive response is triggered and is detrimental to the host by inducing tissue remodeling, resulting in disease. In this review, we will summarize the most recent findings on the inflammation and remodeling events after picornavirus infections of the respiratory airways and cardiac tissue.
Membrane Remodeling
It is well known that picornaviruses divert cellular membranes of infected cells to create replication organelles. This topic has been reviewed recently in [7]. These replication organelles are composed of hijacked intracellular membranes, re-purposed by the viral 3A(B) protein to enhance picornavirus replication [8]. It is thought that they serve a dual purpose of replication complex formation and compartmentalization of the viral RNA [9], allowing the virus to escape from RNA and pathogen intracellular sensing, and subsequently, the immune response of the host. Additionally, members of the enterovirus genus of the picornavirus family have been reported to re-purpose autophagosomes and use them to release progeny virions from the cell in an alternative, non-lytic manner, allowing the viruses to be secreted without killing host cells and to display new properties in dissemination [10]. These mechanisms have been reviewed recently [11, 12].
Airway Remodeling
Respiratory infections by picornaviruses are a common phenomenon, ranging from the common cold induced by human rhinovirus (HRV) to more life-threatening conditions induced by the expanding outbreaks of enterovirus D68 (EV-D68) infections. Here, we will discuss recent findings on the role of picornavirus infections in asthma and cystic fibrosis and the exacerbation of these conditions.
Asthma
Asthma is a chronic respiratory disease characterized by wheezing, shortness of breath, chest tightness, cough, and variable airflow limitations. These symptoms are not continuous and can be caused by a number of factors such as exercise, allergens, irritant exposure, or viral respiratory infections [13]. An ever-increasing number of studies document the involvement of HRV in asthma [14].
Airway remodeling is an important hallmark of asthma, and deposition of increased levels of extracellular matrix (ECM) protein is one of the events leading to that remodeling. HRVs are able to increase deposition of perlecan, an extracellular matrix (ECM) protein, collagen V, and matrix-bound vascular endothelial growth factor in human bronchial epithelial cells [15]. Moreover, it has been reported that HRVs are able to induce the process of epithelial-mesenchymal transition in bronchial cell lines, and are even more efficient when synergizing with TGF-β1, potentially inducing an increased ECM protein deposition [16•]. ECM is known to regulate smooth muscle contraction [17]; the remodeling of this component decreases airway flow and could, in part, explain the role of HRV in asthma. Moreover, infection of monocytes by HRV upregulates ORMDL3 (a sphingolipid biosynthesis regulator), leading to increased levels of IFN-β and the endoplasmic reticulum chaperone BiP (HSPA5). This effect was enhanced in cells harboring genes associated with asthma [18]. Interestingly, it has been shown that IL-6, IL-8, and RANTES levels are lower and TGF-β1 levels are higher when cells are infected by HRV in an atopic asthmatic environment compared with a healthy environment. These results show that in an asthmatic patient, inflammation is decreased, resulting in higher levels of viral replication and increased cell damage [19], providing a plausible explanation for the loss of function of airway cells. A plausible scheme for cellular and tissue remodeling induced during human rhinovirus replication is shown in Fig. 1.
