

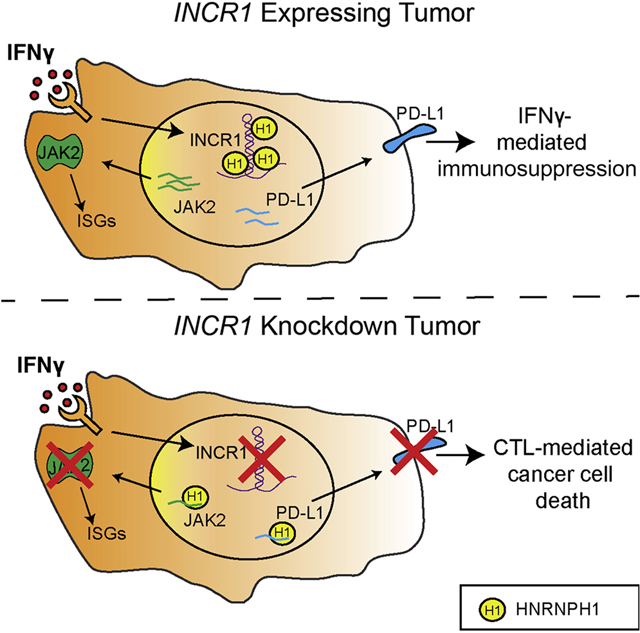
SUMMARY
Tumor interferon signaling promotes PD-L1 expression to suppress T cell-mediated immunosurveillance. We identify the interferon- (IFN)-stimulated non-coding RNA 1 (INCR1) as a long noncoding RNA (lncRNA) transcribed from the PD-L1 locus and show that INCR1 controls IFNγ signaling in multiple tumor types. Silencing INCR1 decreases the expression of PD-L1, JAK2 and several other IFNγ-stimulated genes. INCR1 knockdown sensitizes tumor cells to cytotoxic T cell-mediated killing, improving CAR T cell therapy. We discover that PD-L1 and JAK2 transcripts are negatively regulated by binding to HNRNPH1, a nuclear ribonucleoprotein. INCR1’s primary transcript binds HNRNPH1 to block its inhibitory effects on the neighboring genes PD-L1 and JAK2 enabling their expression. Together, these findings introduce a mechanism of tumor IFNγ signaling regulation mediated by the lncRNA INCR1 and suggest a therapeutic target for cancer immunotherapy.
Graphical Abstract
eTOC Blurb
Mineo et al. investigate the role of lncRNAs in tumor immune evasion. They show that INCR1, a lncRNA expressed in response to interferon stimulation, regulates the expression of several immunosuppressive molecules to promote tumor escape from T cell attack.
INTRODUCTION
Immune checkpoint inhibitors have revolutionized cancer treatment (Pardoll, 2012; Sharma and Allison, 2015). These therapies have been developed based on the ability of cancers to evade anti-tumor immunity by up-regulation of immune checkpoint molecules, such as programmed cell death 1 ligand 1 (PD-L1), in response to stimuli, such as interferon-γ (IFNγ) (Beatty and Gladney, 2015; Garcia-Diaz et al., 2017). Expression of PD-L1 within the tumor microenvironment inhibits the anti-tumor immune response through the binding of the immune checkpoint receptor PD-1 expressed on T cells (Freeman et al., 2000). Immune checkpoint inhibitors that target the PD-1/PD-L1 pathway have been shown to be less toxic than standard chemotherapy and to produce durable tumor regression and overall survival benefits in several tumors including non-small cell lung cancer (NSCLC) and melanoma (Antonia et al., 2017; Larkin et al., 2015; Topalian et al., 2012). However, only a small group of patients respond to these therapies and some of the responders develop acquired resistance (Jenkins et al., 2018). Moreover, immune checkpoint inhibitors have not produced significant benefits in tumors characterized by a highly immunosuppressive microenvironment, such as glioblastoma (GBM) (Jackson et al., 2019). Resistance to immune checkpoint blockade is partly caused by constitutive expression of interferon-stimulated genes (ISGs) in tumors, as a result of a persistent IFNγ signaling (Benci et al., 2016). Therefore, a better understanding of the molecular mechanisms that control IFNγ signaling and PD-L1 expression will benefit the development of alternative and more effective strategies to overcome present therapeutic limitations.
Long noncoding RNAs (lncRNAs), transcripts longer than 200 nucleotides that lack protein coding potential, have emerged as major regulators of a wide range of cellular processes (Geisler and Coller, 2013; Rinn and Chang, 2012). Several thousand lncRNAs are encoded by the human genome, but the function of the large majority of these transcripts remains unexplored (Derrien et al., 2012). Some lncRNAs promote cis or trans regulation of gene expression through transcriptional and post-transcriptional mechanisms via interactions with key regulatory proteins (Atianand et al., 2016; Carpenter et al., 2013; Ramos et al., 2015; Wang et al., 2008; Willingham et al., 2005). It has been challenging to determine the biologic function of several lncRNAs due to mechanisms of action that are not mediated by the lncRNA transcripts themselves. This has led to the concept that many of these transcripts may not be functional (Struhl, 2007). However, more detailed studies have shown that the process of lncRNA transcription itself can promote chromatin accessibility and enhancement of the activity of coding gene promoters (Canzio et al., 2019; Engreitz et al., 2016a; Mowel et al., 2017). Thus, understanding the function of lncRNAs requires a detailed analysis of the mechanisms through which a nascent noncoding transcript can affect proximal genes.
In cancer, aberrant expression of lncRNAs has been associated with tumor development and progression (Gutschner et al., 2013; Kim and Croce, 2018; Prensner and Chinnaiyan, 2011; Prensner et al., 2011; Wu et al., 2018). The lncRNA HIF1A-AS2, for example, was shown to modulate gene expression in response to low oxygen tension, thus promoting cell survival and proliferation in the hypoxic regions of the tumor (Mineo et al., 2016). LncRNA expression can be tissue and cancer-specific, and expression pattern can also distinguish between tumor stage and subtypes, suggesting potential roles as biomarkers (Du et al., 2013). Moreover, the introduction in the clinic of chemically modified antisense oligonucleotide-based therapies renders the targeting of lncRNAs feasible in cancer therapy (Levin, 2019). Although the functional importance of lncRNAs in tumor progression and growth has now been clearly established, their role in immune evasion remains unknown.
In this study, we identify a poorly characterized lncRNA, which we named Interferon-stimulated Non-Coding RNA 1 (INCR1), as a major regulator of IFNγ signaling in tumors by post-transcriptional modulation of PD-L1 and JAK2 expression. INCR1 is transcribed as an antisense RNA from the PD-L1/PD-L2 locus and its expression strongly correlates with PD-L1 but not PD-L2 expression. INCR1 is expressed in human patients and across multiple tumor types, and its levels increase after IFNγ stimulation. We show that silencing INCR1 represses the expression of ISGs, including PD-L1, in both unstimulated and IFNγ-stimulated cells. Furthermore, INCR1 knockdown cells are more susceptible to cytotoxic T cell-mediated killing compared to control cells. In vivo, silencing INCR1 resulted in increased susceptibility to CAR T cell therapy in an experimental tumor model. Finally, we demonstrate that the primary INCR1 transcript, and not the mature lncRNA, modulates the activity of an RNA binding protein, HNRNPH1, to affect PD-L1 and JAK2 levels. This does not occur in trans but “locally”. Together, our data reveal a mechanism of interferon signaling regulation mediated by the lncRNA INCR1.
RESULTS
INCR1 is a lncRNA expressed in tumor cells exposed to IFNγ
To identify tumor lncRNAs with immunomodulatory functions, we performed whole-transcriptome analysis (RNA-seq) of patient-derived glioblastoma (GBM) cell lines (PDGCLs) stimulated with IFNγ (Figure 1A). IFNγ stimulated the transcription of 113 lncRNAs (p<0.01, fold change >2, Table S1) including BANCR, a lncRNA previously shown to be upregulated by IFNγ (Kutty et al., 2018), validating the approach. Among the most upregulated lncRNAs was a poorly characterized lncRNA expressed from the opposite DNA strand of the PD-L1/PD-L2 locus (Figure 1B). Due to its IFN-dependent expression, we named this lncRNA Interferon-stimulated Non-Coding RNA 1 (INCR1). INCR1 expression positively correlated with the expression of 237 other lncRNAs (FDR≤0.25, Figure 1C and Table S2), several of which were transcribed from loci of protein-coding genes known to be IFN regulated (Figure 1D and Table S3). Annotated in Ensembl as ENSG00000286162, the INCR1 gene was predicted to span a genomic region of 172.5 kilobases (kb) located in chr9p24.1 that produces a spliced lncRNA of about 2 kb. Using 5’ and 3’ rapid amplification of cDNA ends (RACE), we identified the 5’ and 3’ ends of the INCR1 transcript (Figure S1A and S1B). RACE sequencing data demonstrated that INCR1 has a canonical polyadenylation signal at the 3’ end whose coordinates are chr9:5,629,748–5,457,434 (Figure S1C and S1D). Moreover, we performed PCR amplification to obtain the full sequence of the INCR1 transcript. Sequencing PCR products from 3 different cell lines revealed that INCR1 is a 2,030 nt long lncRNA, composed of 3 exons (Figure S1E and S1F). The 5’ end of the INCR1 transcript consists of a 182 nt exon located within the first intron of the RIC1 gene. This is followed by a short 94 nt long exon located in the antisense orientation of the second intron of the PD-L2 gene and finally a 3’ end exon of 1,754 nt found in an antisense direction of the third intron of the PD-L1 gene (Figure S1G). Analysis of the INCR1 sequence using PhyloCSF showed the low coding potential of this transcript (Figure 1E). We also confirmed that INCR1 was not translated into protein, using an in vitro transcription/translation assay (Figure 1F). Furthermore, qPCR analysis of cellular fractions showed that INCR1 localized mostly in the nucleus (Figure 1G). Finally, analysis of the INCR1 locus, and the nearby 9p21 cytoband that includes the CDKN2A/B genes, in 32 PDGCLs and 43 long-term GBM cell lines (LTGCLs) revealed a pattern of copy number alterations similar to GBM tumors from the TCGA cohort, suggesting that our experimental cell line models reflected the human disease (Figure 1H).
Figure 1. INCR1 is a lncRNA expressed in tumor cells exposed to IFNγ.
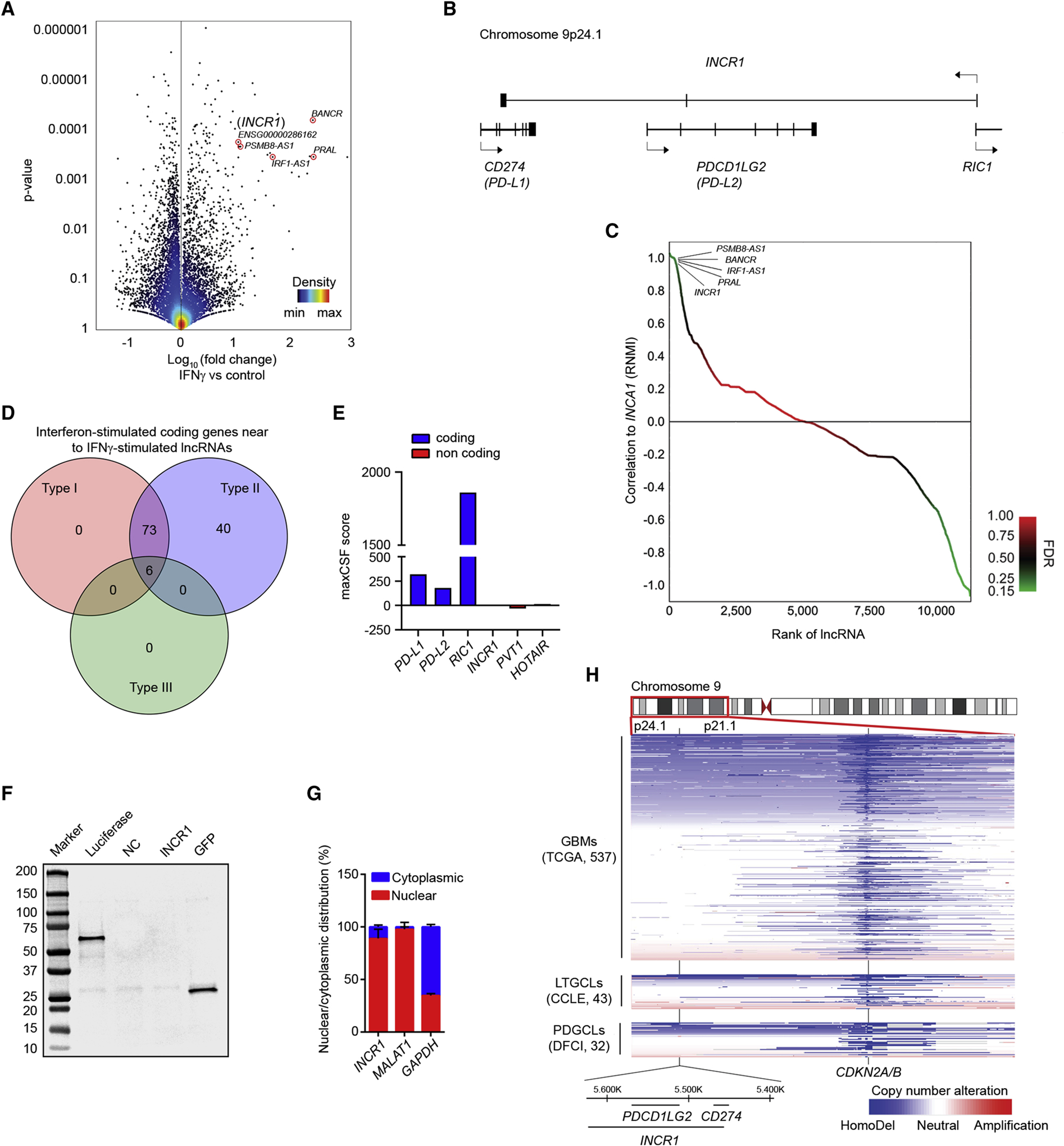
(A) Volcano plot of differentially expressed lncRNAs (RNA-seq) between unstimulated patient-derived GBM cell lines (PDGCLs) and PDGCLs stimulated with 100 U/ml IFNγ for 24 h (n=3 biological replicates). A set of the most up-regulated lncRNAs is annotated. 15,768 lncRNAs were surveyed in the analysis.
(B) Schematic representation of the INCR1 gene and the genes within the same locus but transcribed from the opposite strand.
(C) Approximately 237 lncRNAs were positively correlated and 1,188 negatively correlated (p-value <0.05, FDR<0.25, green color) with INCR1 expression. A set of the correlated lncRNAs is annotated.
(D) The Venn diagram shows the number of coding genes, transcribed from the same loci of lncRNAs correlated with INCR1, whose expression is regulated by one or more IFN types (Type I, II or III). Venn diagram was generated using the Interferome database (www.interferome.org).
(E) PhyloCSF analysis of the maximum CSF score of INCR1 and other known coding (blue) and non-coding (red) genes.
(F) INCR1 was in vitro transcribed and translated and reaction product was analyzed by Western blot (lane 4). Absence of protein product confirms INCR1 as a non-coding RNA. pSP64-Luciferase and pcDNA3.1-GFP vectors were used as positive control (lanes 2 and 5 respectively). No template reaction was used as negative control (NC, lane 3).
(G) qRT-PCR analysis of RNAs extracted from cytoplasmic and nuclear compartments of IFNγ-stimulated patient derived BT333 cells. MALAT1 and GAPDH were used to assess fractionation efficiency.
(H) Heat-map showing copy number losses (blue) and gains (red) in GBM tumors (n=573) and cell lines models (43 LTGCLs and 32 PDGCLs). Horizontal-axis represents genetic markers along cytobands 9p24.1 to 9p21.1 while vertical-axis includes specimens (rows) stratified by groups.
