

Abstract
Celastrol, derived from the roots of the Tripterygium Wilfordi, has attracted interest for its potential anti-inflammatory and lipid-lowering activities. In the present study, the protective effect of celastrol on carbon tetrachloride (CCl4)-induced acute liver injury was investigated. Celastrol improved the increased transaminase activity, inflammation, and oxidative stress induced by CCl4, resulting in improved metabolic disorders found in mice with liver injury. Dual-luciferase reporter assays and primary hepatocyte studies demonstrated that the peroxisome proliferator-activated receptor α (PPARα) signaling mediated the protective effect of celastrol, which was not observed in Ppara-null mice, and co-treatment of wild-type mice with the PPARα antagonist GW6471. Mechanistically, PPARα deficiency potentiated CCl4-induced liver injury through a deoxycholic acid (DCA)-EGR1-inflammatory factor axis. These data demonstrate a novel role for celastrol in protection against acute liver injury through modulating PPARα signaling.
Keywords: Celastrol, Acute liver injury, PPARα, Metabolomics, LC-MS
1. Introduction
Celastrol is a natural compound isolated from the root extracts of Tripterygium wilfordi (thunder god vine). Celastrol shows significant pharmacological activities, including anti-inflammatory, anti-cancer, anti-obesity and treating mesangioproliferative glomerulonephritis [1–3]. It was reported that celastrol could modulate various targets, such as NFκB, Nur77, and the HSF1-PGC1α axis [2,4,5]. Celastrol protects against experimental acetaminophen (APAP)-induced liver injury and α-naphthyl isothiocyanate (ANIT)-induced cholestasis [6,7]. The liver injury induced by APAP, ANIT, triptolide, and sunitinib, disrupts mitochondrial fatty acid β-oxidation and the accumulated serum long-chain acylcarnitines [8–10]. Peroxisome proliferator-activated receptor α (PPARα) regulates mitochondrial fatty acid transport and β-oxidation, bile acid synthesis, and inflammation [11]. PPARα agonist reverses the increase in acylcarnitines levels that result from mitochondrial damage in mouse models of hepatotoxicity [8]. Therefore, PPARα plays an important role in protecting against chemically-induced liver injury.
LC-MS-based metabolomics has been used to identify drug metabolites related to toxicity [12,13], and was applied to investigate the mechanism of liver injury, including cholestasis [8], liver dysfunction [14], and steatohepatitis [15]. Furthermore, metabolomics could determine the roles of nuclear receptors, such as pregnane X receptor (PXR) [16], farnesoid X receptor (FXR) [17], and PPARα [8] in physiology, metabolic diseases and chemically-induced liver toxicities. The results of the present study reveal that celastrol regulates PPARα signaling and significantly attenuates carbon tetrachloride (CCl4)-induced acute liver injury, through modulation of inflammation, oxidative stress, bile acid metabolism and acylcarnitine utilization. The protective effect of celastrol on CCl4-induced liver injury was attenuated in Ppara-null mice (Ppara−/− mice) and co-treatment with PPARα antagonist GW6471 in wild-type (WT) mice. These findings provide a novel role for celastrol in protecting against acute liver injury through the activation of PPARα signaling.
2. Materials and methods
2.1. Chemicals and reagents
Celastrol was provided by Chengdu Mansite Bio-technology Co Ltd (Chengdu, China). CCl4 and corn oil were obtained from the Shanghai Aladdin Bio-Chem Technology (Shanghai, China). Lauroylcarnitine (12:0-carnitine), myristoylcarnitine (14:0-carnitine), palmitoylcarnitine (16:0-carnitine), stearoylcarnitine (18:0-carnitine), deoxycholic acid (DCA), taurocholic acid (TCA), taurohyodeoxycholic acid (THDCA), and taurochenodeoxycholic acid (TCDCA) were ordered from Sigma-Aldrich (St. Louis, Missouri, USA). Tauro-β-muricholic acid (TβMCA) was provided by Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). Tauro-α-muricholic acid (TαMCA) was purchased from Steraloids (Newport, RI, USA). All other chemical reagents and solvents were of the highest grade commercially available.
2.2. Animals
Male WT mice and Ppara−/− mice (6- to 8-weeks-old) on the 129/Sv genetic background were previously described [18]. All mice were maintained in a controlled environment with a standard 12 h light/12 h dark cycle and humidity 50%–60%. Animal experiments were approved by the institutional ethical committee of Kunming Institute of Botany.
Experiment 1: To determine the protective effect of celastrol on CCl4-induced acute liver damage, the WT mice were randomly divided into four groups (n = 5): (1) control group; (2) CCl4 group; (3) CCl4 + celastrol group; (4) celastrol group (Fig. 1D). The Ppara−/− mice were randomly divided into three groups (n = 5): (1) control group; (2) CCl4 group; (3) CCl4 + celastrol group; (4) celastrol group.CCl4 + celastrol and celastrol groups were orally treated with celastrol (10 mg/kg dissolved in 1% DMSO + 2% Tween 80 + 97% water) for five consecutive days [7]. After the mice were treated with celastrol for three days, the mice of CCl4 and CCl4 + celastrol groups were given a single intraperitoneal dose of CCl4 (20% CCl4 solution in corn oil, 1 ml/kg body weight) [19,20].
Fig. 1.
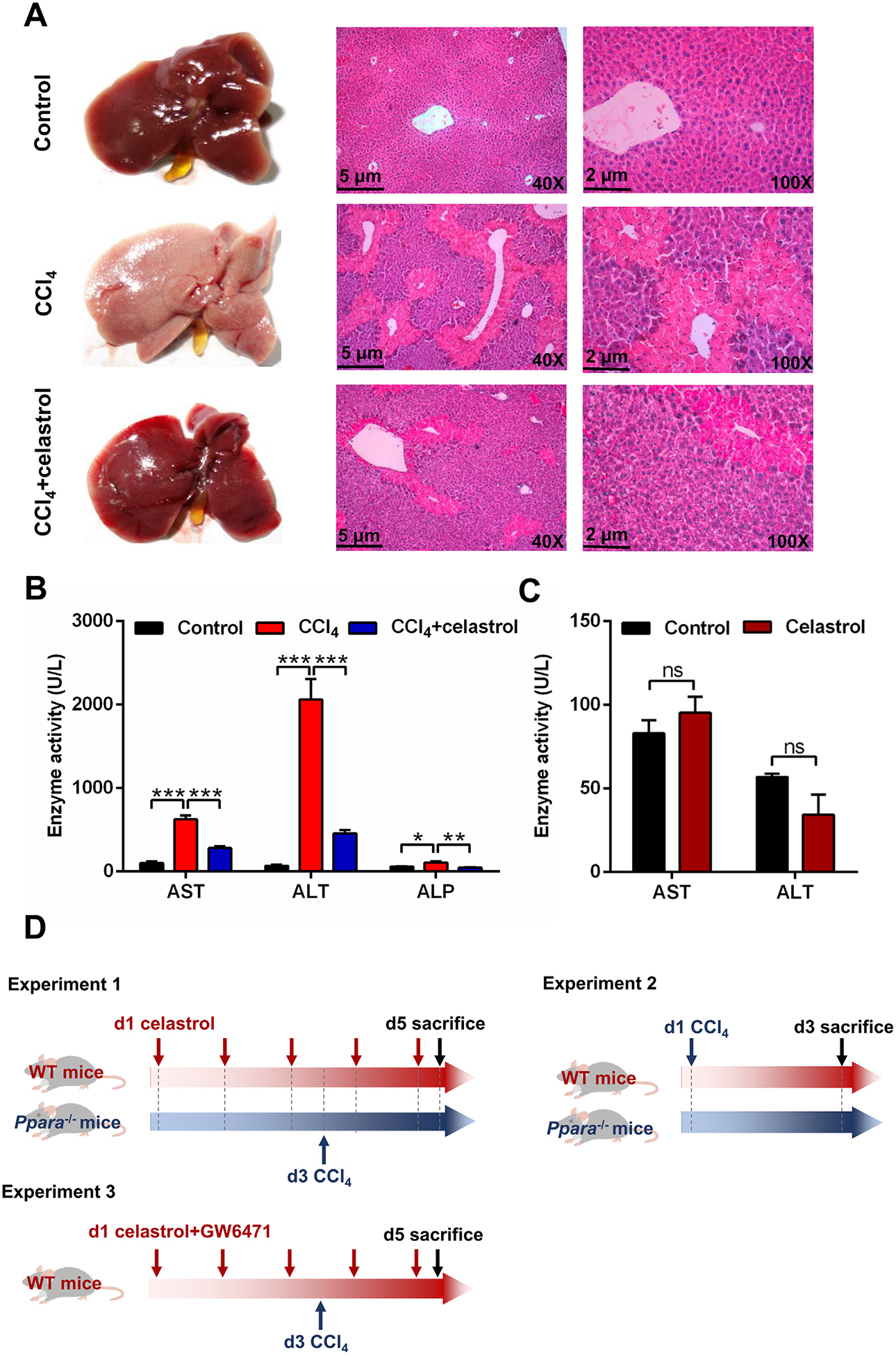
Celastrol attenuated CCl4-induced liver injury in WT mice. (A) Phenotype and H&E staining of liver. (B) Serum AST, ALT, and ALP enzyme activities in control, CCl4, and CCl4 + celastrol groups. (C) Celastrol does not cause hepatotoxicity at the therapeutic dose (10 mg/kg). (D) Experimental scheme for animal experiments. All data are expressed as mean ± SEM (n = 5). *P < 0.05, **P < 0.01, ***P < 0.001, ns = not significant.
Experiment 2: To determine the role of PPARα on CCl4-induced acute liver damage, the WT and Ppara−/− mice were randomly assigned into four groups (n = 5), respectively: (1) control group; (2) CCl4 group; (3) Ppara−/− control group; (4) Ppara−/− CCl4 group (Fig. 1D). CCl4 and Ppara−/− CCl4 groups were given a single intraperitoneal dose of CCl4.
Experiment 3: To investigate the role of PPARα inhibition in the protective effect of celastrol, the WT mice were randomly assigned into four groups (n = 5): (1) control group; (2) CCl4 group; (3) CCl4 + celastrol group; (4) CCl4 + celastrol + GW6471 group (Fig. 1D). The CCl4 + celastrol group was treated with celastrol (10 mg/kg) for 5 consecutive days. The CCl4 + celastrol + GW6471 group was cotreated with GW6471 (10 mg/kg dissolved in 4% DMSO + 2% Tween 80 + 94% normal saline, intraperitoneal administration, 30 min before celastrol) and celastrol for 5 consecutive days. After celastrol treatment for three days, mice in CCl4, CCl4 + celastrol, and CCl4 + celastrol + GW6471 groups were given a single intraperitoneal dose of CCl4.