Fig. 1.
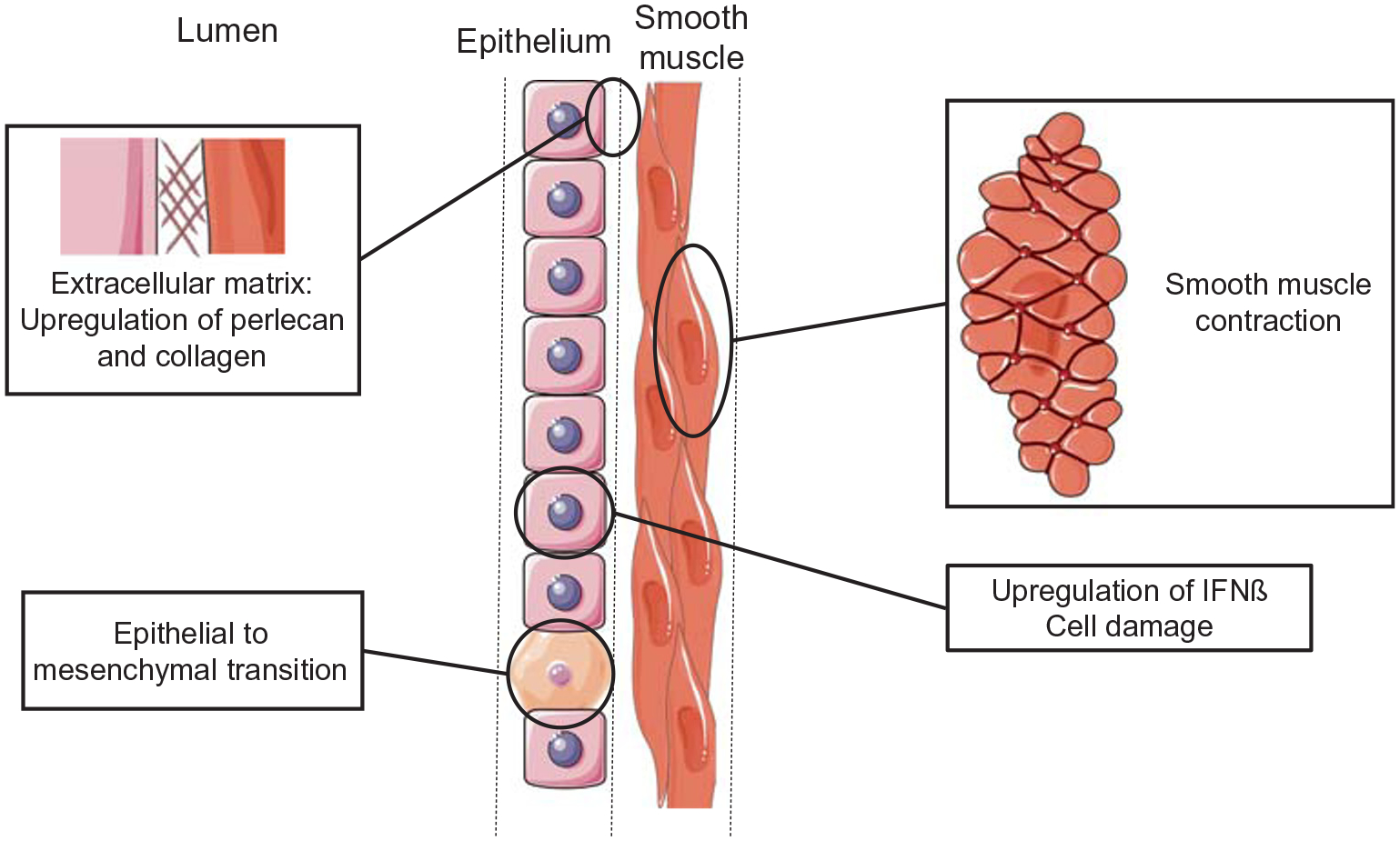
Remodeling of respiratory airway by human rhinoviruses. Following infection and replication, respiratory tissues can undergo changes affecting both cellular morphology and biology as well as the extracellular compartments. The text boxes illustrate the respiratory tissue components that are affected by rhinovirus infections
Cystic Fibrosis
Cystic fibrosis is an autosomal disorder caused by a mutation in the cystic fibrosis transmembrane conductance regulator protein. This mutation induces a change in the activity of chloride and sodium channels of sweat- and mucus-producing cells, resulting in thick and sticky mucus, setting an optimal environment for pathogen replication, especially pseudomonas [20]. It has been reported that HRVs are the most common viruses found in patients suffering from cystic fibrosis [21, 22], present in up to 43% of children under the age of five suffering from cystic fibrosis [23]. However, the precise role of HRV in cystic fibrosis requires further investigation, as it is unclear if chronic infections by these viruses increase the disease sequelae by inducing inflammatory responses or are just a secondary outcome [24].
HRV infection results in the upregulation of the chemokines CXCL10, CXCL11, and CXCL9 in patients suffering from cystic fibrosis, recruiting monocytes to the site of infection [25]. A recent study showed that different groups of rhinoviruses (differentiated by receptor usage) induced different effects on primary isolates of bronchial epithelial cells (BEC) from patients with cystic fibrosis. The so-called major HRV group infection of cystic fibrosis BEC yielded decreased interferon (IFN) responses compared with control BECs. In contrast, minor HRV group infections induced increased levels of IFN as well as increased expression of pattern recognition receptors that act as pathogen sensors [26]. Nevertheless, the association between HRVand cystic fibrosis requires more comprehensive studies since children suffering from this disease appear to be more susceptible to infection and for longer periods of time [27]. It is clear that HRVs are able to profoundly change cellular biology. For example, an in vitro study on alveolar epithelial cell monolayers infected by HRV analyzed differentially expressed genes. During the early steps of infection (up to 6 h post-infection), an upregulation of genes involved in the inflammatory response was observed. Subsequently, these viruses upregulate genes responsible for apoptosis, anti-apoptosis, blood vessel morphogenesis, and wound healing (12 h post-infection). Finally at late times of infection (24 h and 48 h post-infection), a downregulation of genes involved in airway remodeling events is observed [28]. Altogether, these results highlight a potential role for HRV in exacerbating the symptoms of cystic fibrosis.