INCR1 expression correlates with PD-L1 levels in different cancer cells and patient tumors
To validate the RNA-seq data, we performed qPCR analysis using a select number of PDGCLs (n=7; Table S4). In all lines, INCR1 was up-regulated in response to IFNγ stimulation (Figure 2A). IFNγ-treated PDGCLs also expressed high levels of PD-L1 mRNA and protein (Figure 2B and S2A), with only minor differences in RNA copy number compared to INCR1 (Figure S2B). Notably, we observed a highly significant correlation between INCR1 and PD-L1 mRNA and protein (Figure 2C and S2C). Consistent with these findings, INCR1 expression was positively correlated with PD-L1 mRNA levels in patient GBMs, with INCR1-high tumors also expressing higher PD-L1 levels (Figure 2D–F). We also observed up-regulation of PD-L2 in PDGCLs stimulated with IFNγ (Figure S2D). However, there was no significant correlation between PD-L2 and INCR1 expression, in either PDGCLs or GBM patient tumors (Figure S2E–G). In addition, no correlation in expression was observed between INCR1 and its third overlapping gene RIC1 (Figure S1G and S2H). To determine if the correlation between INCR1 and PD-L1 expression extended beyond our PDGCL model, we analyzed the response to IFNγ treatment in cells from other cancers. Our panel included 6 cell lines representing GBM, melanoma, non-small cell lung cancer (NSCLC) and breast cancer (BC). All showed increased expression of INCR1 after IFNγ stimulation (Figure 2G) that also correlated with PD-L1 expression (Figure 2H and 2I). Furthermore, to test if INCR1 expression could be stimulated by other types of interferons, we treated two different PDGCLs with IFNβ and analyzed gene expression. We showed that stimulation with IFNβ was also able to induce the expression of both INCR1 and PD-L1 (Figure S2I). Finally, since it has been shown that STAT1 regulates PD-L1 expression in response to IFNγ (Garcia-Diaz et al., 2017), we analyzed if STAT1 could also regulate transcription of INCR1. Silencing STAT1 resulted in reduced INCR1 levels, suggesting STAT1 dependent regulation of INCR1 expression (Figure S2J). Thus, we have identified a novel lncRNA whose expression is stimulated by interferon and whose expression positively correlates with PD-L1 in tumors and tissue culture cells.
Figure 2. INCR1 expression correlates with PD-L1 levels in different cancer cells and patient tumors.
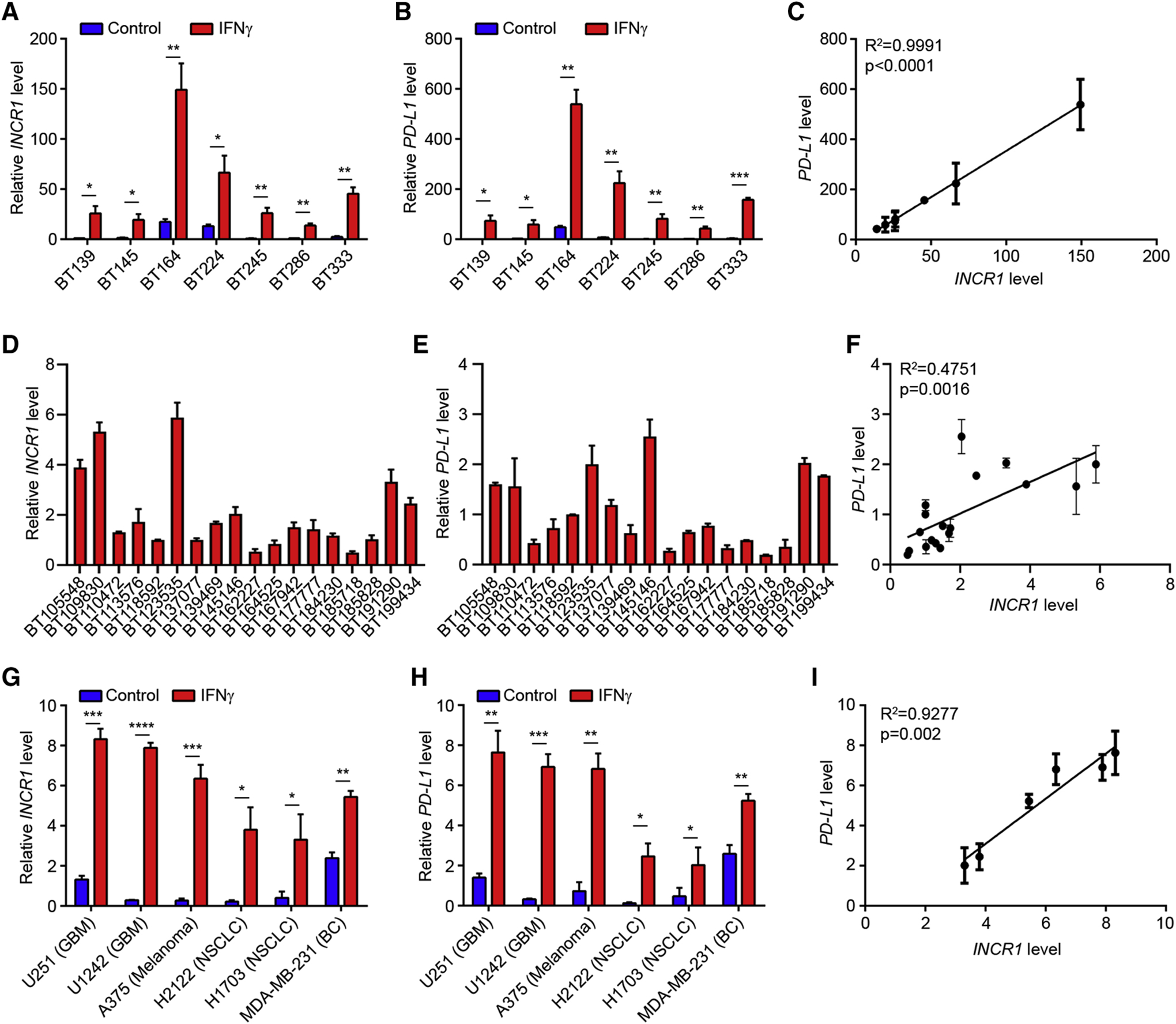
(A-C) qRT-PCR analysis of INCR1 (A) and PD-L1 (B) expression in 7 unstimulated or IFNγ-stimulated (100 U/ml for 24 h) PDGCLs, and correlation of INCR1 expression with PD-L1 expression in IFNγ-stimulated PDGCLs (C). R2=0.9991 calculated using linear regression analysis.
(D-F) Expression of INCR1 (D) and PD-L1 (E) in 18 GBM patient tumor specimens, and correlation of INCR1 expression with PD-L1 expression (F). R2=0.4751 calculated using linear regression analysis.
(G-I) qRT-PCR analysis of INCR1 (G) and PD-L1 (H) expression in 6 unstimulated or IFNγ-stimulated (100 U/ml for 24 h) long term cell lines from different tumor types and correlation of INCR1 expression with PD-L1 expression (I). Tumor cell types include glioblastoma (GBM), melanoma, non-small cell lung cancer (NSCLC) and breast cancer (BC). R2=0.9277 calculated using linear regression analysis.
Data shown as mean ± SD of three biological replicates (A, B, C, G, H and I) and as mean ± SD of three technical replicates (D, E and F). Data were analyzed by unpaired t-test: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
INCR1 regulates tumor IFNγ signaling
Since several lncRNAs act as major modulators of gene expression in response to stimuli (Atianand et al., 2016; Mineo et al., 2016), we hypothesized that INCR1 would regulate the expression of its neighboring genes and the tumor response to IFNγ stimulation. Because primary tumor cells showed low efficiency in maintaining expression of short hairpin RNAs (shRNAs), to test our hypothesis, we first generated stable knockdown of INCR1 in the U251 GBM cell line, and we assessed the impact of INCR1 silencing on global gene expression by RNA-seq. Performing nuclear/cytoplasmic fractionation we validated knockdown of both nuclear and cytoplasmic INCR1 (Figure S3A), whose downregulation did not affect cell viability (Figure S3B). Silencing INCR1 reduced the expression of 938 genes, of which 451 genes were commonly downregulated in unstimulated and IFNγ-stimulated cells (p<0.01, fold change >2, Figure 3A and Table S5). Gene ontology (GO) enrichment analysis showed downregulation of genes involved in immune-related functions, such as IFNγ response, innate immune response and defense response to virus (Figure 3B). Moreover, INCR1 knockdown resulted in reduced expression of 124 ISGs (Figure 3C). Among those genes were important components of the IFNγ signaling pathway (JAK2 and STAT1), as well as major immunosuppressive molecules (PD-L1 and IDO1). We further validated the deregulated expression of selected genes by qPCR. Our data confirmed that silencing INCR1 resulted in reduced mRNA levels of its overlapping genes PD-L1 and PD-L2, both at basal level and in response to IFNγ (Figure 3D–F). Notably, the mRNA of JAK2, whose gene is in the same locus as INCR1 and PD-L1, was significantly downregulated (Figure 3G), along with ISGs from different genomic regions, such as STAT1 and IDO1 (Figure 3H and 3I). Moreover, we showed that silencing INCR1 resulted in downregulation of PD-L1, JAK2, STAT1 and IDO1 protein levels (Figure 3J). Downregulation of total STAT1 was associated with its decreased phosphorylation and nuclear localization in response to IFNγ stimulation (Figure 3J and S3C). In addition, we showed that silencing INCR1 reduced both total PD-L1 protein and the levels of cell surface PD-L1 (Figure 3K). To test if INCR1 could regulate signaling of other cytokines, we treated control and INCR1-knockdown U251 cells with TNFα and analyzed gene expression by qPCR. Our data showed that INCR1 was not significantly induced by TNFα treatment and silencing INCR1 did not affect the expression of TNFα stimulated genes (Figure S3D). Finally, to validate INCR1 regulation of IFNγ signaling in patient derived cells, we targeted INCR1 in BT333 cells using gapmer antisense oligonucleotides (ASO). Gene expression analysis showed that silencing INCR1 in BT333 cells resulted in a response similar to that observed in U251 (Figure S3E), further validating our results.
Figure 3. INCR1 regulates tumor IFNγ signaling.
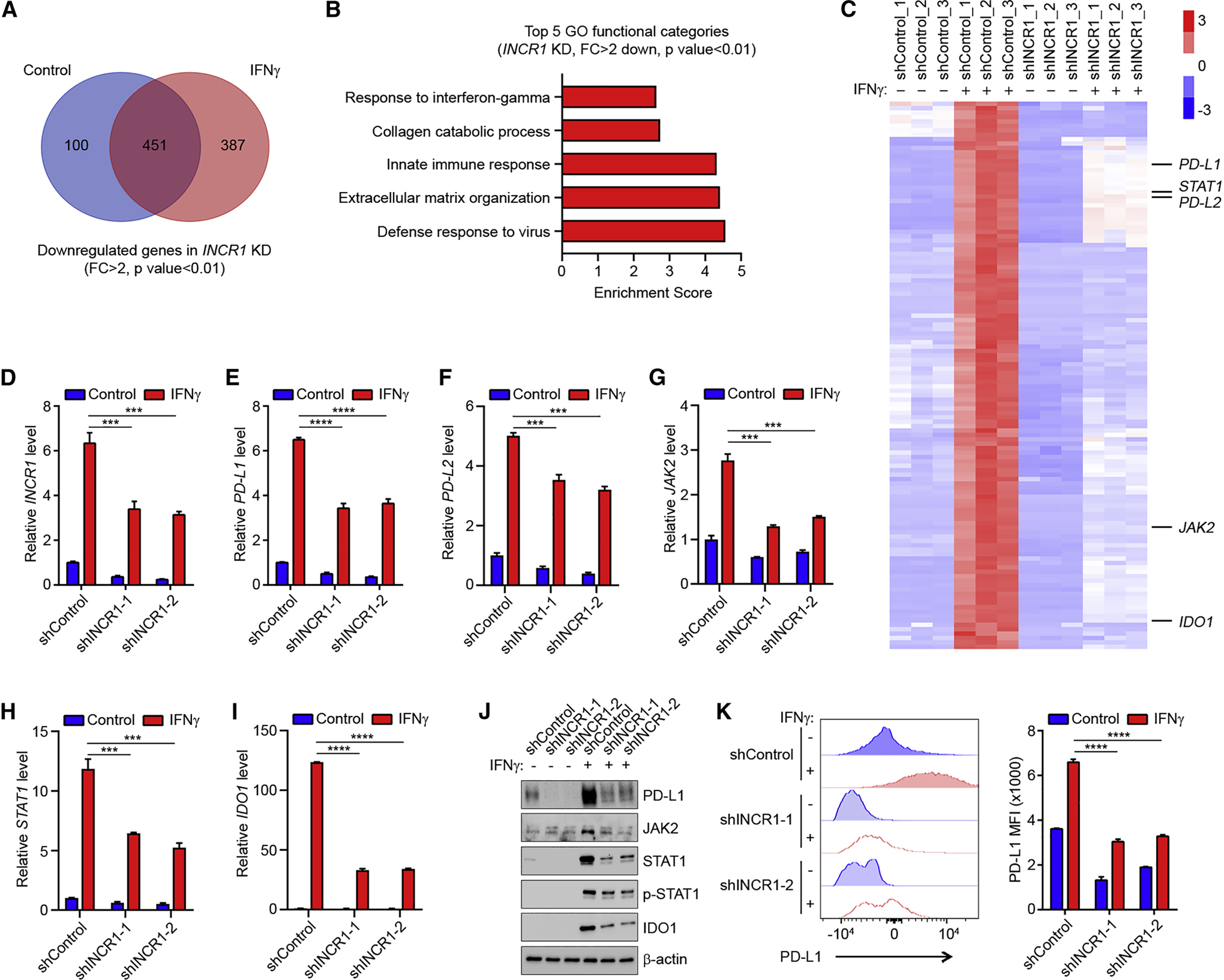
(A) The Venn diagram shows the number of genes (RNA-seq) downregulated in INCR1 knockdown cells relative to control cells unstimulated (Control) or stimulated with 100 U/ml IFNγ for 24 h (n=3 biological replicates).
(B) Gene ontology analysis of genes downregulated in IFNγ-treated INCR1 knockdown cells compared to control cells. Analysis was performed using DAVID 6.8 Tool.
(C) Heatmap of the expression levels of IFNγ-stimulated genes that were significantly downregulated in INCR1 knockdown cells compared to control (n=3 biological replicates).
(D-I) qRT-PCR analysis of INCR1 (D), PD-L1 (E), PD-L2 (F), JAK2 (G), STAT1 (H) and IDO1 (I) expression in control or two independent INCR1-knockdown U251 cells unstimulated or stimulated with 100 U/ml IFNγ for 24 h.
(J) Western blot analysis of PD-L1, JAK2, STAT1, phospho-STAT1 and IDO1 expression in control or two independent INCR1-knockdown U251 cells unstimulated or stimulated with 100 U/ml IFNγ for 24 h.
(K) Flow cytometry analysis of cell surface levels of PD-L1 in control or two independent INCR1-knockdown U251 cells unstimulated or stimulated with 100 U/ml IFNγ for 24 h.
Data are representative of three (D, E, F, G, H, I) or two (J and K) independent experiments. Data shown as mean ± SD of at least three replicates. Data were analyzed by unpaired t-test: ***p < 0.001, ****p < 0.0001.
Since PD-L1 expression significantly contributes to cancer-associated immunosuppression (Beatty and Gladney, 2015), to assess whether INCR1 is able to regulate PD-L1 expression in tumors other than GBM, we selected two non-GBM cell lines with different basal levels of PD-L1 to generate stable INCR1 knockdowns; the A375 melanoma cell line, which showed no detectable basal levels of PD-L1 by immunoblot, and the MDA-MB-231 breast cancer cell line, which exhibited the highest basal levels of PD-L1 among the cell lines analyzed. INCR1 knockdown in A375 cells led to a significant reduction of IFNγ-mediated PD-L1 expression compared to control cells (Figure S3F–H). Silencing INCR1 in MDA-MB-231 cells resulted in a significant decrease of PD-L1 basal levels and a strongly attenuated response to IFNγ stimulation (Figure S3J–L). Moreover, both cell lines showed reduced levels of PD-L1 at their surface when INCR1 was silenced (Fig. S3I and S3M). To further validate the ability of INCR1 to regulate PD-L1 expression, we designed sgRNA targeting the promoter region of INCR1 (that also overlaps the RIC1 gene) to evaluate knockdown in patient-derived cells using CRISPRi. INCR1 silencing caused by the designed sgRNA resulted in reduction of PD-L1 expression, with no effect on RIC1 levels (Figure S3N). Together, these results indicate that INCR1 modulates tumor response to IFNγ treatment by regulation of multiple ISGs in GBM and in other cancer types.
Silencing INCR1 leads to increased T cell-mediated cytotoxicity in vitro and improves CAR T cell efficacy in vivo
CD8+ cytotoxic T lymphocytes (CTLs) act as important effectors of cancer immunoediting (Mittal et al., 2014). Activation of CD8+ CTLs induces the secretion of cytokines, such as IFNγ, which promotes their proliferation and anti-tumor activity (Bhat et al., 2017; Zhang and Bevan, 2011). Because INCR1 silencing reduced the expression of major IFNγ-regulated immune inhibitory molecules in tumor cells, we tested whether silencing INCR1 would increase CD8+ CTL activity. Using a 2D culture system, we found that co-culture of tumor cells with CD8+ CTLs, activated with beads covalently coupled to anti-CD3 and anti-CD28 antibodies, resulted in increased cytotoxicity of INCR1 knockdown cells compared to control, with no significant change in viability of activated T cells (Figure 4A and S4A). We confirmed the greater activity of CD8+ CTLs in killing U251, A375 and MDA-MB-231 cells with silenced INCR1 using live/dead staining and FACS analysis (Figure S4B). Furthermore, using a 3D culture system, we found a significant reduction in the size of INCR1 knockdown tumor spheres compared to control tumor spheres when these were co-incubated with activated CD8+ CTLs (Figure 4B). This was associated with increased IFNγ secretion by CD8+ CTLs, co-cultured with INCR1 knockdown cells compared to CD8+ CTLs co-cultured with control cells (Figure 4C).
