

Abstract
Single-cell RNA-seq (scRNASeq) has become a powerful technique for measuring the transcriptome of individual cells. Unlike the bulk measurements that average the gene expressions over the individual cells, gene measurements at individual cells can be used to study several different tissues and organs at different stages. Identifying the cell types present in the sample from the single cell transcriptome data is a common goal in many single-cell experiments. Several methods have been developed to do this. However, correctly identifying the true cell types remains a challenge. We present a framework that addresses this problem. Our hypothesis is that the meaningful characteristics of the data will remain despite small perturbations of data. We validate the performance of the proposed method on eight publicly available scRNA-seq datasets with known cell types as well as five simulation datasets with different degrees of the cluster separability. We compare the proposed method with five other existing methods: RaceID, SNN-Cliq, SINCERA, SEURAT, and SC3. The results show that the proposed method performs better than the existing methods.
Subject terms: Gene expression analysis, Machine learning
Introduction
Recent advances in single-cell RNA-Seq (scRNASeq) techniques have provided transcriptomes of the large numbers of individual cells (single-cell gene expression data)1–9. In particular, analyzing the diversity and evolution of single cancer cells can enable the advances in early cancer diagnosis, and ultimately choosing the best strategy for cancer treatment10–12. Furthermore, one important analysis on scRNASeq is the identification of cell types that can be achieved by performing an unsupervised clustering method on transcriptome data13–19.
Clustering algorithms such as k-means and density-based spatial clustering of applications with noise (DBSCAN)20 can identify groups of cells given the single-cell gene expression data. However, clusters obtained by these algorithms might not be robust. Such algorithms require non-intuitive parameters13. For instance, given the number of clusters, k-means iteratively assigns data points (cells) to the nearest centroids (cluster center), and recomputes the centroids based on the predefined number of clusters. This algorithm starts with the randomly chosen centroids. Thus, the result of the algorithm depends on the number of clusters (in DBSCAN, the maximum distance between the two data points in the same neighborhood should be determined) and the number of runs.
Another challenge comes from the high dimensionality of data, known as “curse of dimensionality”. Identifying the accurate clusters of data points based on the measured distances between the pairs of data points may fail since those data points become more similar when they are represented in a higher dimensional space13,21. One approach to deal with the curse of high dimensionality is projecting data into a lower dimensional space, known as dimensionally reduction. In this approach, the data is represented in a lower dimensional space while the characteristic(s) (e.g similarities between the data points) of the original data is preserved. Several methods have used different techniques based on this concept (e.g. principal component analysis) to determine the cell types22–26. Another approach to deal with this challenge is feature selection, i.e. eliminating some of the features (genes) that are not informative27. In the following, we provide a brief overview of the related methods that identify the cell types based on the combination of approaches described above.
Methods SC328 and Seurat25 use a combination of feature selection, dimensionality reduction, and clustering algorithms to identify the cell types. Authors of SC3 use a consensus clustering framework that combines clustering solutions obtained by the spectral transformations and k-means clustering based on the complete-linkage hierarchical clustering. They first apply a gene filtering approach on the single-cell gene expression data to remove rare and ubiquitous genes/transcripts. Next, they compute the distance matrices (distance between the cells) using the Euclidean, Pearson, and Spearman metrics. They transform the distance matrices using either principal component analysis (PCA)29, or by computing the eigenvectors of the associated graph Laplacian. Next, they perform a k-means clustering on the first d eigenvectors of the transformed distance matrices. Using the different k-means clustering results, they construct a consensus matrix that represents how often each pair of cells is clustered together. This consensus matrix is used as an input to a hierarchical clustering using a complete linkage and agglomeration strategies30. The clusters are inferred at the k-th level of hierarchy, where k is computed based on the Random Matrix Theory31,32. The accuracy of SC3 is sensitive to the number of eigenvectors (d), chosen for the spectral transformation. The authors report that SC3 performs well when d is between 4% and 7% of the number of cells. The main advantage of SC3 is its high accuracy in identification of cell types. However, it is not scalable33.
Seurat25 is a graph-based clustering method that projects the single cell expression data into the two-dimensional space using the t-distributed stochastic neighbor embedding (t-SNE) technique34. Then, it performs the DBSCAN method20 on the dimensionality-reduced single cell data. Seurat may fail to find the cell types in small datasets (low cell numbers)28. It is reported that this may be due to possible difficulties in estimating the densities when the number of data points is low.
RaceID35 determines the cell types by performing a k-means clustering algorithm. In this method, the gap statistics is used to choose the number of clusters. RaceID does not perform well when the data does not contain rare cell populations but it appears to be the preferred methods when the aim is identification of rare types13,33,36,37.
SNN-Cliq17 uses the shared nearest neighbor (SNN) concept, which considers the effect of the surrounding neighbor data points, to handle the high-dimensional data. The authors of SNN-Cliq compute the similarity between the pairs of data points (the similarity matrix) based on the Euclidean distance, referred as the primary similarity measure. Using the similarity matrix, they list the k-nearest neighbors (KNN) to each data point. They propose a secondary similarity measure that computes the similarity between two data points based on their shared neighborhoods. Consequently, an SNN graph is constructed based on the connectivity between the data points. Then, a graph-based clustering method is applied on the SNN graph in which nodes and weighted edges represent the data points and similarities between the data points, respectively. The main disadvantage of the graph-based methods such as SNN-Cliq is that scRNASeq data is not inherently graph-structured13. Therefore, the accuracy of these methods depends on the graph representation of scRNASeq data.