Myocarditis
Enterovirus are known to be responsible for cardiac diseases, more specifically group B enteroviruses [29, 30]. Following an acute infection, the virus can persist in the cardiac tissue and lead to chronic myocarditis and dilated cardiomyopathy. It is well documented that following enterovirus infection, cardiac tissues become inflamed and remodeling of the heart occurs [31]. More recently, extensive studies on the role of enteroviruses in persistent infections leading to myocarditis have been published (for example [32••],). A model that includes the more recent findings for cardiac tissue remodeling following enterovirus infections is displayed in Fig. 2. As illustrated in the figure, inflammation appears to play an important role in the pathogenesis of enterovirus-mediated heart disease. It has been reported that neutrophils recognize coxsackievirus B3 (CV-B3) and play a role in disease outcome. Although neutrophils can internalize CV-B3, the virus does not replicate well in these cells. Following entry, viruses are detected by endosomal TLR-8 and induce an NFκB response [33]. In a murine model, neutrophil depletion leads to reduced viral replication and cardiomyocyte hypertrophy [34]. Other immune cells are important for the remodeling of cardiac tissue. It has been shown that regulatory T cells (Treg) play a significant role in cardiac fibrosis. During infection, the Treg cell population decreases and fibrosis increases. Further studies with anti-IL-10 antibodies revealed that Treg modulation of fibrosis occurs via IL-10 secretion [35]. Additionally, Tsunoda and colleagues [36] found that TLR4-deficient mice had lower levels of lympho-proliferation, IL-6, and IL-17, all of which were associated with increased myocarditis susceptibility. Similarly, a mutation of Unc93b1, a chaperone protein for TLR3, TLR7, and TLR9, showed that lack of TLR signaling increased viral loads during CV-B3 infection and increased inflammation, necrosis, and fibrosis [37]. A murine model in animals lacking the chemokine receptor CX3CR1 also displayed increased inflammatory cytokines and chemokine expression, leading to a more extensive immune cell infiltrate, cardiac fibrosis, and cardiomyocyte death following CV-B3 infection [38]. Similarly, NKT cell-deficient mice exhibit a higher viral load and an increase in antiviral antibody titers during Theiler’s murine encephalomyelitis virus (TMEV) cardiac infection despite lower levels of lympho-proliferation and reduced IL-4 and IL-10 levels [39]. More recently, the role of IL-22-producing Th22 cells has been described during myocarditis. Following infection by CV-B3, mice expressed IL-22 at higher levels, as well as collagen type I-A1, collagen type III-A1, and matrix metalloproteinase-9, while levels of tissue inhibitor of metalloproteinase-1 (TIMP-1) were decreased. This resulted in cardiac fibrosis, a hallmark of chronic cardiac disease and tissue remodeling [40].
Fig. 2.
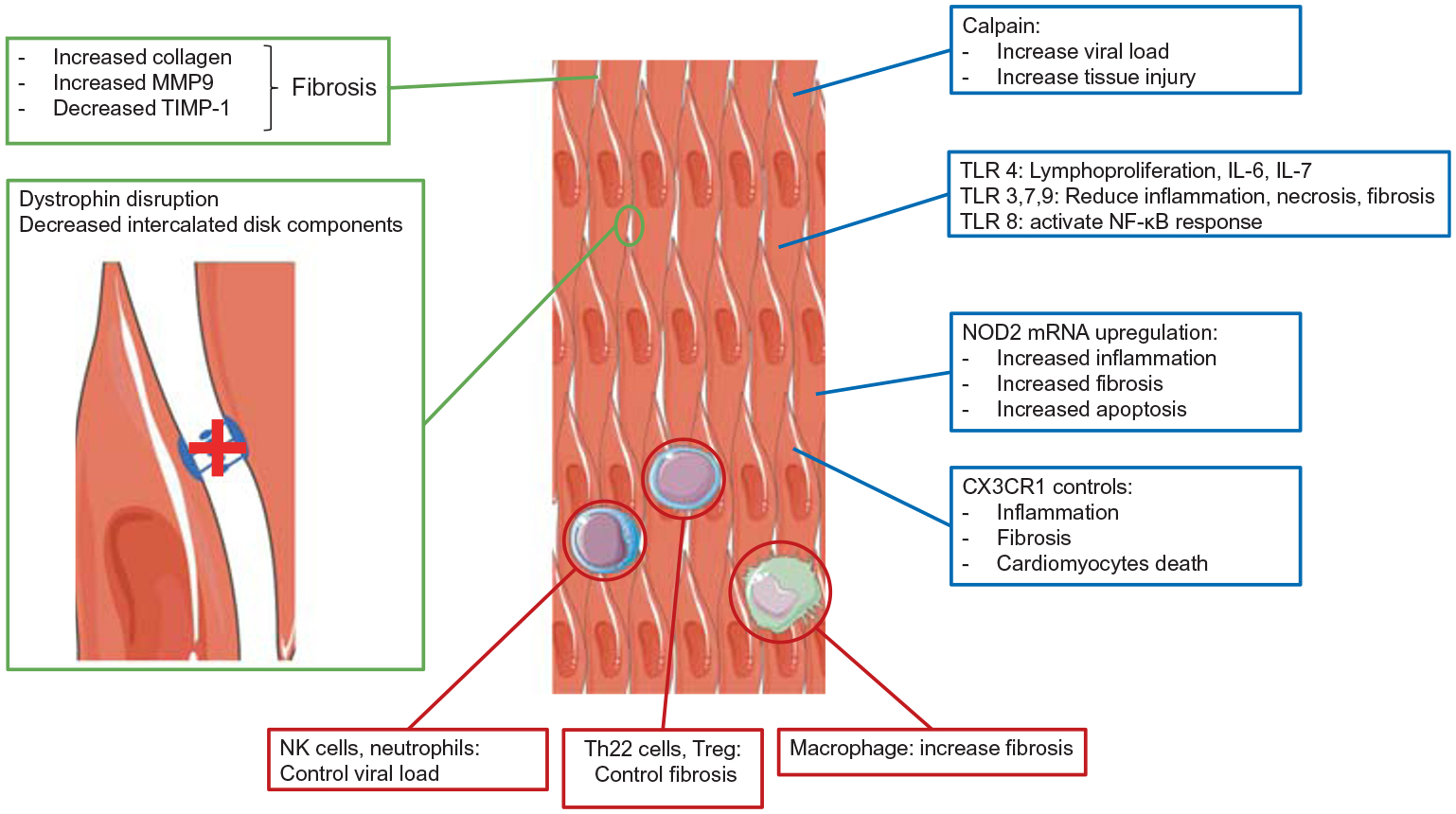
Remodeling of cardiac tissue by enteroviruses. Following infection and replication, cardiac tissues undergo significant changes. There are alterations in cell biology and the expression of many intracellular components, inducing a dysregulation of inflammation and immunity (shown as text in blue boxes). The immune system effectors will then be recruited and can act in an uncontrolled manner, triggering bystander effects (shown as text in red boxes). Ultimately, the extracellular space between cells will undergo drastic changes, impairing systolic function by breaking down the contacts and communication between cardiac cells (shown as text in green boxes)