Figure 4. Silencing INCR1 leads to increased T cell-mediated cytotoxicity in vitro and improves CAR T cell efficacy in vivo.
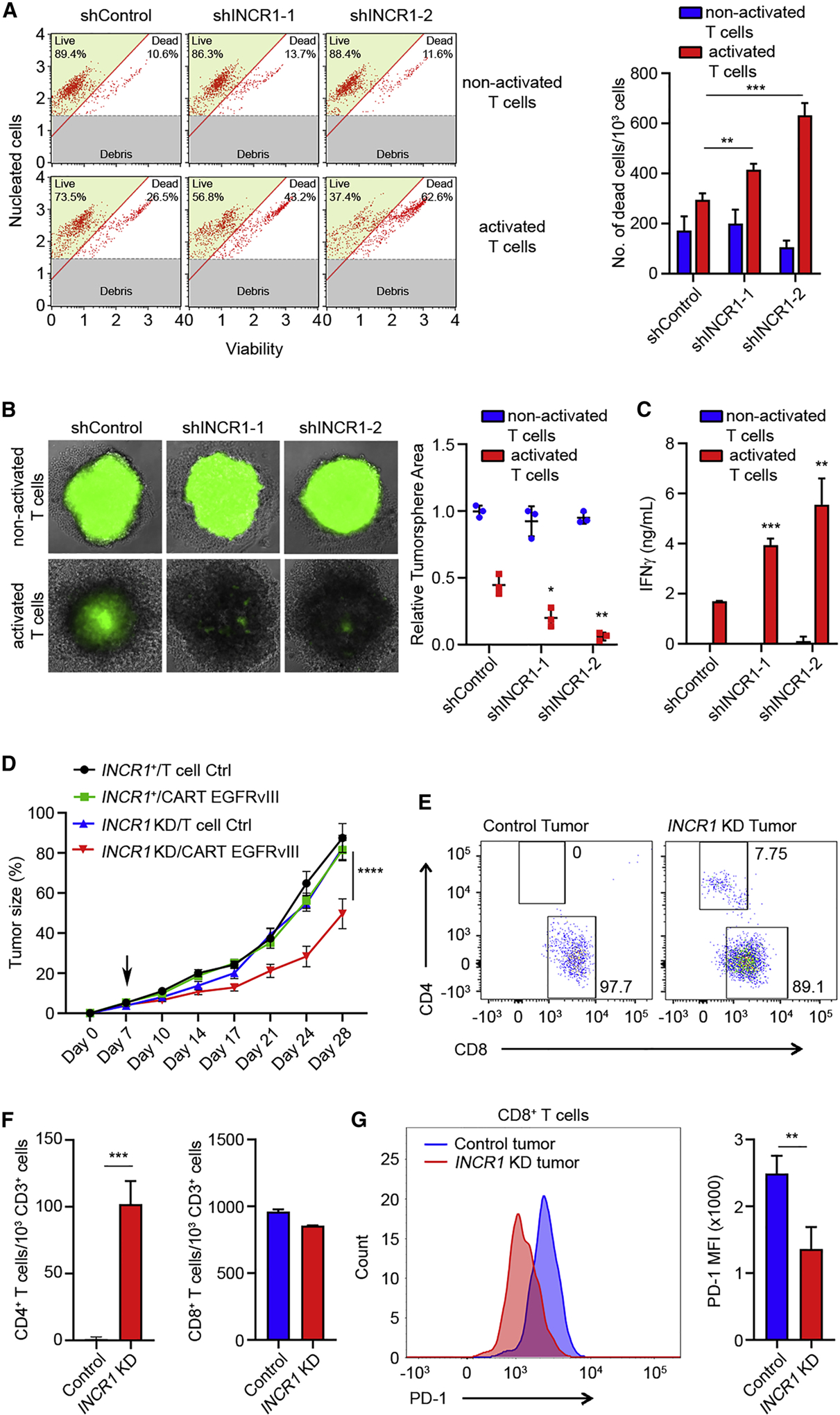
(A) Cell viability analysis of control and two independent INCR1-knockdown U251 cells co-cultured with non-activated and activated CD8+ T cells for 96 h. Cell viability was determined by Muse Cell Analyzer. Shown are representative plots of live and dead cells (left) and number of dead cells (right)
(B) GFP-positive control and two independent INCR1-knockdown U251 tumor spheres were co-cultured with non-activated or activated CD8+ T cells and T cell cytotoxic activity was evaluated at 96 h. Shown are representative fluorescent microscopy pictures (left) and relative tumor sphere area (right).
(C) ELISA analysis of IFNγ secretion from non-activated or activated CD8+ T cells co-cultured with control or two independent INCR1-knockdown U251 cells for 48 h.
(D) NSG mice (n=6 per group) were injected with 2.5 × 106 U251-EGFRvIII shControl (black and green lines) or shINCR1 (blue and red lines) subcutaneously on day 0 and with 1 × 106 T cells intravenously on day 7 (black arrow). Tumor volume was measured over time. Tumor volume data were analyzed by two-way ANOVA: ****p < 0.001.
(E) Flow cytometry analysis of CD4/CD8 composition of EGFRvIII specific CAR T cells infiltrating control (left) and INCR1-knockdown (right) tumors.
(F) Flow cytometry analysis of the number of CD4+ (left) and CD8+ (right) CAR T cells in control and INCR1-knockdown tumors 21 days post intravenous injection of T cells.
(G) Flow cytometry analysis of PD-1 levels in CD8+ CAR T cells isolated from control and INCR1-knockdown tumors.
Data are representative of three (A, B, C) independent experiments. Data shown as mean ± SD of at least three replicates. Data were analyzed by unpaired t-test: *p < 0.05, **p < 0.01, ***p < 0.001.
T cells engineered to express a Chimeric Antigen Receptor (CAR) against a specific tumor antigen are a potential curative therapy for different cancer types, but has produced only modest success in solid tumors mainly due to the highly immunosuppressive microenvironment (D’Aloia et al., 2018). To test if silencing INCR1 could improve CAR T cell function in vivo, U251-EGFRvIII control and INCR1 knockdown tumors were implanted subcutaneously. Seven days after implantation, human T cells expressing GFP alone (control) or the EGFRvIII-directed CAR were injected with a single intravenous dose that is about 1/10 of the dose that is standard (Hillerdal et al., 2014; Song et al., 2015; Wing et al., 2018; Zhang et al., 2019). Under these conditions, mice with control tumors showed no significant response to the CAR T cell therapy. In contrast, CAR T cells significantly reduced tumor growth in mice bearing INCR1 knockdown tumors (Figure 4D). At the end of the study, twenty-one days after T cell injection, tumors were analyzed for the presence of CAR T cells. INCR1 knockdown tumors presented infiltrates of both CD4+ and CD8+ T cells with a predominance of CD8+ T cells. In contrast, no CD4+ T cell infiltrates were observed in control tumors (Figure 4E and 4F). Furthermore, CD8+ T cells in control tumors expressed a significantly higher level of PD-1 compared to those infiltrating tumors with silenced INCR1 (Figure 4G). Taken together, these results show that INCR1 plays a functional role in controlling tumor IFNγ signaling, and that its knockdown leads to increased susceptibility of human tumor cells to T cell-mediated killing.
HNRNPH1 is a binding partner of INCR1
Most lncRNAs have been shown to function through their interaction with proteins, such as transcription factors or heterogeneous nuclear ribonucleoproteins (hnRNPs) (Atianand et al., 2016; Carpenter et al., 2013). To identify the mechanism through which INCR1 regulates PD-L1 expression, we first used RNA Antisense Purification (RAP) to purify proteins in direct contact with endogenous INCR1 in IFNγ-stimulated PDGCLs. We designed probes covering the entire INCR1 sequence, crosslinked RNA-protein complexes by UV irradiation and performed RAP in denaturing conditions to maximize the recovery of specific RNA-protein interactions. We observed greater than 80-fold enrichment in INCR1 RNA compared to control purification (Figure 5A). Mass spectrometry analysis showed HNRNPH1 as the top hit among the proteins identified only in the INCR1 RAP but not in the control RAP (Figure 5B and Table S6). We then validated the interaction between INCR1 and HNRNPH1 by in vivo RNA-immunoprecipitation of UV-crosslinked samples (CLIP). To increase the strength of our validation, we included two lncRNAs previously demonstrated to be binding partners of HNRNPH1 (MALAT1 and NORAD) and one lncRNA known to not bind HNRNPH1 (RMRP) (Uren et al., 2016). We showed that all the tested lncRNAs were highly expressed (Figure S5A) and their localization was mostly nuclear (Figure S5B). qPCR analysis of RNA co-purified with HNRNPH1 confirmed binding of HNRNPH1 to MALAT1 and NORAD and the absence of interaction with RMRP and 18S (Figure S5C–F). Moreover, HNRNPH1 showed significant binding to INCR1 in IFNγ-stimulated cells compared to untreated cells and isotype control (Figure 5C). To identify INCR1 regions bound by HNRNPH1, we first mapped HNRNPH1 binding sites in vivo by enhanced CLIP sequencing (eCLIP-seq). We identified a cluster of peaks in the proximal intron of INCR1 (Figure 5D). To validate sequencing data in vitro, we cloned an INCR1 minigene containing the 5’ and 3’ region of the first intron and used it to generate 7 different in vitro transcribed biotinylated RNA fragments. RNA pull-down assays showed that HNRNPH1 interacted strongly with two RNA fragments (F4 and F6) containing the sequences of the major peaks found in the eCLIP-seq (Figure 5E). We further proved HNRNPH1 binding to the sequence with strongest eCLIP signal by electrophoretic mobility shift assay (EMSA) (Figure S5H). The two fragments bound by HNRNPH1 were enriched in G-stretches, and this motif was found in more than 80% of the HNRNPH1 RNA targets (Figure 5F and Table S7). Gene ontology (GO) enrichment analysis of all the genes identified by eCLIP-seq showed that HNRNPH1, other than bound to INCR1, was also bound to several genes involved in immune system processes, including several IFNγ-stimulated genes, such as PD-L1, JAK2 and STAT1, which expression we showed to be regulated by INCR1 (Figure 5G). However, overexpression of an INCR1 minigene in trans did not produce any significant effect on global ISG expression (Figure S5I), suggesting that INCR1 regulates only genes that are in close proximity (“locally”), and therefore INCR1 effects on the tumor interferon signaling may be mediated by local regulation of the neighboring genes PD-L1 and JAK2. We next validated by RIP HNRNPH1 binding to the ISGs expressed nearby INCR1. Our data showed that HNRNPH1 bound PD-L1 and JAK2 mRNA (Figure 5H and 5I) but did not bind PD-L2 mRNA (Figure S5G). Moreover, to assess if INCR1 was bound to HNRNPH1 in complex with PD-L1 and/or JAK2 mRNA, we performed RAP-RNA analysis to identify RNA-RNA interactions. Using this approach, we did not detect direct or indirect binding between INCR1 and PD-L1 or JAK2 mRNA (Figure S5J). Taken together, these results indicate that INCR1 binding to HNRNPH1 was independent of PD-L1 and JAK2 mRNA binding.
Figure 5. HNRNPH1 is a binding partner of INCR1.
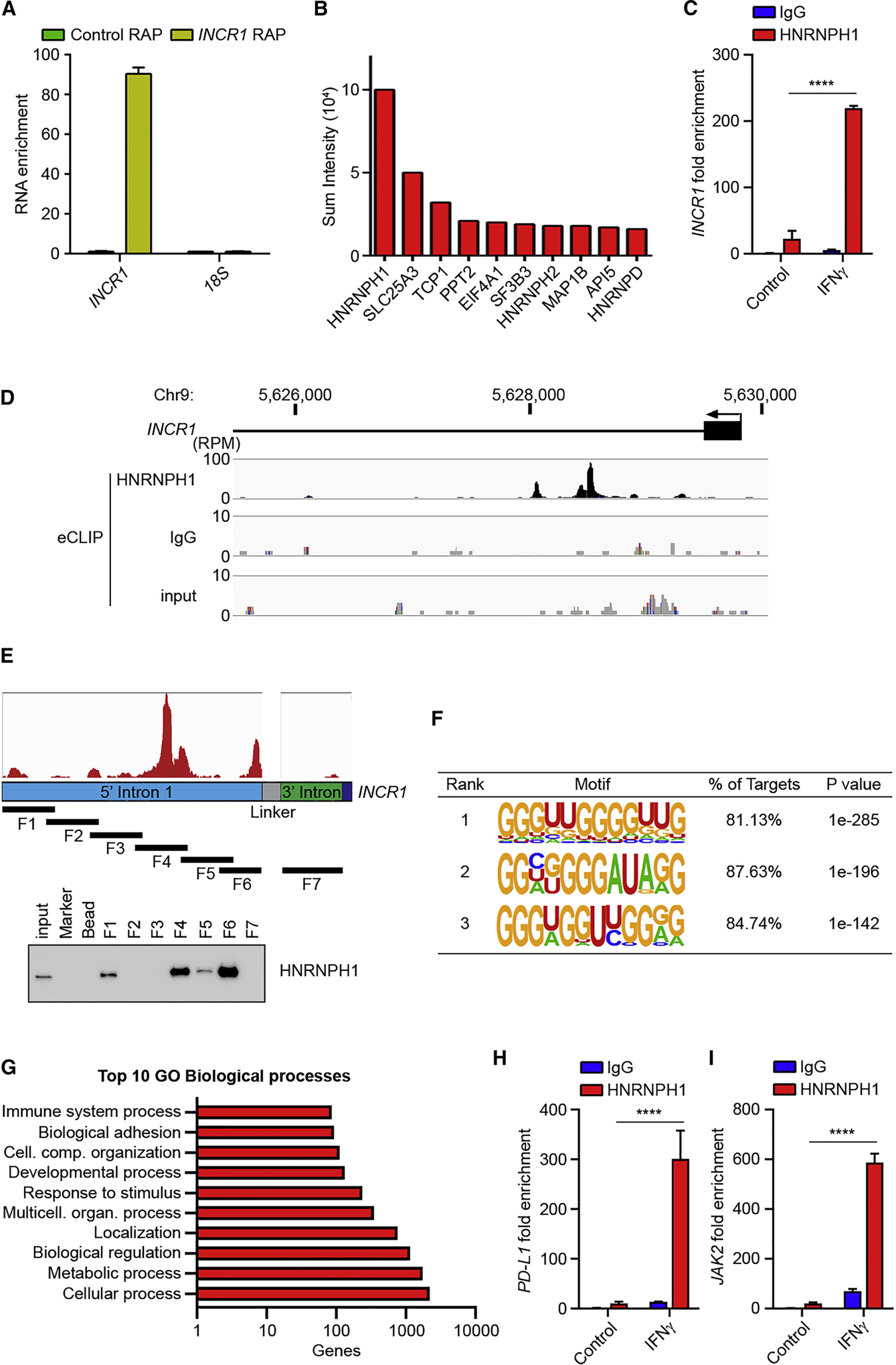
(A) Patient derived BT333 cells were stimulated with 100 U/ml IFNγ for 24 h and RNA captured using biotinylated probes antisense to INCR1 (yellow) or scramble control probe (green) was analyzed by qRT-PCR.
(B) Top 10 proteins co-purified with INCR1 from RNA antisense purification (RAP).
(C) HNRPNH1 RIP (RNA immunoprecipitation) followed by qRT-PCR analysis of co-purified INCR1 in UV-crosslinked patient derived BT164 cells unstimulated or stimulated with 100 U/ml IFNγ for 24 h.
(D) Identification of HNRNPH1 binding sites by eCLIP in A375 cells stimulated with 100 U/ml IFNγ for 6 h. Read density in reads per million (RPM) are shown for HNRNPH1, IgG and input.
(E) Schematic representation of INCR1 minigene with eCLIP reads (red) and RNA fragments (F1–7) covering the 5’ (blue) and 3’ (green) regions of the INCR1 first intron (top); and RNA pull-down validation of INCR1 interaction with HNRNPH1 using the 7 different biotinylated RNA fragments (bottom).
(F) Top motifs identified by de novo motif finding around HNRNPH1 eCLIP sites.
(G) Gene ontology analysis of the genes bound to HNRNPH1 identified by eCLIP. Analysis was performed using PANTHER Classification System.
(H and I) HNRPNH1 RIP followed by qRT-PCR analysis of co-purified PD-L1 (H) and JAK2 (I) in UV-crosslinked patient derived BT164 cells unstimulated or stimulated with 100 U/ml IFNγ for 24 h.
Data are representative of three (C, E, H, I) or two (A, B) independent experiments. Data shown as mean ± SD of at least three replicates. Data were analyzed by unpaired t-test: ****p < 0.0001.