All mice were anesthetized by CO2 and killed 48 h after CCl4 treatment. Whole blood was collected in anti-coagulative tubes. Plasma was got by centrifugation of the whole blood at 2000 g for 5 min at 4 °C. Partial liver tissue was stored at −80 °C, and partial liver tissue was preserved in 10% buffered formalin for histological analysis.
2.3. Biochemical analysis and histological examination
Assay kits for aspartate aminotransferase (AST), alanine aminotransferase (ALT), alkaline phosphatase (ALP), catalase (CAT), and malondialdehyde (MDA) were obtained from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). The procedure was performed following by the according protocols. The liver tissues were fixed in 10% buffer formalin, which was processed by soaking in different alcoholic concentration gradient, cleared in xylene, and embedded in paraffin. Four μm sections were stained with hematoxylin and eosin and examined by light microscopy.
2.4. Sample preparation and metabolomics analysis
Samples of plasma and liver were prepared using a method described previously [21]. The working conditions of the LC-MS system described in a previous report [8]. A 5 μl aliquot of extract was injected into the UPLC-ESI-QTOFMS system. The chromatographic and spectral data were extracted by MassHunter Workstation software (Agilent, Santa Clara, CA, USA). The data matrix was processed using Mass Profinder software (Agilent, Santa Clara, CA, USA) and analyzed by SIMCA-P + 13.0 (Umetrics, Kinnelon, New Jersey, USA) for principal component analysis (PCA) and orthogonal projection to latent structures-discriminant analysis (OPLS-DA). HMDB was assisted to determine the chemical structures of changed metabolites, which were confirmed by comparing retention time and MS/MS fragmentation with authentic standards (Table 1).
Table 1.
Biomarkers recovered by celastrol.
| No. | Identity | Rt (min) | Observed | Error (ppm) | Formula | MS/MS | |
|---|---|---|---|---|---|---|---|
| PR | HILIC | ||||||
| 1 | C8:0-carnitine | 6.3 | - | 288.2169 | 0.3 | C15H29NO4[H+] | 144;85;60 |
| 2 | C10:0-carnitine | 7.4 | - | 316.2483 | −1.3 | C17H33NO4[H+] | 257;144;85 |
| 3a | C12:0-carnitine | 8.4 | - | 344.2783 | −3.2 | C19H37NO4[H+] | 285;183;144;85;60 |
| 4 | C13:0-carnitine | 8.8 | - | 358.2952 | 0.6 | C20H39NO4[H+] | 299;144;85;60 |
| 5a | C14:0-carnitine | 9.4 | - | 372.3109 | 0.5 | C21H41NO4[H+] | 313;211;144;85 |
| 6 | C15:0-carnitine | 9.7 | - | 386.3231 | −8.3 | C22H43NO4[H+] | 327;144;225;85;60 |
| 7a | C16:0-carnitine | 10.3 | - | 400.3423 | 0.7 | C23H45NO4[H+] | 341;239;144;85 |
| 8 | C17:0-carnitine | 10.8 | - | 414.3573 | −0.7 | C24H47NO4[H+] | 355;85;60 |
| 9a | C18:0-carnitine | 11.3 | - | 428.3711 | −5.1 | C25H49NO4[H+] | 369;267;144;85 |
| 10 | C8:1-carnitine | 4.8 | - | 286.2011 | −0.6 | C15H27NO4[H+] | 85;60 |
| 11 | C10:1-carnitine | 6.9 | - | 314.2329 | 1.0 | C17H31NO4[H+] | 85 |
| 12 | C11:1-carnitine | 7.6 | - | 328.2483 | 0.4 | C18H33NO4[H+] | 85;60 |
| 13 | C12:1-carnitine | 7.9 | - | 342.2657 | 5.8 | C19H35NO4[H+] | 283;85;60 |
| 14 | C14:1-carnitine | 8.9 | - | 370.2943 | −1.9 | C21H39NO4[H+] | 311;209;144;85;60 |
| 15 | C16:1-carnitine | 9.7 | - | 398.3261 | −0.5 | C23H43NO4[H+] | 339;237;144;85;60 |
| 16 | C17:1-carnitine | 10.1 | - | 412.3422 | 0.2 | C24H45NO4[H+] | 353 |
| 17 | C18:1-carnitine | 10.6 | - | 426.3573 | −0.7 | C25H47NO4[H+] | 367;265;144;85;60 |
| 18 | C20:1-carnitine | 11.4 | - | 454.3884 | −1.5 | C27H51NO4[H+] | 395;293;144;85;60 |
| 19 | C10:2-carnitine | 6.6 | - | 312.2173 | 1.4 | C17H29NO4[H+] | 85;60 |
| 20 | C12:2-carnitine | 7.4 | - | 340.2481 | −0.2 | C19H33NO4[H+] | 85;60 |
| 21 | C14:2-carnitine | 8.3 | - | 368.2801 | 1.9 | C21H37NO4[H+] | 207;144;85;60 |
| 22 | C15:2-carnitine | 8.7 | - | 382.2965 | 3.5 | C22H39NO4[H+] | 221;85;60 |
| 23 | C16:2-carnitine | 9.2 | - | 396.3109 | 0.3 | C23H41NO4[H+] | 85;60 |
| 24 | C17:2-carnitine | 9.5 | - | 410.3264 | −0.2 | C24H43NO4[H+] | 85;60 |
| 25 | C18:2-carnitine | 10.0 | - | 424.3443 | 5.4 | C25H45NO4[H+] | 365;263;144;85;60 |
| 26 | C20:2-carnitine | 10.8 | - | 452.3720 | −3.1 | C27H49NO4[H+] | 393;144;85;60 |
| 27 | C8:0-OH-carnitine | 4.1 | - | 304.2113 | −1.3 | C15H29NO5[H+] | 85;60 |
| 28 | C10:0-OH-carnitine | 6.3 | - | 332.2430 | 0.0 | C17H33NO5[H+] | 85 |
| 29 | C12:0-OH-carnitine | 7.4 | - | 360.2743 | −0.6 | C19H37NO5[H+] | 199;144;85 |
| 30 | C13:0-OH-carnitine | 8.2 | - | 374.2907 | 1.7 | C20H39NO5[H+] | 85;60 |
| 31 | C14:0-OH-carnitine | 8.4 | - | 388.3052 | −1.0 | C21H41NO5[H+] | 329;85;60 |
| 32 | C16:0-OH-carnitine | 9.3 | - | 416.3367 | −0.5 | C23H45NO5[H+] | 357;85 |
| 33 | C18:0-OH-carnitine | 10.3 | - | 444.3683 | 0.2 | C25H49NO5[H+] | 283;85;60 |
| 34 | C12:1-OH-carnitine | 6.9 | - | 358.2583 | −1.4 | C19H35NO5[H+] | 85;60 |
| 35 | C14:1-OH-carnitine | 8.0 | - | 386.2896 | −1.0 | C21H39NO5[H+] | 327;225;85 |
| 36 | C16:1-OH-carnitine | 8.7 | - | 414.3223 | 2.4 | C23H43NO5[H+] | 253;144;85;60 |
| 37 | C18:1-OH-carnitine | 9.6 | - | 442.3534 | 1.8 | C25H47NO5[H+] | 383;281;144;85;60 |
| 38 | C20:1-OH-carnitine | 10.5 | - | 470.3844 | 0.8 | C27H51NO5[H+] | 85;60 |
| 39a | DCA | 10.2 | - | 391.2857 | 0.8 | C24H40O4[H−] | 373;355;345;327 |
| 40a | Tα/βMCA | 6.3 | - | 514.2825 | −3.5 | C26H45NO7S[H−] | 124;80 |
| 41a | TCA | 7.2 | - | 514.2873 | 5.8 | C26H45NO7S[H−] | 124;80 |
| 42a | TCDCA | 8.1 | - | 498.2890 | −1.0 | C26H48NO6S[H−] | 124;80 |
| 43a | THDCA | 7.1 | - | 498.2893 | −0.4 | C26H48NO6S[H−] | 124;80 |
| 44a | Glutamine | 0.9 | 5.3 | 147.0762 | −1.2 | C5H10N2O3[H+] | 84;56 |
| 45a | Serine | 0.8 | 5.3 | 106.0497 | −1.9 | C3H7NO3[H+] | 70;60;42 |
| 46a | Methionine | 1.0 | 5.2 | 150.0583 | 0.0 | C5H11O2NS[H+] | 133;104;102;56 |
| 47a | Phenylalanine | 1.0 | 5.0 | 166.0865 | 1.2 | C9H11NO2[H+] | 149;131;120;103;93;77 |
| 48a | Proline | 0.9 | 5.5 | 116.0701 | −4.3 | C5H9NO2[H+] | 58;59;70;74 |
| 49a | Threonine | 0.8 | 5.4 | 120.0653 | −1.7 | C4H9NO3[H+] | 102;84;74;56 |
Confirmed by authentic standards.
2.5. Gene expression analysis
Total RNA was extracted from frozen liver tissues or primary hepatocytes using TRIzol reagent (Life technology, Carlsbad, CA, USA). QPCR was carried out using SYBR green PCR master mix (Takara, Dalian, China) in a CFX Connect Real-Time System (Bio-Rad Laboratories, Hercules, CA, USA). QPCR primer sequences were shown in Table 2. All results were normalized to 18S mRNA. Thermal cycling condition was carried according to a previous study [8].
Table 2.
Primer sequences for QPCR.