The mRNA levels for the cytoplasmic pattern recognition receptor NOD2 (nucleotide-binding oligomerization domain 2) have been shown to be upregulated in patients infected by CV-B3 and suffering from myocarditis. Interestingly, NOD2 knockdown decreased inflammatory infiltrate and pro-inflammatory cytokine production, fibrosis, apoptosis, and expression of the coxsackievirus and adenovirus receptor (CAR) [41]. Similarly, the calcium-binding alarmins S100A8 and S100A9, which function as damage-associated molecular patterns, were also upregulated in cardiac tissue of patients suffering from CV-B3-induced myocarditis. In vitro experiments showed that overexpression of these proteins enhanced oxidative stress and CV-B3 replication in cardiomyocytes and stimulated the expression of the chemokine, macrophage inflammatory protein-2 (MIP-2), in macrophages. Moreover, infection of mice deficient in S100A8 and S100A9 by CV-B3 showed improved left ventricular functions as well as a lower level of inflammation and viral replication [42].
Macrophages are known to play a major role in acute inflammation and chronic fibrosis. Mice depleted of macrophages were able to sustain increased levels of CV-B3 replication but had reduced pathology and lower levels of fibrosis. Interestingly, reduced levels of myocarditis and chronic fibrosis were also observed in mice knocked out for galectin 3, a lectin involved in pathogenic cardiovascular remodeling and autoimmune/inflammatory processes [43]; however, in this case viral titers were not increased [44].
The cellular cysteine protease calpain has been reported to act in enterovirus infections [45–47]. Inhibiting its action by using a transgenic mouse model overexpressing calpastatin led to a decrease in tissue injury and viral load. Moreover, the pro-inflammatory factors MPO, perforin, IFNγ, and IL-17 were downregulated as well as the fibrotic factors Smad3 and MMP2 [48]. This study also showed that calpain promotes fibroblast migration in vitro. Interestingly, calpains are also involved in CV-B-induced necrosis of polarized intestinal cells (Caco-2) by mediating tight junctions and actin cytoskeleton rearrangement [49].
Inflammation is the first response to cardiac infection, leading to remodeling of the tissue, but it also triggers changes in the cell itself. It is known that cardiac diseases are linked to changes in cell metabolism (reviewed in [50]), and it has been reported recently that following infection by CV-B3, infiltrating leukocytes activate NFκB signaling, inducing cytokine expression that turns down oxidative gene expression. This deficit impairs energy metabolism in cardiomyocytes as well as their functions [51]. More changes can be seen during myocardial infection of the heart; it is now well known that CV-B3 2A proteinase can disrupt dystrophin and prevent its membrane localization [52] and that expression of 2A alone is sufficient to trigger dilated cardiomyopathy [53]. More recently, it has been shown that the C-terminal fragment of dystrophin (a product of viral 2A proteolytic cleavage) is able to bind the sarcoglycan complex and prevent the natural function of dystrophin. A threshold of 50% of un-cleaved cardiac dystrophin is necessary to prevent cardiac disease [54]. Additionally, by inducing the expression of miR-21, the virus downregulates intercalated disk components, disrupting the connections and communications between cardiac cells, and more specifically by destabilizing desmosomes [55]. CV-B3 infection also promotes the production of collagen I/IV in neonatal rat cardiac fibroblasts, inducing cardiac fibrosis [56]. Interestingly, this effect could be reversed by activating adenosine monophosphate–activated protein kinase. However, it is unlikely that the reduction of fibrosis would be an effective treatment even though acute myocarditis is characterized by inflammation and fibrosis, which decrease during the chronic phase of myocarditis. This reduction does not correlate with a recovery of hemodynamic function in a murine model [57]. More importantly, it has been shown that CV-B3 can infect cardiac tissue from juvenile mice at a subclinical dose, without any detectable disease symptoms. But once they reach adulthood, the mice infected as juveniles are predisposed to experience heart hypertrophy, leading to heart failure. This might be explained by a depletion of cardiac progenitor cell pools following an early differentiation of cardiac progenitor cells induced by CV-B3 [58••].