INCR1 functions as a negative regulator of HNRNPH1 activity
The observed absence of INCR1 binding to PD-L1 and JAK2 transcripts coupled with the observed binding of INCR1, PD-L1 and JAK2 transcripts to HNRNPH1 led us to hypothesize that INCR1 acted as a decoy RNA that competitively inhibits HNRNPH1 function on PD-L1 and JAK2 transcripts. To test our hypothesis, we first investigated the effects of modulating HNRNPH1 levels. Silencing HNRNPH1 increased PD-L1 and JAK2 mRNA levels in both A375 cells (Figure 6A) and patient derived BT139 cells treated with IFNγ (Figure S6A). This was associated with a significant increase in PD-L1 and JAK2 protein in response to IFNγ stimulation (Figure 6B). These data suggested that HNRNPH1 is a negative regulator of PD-L1 and JAK2 expression and that binding of INCR1 is required to prevent HNRNPH1 function. This hypothesis was supported by an inverse correlation observed between HNRNPH1 levels and PD-L1 expression in GBM tumors (TCGA) (Figure 6C). To further prove our hypothesis, we conducted knockdown experiments to silence HNRNPH1 expression in INCR1 knockdown cells. As expected INCR1 knockdown resulted in reduced PD-L1 expression in IFNγ-treated cells. However, silencing HNRNPH1 in those knockdown cells rescued PD-L1 expression (Figure 6D). To confirm that the interaction between INCR1 and HNRNPH1 mediates the regulation of PD-L1 and JAK2, we first evaluated the binding affinity of HNRNPH1 to INCR1 by microscale thermophoresis (MST) and compared it with the binding affinity to PD-L1 and to JAK2. RNA fragments corresponding to the major eCLIP peaks found in the intron 5 of PD-L1 and intron 2 of JAK2 were used to measure binding affinity (Figure S6B). INCR1 RNA fragment showed higher binding affinity to HNRNPH1 compared to PD-L1 and JAK2 RNA fragments (Figure 6E). These data suggest that HNRNPH1 binding to INCR1 may reduce its ability to interact with PD-L1 and JAK2 and thus silencing INCR1 should increase PD-L1 and JAK2 colocalization with HNRNPH1. However, since INCR1 locus is in close proximity to the PD-L1 and JAK2 locus, in situ hybridization studies to analyze PD-L1 and JAK2 RNA colocalization with HNRNPH1 may generate results difficult to interpret because modulation of INCR1 expression is unlikely to change HNRNPH1’s local concentration. Therefore, we compared the affinity of INCR1 with PD-L1 and JAK2 to bind HNRNPH1 in an EMSA competition assay. Using γ−32P-radiolabeled PD-L1 and JAK2 RNA fragments we observed a shift in the RNA:HNRNPH1 complex, confirming the ability of these two sequences to bind HNRNPH1 (Figure 6F). Addition of non-labeled INCR1 RNA fragment at a INCR1:PD-L1 and INCR1:JAK2 ratio of 1:1 efficiently decreased the formation of PD-L1:HNRNPH1 and JAK2:HNRNPH1 complexes (Figure 6F). Competition with JAK2 was more efficient in agreement with MST measurements of dissociation constants that demonstrated weaker affinity between HNRNPH1 and JAK2 than between HNRNPH1 and PD-L1. Increasing the molar ratio to 1:5 and 1:10 led to complete dissociation of the complexes (Figure 6F). Taken together, these results suggest that the affinity of INCR1 to HNRNPH1 is stronger than that to PD-L1 or JAK2, and that INCR1 inhibits HNRNPH1’s interaction with PD-L1 and JAK2.
Figure 6. INCR1 functions as a negative regulator of HNRNPH1 activity.
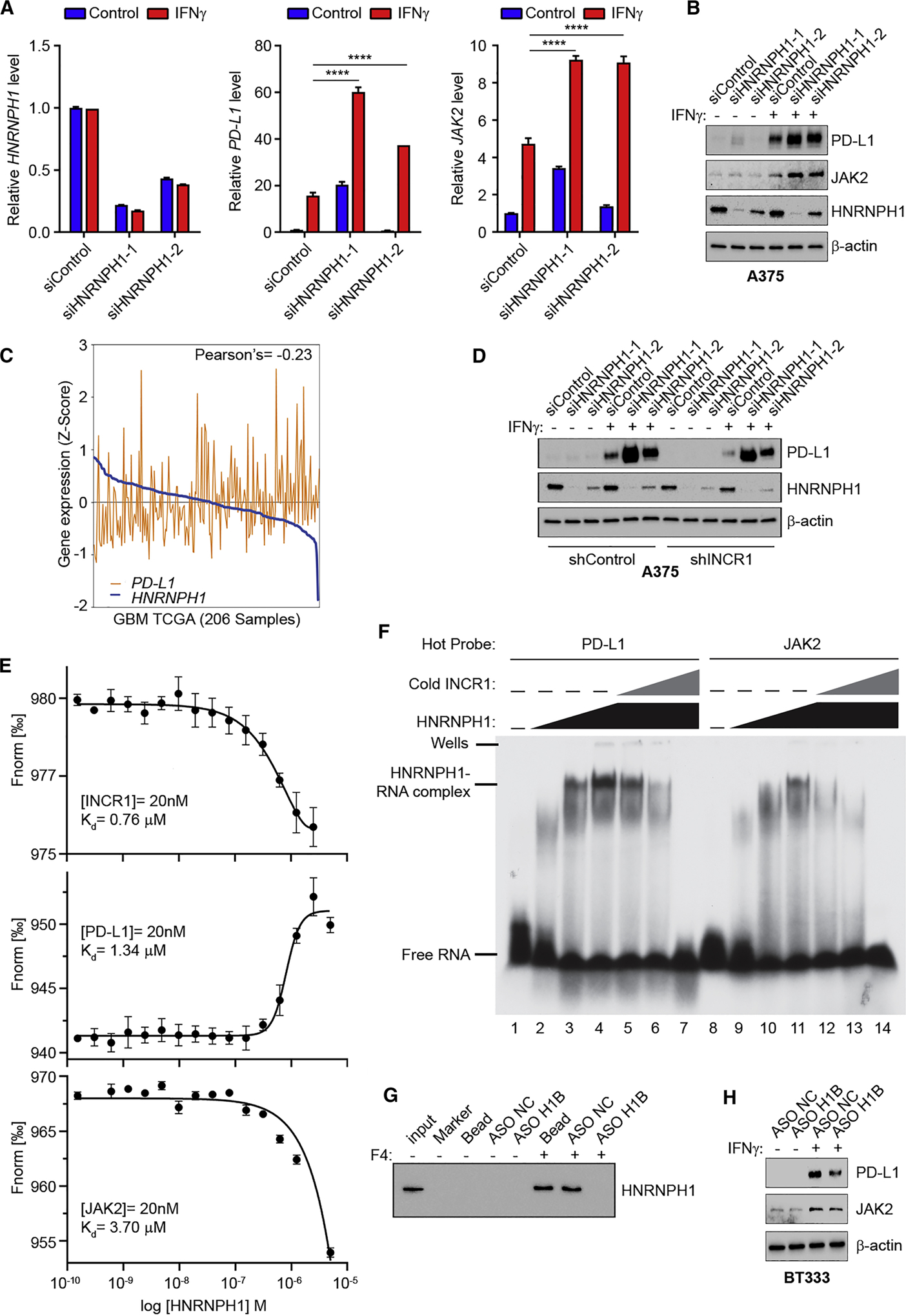
(A) qRT-PCR analysis of HNRNPH1 (left), PD-L1 (center) and JAK2 (right) expression in unstimulated or IFNγ-stimulated (100 U/ml for 24 h) A375 cells transfected with siRNA control or two different siRNAs targeting HNRNPH1.
(B) Western blot analysis of PD-L1, JAK2 and HNRNPH1 expression in control or two independent HNRNPH1-knockdown A375 cells unstimulated or stimulated with 100 U/ml IFNγ for 24 h.
(C) Correlation of HNRNPH1 expression with PD-L1 expression in GBM tumors (n=206).
(D) Control and INCR1-knockdown (shINCR1–2) A375 cells were transfected with siRNA control or two independent siRNAs targeting HNRNPH1. Cells were stimulated with 100 U/ml IFNγ for 24 h and expression of HNRNPH1 and PD-L1 was analyzed by Western blot.
(E) Binding curves of HNRNPH1 interaction with a 50 nucleotide RNA oligonucleotide whose sequence represents the major eCLIP peak of INCR1 (top), PD-L1 (middle) and JAK2 (bottom), demonstrating a specific binding of HNRNPH1 with a Kd of 0.76 μM, 1.34 μM and 3.7 μM respectively.
(F) EMSA analysis of the effect of INCR1 RNA fragment on HNRNPH1 ability to bind to radiolabeled PD-L1 (left) or JAK2 (right) RNA fragments (50 nM). No protein was added to the lane 1 and 8. HNRNPH1 was added at the concentration of 0.65 μM (lane 2 and 9), 3.25 μM (lane 3 and 10) and 6.5 μM (lanes 4–7 and 11–14). INCR1 was added at molar ratio of 1:1 (lane 5 and 12), 1:5 (lane 6 and 13) and 1:10 (lane 7 and 14).
(G) RNA pull-down assay with biotinylated INCR1 fragment 4 (F4) in the presence of antisense oligonucleotide control (ASO NC) or targeting HNRNPH1 binding site (ASO H1B).
(H) Western blot analysis of PD-L1 and JAK2 expression in ASO NC or ASO H1B transfected patient derived BT333 cells unstimulated or stimulated with 100 U/ml IFNγ for 24 h.
Data are representative of three (A, F, G) or two (B, D, H) independent experiments. Data shown as mean ± SD of at least three replicates. Data were analyzed by unpaired t-test: ****p < 0.0001.
To further evaluate how changes in INCR1 interaction with HNRNPH1 can affect PD-L1 and JAK2 expression, we designed an antisense oligonucleotide fully modified with 2’-OMethoxyethyl (2’-MOE) to target the HNRNPH1 binding site (ASO H1B) in the INCR1 RNA. 2’-MOE fully modified ASO, unlike gapmer ASO, do not support RNase H activity thus affecting RNA-protein interaction without inducing RNA cleavage (Khvorova and Watts, 2017). Using ASO H1B we could reduce INCR1 interaction to HNRNPH1 in vitro (Figure 6G), in a dose-dependent manner (Figure S6C). On the contrary, no effect on binding ability was observed using a control ASO (Figure 6G). To study the effect of ASO H1B in vivo, we transfected BT333 patient derived cells and A375 melanoma cells with ASO H1B or control ASO. In both cell lines ASO H1B significantly reduced IFNγ-stimulated PD-L1 and JAK2 protein expression (Figure 6H and_S6D). These results indicate that INCR1 specifically interacts with HNRNPH1 and that blocking this interaction affects PD-L1 and JAK2 expression.
DISCUSSION
IFNγ is a cytokine, mostly secreted by activated T cells, with important roles in regulating immune responses against tumor cells (Ikeda et al., 2002). IFNγ induces MHC class I expression, promotes activation of CTL and inhibits regulatory T cell development. However, in response to increased levels of IFNγ, cancer cells tend to express immune inhibitory molecules (Beatty and Gladney, 2015; Garcia-Diaz et al., 2017; Zaidi and Merlino, 2011). Among them, PD-L1 plays a major role in CTL inhibition within the tumor microenvironment (Freeman et al., 2000). Thus, suppression of IFNγ-mediated tumor resistance is essential for an effective immune response against cancer. Although the molecular mechanisms of interferon signaling in tumors is now emerging, the role of lncRNAs is still poorly understood. Here, we performed lncRNA expression profiling of patient-derived tumor cells stimulated with IFNγ. We showed that treatment of cells with IFNγ (100 units/ml corresponding to a concentration of 5 ng/ml, concentrations comparable to those produced by activated T cells within the tumor microenvironment (Brown et al., 2018; Chiocca et al., 2019; Choi et al., 2019; Johnson et al., 2015), induced the expression of several lncRNAs. The most significantly induced lncRNAs were transcribed from loci of IFNγ-regulated coding genes, suggesting that these genes are co-regulated. Most importantly, among the most up-regulated lncRNAs, we identified INCR1 as a lncRNA transcribed from the PD-L1 locus and we demonstrated that this lncRNA plays an important role in the regulation of PD-L1 expression and IFNγ signaling in tumors. Notably, INCR1 was expressed in all human tumor cell types tested, including patient tumors, suggesting INCR1 expression as a broad mechanism of tumor immune evasion. Due to poor conservation of lncRNAs across species (Marques and Ponting, 2009; Necsulea et al., 2014), we could not test INCR1 expression in mouse cell lines. Therefore, further studies will be required to identify the sequence of the mouse INCR1 and to analyze its expression in mouse derived tumors.
We primarily studied GBM in this study, a tumor reported to express immune checkpoint signaling (such as PD-L1) (Berghoff et al., 2015; Garber et al., 2016; Wilmotte et al., 2005; Wintterle et al., 2003). Escape from immunotherapy in GBM is also characterized by significant elevation in PD-L1 and immune checkpoint signaling (Chiocca et al., 2019; O’Rourke et al., 2017; Speranza et al., 2018) and recent clinical trials of immune checkpoint inhibition in GBM appear to show some encouraging data related to responses (Cloughesy et al., 2019). Therefore, our experimental paradigm of IFNγ stimulation in GBM leading to increased PD-L1 even in GBM is relevant.
INCR1 expression was mostly confined to the nucleus. Nuclear lncRNAs generally are involved in regulation of gene expression by in cis and in trans mechanisms. Cis acting lncRNAs control the expression of one or more genes within the same allelic locus from which they are transcribed (Mowel et al., 2017). In contrast, lncRNAs with in trans function usually control groups of genes involved in a specific biological process (Atianand et al., 2016; Mineo et al., 2016). Analysis of INCR1 expression, compared to genes within its same locus, revealed a strong correlation between INCR1 expression and PD-L1 levels, both in vitro and in patient tumors. This, other than suggesting a common mechanism of regulation between the two genes, led us to hypothesize that INCR1 could play an important role in the regulation of gene expression in response to IFNγ stimulation. Using loss-of-function experiments, we demonstrated INCR1 is a key regulator of PD-L1 expression. Silencing INCR1 in several tumor cell types resulted in reduced levels of PD-L1 both at the mRNA and protein levels. Most importantly, INCR1 knockdown reduced the levels of PD-L1 exposed on the cancer cell surface, thus reducing the possibility of its interaction with PD-1 expressed on immune cells. Silencing INCR1 also reduced the expression of its neighboring gene JAK2 and several other ISGs, including immune inhibitory molecules, such as IDO1 (Liu et al., 2010). Surprisingly, we noticed that, although INCR1 knockdown resulted in reduced JAK2 mRNA levels both in unstimulated and IFNγ-stimulated cells, JAK2 protein levels were only decreased in IFNγ-stimulated cells. This may be because in unstimulated cells INCR1 silencing affected the expression of JAK2 transcript variants which produce protein isoforms not recognized by the antibody used in this study. JAK2 is an important mediator of the response to IFNγ stimulation (Watling et al., 1993). Activated JAK2 phosphorylates the signal transducer and activator of transcription (STAT), which promotes the expression of several ISGs, including immune inhibitory molecules that inactivate antitumor T cell activity (Garcia-Diaz et al., 2017; Schneider et al., 2014). In trans expression of INCR1 did not produce any significant increase in tumor response to IFNγ stimulation, which suggests that global effects of INCR1 on ISG expression are due to a common local mechanism of regulation of PD-L1 and JAK2. Regulation of JAK2 in turn leads to an indirect effect on STAT1 and the global interferon response. Together, these results provide evidence for a key role of INCR1 in the regulation of tumor IFNγ signaling.
Our T cell cytotoxicity studies showed that modulation of INCR1 expression on tumor cells also affects T cell functionality. Stimulation of CD8+ T cells co-cultured with a monolayer of INCR1 knockdown tumor cells resulted in increased secretion of IFNγ by T cells compared to CD8+ T cells co-cultured with control tumor cells, suggesting a more robust T cell activation. This was associated with an increased killing of INCR1 knockdown cells, compared to controls, by activated CD8+ T cells in multiple tumor cell types. One limitation may be the use of tumor cells growing in monolayer, which may not be reflective of physiological cell behavior. In fact, three-dimensional tumor cell cultures were shown to better mimic tumor biology in terms of signaling (Edmondson et al., 2014; Weiswald et al., 2015). Moreover, tumor cell response to therapy changes if cells are grown as tumor spheres compared to monolayer (Breslin and O’Driscoll, 2016; Mathews Griner et al., 2016). Therefore, we validated our results using a 3D system in which CD8+ T cell activity was evaluated when those cells were co-cultured with tumor spheres. We found that, compared to controls, INCR1 knockdown tumor spheres were more susceptible to CD8+ T cell mediated killing.