SINCERA38 performs a hierarchical clustering on the similarity matrix that is computed using the centered Pearson’s correlation. The average linkage approach is used as the default choice for the linkage. Consensus clustering39,40, tight clustering41 and ward linkage42 are provided as alternative clustering approaches. Users can choose a distance threshold or the number of clusters during the visual inspection when the hierarchical clustering is used for the cell cluster identification. SINCERA tends to identify many clusters which likely represent the same cell type13.
One way to identify robust clusters of cells is to resample the cells/genes and compare the original clusters with the ones that are obtained by resampling43. In the current paper, in order to explore the strength of a pattern (cluster of cells) in the data, we analyze the sensitivity of that pattern against small changes in the data. The data is resampled by replacing a certain number of data points with the noise points from a noise distribution. Our hypothesis is that if there is a strong pattern in data, it will remain despite small perturbations44. Here, we develop a stable subtyping (clustering) method that employs the t-distributed stochastic neighbor embedding (t-SNE)34 and k-means clustering to identify the cell types. We add noise and apply a bootstrap method45,46 to identify the stable clusters of cells. We use the Adjusted Rand Index (ARI)47, adjusted mutual information (AMI)48,49, and V-measure50 to evaluate the performance of the clustering result for datasets in which the true cell types are known. We compare the results of our method with five other methods: RaceID35, SNN-Cliq17, SINCERA38, SEURAT25, and SC328 using 8 real datasets with known cell types and 5 simulated datasets. The results of the different methods show that the proposed method performs better than the five methods across different datasets.
Materials and methods
The goal of the proposed method is to identify the cell types present in a mixture of single cells. The input of the method is the single cell gene expression matrix (Mgene×cell) in which rows represent the genes and columns represent the cells. In the following we provide more detail about the input data and different steps of the proposed framework. The overall approach is shown in Fig. 1.
Figure 1.
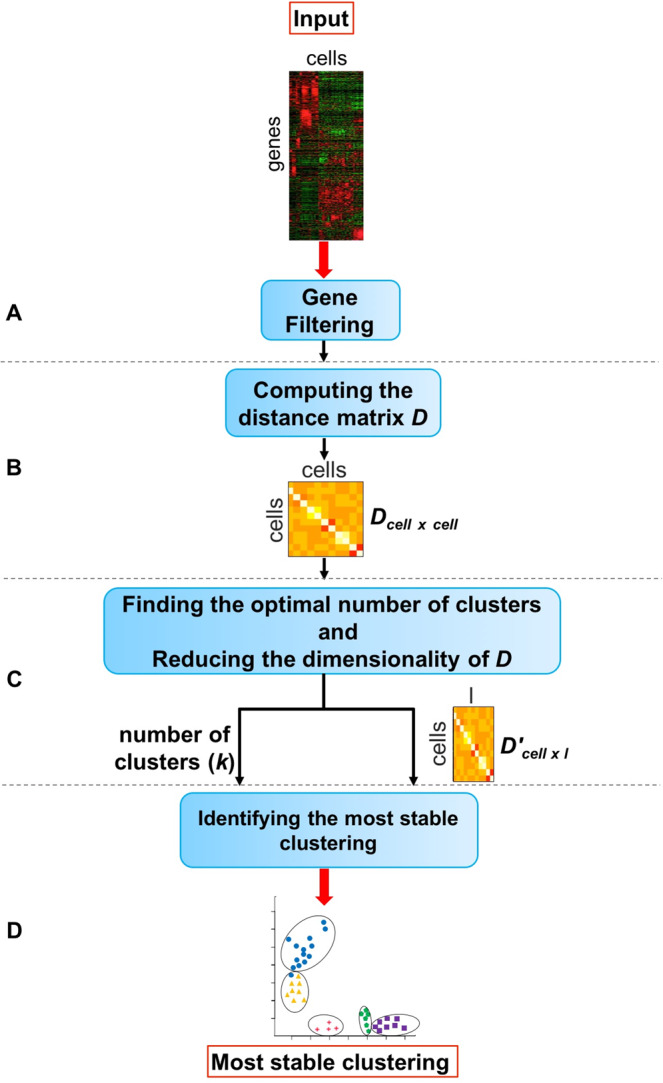
The overall workflow of the proposed method. Given the single cell gene expression matrix, module (A) eliminates the genes that are not expressed in any cell. Using the resulting matrix, module (B) computes the Euclidean distance between the cells. The output of this module is a distance matrix in which the rows and columns are the cells (Dcell×cell). Module (C) reduces the dimensionality of the distance matrix using the t-distributed stochastic neighbor embedding (t-SNE) technique. In this module, an average silhouette method is employed to choose the optimal number of clusters k. Finally in module (D), the lower-dimension distance matrix and the optimal number of clusters k obtained from module (C) are used as the input data to identify the most stable clustering of cells. Figure 2 shows the details of module D.