| Gene | Abbreviation | Sequence |
|---|---|---|
| Choline kinase α | Chkα | AAAGTGCTCTTGCGGCTCTA GACCTCTCTGCAAGAATGGC |
| Chemokine (C-C motif) ligand 2 | Ccl2 | AGGTCCCTGTCATGCTTCTG GGGATCATCTTGCTGGTGAA |
| Carnitine palmitoyltransferase 1 | Cptlb | CCTCTCATGGTGAACAGCAA GGTCCAGTTTACGGCGATAC |
| Carnitine palmitoyltransferase 2 | Cpt2 | CAGCACAGCATCGTACCCA TCCCAATGCCGTTCTCAAAAT |
| Chemokine (C-X-C motif) ligand 1 | Cxcl1 | AACCGAAGTCATAGCCACAC CAGACGGTGCCATCAGAG |
| Chemokine (C-X-C motif) ligand 10 | CxcllO | TCAGCACCATGAACCCAAG CTATGGCCCTCATTCTCACTG GGGAATGCCATTTACTTGGA GTCCGGATATTCAAGGATGC |
| Cholesterol 7α-hydroxylase | Cyp7a1 | |
| Sterol 12α-hydroxylase | Cyp8b1 | TCCTCAGGGTGGTACAGGAG GATAGGGGAAGAGAGCCACC |
| Early growth response 1 | Egrl | ACGACAGCAGTCCCATCTACTCGG GGACTCGACAGGGCAAGCATATGG |
| Glutathione peroxidase 2 | Gpx2 | GGGCTGTGCTGATTGAGA CGGACATACTTGAGGCTGTT |
| Glutathione peroxidase 3 | Gpx3 | GGCTTCCCTTCCAACC AATTTCTGCTCTTTCTCCC |
| Glutathione peroxidase 4 | Gpx4 | ACGATGCCCACCCACT CCACGCAGCCGTTCTT |
| Glutathione S-transferase α2 | Gsta2 | TTATGTCCCCCAGACCAAAG CCTGTTGCCCACAAGGTAGT |
| Glutathione S-transferase α4 | Gsta4 | AGACCACGGAGAGGCT CCTGACCACCTCAACATAGGG |
| Hydroxyacyl-CoA dehydrogenase | Hadha | AAGGGGATGTGGCAGTTATT ACTCCTGATTTGGTCGTTGG |
| Interleukin 1 | Mb | CCCTGCAGCTGGAGAGTGTGGA TGTGCTCTGCTTGTGAGGTGCTG |
| Interleukin 6 | H6 | TGATGCACTTGCAGAAAACA ACCAGAGGAAATTTTCAATAGGC |
| Lysophosphatidylcholine acyltransferase 1 | Lpcatl | CACGAGCTGCGACTGAGC ATGAAAGCAGCGAACAGGAG |
| Lysophosphatidylcholine acyltransferase 4 | Lpcat4 | GAGTTACACCTCTCCGGCCT GGCCAGAGGAGAAAGAGGAC |
| Medium-chain acyl-CoA dehydrogenase | Mcad | GCGAGCAGAAATGAAACTCC AGCTCTAGACGAAGCCACGA |
| Sodium taurocholate contransporting polypeptide | Ntcp | AGGGGGACATGAACCTCAG TCCGTCGTAGATTCCTTTGC |
| Organic anion transporting protein 1 | Oatpl | ACTCCCATAATGCCCTTGG TAATCGGGCCAACAATCTTC |
| Organic anion transporting protein 4 | Oatp4 | ACCAAACTCAGCATCCAAGC TAGCTGAATGAGAGGGCTGC |
| Phosphate cytidylyltransferase 1 α | Pcyt1α | AGCCCTATGTCAGGGTGACT GGCATGACCAGAGTGAAACA |
| Phosphate cytidylyltransferase 1 β | Pcytlb | ATAGAGCACACATGCCCACA GGCAACGGTCAGTTTTTCAT |
| Phospholipase D1 | Pldl | CTGCATCCTCAAACGGAAAG GCTTGCTGTACTCGCTGTTG |
| Peroxisome proliferator-activated receptor α | Pparα | CCCAAGGGAGGAATAGCTTCT CTCTGCGATGCGGTTCCAA |
| Peroxisome proliferator-activated receptor γ | Pparg | CCACCAACTTCGGAATCAGCT TTTGTGGATCCGGCAGTTAAGA |
| Sphingomyelinphosphodiesterase 3 | Smpd3 | CCTGACCAGTGCCATTCTTT AGAAACCCGGTCCTCGTACT |
| Tumour necrosis factor α | Tnfa | CCACCACGCTCTTCTGTCTAC AGGGTCTGGGCCATAGAACT |
| 18S ribosomal RNA | 18S | ATTGGAGCTGGAATTACCGC CGGCTACCACATCCAAGGAA |
2.6. Primary mouse hepatocytes cultures and luciferase reporter assays
Primary mouse hepatocytes were isolated from 6-week-old 129/Sv mice as described previously [8]. To evaluate the effect of celastrol on PPARα signaling, hepatocytes were harvested after incubation with celastrol (30, 60, and, 120 nM) for 24 h [7,22]. To evaluate the function of DCA, TCA, THDCA, and TCDCA, primary mouse hepatocytes were harvested after incubation with celastrol (120 nM), DCA (50, 100, and 200 μM), TCA (100 μM), THDCA (100 μM), and TCDCA (100 μM) for 24 h [7,22,23].
For luciferase reporter gene assays, HEK293 cells were transfected with PPARα, PPRE-luciferase, and renilla-luciferase [24]. The transfection procedure was detailed in the Lipofectamine 2000 instruction manual (Invitrogen, Grand Island, NY). Transfected cells were treated with celastrol (30, 60, and, 120 nM) and fenofibrate (50 μM) for 24 h [7,22]. Luciferase activity was assayed in a Dual-luciferase Reporter Assay System.
2.7. Data analysis
All data were expressed as mean ± SEM. Statistical analysis was performed using the one-way ANOVA followed by Dunnett’s test. P value < 0.05 was considered statistically significant.
3. Results
3.1. Celastrol protected against CCl4-induced acute liver injury
Celastrol, a pentacyclic triterpene isolated from the roots of the Tripterygium Wilfordi, has anti-inflammatory effects against various inflammatory diseases [1]. Usually, liver injury is accompanied by inflammatory infiltration. Therefore, it was predicted that celastrol could protect mice from acute liver injury induced by CCl4. The hepatic phenotype revealed that the histology of the CCl4 + celastrol group was similar to the control group. Celastrol alleviated the periportal parenchymal necrosis induced by CCl4 (Fig. 1A). The increased AST, ALT, and ALP by CCl4 in mice were significantly decreased by celastrol treatment (Fig. 1B). These results showed that celastrol could protect against CCl4-induced acute liver injury.
3.2. Metabolic disorder of serum and hepatic metabolites was improved by celastrol
PCA modeling was used to analyze the serum data sets from the control, CCl4, and CCl4 + celastrol groups. The CCl4 group was separated from the control and CCl4 + celastrol groups, indicating that celastrol treatment significantly normalized the metabolites changed by CCl4 (Fig. 2A). Three ions m/z 344.2794+, 372.3107+, and 398.3263+ were found to deviate from the ions cloud in OPLS-DA S-plot compared the control group with the CCl4 group (Fig. 2B). Chemical formula calculation showed these three ions corresponded to C19H37NO4, C21H41NO4, and C23H43NO4. These ions m/z 344.2794+ (Rt = 8.426), 372.3107+ (Rt = 9.376), and 398.3263+ (Rt = 9.677) were identified as C12:0-carnitine, C14:0-carnitine, and C16:1-carnitine based on their MS/MS fragmentation, respectively (Fig. 2B and Table 1). Targeted metabolomic analysis showed that celastrol decreased the levels of 29 long-chain acylcarnitines that were increased by CCl4 (Fig. 2C). A previous study found that the increased long-chain acylcarnitine resulted from mitochondrial dysfunction and the inability of efficiently metabolize fatty acids [8]. The mRNA levels of carnitine palmitoyltransferase 1b (Cpt1b) and Cpt2 were increased by CCl4, however, the levels of Ppara, medium-chain acyl-CoA dehydrogenase (Mcad), and hydroxyacyl-CoA dehydrogenase (Hadha) mRNAs remained unchanged in the CCl4 group compared with the control group (Fig. 2D). Using non-targeted metabolomics analysis in positive and negative modes, other metabolites, such as six amino acids and five bile acids, were improved after celastrol treatment (Fig. 2C). The expression level of mRNAs encoded by genes involved in bile acid synthesis (sterol 12α-hydroxylase (Cyp8b1) and cholesterol 7α-hydroxylase (Cyp7a1)) and transport (sodium taurocholate cotransporting polypeptide (Ntcp), organic anion transporting polypeptide 1 (Oatp1), and Oatp4) were measured [8], and also improved by celastrol treatment (Fig. 2E). These results showed that celastrol could improve serum metabolites influenced by CCl4.
Fig. 2.
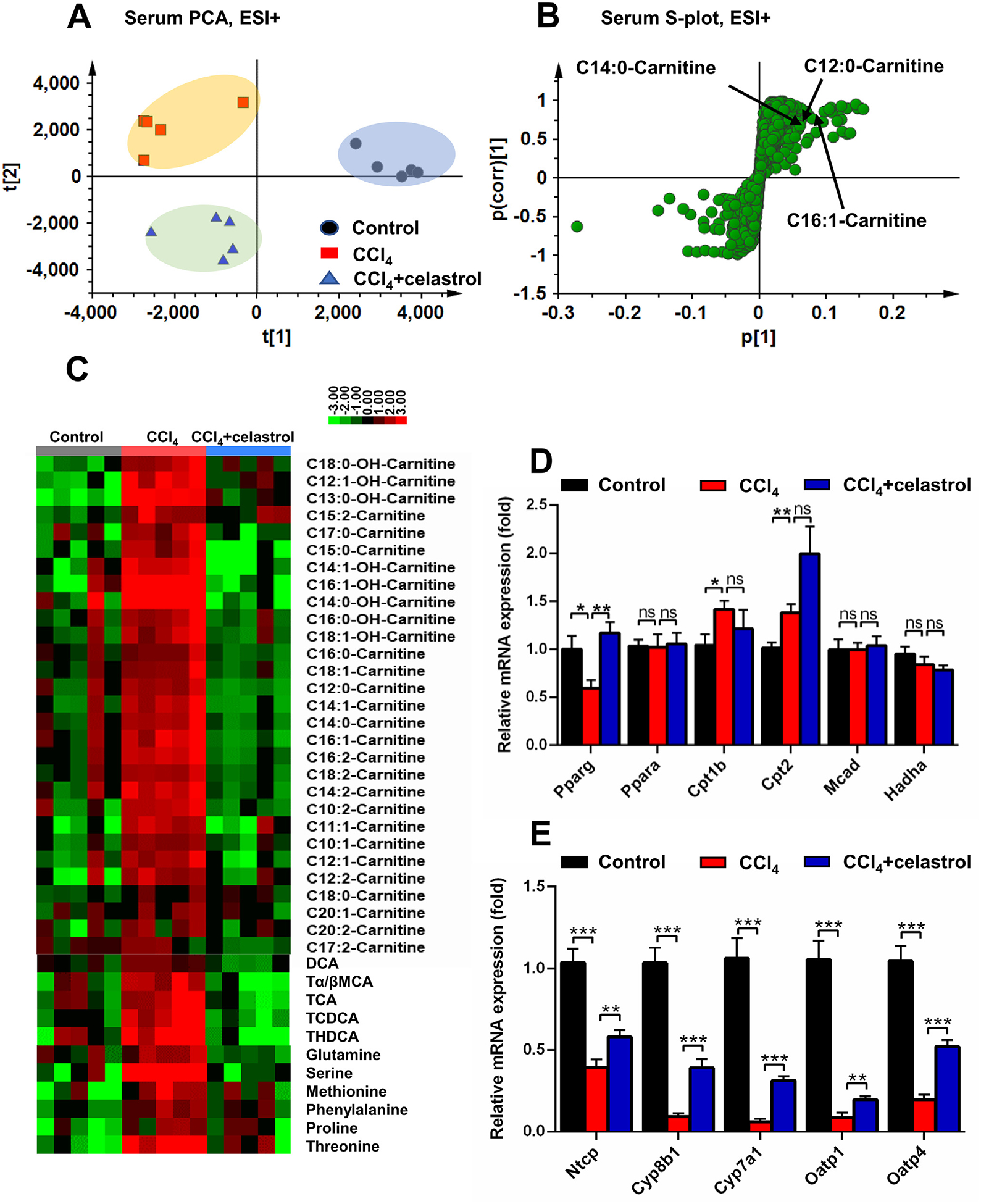
Celastrol decreased the accumulation of acylcarnitines and bile acids induced by CCl4 in serum. PCA score plot (A) and OPLS-DA S-plot (B) derived from LCMS data of serum ions in positive mode. Each point represented an individual mouse serum sample (-left) and an ion in the samples (-right). Metabolites were labeled in the S-plot (●, control group; ■, CCl4 group; ▲, CCl4 + celastrol group). (C) Heat map analysis of the relative abundance of long-chain acylcarnitines, amino acids, and bile acids in serum of control, CCl4, and CCl4 + celastrol groups. QPCR analysis was performed to measure the mRNAs coded by acylcarnitine-related genes and Pparg (D) and bile acid-related genes in liver (E). All data were repressed as mean ± SEM (n = 5). Value represents fold change after normalization to control. *P < 0.05, **P < 0.01, ***P < 0.001, ns = not significant.