Hand, Foot, and Mouth Disease
Hand, foot, and mouth disease (HFMD) is characterized by sores in the mouth and blisters on the hands, feet, and legs. This disease is known to be caused by viral several enterovirus strains (e.g., EV-A71, EV-A68, CV-A6, CV-A16). The disease can be recapitulated in non-obese diabetic/severe combined immuno-deficient (NOD/SCID) mice, gamma interferon receptor (ifngr) knockout mice, and stat-1 knockout mice [59]. These infection models produced disease symptoms, including paralysis, and death rates that can be used for further studies. In addition to causing hand, foot, and mouth disease, enteroviruses like EV-A71 can infect the CNS. It has also been demonstrated that EV-A71 can infect and replicate in human microvascular endothelial cells and can be shed on both sides of these polarized cells. Moreover, actin cytoskeleton destruction, membrane remodeling, and cell death were observed along with an increase in permeability in a model of the blood-brain barrier [60]. Finally, another enterovirus (CV-A16) can upregulate the transcription of its receptor, scavenger receptor class B member 2 (SCARB2), in 293 T cells. This results in enhancement of subsequent rounds of infection, potentially facilitating co-infection and possible recombination with EV-A71 [61].
Conclusions
In this review, we have summarized recent findings on inflammation and remodeling of airway and cardiac tissues during picornaviruses infections. Although most picornavirus infections appear to be asymptomatic or lead to mild disease syndromes such as the common cold, the outcomes of these infections can lead to long-lasting effects in susceptible individuals, potentially life threatening, with or without chronic infections. These long-lasting effects can happen months or even years after the initial infection without any symptoms observed before the final stages, making it hard to diagnose and treat in the early phases when the pathology is mild. As a result, the importance of enterovirus infections in chronic diseases has been underestimated for a long time and has only more recently been acknowledged in disease syndromes such as cardiomyopathy [30, 32••] and diabetes [62]. Nevertheless, due to the asymptomatic nature of some acute infections, it is unlikely that we can diagnose these pathologies in the early stages, as healthy populations will never get sampled. The therapeutic options are to develop preventive vaccination, as explained by Dunne et al. [63], or to find ways to reverse tissue damage and remodeling in patients. For both options, a comprehensive understanding of cell-specific replication determinants and the pathogenic effects on infected tissues is critical to better anticipate these events and design therapeutic strategies.
Funding Information
Research in the authors’ laboratory was supported by U.S. Public Health Service grants AI022693 and AI026765 from the National Institutes of Health (to BLS) and by a postdoctoral fellowship from the George E. Hewitt Foundation for Medical Research (to AB).
Footnotes
Data Availability The figures presented in this review were designed by adapting the original Servier Medical Art website material. The original documents can be found at https://smart.servier.com/.
Conflict of Interest The authors declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent This article does not contain any studies with human or animal subjects performed by any of the authors.
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
- 1.Walker PJ, Siddell SG, Lefkowitz EJ, Mushegian AR, Dempsey DM, Dutilh BE, et al. Changes to virus taxonomy and the international code of virus classification and nomenclature ratified by the international committee on taxonomy of viruses (2019). Arch Virol. 2019;164:2417–29. [DOI] [PubMed] [Google Scholar]
- 2.Zell R, Delwart E, Gorbalenya AE, Hovi T, King AMQ, Knowles NJ, et al. ICTV virus taxonomy profile: Picornaviridae. J Gen Virol. 2017;98:2421–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Feng Q, Hato SV, Langereis MA, Zoll J, Virgen-Slane R, Peisley A, et al. MDA5 detects the double-stranded RNA replicative form in picornavirus-infected cells. Cell Rep. 2012;2:1187–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–5. [DOI] [PubMed] [Google Scholar]
- 5.Barral PM, Sarkar D, Fisher PB, Racaniello VR. RIG-I is cleaved during picornavirus infection. Virology. 2009;391:171–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Omura S, Kawai E, Sato F, Martinez NE, Minagar A, Al-Kofahi M, et al. Theiler’s virus-mediated immunopathology in the CNS and heart: roles of organ-specific cytokine and lymphatic responses. Front Immunol. 2018;9:2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van der Schaar HM, Dorobantu CM, Albulescu L, Strating J, van Kuppeveld FJM. Fat(al) attraction: picornaviruses usurp lipid transfer at membrane contact sites to create replication organelles. Trends Microbiol. 2016;24:535–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang J, Ptacek JB, Kirkegaard K, Bullitt E. Double-membraned liposomes sculpted by poliovirus 3AB protein. J Biol Chem. 2013;288:27287–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.den Boon JA, Ahlquist P. Organelle-like membrane compartmentalization of positive-strand RNA virus replication factories. Annu Rev Microbiol. 2010;64:241–56. [DOI] [PubMed] [Google Scholar]
