

Abstract
Purpose:
Enhancer of zeste homolog 2 (EZH2) activity is dysregulated in many cancers.
Patients and Methods:
This phase I study determined the safety, maximum-tolerated dose (MTD), pharmacokinetics, and pharmacodynamics of the intravenously administered, highly selective EZH2 inhibitor, GSK2816126, (NCT02082977). Doses of GSK2816126 ranged from 50 to 3,000 mg twice weekly, and GSK2816126 was given 3-weeks-on/1-week-off in 28-day cycles. Eligible patients had solid tumors or B-cell lymphomas with no available standard treatment regimen.
Results:
Forty-one patients (21 solid tumors, 20 lymphoma) received treatment. All patients experienced ≥ 1 adverse event (AE). Fatigue [22 of 41 (53.7%)] and nausea [20 of 41 (48.8%)] were the most common toxicity. Twelve (32%) patients experienced a serious AE. Dose-limiting elevated liver transaminases occurred in 2 of 7 patients receiving 3,000 mg of GSK2816126; 2,400 mg was therefore established as the MTD. Following intravenous administration of 50 to 3,000 mg twice weekly, plasma GSK2816126 levels decreased biexponentially, with a mean terminal elimination half-life of approximately 27 hours. GSK2816126 exposure (maximum observed plasma concentration and area under the plasma-time curve) increased in a dose-proportional manner. No change from baseline in H3K27me3 was seen in peripheral blood mononuclear cells. Fourteen of 41 (34%) patients had radiological best response of stable disease, 1 patient with lymphoma achieved a partial response, 21 of 41 (51%) patients had progressive disease, and 5 patients were unevaluable for antitumor response.
Conclusions:
The MTD of GSK2816126 was established at 2,400 mg, but the dosing method and relatively short half-life limited effective exposure, and modest anticancer activity was observed at tolerable doses.
Introduction
Enhancer of zeste homolog 2 (EZH2) is the catalytic subunit of the polycomb repressive complex 2 (PRC2) responsible for maintaining transcriptional repression of its target genes through trimethylation of histone H3 on lysine 27 (H3K27me3; ref. 1). PRC2 activity is essential for maintaining the self-renewal capacity of embryonic and adult stem cells, by epigenetic repression of target genes controlling cell-cycle arrest and terminal differentiation (2).
EZH2 activity and H3K27me3 levels are dysregulated in many cancers through numerous pathways including gain-of-function heterozygous mutations, EZH2 overexpression, and inactivating mutations in UTX, an H3K27 demethylase that acts in opposition to EZH2 (3). Mutations have been identified in approximately 14% to 22% of germinal center B-cell (GCB) diffuse large B-cell lymphomas (DLBCL) and 7% to 22% of follicular lymphomas (FL; refs. 4–7)
Biochemical studies have demonstrated that Y641, A677, and A687 mutants exhibit an altered substrate preference and catalytic efficiency to enhance the generation of H3K27me3 (8–10). Consistent with these data, primary lymphomas and lymphoma cell lines harboring these EZH2 mutations have elevated levels of H3K27me3 (3, 9, 10). EZH2 overexpression in numerous other solid tumors, including prostate and endometrial cancers, correlates with increased tumor aggressiveness and poor prognosis (11, 12).
GSK2816126 is a highly selective and potent inhibitor of wild-type (WT) and mutant (Y641N, A677G, and A687V) EZH2, over 150-fold selective over EZH1, and more than 1,000-fold selective versus 20 other Su(var) 3–9, Enhancer-of-zeste and Trithorax (SET) and non-SET domain-containing human methyl transferases (13).
In cell culture, GSK2816126 induced loss of H3K27me3 in both EZH2 WT and mutant cell lines from a diverse panel of lymphoma cell lines independent of EZH2 mutation status, and sensitivity to GSK2816126 was not dependent on B-cell lymphoma 2 (BCL2), apoptosis regulator, translocation, or TP53 mutations (13). In proliferation assays using a panel of B-cell lymphoma lines, those of DLBCL origin with EZH2 activating mutations were the most sensitive to GSK2816126 (13). In mice bearing KARPAS-422 xenograft tumors, marked tumor regression was noted after treatment with GSK2816126. The treatment in mice was well tolerated, with no significant changes in blood cell counts or decrease in body weight. Several other tumor types including SMARCB1-mutant malignant rhabdoid tumors, and melanoma were also sensitive to EZH2 inhibition in preclinical studies (14–16). The SMARCB1/INI1 gene is a tumor suppressor gene that codes for a subunit of the chromatin remodeling complex regulating cell-cycle activity. Its loss results in uncontrolled cell-cycle progression through several mechanisms including elevated expression of EZH2 (17, 18).
Pharmacokinetic studies indicated that GSK2816126 is not orally bioavailable; however, pharmacokinetic/pharmacodynamic modeling predicted that a therapeutically effective dose could be achieved with twice weekly intravenous dosing of 950 to 2,700 mg. These data, together with preclinical efficacy studies, suggest potential efficacy of GSK2816126 in patients with advanced solid and hematologic cancers after standard-of-care treatment options have failed.
This study was a first-in-human, open-label, dose-escalation study of GSK2816126 to assess safety, pharmacokinetics, pharmacodynamics, and preliminary clinical activity in patients with non-Hodgkin lymphoma (NHL), metastatic solid tumors, and multiple myeloma with relapsed or refractory disease.
Materials and Methods
Study design
This was an open-label, phase I, dose-escalation study initially planned as 2 parts: a dose escalation study (part 1) of GSK2816126 (Fig. 1) and a cohort expansion study (part 2) to understand the clinical activity of GSK2816126. Data analysis at the end of part 1 determined that dosing challenges would prevent adequate exposure for significant benefit, and part 2 was not initiated. The study was initiated on April 24, 2014, and terminated on June 20, 2017. The complete study protocol can be found at https://www.gsk-studyregister.com/study/4973.
Figure 1.
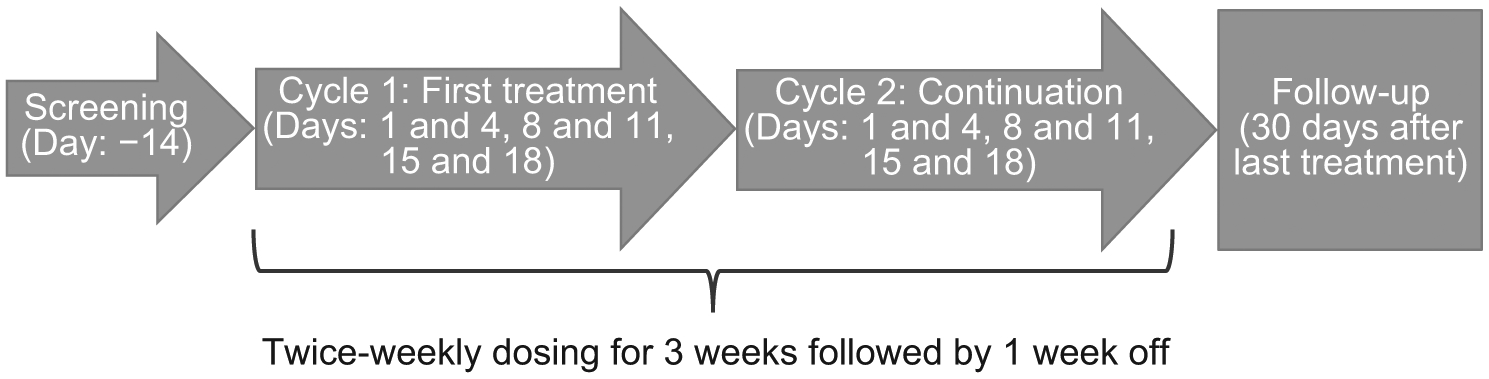
Study design. Patients could continue treatment in the study until the occurrence of disease progression, unacceptable toxicity, or withdrawal of consent. In part 1, the dose was escalated based on all available data.
The primary objective was to determine safety and tolerability and establish a recommended phase II dose for intravenously administered GSK2816126. The secondary objective was to describe the pharmacokinetics after single- and repeated-dose administration, as well as the pharmacodynamics and clinical activity of GSK2816126.
In part 1, accelerated dose escalation was used, with one patient enrolled per dose level beginning with a starting dose of 50 mg given as an intravenous infusion over 2 to 4 hours. GSK2816126 was administered twice weekly in a 3-weeks-on/1-week-off, 28-day cycle. Twice-weekly intravenous administration was selected based on preclinical pharmacokinetic murine and canine studies, and efficacy studies in mice, which showed similar tumor growth inhibition for doses ranging from daily to twice weekly, including intermittent twice-weekly dosing with 2-weeks-on/1-week-off. Dose escalation continued until a maximum-tolerated dose (MTD), or a dose of 3,000 mg twice weekly was reached.
Accelerated dose escalation was used until the first instance of a ≥ grade 2 drug-related, nonhematologic toxicity or until a dose-limiting toxicity (DLT) was observed. The dose of the next cohort was determined based on the evaluation of at least 1 patient who completed 1 cycle of treatment. In the absence of grade ≥ 2 nonhematologic toxicity or a DLT, subsequent cohorts were allowed up to 100% dose escalations (up to 500 mg). Subsequent dose-escalation steps were planned up to a maximum of 50% at each step. Following the initial occurrence of a grade 2 toxicity or DLT in a patient during the first cycle, accelerated dose escalation transitioned to a standard 3+3 dose escalation.
During the 3+3 dose escalation, 3 patients were enrolled per dose level and observed for any DLT during the first 28 days of treatment. Dosing proceeded to the next higher dose level if no DLTs were observed in any of the patients (similar to accelerated dose-escalation phase, ≤ 100% increase in dose up to 500 mg, ≤ 50% increase in dose thereafter). An additional 3 patients were enrolled at this dose level if 1 of 3 patients experienced a DLT. Patients were entered into the study using a staggered approach, with at least 7 days between each patient to minimize the risk of inadvertently exceeding the MTD in multiple patients. Any dose level could be expanded up to 12 patients to collect adequate data on safety, pharmacokinetics, or pharmacodynamics, with no fixed number prespecified for expansion. Because of elevated toxicity in the 3,000-mg cohort, the expansion of this cohort (3,000 mg) was stopped after only 7 patients had been enrolled to ensure minimal impact on patient safety, and additional patients were assessed at the 2,400-mg dose level. Intrapatient dose escalations were considered on a case-by-case basis provided that the patient did not experience a ≥ grade 2 drug-related, nonhematologic toxicity or DLT.
Eligibility criteria
Patients with DLBCL or transformed FL, who had relapsed after or were refractory to ≥ 1 prior line of therapy and not a candidate for standard salvage regimens or autologous stem cell transplantation or who had relapsed after or were refractory to ≥ 2 prior chemotherapy regimens were eligible for the study. Patients with other NHLs who had failed ≥ 1 prior line of therapy and for which there was no standard salvage regimen; patients with relapsed/refractory multiple myeloma; and patients with solid tumors with lesions evaluable by Response Evaluation Criteria in Solid Tumors (RECIST) criteria [castrate resistant prostate cancer (CRPC) or patients with CRPC with bone-only disease], who had received ≥ 1 and <3 standard chemotherapy regimens were also eligible. Finally, patients with solid tumor types with no approved therapy or for which standard therapy was refused or declined were eligible. EZH2 mutation status was not an entry criterion but was evaluated when tumor tissue was available.
All patients were required to have adequate organ function and an Eastern Cooperative Oncology Group performance status of 0 or 1. A fresh biopsy sample or archival tumor tissue was also required prior to study enrollment.
Patients with uncontrolled diabetes or any other medical condition that could interfere with safety assessments were excluded. Concomitant administration of drugs known to prolong QT were to be avoided beginning 14 days prior to the first dose of study drug. Patients currently receiving chemotherapy, radiation therapy, immunotherapy, or biologic therapy; those who had received an investigational anticancer drug within 4 weeks, or within 5 half-lives (whichever is shorter) of the first dose of study drug; or patients who had unresolved grade >1 toxicity [National Cancer Institute – Common Terminology Criteria for Adverse Events (NCI-CTCAE) version 4] from previous anticancer therapy, with the exception of alopecia and peripheral neuropathy, were also excluded. Patients could not have undergone any major surgery, radiotherapy, or immunotherapy within the 4 weeks prior to the first dose of study drug or palliative radiotherapy to a single symptomatic lesion within the 2 weeks prior to the first dose of study drugs. Those who previously underwent an autologous stem cell transplant were allowed to enroll if a minimum of 100 days had elapsed from the time of transplant and the patient had recovered from transplant-associated toxicities prior to the first dose of GSK2816126.
Complete eligibility criteria are included in the Supplementary Materials and Methods.
Study assessments
A physical examination, electrocardiogram, assessment of vital signs, biomarker assessments, coagulation testing, and hematology were performed at screening, day 1, and weekly thereafter.
Safety data were evaluated prior to defining a new dose and starting the next cohort. Subsequently, the dose was escalated up to the next level.
Pharmacokinetic assessments
Blood samples were analyzed by Bioanalytical Science and Toxicokinetics at GlaxoSmithKline, King of Prussia, Pennsylvania. Analysis of pharmacokinetic exposure in blood [i.e., dose, concentration, maximum concentration [Cmax], or area under the plasma–time curve (AUC)], safety and efficacy responses, and population pharmacokinetic parameters [i.e., clearance (CL), volume of distribution (Vd)], with relevant covariates that may have influenced exposure (i.e., age, weight, or disease-related covariates) of GSK2816126, were conducted using noncompart-mental methods and Phoenix WinNonLin v6.3 (Certara).
Each pharmacokinetic sample was collected as close as possible to the planned time relative to the dose administered (i.e., time 0). The actual date and time of each blood sample collection was recorded. Blood sampling for pharmacokinetics was performed on day 1 and day 4 predose and at the end of the infusion; day 8 and day 11 soon after electrocardiogram (ECG) and pharmacodynamic biomarker collection; day 15 and day 21 at the time of pharmacodynamic biomarker collection. During cycles 2, 4, 6, and 12, samples were taken predose and within 5 minutes prior to the end of the infusion on day 4.
The pharmacokinetic sampling during the first treatment cycle (days 1 and 15) was done predose; 0.5, 1, and 2 hours following the start of infusion; immediately prior to the end of the infusion; 0.5, 1, 2, 3, 4, and 6 hours following the end of infusion; and 12, 18, 24, and 72 or 96 hours following the start of infusion. An additional end of infusion sample was taken on day 4 of the first treatment cycle.
Pharmacodynamic assessments
Samples from tumor or surrogate tissue/body fluid were obtained pre- and posttreatment for pharmacodynamic analysis. Changes from baseline in H3K27me3 were recorded to provide evidence of target engagement.
Blood sampling for pharmacodynamic biomarkers was performed during the first treatment cycle [day 1 (predose) and days 4, 8, 11, 15, 18, 21 and 28]. Further, samples were obtained 2 hours after the end of infusion on days 1, 4, 8, 11, 15, and 18 and on days 21 and 28. A tumor biopsy for pharmacodynamic analysis (required for part 1 pharmacokinetic/pharmacodynamic cohort at MTD) was obtained during the first treatment cycle on day 1 and day 18 (after the 6th dose). The biopsy was performed according to individual institutional standards.
Antitumor assessments
Responses for patients with lymphoma were assessed using the Revised Response Criteria for Malignant Lymphoma (19), whereas responses for patients with solid tumors were assessed according to RECIST (20, 21). Baseline disease assessment for patients with lymphomas were completed within 4 weeks of the first dose of GSK2816126. Disease assessments were done 8 weeks after dosing was initiated, every 12 weeks thereafter, and at the final study visit. Disease assessments for patients with solid tumors were completed 8 weeks after the first dose of GSK2816126, repeated every 8 weeks thereafter, and at the final study visit.
Ethics
This study was conducted in accordance with the International Conference on Harmonization, Good Clinical Practice guidelines, and the ethical principles outlined in the Declaration of Helsinki 2008. The study protocol, any amendments, the informed consent, and other information that required pre-approval were reviewed and approved by a national, regional, or investigational center ethics committee or institutional review board, in accordance with the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use Good Clinical Practice and applicable country-specific requirements, including US 21 Code of Federal Regulations 312.3(b) for constitution of independent ethics committees. Ethics committee or institutional review board approvals are maintained in the sponsor’s study file. All participants provided written informed consent prior to study entry.
Statistical analyses
Results were summarized using descriptive statistics, and no formal statistical hypotheses were tested. The sample size for dose escalation in part 1 was not driven by statistical considerations.
Results
Patient disposition
In total, 42 patients from the United Kingdom, United States, and France were enrolled in the study across dose levels. Of these 42 patients, 1 patient withdrew consent and did not receive any study drug, 14 patients withdrew from the study (9 on investigator advice, 2 withdrew consent, 2 were withdrawn due to study termination, and 1 was lost to follow-up), and 5 patients died. No patient withdrew due to an AE or serious AE (SAE). Of the 9 withdrawn on investigator advice, 7 had disease progression, 1 refused follow up, and 1 reported “alteration of general state.”
Patient characteristics
Sixty-one percent of patients were men (25/41) and the mean age was 58.2 years (Table 1); 26 patients were between 18 and 64 years of age, 12 were 65 to 74 years of age, and 4 were >74 years of age. Twenty-one patients had stage III or IV solid tumors, and 20 had lymphomas (11/20 were Ann Arbor stage IVA). Of the 21 patients with solid tumors, all had measurable disease, 9 had visceral disease, 4 had nonvisceral disease, and 8 had both visceral and nonvisceral disease. The primary tumor type at initial diagnosis was prostate cancer in 5 patients; head and neck cancer in 2; ovarian cancer in 2; pancreatic cancer in 2; liposarcoma in 2; and one case each of epithelioid sarcoma, endometrial cancer, colorectal cancer gastric cancer, mesothelioma, thymic carcinoma, cholangiocarcinoma, and adrenocortical carcinoma. EZH2 mutation status was determined for 6 patients with GCB-DLBCL; 5 had no mutation and 1 had an Y641H point mutation. Lack of tissue or poor tissue quality precluded assessment of EZH2 mutation status in other lymphoma samples. EZH2 mutation status was not assessed in solid tumors owing to the low incidence of EZH2 mutations in these tumor types.
Table 1.
Patient characteristics
| Category | Patients N = 41 |
|---|---|
| Age, years, median (range) | 57.0 (21–80) |
| Females, n (%) | 16 (39) |
| ≥1 past and/or current medical condition at baseline (%) | 31 (76) |
| ECOG PS, n (%) | |
| 0 | 16 (39) |
| 1 | 25 (61) |
| Solid tumor, n (%) | 21 (51.2) |
| Tumor type | |
| Prostate | 5 (12.2) |
| Head and neck | 2 (4.9) |
| Ovarian | 2 (4.9) |
| Pancreas | 2 (4.9) |
| Soft tissue sarcoma | 2 (4.9) |
| Endometrial/uterine, mesothelioma, gastric, colona | 4 (9.8) |
| Other | 4 (9.8) |
| Stage, n (%) | |
| III | 2 (4.9) |
| IV | 19 (89.5) |
| Lymphoma (NHL), n (%) | 20 (48.7) |
| DLBCL | 15 (75) |
| GCBb | 14 (53)d |
| Follicular | 4(20) |
| Transformedc | 3 (75) |
| MZL | 1 (5) |
| Ann Arbor stage, n (%) | |
| IA | 1 (2.4) |
| IIA | 2 (4.9) |
| IIIA | 5 (12.2) |
| IVA | 11 (26.8) |
| IVB | 1 (2.4) |
Abbreviations: ECOG, Eastern Cooperative Oncology Group; MZL, marginal zone lymphoma; NHL, non-Hodgkin lymphoma; PS, performance status.
One of each tumor type.
Percentage calculated as a proportion of all DLBCL.
Calculated as a percentage of all FL.
Eight confirmed centrally by immunohistochemistry; six could not be confirmed centrally due to limited availability of sample or inconclusive results.
Dose distribution and intensity
Of the 41 patients who were treated, 2 patients (4.9%) received the 50 mg dose, 1 each (2.4%) received the 100, 200, and 400 mg doses, 3 (7.3%) received the 800 mg dose, and 4 (9.8%) received the 1,200 mg dose (Table 2). Ten patients (24.4%) were in the 1,800-mg dose cohort, 12 (29.3%) were in the 2,400-mg dose cohort, and 7 (17.1%) were in the 3,000-mg dose cohort.
Table 2.
Summary of exposure to study treatment
| 50 mg (N = 2) | 100 mg (N = 1) | 200 mg (N = 1) | 400 mg (N = 1) | 800 mg (N = 3) | 1,200 mg (N = 4) | 1,800 mg (N = 10) | 2,400 mg (N = 12) | 3,000 mg (N = 7) | Total (N = 41) | |
|---|---|---|---|---|---|---|---|---|---|---|
| Dose intensity (mg/cycle)a | ||||||||||
| N | 2 | 1 | 1 | 1 | 3 | 4 | 10 | 12 | 7 | 41 |
| Mean | 183.3 | 680.0 | 1,200.0 | 2,400.0 | 4,932.6 | 6,850.0 | 9,906.4 | 1,2393.3 | 1,2678.6 | 9,350.7 |
| SD | 47.14 | - | - | - | 1,284.58 | 412.31 | 1,473.89 | 1,856.04 | 5,817.92 | 4,737.17 |
| Median | 183.3 | - | - | - | 4,400.0 | 6,900.0 | 10,277.1 | 12,600.0 | 1,4250.0 | 9,900.0 |
| Min | 150 | - | - | - | 4,000 | 6,400 | 6,300 | 9,600 | 3,000 | 150 |
| Max | 217 | - | - | - | 6,398 | 7,200 | 11,550 | 14,400 | 18,000 | 18,000 |
| Number of cycles | ||||||||||
| N | 2 | 1 | 1 | 1 | 3 | 4 | 10 | 12 | 7 | 41 |
| Mean | 2.0 | 5.0 | 2.0 | 2.0 | 3.0 | 2.3 | 3.8 | 2.3 | 2.4 | 2.8 |
| SD | 1.41 | - | - | - | 2.65 | 0.50 | 1.87 | 1.42 | 1.90 | 1.70 |
| Median | 2.0 | - | - | - | 2.0 | 2.0 | 3.5 | 2.0 | 2.0 | 2.0 |
| Min | 1 | - | - | - | 1 | 2 | 2 | 1 | 1 | 1 |
| Max | 3 | - | - | - | 6 | 3 | 7 | 5 | 6 | 7 |
| Number of cycles | ||||||||||
| ≤2 | 1 (50%) | 0 | 1 (100%) | 1 (100%) | 2 (67%) | 3 (75%) | 4 (40%) | 8 (67%) | 5 (71%) | 25 (61%) |
| 3–5 | 1 (50%) | 1 (100%) | 0 | 0 | 0 | 1 (25%) | 4 (40%) | 4 (33%) | 1 (14%) | 12 (29%) |
| >5 | 0 | 0 | 0 | 0 | 1 (33%) | 0 | 2 (20%) | 0 | 1 (14%) | 4 (10%) |
Dose intensity (mg/cycle) is the average dose per cycle and is multiplied by the number of doses planned for each cycle.
For the 28-day cycles, the mean dose intensity of GSK2816126 was 9,350.7 mg/cycle, and the median was 9,900.0 mg/cycle (range, 150–18,000 mg/cycle). The mean number of cycles given to patients was 2.8 (range, 1–7 cycles). Of the 41 patients, 25 (61%) had ≤ 2 cycles, 12 (29%) had 3 to 5 cycles, and 4 (10%) had >5 cycles. Two of the 41 patients treated (5%) had ≥ 1 dose reduction (800 and 3,000 mg dose cohorts), and 10 (24%) had ≥ 1 dose interruption. Of the 16 total dose interruptions, each of which lasted 1 to 5 days, 8 were due to an AE.
Adverse events
All patients experienced at least 1 AE during the study. Fatigue [22/41 (53.7%)] and nausea [20/41 (48.8%)] were the most common AEs (Table 3). One or more grade ≥ 3 AEs were reported in 18 (44%) patients (16 grade 3, and 1 each of grade 4 and 5). Most grade ≥ 3 events occurred in 1 or 2 patients each; however, grade 3 alanine aminotransferase (ALT) increase occurred in 4 patients. A grade 4 aspartate aminotransferase increase (DLT, study drug related) occurred in 1 patient in the 3,000-mg dose cohort 2 days after receiving the first dose of study drug and a grade 5 respiratory tract infection (nondrug related) occurred in 1 patient in the 50-mg dose cohort 11 days after the initial dose. Investigator assessment of the grade 5 respiratory tract infection for the patient was secondary to extensive disease in the chest with lung parenchyma infiltration, collapsed right lung segments, and extensive pleural disease with effusion.
Table 3.
Summary of treatment-related and serious adverse events
| 50 mg (N = 2) | 100 mg (N = 1) | 200 mg (N = 1) | 400 mg (N = 1) | 800 mg (N = 3) | 1,200 mg (N = 4) | 1,800 mg (N = 10) | 2,400 mg (N = 12) | 3,000 mg (N = 7) | Total (N = 41) | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Any event (all patients) | 2 (100) | 1 (100) | 1 (100) | 1 (100) | 3 (100) | 4 (100) | 10 (100) | 12 (100) | 7 (100) | 41 (100) | |
| Treatment-related AEs | |||||||||||
| Any event | 1 (50) | 1 (100) | 1 (100) | 1 (100) | 3 (100) | 3(75) | 9 (90) | 11 (92) | 7 (100) | 37 (90) | |
| Fatigue | 0 | 1 (100) | 1 (100) | 1 (100) | 2(67) | 1 (25) | 6 (60) | 6 (50) | 2 (29) | 20 (49) | |
| Nausea | 1 (50) | 0 | 0 | 0 | 1 (33) | 0 | 3 (30) | 8(67) | 3 (43) | 16 (39) | |
| Vomiting | 0 | 0 | 0 | 0 | 1 (33) | 1 (25) | 2 (20) | 2 (17) | 3 (43) | 9 (22) | |
| Anemia | 0 | 0 | 0 | 0 | 2 (67) | 0 | 1 (10) | 4 (34) | 1 (14) | 8 (20) | |
| Alanine aminotransferase increased | 0 | 0 | 0 | 0 | 0 | 0 | 1 (10) | 3 (25) | 3 (43) | 7(17) | |
| Infusion-related reaction | 0 | 0 | 0 | 0 | 1 (33) | 1 (25) | 0 | 2 (17) | 3 (43) | 7 (17) | |
| Oral paresthesia | 0 | 0 | 0 | 0 | 1 (33) | 0 | 1 (10) | 2 (17) | 1 (14) | 5 (12) | |
| Diarrhea | 0 | 0 | 0 | 0 | 0 | 1 (25) | 0 | 2 (17) | 1 (14) | 4 (10) | |
| Pruritus | 0 | 1 (100) | 0 | 0 | 1 (33) | 0 | 0 | 1 (8) | 1 (14) | 4 (10) | |
| Aspartate aminotransferase increased | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (8) | 2 (29) | 3 (7) | |
| Blood alkaline phosphatase increased | 0 | 0 | 0 | 0 | 1 (33) | 0 | 0 | 1 (8) | 1 (14) | 3 (7) | |
| Constipation | 1 (50) | 0 | 0 | 0 | 1 (33) | 0 | 0 | 0 | 1 (14) | 3 (7) | |
| Serious AEs (≥Grade 3) | |||||||||||
| Any event | 1 (50) | 0 | 0 | 0 | 1 (33) | 2 (50) | 1 (10) | 5 (42) | 3 (43) | 13 (32) | |
| Pyrexia | 0 | 0 | 0 | 0 | 0 | 1 (25) | 0 | 1 (8) | 1 (14) | 3 (7) | |
| Respiratory tract infection | 1 (50) | 0 | 0 | 0 | 0 | 0 | 0 | 1 (8) | 0 | 2 (5) | |
| Back pain | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (14) | 1 (2) | |
| Bronchitis | 0 | 0 | 0 | 0 | 0 | 1 (25) | 0 | 0 | 0 | 1 (2) | |
| Campylobacter infection | 0 | 0 | 0 | 0 | 1 (33) | 0 | 0 | 0 | 0 | 1 (2) | |
| Enterocolitis | 0 | 0 | 0 | 0 | 1 (33) | 0 | 0 | 0 | 0 | 1 (2) | |
| Hemoptysis | 0 | 0 | 0 | 0 | 0 | 0 | 1 (10) | 0 | 0 | 1 (2) | |
| Hypokalemia | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (8) | 0 | 1 (2) | |
| Intestinal obstruction | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (8) | 0 | 1 (2) | |
| Pneumonia | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (8) | 0 | 1 (2) | |
| Skin bacterial infection | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (8) | 0 | 1 (2) | |
| Tumor pain | 0 | 0 | 0 | 0 | 1 (33) | 0 | 0 | 0 | 0 | 1 (2) | |
| Vomiting | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (14) | 1 (2) | |
Hematology results and laboratory values were unchanged or returned to normal after treatment in 68% of patients. Grade 3 or 4 increases in ALT and AST occurred in 4 of 41 patients (10%) and 2 of 41 patients (5%), respectively, all of which were reversible. There were not grade 3 or 4 changes in direct bilirubin in any patient.
In most patients, vital signs were numerically similar to those observed at baseline, and most patients had ECG results that were unchanged relative to baseline. ECGs were performed pre-infusion, at the end of infusion, before bed, and before discharge the next morning. Seventeen percent and 10% of patients had a maximum increase in QTc (relative to baseline) to grade 2 (481–500 milliseconds) or grade 3 (≥ 501 milliseconds), respectively, using Bazett’s method and 2% and 0%, respectively, using Fridericia’s method. No AEs of QT prolongation were considered serious by the investigator. One (2%) AE of QT prolongation was considered to be treatment-related by the investigator. Four patients (10%) had both abnormal and clinically significant post-baseline QT prolongation results. All 4 patients with grade 3 (>500 milliseconds) clinically significant QT prolongations returned to baseline using Bazett’s method before discharge. The potential underlying etiology of QT prolongation was unknown but was suspected to be cancer-related or due to concomitant medications, which were subsequently discontinued. One of these patients discontinued study treatment shortly after infusion due to disease progression. No grade 3 QT prolongation events were noted with Fridericia’s method.
Serious AEs
Thirteen patients (32%) experienced a total of 16 SAEs, all of which were grade 3 or less, and none were considered related to the study drug. SAEs reported included various infections (n = 7) and pyrexia (n = 3). The remaining SAEs were tumor pain, hemoptysis, intestinal obstruction, hypokalemia, back pain, and vomiting, which occurred in 1 patient each. One patient experienced an SAE of respiratory tract infection that resulted in death. There were no treatment-related SAEs other than the DLTs noted below.
Treatment-related AEs
Of the 41 patients treated, 37 (90%) experienced at least 1 AE that was considered to be treatment related. The most frequent treatment-related AEs were fatigue and nausea, which were observed in 20 (49%) and 16 (39%) patients, respectively. Other AEs are listed in Table 3.
Dose-limiting toxicity
Two of 7 patients in the 3,000-mg dose cohort experienced a grade 2 or higher ALT increase. One patient had both a grade 3 ALT increase and grade 4 AST increase, and the other patient had a grade 2 ALT increase.
Pharmacokinetic and pharmacodynamic results
Following intravenous administration of 50 to 3,000 mg twice weekly, GSK28616126 levels decreased in a bi-exponential manner with a mean terminal elimination half-life of approximately 27 hours (Fig. 2). GSK2816126 exposure (Cmax and AUC) increased in a dose-proportional manner over the 50- to 3,000-mg dose levels. There was moderate- to-high between-patient variability (Table 4). Up to the 2,400 mg dose, the trough value at day 15 was below the in vitro protein-binding adjusted IC50 value of 475 ng/mL for H3K27me3 in KARPAS-422 cells (Fig. 2). On day 15, the trough plasma concentration (Ctrough; μg/mL) was nonquantifiable for the 50- to 400-mg dose levels, and the geometric mean Ctrough ranged from 0.1 μg/mL for the 800 mg dose to 0.41 μg/mL for the 3,000 mg dose.
Figure 2.
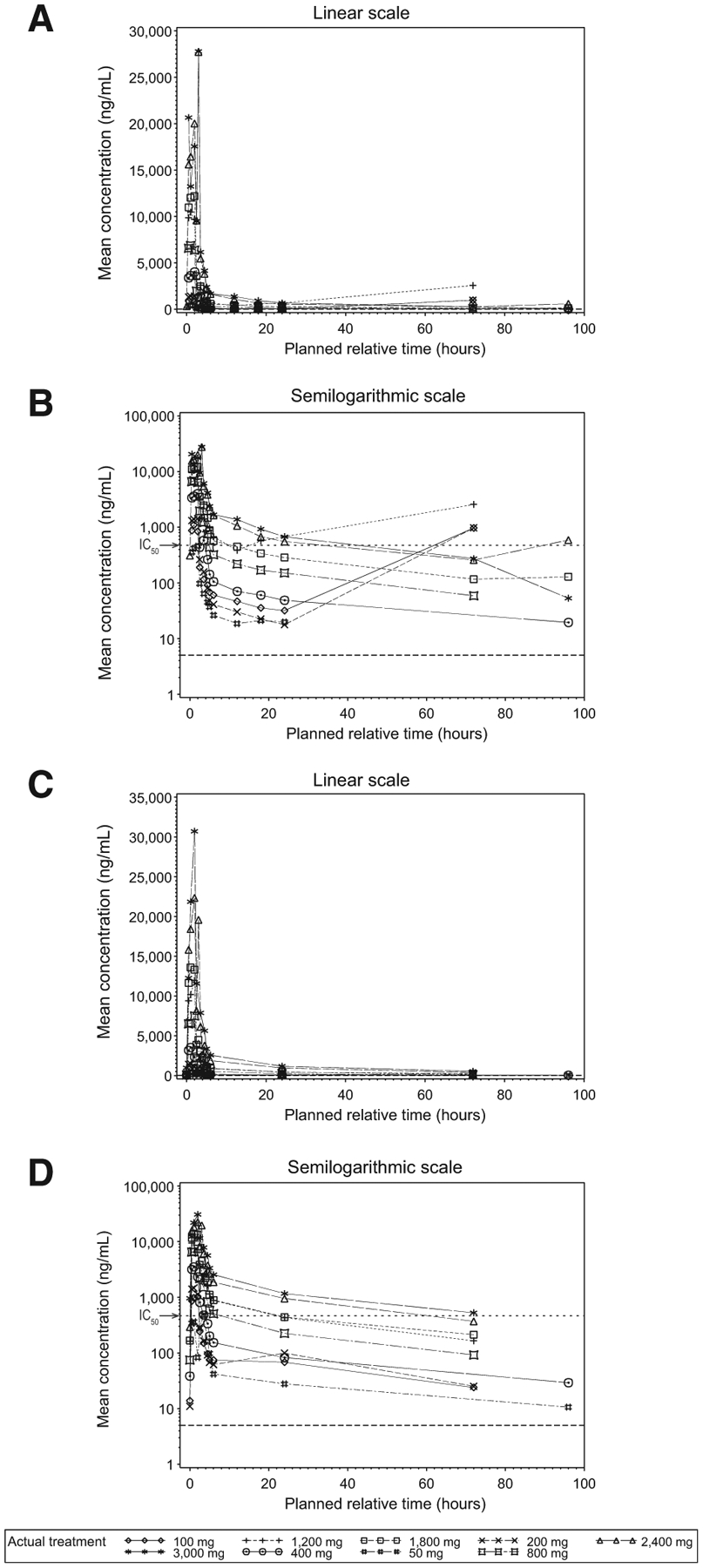
Mean plasma concentration-time plots (linear and semilogarithmic) for cycle 1, day 1 and day 15. Mean plasma GSK2816126 concentration–time plots are shown for cycle 1, day 1 visit (A and B) and cycle 1, day 15 visit (C and D) by nominal time on a linear scale (A and C) and a semilogarithmic scale (B and D). The dotted line in panels B and D represents the lower limit of quantitation (5 ng/mL). The protein-binding adjusted IC50 for H3K27me3 (475 ng/mL) is indicated by an arrow on B and D.
Table 4.
Plasma GSK2816126 pharmacokinetic parameters based on actual sampling times for cycle 1, day 1 and cycle 1, day 15
| PK Para | Day | 50 mg | 100 mg | 200 mg | 400 mg | 800 mg |
|---|---|---|---|---|---|---|
| AUC(0–∞) (h*μg/mL) | 1 | 1.33 (82), n = 2 | NA (NA), n = 0 | NA (NA), n = 0 | 13.2 (NA), n = 1 | 26.4 (12), n = 3 |
| 15 | 3.90 (NA), n = 1 | 7.00 (NA), n = 1 | NA (NA), n = 0 | 14.8 (NA), n = 1 | 33.3 (24), n = 2 | |
| AUC(0–t) (h*μg/mL) | 1 | 1.20 (62), n = 2 | 27.2 (NA), n = 1 | 27.7 (NA), n = 1 | 11.8 (NA), n = 1 | 24.2 (10), n = 3 |
| 15 | 3.20 (NA), n = 1 | 5.70 (NA), n = 1 | 7.10 (NA), n = 1 | 13.1 (NA), n = 1 | 25.8 (30), n = 3 | |
| Cmax (μg/mL) | 1 | 0.42 (52), n = 2 | 1.10 (NA), n = 1 | 1.40 (NA), n = 1 | 4.00 (NA), n = 1 | 7.31 (16), n = 3 |
| 15 | 0.50 (NA), n = 1 | 1.00 (NA), n = 1 | 1.50 (NA), n = 1 | 3.50 (NA), n = 1 | 7.46 (13), n = 3 | |
| tmax (hours) | 1 | 1.50 (1.0, 2.0), n = 2 | 1.00 (1.0, 1.0), n = 1 | 2.00 (2.0, 2.0), n = 1 | 1.90 (1.9, 1.9), n = 1 | 1.00 (0.5, 1.9), n = 3 |
| 15 | 2.7 (2.7, 2.7), n = 1 | 2.0 (2.0, 2.0), n = 1 | 1.0 (1.0, 1.0), n = 1 | 1.0 (1.0, 1.0), n = 1 | 2.0 (1.9, 2.0), n = 3 | |
| t1/2 (hours) | 1 | 7.62 (96), n = 2 | NA (NA), n = 0 | NA (NA), n = 0 | 49.1 (NA), n = 1 | 26.5 (31), n = 3 |
| 15 | 46.50 (NA), n = 1 | 36.80 (NA), n = 1 | NA (NA), n = 0 | 39.70 (NA) | 27.35 (9), n = 2 | |
| CL (L/h) | 1 | 37.39 (73), n = 2 | NA (NA), n = 0 | NA (NA), n = 0 | 35.30 (NA), n = 1 | 32.49 (9), n = 3 |
| 15 | 17.2 (NA), n = 1 | 17.5 (NA), n = 1 | NA (NA), n = 0 | 32.5 (NA), n = 1 | 26.8 (23), n = 2 | |
| V (L) | 1 | 194 (37), n = 2 | NA (NA), n = 0 | NA (NA), n = 0 | 722 (NA), n = 1 | 467 (15), n = 3 |
| 15 | 757 (NA), n = 1 | 598 (NA), n = 1 | NA (NA), n = 0 | 887 (NA), n = 1 | 601 (26), n = 2 | |
| PK Para | Visit Day | 1,200 mg | 1,800 mg | 2,400 mg | 3,000 mg | |
| AUC(0–∞) (h*μg/mL) | 1 | 60.9 (NA), n = 1 | 51.2 (23), n = 10 | 102 (28), n = 10 | 103 (48), n = 7 | |
| 15 | 60.2 (10), n = 1 | 81.7 (70), n = 9 | 123 (27), n = 11 | 183 (32), n = 4 | ||
| AUC(0–t) (h*μg/mL) | 1 | 85.7 (91), n = 4 | 45.9 (23), n = 10 | 92.4 (26), n = 12 | 94.0 (48), n = 7 | |
| 15 | 53.4 (14), n = 4 | 68.7 (75), n = 9 | 105 (29), n = 11 | 158 (35), n = 4 | ||
| Cmax (μg/mL) | 1 | 10.9 (10), n = 4 | 13.0 (27), n = 10 | 22.3 (35), n = 12 | 23.8 (21), n = 7 | |
| 15 | 11.6 (27), n = 4 | 14.2 (27), n = 9 | 21.6 (27), n = 11 | 30.5 (14), n = 4 | ||
| tmax (hours) | 1 | 2.05 (1.1, 72), n = 4 | 1.95 (1.0, 2.9), n = 10 | 2.00 (0.8, 2.8), n = 12 | 2.00 (0.5, 3.3), n = 7 | |
| 15 | 2.05 (2.0, 2.1), n = 4 | 1.90 (0.5, 2.7), n = 9 | 2.00 (1.0, 3.0), n = 11 | 2.25 (2.0, 3.0), n = 4 | ||
| t1/2 (hours) | 1 | 14.1 (NA), n = 1 | 31.8 (17), n = 10 | 33.7 (20), n = 10 | 22.5 (56), n = 7 | |
| 15 | 27.30 (18), n = 4 | 29.88 (50), n = 9 | 22.75 (44), n = 11 | 27.25 (44), n = 4 | ||
| CL (L/h) | 1 | 20.50 (NA), n = 1 | 39.01 (24), n = 10 | 26.06 (27), n = 10 | 28.60 (32), n = 7 | |
| 15 | 22.6 (14), n = 4 | 30.9 (25), n = 9 | 22.2 (25), n = 11 | 19.0 (36), n = 4 | ||
| V (L) | 1 | 284 (NA), n = 1 | 702 (50), n = 10 | 482 (31), n = 10 | 434 (74), n = 7 | |
| 15 | 499 (37), n = 4 | 918 (111), n = 9 | 424 (33), n = 11 | 480 (72), n = 4 |
Abbreviations: NA, not applicable; Par, parameter; PK, pharmacokinetic; t1/2, apparent terminal phase half-life; tmax, time to Cmax; V, volume.
Values denote geometric mean (CVb%) except for tmax, which is presented as median (range).
Peripheral blood mononuclear cells (PBMC) showed no evidence of global changes in H3K27me3 ratios compared with baseline. Of the paired tumor biopsies obtained from 4 patients, one tumor biopsy pair was evaluable; however the results were inconclusive due to low baseline H3K27me3 levels in predose sample. Given that a pharmacodynamic response was not observed or could not be assessed, a relationship between pharmacokinetic and pharmacodynamic parameters could not be determined.
Antitumor activity
Investigator-assessed best response in patients with solid tumors included 11 of 21 (52%) with progressive disease, 8 of 21 (38%) with stable disease, and 2 who were not evaluable. In the 20 patients with lymphoma, 1 patient with germinal cell B (GCB)-DLBCL as determined by local subtyping (5%) achieved a partial response (Fig. 3); 6 patients (30%) had stable disease (SD); and 10 patients (50%) had progressive disease. One patient with an EZH2 mutation had progressive disease.
Figure 3.
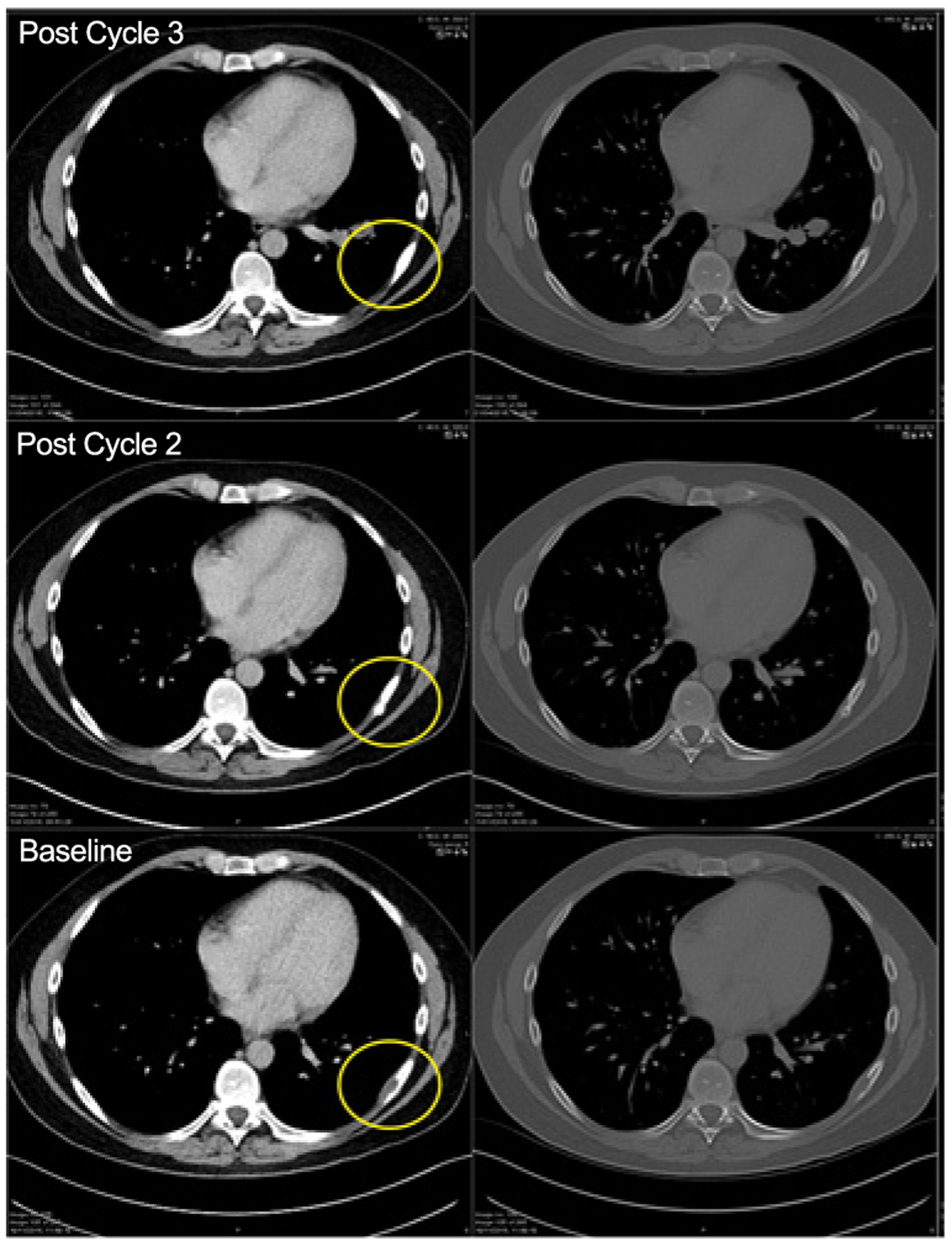
Computed tomography scans of a patient with a partial response. The encircled area represents the soft tissue component associated with a rib lesion.
The patient with a partial response was treated at the 1,800 mg dose and the response lasted for 3 cycles. No tumor was present in this patient’s tissue sample; thus EZH2 mutation status could not be assessed. Three additional patients were not evaluable. Progression-free survival was not assessed in either patients with solid tumors or lymphoma.
Among the 6 patients with lymphoma with SD, 1 had FL, 1 had activated B-cell–like (ABC)-DLBCL, and 4 had GCB-DLBCL. GCB status was confirmed by central testing in these 4 patients. There was insufficient tumor sample from the patient with ABC-DLBCL to confirm subtype status centrally. The duration of SD in 5 of these patients ranged from 34 to 118 days but was not assessed in 1 patient.
The percentage change from baseline in tumor measurements for patients with solid tumors and patients with lymphoma is shown in Supplementary Fig. S1.
Discussion
Here, we report the results for a phase I study of GSK2816126, an EZH2 inhibitor, in patients with relapsed/refractory solid tumors and hematologic malignancies. Although GSK2816126 is a potent, selective inhibitor of EZH2 methyltransferase activity, the lack of oral bioavailability necessitated intravenous administration of GSK2816126, thus limiting the frequency of dosing. Based on modeling using available preclinical pharmacokinetic data from murine and canine studies, and efficacy data from EZH2 mutant tumor xenograft studies in mice, tumor eradication with GSK2816126 was predicted to be achieved in humans at doses ranging between 950 and 2,700 mg when administered twice weekly, suggesting potential clinical utility of GSK2816126. This study identified the MTD for GSK2816126 as 2,400 mg; however, given the limitation of twice weekly intravenous dosing and the observed pharmacokinetic profile for GSK2816126, biologically effective exposure was not achieved at tolerated doses and anticancer activity was minimal. Thus, the planned portion of this trial, in which efficacy would have been evaluated in relationship to EZH2 mutation status in DLBCL was not opened.
Historically, the translation of anticancer efficacy from non-clinical models to the clinic has been very challenging. Even though mouse models of tumor inhibition lie at the center of translational efforts in oncology, the results from this study suggest that a more comprehensive approach with validated biomarkers in surrogate tissues is required to strengthen the probability of success.
Pharmacokinetic studies using dose proportional assessment have shown that exposure to GSK2816126 increased proportionally to the increase in dose. However, trough values at day 15 for all doses up to 2,400 mg were below the in vitro protein binding adjusted IC50 for inhibition of tri-methylation (H3K27me3). PK/PD modeling predicted that maintenance of plasma concentrations above the protein-adjusted IC50 for inhibition of methylation would be required for clinical efficacy; therefore, these data suggest that serum concentrations are too low to be clinically effective at doses that could be achieved with the chosen schedule of administration. Although a more frequent dosing schedule would result in higher serum concentrations, more intravenous doses were considered too burdensome for patients to justify evaluation. Thus, the interplay between the lack of oral bioavailability and the desire to achieve a patient-feasible interdose interval in an intravenous setting may have resulted in suboptimal levels of target engagement.
With regard to the relationship between GSK2816126 exposure and pharmacodynamic parameters, there was no detectable relationship between GSK2816126 dose and time after exposure for histone H3K27me3 in PBMCs. Although this may be due to the GSK2816126 exposure, preclinical studies evaluating the effects of EZH2 inhibition on H3K27me3 in individual blood cell populations have since demonstrated that H3K27me3 reduction is most pronounced in granulocytes (data not shown). For this reason, the inability to detect a reduction in H3K27me3 may be due to lack of sensitivity in PBMCs. An effect on the histone marker in paired pre-/posttherapy tumor biopsy specimens could not be assessed for technical reasons.
In our study, one patient with GCB-DLBCL treated at the 1,800 mg dose had a partial response lasting 91 days, and 6 patients achieved SD (5 DLBCL and 1 FL). Among 21 patients with solid tumors, 8 (38%) showed SD, with no complete or partial responses. In comparison, an early clinical study of the orally available EZH2 inhibitor, tazemetostat, showed a more favorable pharmacokinetic profile, with the recommended dose determined to be 800 mg twice daily (22). The objective response rate in this dose-escalation study with tazemetostat was 38% in patients with B-cell lymphomas and 5% in patients with solid tumors. In patients with solid tumors, responses with tazemetostat were only seen in patients with INI1-negative or SMARCA4-negative tumors. These findings highlight the viability of EZH2 as a target for anticancer therapies. However, additional research is needed to identify better EZH2 inhibitors with optimized pharmacokinetic profiles and demonstrated activity against less-sensitive tumor types.
In conclusion, this study showed that GSK2816126 is not a viable drug to target EZH2 in patients with refractory/relapsed solid and hematologic malignancies, despite preclinical data showing sensitivity of multiple solid tumor and lymphoma cell lines (13). The study defined the MTD of GSK2816126 as 2,400 mg, but at this level, the drug showed inadequate clinical activity, with off-target DLTs precluding further dose escalation. EZH2 is, nonetheless, a suitable target for therapy as demonstrated by initial clinical experience with tazemetostat (22).
Supplementary Material
Translational Relevance.
Dysregulation of epigenetics is a feature of many malignancies. Preclinical studies indicated that GSK2816126 is a potent inhibitor of enhancer of zeste homolog 2 (EZH2), with marked growth inhibitory effects in malignant cells carrying a mutation in EZH2. This trial tested the safety and clinical efficacy of GSK2816126 given by intravenous infusion in a dose-escalation study of patients with solid tumors or lymphoma. The maximum-tolerated dose was determined as 2,400 mg, and liver transaminitis was the dose-limiting toxicity. Minimal anticancer activity was seen due to limited exposure resulting from the challenges of twice-weekly intravenous dosing and the pharmacokinetic profile of GSK2816126. A patient with lymphoma achieved a radiologic partial response.
Acknowledgments
All listed authors meet the criteria for authorship set forth by the International Committee for Medical Journal Editors. Editorial support (assembling tables and figures, collating author comments, copyediting, fact checking, and referencing) and graphic services were provided by AOI Communications, L.P., and were funded by GlaxoSmithKline. The authors would like to thank Fabio Rigat for his contribution to the analysis of the PBMC H3K27me3 data and Uma Kamasani for her contribution to the analysis and reporting and with generating figures. Funding for this study (ClinicalTrials.gov Identifier: NCT02082977, www.clinicaltrials.gov) was provided by GlaxoSmithKline.
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/)
Disclosure of Potential Conflicts of Interest
T. Yap is an employee/paid consultant for Aduro, Almac, AstraZeneca, Atrin, Bayer, Bristol-Myers Squibb, Calithera, Clovis, Cybrexa, EMD Serono, Ignyta, Jansen, Merck, Pfizer, Roche, Seattle Genetics, and Vertex Pharmaceuticals; reports commercial research funding from AstraZeneca, Bayer, Pfizer, Tesaro, Jounce, Eli Lilly, Seattle Genetics, Kyowa, Constellation, and Vertex Pharmaceuticals; and reports honoraria from AstraZeneca, Merck, Pfizer, and Tesaro. J.N. Winter is an employee/paid consultant for AstraZeneca, Merck, Bayer, Gilead, and Adicet Bio. L. Giulino-Roth is an employee/paid consultant for Janssen, Celgene, and ADC Therapeutics, and is an unpaid consultant/advisory board member for Merck. J. Lopez reports receiving commercial research grants from Basilea, Genmab, and Roche Genentech. J. Michot is an employee/paid consultant for Atheneum; holds ownership interest (including patents) in Celgene; and reports receiving other remuneration from Novartis. J.P. Leonard is an employee/paid consultant for Sutro, Bayer, Gilead, Celgene, Merck, Mophosys, Beigene, Nordic Nanovector, Roche/Genentech, ADC Therapeutics, Sandoz, Karyopharm, Miltenyi, Akcea Therapeutics, and Epizyme, and reports receiving commercial research grants from GlaxoSmithKline. V. Ribrag is an employee/paid consultant for Infinity Pharmaceuticals, Bristol-Myers Squibb, PharmaMar, Gilead Sciences, NanoString Technologies, Incyte, Bristol-Myers Squibb, MSD, Roche/Genentech, Epizyme, Immune Design, and Roche; receives commercial research grants from arGEN-X BVBA and his institution receives research grants from Epizyme; receives honoraria from Infinity Pharmaceuticals, Bristol-Myers Squibb, Eisai, PharmaMar, Gilead Sciences, AZD, Epizyme, Incyte, MSD, and SERVIER; reports receving other remuneration from SERVIER, Roche, Bristol-Myers Squibb, and AZD. M.T. McCabe is an employee/paid consultant for and holds ownership interest (including patents) in GlaxoSmithKline. C.L. Creasy is an employee/paid consultant for and holds ownership interest (including patents) in GlaxoSmithKline. M. Stern is an employee/paid consultant for GlaxoSmithKline. T. Pene Dumitrescu is an employee/paid consultant for GlaxoSmithKline. X. Wang is an employee/paid consultant for GlaxoSmithKline. S. Frey is an employee/paid consultant for and holds ownership interest (including patents) in GlaxoSmithKline. J. Carver is an employee/paid consultant for and holds ownership interest (including patents) in GlaxoSmithKline. T. Horner is an employee/paid consultant for and holds ownership interest (including patents) in GlaxoSmithKline. C. Oh is an employee/paid consultant for GlaxoSmithKline. A. Khaled is an employee/paid consultant for and holds ownership interest (including patents) in GlaxoSmithKline. A. Dhar is an employee/paid consultant for and holds ownership interest (including patents) in GlaxoSmithKline. P.W.M. Johnson is an employee/paid consultant for Takeda, Bristol-Myers Squibb, Novartis, Celgene, Kite Pharma, Genmab, Incyte, Morphosys, Kymera, Janssen, and Oncimmune, and reports receiving commercial research grants from Epizyme and Janssen. No potential conflicts of interest were disclosed by the other authors.
Data Sharing Statement
Anonymized individual participant data and study documents can be requested for further research from www.clinicalstudydatarequest.com.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
References
- 1.Margueron R, Reinberg D. The polycomb complex PRC2 and its mark in life. Nature 2011;469:343–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Richly H, Aloia L, Di Croce L. Roles of the polycomb group proteins in stem cells and cancer. Cell Death Dis 2011;2:e204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McCabe MT, Creasy CL. EZH2 as a potential target in cancer therapy. Epigenomics 2014;6:341–51. [DOI] [PubMed] [Google Scholar]
- 4.Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet 2010;42:181–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pasqualucci L, Trifonov V, Fabbri G, Ma J, Rossi D, Chiarenza A, et al. Analysis of the coding genome of diffuse large B-cell lymphoma. Nat Genet 2011;43:830–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bodor C, Grossmann V, Popov N, Okosun J, O’Riain C, Tan K, et al. EZH2 mutations are frequent and represent an early event in follicular lymphoma. Blood 2013;122:3165–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ryan RJ, Nitta M, Borger D, Zukerberg LR, Ferry JA, Harris NL, et al. EZH2 codon 641 mutations are common in BCL2-rearranged germinal center B cell lymphomas. PLoS One 2011;6:e28585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McCabe MT, Graves AP, Ganji G, Diaz E, Halsey WS, Jiang Y, et al. Mutation of A677 in histone methyltransferase EZH2 in human B-cell lymphoma promotes hypertrimethylation of histone H3 on lysine 27 (H3K27). Proc Natl Acad Sci U S A 2012;109:2989–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ott HM, Graves AP, Pappalardi MB, Huddleston M, Halsey WS, Hughes AM, et al. A687V EZH2 is a driver of histone H3 lysine 27 (H3K27) hypertrimethylation. Mol Cancer Ther 2014;13:3062–73. [DOI] [PubMed] [Google Scholar]
- 10.Yap DB, Chu J, Berg T, Schapira M, Cheng SW, Moradian A, et al. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood 2011;117:2451–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bachmann IM, Halvorsen OJ, Collett K, Stefansson IM, Straume O, Haukaas SA, et al. EZH2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate, and breast. J Clin Oncol 2006;24:268–73. [DOI] [PubMed] [Google Scholar]
- 12.Yu J, Yu J, Rhodes DR, Tomlins SA, Cao X, Chen G, et al. A polycomb repression signature in metastatic prostate cancer predicts cancer outcome. Cancer Res 2007;67:10657–63. [DOI] [PubMed] [Google Scholar]
- 13.McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 2012;492:108–12. [DOI] [PubMed] [Google Scholar]
- 14.Knutson SK, Warholic NM, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A 2013;110:7922–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koppens MA, Bounova G, Cornelissen-Steijger P, de Vries N, Sansom OJ, Wessels LF, et al. Large variety in a panel of human colon cancer organoids in response to EZH2 inhibition. Oncotarget 2016;7:69816–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zingg D, Debbache J, Schaefer SM, Tuncer E, Frommel SC, Cheng P, et al. The epigenetic modifier EZH2 controls melanoma growth and metastasis through silencing of distinct tumour suppressors. Nat Commun 2015;6: 6051. [DOI] [PubMed] [Google Scholar]
- 17.Roberts CW, Biegel JA. The role of SMARCB1/INI1 in development of rhabdoid tumor. Cancer Biol Ther 2009;8:412–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilson BG, Wang X, Shen X, McKenna ES, Lemieux ME, Cho YJ, et al. Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell 2010;18:316–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheson BD, Pfistner B, Juweid ME, Gascoyne RD, Specht L, Horning SJ, et al. Revised response criteria for malignant lymphoma. J Clin Oncol 2007;25:579–86. [DOI] [PubMed] [Google Scholar]
- 20.Chung WH, Hung SI, Chen YT. Genetic predisposition of life-threatening antiepileptic-induced skin reactions. Expert Opin Drug Saf 2010;9:15–21. [DOI] [PubMed] [Google Scholar]
- 21.Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 2009;45:228–47. [DOI] [PubMed] [Google Scholar]
- 22.Italiano A, Soria JC, Toulmonde M, Michot JM, Lucchesi C, Varga A, et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: a first-in-human, open-label, phase 1 study. Lancet Oncol 2018;19:649–59. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.