PCA and OPLS-DA models were then used to analyze metabolites from livers of control, CCl4, and CCl4 + celastrol groups. Significant differences in the hepatic metabolites between the CCl4 and control groups were found, including increased acylcarnitines, lyso-phosphocholine 18:2 (LPC18:2), and lyso-phosphatidylethanolamine 22:6 (LPE22:6) that largely contributed to the separation (Fig. 3A,B). Further acylcarnitine targeted analysis indicated that the levels of 34 medium-and long-chain acylcarnitines that were increased in the CCl4 group were significantly decreased after celastrol treatment (Fig. 3C). The levels of LPE22:6, LPC16:0, and LPC18:3 were recovered by celastrol (Fig. 3D). The mRNAs associated with LPC metabolism (lysophosphatidylcholine acyltransferase 1 (Lpcat1) and Lpcat4), PC synthesis (choline kinase α (Chka) and phosphate cytidylyltransferase 1 α (Pcyt1a)), PC metabolism (phospholipase D1 (Pld1)), and SM metabolism (sphingomyelin phosphodiesterase 3 (Smpd3)) also improved by celastrol (Fig. 3E). These results showed that celastrol improved hepatic metabolites influenced by CCl4.
Fig. 3.
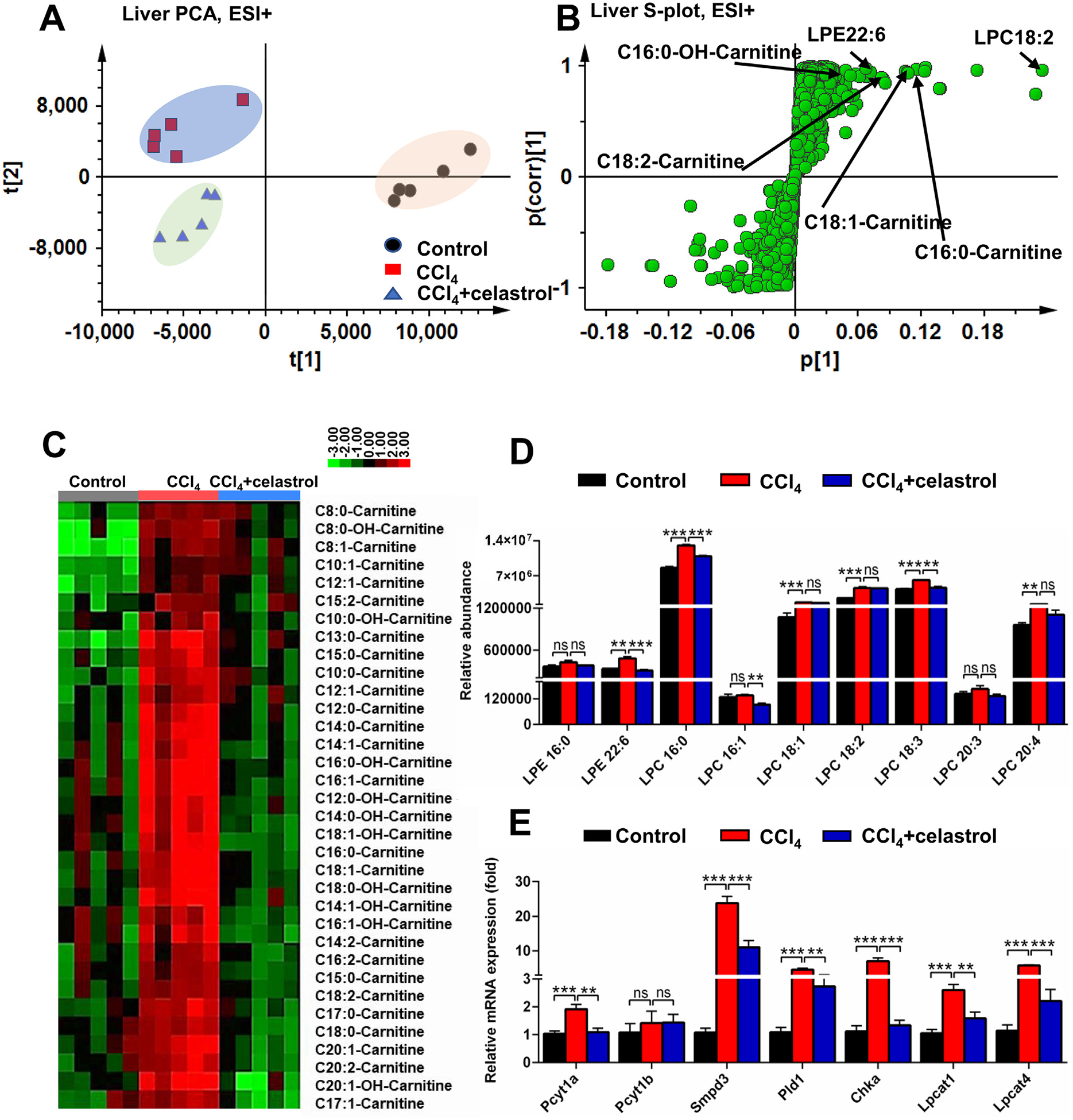
Celastrol decreased the accumulation of acylcarnitines and lipids induced by CCl4 in liver. PCA score plot (A) and OPLS-DA S-plot (B) derived from LC-MS data of hepatic ions in positive mode. Each point represented an individual mouse hepatic sample (-left) and an ion in the samples (-right). Metabolites were labeled in the S-plot (●, control group; ■, CCl4 group; ▲, CCl4 + celastrol group). (C) Heap map analysis of the relative abundance of medium- and long-chain acylcarnitines in liver of control, CCl4, and CCl4 + celastrol groups. (D) Celastrol decreased LPEs and LPCs levels in liver. (E) Lipid-related mRNAs were attenuated after celastrol treatment in liver. All data are expressed as mean ± SEM (n = 5). Values represent fold change after normalization to control. **P < 0.01, ***P < 0.001, ns = not significant.
3.3. Inflammatory cytokine and oxidative stress in acute liver injury were decreased by celastrol
Increased serum acylcarnitines is an indication of mitochondrial dysfunction, which induces oxidative stress in vitro [8], suggesting that the increase of acylcarnitines in CCl4-induced liver injury resulted in increased oxidative stress. Therefore, oxidative stress was evaluated. Hepatic CAT and MDA that were increased in the CCl4 group, were decreased after celastrol treatment (Fig. 4A). The expression levels of several anti-oxidative gene mRNAs that were increased in the CCl4 group, were lower after celastrol treatment, including glutathione peroxidases (glutathione peroxidase 2 (Gpx2), Gpx3, and Gpx4) and glutathione S-transferases (glutathione S-transferase α2 (Gsta2) and Gsta4) (Fig. 4B). Early growth response 1 (EGR1) pathway analysis indicated that the up-regulated Egr1 mRNA and its downstream inflammatory cytokines (chemokine (C-X-C motif) ligand 1 (Cxcl1), chemokine (C–C motif) ligand 2 (Ccl2), Cxcl10, tumor necrosis factor α (Tnfa), and interleukin 6 (Il6)) mRNAs in the CCl4 group were improved by celastrol treatment (Fig. 4C). After celastrol treatment, the expression of Egr1, Cxcl1, Ccl2, Cxcl10, Tnfa, and Il6 mRNAs were reduced 43.5%, 65.2%, 76.2%, 92.8%, 52.4%, and 53.1%, respectively, compared with the CCl4 group. Celastrol did not reverse the expression of Il1b mRNA (Fig. 4C), which was observed in ANIT-induced cholestasis [7]. Whether celastrol can directly combined with these inflammatory cytokines, needs further studies. These results showed that celastrol reduced inflammatory cytokine expression and oxidative stress induced by CCl4.
Fig. 4.
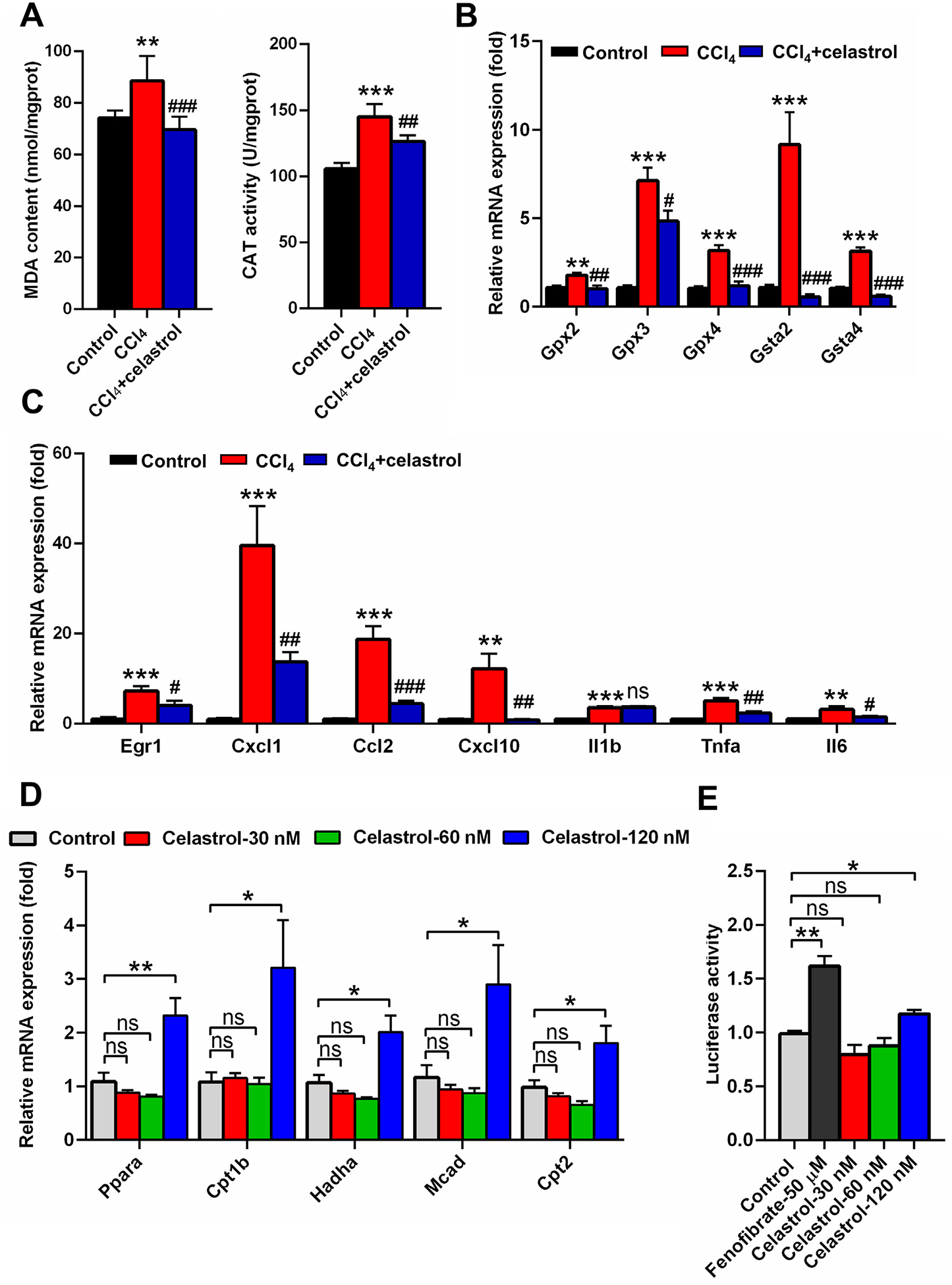
Celastrol eliminated oxidative stress and activated the PPARα signaling pathway. (A) Hepatic MDA and CAT levels in control, CCl4, and CCl4 + celastrol groups. (B) QPCR analysis of the mRNA expression of hepatic Gpx and Gst isoforms. (C) QPCR analysis of the mRNA expression of Egr1 and its downstream genes in liver. **P < 0.01 and ***P < 0.001 verse control; #P < 0.05, ##P < 0.01, ###P < 0.001, and ns means not significant verse CCl4. (D) QPCR analysis of the gene expression of PPARα and its target genes in primary mouse hepatocyte after celastrol treatment for 24 h in vitro. (E) Luciferase assays of the activation of PPARα in HEK293 cells. All data are expressed as mean ± SEM (n = 5). Values represent fold change after normalization to control. *P < 0.05, **P < 0.01, ns = not significant.
3.4. Celastrol activates PPARα signaling pathway
Since the levels of acylcarnitine and lipids were modulated by celastrol, and the PPARα signaling pathway participates in the metabolism of acylcarnitines and lipids [11], the effect of celastrol on PPARα signaling was investigated. Low concentrations of celastrol (120 nM) could activate PPARα and increase its target gene mRNAs Cpt1b, Cpt2, Mcad, and Hadha in primary mouse hepatocytes after a 24 h exposure (Fig. 4D). Dual-luciferase reporter gene assays performed with HEK293 cells co-transfected with PPARα and PPRE-luciferase expression plasmid, demonstrated that 120 nM celastrol significantly increased the luciferase reporter gene activity (Fig. 4E). These results demonstrated a positive regulatory role of celastrol on PPARα signaling.
Furthermore, the role of PPARα in the protective effects of celastrol in the CCl4-induced liver damage was explored using Ppara−/− mice and a PPARα antagonist. H&E staining revealed that the CCl4-induced liver injury was not attenuated by celastrol in Ppara−/− mice (Fig. 5A). The decreased AST, ALT, and ALP levels after celastrol treatment in WT mice were not observed in the Ppara−/− mice (Fig. 5B). Serum and hepatic metabolomics analysis in positive and negative modes showed that the levels of metabolites, such as acylcarnitines, bile acids, and amine acids, in the CCl4 + celastrol group were similar to the CCl4 group metabolites in Ppara−/− mice (Figs. 5C–E and 6A–E). Although celastrol increased AST and ALT levels in Ppara−/− mice (Fig. 5B), the ALP levels, histologic injury, and level of metabolites were not increased by celastrol (Figs. 5 and 6A–E). Hepatotoxicity was also not observed in WT mice treated for 5 days with celastrol (Fig. 1C). These data suggested that the levels of ALT and AST were more sensitive to celastrol exposure when PPARα signaling is low under pathological conditions. At the same time, the protective effect of celastrol was attenuated in WT mice after cotreatment with the PPARα antagonist GW6471 (Fig. 6F). These results showed that the protective effect of celastrol on liver injury was via activation of PPARα signaling.
Fig. 5.
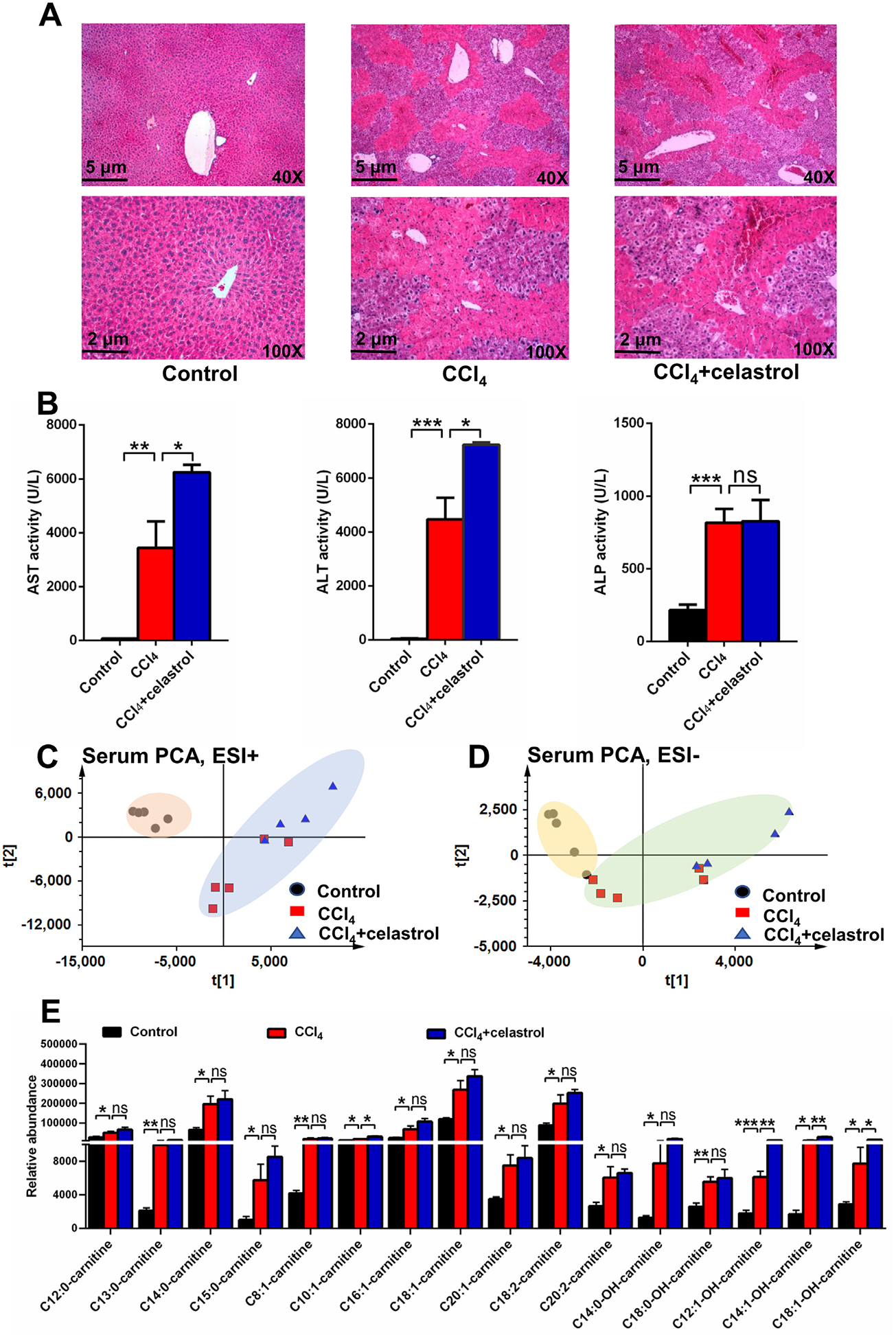
Role of PPARα in celastrol protected against liver injury using Ppara−/− mice. (A) H&E staining of liver. (B) Serum AST, ALT, and ALP enzyme activities. PCA score plot derived from LC-MS data of serum ions in both positive (C) and negative (D) modes. Each point represents an individual mouse serum sample (●, control group; ■, CCl4 group; ▲, CCl4 + celastrol group). (E) Serum acylcarnitine levels in Ppara−/− mice. All data are expressed as mean ± SEM (n = 5). *P < 0.05, **P < 0.01, ***P < 0.001, ns = not significant.
Fig. 6.
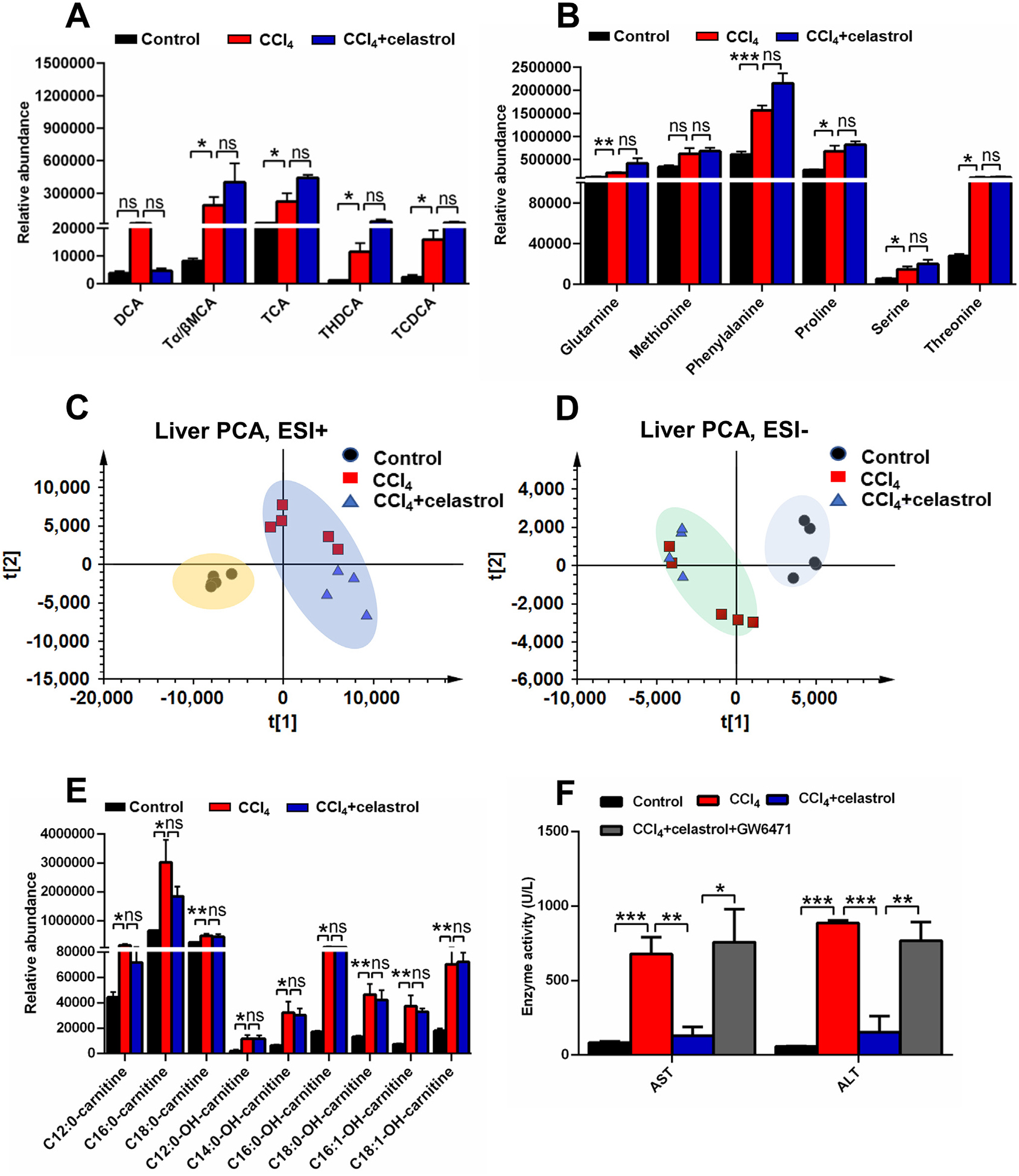
Role of celastrol was dependent on PPARα using Ppara−/− mice and PPARα inhibition GW6471. Serum bile acids (A) and amino acids (B) levels in Ppara−/− mice. PCA score plot derived from LC-MS data of liver ions in both positive (C) and negative (D) modes. Each point represented an individual mouse serum sample (●, control group; ■, CCl4 group; ▲, CCl4 + celastrol group). (E)Hepatic acylcarnitine levels in Ppara−/− mice. (F) The protective effect of celastrol was attenuated after GW6471 cotreatment. All data are expressed as mean ± SEM (n = 5). *P < 0.05, **P < 0.01, ***P < 0.001, ns = not significant.
3.5. Deficiency of PPARα increased CCl4-induced liver injury
Ppara−/− mice were used to evaluate the role of PPARα in CCl4-induced acute liver injury. Histology analysis showed that CCl4 induced obvious parenchymal necrosis in WT mice, and the parenchymal necrosis was more severe in Ppara−/− mice (Fig. 7A). The levels of AST, ALT, and ALP in CCl4-induced liver injury were higher in Ppara−/− mice compared with WT mice (Fig. 7B). Serum metabolomics was used to determine the differences among control, CCl4, Ppara−/− control, and Ppara−/− CCl4 groups (Fig. 7C). The four top increased ions 391.2854−, 498.2895−, 514.2843−, and 514.2843− were observed in control group compared with CCl4 group (Fig. 7D). These ions were identified as DCA, THDCA, TCA, and Tα/βMCA, respectively. Bile acid analysis revealed that the increase of bile acids in the CCl4 group was further increased in the Ppara−/− CCl4 group, especially DCA (Fig. 7E). Other bile acids such as Tα/βMCA, TCA, THDCA, and TCDCA were not significantly increased in the Ppara−/− CCl4 group compared with the CCl4 group (Fig. 7E). The mRNA levels produced by bile acids synthesis and transport genes were further decreased in Ppara−/− mice treated with CCl4 (Fig. 7F). DCA, THDCA and TCDCA could increase the expression of Egr1 mRNA and its downstream inflammatory cytokine Cxcl10 mRNA in mouse primary hepatocytes (Fig. 8A,B). Celastrol reversed the down-regulation of cell viability induced by DCA (Fig. 8C), and inhibited the increase of Egr1 and Il6 mRNA expression (Fig. 8D). These results indicated that PPARα plays an important role in CCl4-induced liver injury, and the potentiation of CCl4-induced liver injury in Ppara−/− mice might be due to DCA-EGR1-inflammatory factor pathway.
Fig. 7.
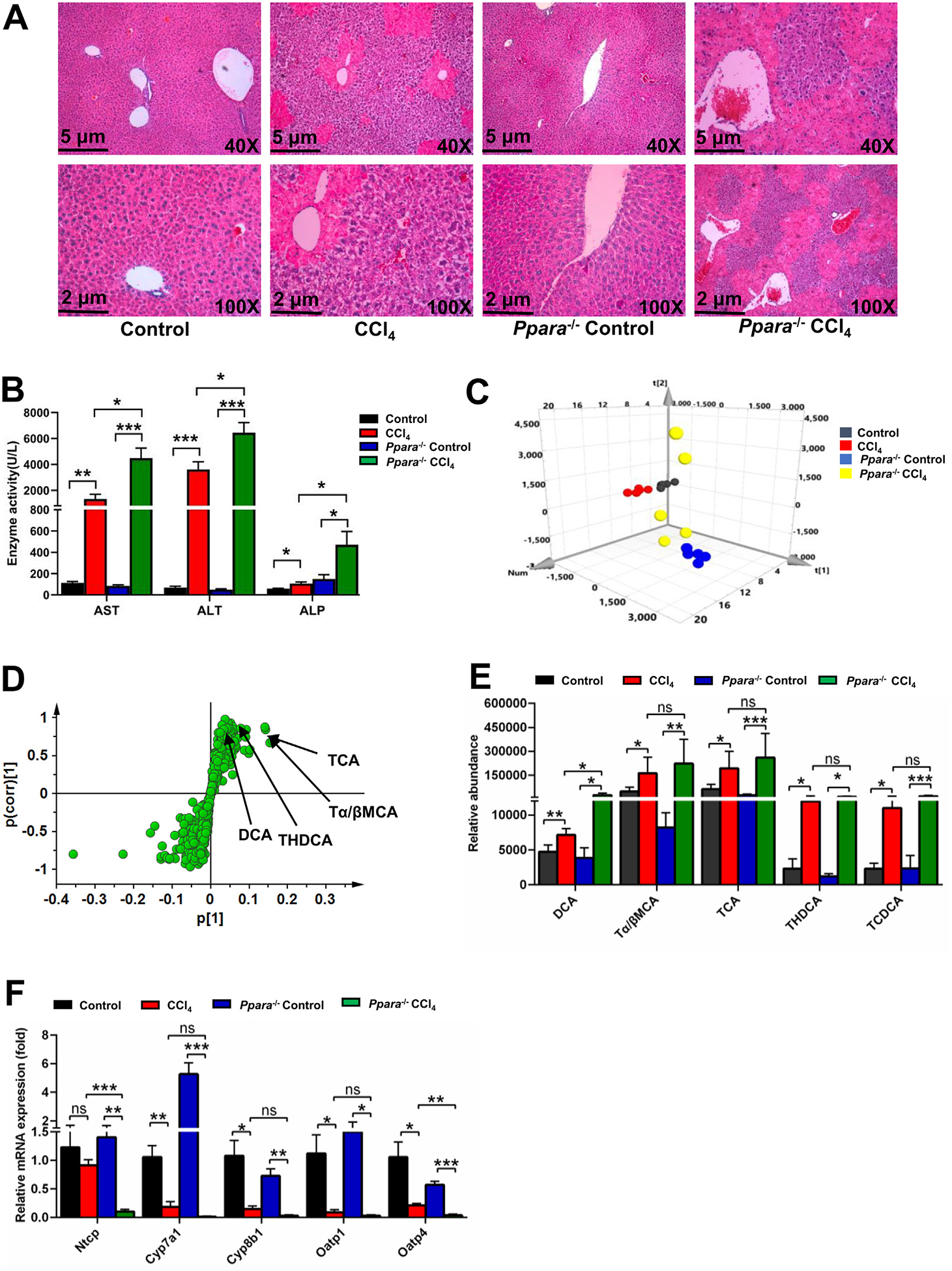
Role of PPARα in CCl4-induced liver injury using Ppara−/− mice. (A) H&E staining of liver. (B) Serum AST, ALT, and ALP enzyme activities. PCA score plot (C) and OPLS-DA S-plot (D) derived from LC-MS data of serum ions. Each point represented an individual mouse serum sample (-up) and an ion in the sample (-down). Metabolites were labeled in the OPLS-DA S-plot. (E) Serum bile acids levels in CCl4-induced liver injury. (F) QPCR analysis of the hepatic mRNA expression of bile acids synthesis and basolateral uptake transporters. All data are expressed as mean ± SEM (n = 5). Values represent fold change after normalization to control. *P < 0.05, **P < 0.01, ***P < 0.001, ns = not significant.
Fig. 8.
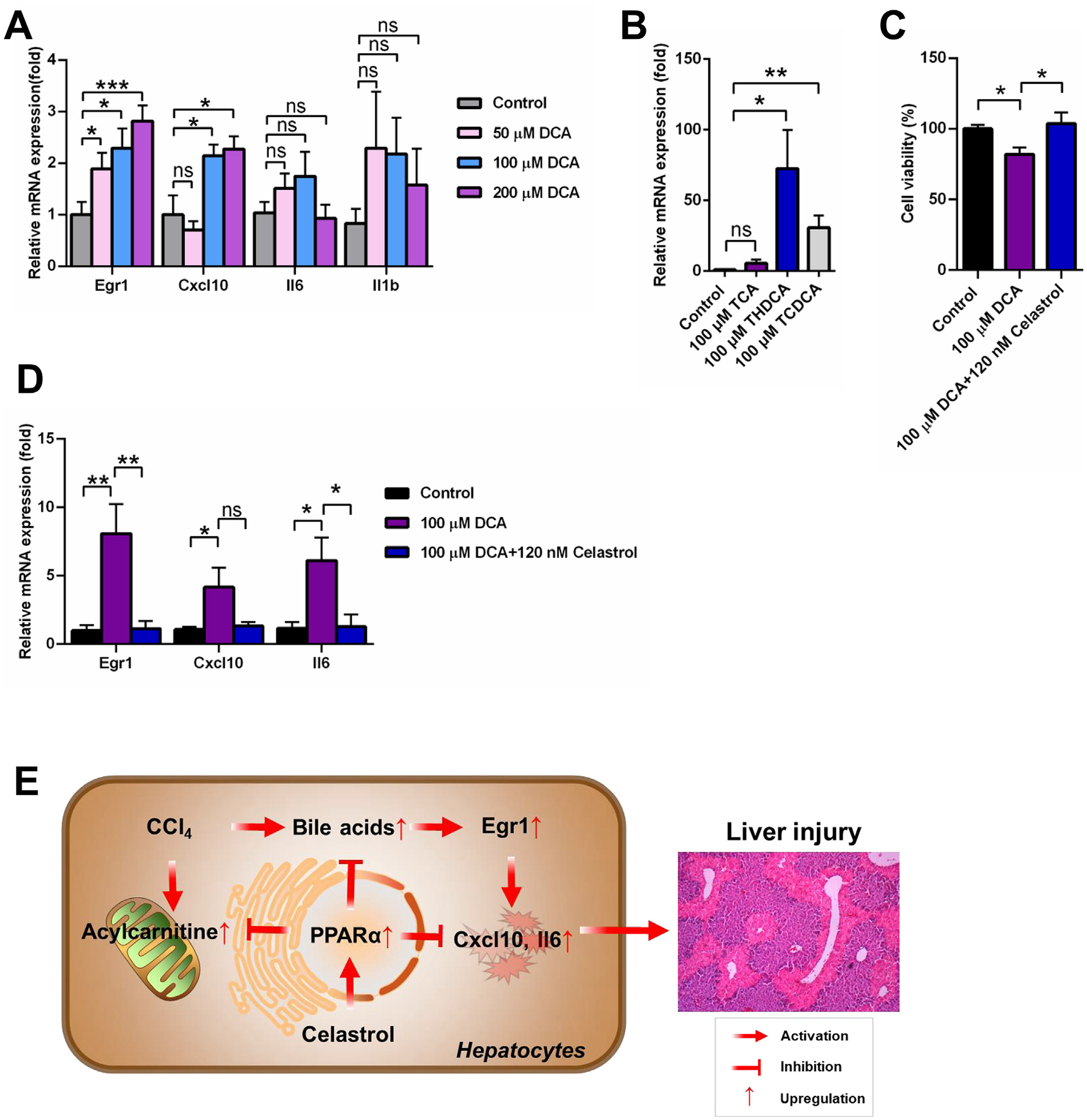
Bile acids, especially DCA, activated EGR1-inflammatory factor axis. (A) DCA increased Egr1 mRNA and its downstream mRNAs Cxcl10 in primary mouse hepatocyte. (B) THDCA and TCDCA increased Egr1 mRNA in primary mouse hepatocyte. (C) Celastrol reversed the down-regulation of cell viability induced by DCA in primary mouse hepatocyte. (D) Celastrol reversed the increase of Egr1 and its downstream genes in primary mouse hepatocyte. (E) Proposed mechanism of hepatoprotective effect of celastrol against liver injury. All data are expressed as mean ± SEM (n = 5). Value represented fold change after normalization to control. *P < 0.05, **P < 0.01, ***P < 0.001, ns = not significant.
4. Discussion
The present study demonstrated a protective role for celastrol in CCl4-induced acute liver injury. The CCl4 model is frequently-used to study hepatotoxicity and liver fibrosis and to evaluate the hepatoprotective effects of drugs or natural products. One of the intriguing findings in this study was that celastrol could activate PPARα in primary hepatocytes and luciferase reporter gene assays. Further studies using Ppara−/− mice revealed a protective role for celastrol dependent on PPARα activation (Fig. 8E).
Long-chain acylcarnitines are associated with the cellular stress response and activate some receptors, such as TLR2, NFκB, and JNK [25]. A previous study found that elevated long-chain acylcarnitines protected TP-induced liver injury through activation of the NOTCH-NRF2 pathway and induced a defense response against liver redundant line feed injury in vitro and in vivo [9]. More importantly, acylcarnitines that result from impaired mitochondrial function and fatty acid β-oxidation, were common biomarkers for liver injury. Acylcarnitines levels were increased and mitochondrial fatty acid β-oxidation was inhibited in triptolide-, ANIT-, APAP-, and sunitinib-induced liver injury [8–10,26]. Serum acylcarnitines levels were used as clinical biomarkers for screening inborn genetic defects in fatty acid β-oxidation [27]. The current study found that acylcarnitine levels were significantly increased after CCl4 treatment, indicating that mitochondrial fatty acid β-oxidation was disrupted. It was reported that PPARα activation could reduce the accumulation of acylcarnitines and increased mitochondrial fatty acid β-oxidation in mice administered ANIT and cocaine [8,28]. Therefore, PPARα activation may be considered as a therapeutic target for the treatment of chemical-induced liver injury.
PPARα is expressed in metabolically-active tissues and regulates genes involved in mitochondrial and peroxisomal fatty acid β-oxidation, bile acid and amino acid metabolism, and inflammation [11]. PPARα also plays an important role in liver injury. PPARα deficiency potentiates chemically-induced hepatic injury. PPARα expression was lower in patients with hepatitis C virus (HCV) infection and steatohepatitis [29,30]. Ppara−/− mice treated with cholic acid had disrupted bile acid and phospholipids homeostasis, indicating that PPARα was an essential regulator of bile acid synthesis and secretion [31]. Activation of PPARα may protect against ANIT-induced cholestasis [8], sunitinib-induced liver injury [10], concanavalin A- or diet-induced hepatitis [32,33], triptolide-induced liver injury [9], and pyrazinamide-induced hepatotoxicity [34]. Many traditional Chinese medicines protect against liver injury through activating PPARα signaling. For example, formononetin could activate PPARα, thereby inhibiting hepatic inflammation in cholestasis [35]. Nuciferine attenuated hepatic steatosis by activating the PPARα/PGC1α pathway [36]. In the present study, in vitro data demonstrated that celastrol was a PPARα agonist. The protective role of celastrol disappeared in CCl4-induced liver injury when celastrol was co-treated with the PPARα antagonist GW6471. More importantly, celastrol could not protect against CCl4-induced liver injury in Ppara−/− mice, demonstrating the actual role of PPARα in the protective effect of celastrol. The activation of the Cpt1b and Cpt2 expression by celastrol would contribute to the elimination of acylcarnitines and improve the disruption of mitochondrial fatty acid β-oxidation by CCl4. Therefore, this study provided evidences that the PPARα signaling has an important role in the protective effects of celastrol against chemically-induced liver injury.
Generally, bile acids facilitate the digestion and absorption of fat. But excessive accumulation of bile acids can result in apoptosis and inflammation in vivo and in vitro. It was reported that DCA could induce apoptosis in hepatocytes upon activation of death receptors [37]. DCA enhanced miR-34a/SIRT1/p53 proapoptotic signaling in a dose and time-dependent manner [23]. Furthermore, bile acids, such as DCA, chenodeoxycholic acid (CDCA), and TCA, up-regulated EGR1, which then regulated production of inflammatory mediators [38]. A previous study found that celastrol modulated the expression of pro-inflammatory cytokines [1]. Therefore, we hypothesized that celastrol could protect DCA-induced inflammatory. Indeed, DCA elevated the expression of Egr1 mRNA and the inflammatory factors Cxcl10 mRNAs. Celastrol improved the hepatic EGR1-inflammatory factor pathway that was elevated by CCl4 treatment. Several bile acids, including DCA, THDCA, and TCDCA, could activate EGR1-infalmmaroty factor pathway in the present study. Compared with the WT mice, the increase of DCA was higher than other bile acid metabolites in Ppara−/− mice after CCl4 treatment, suggesting that DCA likely contributed to increased inflammation.
In this study, the protective role of celastrol in acute liver injury induced by CCl4 exposure was demonstrated by histological and biochemical analysis, and the mechanism was deciphered using UPLC-ESI-QTOFMS-based metabolomics, primary hgepatocyte cultures, reporter gene assays, and Ppara−/− mice. Finally, celastrol decreased proinflammatory cytokines and oxidative stress, and recovered the bile acid and acylcarnitine homeostasis from mice treated with CCl4. This study revealed that PPARα activation by celastrol significantly attenuated CCl4-induced liver injury.
Acknowledgements
This work was supported by the National Key Research and Development Program of China (2017YFC1700906, 2017YFC1702900), Double Thousand Program of Jiangxi Province (jxsq2018102022), CAS “Light of West China” Program (Y72E8211W1).
Abbreviations:
- ANIT
α-naphthyl isothiocyanate
- ALP
alkaline phosphatase
- ALT
alanine aminotransferase
- APAP
acetaminophen
- AST
aspartate aminotransferase
- CAT
catalase
- Ccl2
chemokine (C-C motif) ligand 2
- CCl4
carbon tetrachloride
- Chka
choline kinase α
- Cpt2
carnitine palmitoyltransferase 2
- Cxcl1
chemokine (C-X-C motif) ligand 1
- Cyp7a1
cholesterol 7α-hydroxylase
- Cyp8b1
sterol 12α-hydroxylase
- DCA
deoxycholic acid
- Egr1
early growth response 1
- Gpx2
glutathione peroxidase 2
- Gsta2
glutathione S-transferase α2
- Hadha
hydroxyacyl-CoA dehydrogenase
- Il6
interleukin 6
- Lpcat1
lysophosphatidylcholine acyltransferase 1
- Mcad
medium-chain acyl-CoA dehydrogenase
- MDA
malondialdehyde
- Ntcp
sodium taurocholate cotransporting polypeptide
- Oatp1
organic anion transporting polypeptide 1
- OPLS-DA
orthogonal projection to latent structures-discriminant analysis
- PCA
principal component analysis
- Pcyt1a
phosphate cytidylyltransferase 1α
- Pld1
phospholipase D1
- Ppara-null mice
Ppara−/− mice
- PPARα
peroxisome proliferator-activated receptor α
- Smpd3
sphingomyelin phosphodiesterase 3
- TCA
taurocholic acid
- TCDCA
taurochenodeoxycholic acid
- THDCA
taurohyodeoxycholic acid
- Tnfa
tumour necrosis factor α
- WT
wild type
- 12:0-carnitine
lauroylcarnitine
- 14:0-carnitine
myristoylcarnitine
- 16:0-carnitine
palmitoylcarnitine
- 18:0-carnitine
stearoylcarnitine
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Compliance with ethical standards
All procedures performed in studies involving animals were approval by Animal Experimental Ethics Committee, Kunming Institute of Botany, Chinese Academy of Sciences, Kunming, China.
References
- [1].Kannaiyan R, Shanmugam MK, Sethi G, Molecular targets of celastrol derived from Thunder of God Vine: Potential role in the treatment of inflammatory disorders and cancer, Cancer Lett. 303 (2011) 9–20. [DOI] [PubMed] [Google Scholar]
- [2].Hu M, Luo Q, Alitongbieke G, Chong S, Xu C, Xie L, Chen X, Zhang D, Zhou Y,Wang Z, Ye X, Cai L, Zhang F, Chen H, Jiang F, Fang H, Yang S, Liu J,Diaz-Meco MT, Su Y, Zhou H, Moscat J, Lin X, Zhang XK, Celastrol-induced Nur77 interaction with TRAF2 alleviates inflammation by promoting mitochondrial ubiquitination and autophagy, Mol. Cell 66 (2017) 141–153.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Guo L, Luo S, Du Z, Zhou M, Li P, Fu Y, Sun X, Huang Y, Zhang Z, Targeted delivery of celastrol to mesangial cells is effective against mesangioproliferative glomerulonephritis, Nat. Commun 8 (2017) 878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Liu X, Cai F, Zhang Y, Yang A, Liu L, Celastrol, an NF-κB inhibitor, ameliorates hypercalciuria and articular cartilage lesions in a mouse model of secondary osteoporosis, J. Pharmacol. Sci 130 (2016) 204–211. [DOI] [PubMed] [Google Scholar]
- [5].Ma X, Xu L, Alberobello AT, Gavrilova O, Bagattin A, Skarulis M, Liu J,Finkel T, Mueller E, Celastrol protects against obesity and metabolic dysfunction through activation of a HSF1-PGC1alpha transcriptional axis, Cell Metab. 22 (2015) 695–708. [DOI] [PubMed] [Google Scholar]
- [6].Abdelaziz HA, Shaker ME, Hamed MF, Gameil NM, Repression of acetaminophen-induced hepatotoxicity by a combination of celastrol and brilliant blue G, Toxicol. Lett 275 (2017) 6–18. [DOI] [PubMed] [Google Scholar]
- [7].Zhao Q, Liu F, Cheng Y, Xiao XR, Hu DD, Tang YM, Bao WM, Yang JH,Jiang T, Hu JP, Gonzalez FJ, Li F, Celastrol protects from cholestatic liver injury through modulation of SIRT1-FXR signaling, Mol. Cell. Proteomics 18 (2019) 520–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zhao Q, Yang R, Wang J, Hu DD, Li F, PPARα activation protects against cholestatic liver injury, Sci. Rep 7 (2017) 9967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Zhu X, Wang YK, Yang XN, Xiao XR, Zhang T, Yang XW, Qin HB, Li F, Metabolic activation of myristicin and its role in cellular toxicity, J. Agr. Food Chem 67 (2019) 4328–4336. [DOI] [PubMed] [Google Scholar]
- [10].Hu DD, Zhao Q, Cheng Y, Xiao XR, Huang JF, Qu Y, Li X, Tang YM, Bao WM, Yang JH, Jiang T, Hu JP, Gonzalez FJ, Li F, The protective roles of PPARα activation in triptolide-induced liver injury, Toxicol. Sci 171 (2019) 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Mandard S, Muller M, Kersten S, Peroxisome proliferator-activated receptor α target genes, Cell. Mol. Life Sci 61 (2004) 393–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Li F, Patterson AD, Krausz KW, Dick B, Frey FJ, Gonzalez FJ, Idle JR, Metabolomics reveals the metabolic map of procainamide in humans and mice, Biochem. Pharmacol 83 (2012) 1435–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Li F, Patterson AD, Hofer CC, Krausz KW, Gonzalez FJ, Idle JR, A comprehensive understanding of thioTEPA metabolism in the mouse using UPLC-ESIQTOFMS-based metabolomics, Biochem. Pharmacol 81 (2011) 1043–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Li F, Patterson AD, Krausz KW, Jiang C, Bi H, Sowers AL, Cook JA,Mitchell JB, Gonzalez FJ, Metabolomics reveals that tumor xenografts induce liver dysfunction, Mol. Cell. Proteomics 12 (2013) 2126–2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Tanaka N, Takahashi S, Hu X, Lu Y, Fujimori N, Golla S, Fang Z-Z, Aoyama T, Krausz KW, Gonzalez FJ, Growth arrest and DNA damage-inducible 45α protects against nonalcoholic steatohepatitis induced by methionine- and choline-deficient diet, BBA-Mol. Basis Dis 2017 (1863) 3170–3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Li F, Lu J, Cheng J, Wang L, Matsubara T, Csanaky IL, Klaassen CD,Gonzalez FJ, Ma X, Human PXR modulates hepatotoxicity associated with rifampicin and isoniazid co-therapy, Nat. Med 19 (2013) 418–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Li F, Jiang C, Krausz KW, Li Y, Albert I, Hao H, Fabre KM, Mitchell JB, Patterson AD, Gonzalez FJ, Microbiome remodelling leads to inhibition of intestinal farnesoid X receptor signalling and decreased obesity, Nat. Commun 4 (2013) 2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lee SS, Pineau T, Drago J, Lee EJ, Owens JW, Kroetz DL, Fernandez-Salguero PM, Westphal H, Gonzalez FJ, Targeted disruption of the α isoform of the peroxisome proliferator-activated receptor gene in mice results in abolishment of the pleiotropic effects of peroxisome proliferators, Mol. Cell. Biol 15 (1995) 3012–3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lee BH, Huang YY, Duh PD, Wu SC, Hepatoprotection of emodin and Polygonum multiflorum against CCl4-induced liver injury, Pharm. Biol 50 (2012) 351–359. [DOI] [PubMed] [Google Scholar]
- [20].Takahashi S, Tanaka N, Golla S, Fukami T, Krausz KW, Polunas MA, Weig BC,Masuo Y, Xie C, Jiang C, Gonzalez FJ, Editor’s highlight: farnesoid X receptor protects against low-dose carbon tetrachloride-induced liver injury through the taurocholate-JNK pathway, Toxicol. Sci 158 (2017) 334–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Yang XN, Liu XM, Fang JH, Zhu X, Yang XW, Xiao XR, Huang JF,Gonzalez FJ, Li F, PPARα mediates the hepatoprotective effects of nutmeg, J. Proteome Res 17 (2018) 1887–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhang T, Zhao Q, Xiao X, Yang R, Hu D, Zhu X, Gonzalez FJ, Li F, Modulation of lipid metabolism by celastrol, J. Proteome Res 18 (2019) 1133–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ferreira DM, Afonso MB, Rodrigues PM, Simao AL, Pereira DM,Borralho PM, Rodrigues CM, Castro RE, c-Jun N-terminal kinase 1/c-Jun activation of the p53/microRNA 34a/sirtuin 1 pathway contributes to apoptosis induced by deoxycholic acid in rat liver, Mol. Cell. Biol 34 (2014) 1100–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Malliou F, Andreadou I, Gonzalez FJ, Lazou A, Xepapadaki E, Vallianou I,Lambrinidis G, Mikros E, Marselos M, Skaltsounis AL, Konstandi M, The olive constituent oleuropein, as a PPARα agonist, markedly reduces serum triglycerides, Nutr. Biochem 59 (2018) 17–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].McCoin CS, Knotts TA, Adams SH, Acylcarnitines-old actors auditioning for new roles in metabolic physiology, Nat. Rev. Endorinol 11 (2015) 617–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Chen C, Krausz KW, Shah YM, Idle JR, Gonzalez FJ, Serum metabolomics reveals irreversible inhibition of fatty acid β-oxidation through the suppression of PPARα activation as a contributing mechanism of acetaminophen-induced hepatotoxicity, Chem. Res. Toxicol 22 (2009) 699–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Santra S, Hendriksz C, How to use acylcarnitine profiles to help diagnose inborn errors of metabolism, Arch. Dis. Childhood-E 95 (2010) 151–156. [DOI] [PubMed] [Google Scholar]
- [28].Shi X, Yao D, Gosnell BA, Chen C, Lipidomic profiling reveals protective function of fatty acid oxidation in cocaine-induced hepatotoxicity, J. Lipid Res 53 (2012) 2318–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Dharancy S, Malapel M, Perlemuter G, Roskams T, Cheng Y, Dubuquoy L,Podevin P, Conti F, Canva V, Philippe D, Gambiez L, Mathurin P, Paris JC,Schoonjans K, Calmus Y, Pol S, Auwerx J, Desreumaux P, Impaired expression of the peroxisome proliferator-activated receptor α during hepatitis C virus infection, Gastroenterology 128 (2005) 334–342. [DOI] [PubMed] [Google Scholar]
- [30].Francque S, Verrijken A, Caron S, Prawitt J, Paumelle R, Derudas B, Lefebvre P, Taskinen MR, Van Hul W, Mertens I, Hubens G, Van Marck E, Michielsen P,Van Gaal L, Staels B, PPARα gene expression correlates with severity and histological treatment response in patients with non-alcoholic steatohepatitis, J. Hepatol 63 (2015) 164–173. [DOI] [PubMed] [Google Scholar]
- [31].Li F, Patterson AD, Krausz KW, Tanaka N, Gonzalez FJ, Metabolomics reveals an essential role for peroxisome proliferator-activated receptor α in bile acid homeostasis, J. Lipid Res 53 (2012) 1625–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Mohamed DI, Elmelegy AA, El-Aziz LF, Abdel Kawy HS, El-Samad AA, El-Kharashi OA, Fenofibrate a peroxisome proliferator activated receptor-α agonist treatment ameliorates concanavalin A-induced hepatitis in rats, Eur. J. Pharmacol 721 (2013) 35–42. [DOI] [PubMed] [Google Scholar]
- [33].Rajamoorthi A, Arias N, Basta J, Lee RG, Baldan A, Amelioration of diet-induced steatohepatitis in mice following combined therapy with ASO-Fsp27 and fenofibrate, J. Lipid Res 58 (2017) 2127–2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zhang Y, Guo H, Hassan HM, Ding PP, Su Y, Song Y, Wang T, Sun L, Zhang L,Jiang Z, Pyrazinamide induced hepatic injury in rats through inhibiting the PPARα pathway, J. Appl. Toxicol JAT 36 (2016) 1579–1590. [DOI] [PubMed] [Google Scholar]
- [35].Yang S, Wei L, Xia R, Liu L, Chen Y, Zhang W, Li Q, Feng K, Yu M, Zhang W,Qu J, Xu S, Mao J, Fan G, Ma C, Formononetin ameliorates cholestasis by regulating hepatic SIRT1 and PPARα, Biochem. Res. Commun 512 (2019) 770–778. [DOI] [PubMed] [Google Scholar]
- [36].Zhang C, Deng J, Liu D, Tuo X, Xiao L, Lai B, Yao Q, Liu J, Yang H, Wang N, Nuciferine ameliorates hepatic steatosis in high-fat diet/streptozocin-induced diabetic mice through a PPARα/PPARγ coactivator-1α pathway, Brit. J. Pharmacol 175 (2018) 4218–4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Higuchi H, Gores GJ, Bile acid regulation of hepatic physiology: IV. Bile acids and death receptors, Am. J. Physiol. Gastrointestinal and liver physiology 284 (2003) G734–G738. [DOI] [PubMed] [Google Scholar]
- [38].Allen K, Jaeschke H, Copple BL, Bile acids induce inflammatory genes in hepatocytes: A novel mechanism of inflammation during obstructive cholestasis, Am. J. Pathol 178 (2011) 175–186. [DOI] [PMC free article] [PubMed] [Google Scholar]