Inhibition of tumor immune evasion by targeting the IFNγ signaling is also associated with reduced expression of genes important for antitumor immunity, such as antigen presenting molecule MHC I, which in turn can potentially reduce immune cell function (Castro et al., 2018; Manguso et al., 2017). Although further studies will be required to determine the effect of INCR1 targeting as single agent on tumor immune composition and function in vivo, it has been shown that blocking tumor interferon signaling in combination with immunotherapy produces durable tumor response (Benci et al., 2016). Using a subcutaneous tumor model, we showed improved efficacy of CAR T cell therapy in tumors with knockdown of INCR1. CAR T cell therapy has demonstrated efficacy in different hematologic cancers but has had modest success in solid tumors (D’Aloia et al., 2018; Park et al., 2016). This is mostly due to immunosuppression and poor tumor penetration by T cells. Researchers have been trying different approaches to enhance the efficacy of CAR T therapy for solid tumors, including local delivery and multiple rounds of CAR T injection (Brown et al., 2016). However, the most commons side effect of CAR T cells is cytokine release syndrome (CRS), a systemic inflammation response caused by cytokines released by infused CAR T cells (Brudno and Kochenderfer, 2016). It has been shown that higher levels of CAR T cells in blood were associated with increased CRS grade (Hay et al., 2017; Lee et al., 2015; Porter et al., 2015). Thus, injection of a high dose of CAR T cells or multiple injections of low doses may produce increased toxicity. In our animal studies, the use of a single intravenous injection of a low dose of CAR T cells did not produce a significant effect on control tumors. However, the same low dose of CAR T cells significantly inhibited the growth of INCR1 knockdown tumors. Notably, analysis of CAR T cells in tumors showed higher PD-1 expression on CAR T cells infiltrating control tumors compared to those in tumors with silenced INCR1, suggesting a more exhausted phenotype (Freeman et al., 2006). Moreover, only CD8+ infiltrates were found in control tumors compared to INCR1 knockdown tumors in which both CD4+ and CD8+ infiltrates were detected. These results suggest that the observed increased CAR T cell efficacy is due to a more permissive microenvironment in INCR1 knockdown tumors, in which coexistence of CD4+ and CD8+ CAR T populations is important for long-term antitumor activity (Turtle et al., 2016; Wang et al., 2018).
In addition, our findings provide insights on the molecular mechanism through which INCR1 regulates ISG expression. Most lncRNAs with known functions have been shown to act through the binding of protein partners (Engreitz et al., 2016b; Ramos et al., 2015). Several of these lncRNAs interact with heterogeneous nuclear ribonucleoproteins (hnRNPs) to modulate different biological processes, including gene expression and RNA metabolism. For example, it has been shown that lincRNA-EPS interacts with HNRNPL to control inflammatory responses by suppressing the transcription of immune genes (Atianand et al., 2016). On the contrary, lincRNA-Cox2 activates gene expression after TLR signaling trough the binding of HNRNPA/B or A2/B1 (Carpenter et al., 2013). Our results identify HNRNPH1 as the protein partner of INCR1. This protein is mostly involved in pre-mRNA splicing and stability. Interestingly, we found that HNRNPH1 was also able to bind PD-L1 and JAK2 mRNA and it functions as a negative regulator of their expression. In response to IFNγ treatment, HNRNPH1 knockdown cells showed increased levels of PD-L1 and JAK2 mRNA and protein compared to control cells. This was in accordance with TCGA data from GBM patients showing an inverse correlation between HNRNPH1 and PD-L1 expression. These findings suggest that binding of HNRNPH1 to PD-L1 and JAK2 transcripts may result in changes in gene splicing and/or reduced mRNA stability, that in turn impairs protein production. Further studies will be required to determine the exact mechanisms through which HNRNPH1 counteracts PD-L1 and JAK2 expression. One possibility is that HNRNPH1 binding to those genes alter the proper pre-mRNA splicing thus promoting mRNA decay. It has been previously reported that HNRNPH1 can act as a splicing suppressing factor. For instance, HNRNPH1 binding to exon 7 of the TRF2 pre-mRNA suppresses splicing of this exon, thus preventing the production of the short isoform of TFR2 mRNA, that in turn results in the inhibition of neuronal differentiation (Grammatikakis et al., 2016). Moreover, HNRNPH1 binding to COLQ pre-mRNA antagonizes SRSF1 ability to splice exon 16. This produces a mutated form of COLQ that promotes acetylcholinesterase (AChE) deficiency in patients (Rahman et al., 2015). Finally, it has been shown that HNRNPH1 can also bind the pre-mRNA of U11–48K promoting the formation of an unstable splice variant and mRNA decay (Turunen et al., 2013).
Our results show that binding of INCR1, PD-L1 and JAK2 transcripts to HNRNPH1 was mutually exclusive. Moreover, silencing HNRNPH1 in INCR1 knockdown cells was sufficient to rescue PD-L1 expression levels in response to IFNγ treatment. These data suggested that INCR1 could bind HNRNPH1 to impair its interaction with PD-L1 and JAK2. As shown by Denzler and colleagues, this model of action would be plausible if INCR1 binding site abundance was similar to HNRNPH1 copy number (Denzler et al., 2014; Denzler et al., 2016). However, since INCR1 acts by regulating the neighboring genes PD-L1 and JAK2, measuring local concentration of HNRNPH1 at the INCR1 locus would be challenging due to the ubiquitous expression of HNRNPH1 in the nucleus. CLIP-seq analysis identified multiple HNRNPH1 binding sites in the proximal intron of INCR1 containing poly-G runs of varying length. We validated in vitro the presence of two main HNRNPH1 binding sites in the INCR1 transcript, whose binding affinity to HNRNPH1 was higher compared to PD-L1 and JAK2. Moreover, our binding competition experiment showed that INCR1 could affect the ability of HNRNPH1 to interact with PD-L1 and JAK2. Blocking the interaction between INCR1 and HNRNPH1 in vivo using chemically modified antisense oligonucleotides resulted in reduced PD-L1 and JAK2 protein levels. Thus, our studies identified the INCR1 primary transcript and its sequence that binds HNRNPH1 in response to IFNγ. INCR1 is co-transcribed with PD-L1 to impair HNRNPH1 function and allow PD-L1 and JAK2 expression (Figure 7). Therefore, INCR1 acts as a critical component of the tumor interferon signaling circuit with important implications in the modulation of immune checkpoints and tumor immune evasion.
Figure 7. INCR1 interaction with HNRNPH1 regulates PD-L1 expression and IFNγ signaling.
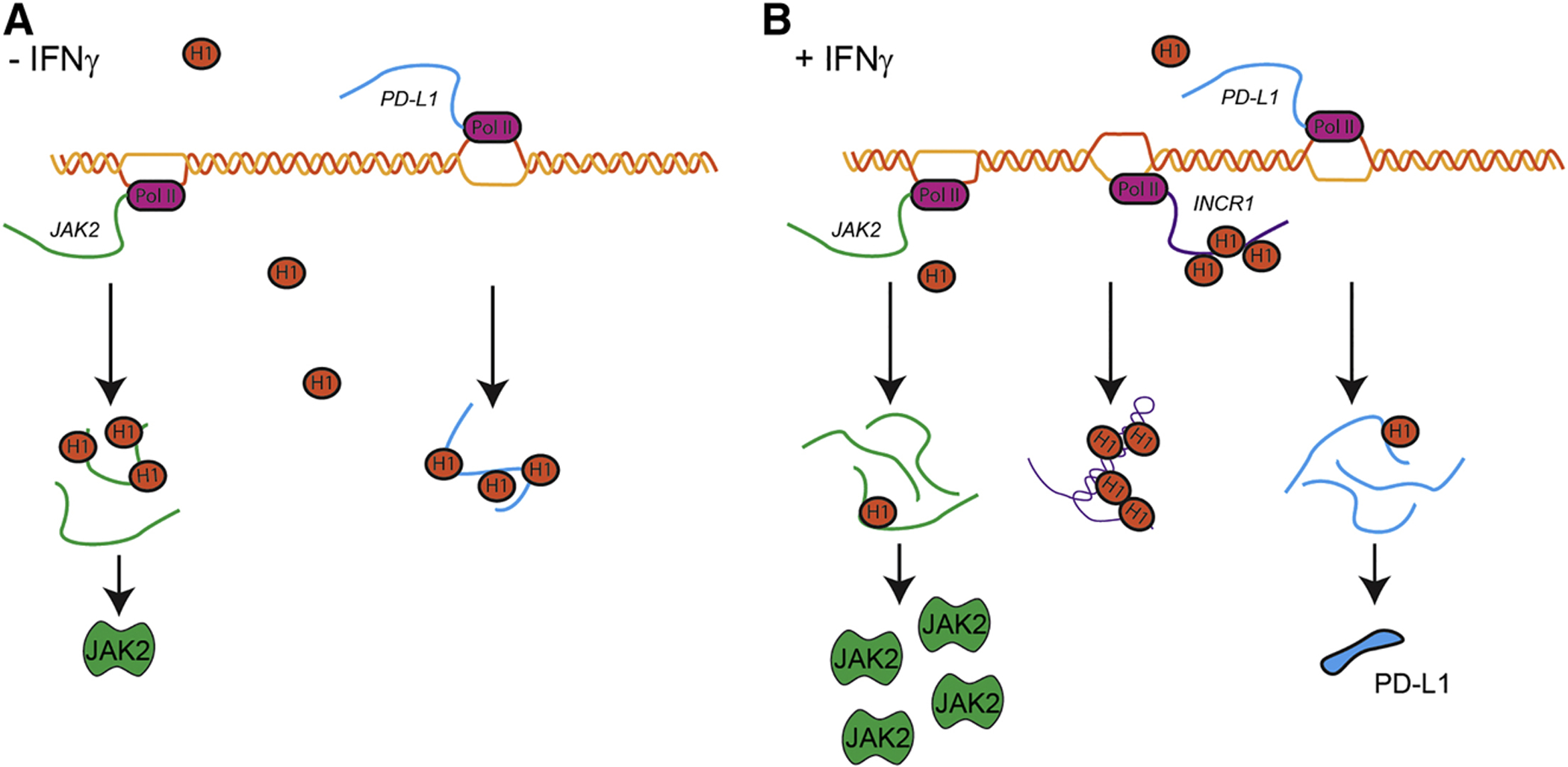
(A) In cells not stimulated with IFNγ, HNRNPH1 (H1) binds PD-L1 (blue) and JAK2 (green) transcripts to negatively regulate their expression.
(B) In response to IFNγ stimulation, INCR1 is transcribed from the PD-L1 locus. INCR1 binds HNRNPH1 to enable PD-L1 (blue) and JAK2 (green) expression and promote IFNγ-mediated immunosuppression.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, E. Antonio Chiocca (eachiocca@bwh.harvard.edu)
Materials Availability
All unique reagents generated in this study are available from the corresponding authors with a completed Material Transfer Agreement.
Data and Code Availability
The datasets generated during this study are available at Gene Expression Omnibus (GEO) GSE137489
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Male and female mice aged 6 to 8 weeks were used for all the experiments. NOD.Cg-Prkdcscid B2mtm1Unc Il2rgtm1Wjl/SzJ mice were purchased from The Jackson Laboratory. Animals were bred and maintained under pathogen-free condition at the BWH Center for Comparative Medicine, Brigham and Women’s Hospital. Mice studies were conducted according to the protocols approved by the Institutional Animal Care and Use Committee (IACUC).
Cell Lines
Patient-derived primary GBM cells (PDGCLs, BT cell lines) were generated as previously described (Stevens et al., 2016). U251 cells were obtained from the NCI-DTP. U1242 cells were obtained from James Van Brocklyn (Ohio State University). A375 cells were obtained from Frank Stephen Hodi (Dana-Farber Cancer Institute). H2122 and H1703 cells were obtained from Sandro Santagata (Brigham and Women’s Hospital). MDA-MB-231 cells were obtained from David Walt (Brigham and Women’s Hospital). BT cell lines were cultured as neurospheres in stem cell conditions using Neurobasal (Thermo Fisher Scientific) supplemented with Glutamine (Thermo Fisher Scientific), B27 (Thermo Fisher Scientific), 20 ng/ml epidermal growth factor (EGF) and fibroblast growth factor (FGF)-2 (PrepoTech). U251, U1242, A375, H2122 and H1703 cells were cultured in DMEM (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (FBS, Sigma-Aldrich) and 100 U/ml penicillin-streptomycin (Thermo Fisher Scientific). MDA-MB-231 cells were cultured in RPMI (Thermo Fisher Scientific) supplemented with 10% FBS and 100 U/ml penicillin-streptomycin. All cell lines were maintained in humidified 5% CO2 incubator at 37°C.
METHOD DETAILS
Cell culture and transfection
Unless otherwise specified, IFNγ (PeproTech) stimulation was performed at 100 U/ml IFNγ for a period of 24 h. IFNβ and TNFα were obtained from PeproTech. Stable U251, A375 and MDAMB-231 knockdown were obtained by infecting cells with shRNA 1 (clone: CS-SH128T-3-LVRU6GP; target sequence: GCCATTGCAGGAAATATAAGA, GeneCopoeia) and shRNA 2 (clone: CS-SH128T-6-LVRU6GP; target sequence: CAGCTCTCAATTCTGTGAAACTCAA, GeneCopoeia). LAN GapmeRs (Exiqon) knockdown experiments were performed transfecting BT cells with 50 nM of GapmeR (TTACATGATGACCTTT) using Lipofectamine 2000 (Thermo Fisher Scientific). CRISPRi experiments were performed by transfecting dCAS9-KRAB expressing patient-derived cells with sgRNA control or sgRNA targeting the INCR1 promoter. Stable U251-EGFRvIII were obtained by infecting cells with pLV-IRES-mCherry-EGFRvIII vector. STAT1 and HNRNPH1 knockdown was performed transfecting 50 pmol/well of Duplex siRNAs (hs.Ri.STAT1.13.1 and hs.Ri.STAT1.13.2; hs.Ri.HNRNPH1.13.1 and hs.Ri.HNRNPH1.13.2, Integrated DNA Technologies) for 6 well plate using Lipofectamine RNAiMAX (Thermo Fisher Scientific). HNRNPH1 binding site blocking experiments were performed transfecting cells with 100 nM of fully 2’-O-Methoxyethyl (2’-MOE) and phosphorothioate bond modified antisense oligonucleotide control (ASO NC, GCGACTATACGCGCAATATG) or targeting HNRNPH1 binding site on the INCR1 gene (ASO H1B, CTCCAGCTCCCCCCGGCAAC) (Integrated DNA Technologies).
Human specimens
Tumor tissue samples were obtained as approved by the Institutional Review Board (IRB) at the Dana-Farber Cancer Institute. Patient samples were processed for extraction of total RNA.
RNA-Seq and analysis of RNA-Seq data
BT333, U251 shControl and U251 shINCR1 cells were stimulated with IFNγ for 24 h. RNA was extracted using RNeasy kit (QIAGEN). RNA sequencing on BT333 patient derived cell lines was performed by Admera Health. 1μg of total RNA from unstimulated and IFNγ-stimulated cells was used and ribosomal RNA was removed using Ribo-Zero rRNA Removal kit (Illumina). RNA libraries were prepared using NEBNext Ultra II RNA Library Prep Kit for Illumina (New England BioLabs). Paired-end reads were sequences on a HiSeq System (Illumina) to achieve at least 40 million reads per sample. RNA sequencing on INCR1 knockdown U251 cells was performed by Psomagen. 1μg of total RNA from unstimulated and IFNγ-stimulated cells was used to prepare RNA libraries using TruSeq Stranded mRNA Library Kit (Illumina). Paired-end reads were sequences on a NovaSeq6000 System (Illumina) to achieve at least 40 million reads per sample. Libraries prepared from three independent experiments were analyzed. Raw reads were examined for quality issues using FastQC. Trimmed reads were aligned to the UCSC build 38 of the human genome (h38), augmented with transcript information from Ensembl release GRCh38 using STAR. Count of reads aligning known genes was generated by HT-Seq. Differential expression at the gene level was calculated with DESeq. To identify lncRNAs associated with INCR1 expression, we used PARIS algorithm, a Genepattern module that uses a mutual information-based metric (RNMI) to rank genetic features based on the degree of correlation to the target profile (INCR1).
5’ Rapid amplification of cDNA ends
5 μg of total RNA was incubated with 1 μl of 50 ng/μl GSP-RT primer in a total volume of 12 μl. The mixture was incubated at 80°C for 3 minutes and then placed on ice. To this mixture, 4 μl of 5x buffer, 1 μl of 10 mM dNTPs, 2 μl of 0.1 M DTT and 1 μl of superscript III (Invitrogen) was added. This mixture was incubated at 42°C for 1 hour and then at 50°C for 10 minutes. The reverse transcriptase was inactivated by heating at 70°C for 15 minutes and then the RNA template was destroyed by adding 0.75 μl of RNAse H (NEB) for 20 minutes at 37°C. This reaction mixture was diluted to 400 μl with TE and then purified using QiaQuick PCR cleanup column (Qiagen). A polyadenosine tail was added using terminal deoxytransferase (NEB) by mixing 30 μl of cDNA with 5 μl of 10X buffer, 2.5 μl of CoCl2 (2.5 M), 1 μl of dATP (10 mM) and 1 μl of Tdt, 10.5 μl of dH2O and incubating at 37°C for 5 minutes. The reaction was inactivated by heating to 65°C for 10 minutes. The reaction was diluted to 500 μl with TE. For amplification by PCR, 1 μl of tailed cDNA was mixed with 25 pmol of 5’GSP1 primer, QO primer and QT primer and amplified with Accuprime G-C rich Polymerase (Invitrogen). Upon completion, the PCR reaction was diluted 1:20 and 1 μl was used in a PCR reaction using 25 pmol of 5’GSP2 and QI primers with Accuprime G-C Rich polymerase. PCR amplicons were cleaned up with Qiagen PCR cleanup kit and were directly sequenced. The primers used for 5’ Rapid amplification of cDNA ends are listed in Table S8.
3’ Rapid amplification of cDNA ends
cDNA was generated from 5 μg of RNA according to manufacturer’s instructions using Superscript III (Invitrogen) and QT primer. RNA template was destroyed by adding 0.75 μl of RNAse H (NEB) for 10 minutes at 37°C. The reaction mixture was diluted to 500 μl. For amplification of INCR1 3’ end, PCR reactions were carried out with 1 μl of cDNA using 25 pmol of 3’GSP1 and QO primer with Accuprime G-C Rich polymerase (Invitrogen). Upon completion, this reaction mixture was diluted 1:20 and 1 μl was used for a second round of amplification with 3’GSP2 and QI Primer. The resultant amplicons were cleaned up with Qiagen PCR cleanup kit and directly sequenced. The primers used for 3’ Rapid amplification of cDNA ends are listed in Table S8.
Determination of Somatic Copy Number Alterations
Somatic copy-number alterations (SCNAs) were determined from whole-exome sequencing data (PDGCLs) and SNP 6.0 Affymetrix microarray data (LTGCLs and TCGA samples) using Genomic Identification of Significant Targets in Cancer (GISTIC 2.0) (Mermel et al., 2011). Copy number calls for GBM cell lines (PDGCLs and LTGCLs) are described in Ferrer-Luna R. et al. manuscript under preparation. In brief, SNP array data from the LTGCLs and 187 normal control samples were downloaded from the CCLE data portal (http://www.broadinstitute.org/ccle/data) on 29 September 2012 and analyzed using the GenePattern Copy Number Inference pipeline (Beroukhim et al., 2010) to generate raw copy-number estimates. These estimates were further refined using Tangent normalization (Beroukhim et al., 2010) against 3,000 normal samples profiled by TCGA. Copy-number calls were made using circular binary segmentation (CBS) (Snijders et al., 2001). Copy-number calls for TCGA samples were generated as previously described (Brennan et al., 2013; Zack et al., 2013). Copy-number calls were made from PDGCLs using ReCapSeg (https://gatkforums.broadinstitute.org/gatk/categories/recapseg-documentation). For cell lines (PDGCLs and LTGCLs), GISTIC 2.0 algorithm was calculated at gene level using next parameters: Amplification/Deletion threshold = 0.3, joint segment size = 10, Significance threshold (q-value) = 0.25. For each gene, GISTIC provided the following SCNA calls: High Level Gain (Amplifications), Low Level Gains, Low Level Loss, and High Level Loss (Homozygous Deletions). Amplifications and Low Level Gains represent copy-number log2ratios of >0.9 and between 0.9 and 0.3, respectively; Homozygous Deletions and Low Level Losses represent copy-number ratios of <−1.3 and between −1.3 and 0.3, respectively.
Quantitative Real-Time PCR analysis
Total RNA from cell lines and patients’ tissues was extracted using TRIzol (Thermo Fisher Scientific). Nuclear/cytoplasmic fractionation was performed as previously described (Mineo et al., 2016). RNA was reverse-transcribed using iScript cDNA Synthesis Kit (BioRad) and quantitative real-time PCR was performed using SYBR Green Master Mix (Applied Biosystem). 18S expression levels were used as control. For copy number analysis, absolute quantification of INCR1 and PD-L1 RNA was performed using the standard-curve method. The primers used in this study are listed in Table S8.
Immunoblot analysis and antibodies
Immunoblotting was performed as previously described (Mineo et al., 2016). The following antibodies were used: anti-PD-L1, anti-IDO, anti-JAK2, anti-STAT1, anti-phospho-STAT1 and anti-β-Actin (13684, 86630, 3230, 9172, 9167 and 3700, respectively, Cell Signaling Technology); anti-hnRNP-H (A300–511A, Bethyl Laboratories).
In vitro transcription/translation assay
In vitro transcription and translation was performed using TnT Quick Coupled Transcription/Translation System and Transcend Non-Radioactive Translation Detection System (Promega) following the manufacturer’s instructions. The lncRNA was transcribed from a T7 promoter of a pcDNA3.1 plasmid (Thermo Fisher Scientific). The pSP64-Luciferase plasmid supplied with the TnT Quick Coupled Transcription/Translation System and a GFP cloned in pcDNA3.1 plasmid were used as a positive control.
Immunofluorescence
Cells were starved overnight and treated with 100 U/ml IFNγ for 3 h. Cells were fixed with 4% paraformaldehyde for 10 min, permeabilized for 5 min with 0.3% Triton-X100, blocked with 2% BSA for 1 h. Cells were incubated with STAT1 antibody (14994, Cell Signaling Technology) overnight after which they were incubated for 1 h at RT to secondary anti-rabbit AlexaFluor 488 (Jackson ImmunoResearch), followed by incubation with Hoechst 33342 (Thermo Fisher Scientific) for 30 min. Confocal images were acquired with ZEN software on a Zeiss LSM 710 Confocal system (Carl Zeiss Inc.).
CD8+ T cell isolation
Peripheral blood mononuclear cells (PBMCs) were obtained from healthy human donors as approved by the IRB at the Brigham and Women’s Hospital. PBMCs were isolated using Ficoll Paque Plus (GE Healthcare Life Sciences) following the manufacturer’s instructions. CD8+ T cells were isolated by negative selection using the CD8+ T Cell Isolation Kit (Miltenyi Biotec). Isolation was performed according to the manufacturer’s recommendations.
Flow cytometry
Cells were harvested at the indicated time-points and washed with FACS buffer (PBS supplemented with 2% FBS). Cells were stained incubating with the indicated antibodies diluted in FACS buffer. After staining cells were fixed 2% paraformaldehyde (PFA). Flow cytometry was performed on a BD LSR II (BD Biosciences) and data analyzed using FlowJo. The following antibodies were used: anti-CD274, anti-PD1, anti-CD8, anti-CD4, anti-CD3 (329706, 329919, 344710, 317442, 300407 BioLegend); LIVE/DEAD Fixable Near-IR Dead Cell Stain Kit (L10119, Thermo Fisher Scientific).
ELISA assay
Control and INCR1 knockdown tumor cells were co-cultured at a 1:1 ratio with non-stimulated CD8+ T cells or CD8+ T cells stimulated with Dynabeads Human T-Activator CD3/CD28 (Thermo Fisher Scientific). Culture media was collected and secreted levels of IFNγ were analyzed at 48 h using Human IFNγ ELISA MAX Deluxe (BioLegend). The absorbance was read at 450 nm using a microplate reader. Secreted IFNγ was quantified based on the standard curve
T cell cytotoxicity assay
For 2D assays, control or INCR1 knockdown tumor cells were co-cultured for 96 h at a 1:1 ratio with non-stimulated CD8+ T cells or CD8+ T cells stimulated with Dynabeads Human T-Activator CD3/CD28 (Thermo Fisher Scientific) and 10 ng/ml interleukin-2 (PeproTech). T cells were then removed washing with PBS. Cell viability was determined using Muse count and viability kit (Millipore). Alternatively, after incubation, tumor cells were harvested and stained using LIVE/DEAD staining. Percent of dead cells was determined by flow cytometry. For 3D assays, 750 GFP positive control or INCR1-knockdown tumor cells were seeded in a round bottom low-attachment 96 well plate. Cells were allowed to form tumorspheres for 72h. After tumorspheres were formed, two thousand non-stimulated CD8+ T cells or CD8+ T cells stimulated with Dynabeads Human T-Activator CD3/CD28 and 10 ng/ml interleukin-2 were added. Tumorspheres and CD8+ T cells were co-cultured for 96 h and changes in GFP intensity were measured using ImageJ.
In vitro T cells transduction
Generation of T cells expressing chimeric antigen receptor (CAR) against EGFRvIII is described in Khalsa JK et al., manuscript under preparation. In brief, the EGFRvIII CAR was constructed as described previously (Johnson et al., 2015) using the self-inactivating lentiviral transfer vector pRRL.PPT.EFS bearing an IRS-GFP cassette and packaged as described previously (Shah et al., 2008). pRRL.PPT.EFS-GFP vector served as control. T cells were isolated from PBMCs by EasySep Human T cell isolation kit (Stem Cell Technology). Isolated T cells were counted and cultured at 1:1 ratio with Dynabeads human T activator CD3/CD28 (Thermo Fisher Scientific) in X-vivo15 medium supplemented with 30U/ml IL-2. Next day, 1.5 million T cells/ml were transduced with EGFRvIII-CAR or control lentivirus at MOI 10 and 6μg/ml polybrene in 6 well plates. Medium was replaced next morning and GFP expression was checked 48 hours post-infection. Before injecting T cells in mice, the ability of EGFRvIII-specific CAR T cells to kill target cells was tested in vitro by 3D T cell cytotoxicity assay.
In vivo studies
2.5 × 106 U251 EGFRvIII shControl or shINCR1 were injected subcutaneously into 6- to 8-week-old male and female NSG mice (Jackson), with 6 mice per group (n=2 males and n=4 females). Seven days after tumor implantation, 1 × 106 CAR T cells or GFP transduced T cells were injected intravenously via tail vein in 200 μl of PBS. Tumor size was measured by calipers in three dimensions, LxWxH, for the duration of the experiments. Tumor growth was followed for 4 weeks, or until predetermined IACUC-approved endpoint was reached. Twenty-one days post CAR T cell injection, tumors were resected and tissues digested using Tumor Dissociation Kit (Miltenyi Biotec) according to the manufacturer protocol. Tumor single cell suspension was then processed immediately to isolate tumor infiltrating CAR T cells using Percoll density gradient medium (GE Healthcare). Tumor cell suspension was mixed with 70% Percoll solution, then a layer of 37% Percoll was added on top of the suspension. Samples were centrifugated at 500 g for 40 min at room temperature. Mononuclear cell layer was transferred to a separate tube and washed with PBS. Cells were used immediately for flow cytometry analysis.
RNA antisense purification (RAP)
RAP was performed as previously described with some modifications (McHugh et al., 2015). For RAP-MS (Mass Spectrometry), BT cells were stimulated with IFNγ and nuclear extracts incubated with biotinylated probes against INCR1 (see Table S8) at 67°C for 2 h. Scrambled biotinylated probe was used as control. RNA was purified using streptavidin agarose beads (Thermo Fisher Scientific). Co-purified proteins were analyzed by Mass Spectrometry as previously described (Mineo et al., 2016). For RAP-RNA, proteins were digested using Proteinase K (Thermo Fisher Scientific) and RNA was extracted using TRIzol.
UV-crosslink RNA immunoprecipitation
UV-crosslink RNA immunoprecipitation assay was performed as previously described with some modifications (Mineo et al., 2016). Briefly, cells were UV irradiated at 400 mJ/cm2 and nuclear extracts were prepared by incubating cells in RLN Buffer (50 mM Tris, 1.5 mM MgCl2, 150 mM NaCl, 0.5% NP-40, protease inhibitors) for 5 min. Nuclei were pelleted by centrifuging at 1,450 × g for 2 min and lysed for 10 min in CLIP Buffer (50 mM Tris, 150 mM NaCl, 1% NP-40, 0.1% Sodium Deoxycholate, phosphatase and protease inhibitors, 100 U/ml RNase inhibitor [New England BioLabs]). Samples were sonicated with microtip, 5 watts power (25% duty) for 60 seconds total in pulses of 1 second on followed by 3 seconds off. DNA was digested incubating samples for 15 min at 37°C in 1X DNase salt solution (2.5 mM MgCl2, 0.5 mM CaCl2) with 30 U TurboDNase. EDTA was added to the samples to a final concentration of 4 mM and samples centrifuged at 16,000 × g for 10 min. Nuclear extracts were precleared with Protein A/G Plus Agarose beads (Thermo Fisher Scientific) and incubated with primary antibody (anti-hnRNP-H) or rabbit IgG control (Bethyl Laboratories) overnight at 4°C. Protein/RNA complexes were precipitated using Protein A/G Plus Agarose beads (Thermo Fisher Scientific). Beads were washed and incubated with Proteinase K (Thermo Fisher Scientific) and RNA was extracted using TRIzol.
Enhanced CLIP (eCLIP)
A375 cells were stimulated with IFNγ for 6 h and UV irradiated at 400 mJ/cm2. eCLIP was performed by EclipseBioInnovations as previously described (Van Nostrand et al., 2016).
Expression and purification of HNRNPH1
HNRNPH1 isoform A was cloned in pET21-His-Smt3 and protein expressed by transformation of Rosetta-2 (DE3) pLys(S) E. coli (EMD Millipore). Cells were lysed in 20 ml of 50 mM Tris [pH 8.0], 300 mM KCl, 0.02% NP-40, 10 mM Imidazol, 10% Glycerol, 0.1 mM EDTA, 0.1 mM DTT, 0.1 μg/μl lysozyme. Re-suspended cells were incubated on ice for 20 min. Cells were further disrupted and DNA was sheared by sonication (3, 20 second bursts with 20 seconds rests). Insoluble material was pelleted by centrifugation (30 min at 20,000 × g at 4°C). Soluble material was decanted. Insoluble pellet was resolubilized in 50 mM Tris [pH 8.0], 300 mM KCl, 0.02% NP-40, 10 mM Imidazol, 10% Glycerol, 0.1 mM EDTA 0.1 mM DTT, 6 M Urea followed by sonication. Remaining insoluble material was pelleted by centrifugation (30 min at 20,000 × g at 4°C). Soluble material was decanted to new tube. Expression was analyzed by Coomassie staining. 1 ml of TALON resin (Clontech) was equilibrated in respective lysis buffers and added to lysates. Beads and lysates were tumbled at 4°C for 2 h. Beads were washed 2 times in 50 ml lysis buffer and loaded onto column. Column was washed with 10 ml of lysis buffer and then eluted in Lysis buffer containing 300 mM Imidazole. 10 fractions of 1.5 mL were collected and analyzed by Coomassie staining. Protein purified under denaturing conditions was dialyzed against 50 mM Tris [pH 8.0], 300 mM KCl, 0.02% NP-40, 10 mM Imidazole, 10% Glycerol, 0.1 mM EDTA 0.1 mM DTT, 4 M Urea overnight. The following day, protein was dialyzed against 50 mM Tris [pH 8.0], 300 mM KCl, 0.02% NP-40, 10 mM Imidazole, 10% Glycerol, 0.1 mM EDTA 0.1 mM DTT, 2 M Urea for 4 h and then 50 mM Tris pH 8.0, 300 mM KCl, 0.02% NP-40, 10 mM Imidazole, 10% Glycerol, 0.1 mM EDTA 0.1 mM DTT, 1 M Urea for 4 h. Finally, the protein was dialyzed against 50 mM Tris [pH 8.0], 300 mM KCl, 0.02% NP-40, 10% Glycerol, 0.1 mM EDTA 0.1 mM DTT, 0 M Urea overnight. Dialyzed protein was clarified by centrifugation (30 min at 20,000 × g at 4°C). Purity of protein was analyzed by Coomassie staining.
Biotinylated RNA pulldown assay
Genomic DNA was extracted from cell cultures to generate amplicons corresponding the 5’ and 3’ ends of the INCR1 intron 1. PCR products were cloned in pCR2.1-TOPO (Thermo Fisher Scientific) to generate pCR2.1-INCR1 Intron 1 (5’ half) and pCR2.1-INCR1 Intron 1 (3’ half). PCR products from pCR2.1-INCR1 Intron 1 (3’ half) were generated to add HindIII linkers to the 3’ end and this fragment was cloned between SpeI and HindIII in pCR2.1-INCR1 Intron1 (5’ half) to generate a pCR2.1-INCR1 minigene. In vitro transcripts of biotinylated RNA were generated by PCR and numbered fragment 1 – 7 in a 5’ to 3’ direction. Each fragment allowed for transcription of a 300 nucleotide RNA, each with a 50 nucleotide overlap to the adjacent fragment. T7 promoter sequence was added by PCR. In vitro transcription reactions were performed using T7 HiScribe (New England Biolabs) according to manufacturer’s instructions, except the final concentration of UTP was reduced to 7.5 μM and instead supplemented with 2.5 μM Biotin-16-UTP (Sigma Aldrich). Transcribed RNAs were extracted by acidic phenol chloroform extraction (Thermo Fisher Scientific) and precipitated with ammonium acetate. Unincorporated nucleotides from resuspended RNAs were removed by gel filtration chromatography through Illustra Microspin G-25 columns (GE Healthcare). Concentrations of each RNA was brought to 4 μM with DEPC-treated H2O (Thermo Fisher Scientific). 1 μl of 4 μM biotinylated in vitro transcribed RNA was added to cell lysates and protein complexes allowed to assemble for 2 h at 4°C. After incubation, 10 μl of streptavidin-agarose (Thermo Fisher Scientific) were added and tumbled for an additional hour. Beads were washed 4 times with lysis buffer and complexes were eluted with 2x SDS loading buffer. Eluted proteins were resolved on 4 – 20% gradient gel (Bio-Rad) and assayed by western blotting. For RNA pulldown assays with blocking oligos, prior to preforming pulldown assay, 4 pmol of biotinylated RNA was incubated with indicated amount of blocking oligo in 20 μl of binding buffer (10 mM Tris [pH 7.9], 50 mM NaCl, 10 mM MgCl2 1 mM DTT). RNA/oligo mixture was incubated at 90°C for 5 minutes and allowed to cool to room temperature for 30 minutes to facilitate annealing. The primers used in this assay are listed in Table S8.
RNA electrophoretic mobility shift assay
Synthetic RNA was obtained from IDT and radiolabeled with γ−32P ATP (6000 Ci/mmol, Perkin-Elmer) using T4 Polynucleotide Kinase (New England Biolabs). Unincorporated nucleotides were removed by gel filtration chromatography through Illustra Microspin G-25 columns (GE Healthcare). RNA/protein complexes were allowed to form at room temperature by adding indicated amount of protein to 1 pmol of radiolabeled RNA in 20 μl reaction containing 50 mM Tris [pH 8.0], 300 mM KCl, 0.02% NP-40, 10% Glycerol, 0.5 μg/μl Heparin for 10 minutes. Complexes were loaded onto native polyacrylamide gels and ran for 3 h at 150 V. Gels were dried and visualized by autoradiography. The synthetic RNAs used in this assay are listed in Table S8.
Microscale thermophoresis
MicroScale Thermophoresis experiments were performed according to the NanoTemper technologies protocol in a Monolith NT.115Pico (red/blue) instrument (NanoTemper Technologies). Serial dilutions of HNRNPH1 were done using a buffer containing 50 mM Tris [pH 8.0], 300 mM KCl, 0.02% NP-40, 10% Glycerol, and 0.5 mg/ml Heparin. RNA oligos were labelled with a FAM moiety at their 3’ ends (IDT). The RNA concentration was kept constant at 20 nM throughout the experiments. The RNA-protein mixture was incubated at room temperature for 15 mins before running into the MST instrument. The experiments were performed using 40% and 60% MST power and between 20–80% LED power at 22 °C. The MST traces were recorded using the standard parameters: 5 s MST power off, 30 s medium MST power on and 5 s MST power off. The reported measurement values are the combination of two effects: the fast, local environment dependent responses of the fluorophore to the temperature jump and the slower diffusive thermophoresis fluorescence changes. The data presented here are the average of 3 independent experiments. Average normalized fluorescence (%) was plotted against HNRNPH1 concentration to determine the binding constant (Kd). Ligand depletion model with one binding site was used (Using GraphPad Prism 8) to fit the binding which follows the following model:
Y = Bmax*X/(Kd + X). The synthetic RNAs used in this assay are listed in Table S8.
QUANTIFICATION AND STATISTICAL ANALYSIS
Graphs were generated and statistical analysis was performed using Prism (GraphPad). Statistical details of experiments, including number of experiments, statistical test and statistical significance (p value) are reported in the figure legends. Independent experiments were performed to define the reproducibility of the results.
Supplementary Material
Table S1. RNA-seq analysis of lncRNA expression in control and IFNγ-stimulated BT333 cells. Related to Figure 1
Table S2. LncRNAs correlated and anti-correlated with INCR1. Related to Figure 1
Table S3. Coding genes neighbor of lncRNAs correlated with INCR1. Related to Figure 1
Table S4. Patient demographics and MGMT promoter methylation status of PDGCLs. Related to Figure 2
Table S5. RNA-seq analysis of gene expression in control and IFNγ-stimulated U251 shControl and shINCR1 cells. Related to Figure 3
Table S6. Mass spectrometry data of proteins identified in the INCR1 RAP but not in control RAP. Related to Figure 5
Table S7. Genes and binding sites identified by HNRNPH1 eCLIP-seq. Related to Figure 5
Table S8. Oligonucleotides Used. Related to STAR Methods
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-PD-L1 | Cell Signaling Technology | Cat# 13684; RRID: AB_2687655 |
| Anti-IDO | Cell Signaling Technology | Cat# 86630; RRID: AB_2636818 |
| Anti-JAK2 | Cell Signaling Technology | Cat# 3230; RRID: AB_2128522 |
| Anti-STAT1 (WB) | Cell Signaling Technology | Cat# 9172; RRID: AB_2198300 |
| Anti-STAT1 (IF) | Cell Signaling Technology | Cat# 14994; RRID: AB_2737027 |
| Anti-STAT1, phospho (Tyr701) | Cell Signaling Technology | Cat# 9167; RRID: AB_561284 |
| Anti-β-Actin | Cell Signaling Technology | Cat# 3700; RRID: AB_2242334 |
| Anti-hnRNP-H | Bethyl | Cat# A300–511A; RRID: AB_203269 |
| PE anti-human CD274 | BioLegend | Cat# 329706; RRID: AB_940368 |
| Brilliant Violet 421™ anti-human CD279 | BioLegend | Cat# 329919; RRID: AB_10900818 |
| PerCP/Cyanine5.5 anti-human CD8 | BioLegend | Cat# 344710; RRID: AB_2044010 |
| Brilliant Violet 785™ anti-human CD4 | BioLegend | Cat# 317442; RRID: AB_2563242 |
| PE anti-human CD3 | BioLegend | Cat# 300407; RRID: AB_314061 |
| Sheep Anti-Mouse | GE Healthcare | Cat# NA931; RRID: AB_772210 |
| Donkey Anti-Rabbit | GE Healthcare | Cat# NA934; RRID: AB_772206 |
| Alexa Fluor 488 AffiniPure Donkey Anti-Rabbit IgG | Jackson ImmunoResearch | Cat# 711-545-152; RRID: AB_2313584 |
| Bacterial and Virus Strains | ||
| One Shot™ TOP10 Chemically Competent E. coli | Thermo Fisher Scientific | Cat# C404010 |
| Rosetta 2(DE3) pLysS Competent Cells | Millipore | Cat# 71403-M |
| Biological Samples | ||
| Human brain tumor tissues | Brigham and Women’s Biorepository Core | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant Human IFN-γ | PeproTech | Cat# 300–02 |
| Recombinant Human IFN-β | PeproTech | Cat# 300–02BC |
| Recombinant Human TNF-α | PeproTech | Cat# 300–01A |
| Recombinant Human IL-2 | PeproTech | Cat# 200–02 |
| Trizol | Thermo Fisher Scientific | Cat# 15596018 |
| Critical Commercial Assays | ||
| Lipofectamine 2000 | Thermo Fisher Scientific | Cat# 11668019 |
| Lipofectamine RNAiMAX | Thermo Fisher Scientific | Cat# 13778500 |
| iScript cDNA Synthesis kit | Bio-Rad | Cat# 1708891 |
| Power SYBR Green PCR Master Mix | Thermo Fisher Scientific | Cat# 4368577 |
| RNeasy Mini Kit | Qiagen | Cat# 74104 |
| TnT Quick Coupled Transcription/Translation System | Promega | Cat# L1171 |
| LIVE/DEAD Fixable Near-IR Dead Cell Stain Kit | Thermo Fisher Scientific | Cat# L10119 |
| Dynabeads Human T-Activator CD3/CD28 | Thermo Fisher Scientific | Cat# 11161D |
| Muse Count & Viability Kit | Millipore | Cat# MCH100102 |
| DsiRNA STAT1–1 | Integrated DNA Technologies | hs.Ri.STAT1.13.1 |
| DsiRNA STAT1–2 | Integrated DNA Technologies | hs.Ri.STAT1.13.2 |
| DsiRNA HNRNPH1–1 | Integrated DNA Technologies | hs.Ri.HNRNPH1.13.1 |
| DsiRNA HNRNPH1–2 | Integrated DNA Technologies | hs.Ri.HNRNPH1.13.2 |
| SuperScript III Reverse Transcriptase | Thermo Fisher Scientific | Cat# 18080044 |
| QIAquick PCR Purification Kit | Qiagen | Cat# 28104 |
| Ficoll Paque Plus | GE Healthcare | Cat# 17-1440-02 |
| CD8+ T Cell Isolation Kit, human | Miltenyi Biotec | Cat# 130-096-495 |
| ELISA MAX Deluxe Set Human IFN-γ | BioLegend | Cat# 430104 |
| EasySep Human T Cell Isolation Kit | Stemcell Technologies | Cat# 17951 |
| Tumor Dissociation Kit, mouse | Miltenyi Biotec | Cat# 130-096-730 |
| HiScribe T7 High Yield RNA Synthesis Kit | NEB | Cat# E2040S |
| Microspin G-25 Columns | GE Healthcare | Cat# 27-5325-01 |
| Deposited Data | ||
| Raw and analyzed sequencing data | This study; See Table S1, Table S5, Table S7 | GSE137489 |
| Experimental Models: Cell Lines | ||
| Patient derived BT cell lines | Ligon Lab | https://www.dana-farber.org/research/departments-centers-and-labs/integrative-research-centers/center-for-patient-derived-models/ |
| Human: U1242 cells | Van Brocklyn Lab | N/A |
| Human: A375 cells | Hodi Lab | N/A |
| Human: H2122 cells | Santagata Lab | N/A |
| Human: H1703 cells | Santagata Lab | N/A |
| Human: MDA-MB-231 cells | Walt Lab | N/A |
| Human: U251 cells | NCI-DTP | Cat# U-251; RRID: CVCL_0021 |
| Human: CAR T cells | Shah Lab | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: NOD.Cg-B2mtm1Unc Prkdcscid Il2rgtm1Wjl/SzJ | The Jackson Laboratory | JAX: 010636 |
| Oligonucleotides | ||
| Please refer to Table S8 for details. | This study | N/A |
| Recombinant DNA | ||
| pcDNA3.1 | Thermo Fisher Scientific | Cat# V79020 |
| pcDNA3.1-INCR1 | This study | N/A |
| pcDNA3.1-INCR1 mini-gene | This study | N/A |
| pCR2.1-TOPO | Thermo Fisher Scientific | Cat# K450002 |
| pET21-His-Smt3 | Lyons Lab | N/A |
| psi-LVRU6GP-shINCR1–1 | GeneCopoeia | CS-SH128T-6-LVRU6GP |
| psi-LVRU6GP-shINCR1–2 | GeneCopoeia | CS-SH128T-3-LVRU6GP |
| pLV-IRES-mCherry-EGFRvIII | Shah Lab | N/A |
| pRRL.PPT.EFS-GFP | Shah Lab | N/A |
| pHR-SFFV-KRAB-dCas9-P2A-mCherry | Gilbert et al., 2014 | Addgene Plasmid # 60954 |
| lentiGuide-Puro | Sanjana et al., 2014 | Addgene Plasmid # 52963 |
| Software and Algorithms | ||
| Copy Number Inference pipeline | Beroukhim et al., 2010 | https://genepattern.broadinstitute.org |
| GISTIC2.0 | Mermel et al., 2011 | https://genepattern.broadinstitute.org |
| Circular Binary Segmentation | Snijders et al., 2001 | https://genepattern.broadinstitute.org |
| ReCapSeg | GATK | https://gatkforums.broadinstitute.org/gatk/categories/recapseg-documentation |
| GISTIC2.0 | Mermel et al., 2011 | https://genepattern.broadinstitute.org |
| GenePattern | Reich et al., 2006 | https://genepattern.broadinstitute.org |
| Fiji - ImageJ | https://imagej.net/Fiji | RRID: SCR_002285 |
| FlowJo | https://www.flowjo.com/ | RRID: SCR_008520 |
| Graphpad prism version 8 | https://www.graphpad.com/scientific-software/prism/ | RRID: SCR_002798 |
| ZEN Digital Imaging for Light Microscopy | http://www.zeiss.com/microscopy/en_us/products/microscope-software/zen.html#introduction | RRID: SCR_013672 |
| Other | ||
| Copy number LTGCL dataset (CCLE, 29-Sept-2012) | Cancer Cell Line Encyclopedia project | https://portals.broadinstitute.org/ccle |
| Somatic genomic alterations of GBM | The Cancer Genome Atlas project | https://tcga-data.nci.nih.gov/docs/publications/gbm_2013/ |
Highlights.
INCR1 is expressed from the PD-L1 locus in interferon stimulated tumor cells
INCR1 regulates tumor interferon signaling
Silencing INCR1 sensitizes tumor cells to T cell-mediated killing
INCR1 binds HNRNPH1 to promote PD-L1 and JAK2 expression
ACKNOWLEDGMENTS
This work was supported by NIH 2P01CA163205 and CA069246-20 (to E.A.C.), the Brigham Research Institute microgrant (to M. M.), NIH K99GM124458 (to S.M.L.), NIHR01GM126150 (to P.I.), NIH R35GM126901 (to P.J.A.), Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) - Research Fellowship Grant No. 400975596 (to N.vS.), the American Brain Tumor Association Basic Research Fellowship (to C.P.), the Howard Hughes Medical Institute Medical Research Fellow Program (to A.G.L.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
M.M. and E.A.C. are inventors on a patent application covering the use of INCR1 as therapeutic and diagnostic target.
REFERENCES
- Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, Hui R, Yokoi T, Chiappori A, Lee KH, de Wit M, et al. (2017). Durvalumab after Chemoradiotherapy in Stage III Non-Small-Cell Lung Cancer. N Engl J Med 377, 1919–1929. [DOI] [PubMed] [Google Scholar]
- Atianand MK, Hu W, Satpathy AT, Shen Y, Ricci EP, Alvarez-Dominguez JR, Bhatta A, Schattgen SA, McGowan JD, Blin J, et al. (2016). A Long Noncoding RNA lincRNA-EPS Acts as a Transcriptional Brake to Restrain Inflammation. Cell 165, 1672–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty GL, and Gladney WL (2015). Immune escape mechanisms as a guide for cancer immunotherapy. Clin Cancer Res 21, 687–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, Twyman-Saint Victor C, Cucolo L, Lee DSM, Pauken KE, Huang AC, et al. (2016). Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell 167, 1540–1554 e1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berghoff AS, Kiesel B, Widhalm G, Rajky O, Ricken G, Wohrer A, Dieckmann K, Filipits M, Brandstetter A, Weller M, et al. (2015). Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro Oncol 17, 1064–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, et al. (2010). The landscape of somatic copy-number alteration across human cancers. Nature 463, 899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat P, Leggatt G, Waterhouse N, and Frazer IH (2017). Interferon-gamma derived from cytotoxic lymphocytes directly enhances their motility and cytotoxicity. Cell Death Dis 8, e2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, et al. (2013). The somatic genomic landscape of glioblastoma. Cell 155, 462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breslin S, and O’Driscoll L (2016). The relevance of using 3D cell cultures, in addition to 2D monolayer cultures, when evaluating breast cancer drug sensitivity and resistance. Oncotarget 7, 45745–45756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CE, Aguilar B, Starr R, Yang X, Chang WC, Weng L, Chang B, Sarkissian A, Brito A, Sanchez JF, et al. (2018). Optimization of IL13Ralpha2-Targeted Chimeric Antigen Receptor T Cells for Improved Anti-tumor Efficacy against Glioblastoma. Mol Ther 26, 31–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, Ostberg JR, Blanchard MS, Kilpatrick J, Simpson J, et al. (2016). Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N Engl J Med 375, 2561–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brudno JN, and Kochenderfer JN (2016). Toxicities of chimeric antigen receptor T cells: recognition and management. Blood 127, 3321–3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canzio D, Nwakeze CL, Horta A, Rajkumar SM, Coffey EL, Duffy EE, Duffie R, Monahan K, O’Keeffe S, Simon MD, et al. (2019). Antisense lncRNA Transcription Mediates DNA Demethylation to Drive Stochastic Protocadherin alpha Promoter Choice. Cell 177, 639–653 e615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter S, Aiello D, Atianand MK, Ricci EP, Gandhi P, Hall LL, Byron M, Monks B, Henry-Bezy M, Lawrence JB, et al. (2013). A long noncoding RNA mediates both activation and repression of immune response genes. Science 341, 789–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro F, Cardoso AP, Goncalves RM, Serre K, and Oliveira MJ (2018). Interferon-Gamma at the Crossroads of Tumor Immune Surveillance or Evasion. Front Immunol 9, 847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiocca EA, Yu JS, Lukas RV, Solomon IH, Ligon KL, Nakashima H, Triggs DA, Reardon DA, Wen P, Stopa BM, et al. (2019). Regulatable interleukin-12 gene therapy in patients with recurrent high-grade glioma: Results of a phase 1 trial. Sci Transl Med 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi BD, Yu X, Castano AP, Bouffard AA, Schmidts A, Larson RC, Bailey SR, Boroughs AC, Frigault MJ, Leick MB, et al. (2019). CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat Biotechnol 37, 1049–1058. [DOI] [PubMed] [Google Scholar]
- Cloughesy TF, Mochizuki AY, Orpilla JR, Hugo W, Lee AH, Davidson TB, Wang AC, Ellingson BM, Rytlewski JA, Sanders CM, et al. (2019). Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med 25, 477–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Aloia MM, Zizzari IG, Sacchetti B, Pierelli L, and Alimandi M (2018). CAR-T cells: the long and winding road to solid tumors. Cell Death Dis 9, 282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denzler R, Agarwal V, Stefano J, Bartel DP, and Stoffel M (2014). Assessing the ceRNA hypothesis with quantitative measurements of miRNA and target abundance. Mol Cell 54, 766–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denzler R, McGeary SE, Title AC, Agarwal V, Bartel DP, and Stoffel M (2016). Impact of MicroRNA Levels, Target-Site Complementarity, and Cooperativity on Competing Endogenous RNA-Regulated Gene Expression. Mol Cell 64, 565–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles DG, et al. (2012). The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res 22, 1775–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Z, Fei T, Verhaak RG, Su Z, Zhang Y, Brown M, Chen Y, and Liu XS (2013). Integrative genomic analyses reveal clinically relevant long noncoding RNAs in human cancer. Nat Struct Mol Biol 20, 908–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmondson R, Broglie JJ, Adcock AF, and Yang L (2014). Three-dimensional cell culture systems and their applications in drug discovery and cell-based biosensors. Assay Drug Dev Technol 12, 207–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engreitz JM, Haines JE, Perez EM, Munson G, Chen J, Kane M, McDonel PE, Guttman M, and Lander ES (2016a). Local regulation of gene expression by lncRNA promoters, transcription and splicing. Nature 539, 452–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engreitz JM, Ollikainen N, and Guttman M (2016b). Long non-coding RNAs: spatial amplifiers that control nuclear structure and gene expression. Nat Rev Mol Cell Biol 17, 756–770. [DOI] [PubMed] [Google Scholar]
- Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC, et al. (2000). Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med 192, 1027–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman GJ, Wherry EJ, Ahmed R, and Sharpe AH (2006). Reinvigorating exhausted HIV-specific T cells via PD-1-PD-1 ligand blockade. J Exp Med 203, 2223–2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garber ST, Hashimoto Y, Weathers SP, Xiu J, Gatalica Z, Verhaak RG, Zhou S, Fuller GN, Khasraw M, de Groot J, et al. (2016). Immune checkpoint blockade as a potential therapeutic target: surveying CNS malignancies. Neuro Oncol 18, 1357–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, Zaretsky JM, Sun L, Hugo W, Wang X, et al. (2017). Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep 19, 1189–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler S, and Coller J (2013). RNA in unexpected places: long non-coding RNA functions in diverse cellular contexts. Nat Rev Mol Cell Biol 14, 699–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grammatikakis I, Zhang P, Panda AC, Kim J, Maudsley S, Abdelmohsen K, Yang X, Martindale JL, Motino O, Hutchison ER, et al. (2016). Alternative Splicing of Neuronal Differentiation Factor TRF2 Regulated by HNRNPH1/H2. Cell Rep 15, 926–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutschner T, Hammerle M, Eissmann M, Hsu J, Kim Y, Hung G, Revenko A, Arun G, Stentrup M, Gross M, et al. (2013). The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res 73, 1180–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay KA, Hanafi LA, Li D, Gust J, Liles WC, Wurfel MM, Lopez JA, Chen J, Chung D, Harju-Baker S, et al. (2017). Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor-modified T-cell therapy. Blood 130, 2295–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillerdal V, Ramachandran M, Leja J, and Essand M (2014). Systemic treatment with CAR-engineered T cells against PSCA delays subcutaneous tumor growth and prolongs survival of mice. BMC Cancer 14, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda H, Old LJ, and Schreiber RD (2002). The roles of IFN gamma in protection against tumor development and cancer immunoediting. Cytokine Growth Factor Rev 13, 95–109. [DOI] [PubMed] [Google Scholar]
- Jackson CM, Choi J, and Lim M (2019). Mechanisms of immunotherapy resistance: lessons from glioblastoma. Nat Immunol. [DOI] [PubMed] [Google Scholar]
- Jenkins RW, Barbie DA, and Flaherty KT (2018). Mechanisms of resistance to immune checkpoint inhibitors. Br J Cancer 118, 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LA, Scholler J, Ohkuri T, Kosaka A, Patel PR, McGettigan SE, Nace AK, Dentchev T, Thekkat P, Loew A, et al. (2015). Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci Transl Med 7, 275ra222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khvorova A, and Watts JK (2017). The chemical evolution of oligonucleotide therapies of clinical utility. Nat Biotechnol 35, 238–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T, and Croce CM (2018). Long noncoding RNAs: Undeciphered cellular codes encrypting keys of colorectal cancer pathogenesis. Cancer Lett 417, 89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutty RK, Samuel W, Duncan T, Postnikova O, Jaworski C, Nagineni CN, and Redmond TM (2018). Proinflammatory cytokine interferon-gamma increases the expression of BANCR, a long non-coding RNA, in retinal pigment epithelial cells. Cytokine 104, 147–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, et al. (2015). Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med 373, 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, Fry TJ, Orentas R, Sabatino M, Shah NN, et al. (2015). T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 385, 517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin AA (2019). Treating Disease at the RNA Level with Oligonucleotides. N Engl J Med 380, 57–70. [DOI] [PubMed] [Google Scholar]
- Liu X, Shin N, Koblish HK, Yang G, Wang Q, Wang K, Leffet L, Hansbury MJ, Thomas B, Rupar M, et al. (2010). Selective inhibition of IDO1 effectively regulates mediators of antitumor immunity. Blood 115, 3520–3530. [DOI] [PubMed] [Google Scholar]
- Manguso RT, Pope HW, Zimmer MD, Brown FD, Yates KB, Miller BC, Collins NB, Bi K, LaFleur MW, Juneja VR, et al. (2017). In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 547, 413–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques AC, and Ponting CP (2009). Catalogues of mammalian long noncoding RNAs: modest conservation and incompleteness. Genome Biol 10, R124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews Griner LA, Zhang X, Guha R, McKnight C, Goldlust IS, Lal-Nag M, Wilson K, Michael S, Titus S, Shinn P, et al. (2016). Large-scale pharmacological profiling of 3D tumor models of cancer cells. Cell Death Dis 7, e2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh CA, Chen CK, Chow A, Surka CF, Tran C, McDonel P, Pandya-Jones A, Blanco M, Burghard C, Moradian A, et al. (2015). The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature 521, 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, and Getz G (2011). GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol 12, R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mineo M, Ricklefs F, Rooj AK, Lyons SM, Ivanov P, Ansari KI, Nakano I, Chiocca EA, Godlewski J, and Bronisz A (2016). The Long Non-coding RNA HIF1A-AS2 Facilitates the Maintenance of Mesenchymal Glioblastoma Stem-like Cells in Hypoxic Niches. Cell Rep 15, 2500–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal D, Gubin MM, Schreiber RD, and Smyth MJ (2014). New insights into cancer immunoediting and its three component phases--elimination, equilibrium and escape. Curr Opin Immunol 27, 16–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mowel WK, McCright SJ, Kotzin JJ, Collet MA, Uyar A, Chen X, DeLaney A, Spencer SP, Virtue AT, Yang E, et al. (2017). Group 1 Innate Lymphoid Cell Lineage Identity Is Determined by a cis-Regulatory Element Marked by a Long Non-coding RNA. Immunity 47, 435–449 e438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Necsulea A, Soumillon M, Warnefors M, Liechti A, Daish T, Zeller U, Baker JC, Grutzner F, and Kaessmann H (2014). The evolution of lncRNA repertoires and expression patterns in tetrapods. Nature 505, 635–640. [DOI] [PubMed] [Google Scholar]
- O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, Martinez-Lage M, Brem S, Maloney E, Shen A, et al. (2017). A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardoll DM (2012). The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 12, 252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JH, Geyer MB, and Brentjens RJ (2016). CD19-targeted CAR T-cell therapeutics for hematologic malignancies: interpreting clinical outcomes to date. Blood 127, 3312–3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, Bagg A, Marcucci KT, Shen A, Gonzalez V, et al. (2015). Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med 7, 303ra139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prensner JR, and Chinnaiyan AM (2011). The emergence of lncRNAs in cancer biology. Cancer Discov 1, 391–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prensner JR, Iyer MK, Balbin OA, Dhanasekaran SM, Cao Q, Brenner JC, Laxman B, Asangani IA, Grasso CS, Kominsky HD, et al. (2011). Transcriptome sequencing across a prostate cancer cohort identifies PCAT-1, an unannotated lincRNA implicated in disease progression. Nat Biotechnol 29, 742–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman MA, Azuma Y, Nasrin F, Takeda J, Nazim M, Bin Ahsan K, Masuda A, Engel AG, and Ohno K (2015). SRSF1 and hnRNP H antagonistically regulate splicing of COLQ exon 16 in a congenital myasthenic syndrome. Sci Rep 5, 13208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos AD, Andersen RE, Liu SJ, Nowakowski TJ, Hong SJ, Gertz C, Salinas RD, Zarabi H, Kriegstein AR, and Lim DA (2015). The long noncoding RNA Pnky regulates neuronal differentiation of embryonic and postnatal neural stem cells. Cell Stem Cell 16, 439–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinn JL, and Chang HY (2012). Genome regulation by long noncoding RNAs. Annu Rev Biochem 81, 145–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider WM, Chevillotte MD, and Rice CM (2014). Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol 32, 513–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah K, Hingtgen S, Kasmieh R, Figueiredo JL, Garcia-Garcia E, Martinez-Serrano A, Breakefield X, and Weissleder R (2008). Bimodal viral vectors and in vivo imaging reveal the fate of human neural stem cells in experimental glioma model. J Neurosci 28, 4406–4413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, and Allison JP (2015). The future of immune checkpoint therapy. Science 348, 56–61. [DOI] [PubMed] [Google Scholar]
- Snijders AM, Nowak N, Segraves R, Blackwood S, Brown N, Conroy J, Hamilton G, Hindle AK, Huey B, Kimura K, et al. (2001). Assembly of microarrays for genome-wide measurement of DNA copy number. Nat Genet 29, 263–264. [DOI] [PubMed] [Google Scholar]
- Song DG, Ye Q, Poussin M, Liu L, Figini M, and Powell DJ Jr. (2015). A fully human chimeric antigen receptor with potent activity against cancer cells but reduced risk for off-tumor toxicity. Oncotarget 6, 21533–21546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speranza MC, Passaro C, Ricklefs F, Kasai K, Klein SR, Nakashima H, Kaufmann JK, Ahmed AK, Nowicki MO, Obi P, et al. (2018). Preclinical investigation of combined gene-mediated cytotoxic immunotherapy and immune checkpoint blockade in glioblastoma. Neuro Oncol 20, 225–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens MM, Maire CL, Chou N, Murakami MA, Knoff DS, Kikuchi Y, Kimmerling RJ, Liu H, Haidar S, Calistri NL, et al. (2016). Drug sensitivity of single cancer cells is predicted by changes in mass accumulation rate. Nat Biotechnol 34, 1161–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struhl K (2007). Transcriptional noise and the fidelity of initiation by RNA polymerase II. Nat Struct Mol Biol 14, 103–105. [DOI] [PubMed] [Google Scholar]
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. (2012). Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 366, 2443–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, Sommermeyer D, Melville K, Pender B, Budiarto TM, et al. (2016). CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest 126, 2123–2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turunen JJ, Verma B, Nyman TA, and Frilander MJ (2013). HnRNPH1/H2, U1 snRNP, and U11 snRNP cooperate to regulate the stability of the U11–48K pre-mRNA. RNA 19, 380–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uren PJ, Bahrami-Samani E, de Araujo PR, Vogel C, Qiao M, Burns SC, Smith AD, and Penalva LO (2016). High-throughput analyses of hnRNP H1 dissects its multi-functional aspect. RNA Biol 13, 400–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Nostrand EL, Pratt GA, Shishkin AA, Gelboin-Burkhart C, Fang MY, Sundararaman B, Blue SM, Nguyen TB, Surka C, Elkins K, et al. (2016). Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat Methods 13, 508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Aguilar B, Starr R, Alizadeh D, Brito A, Sarkissian A, Ostberg JR, Forman SJ, and Brown CE (2018). Glioblastoma-targeted CD4+ CAR T cells mediate superior antitumor activity. JCI Insight 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Arai S, Song X, Reichart D, Du K, Pascual G, Tempst P, Rosenfeld MG, Glass CK, and Kurokawa R (2008). Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature 454, 126–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watling D, Guschin D, Muller M, Silvennoinen O, Witthuhn BA, Quelle FW, Rogers NC, Schindler C, Stark GR, Ihle JN, et al. (1993). Complementation by the protein tyrosine kinase JAK2 of a mutant cell line defective in the interferon-gamma signal transduction pathway. Nature 366, 166–170. [DOI] [PubMed] [Google Scholar]
- Weiswald LB, Bellet D, and Dangles-Marie V (2015). Spherical cancer models in tumor biology. Neoplasia 17, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willingham AT, Orth AP, Batalov S, Peters EC, Wen BG, Aza-Blanc P, Hogenesch JB, and Schultz PG (2005). A strategy for probing the function of noncoding RNAs finds a repressor of NFAT. Science 309, 1570–1573. [DOI] [PubMed] [Google Scholar]
- Wilmotte R, Burkhardt K, Kindler V, Belkouch MC, Dussex G, Tribolet N, Walker PR, and Dietrich PY (2005). B7-homolog 1 expression by human glioma: a new mechanism of immune evasion. Neuroreport 16, 1081–1085. [DOI] [PubMed] [Google Scholar]
- Wing A, Fajardo CA, Posey AD Jr., Shaw C, Da T, Young RM, Alemany R, June CH, and Guedan S (2018). Improving CART-Cell Therapy of Solid Tumors with Oncolytic Virus-Driven Production of a Bispecific T-cell Engager. Cancer Immunol Res 6, 605–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wintterle S, Schreiner B, Mitsdoerffer M, Schneider D, Chen L, Meyermann R, Weller M, and Wiendl H (2003). Expression of the B7-related molecule B7–H1 by glioma cells: a potential mechanism of immune paralysis. Cancer Res 63, 7462–7467. [PubMed] [Google Scholar]
- Wu X, Tudoran OM, Calin GA, and Ivan M (2018). The Many Faces of Long Noncoding RNAs in Cancer. Antioxid Redox Signal 29, 922–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, Lawrence MS, Zhsng CZ, Wala J, Mermel CH, et al. (2013). Pan-cancer patterns of somatic copy number alteration. Nat Genet 45, 1134–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi MR, and Merlino G (2011). The two faces of interferon-gamma in cancer. Clin Cancer Res 17, 6118–6124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, and Bevan MJ (2011). CD8(+) T cells: foot soldiers of the immune system. Immunity 35, 161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Jiang D, Yang H, He Z, Liu X, Qin W, Li L, Wang C, Li Y, Li H, et al. (2019). Modified CAR T cells targeting membrane-proximal epitope of mesothelin enhances the antitumor function against large solid tumor. Cell Death Dis 10, 476. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. RNA-seq analysis of lncRNA expression in control and IFNγ-stimulated BT333 cells. Related to Figure 1
Table S2. LncRNAs correlated and anti-correlated with INCR1. Related to Figure 1
Table S3. Coding genes neighbor of lncRNAs correlated with INCR1. Related to Figure 1
Table S4. Patient demographics and MGMT promoter methylation status of PDGCLs. Related to Figure 2
Table S5. RNA-seq analysis of gene expression in control and IFNγ-stimulated U251 shControl and shINCR1 cells. Related to Figure 3
Table S6. Mass spectrometry data of proteins identified in the INCR1 RAP but not in control RAP. Related to Figure 5
Table S7. Genes and binding sites identified by HNRNPH1 eCLIP-seq. Related to Figure 5
Table S8. Oligonucleotides Used. Related to STAR Methods
Data Availability Statement
The datasets generated during this study are available at Gene Expression Omnibus (GEO) GSE137489