Data source
The eight publicly available scRNA-seq datasets as well as the five simulation datasets we used in our analysis are included in the Supplementary Materials. Among the eight real datasets, all but three (Klein51, Patel52, Treutlein53) are considered as’gold standard’ since the labels of the cells are known in a definitive way. Patel52 and Treutlein53 are referred as'silver standard’ by Kiselev et al.28 since their cell labels are determined based on the computational methods and the authors’ knowledge of the underlying biology.
We obtained the processed data from Hemberg lab's website (https://hemberg-lab.github.io/scRNA.seq.datasets). Hemberg et al.54 use the SingleCellExperiment Bioconductor S4 class55 to store the data, and the scater package56 for the quality control and plotting purposes. The normalized data is deposited as a SingleCellExperiment object (.RData file) and the cell type information is accessed in the cell_type1 column of the “colData” slot of this object. The gene expression values of the cells are organized as a matrix in which rows are cells and columns are the genes. In our analysis, genes (features) that are not expressed in any cells are removed. We did not filter any cell in this analysis.
Gene filtering
As shown in Fig. 1A, we remove the genes/transcripts that are not expressed in any cell (expression value is zero in all cells). Such genes cannot provide useful information that can differentiate between cell types57. The result of performing the filtering method on the single cell gene expression matrix (Mgene×cell) is used as the input to the second module of the proposed framework.
Measuring the dissimilarity between the cells
The distance between the cells is calculated using the Euclidean metric (Fig. 1B). The output of this step is the distance (dissimilarity) matrix Dcell×cell. We reduce the dimension of D by performing the t-distributed stochastic neighbor embedding (t-SNE)34,58, the nonlinear dimensionality reduction/visualization technique (Fig. 1C). We will refer to the output as D′cell×l, where 2 ≤ l ≤ cell. In this study, the number of dimensions is 2.
Clustering
Identification of the optimal number of clusters
This section describes the third module of the proposed method (Fig. 1C). In this analysis, the t-SNE is repeatedly (n = 50) applied on the distance matrix Dcell×cell to obtain the dimensionality-reduced distance matrix D′cell×l. Each time, the optimal number of clusters is calculated based on the average silhouette method using the dimensionality reduced distance matrix D′. In order to find the optimal number of clusters k, the k-means clustering is applied on the D′ matrix using a range value (default = 2:20), and the k that maximizes the average silhouette measure is selected. Finally, the average of the selected numbers k across different repeats (n = 50) (rounded to the nearest integer) is considered as the final optimal number of clusters.
The silhouette evaluates the quality of that clustering based on how well its data points are clustered. A silhouette measure is assigned to each data point representing how close a data point is to its own cluster in comparison to other clusters. For each data point i, this measure is calculated as follows:
where a(i) is the average distance between the data point i and all other data points within the same cluster. b(i) is the smallest average distance of i to all points in any other cluster of which i is not a member. s(i) takes values from −1 to 1, where a high positive score shows that the given data point is well clustered (close to other points in its own cluster and far from points in the other clusters). Conversely, a high negative score shows that data point is poorly clustered.
k-means clustering based on the resampling method
This section describes the detail of the last module of the proposed method. As shown in Fig. 2, using the dimensionality reduced distance matrix D′ and the chosen number of clusters k from the previous step, we identify the most stable clustering by generating different clustering solutions (clusteringi (i ∈ [1..n])) and measure the stability of each clustering solution based on a resampling method. The stability measure assigned to each particular clustering (denoted as clusteringi) represents how often the k clusters belonging to that clustering are preserved when the input data (D′) is resampled several times. The resampled datasets are generated from D′ by randomly replacing 5% of data points (cells) with noise. These noisy datasets are then used as the input to k-means algorithm. Hence, several clusterings (clusteringi,j, j ∈ [1..m]) are generated from the resampled data (resampled versions of clusteringi).
Figure 2.

Identifying the most stable clustering. In this analysis, given the lower-dimension distance matrix D′cell×l and the optimal number of clusters k, we calculate n different clusterings (clustering1, ..., clusteringn) using the k-means clustering algorithm. Then, the stability of each clustering is assessed based on a resampling approach (grey box). A stability score is assigned to each clustering based on how often its clusters are recovered when the input data is perturbed (resampled). A clustering with the maximum stability score is selected as the final solution.
In order to assess the stability of each cluster c in the clusteringi (original clustering), the cluster c is compared to all the clusters in the clustering that is obtained from the resample data (clusteringi,j) based on the Jaccard distance. The Jaccard coefficient59, a similarity measure between sets, is used to compute the similarity between two clusters as follows:
where the term A and B are two clusters, consisting of some data points in X = {x1, …, xN}.
If the Jaccard similarity between the cluster c (from the original clustering clusteringi) and the most similar cluster in the resampled clustering is equal or greater than 0.75, that cluster is considered stable (preserved). Thus, the stability of each cluster in clusteringi is calculated as the percentage of the times that cluster is preserved (Jaccard coefficient ≥ 0.75) across the m different resamplings.
We then average the stability measures of the k clusters belonging to clusteringi, and consider it as the overall stability measure of clusteringi. Among n different clustering solutions (clusteringi (i ∈ [1..n])), we select the clustering solution with the maximum stability measure as the final clustering solution.
Figure 3 shows the detail of the resampling method we performed to compute the stability measure for each clustering. The clusters that are obtained by applying k-mean on the resampled dataset are compared with the clusters from the original input data only based on the non-noise points (the noise data points are excluded when two clusters are compared based on the Jaccard similarity metric.
Figure 3.

The resampling framework to compute the stability measure for each clustering. The input includes N data points X = {x1, ..., xN}, the number of clusters k, the number of resamplings m, and the clustering C that is obtained by applying k-means on X. This analysis generates m resampling data by randomly replacing 5% of data points with the noise, and computes m resampled clusterings based on k-means clustering. Each cluster c in C is compared with the most similar cluster in the resampling clustering, and the Jaccard coefficient between the two clusters is computed, while the noise points are excluded. The percentage of the times that Jaccard coefficients are larger than 0.75 is considered the stability measure for cluster c. The average of stability measures for all clusters belonging to clustering C is calculated and considered as the overall stability measure for clustering C.
Validation methods
We use 13 different datasets in which the cell types (labels) are known. To measure the level of similarity between the reference labels and the inferred labels that are obtained by each clustering method, we use three different metrics: adjusted rand index (ARI), adjusted mutual information (AMI), and V-measure as explained in the following.
Adjusted rand index
Given the cell labels, the Adjusted Rand Index (ARI)47 is used to assess the similarity between the inferred clustering and the true clustering. ARI ranges from 0, for poor matching (a random clustering), to 1 for a perfect agreement with the true clustering. For a set of n data points, the contingency table is constructed based on the shared number of data points between two clusters. Suppose X = {X1, X2, ..., XR} and Y = {Y1, Y2, ..., YC} represent two different clusterings with R and C clusters, respectively. The overlap between X and Y can be summarized as a contingency table MR×C = [nij], where i = 1...R, j = 1...C. Xi and Yj denote a cluster in clusterings X and Y, and i and j refer to the row number and the column number of the contingency table, respectively. The ARI is defined as follow:
| 1 |
where nij denotes the number of shared data points between clusters Xi and Yj (nij = |Xi∩Yj|), and (the sum of the ith row of the contingency table), and (the sum of the jth column of the contingency table).
Adjusted mutual information
The adjusted mutual information (AMI)48,49 is a variation of mutual information that corrects for random partitioning, similar to the way the ARI corrects the rand index. As explained in the previous section, given two different clusterings X = {X1, X2, ..., XR} and Y = {Y1, Y2, ..., YC} of n data points with R and C clusters, respectively, the mutual information of cluster overlap between X and Y can be summarized as a contingency table MR×C = [nij], where i = 1...R, j = 1...C, and nij represents the number of common data points between clusters Xi and Yj. Suppose a data point is picked at random from X, the probability that the data point falls into cluster Xi is . The entropy60 associated with the clustering X is calculated as follows:
| 2 |
H(X) is non-negative and takes the value 0 only when there is no uncertainty determining a data point's cluster membership (there is only one cluster). The mutual information (MI) between two clusterings X and Y is calculated as follows:
| 3 |
where P(i, j) denotes the probability that a data point belongs to both the cluster Xi in X and the cluster Yj in Y:
| 4 |
MI is a non-negative quantity upper bounded by the entropies H(X) and H(Y). It quantifies the information shared by the two clusterings and therefore can be considered as a clustering similarity measure. The adjusted measure for the mutual information is defined as follows:
| 5 |
where the expected mutual information between two random clusterings is:
| 6 |
where the ai and bj are the partial sums of the contingency table: and .
The adjusted mutual information (AMI) takes a value of 1 when the two clusterings are identical and 0 when the MI between two partitions equals the value expected due to chance alone.
V-measure
The V-measure50 is the harmonic mean between two measures: homogeneity and completeness. The homogeneity criteria is satisfied if a clustering assigns only those data points that are members of a single class (true cluster) to a single cluster. Thus, the class distribution within each cluster should be skewed to a single class (zero entropy). To determine how close a given clustering is to this ideal, the conditional entropy of the class distribution given the identified clustering is computed as H(C|K), where C = {C1, C2, ..., Cl} is a set of classes and K is a clustering K = {K1, K2, ..., Km}. In the perfectly homogeneous case, this value is 0. However, this value is dependent on the size of the dataset and the distribution of class sizes. Thus, this conditional entropy is normalized by the maximum reduction in entropy the clustering information could provide, H(C). Therefore, the homogeneity is defined as follows:
| 7 |
The completeness is symmetrical to homogeneity50. In order to satisfy the completeness criteria, a clustering must assign all of those data points that are members of a single class to a single cluster. To measure the completeness, the distribution of cluster assignments within each class is assessed. In a perfectly complete clustering solution, each of these distributions will be completely skewed to a single cluster.
Given the homogeneity h and completeness c, the V-measure is computed as the weighted harmonic mean of homogeneity and completeness:
| 8 |
if β is greater than 1, completeness is weighted more strongly in the calculation. If β is less than 1, homogeneity is weighted more strongly. Since the computations of homogeneity, completeness and V-measure are completely independent of the number of classes, the number of clusters, the size of the dataset and the clustering algorithm, these measures can be employed for evaluating any clustering solution.
Results
Tables 1–3 shows the comparison between the proposed method and five other methods: RaceID35, SC328, SEURAT25, SINCERA38, and SNN-Cliq17 using the three metrics: ARI, AMI, and V-measures, respectively.
Table 2.
A comparison between the results of six methods: proposed, RaceID, SC3, Seurat, SINCERA, and SNN-Cliq.
| Dataset | #cell types | Proposed | RaceID | SC3 | SINCERA | SNN-Cliq | Seurat | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| K (mean ± sd) | AMI (mean ± sd) | K (mean ± sd) | AMI (mean ± sd) | K (mean ± sd) | AMI (mean ± sd) | K | AMI | K | AMI | K | AMI | ||
| Biase | 3 | 3 ± 0 | 0.92 ± 0.02 | 3.14 ± 0.6 | 0.85 ± 0.23 | 3 ± 0 | 0.92 ± 0 | 6 | 0.64 | 6 | 0.62 | 4 | 0.74 |
| Deng | 10 | 10 ± 0 | 0.73 ± 0.01 | 1 ± 0 | 0 ± 0 | 9 ± 0 | 0.81 ± 0.006 | 3 | 0.48 | 17 | 0.6 | 6 | 0.59 |
| Goolam | 5 | 3 ± 0 | 0.73 ± 0.04 | 1 ± 0 | 0 ± 0 | 6 ± 0 | 0.69 ± 0 | 13 | 0.4 | 17 | 0.42 | 3 | 0.11 |
| Klein | 4 | 6 ± 0 | 0.67 ± 0.06 | 2.98 ± 0.14 | 0.51 ± 0.05 | 19 ± 0 | 0.53 ± 0.006 | 43 | 0.52 | 265 | 0.21 | 3 | 0.06 |
| Patel | 5 | 5 ± 0 | 0.86 ± 0.01 | 7.44 ± 1.88 | 0.66 ± 0.1 | 17 ± 0 | 0.93 ± 0 | 10 | 0.73 | 26 | 0.31 | 5 | 0.68 |
| Pollen | 11 | 8 ± 0 | 0.72 ± 0.01 | 8.36 ± 2.27 | 0.68 ± 0 | 10 ± 0 | 0.53 ± 0.01 | 10 | 0.91 | 22 | 0.74 | 8 | 0.87 |
| Treutlein | 5 | 3 ± 0 | 0.54 ± 0.03 | 1 ± 0 | 0 ± 0 | 3 ± 0 | 0.62 ± 0 | 7 | 0.46 | 5 | 0.51 | 1 | 0 |
| Yan | 8 | 5 ± 0 | 0.78 ± 0.01 | 5.5 ± 2.34 | 0.61 ± 0.17 | 4 ± 0 | 0.72 ± 0 | 8 | 0.72 | 13 | 0.76 | 3 | 0.58 |
| sim3 | 3 | 3 ± 0 | 1 ± 0 | 1 ± 0 | 0 ± 0 | 3 ± 0 | 1 ± 0 | 120 | 0.23 | 147 | 0.21 | 3 | 1 |
| sim4 | 4 | 4 ± 0 | 0.99 ± 0.007 | 1 ± 0 | 0 ± 0 | 4 ± 0 | 0.99 ± 0.001 | 464 | 0.21 | 437 | 0.2 | 3 | 0.66 |
| sim6 | 6 | 7.9 ± 0.3 | 0.64 ± 0.02 | 1 ± 0 | 0 ± 0 | 3 ± 0 | 0.51 ± 0.004 | 68 | 0.42 | 143 | 0.3 | 6 | 1 |
| sim8 | 8 | 9.34 ± 0.47 | 0.85 ± 0.01 | 1 ± 0 | 0 ± 0 | 4 ± 0 | 0.56 ± 0.007 | 68 | 0.51 | 290 | 0.31 | 8 | 1 |
| sim_Tung | 8 | 8 ± 0 | 0.51 ± 0.008 | 1 ± 0 | 0 ± 0 | 8 ± 0 | 0.006 ± 0 | 17 | 0.04 | 77 | 0.13 | 8 | 0 |
The adjusted mutual information (AMI)48,49, is used to evaluate the performance of each clustering method. The proposed method, RaceID, and SC3 are performed 50, 50, and 5 times on each dataset, respectively. The average AMIs across different runs are computed for the proposed method, SC3, and RaceID. Since SNN-Cliq, SINCERA and SEURAT are deterministic, they are performed only once.
Table 1.
A comparison between the results of six methods: proposed, RaceID, SC3, Seurat, SINCERA, and SNN-Cliq.
| Dataset | #cell types | Proposed | RaceID | SC3 | SINCERA | SNN-Cliq | Seurat | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| K (mean ± sd) | ARI (mean ± sd) | K (mean ± sd) | ARI (mean ± sd) | K (mean ± sd) | ARI (mean ± sd) | K | ARI | K | ARI | K | ARI | ||
| Biase | 3 | 3 ± 0 | 0.94 ± 0.01 | 3.14 ± 0.6 | 0.84 ± 0.25 | 3 ± 0 | 0.94 ± 0 | 6 | 0.71 | 6 | 0.66 | 4 | 0.78 |
| Deng | 10 | 10 ± 0 | 0.58 ± 0.02 | 1 ± 0 | 0 ± 0 | 9 ± 0 | 0.65 ± 0.002 | 3 | 0.42 | 17 | 0.4 | 6 | 0.45 |
| Goolam | 5 | 3 ± 0 | 0.80 ± 0.09 | 1 ± 0 | 0 ± 0 | 6 ± 0 | 0.59 ± 0 | 13 | 0.19 | 17 | 0.2 | 3 | 0.05 |
| Klein | 4 | 6 ± 0 | 0.69 ± 0.01 | 2.98 ± 0.14 | 0.48 ± 0.001 | 19 ± 0 | 0.44 ± 0.01 | 43 | 0.45 | 265 | 0.11 | 3 | 0 |
| Patel | 5 | 5 ± 0 | 0.66 ± 0.09 | 7.44 ± 1.88 | 0.66 ± 0.08 | 17 ± 0 | 0.45 ± 0.01 | 10 | 0.78 | 26 | 0.14 | 5 | 0.63 |
| Pollen | 11 | 8 ± 0 | 0.86 ± 0.02 | 8.36 ± 2.27 | 0.55 ± 0.11 | 10 ± 0 | 0.93 ± 0 | 10 | 0.9 | 22 | 0.71 | 8 | 0.85 |
| Treutlein | 5 | 3 ± 0 | 0.72 ± 0.03 | 1 ± 0 | 0 ± 0 | 3 ± 0 | 0.66 ± 0 | 7 | 0.35 | 5 | 0.62 | 1 | 0 |
| Yan | 8 | 5 ± 0 | 0.81 ± 0.02 | 5.5 ± 2.34 | 0.55 ± 0.17 | 4 ± 0 | 0.76 ± 0 | 8 | 0.59 | 13 | 0.79 | 3 | 0.56 |
| sim3 | 3 | 3 ± 0 | 1 ± 0 | 1 ± 0 | 0 ± 0 | 3 ± 0 | 1 ± 0 | 120 | 0.12 | 147 | 0.03 | 3 | 1 |
| sim4 | 4 | 4 ± 0 | 0.99 ± 0.005 | 1 ± 0 | 0 ± 0 | 4 ± 0 | 0.99 ± 0.0005 | 464 | 0.08 | 437 | 0.01 | 3 | 0.57 |
| sim6 | 6 | 7.9 ± 0.3 | 0.56 ± 0.03 | 1 ± 0 | 0 ± 0 | 3 ± 0 | 0.53 ± 0.005 | 68 | 0.25 | 143 | 0.06 | 6 | 1 |
| sim8 | 8 | 9.34 ± 0.47 | 0.77 ± 0.03 | 1 ± 0 | 0 ± 0 | 4 ± 0 | 0.53 ± 0.04 | 68 | 0.35 | 290 | 0.05 | 8 | 1 |
| sim_Tung | 8 | 8 ± 0 | 0.42 ± 0 | 1 ± 0 | 0 ± 0 | 8 ± 0 | 0 ± 0 | 17 | 0.001 | 77 | 0.001 | 8 | 0 |
The adjusted rand index (ARI)47 is used to evaluate the performance of each clustering method. The proposed method, RaceID, and SC3 are performed 50, 50, and 5 times on each dataset, respectively. SC3 was performed only 5 times because it is very stable (standard deviation of zero for all datasets). The average ARIs across different runs are computed for the proposed method, SC3, and RaceID. Since SNN-Cliq, SINCERA and SEURAT are deterministic, they are performed only once. The proposed method was the best for 8 out of the 13 datasets. The proposed method also yielded the best average ARI, as shown in Fig. 4.
Table 3.
A comparison between the results of six methods: proposed, RaceID, SC3, Seurat, SINCERA, and SNN-Cliq.
| Dataset | #cell types | Proposed | RaceID | SC3 | SINCERA | SNN-Cliq | Seurat | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| K (mean ± sd) | V-measure (mean ± sd) | K (mean ± sd) | V-measure (mean ± sd) | K (mean ± sd) | V-measure (mean ± sd) | K | V-measure | K | V-measure | K | V-measure | ||
| Biase | 3 | 3 ± 0 | 0.93 ± 0.03 | 3.14 ± 0.6 | 0.87 ± 0.2 | 3 ± 0 | 0.93 ± 0 | 6 | 0.72 | 6 | 0.7 | 4 | 0.73 |
| Deng | 10 | 10 ± 0 | 0.72 ± 0.01 | 1 ± 0 | 0 ± 0 | 9 ± 0 | 0.74 ± 0.001 | 3 | 0.93 | 17 | 0.64 | 6 | 0.93 |
| Goolam | 5 | 3 ± 0 | 0.82 ± 0.04 | 1 ± 0 | 0 ± 0 | 6 ± 0 | 0.98 ± 0 | 13 | 0.71 | 17 | 0.65 | 3 | 0.66 |
| Klein | 4 | 6 ± 0 | 0.38 ± 0.01 | 2.98 ± 0.14 | 0.4 ± 0.06 | 19 ± 0 | 0.31 ± 0.002 | 43 | 0.36 | 265 | 0.29 | 3 | 0.46 |
| Patel | 5 | 5 ± 0 | 0.56 ± 0.02 | 7.44 ± 1.88 | 0.54 ± 0.04 | 17 ± 0 | 0.46 ± 0.002 | 10 | 0.55 | 26 | 0.44 | 5 | 0.62 |
| Pollen | 11 | 8 ± 0 | 0.95 ± 0.01 | 8.36 ± 2.27 | 0.76 ± 0.03 | 10 ± 0 | 0.93 ± 0 | 10 | 0.94 | 22 | 0.72 | 8 | 0.93 |
| Treutlein | 5 | 3 ± 0 | 0.96 ± 0 | 1 ± 0 | 0 ± 0 | 3 ± 0 | 0.89 ± 0 | 7 | 0.93 | 5 | 0.92 | 1 | 0 |
| Yan | 8 | 5 ± 0 | 0.83 ± 0.02 | 5.5 ± 2.34 | 0.68 ± 0.07 | 4 ± 0 | 0.81 ± 0 | 8 | 0.65 | 13 | 0.78 | 3 | 0.73 |
| sim3 | 3 | 3 ± 0 | 1 ± 0 | 1 ± 0 | 0 ± 0 | 3 ± 0 | 1 ± 0 | 120 | 0.95 | 147 | 0.95 | 3 | 1 |
| sim4 | 4 | 4 ± 0 | 0.99 ± 0.0002 | 1 ± 0 | 0 ± 0 | 4 ± 0 | 0.99 ± 0.00003 | 464 | 0.97 | 437 | 0.97 | 3 | 0.96 |
| sim6 | 6 | 7.9 ± 0.3 | 0.98 ± 0 | 1 ± 0 | 0 ± 0 | 3 ± 0 | 0.97 ± 0.0004 | 68 | 0.97 | 143 | 0.97 | 6 | 1 |
| sim8 | 8 | 9.34 ± 0.47 | 0.99 ± 0 | 1 ± 0 | 0 ± 0 | 4 ± 0 | 0.98 ± 0.004 | 68 | 0.98 | 290 | 0.98 | 8 | 1 |
| sim_Tung | 8 | 8 ± 0 | 0.96 ± 0.03 | 1 ± 0 | 0 ± 0 | 8 ± 0 | 0.66 ± 0 | 17 | 0.82 | 77 | 0.8 | 8 | 0.66 |
The V-measure50 is used to evaluate the performance of each clustering method. The proposed method, RaceID, and SC3 are performed 50, 50, and 5 times on each dataset, respectively. The average V-measures across different runs are computed for the proposed method, SC3, and RaceID. Since SNN-Cliq, SINCERA and SEURAT are deterministic, they are performed only once.
We used the R package fpc61 to compute the k-means clustering based on the resampling method. We generated 20 different clusterings, and for each clustering we computed 1,000 clusterings based on the resampled datasets to find the most meaningful clustering. We used the log-transformation (M′ = log2(M + 1)) for all methods except SINCERA. For SINCERA we followed the authors instructions38 and used the original z-score normalization instead of the log-transformation. In order to generate SC3 results, we used the R package SC3 (http://bioconductor.org/packages/SC3, v.1.8.0). We applied the same gene filtering approach that authors proposed in their study (parameter gene_filter=TRUE).
For SEURAT we used the Seurat R package (v.2.3.4)62. We performed the t-SNE using the Rtsne R package with the default parameters, and we used DBSCAN algorithm for clustering. We ran SNN-cliq with the default parameters that are provided by the authors17. For RaceID, we used the R code provided by the authors35 (https://github.com/dgrun/RaceID).
As shown in Fig. 4, the proposed method performs better than the five methods across 13 different datasets. In this figure, the three boxplots shows the the performance of each method on these 13 datasets based on the adjusted rand index (ARI), adjusted mutual information (AMI), and V-measure. We performed the proposed method, SC3 and RaceID on each dataset for 50, 5, and 50 times, respectively. In these three methods, we calculated the average of ARIs, AMIs, and V-measures over different runs. Since SC3 is reported as a stable method by the authors28, we run it only 5 times. Indeed, we have observed the results with a very small standard deviation in all 5 runs for all 13 datasets confirming the claims of the authors. The other clustering methods SEURAT, SINCERA, and SNN-Cliq were run only once since they are deterministic.
Figure 4.

The performance comparison using 13 single cell datasets based on three metrics: the adjusted rand index (ARI), adjusted mutual information (AMI), and V-measure.The proposed method and RaceID were applied 50 times on each dataset. SC3 was used only 5 times on each dataset because it is very stable. The average ARIs, AMIs, and V-measures across different runs are computed for the proposed method, RaceID, and SC3. Since SNN-Cliq, SINCERA, and SEURAT are deterministic, they are run only once for each dataset.
Discussion
The results shown in Tables 1–3 merit some discussion. The Goolam dataset, for instance, includes 5 true cell types. On this dataset, the proposed algorithm identifies 3 clusters, while SC3 identifies 6, RaceID 1, Seurat 2, SINCERA 13 and SNN-Cliq 17 types. Even though the number of clusters closest to the number of true types is 6, as yielded by SC3, the membership of various cells in these clusters is not correct since the ARI index associated to these 6 clusters is only 0.59 compared to the ARI index of 0.8 associated to the 3 clusters constructed by the proposed method.
Conversely, for the Patel dataset that includes 5 cell types, the proposed method was able to correctly estimate the number of clusters (k = 5). However, the distribution of the individual cells across these five clusters is not perfect, as illustrated by the lower ARI value of 0.66, compared to the 0.78 ARI associated with the SINCERA results.
As another observation, the Pollen dataset includes 11 cell types. Using this dataset, the number of clusters (k = 10) determined by SINCERA is close to the correct number of cell types. However, SC3 achieved better clustering (ARI = 0.93) in contrast to the five other methods. SC3 identified 17 different clusters using this dataset.
Two conclusions may be drawn from these observations. First, results should not be assessed based on the agreement between the number of clusters found and the number of known cell types – the assignment of each cell to a given type is more important. Second, larger number of clusters reported will be associated with larger values of ARI. Therefore, results that include very large number of clusters should be regarded with caution.
RaceID and Seurat both were not able to find a meaningful clustering for the Treutlein dataset. The identified number of clusters by both RaceID and Seurat is 1 (k = 1), while this dataset includes 5 different cell types. As a result, the clusterings obtained by these two methods are poorly matched to the reference clustering. In Deng dataset, the best ARI of 0.65 is obtained by SC3 but this value is not very high. The poor results obtained by all 6 methods using this dataset might be due to noisy data.
We also assessed the reproducibility/stability of the stochastic methods: proposed, RaceID, and SC3 by running each method several times. Although SC3’s consensus pipeline provides a very stable solution (very low standard deviation for the three metrics and k across all datasets), it is computationally more costly than other methods. In summary, one key advantage of our proposed method is that we produce consistent clustering across different datasets.
The run time for each method using 13 different datasets is shown in Fig. 5. It is notable that RaceID, the proposed method, and SC3 have a non-linear increase in run time. At this time, it appears that it is unfeasible to perform this method on large datasets consisting of thousands of cells. The fastest method among all the methods is Seurat, which is a graph-based method. The graph-based methods often return only a single clustering solution with a faster run time and they do not require the user to provide the number of clusters33. Seurat is a popular choice for the large data sets based on the its optimal speed and scalability. However, it has been shown that Seurat does not provide an accurate solution for smaller datasets33. The details of the run times are included in Supplementary Materials.
Figure 5.

The run time of the different methods using 13 single cell datasets.
More generally, finding an optimal clustering method that provides stable solutions for all situations may not be possible. In fact, because no method can perform well for all situations, a comparative analysis of methods based on a set of criteria should be employed33.
Conclusion
Recent advances in single-cell RNA-Seq (scRNASeq) provide the opportunity to perform single-cell transcriptome analysis. In this paper, we develop a pipeline to cluster the individual cells based on their gene expression values such that each cluster consisting of cells with specific functions or distinct developmental stages. We first filter genes that are not expressed in any cell. Then, we compute the distance between the cells using the Euclidean distance. We reduce the dimensions of the distance matrix data using the t-distributed stochastic neighbor embedding (t-SNE) technique. Based on the dimensionality reduced distance matrix, we explore strong patterns (clusters) of cells by randomly drawing a percentage of the data points without replacement, and replacing them with points from a noise distribution. We apply the proposed method on 13 different single cell datasets, and we compare it with five related methods: RaceID, SC3, Seurat, SINCERA, and SNN-Cliq. The results of the evaluation on datasets demonstrate that the proposed method yields better clustering results in comparison to the existing methods.
Supplementary information
Acknowledgements
Any opinions, findings and conclusions or recommendations expressed in this manuscript are those of the authors and do not necessarily reflect the views of any of the funding agencies. NIH/NIDDK (1R01DK107666-01); National Science Foundation (SBIR 1853207); and by the Robert J. Sokol M.D. Endowment in Systems Biology.
Author contributions
A.P. and S.D. conceived and designed the project. A.P. implemented the workflow and performed the data analysis and all computational experiments. A.S. and N.S. helped A.P. to perform the data analysis. A.P. and S.D. wrote the manuscript. All authors reviewed the manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
is available for this paper at 10.1038/s41598-020-66848-3.
References
- 1.Kalisky T, Quake SR. Single-cell genomics. Nature Methods. 2011;8:311. doi: 10.1038/nmeth0411-311. [DOI] [PubMed] [Google Scholar]
- 2.Trapnell C. Defining cell types and states with single-cell genomics. Genome Research. 2015;25:1491–1498. doi: 10.1101/gr.190595.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Navin NE. The first five years of single-cell cancer genomics and beyond. Genome Research. 2015;25:1499–1507. doi: 10.1101/gr.191098.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang Y, Navin NE. Advances and applications of single-cell sequencing technologies. Molecular Cell. 2015;58:598–609. doi: 10.1016/j.molcel.2015.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haque A, Engel J, Teichmann SA, Lönnberg T. A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications. