

Abstract
Objective:
Vascular calcification is a cardiovascular risk factor and accelerated in diabetes. Previous work has established a role for calcification-prone extracellular vesicles (EVs) in promoting vascular calcification. However, the mechanisms by which diabetes provokes cardiovascular events remain incompletely understood. Our goal was to identify that increased S100A9 promotes the release of calcification-prone EVs from human macrophages in diabetes.
Approach and Results:
Human primary macrophages exposed to high glucose (25mmol/L) increased S100A9 secretion and the expression of receptor for advanced glycation end products (RAGE) protein. Recombinant S100A9 induced the expression of pro-inflammatory and osteogenic factors, as well as the number of EVs with high calcific potential (alkaline phosphatase activity, p<0.001) in macrophages. Treatment with a RAGE antagonist or silencing with S100A9 siRNA in macrophages abolished these responses, suggesting that stimulation of the S100A9-RAGE axis by hyperglycemia favors a pro-calcific environment. We further showed that an imbalance between nuclear factor-2 erythroid related factor-2 (Nrf2) and NF-κB pathways contributes to macrophage activation and promotes a pro-calcific environment. In addition, streptozotocin-induced diabetic Apoe−/−S100a9−/− mice and mice treated with S100a9 siRNA encapsulated in macrophage-targeted lipid nanoparticles (LNPs) showed decreased inflammation and microcalcification in atherosclerotic plaques, as gauged by molecular imaging and comprehensive histological analysis. In human carotid plaques, comparative proteomics in diabetic patients and histological analysis showed that the S100A9-RAGE axis associates with osteogenic activity and the formation of microcalcification.
Conclusions:
Under hyperglycemic conditions, macrophages release calcific EVs through mechanisms involving the S100A9-RAGE axis, thus contributing to the formation of microcalcification within plaques.
Keywords: Diabetes, macrophages, extracellular vesicles, atherosclerosis, vascular calcification, S100A9, RAGE
Subject codes: Basic, Translational, Clinical Research; Vascular Biology
Graphical Abstract
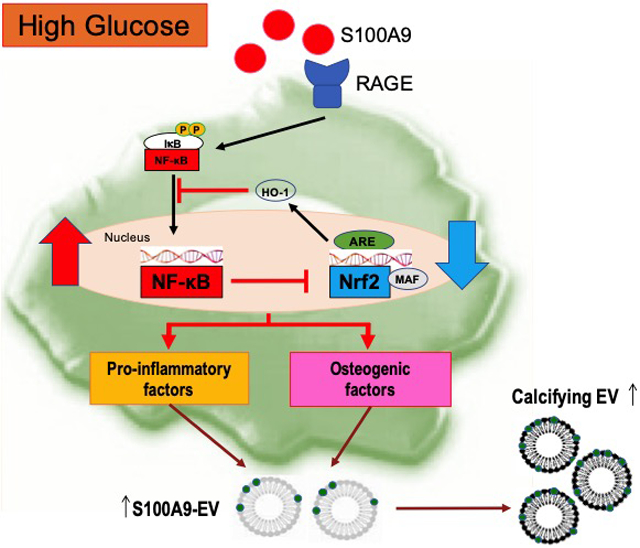
Introduction
Cardiovascular diseases (CVD) are a leading cause of death in the Western world1, and the majority of fatal myocardial infarctions result from rupture of atherosclerotic plaques. Patients with diabetes, an independent risk factor for CVD, have accelerated vascular calcification and plaque progression, coupled with an increased risk of cardiovascular diseases, including death, myocardial infarction, and stroke2. Despite the vast clinical burden, mechanisms by which diabetes provokes cardiovascular events remain incompletely understood.
Coronary artery calcification (CAC) predicts acute cardiovascular events. Cardiovascular risk increases with CAC score and correlates inversely with calcification density3,4. Microcalcification, a process involving the formation of early calcium deposits in close proximity that are aligned along the axis of the cap, can increase the local tissue stresses more than a factor of five in the tissue space between the microcalcifications and trigger fibrous cap rupture, as assessed by computational modeling and high-resolution imaging5,6,7.
In the pro-inflammatory milieu, smooth muscle cells (SMCs) undergo phenotypic modulation into osteoblast-like cells through the induction of specific signaling pathways8,9. We previously reported that SMC-derived extracellular vesicles (EVs) initiate microcalcification in atherosclerotic plaques7. However, vulnerable plaques containing microcalcifications tend to have more macrophages and less SMCs than stable plaques. In addition, our molecular imaging studies showed co-localization of macrophages and microcalcification; thereby, suggesting a direct role of macrophages in calcification10. While the role of SMC-derived EVs in microcalcification is well-established7,11,12, macrophage-derived calcified EVs remain under-explored.
S100A9, a calcium-binding protein and also referred to as myeloid-related protein-14 (MRP-14), was identified as a biomarker for acute cardiovascular events13. S100A9 mediates macrophage recruitment to atheroma14, and macrophages contain higher levels of S100A9 in type 1 diabetes15. Moreover, the S100 family associates with vascular calcification, and human atheroma contains S100A9-positive EVs16,17. Previous studies demonstrated that multiple members of the S100 family, including S100A9, signal through the receptor for advanced glycation end products (RAGE)18, 19.
Our present in vitro and in vivo studies demonstrate that when exposed to high glucose, pro-inflammatory macrophages release calcific EVs, resulting in the excessive formation of atherosclerotic microcalcification via the S100A9-RAGE axis.
Materials and Methods
Detailed methods of all procedures are provided in the online-only Data Supplement. The data, analytic methods, and study materials will be made available to other researchers upon request for purposes of reproducing the results or replicating the procedures. Requests can be made to the corresponding author who manages the information.
Culture and stimulation of human primary macrophages
Human peripheral blood mononuclear cells (PBMC) cultured with 5% non-depleted human serum underwent differentiation into macrophages in vitro. PBMC were isolated from the buffy coat derived from de-identified healthy donors (Research Blood Components, LLC., Boston, MA). The company recruited donors under a New England Institutional Review Board-approved protocol for the Collection of White Blood for Research Purposes (NEIRB#04–144). We had no access to the information about donors. Confluent macrophages were starved for 24 hours in 0.1% human serum media with 5mM D-glucose (normal glucose condition) or 25mM D-glucose (high glucose condition), and endotoxin-free recombinant human S100A9 (R&D Systems Inc., Minneapolis, MN) was used to stimulate human primary macrophages or the human macrophage-like cell line THP-1 cells.
Isolation and quantification of EVs from human primary macrophages
Culture medium (0.1% EV-depleted human serum) conditioned by PBMC for 24 hours underwent centrifugation at 1,000 g for 5 minutes to remove cell debris. The EV fraction was harvested from the media by ultracentrifugation at 100,000 g for 40 minutes at 4°C (Optima Max Ultracentrifuge, Beckman Coulter). Nanoparticle Tracking Analysis using a Nanosight LM10 measured EVs between 30–300 nm. For protein isolation and alkaline phosphatase (ALP) activity, EVs were washed with PBS followed by another ultracentrifugation at 100,000 g for 40 minutes at 4°C.
Animal procedures
All animal experiments were approved by the Brigham and Women’s Hospital’s Animal Welfare Assurance (protocol 2016N000178). All mice had a congenic C57BL/6J background and consumed a high-fat, high-cholesterol diet (HCD) (1.25% cholesterol, D12108C, Research Diets, Inc., New Brunswick, NJ, USA). “The data from both male and female mice were pooled in this study due to the limited sample size in female mice.” To induce hyperglycemia in mice, 42 mg/kg streptozotocin (STZ; Sigma-Aldrich) was injected intraperitoneally for 5 consecutive days. Blood glucose was measured in the beginning and every week after STZ injection with the OneTouch glucometer (Contour, Bayer HealthCare, Mishawaka, IN), and the mice with blood glucose more than 300 mg/dL were considered to have diabetes. For in vivo silencing experiments, S100a9 siRNA and control siRNA were encapsulated in macrophage-targeted C12–200 lipid nanoparticles, as previously described20,21, and injected via tail vein (0.5 mg/kg twice a week) in Apoe−/− mice. At the end of the experiments, all mice were imaged with intra-vital microscopy (IVM). Aortas then were isolated and imaged using fluorescent reflection imaging (FRI; Kodak). For histology, samples (aortic roots, aortic arches, and pancreas) were embedded in OCT compound (VWR) and stored at −80℃ until use.
Human studies
Proteomics of human carotid plaque specimens involved label-free proteomics sample preparation and liquid chromatography tandem mass spectrometry (LC- MS/MS) by the Orbitrap Fusion Lumos (Thermo Scientific). Carotid plaque specimens were obtained from patients undergoing carotid endarterectomy procedures approved by the ethics boards of Brigham and Women’s Hospital, the University of Saskatchewan, Regina Qu’Appelle Health Region, and the University of Regina (protocol# REB-13–115). All patients gave written informed consent to participate. For immunohistochemistry and immunofluorescence experiments, specimens of discarded human carotid plaques (n=25) were obtained at endarterectomy by protocols approved by the Human Investigation Review Committee at the Brigham and Women’s Hospital.
Statistics
Statistical analyses were performed using GraphPad Prism 5.0 (GraphPad Software). The unpaired Student’s t-test was used for comparisons between two groups, or one-way ANOVA followed by Bonferroni post-hoc test or two-way ANOVA followed by the Bonferroni test was used for comparisons of multiple groups. All data are expressed as mean ± SEM for continuous variables. Data have been analyzed for normality and equal variance as a justification for utilizing parametric or nonparametric analyses using Prism software. P values of <0.05 were considered statistically significant.
Results
S100A9-RAGE axis regulates macrophage activation in human primary macrophages subjected to high glucose
Although a few studies suggested that monocytes/macrophages secrete high levels of S100A9 in mice with type 1 diabetes15,22, the mechanisms of its action, particularly in human macrophages, has not been demonstrated. This study used human primary macrophages isolated from peripheral blood mononuclear cells (PBMC) in most in vitro experiments (n=40 PBMC donors), unless otherwise stated. To examine whether high glucose induces S100A9, we cultured macrophages with various concentrations of glucose (5, 15, and 25 mmol/L), and performed real-time qPCR analysis at 6 hours after glucose exposure. The mRNA levels of S100A9 increased in a concentration-dependent manner (Supplemental Figure Ia), and high glucose (25 mmol/L) significantly elevated the mRNA levels of S100A9 as compared to normal glucose (5 mmol/L; p<0.0001) (Figure 1A, left panel). This increase was not seen in an isoosmotic control (mannitol) (Supplemental Figure Ib). High glucose condition enhanced S100A9 protein expression in macrophages as compared to normal glucose condition (Western blot analysis; Figure 1A, right panel). High glucose promoted macrophage secretion of S100A9 into the media more than normal glucose (ELISA; 638.6±77.9 pg/mL vs 497.8±61.0 pg/mL; p<0.001) (Figure 1B), as a similar increased secretion of the S100A8/A9 heterodimer (1977.3±282.2 pg/mL vs 1154.4±147.3 pg/mL; p<0.001) (Supplemental Figure Ic).
Figure 1. S100A9-RAGE axis regulates macrophage activation in human primary macrophages exposed to high glucose (25mmol/L glucose).
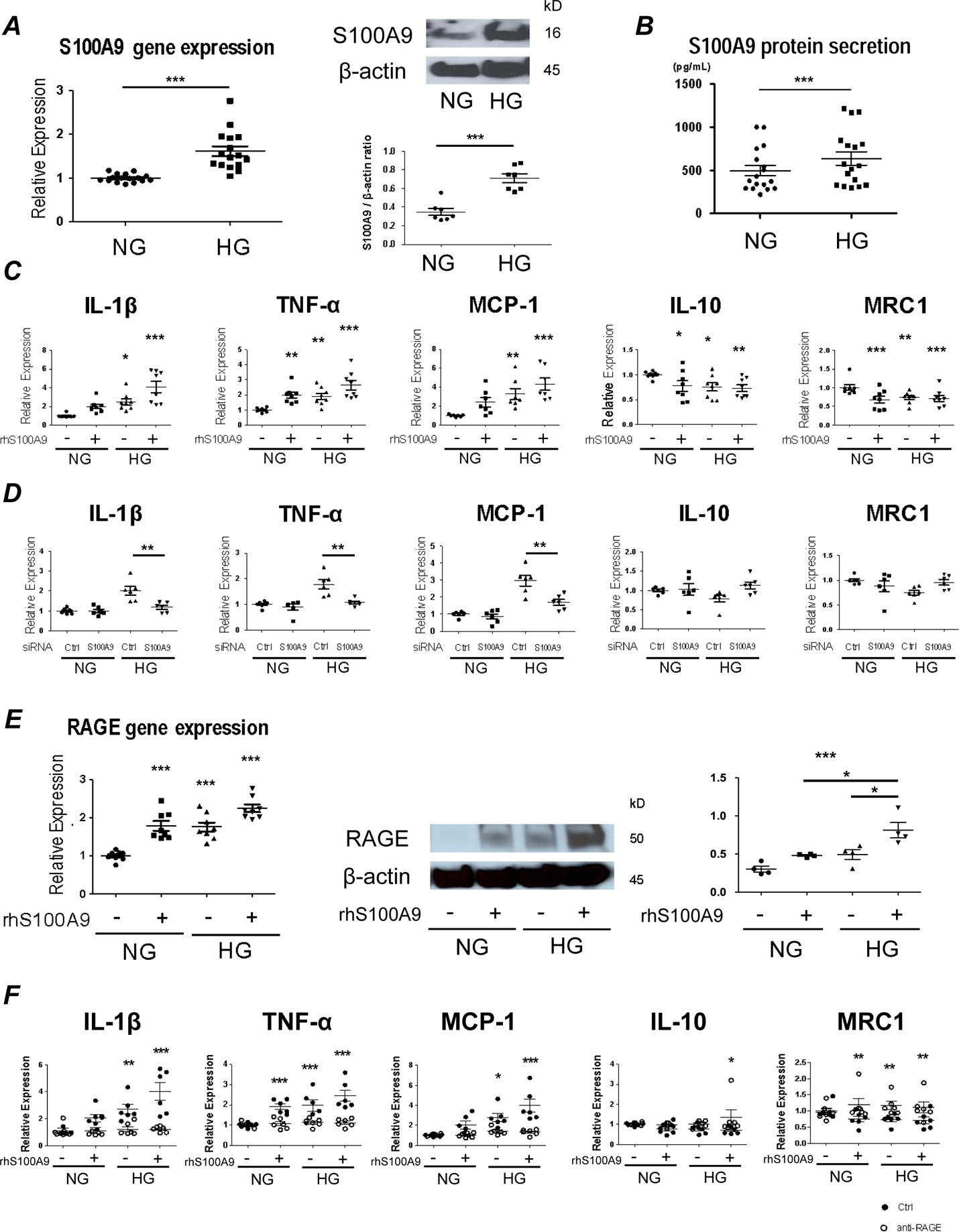
(A) mRNA and protein expression of S100A9 were measured in human primary macrophages after administration with normal glucose (NG; 5 mmol/L glucose) and high glucose (HG; 25 mmol/L glucose) by real-time quantitative polymerase chain reaction (qPCR) (left, administered for 6 hours, n = 16 PBMC donors) and Western blot (right, administered for 24 hours). These blots from one donor are representative of the 7 PBMC donors. The graph shows the signal intensity of S100A9 / β-actin ratio (n = 7 PBMC donors). (B) S100A9 levels in supernatants of human primary macrophages were measured by ELISA after administration with NG and HG condition for 24 hours (n = 17 PBMC donors). (C) mRNA expression of pro-inflammatory factors, IL-1β, TNF-α and MCP-1, and anti-inflammatory factors, IL-10 and MRC1 was measured in human primary macrophages after stimulation with HG and recombinant human S100A9 (rhS100A9) for 6 hours (n=8 PBMC donors). (D) mRNA expression was measured in human primary macrophages after suppression by S100A9 siRNA for 48 hours and administration with HG for 6 hours (n = 6 PBMC donors). (E) mRNA and protein expression of RAGE were measured in human primary macrophages after stimulation with HG and rhS100A9 by real-time qPCR (left, stimulated for 6 hours, n = 8 PBMC donors) and western blot (right, stimulated for 24 hours). These blots from one donor are representative of the 4 PBMC donors. The graph shows the signal intensity of RAGE / β-actin ratio (n = 4 PBMC donors). (F) mRNA expression of pro-inflammatory factors, IL-1β, TNF-α and MCP-1, and anti-inflammatory factors, IL-10 and MRC1 was measured in human primary macrophages after pretreatment with RAGE-antagonist (FPS-ZM1, 10 μg/mL) for 4 hours, and stimulation with HG and rhS100A9 for 6 hours (n = 6 PBMC donors). P value was calculated by unpaired student’s t-test or one-way ANOVA, based on a comparison with NG, or two-way ANOVA followed by Bonferroni test. *P < 0.05, **P < 0.01, ***P < 0.001. Error bars indicate ± SEM.
To evaluate the effects of S100A9 on macrophage activation, differentiated macrophages from human PBMCs were treated with various concentrations of endotoxin-free recombinant human S100A9 (5, 10, 50, and 100 ng/mL). After 6 hours, the mRNA levels of pro-inflammatory factors IL-1β, TNF-α, and MCP-1 were analyzed. S100A9 (10 ng/mL) significantly increased pro-inflammatory factors, to the same extent as 50 and 100 ng/mL (Supplemental Figure IIa). Therefore, we treated the macrophages with recombinant human S100A9 at 10 ng/mL in subsequent experiments. The combination of high glucose and recombinant human S100A9 further elevated mRNA expression of pro-inflammatory IL-1β, TNF-α, and MCP-1; whereas, the anti-inflammatory mediators IL-10 and MRC1 decreased (Figure 1C). Furthermore, stimulation with heat-inactivated rhS100A9 (10ng/ml) under normal glucose condition did not show any changes in the mRNA levels of pro-inflammatory IL-1β, TNF-α, and MCP-1 (Supplemental Figure III a), and the anti-inflammatory factors IL-10 and MRC1, demonstrating that the effect is not driven by endotoxin contamination (Supplemental Figure III b). S100A9 also exists as a heterodimer with S100A8 (S100A8/A9)23. The effects that S100A8/A9 has on macrophage activation were evaluated utilizing treatment with rhS100A8/A9 (1 μg/ml), which increased IL-1β and TNF-α (Supplemental Figure IV a). These results suggest that the heterodimer has the potential to induce a downstream effect. Our loss-of-function experiments sought to establish whether S100A9 plays a causal role in pro-inflammatory macrophage activation. Silencing macrophages with siS100A9 achieved up to 50% reduction of S100A9 mRNA expression (Supplemental Figure IIb), and reduced mRNA expression of pro-inflammatory IL-1β, TNF-α, and MCP-1 when exposed to high glucose (Figure 1D).
The RAGE axis was investigated to determine what mediates S100A9-induced macrophage activation. RAGE is a key player, as it can modulate innate immunity and inflammatory processes, and S100A9 binds to RAGE18,19. Macrophages stimulated with high glucose and recombinant human S100A9 showed a significant increase in RAGE mRNA and protein (Figure 1E). To test the involvement of RAGE in S100A9-induced macrophage activation, cells were pretreated with FPS-ZM1 (a high affinity RAGE-specific inhibitor; 10 μg/mL) for 1 hour before stimulation. FPS-ZM1 significantly suppressed the mRNA levels of pro-inflammatory IL-1β, TNF-α, and MCP-1; whereas, the anti-inflammatory factors, IL-10 and MRC1, significantly increased in that condition (Figure 1F). The pro-inflammatory effects that AGE-BSA (100μM), another RAGE activator, has on macrophages under normal glucose condition were evaluated to determine if there was a comparable effect on the RAGE axis. Stimulation with AGE-BSA increased the mRNA levels of the pro-inflammatory genes, IL-1β, TNF-α, and MCP-1 (Supplemental Figure V a), and tended to decrease the mRNA levels of anti-inflammatory MRC1 (Supplemental Figure V b). This further demonstrates that RAGE activation induces pro-inflammatory responses in macrophages.
High glucose accelerates S100A9-mediated release and calcific potential of macrophage-derived EVs in vitro
To determine that S100A9 promotes EV release from pro-inflammatory macrophages and their calcific potential in diabetes, nanoparticle tracking analysis (NTA) that can measure EV numbers and size was utilized. NTA revealed that EV secretion from macrophages treated with high glucose and recombinant human S100A9 for 24 hours is markedly higher, but their size distribution did not differ (Figure 2A). An investigation into the role of S100A9-RAGE axis on EV secretion and calcific potential followed. Silencing with siS100A9 and addition of RAGE inhibitor (FPS-ZM1; 10 μg/mL) reduced EV secretion in macrophages exposed to high glucose and recombinant human S100A9 (Figures 2B and 2C). Western blot analysis of EVs isolated from macrophages stimulated with high glucose and recombinant human S100A9 showed enhanced S100A9 signals when compared to a normal glucose condition (Figure 2D and Supplemental Figure IIc). Of note, high glucose and recombinant human S100A9 increased ALP activity in EVs (Figure 2E), suggesting that the loading of S100A9 into the EVs promotes their calcific potential as we previously revealed7.
Figure 2. S100A9 regulates secretion and calcific potential of extracellular vesicle (EV) from human primary macrophages.
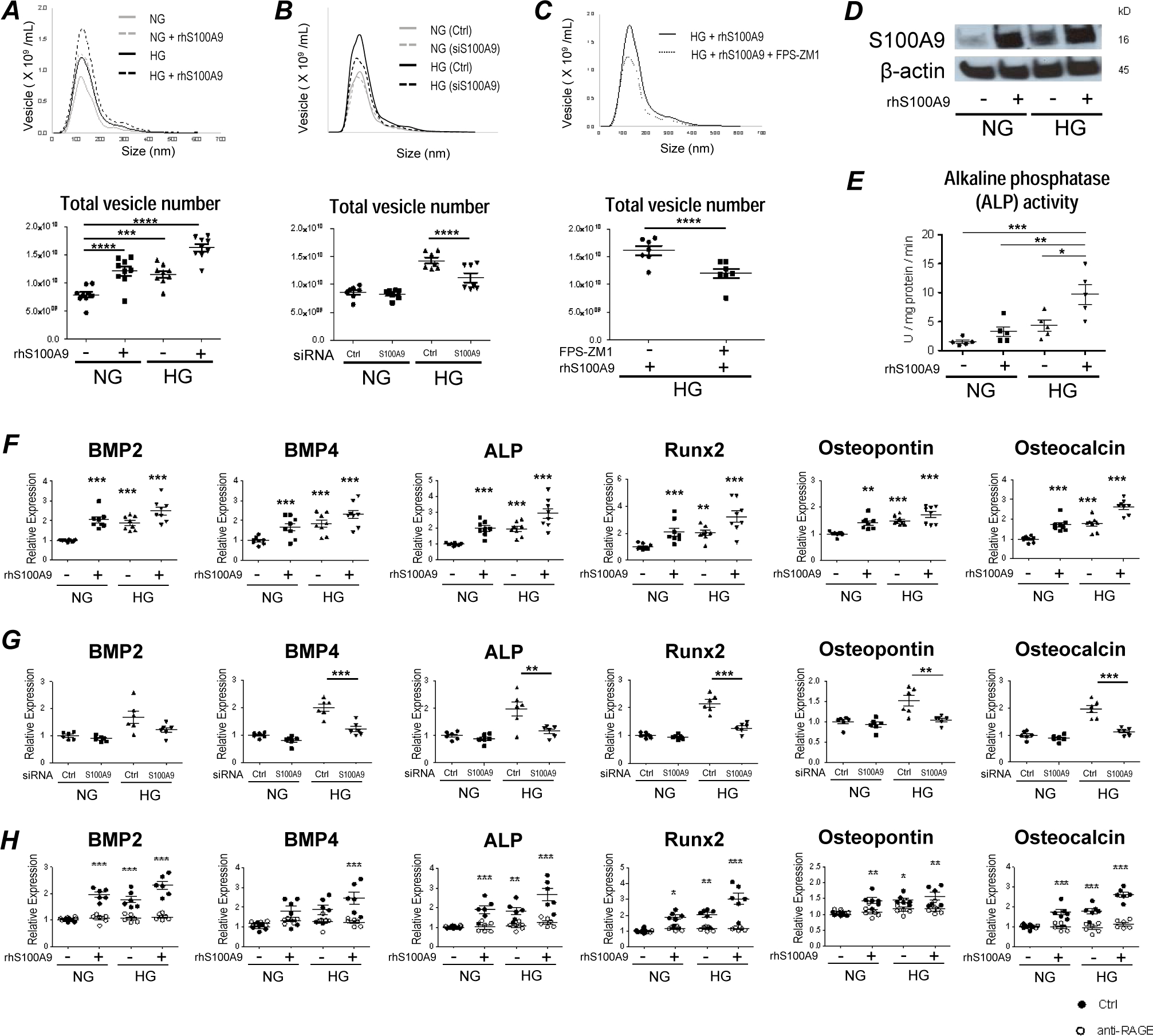
(A) Size distribution and quantification of EVs derived from human primary macrophages were measured after stimulation with HG and rhS100A9 for 24 hours by nanoparticle tracking analysis (n = 9 PBMC donors). (B) Size distribution and quantification of EVs derived from human primary macrophages were measured after suppression by S100A9 siRNA for 48 hours and administration with NG and HG for 24 hours (n = 7 PBMC donors). (C) Size distribution and quantification of EVs derived from human primary macrophages were measured after pretreatment with RAGE-antagonist (FPS-ZM1, 10 μg/mL) for 4 hours, and stimulation with HG and rhS100A9 for 24 hours (n = 7 PBMC donors). (D) The expression of S100A9 was measured within EVs derived from human primary macrophages by western blotting after stimulation with HG and rhS100A9 for 24 hours. These blots from one donor are representative of the 4 PBMC donors. (E) Calcific potential of EVs derived from human primary macrophages was measured by alkaline phosphatase (ALP) activity after stimulation with HG and rhS100A9 for 24 hours (n = 5 PBMC donors). (F) mRNA expression of the osteogenic factors, BMP2, BMP4, ALP, Runx2, osteopontin, and osteocalcin was measured in human primary macrophages after stimulation with HG and rhS100A9 for 12 hours (n = 8 PBMC donors). (G) mRNA expression of osteogenic factors was measured in human primary macrophages after suppression by S100A9 siRNA for 48 hours and administration with HG for 12 hours (n = 6 PBMC donors). (H) mRNA expression of osteogenic factors was measured in human primary macrophages after pretreatment with RAGE-antagonist (FPS-ZM1, 10 μg/mL) for 4 hours, and stimulation with HG and rhS100A9 for 12 hours (n = 6 PBMC donors). P value was calculated by unpaired student’s t-test or one-way ANOVA, based on a comparison with NG, or two-way ANOVA followed by Bonferroni test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Error bars indicate ± SEM.
To understand how macrophages promote EV calcification, we first examined whether high glucose induces the osteogenic factors in these inflammatory cells. Macrophages stimulated with high glucose and recombinant human S100A9 showed increased mRNA expression of osteogenic BMP2, BMP4, ALP, Runx2, osteopontin, and osteocalcin (Figure 2F). Similar to pro-inflammatory effects, these osteogenic responses were not shown when treating human PBMCs with heat-inactivated rhS100A9 under NG condition (Supplemental Figure IIIc). These effects were then inhibited with siS100A9 treatment or RAGE inhibition by FPS-ZM1 (10 μg/mL) (Figures 2G and 2H). Additionally, the osteogenic effects that rhS100A8/A9 have on macrophages was assessed. Stimulation with high glucose and rhS100A8/A9 did not have a significant effect on the mRNA levels of the osteogenic factors, except for Runx2 (Supplemental Figure IV b), supporting previous findings where the S100A9 homodimer showed to have a greater binding affinity to RAGE than the S100A8/A9 heterodimer23. Furthermore, the effect that AGE-BSA has on vesicle secretion and osteogenic gene expression was investigated. Treatment with AGE-BSA under normal glucose condition increased vesicle secretion (Supplemental Figure V c); however, it did not have an effect on the mRNA levels of the osteogenic genes, BMP2, BMP4, ALP, Runx2, osteopontin, and osteocalcin (Supplemental Figure V d). These results suggest that the S100A9-RAGE axis has the potential to mediate osteogenic responses in macrophages; thereby, triggering the release of EVs with high calcific potential.
High glucose contributes to imbalance of Nrf2 and NF-κB pathways and increases macrophage activation
Next, we explored mechanisms that link high glucose and S100A9 to its downstream pro-inflammatory and pro-osteogenic responses. We investigated Nrf2 and NF-κB as potential regulators of macrophage activation by high glucose and S100A9. It has been demonstrated that coordination of Nrf2 and NF-κB favors redox homeostasis in healthy cells24. Hyperglycemia, however, triggers Nrf2-NF-κB imbalance, and enhanced NF-κB signaling results in excess production of pro-inflammatory cytokines24. S100A9 induces the secretion of pro-inflammatory cytokines in monocytes/macrophages via the NF-kB pathway. We first examined the activation of NF-κB pathway in the human monocyte cell line, THP-1. Western blot analysis indicated that high glucose induces Ser536 phosphorylation of p65 within 60 minutes, and recombinant human S100A9 increases Ser536 phosphorylation of p65 within 15 minutes (Figure 3A). Ser536 of the p65 subunit represents the site most subject to inducible phosphorylation in response to inflammatory stimuli25. We then examined whether high glucose and S100A9 activate the Nrf2 signaling. The transcription factor Nrf2 can limit inflammatory responses. High glucose and S100A9 significantly decreased nuclear Nrf2 from THP-1 cells (Figure 3B). We also tested the mRNA expression of the Nrf2-dependent antioxidant enzymes, hemeoxygenase-1 (HO-1) and NADPH quinone oxidoreductase (NQO1), which associate with anti-inflammatory properties26,27. HO-1 and NQO1 mRNA levels decreased in macrophages stimulated with high glucose and S100A9 (Figure 3B). These data indicate that high glucose and S100A9 promote disturbance of Nrf2 and NF-κB pathways.
Figure 3. Imbalance of the Nrf2 and NF-κB pathways, induced by high glucose and S100A9, contribute to increased pro-inflammatory and osteogenic activity in human primary macrophages.
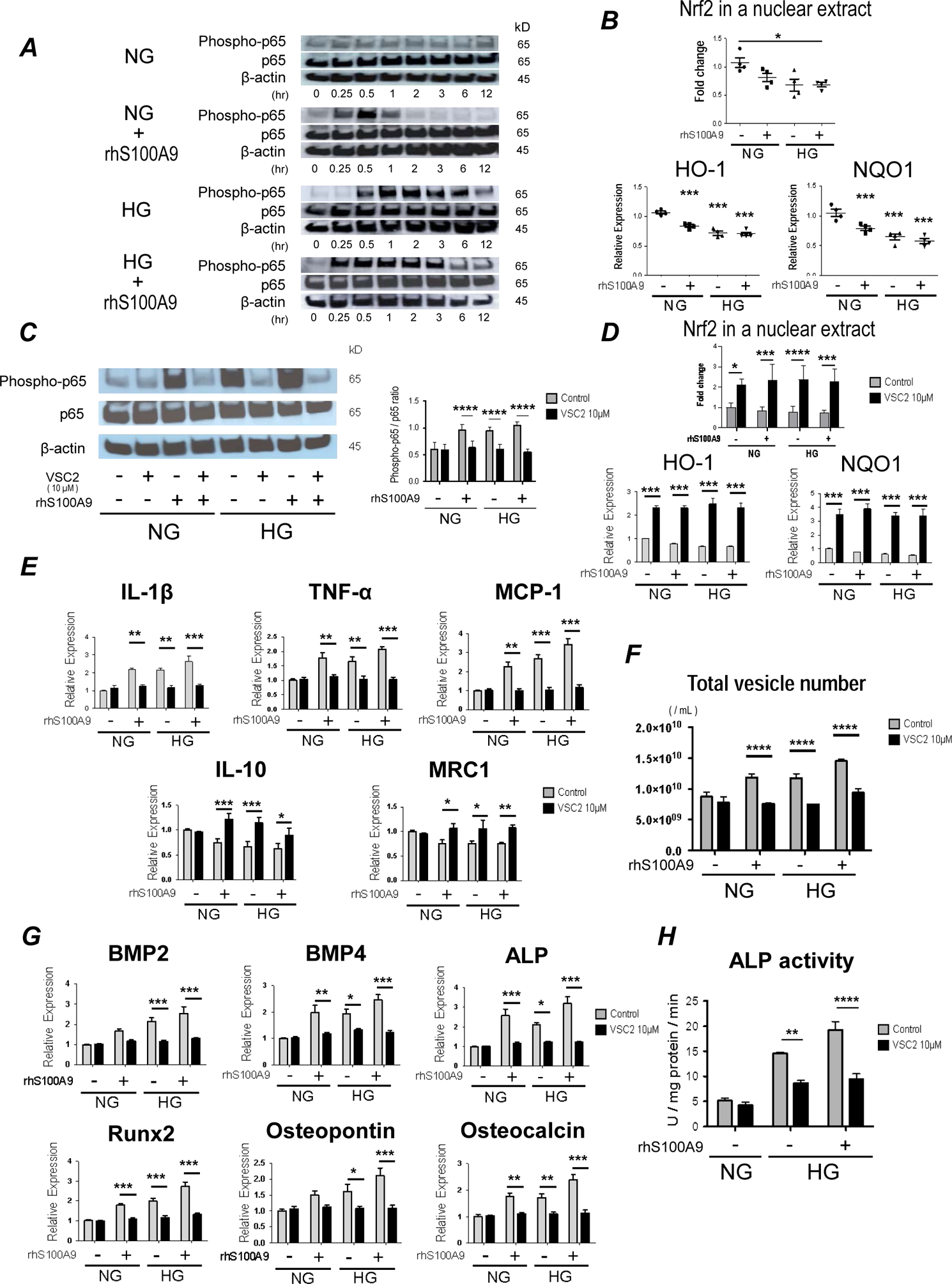
(A) THP-1 cells were cultured in either NG or HG, and then stimulated with rhS100A9 for the indicated times before being analyzed by Western blot with antibodies specific for the indicated targets. Data representative of the 3 different experiments. (B) Nrf2 in a nuclear extract of THP-1 cells (top panel, treated for 24 hours, 4 different experiments) was measured by Western blot and mRNA expression of detoxifying enzymes, hemeoxygenase-1 (HO-1) and NADPH quinone oxidoreductase 1 (Nqo1) (bottom panel, treated for 6 hours) were measured after stimulation with HG and rhS100A9 (n = 4 PBMC donors). (C) The expression of Ser536 phosphorylation of p65 was measured in human primary macrophages by Western blot after pretreatment with VSC2 (10 μM) for 1 hour and stimulation with HG and rhS100A9 for 24 hours. The graph shows the signal intensity of phosphorylated p-65 / p65 ratio (n = 4 PBMC donors). (D) Nrf2 in a nuclear extract of THP-1 cells (top panel, treated for 24 hours, 3 different experiments) was measured by Western blot and mRNA expression of HO-1 and Nqo1 (bottom panel, treated for 6 hours, n = 4 PBMC donors) was measured after pretreatment with VSC2 (10 μM) for 1 hour and stimulation with HG and rhS100A9. (E) mRNA expression of pro-inflammatory factors, IL-1β, TNF-α and MCP-1, and anti-inflammatory factors, IL-10 and MRC1 was measured in human primary macrophages after pretreatment with VSC2 (10 μM) for 1 hour, and stimulation with HG and rhS100A9 for 6 hours (n = 4 PBMC donors). (F) The number of EVs derived from human primary macrophages were measured after pretreatment with VSC2 (10 μM) for 1 hour, and stimulation with HG and rhS100A9 for 24 hours (n = 4 PBMC donors). (G) mRNA expression of osteogenic factors was measured in human primary macrophages after pretreatment with VSC2 (10 μM) for 1 hour, and stimulation with HG and rhS100A9 for 12 hours (n = 4 PBMC donors). (H) Calcific potential of EVs derived from THP-1 cells was measured by ALP activity after pretreatment with VSC2 (10 μM) for 1 hour, and stimulation with HG and rhS100A9 for 24 hours (3 different experiments). P value was calculated by one-way ANOVA, based on a comparison with NG, or two-way ANOVA followed by Bonferroni test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Error bars indicate ± SEM.
Activation of Nrf2 signaling by VSC2 inhibits macrophage activation when treated with high glucose and S100A9
The vinyl sulfone compound, VSC2, previously shown to activate the Nrf2 signaling pathway28, was used to determine whether the crosstalk between Nrf2 and NF-κB pathways mediates the pro-inflammatory effects of high glucose and S100A9. Western blot analysis indicated that treatment with VSC2 (10μM) decreases Ser536 phosphorylation of p65 in macrophages stimulated with high glucose and S100A9 (Figure 3C). Furthermore, mRNA levels of HO-1 and NQO1 in macrophages and nuclear Nrf2 in THP-1 cells were significantly increased by VSC2 in this pro-diabetic condition (Figure 3D). The mRNA levels of the antioxidant genes, PRDX1, GCLC, and SOD1, increased by VSC2, providing further evidence that VSC2 is related to redox state (Supplemental figure VI a). These results indicate that VSC2 may reset the imbalance of Nrf2 and NF-κB pathways through activation of the Nrf2, thereby suppressing NF-κB signaling.
We then examined the role of VSC2 on mediators and modulators of inflammation. VSC2 decreased mRNA expression of the pro-inflammatory cytokines IL-1β, TNF-α, and MCP-1; whereas, the anti-inflammatory factors, IL-10 and MRC1, increased in macrophages stimulated with high glucose and S100A9 (Figure 3E).
We further explored other potential effects of Nrf2 signaling. Activation of Nrf2 may decrease vascular calcification29. We thus examined whether VSC2 inhibits the induction of osteogenic factors in high glucose-S100A9 stimulated human primary macrophages. VSC2 inhibited the effects of high glucose and S100A9 on the mRNA expression of pro-osteogenic BMP2, BMP4, ALP, Runx2, osteopontin, and osteocalcin (Figure 3G). Likewise, in EVs derived from high glucose-S100A9 stimulated macrophages, VSC2 treatment substantially suppressed EV secretion (Figure 3F), resulting in decreased ALP activity (Figure 3H). Additionally, it decreased the mRNA levels of the osteogenic factors BMP4, ALP, and osteopontin (Supplemental Figure VIIa). These results suggest that imbalance of the Nrf2 and NF-κB pathways, induced by high glucose and S100A9, may foster a pro-inflammatory and pro-osteogenic milieu in macrophages.
Diabetic mice have accelerate inflammation and calcification in atherosclerotic lesions
To examine the effect of diabetes on atherosclerotic lesion formation, Apoe−/− mice were injected with streptozotocin (STZ) and fed a HCD for 8 weeks. Both male and female mice were used (n=10 per group). Diabetic mice showed a significant increase in blood glucose and decrease in body weight, as compared to non-diabetic controls (Supplemental Figure VIIIa). Additionally, a decrease in the size of Langerhans islet cells were seen in STZ-induced pancreas cells (Supplemental figure VIIIb). Atherosclerotic lesions were evaluated in the aortic arch and brachiocephalic artery (BCA), an artery prone to developing advanced lesions30. In diabetic mice, von Kossa staining revealed extensive calcified lesions in lesser curvature of the aortic arch and BCA (Figure 4A). Moreover, a near-infrared fluorescent (NIRF) calcium tracer showed a significant increase in microcalcifications (yellow arrows) trapped within the plaque and fibrous cap of diabetic Apoe−/− mice compared to those of non-diabetic mice (Figure 4B). These results suggest that diabetes accelerates vascular microcalcification and formation of rupture-prone lesions in Apoe−/− mice.
Figure 4. S100A9 deficiency attenuates calcific potential in atherosclerotic plaques in diabetic Apoe−/− mice.
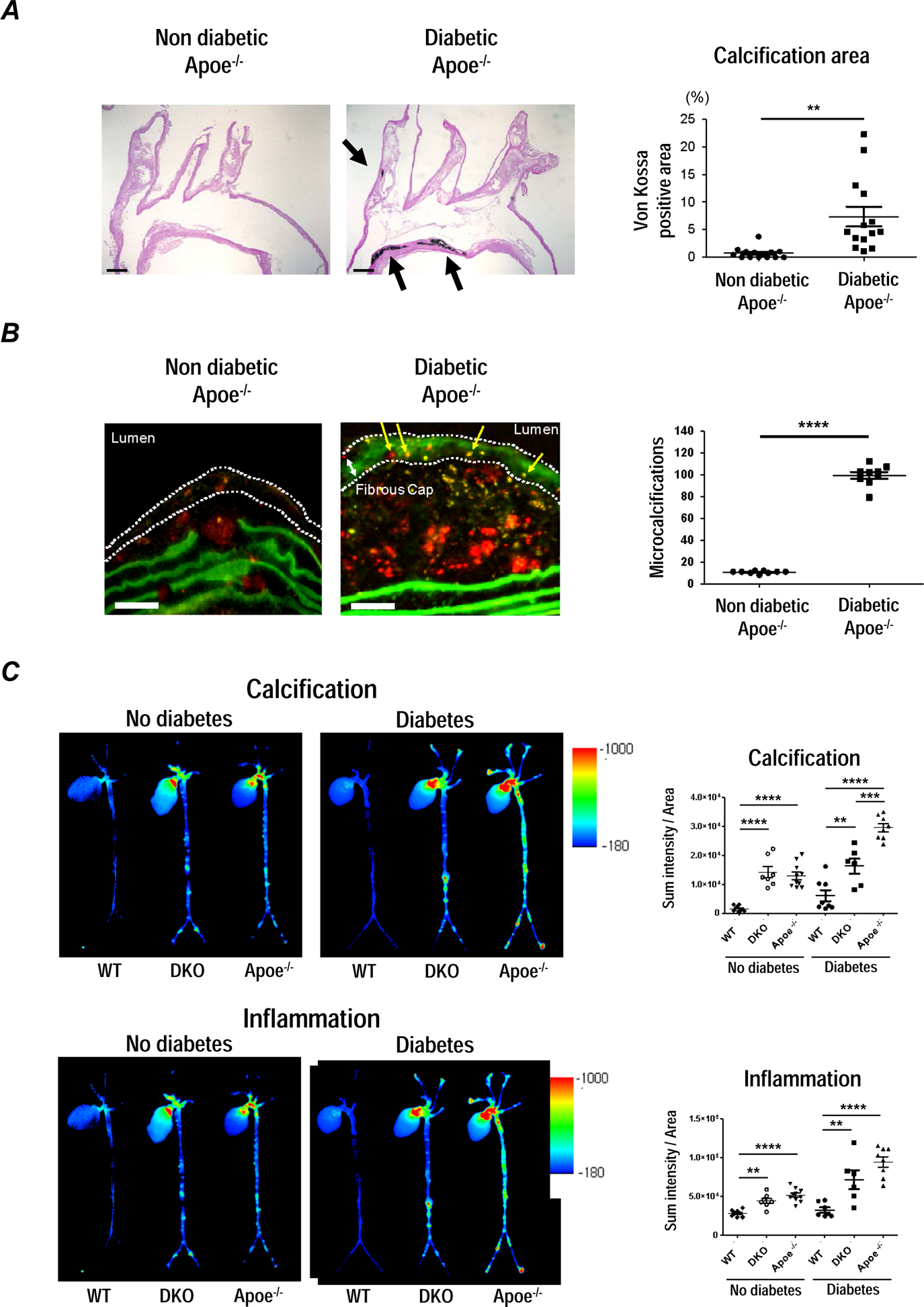
Apoe−/− mice fed a HCD for 18 weeks. After 10 weeks, streptozotocin (42 mg/kg/day) was intraperitoneally injected to diabetic group for 5 days, and fed a HCD for 8 weeks. (A) Von Kossa staining on longitudinal sections of the aortic arch; one of 14 animals per group is shown. Scale bars: 200 μm. The graph shows the percentage of positive area in the plaques. (B) Microcalcifications (yellow arrows) within the fibrous cap (white dotted line) were detected by Near-infrared fluorescent (NIRF) calcium tracer (OS680). The graph shows the number of microcalcifications (n=9). Scale bars: 50 μm. (C) Molecular imaging of aortic vascular calcification assessed by OsteoSense750 and vascular inflammation assessed by proteolytic activity, ProSense680. (n=6–10). The graph shows the SUM intensity related to total aortic area for fluorescent reflection imaging (FRI) in Apoe+/+S100a9+/+ (wild type; WT), Apoe−/− S100a9−/− (double knockout; DKO), and Apoe−/− S100a9+/+ (Apoe−/−) mice with diabetes and non-diabetic control. P value was calculated by paired student’s t-test or two-way ANOVA followed by Bonferroni test. **P < 0.01, ***P < 0.001, ****P < 0.0001. Error bars indicate ± SEM.
Systemic S100A9 deficiency attenuates calcific potential in atherosclerotic plaques in diabetic mice
We then examined the effects of systemic deleting S100A9 in diabetic Apoe−/− mice consuming a HCD for 8 weeks. Apoe+/+S100a9+/+, Apoe−/− S100a9+/+, and Apoe−/−S100a9−/− mice were randomly assigned to diabetic and non-diabetic groups (n=6–10 per group). Animals were co-injected with two spectrally different molecular imaging agents: OsteoSense750 for osteogenic activity, indicating calcification/microcalcification and ProSense680 for proteolytic activity, indicating inflammation. Ex vivo NIRF molecular imaging assessed the effects of S100A9 deficiency on macrophage and osteogenic activity in the aorta of diabetic mice. In diabetes, Apoe−/−S100a9−/−double knockout (DKO) mice exhibited reduced calcification and inflammation when compared with Apoe+/+S100a9+/+ and Apoe−/−S100a9+/+ littermates, as demonstrated by quantitative fluorescence reflectance imaging (FRI) (Figure 4C). These results indicated that suppression of S100A9 attenuates vascular calcification and inflammation in atherosclerotic plaques of diabetic mice.
Macrophage-targeted S100A9 silencing inhibits calcification in atherosclerotic plaques of diabetic mice
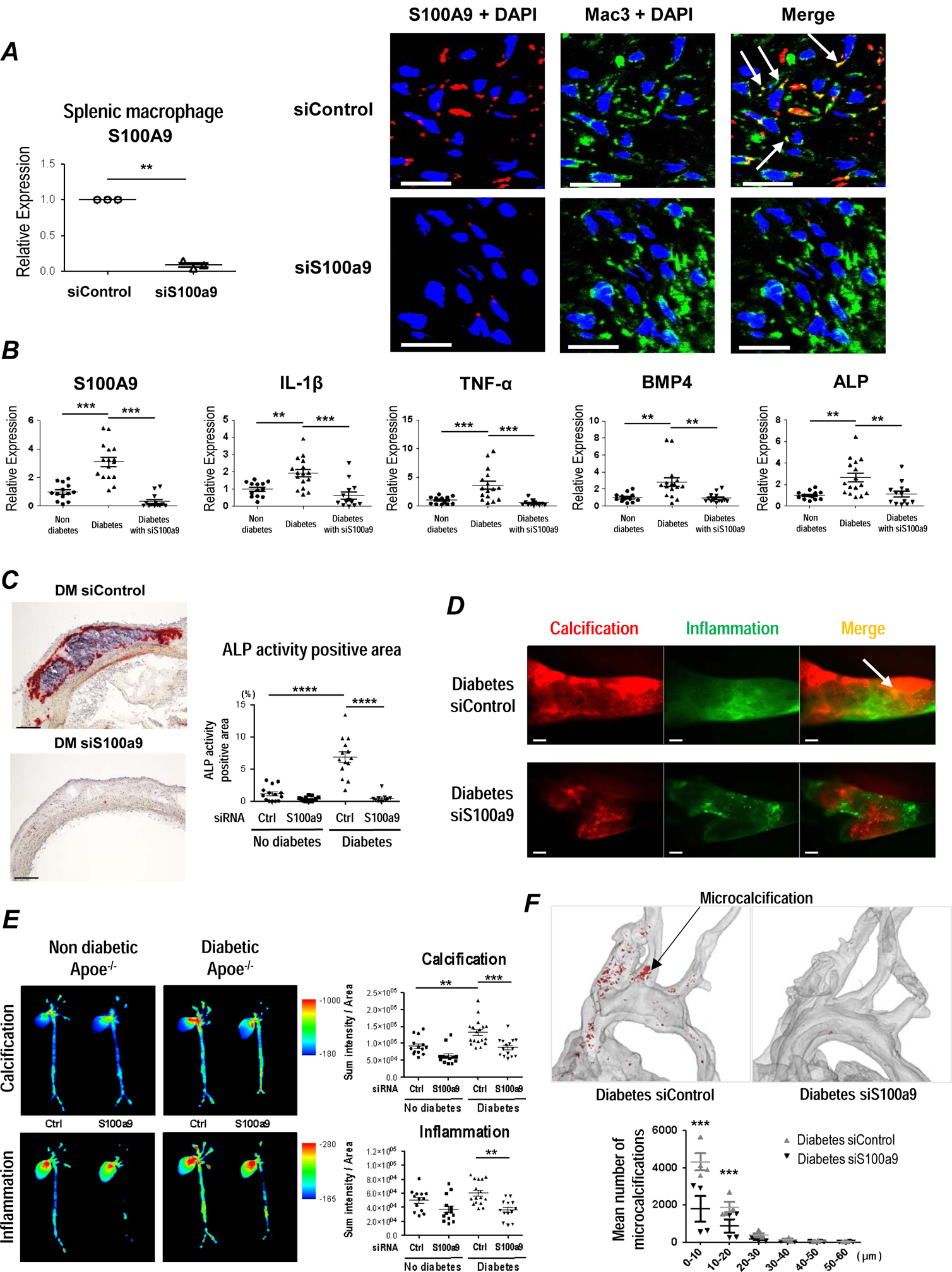
While systemic S100A9-deficient mice provide important evidence that S100A9 deletion reduces calcification, the relative contribution of macrophage-derived S100A9 remains unknown. S100a9 siRNA was encapsulated in macrophage-targeted lipid nanoparticles C12–200. The efficacy of this method on in vivo siRNA delivery to macrophages was extensively validated31. A single injection of 0.5 mg/kg C12–200-siS100a9 achieved a 91% reduction of S100A9 mRNA in splenic macrophages within 72 hours (Figure 5A). To validate the selectivity of S100A9 silencing to macrophage in vivo, we administered 0.5 mg/kg C12–200-siS100a9 twice during a period of 18 weeks after initiation of HCD. Fluorescent in situ hybridization demonstrated that plaque macrophages elaborated S100A9 mRNA signal in control mice, while few cells were positive in C12–200-siS100a9 mice (Figure 5A).
Figure 5. S100A9 expressed by macrophages regulates vascular calcification in diabetic Apoe−/− mice.
(A) Macrophage-targeted LNP containing control siRNA (siControl) or S100a9 siRNA (siS100a9) were injected via tail vein, and F4/80 positive splenic macrophages were isolated. S100A9 mRNA was quantified by real-time PCR. (left panel, n = 3 per each group). Fluorescent in situ hybridization shows less S100A9 mRNA expression localized to macrophage-positive cells in Apoe−/− mice injected with macrophage-targeted LNP containing siS100a9. White arrows show the co-localization of S100A9 mRNA in macrophage-positive cells. One of 4 animals per group is shown (right panel). Scale bars: 20 μm. (B) mRNA expression of S100A9, inflammatory cytokines, and osteogenic factors was measured in splenic macrophages from diabetic and non-diabetic Apoe−/− mice injected with macrophage-targeted LNP containing siControl (No diabetes and Diabetes) and siS100a9 (Diabetes with siS100a9) (n = 13, No diabetes and Diabetes with siS100a9; n = 16, Diabetes). (C) Immunohistochemical evaluation of ALP activity in aortic plaques from diabetic and non-diabetic Apoe−/− mice injected with macrophage-targeted LNP containing siControl and siS100a9 (n = 14, No diabetes and Diabetes with siControl; n = 13, No diabetes with siControl; n = 11, Diabetes with siS100a9). Scale bars: 100 μm. (D) In vivo molecular imaging of carotid arteries from diabetic Apoe−/− mice. Vascular calcification assessed by osteogenic activity, OsteoSense680 (red) and vascular inflammation assessed by proteolytic activity, ProSense750 (green). White arrow shows the co-localization of calcification and inflammation, indicating the formation of microcalcification (yellow). The image represents 7–10 mice per group. Scale bar = 200 μm. (E) Molecular imaging of aortic vascular calcification assessed by OsteoSense680 and vascular inflammation assessed by ProSense750. (n= 13–18). The graph shows the SUM intensity related to total aortic area for FRI in diabetic and non-diabetic Apoe−/− mice treated with macrophage-targeted LNP containing control siRNA (siControl) or S100a9 siRNA (siS100a9). (F) 3D micro-CT scanning of aorta from diabetic Apoe−/− mice injected with macrophage-targeted LNP containing siControl and siS100a9 (n = 4, per each group). The graph shows the mean number of microcalcifications assessed by size contribution. P value was calculated by paired student’s t-test or two-way ANOVA followed by Bonferroni test. **P < 0.01, ***P < 0.001, ****P < 0.0001. Error bars indicate ± SEM.
S100a9 siRNA or control siRNA encapsulated in C12–200 were then injected at 0.5 mg/kg, twice a week, for 8 weeks after STZ-injection, in diabetic and non-diabetic Apoe−/− both male and female mice (n=13–18 per group). S100a9 siRNA effectively reduced the protein production in splenic macrophage, and aortic lesions (Figure 5B and Supplemental Figure IXa). C12–200-siS100a9 reduced mRNA expression of IL-1β and TNF-α in splenic macrophages (Figure 5B), consistent with a statistically significant reduction of macrophage accumulation in aortic atherosclerotic plaques from diabetic Apoe−/− mice detected by Mac3 (Supplemental Figure IXb). In addition, C12–200-siS100a9 also reduced mRNA expression of pro-osteogenic factors, BMP4 and ALP in splenic macrophages from diabetic Apoe−/− mice (Figure 5B). In diabetic mouse plaques C12–200-siS100a9 reduced ALP activity, consistent with the reduction of calcification detected by von Kossa staining (Figure 5C and Supplemental Figure IXc).
Furthermore, NIRF intravital microscopy revealed that C12–200-siS100a9 reduced calcification burden and formation of microcalcifications (Osteosense750) and inflammation (Prosense680) in carotid arteries of diabetic Apoe−/− mice (Figure 5D and Supplemental Figure IXd). Ex vivo FRI of excised aortas showed decreased osteogenic activity in C12–200-siS100a9 treated mice, as compared with untreated diabetic mice (Figure 5E and Supplemental Figure IXe). In addition, 3D micro-CT scanning of aorta showed decreased microcalcification in C12–200-siS100a9 treated mice, consistent with the results of von Kossa staining (Figure 5F). Of note, serum biochemical measurements including renal function, lipid, and cations did not differ between the mice treated C12–200-siS100a9 or C12–200-control siRNA (Supplemental Table 1). These results indicate that macrophage-derived S100A9 indeed accelerates calcific potential in atherosclerotic plaques in diabetic mice, which results from a series of events involving the initiation of microcalcifications and subsequent formation of large calcification areas.
Increased S100A9 in macrophages in calcified human atheroma
We evaluated the association of S100A9 and calcification with RAGE and AGE, key molecules associated with diabetes32 in human carotid endarterectomy specimens (n=25) stained with anti-S100A9, anti-RAGE and anti-AGE antibodies. The plaques were divided into two groups, including low- (less than 50% mean) and high- (more than 50% mean) positive expression area. Groups with a high expression of S100A9 and AGE tended to have more RAGE staining than plaque groups with low S100A9 and AGE expression (Supplemental Figure Xa). In addition, S100A9 percent positive area colocalized with ALP activity and RAGE (Figure 6A). Macrophage (CD68) and smooth muscle cell (α-SMA) content did not differ among these groups, indicating lack of change in plaque cellular composition (Supplemental Figure Xb). Furthermore, S100A9 expression colocalized with RAGE- and ALP-positive cells, as demonstrated by immunofluorescence (Figure 6B). Prior studies identified S100A9-positive EVs were in human calcified plaques17. Consistent with these reports, electron microscopy–based immunogold staining detected S100A9 in calcified EVs of human atheroma (Figure 6C). These results indicate that S100A9-RAGE axis associates with osteogenic activity and the formation of microcalcification in human plaques.
Figure 6. The important role of S100A9 and RAGE in human diabetic calcified carotid arteries.
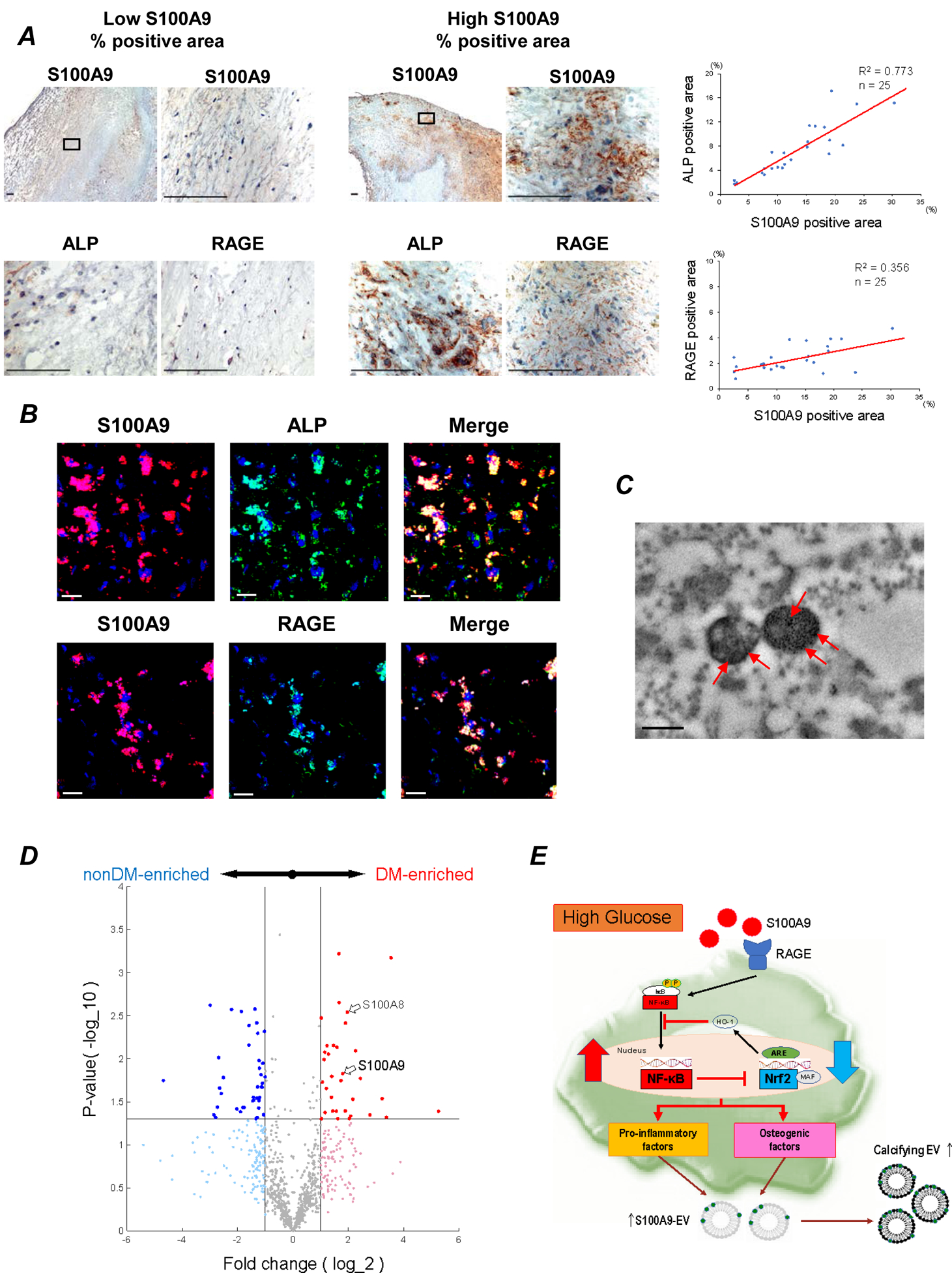
(A) 25 human atheroma obtained from carotid endarterectomy. Staining for S100A9, RAGE, and ALP in human atheroma (n=25). One of low S100A9 % positive area (left panel) and high S100A9 % positive area (right panel) are shown. Scale bars: 100 μm. Each graph shows the correlation between percentage of S100A9 positive area and ALP- and RAGE-positive area. The average of 5–10 high power fields was used for the analysis. (B) Serial section of human atheroma double-immune stained with anti-S100A9 (red) to anti-RAGE or anti-ALP (green) antibodies. Yellow area in Merge indicate S100A9 positive cells for either RAGE or ALP. Scale bars: 20 μm. The data represent 5 donors that showed similar results. (C) Transmission electron microscopy–based immunogold staining of S100A9 (red arrows) in EVs from human carotid plaques. One of 3 plaques is shown. Scale bar: 300 nm. (D) Carotid plaque specimens were obtained from diabetic (n = 4) and non-diabetic patients (n = 4) undergoing carotid endarterectomy. Volcano plot for the proteomics data. Red and blue markers indicate the significantly enriched (p<0.05) proteins in diabetic patient group and non-diabetic patient group, respectively, with a fold change (FC) cutoff of 2.0. (E) The schematic of the potential mechanism for macrophage-mediated EV microcalcification in diabetes. S100A9-RAGE axis may regulate pro-inflammatory and pro-osteogenic macrophage activation via Nrf2 and NF-κB pathways.
Furthermore, we took a proteomics approach to assess S100A9 levels in carotid plaque specimens obtained from diabetic (n = 4) and non-diabetic patients (n = 4) undergoing carotid endarterectomy for stroke prevention. We quantified 984 proteins of which 76 proteins either increased (33 proteins) or decreased (43 proteins) 2-fold between diabetic and non-diabetic patient groups (Supplemental Table 2). S100A9 was enriched in the diabetic patient group compared with the non-diabetic patient group, as was S100A8 (Figure 6D). The proteomics data confirm the increased association of S100A9 in atherosclerotic plaques isolated from diabetic versus non-diabetic patients.
Discussion
This study provides both in vitro and in vivo evidence that diabetes promotes pro-inflammatory and osteogenic activity in macrophages via a mechanism mediated by the S100A9-RAGE axis. These findings further implicate macrophage-mediated EV microcalcification in the development of atherosclerosis in the context of diabetes. The key results demonstrate that (1) S100A9 in the presence of high glucose induces pro-inflammatory and osteogenic activity in human primary macrophages via S100A9-RAGE signaling; (2) S100A9, in the presence of high glucose, promotes the release of calcific EVs from activated human primary macrophages; (3) macrophage-derived EVs are enriched in S100A9; (4) the crosstalk between the Nrf2 and NF-κB pathways contributes to the underlying mechanisms of macrophage activation in a pro-calcific environment; (5) diabetic Apoe−/− mice lacking S100A9 and mice treated with macrophage-targeted lipid nanoparticles encapsulating S100a9 siRNA develop less arterial calcification/microcalcification consistent with the decrease of inflammation, as gauged by micro-CT, molecular imaging, and histological analyses; and (6) in diabetic patients, high levels of S100A9 were detected by proteomics and colocalized with ALP activity and RAGE by immunostaining. These findings point to macrophage S100A9 as a key regulator of vascular calcification in diabetes.
Arterial calcification is a prominent risk marker and a common concomitant of diabetes, chronic kidney disease, and aging33. Patients with diabetic nephropathy are particularly prone to vascular calcification34. Arterial calcification in diabetes associates with high cardiovascular mortality, future coronary heart disease events, stroke and amputation35,36. In patients with chronic kidney disease, increased levels of calcium and inorganic phosphate promote osteogenic differentiation of vascular smooth muscle cells36. Patients with diabetes have more coronary artery calcification than non-diabetic individuals, even in the initial stages of renal impairment34. The present study demonstrated that hyperglycemia induces vascular calcification not via increased levels of calcium and phosphate, but rather at least partially through a direct action of glucose on macrophages and subsequent activation of the S100A9-RAGE axis.
We previously reported that calcification links with inflammation in early atherosclerosis37,38. Macrophages play a central role in atherogenesis and may accelerate cardiovascular disease in diabetes39. Histopathology evaluation of coronary atherectomy specimens demonstrated that patients with diabetes exhibit a greater area occupied by macrophages and higher prevalence of thrombosis, and increased S100 family promote damaging features in the plaque39,40. The present study from both mice and humans demonstrated that macrophages associate with the pro-inflammatory and pro-thrombotic mediator S100A9.
This study further revealed that the S100A9-RAGE axis promotes not only pro-inflammatory functions of macrophages, but also their pro-calcific pathways when exposed to high glucose shown by increased levels of common osteogenic factors, including osteocalcin, osteopontin, BMP2/4, ALP and Runx2. Accumulating evidence suggests that hyperglycemia-dependent overproduction of mitochondrial superoxide results in NF-κB activation41, and the NF-kB pathway induces the expression of S100A942. Furthermore, under conditions of high glucose, advanced glycation end products (AGEs), including S100A9, are formed. The binding of AGEs to RAGE also activates NF-kB via the mitogen-activated protein (MAP) kinase-signaling pathway43. NF-kB induces expression of RAGE, which assembles on the cell surface43. Thus, S100A9 binding to RAGE activates the NF-kB pathway, resulting in the induction of a positive feedback loop44. While macrophages themselves do not calcify, they release calcific EVs that instigate local calcification. We previously proposed that EVs participate actively in both the initiation and progression of microcalcification45. We also demonstrated that S100A9 nucleates microcalcifications derived from mouse RAW264.7 cell EVs46. Consistent with previous observations, we showed that high glucose accelerates the release of EVs enriched in S100A9 and ALP activity from human primary macrophages, supporting the contribution of macrophage-derived EVs to the formation of microcalcifications implicated in plaque destabilization.
The molecular mechanisms by which S100A9 in macrophages promote arterial calcification in diabetes are unknown. Our results demonstrate that decreased Nrf2 activation, in tandem with increased NF-κB activation in macrophages, contributes to the augmented osteogenic phenotype in the setting of diabetes. Nrf2 inhibits osteoblastogenesis by interfering with RUNX2-dependent transcriptional activation29,47. In normal conditions, the balance between Nrf2 and NF-κB maintains redox homeostasis, an equilibrium subject to disturbance in disease24. The Nrf2 pathway inhibits the activation of the NF-κB signaling by amplifying antioxidant defenses, such as HO-1 expression, neutralizing ROS and inhibiting ROS-mediated NF-κB activation48. On the other hand, NF-κB can respond to the cellular redox status since a reducing environment prevents its activation; whereas, oxidative/nitrosative stress promotes phosphorylation and degradation of IκB49. Cells overexpressing NF-κB express less HO-1 and thus NF-κB activation represses Nrf2 transcriptional activity24. S100A9 can induce the secretion of pro-inflammatory cytokines through mechanisms dependent on ROS production and the activation of ROS-sensitive transcription factors, such as NF-kB50. Moreover, hyperglycemia itself also elevates oxidative stress by increasing citric acid cycle flux41, activating NF-kB. These functions of NF-kB thus perturb the Nrf2 and NF-kB pathways, resulting in suppression of Nrf2 activity. Our observations show that, in concert with increased NF-κB activation, Nrf2 activity decreases in macrophages, which may contribute to the increased pro-inflammatory and pro-calcific factors seen in macrophages exposed to elevated glucose. Indeed, activation of Nrf2 induced by VSC2 recovered the imbalance of the Nrf2 and NF-κB pathways, and suppressed the effect of high glucose-S100A9 on the functions of inflammation in calcification. Moreover, we showed that ameliorating the NF-κB-Nrf2 imbalance, caused by high glucose-S100A9 with VSC2, reduced the mRNA levels of the osteogenic genes BMP4, ALP, and osteopontin within macrophage-derived EVs. In the setting of diabetes, macrophages may thus serve as parental cells to convey osteogenic information to another cell by releasing EVs that contain osteogenic mRNA cargo. These results provide evidence that the imbalance of the Nrf2 and NF-κB axis promotes generation of macrophage-derived EVs microcalcifications, which further links the association of inflammation with the formation of microcalcifications.
The present study used in vivo molecular imaging to show that macrophage-specific S100A9 deficiency in diabetic mice significantly decreased macrophage and microcalcification burdens in atherosclerotic plaques, consistent with the decrease in systemic S100A9 deficiency mice. The selectivity of the protective effect of S100A9 deficiency suggests that the inflammatory state of macrophages and calcification in the setting of hyperglycosemia may be differing from that of vascular calcification in the absence of diabetes. Our observations indicate that the inhibitory effect of S100A9 deficiency on the inflammatory phenotype of macrophages associated with diabetes is likely to explain its pro-calcific effects in diabetic mice and suggest that hyperglycosemia-accelerated arterial calcification results from increased inflammation characterized by increased S100A9 expression in macrophages. This study identifies a key step in the mechanism of diabetes-induced macrophage inflammatory changes and vascular calcification.
There are limitations of this study that should be addressed in future research. The study focused on the involvement of the S100A9-RAGE axis in macrophage-mediated EV microcalcification in diabetes. We previously reported that EVs containing ALP and S100A9 aggregate and form microcalcifications7, 46. However, it was not possible to identify the S100A9-RAGE axis at the site where local microcalcification formed while evaluating these associations in human carotid artery samples with currently available research tools. Furthermore, we did not address the effects of macrophage-mediated EVs on SMCs in vascular calcification. In addition, the study demonstrated S100A9-RAGE axis is related to vascular calcification in diabetes, including human primary macrophages, diabetic Apoe−/− mice lacking S100A9 and treated with macrophage-targeted lipid nanoparticles encapsulating S100a9 siRNA, and human carotid endarterectomy specimens. However, we could not address the effects of confounding factors in our human samples. Future studies that are able to overcome these limitations are needed in order to solidify the mechanisms underlying the association between S100A9-RAGE axis and vascular calcification via macrophage-mediated EVs.
In summary, we demonstrated several lines of novel evidence that S100A9-RAGE axis in diabetes induces macrophage-derived EVs microcalcifications via the crosstalk between Nrf2 and NF-κB signaling pathways. No current therapies effectively prevent or treat arterial calcification. The present study may expand mechanistic understanding of this process that may aid the development of such treatments.
Supplementary Material
Highlights.
Macrophage S100A9 plays a key regulator of vascular calcification in diabetes.
Diabetes promotes pro-inflammatory and osteogenic activity in macrophages via a S100A9-RAGE axis-mediated pathway.
Pro-inflammatory macrophages induced by S100A9-RAGE axis release EVs with high calcific potential in diabetic milieu.
Our study reveals a novel mechanism by which the imbalance between the Nrf2 and NF-κB contributes to macrophage activation in pro-calcific environment.
Acknowledgments
We thank Toshiaki Nakano, Gabriel Guimaraes, Whitney S. Golden, Eugenia Shvartz, Shinsuke Ito, Lang H. Lee, Fabrizio Buffolo, and Florian Schlotter for their expert assistance.
Sources of Funding
This study was supported by the National Institutes of Health Grants R01HL136431, R01HL147095 and R01HL141917 to Elena Aikawa, and the International Research Fund for Subsidy of Kagawa University School of Medicine Alumni to Ryo Kawakami.
Non-standard Abbreviations and Acronyms
- AGE
advanced glycation end product
- ALP
alkaline phosphatase
- ApoE
apolipoprotein E
- BCA
brachiocephalic artery
- BMP2
bone morphogenetic protein-2
- BMP4
bone morphogenetic protein-4
- CAC
coronary arterial calcification
- CVD
cardiovascular disease
- EVs
extracellular vesicles
- FRI
fluorescent reflection imaging
- HCD
high-cholesterol diet
- HO-1
hemeoxygenase-1
- IL-1β
interleukin-1 beta
- IL-10
interleukin-10
- IVM
intra-vital microscopy
- LC-MS/MS
liquid chromatography tandem mass spectrometry
- LNPs
lipid nanoparticles
- MCP-1
monocyte chemotactic protein-1
- MRC1
mannose receptor C-type 1
- MRP-14
myeloid-related protein-14
- NF-κB
nuclear factor-kappa B
- NIRF
near-infrared fluorescent
- NQO1
NADPH quinone oxidoreductase 1
- Nrf2
nuclear factor-2 erythroid related factor-2
- NTA
nanoparticle tracking analysis
- PBMC
human peripheral blood mononuclear cells
- qPCR
quantitative polymerase chain reaction
- Runx2
runt-related transcription factor 2
- RAGE
receptor for advanced glycation end product
- SMC
smooth muscle cell
- STZ
streptozotocin
- TNF-α
tumor necrosis factor-alpha
- VSC2
a vinyl sulfone compounds with a methoxy group at the 2′ position
Footnotes
Disclosures
None.
References
- 1.Roth GA, Johnson C, Abajobir A, et al. Global, Regional, and National Burden of Cardiovascular Diseases for 10 Causes, 1990 to 2015. J Am Coll Cardiol. 2017;70:1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cavender MA, Steg PG, Smith SC Jr, Eagle K, Ohman EM, Goto S, Kuder J, Im K, Wilson PW, Bhatt DL; REACH Registry Investigators. Impact of Diabetes Mellitus on Hospitalization for Heart Failure, Cardiovascular Events, and Death: Outcomes at 4 Years From the Reduction of Atherothrombosis for Continued Health (REACH) Registry. Circulation. 2015;132:923–931. [DOI] [PubMed] [Google Scholar]
- 3.Martin SS, Blaha MJ, Blankstein R, Agatston A, Rivera JJ, Virani SS, Ouyang P, Jones SR, Blumenthal RS, Budoff MJ, Nasir K. Dyslipidemia, coronary artery calcium, and incident atherosclerotic cardiovascular disease: implications for statin therapy from the multi-ethnic study of atherosclerosis. Circulation. 2014;129:77–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Criqui MH, Denenberg JO, Ix JH, McClelland RL, Wassel CL, Rifkin DE, Carr JJ, Budoff MJ, Allison MA. Calcium density of coronary artery plaque and risk of incident cardiovascular events. JAMA. 2014;311:271–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vengrenyuk Y, Carlier S, Xanthos S, Cardoso L, Ganatos P, Virmani R, Einav S, Gilchrist L, Weinbaum S. A hypothesis for vulnerable plaque rupture due to stress-induced debonding around cellular microcalcifications in thin fibrous caps. Proc Natl Acad Sci U S A. 2006;103:14678–14683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hutcheson JD, Maldonado N, Aikawa E. Small entities with large impact: microcalcifications and atherosclerotic plaque vulnerability. Curr Opin Lipidol. 2014;25:327–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hutcheson JD, Goettsch C, Bertazzo S, Maldonado N, Ruiz JL, Goh W, Yabusaki K, Faits T, Bouten C, Franck G, Quillard T, Libby P, Aikawa M, Weinbaum S, Aikawa E. Genesis and growth of extracellular-vesicle-derived microcalcification in atherosclerotic plaques. Nat Mater. 2016;15:335–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tintut Y, Patel J, Parhami F, Demer LL. Tumor necrosis factor-alpha promotes in vitro calcification of vascular cells via the cAMP pathway. Circulation. 2000;102:2636–2642. [DOI] [PubMed] [Google Scholar]
- 9.Shao JS, Aly ZA, Lai CF, Cheng SL, Cai J, Huang E, Behrmann A, Towler DA. Vascular Bmp Msx2 Wnt signaling and oxidative stress in arterial calcification. Ann N Y Acad Sci. 2007;1117:40–50. [DOI] [PubMed] [Google Scholar]
- 10.New SE, Aikawa E. Molecular imaging insights into early inflammatory stages of arterial and aortic valve calcification. Circ Res. 2011;108:1381–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kapustin AN, Davies JD, Reynolds JL, McNair R, Jones GT, Sidibe A, Schurgers LJ, Skepper JN, Proudfoot D, Mayr M, Shanahan CM. Calcium regulates key components of vascular smooth muscle cell-derived matrix vesicles to enhance mineralization. Circ Res. 2011;109:e1–12. [DOI] [PubMed] [Google Scholar]
- 12.Kapustin AN, Chatrou ML, Drozdov I, et al. Vascular smooth muscle cell calcification is mediated by regulated exosome secretion. Circ Res. 2015;116:1312–1323. [DOI] [PubMed] [Google Scholar]
- 13.Healy AM, Pickard MD, Pradhan AD, et al. Platelet expression profiling and clinical validation of myeloid-related protein-14 as a novel determinant of cardiovascular events. Circulation. 2006. ;113:2278–2284. [DOI] [PubMed] [Google Scholar]
- 14.Croce K, Gao H, Wang Y, Mooroka T, Sakuma M, Shi C, Sukhova GK, Packard RR, Hogg N, Libby P, Simon DI. Myeloid-related protein-8/14 is critical for the biological response to vascular injury. Circulation. 2009;120:427–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johansson F, Kramer F, Barnhart S, Kanter JE, Vaisar T, Merrill RD, Geng L, Oka K, Chan L, Chait A, Heinecke JW, Bornfeldt KE. Type 1 diabetes promotes disruption of advanced atherosclerotic lesions in LDL receptor-deficient mice. Proc Natl Acad Sci U S A. 2008;105:2082–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hofmann Bowman MA, Gawdzik J, Bukhari U, Husain AN, Toth PT, Kim G, Earley J, McNally EM. S100A12 in vascular smooth muscle accelerates vascular calcification in apolipoprotein E-null mice by activating an osteogenic gene regulatory program. Arterioscler Thromb Vasc Biol. 2011;31:337–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McCormick MM, Rahimi F, Bobryshev YV, Gaus K, Zreiqat H, Cai H, Lord RS, Geczy CL. S100A8 and S100A9 in human arterial wall. Implications for atherogenesis. J Biol Chem. 2005;280:41521–41529. [DOI] [PubMed] [Google Scholar]
- 18.Hofmann MA, Drury S, Fu C, et al. RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell. 1999;97:889–901. [DOI] [PubMed] [Google Scholar]
- 19.Boyd JH, Kan B, Roberts H, Wang Y, Walley KR. S100A8 and S100A9 mediate endotoxin-induced cardiomyocyte dysfunction via the receptor for advanced glycation end products. Circ Res. 2008;102:1239–1246. [DOI] [PubMed] [Google Scholar]
- 20.Leuschner F, Dutta P, Gorbatov R, et al. Therapeutic siRNA silencing in inflammatory monocytes in mice. Nat Biotechnol. 2011;29:1005–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koga J, Nakano T, Dahlman JE, Figueiredo JL, Zhang H, Decano J, Khan OF, Niida T, Iwata H, Aster JC, Yagita H, Anderson DG, Ozaki CK, Aikawa M. Macrophage Notch Ligand Delta-Like 4 Promotes Vein Graft Lesion Development: Implications for the Treatment of Vein Graft Failure. Arterioscler Thromb Vasc Biol. 2015;35:2343–2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bouma G, Coppens JM, Lam-Tse WK, Luini W, Sintnicolaas K, Levering WH, Sozzani S, Drexhage HA, Versnel MA. An increased MRP8/14 expression and adhesion, but a decreased migration towards proinflammatory chemokines of type 1 diabetes monocytes. Clin Exp Immunol. 2005;141:509–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Björk P, Björk A, Vogl T, Stenström M, Liberg D, Olsson A, Roth J, Ivars F, Leanderson T. Identification of human S100A9 as a novel target for treatment of autoimmune disease via binding to quinoline-3-carboxamides. PLoS Biol. 2009;7:e97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ganesh Yerra V, Negi G, Sharma SS, Kumar A. Potential therapeutic effects of the simultaneous targeting of the Nrf2 and NF-κB pathways in diabetic neuropathy. Redox Biol. 2013;1:394–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ghosh S, Karin M. Missing pieces in the NF-kappaB puzzle. Cell. 2002;109:S81–S96. [DOI] [PubMed] [Google Scholar]
- 26.Paine A, Eiz-Vesper B, Blasczyk R, Immenschuh S. Signaling to heme oxygenase-1 and its anti-inflammatory therapeutic potential. Biochem Pharmacol. 2010;80:1895–1903. [DOI] [PubMed] [Google Scholar]
- 27.Rushworth SA, MacEwan DJ, O’Connell MA. Lipopolysaccharide-induced expression of NAD(P)H: quinone oxidoreductase 1 and heme oxygenase-1 protects against excessive inflammatory responses in human monocytes. J Immunol. 2008;181:6730–6737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee JA, Kim JH, Woo SY, Son HJ, Han SH, Jang BK, Choi JW, Kim DJ, Park KD, Hwang O. A novel compound VSC2 has anti-inflammatory and antioxidant properties in microglia and in Parkinson’s disease animal model. Br J Pharmacol. 2015;172:1087–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ha CM, Park S, Choi YK, Jeong JY, Oh CJ, Bae KH, Lee SJ, Kim JH, Park KG, Jun do Y, Lee IK. Activation of Nrf2 by dimethyl fumarate improves vascular calcification. Vascul Pharmacol. 2014;63:29–36. [DOI] [PubMed] [Google Scholar]
- 30.Renard CB, Kramer F, Johansson F, Lamharzi N, Tannock LR, von Herrath MG, Chait A, Bornfeldt KE. Diabetes and diabetes-associated lipid abnormalities have distinct effects on initiation and progression of atherosclerotic lesions. J Clin Invest. 2004;114:659–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Novobrantseva TI, Borodovsky A, Wong J, et al. Systemic RNAi-mediated Gene Silencing in Nonhuman Primate and Rodent Myeloid Cells. Mol Ther Nucleic Acids. 2012;1:e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goldin A, Beckman JA, Schmidt AM, Creager MA. Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation. 2006;114:597–605. [DOI] [PubMed] [Google Scholar]
- 33.Edmonds ME. Medial arterial calcification and diabetes mellitus. Z Kardiol. 2000;89:101–104. [DOI] [PubMed] [Google Scholar]
- 34.Mehrotra R, Budoff M, Christenson P, Ipp E, Takasu J, Gupta A, Norris K, Adler S. Determinants of coronary artery calcification in diabetics with and without nephropathy. Kidney Int. 2004;66:2022–2031. [DOI] [PubMed] [Google Scholar]
- 35.Lehto S, Niskanen L, Suhonen M, Rönnemaa T, Laakso M. Medial artery calcification. A neglected harbinger of cardiovascular complications in non-insulin-dependent diabetes mellitus. Arterioscler Thromb Vasc Biol. 1996;16:978–983. [DOI] [PubMed] [Google Scholar]
- 36.Jono S, McKee MD, Murry CE, Shioi A, Nishizawa Y, Mori K, Morii H, Giachelli CM. Phosphate regulation of vascular smooth muscle cell calcification. Circ Res. 2000;87:E10–7. [DOI] [PubMed] [Google Scholar]
- 37.Aikawa E, Nahrendorf M, Figueiredo JL, Swirski FK, Shtatland T, Kohler RH, Jaffer FA, Aikawa M, Weissleder R. Osteogenesis associates with inflammation in early-stage atherosclerosis evaluated by molecular imaging in vivo. Circulation. 2007;116:2841–2850.. [DOI] [PubMed] [Google Scholar]
- 38.Aikawa E, Aikawa M, Libby P, Figueiredo JL, Rusanescu G, Iwamoto Y, Fukuda D, Kohler RH, Shi GP, Jaffer FA, Weissleder R. Arterial and aortic valve calcification abolished by elastolytic cathepsin S deficiency in chronic renal disease. Circulation. 2009;119:1785–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Burke AP, Kolodgie FD, Zieske A, Fowler DR, Weber DK, Varghese PJ, Farb A, Virmani R. Morphologic findings of coronary atherosclerotic plaques in diabetics: a postmortem study. Arterioscler Thromb Vasc Biol. 2004;24:1266–1271. [DOI] [PubMed] [Google Scholar]
- 40.Moreno PR, Murcia AM, Palacios IF, Leon MN, Bernardi VH, Fuster V, Fallon JT. Coronary composition and macrophage infiltration in atherectomy specimens from patients with diabetes mellitus. Circulation. 2000;102:2180–2184. [DOI] [PubMed] [Google Scholar]
- 41.Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, Giardino I, Brownlee M. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404:787–790. [DOI] [PubMed] [Google Scholar]
- 42.Gebhardt C, Németh J, Angel P, Hess J. S100A8 and S100A9 in inflammation and cancer. Biochem Pharmacol. 2006;72:1622–1631. [DOI] [PubMed] [Google Scholar]
- 43.Bierhaus A, Humpert PM, Morcos M, Wendt T, Chavakis T, Arnold B, Stern DM, Nawroth PP. Understanding RAGE, the receptor for advanced glycation end products. J Mol Med (Berl). 2005;83:876–886. [DOI] [PubMed] [Google Scholar]
- 44.Schmidt AM, Yan SD, Yan SF, Stern DM. The biology of the receptor for advanced glycation end products and its ligands. Biochim Biophys Acta. 2000;1498:99–111. [DOI] [PubMed] [Google Scholar]
- 45.Goettsch C, Hutcheson JD, Aikawa M, et al. Sortilin mediates vascular calcification via its recruitment into extracellular vesicles. J Clin Invest. 2016;126:1323–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.New SE, Goettsch C, Aikawa M, Marchini JF, Shibasaki M, Yabusaki K, Libby P, Shanahan CM, Croce K, Aikawa E. Macrophage-derived matrix vesicles: an alternative novel mechanism for microcalcification in atherosclerotic plaques. Circ Res. 2013;113:72–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hinoi E, Takarada T, Fujimori S, Wang L, Iemata M, Uno K, Yoneda Y. Nuclear factor E2 p45-related factor 2 negatively regulates chondrogenesis. Bone. 2007;40:337–344. [DOI] [PubMed] [Google Scholar]
- 48.Soares MP, Seldon MP, Gregoire IP, Vassilevskaia T, Berberat PO, Yu J, Tsui TY, Bach FH. Heme oxygenase-1 modulates the expression of adhesion molecules associated with endothelial cell activation. J Immunol. 2004;172:3553–3563. [DOI] [PubMed] [Google Scholar]
- 49.Banning A, Brigelius-Flohé R. NF-kappaB, Nrf2, and HO-1 interplay in redox-regulated VCAM-1 expression. Antioxid Redox Signal. 2005;7:889–899. [DOI] [PubMed] [Google Scholar]
- 50.Simard JC, Cesaro A, Chapeton-Montes J, Tardif M, Antoine F, Girard D, Tessier PA. S100A8 and S100A9 induce cytokine expression and regulate the NLRP3 inflammasome via ROS-dependent activation of NF-κB(1.). PLoS One. 2013;8:e72138. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.