- 10.Bird SW, Kirkegaard K. Escape of non-enveloped virus from intact cells. Virology. 2015;479–480:444–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Corona AK, Mohamud Y, Jackson WT, Luo H. Oh, SNAP! How enteroviruses redirect autophagic traffic away from degradation. Autophagy. 2018;14:1469–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mutsafi Y, Altan-Bonnet N. Enterovirus transmission by secretory autophagy. Viruses. 2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Flather D, Semler BL. Picornaviruses and nuclear functions: targeting a cellular compartment distinct from the replication site of a positive-strand RNA virus. Front Microbiol. 2015;6:594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jartti T, Gern JE. Role of viral infections in the development and exacerbation of asthma in children. J Allergy Clin Immunol. 2017;140:895–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuo C, Lim S, King NJ, Bartlett NW, Walton RP, Zhu J, et al. Rhinovirus infection induces expression of airway remodelling factors in vitro and in vivo. Respirology. 2011;16:367–77. [DOI] [PubMed] [Google Scholar]
- 16.•.Minor DM, Proud D. Role of human rhinovirus in triggering human airway epithelial-mesenchymal transition. Respir Res. 2017;18:110. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study provides evidence that both human rhinovirus type 16 and human rhinovirus type 1A are able to induce physical changes in bronchial epithelial cells that are associated with the process of epithelial-mesenchymal transition, which may contribute to tissue pathology in airways of patients with asthma. The data presented in this manuscript provide additional evidence that recurring human rhinovirus infections can contribute to airway remodeling in asthma.
- 17.Polio SR, Stasiak SE, Jamieson RR, Balestrini JL, Krishnan R, Parameswaran H. Extracellular matrix stiffness regulates human airway smooth muscle contraction by altering the cell-cell coupling. Sci Rep. 2019;9:9564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu YP, Rajamanikham V, Baron M, Patel S, Mathur SK, Schwantes EA, et al. Association of ORMDL3 with rhinovirus-induced endoplasmic reticulum stress and type I interferon responses in human leucocytes. Clin Exp Allergy. 2017;47:371–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xatzipsalti M, Psarros F, Konstantinou G, Gaga M, Gourgiotis D, Saxoni-Papageorgiou P, et al. Modulation of the epithelial inflammatory response to rhinovirus in an atopic environment. Clin Exp Allergy. 2008;38:466–72. [DOI] [PubMed] [Google Scholar]
- 20.Rafeeq MM, Murad HAS. Cystic fibrosis: current therapeutic targets and future approaches. J Transl Med. 2017;15:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goffard A, Lambert V, Salleron J, Herwegh S, Engelmann I, Pinel C, et al. Virus and cystic fibrosis: rhinoviruses are associated with exacerbations in adult patients. J Clin Virol. 2014;60:147–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rowan NR, Wang EW, Kanaan A, Sahu N, Williams JV, Phillips CD, et al. Respiratory viral detection in the paranasal sinuses of patients with cystic fibrosis. Am J Rhinol Allergy. 2017;31:105–8. [DOI] [PubMed] [Google Scholar]
- 23.Stelzer-Braid S, Liu N, Doumit M, D’Cunha R, Belessis Y, Jaffe A, et al. Association of rhinovirus with exacerbations in young children affected by cystic fibrosis: preliminary data. J Med Virol. 2017;89:1494–7. [DOI] [PubMed] [Google Scholar]
- 24.Flight WG, Bright-Thomas RJ, Tilston P, Mutton KJ, Guiver M, Webb AK, et al. Chronic rhinovirus infection in an adult with cystic fibrosis. J Clin Microbiol. 2013;51:3893–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ramirez IA, Caverly LJ, Kalikin LM, Goldsmith AM, Lewis TC, Burke DT, et al. Differential responses to rhinovirus- and influenza-associated pulmonary exacerbations in patients with cystic fibrosis. Ann Am Thorac Soc. 2014;11:554–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schogler A, Stokes AB, Casaulta C, Regamey N, Edwards MR, Johnston SL, et al. Interferon response of the cystic fibrosis bronchial epithelium to major and minor group rhinovirus infection. J Cyst Fibros. 2016;15:332–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dijkema JS, van Ewijk BE, Wilbrink B, Wolfs TF, Kimpen JL, van der Ent CK. Frequency and duration of rhinovirus infections in children with cystic fibrosis and healthy controls: a longitudinal cohort study. Pediatr Infect Dis J. 2016;35:379–83. [DOI] [PubMed] [Google Scholar]
- 28.Reza Etemadi M, Ling KH, Zainal Abidin S, Chee HY, Sekawi Z. Gene expression patterns induced at different stages of rhinovirus infection in human alveolar epithelial cells. PLoS One. 2017;12: e0176947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dennert R, Crijns HJ, Heymans S. Acute viral myocarditis. Eur Heart J. 2008;29:2073–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Knowlton KU. Dilated cardiomyopathy. Circulation. 2019;139: 2339–41. [DOI] [PubMed] [Google Scholar]
- 31.Gomez RM, Castagnino CG, Berria MI. Extracellular matrix remodelling after coxsackievirus B3-induced murine myocarditis. Int J Exp Pathol. 1992;73:643–53. [PMC free article] [PubMed] [Google Scholar]
- 32.••.Bouin A, Gretteau PA, Wehbe M, Renois F, N’Guyen Y, Leveque N, et al. Enterovirus persistence in cardiac cells of patients with idiopathic dilated cardiomyopathy is linked to 5′ terminal genomic RNA-deleted viral populations with viral-encoded proteinase activities. Circulation. 2019;139:2326–38 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reveals the nature of enterovirus genomic RNAs associated with persistent cardiac infections in human patients. It also describes how viral proteinases encoded by these altered genomic RNAs may contribute to the pathology associated with such infections.
- 33.Triantafilou K, Orthopoulos G, Vakakis E, Ahmed MA, Golenbock DT, Lepper PM, et al. Human cardiac inflammatory responses triggered by Coxsackie B viruses are mainly toll-like receptor (TLR) 8-dependent. Cell Microbiol. 2005;7:1117–26. [DOI] [PubMed] [Google Scholar]
- 34.Rivadeneyra L, Charo N, Kviatcovsky D, de la Barrera S, Gomez RM, Schattner M. Role of neutrophils in CVB3 infection and viral myocarditis. J Mol Cell Cardiol. 2018;125:149–61. [DOI] [PubMed] [Google Scholar]
- 35.Cao Y, Xu W, Xiong S. Adoptive transfer of regulatory T cells protects against coxsackievirus B3-induced cardiac fibrosis. PLoS One. 2013;8:e74955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sato F, Omura S, Kawai E, Martinez NE, Acharya MM, Reddy PC, et al. Distinct kinetics of viral replication, T cell infiltration, and fibrosis in three phases of myocarditis following Theiler’s virus infection. Cell Immunol. 2014;292:85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lafferty EI, Wiltshire SA, Angers I, Vidal SM, Qureshi ST. Unc93b1-dependent endosomal toll-like receptor signaling regulates inflammation and mortality during coxsackievirus B3 infection. J Innate Immun. 2015;7:315–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Muller I, Pappritz K, Savvatis K, Puhl K, Dong F, El-Shafeey M, et al. CX3CR1 knockout aggravates coxsackievirus B3-induced myocarditis. PLoS One. 2017;12:e0182643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kawai E, Sato F, Omura S, Martinez NE, Reddy PC, Taniguchi M, et al. Organ-specific protective role of NKT cells in virus-induced inflammatory demyelination and myocarditis depends on mouse strain. J Neuroimmunol. 2015;278:174–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guo Y, Wu W, Cen Z, Li X, Kong Q, Zhou Q. IL-22-producing Th22 cells play a protective role in CVB3-induced chronic myocarditis and dilated cardiomyopathy by inhibiting myocardial fibrosis. Virol J. 2014;11:230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tschope C, Muller I, Xia Y, Savvatis K, Pappritz K, Pinkert S, et al. NOD2 (nucleotide-binding oligomerization domain 2) is a major pathogenic mediator of Coxsackievirus B3-induced myocarditis. Circ Heart Fail. 2017;10(9). [DOI] [PubMed] [Google Scholar]
- 42.Muller I, Vogl T, Pappritz K, Miteva K, Savvatis K, Rohde D, et al. Pathogenic role of the damage-associated molecular patterns S100A8 and S100A9 in coxsackievirus B3-induced myocarditis. Circ Heart Fail. 2017;10. [DOI] [PubMed] [Google Scholar]
- 43.Dong R, Zhang M, Hu Q, Zheng S, Soh A, Zheng Y, et al. Galectin-3 as a novel biomarker for disease diagnosis and a target for therapy (review). Int J Mol Med. 2018;41:599–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jaquenod De Giusti C, Ure AE, Rivadeneyra L, Schattner M, Gomez RM. Macrophages and galectin 3 play critical roles in CVB3-induced murine acute myocarditis and chronic fibrosis. J Mol Cell Cardiol. 2015;85:58–70. [DOI] [PubMed] [Google Scholar]
- 45.Yoon SY, Ha YE, Choi JE, Ahn J, Lee H, Kweon HS, et al. Coxsackievirus B4 uses autophagy for replication after calpain activation in rat primary neurons. J Virol. 2008;82:11976–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Upla P, Marjomaki V, Nissinen L, Nylund C, Waris M, Hyypia T, et al. Calpain 1 and 2 are required for RNA replication of echovirus1. J Virol. 2008;82:1581–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li M, Wang X, Yu Y, Yu Y, Xie Y, Zou Y, et al. Coxsackievirus B3-induced calpain activation facilitates the progeny virus replication via a likely mechanism related with both autophagy enhancement and apoptosis inhibition in the early phase of infection: an in vitro study in H9c2 cells. Virus Res. 2014;179:177–86. [DOI] [PubMed] [Google Scholar]
- 48.Li M, Su Y, Yu Y, Yu Y, Wang X, Zou Y, et al. Dual roles of calpain in facilitating coxsackievirus B3 replication and prompting inflammation in acute myocarditis. Int J Cardiol. 2016;221:1123–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bozym RA, Patel K, White C, Cheung KH, Bergelson JM, Morosky SA, et al. Calcium signals and calpain-dependent necrosis are essential for release of coxsackievirus B from polarized intestinal epithelial cells. Mol Biol Cell. 2011;22:3010–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Doenst T, Nguyen TD, Abel ED. Cardiac metabolism in heart failure: implications beyond ATP production. Circ Res. 2013;113:709–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Remels AHV, Derks WJA, Cillero-Pastor B, Verhees KJP, Kelders MC, Heggermont W, et al. NF-kappaB-mediated metabolic remodelling in the inflamed heart in acute viral myocarditis. Biochim Biophys Acta Mol basis Dis. 2018;1864:2579–89. [DOI] [PubMed] [Google Scholar]
- 52.Badorff C, Lee GH, Lamphear BJ, Martone ME, Campbell KP, Rhoads RE, et al. Enteroviral protease 2A cleaves dystrophin: evidence of cytoskeletal disruption in an acquired cardiomyopathy. Nat Med. 1999;5:320–6. [DOI] [PubMed] [Google Scholar]
- 53.Xiong D, Yajima T, Lim BK, Stenbit A, Dublin A, Dalton ND, et al. Inducible cardiac-restricted expression of enteroviral protease 2A is sufficient to induce dilated cardiomyopathy. Circulation. 2007;115: 94–102. [DOI] [PubMed] [Google Scholar]
- 54.Barnabei MS, Sjaastad FV, Townsend D, Bedada FB, Metzger JM. Severe dystrophic cardiomyopathy caused by the enteroviral protease 2A-mediated C-terminal dystrophin cleavage fragment. Sci Transl Med. 2015;7:294ra106. [DOI] [PubMed] [Google Scholar]
- 55.Ye X, Zhang HM, Qiu Y, Hanson PJ, Hemida MG, Wei W, et al. Coxsackievirus-induced miR-21 disrupts cardiomyocyte interactions via the downregulation of intercalated disk components. PLoS Pathog. 2014;10:e1004070.24722419 [Google Scholar]
- 56.Jiang S, Jiang D, Zhao P, He X, Tian S, Wu X, et al. Activation of AMP-activated protein kinase reduces collagen production via p38 MAPK in cardiac fibroblasts induced by coxsackievirus B3. Mol Med Rep. 2016;14:989–94. [DOI] [PubMed] [Google Scholar]
- 57.Becher PM, Gotzhein F, Klingel K, Escher F, Blankenberg S, Westermann D, et al. Cardiac function remains impaired despite reversible cardiac remodeling after acute experimental viral myocarditis. J Immunol Res. 2017;2017:6590609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.••.Sin J, Puccini JM, Huang C, Konstandin MH, Gilbert PE, Sussman MA, et al. The impact of juvenile coxsackievirus infection on cardiac progenitor cells and postnatal heart development. PLoS Pathog. 2014;10:e1004249. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study describes a mouse model of juvenile infection with a dose of coxsackievirus B3 that is subclinical for adult animals. Following induced cardiac stress, juvenile-infected adult mice were found to have cardiac hypertrophy and dilation, hallmarks of progression to heart failure. Cardiac progenitor cell numbers were reduced in the hearts of these mice, suggesting that the exhausted cardiac progenitor cell pool following juvenile coxsackievirus infection may impair the heart’s ability to adapt to increased load.
- 59.Liao CC, Liou AT, Chang YS, Wu SY, Chang CS, Lee CK, et al. Immunodeficient mouse models with different disease profiles by in vivo infection with the same clinical isolate of enterovirus 71. J Virol. 2014;88:12485–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Volle R, Archimbaud C, Couraud PO, Romero IA, Weksler B, Mirand A, et al. Differential permissivity of human cerebrovascular endothelial cells to enterovirus infection and specificities of sero-type EV-A71 in crossing an in vitro model of the human blood-brain barrier. J Gen Virol. 2015;96:1682–95. [DOI] [PubMed] [Google Scholar]
- 61.Jin J, Li R, Jiang C, Zhang R, Ge X, Liang F, et al. Transcriptome analysis reveals dynamic changes in coxsackievirus A16 infected HEK 293T cells. BMC Genomics. 2017;18:933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tracy S, Smithee S, Alhazmi A, Chapman N. Coxsackievirus can persist in murine pancreas by deletion of 5′ terminal genomic sequences. J Med Virol. 2015;87:240–7. [DOI] [PubMed] [Google Scholar]
- 63.Dunne JL, Richardson SJ, Atkinson MA, Craig ME, Dahl-Jorgensen K, Flodstrom-Tullberg M, et al. Large enteroviral vaccination studies to prevent type 1 diabetes should be well founded and rely on scientific evidence. Reply to Skog O, Klingel K, Roivainen M et al. [letter]. Diabetologia. 2019;62:1100–3. [DOI] [PubMed] [Google Scholar]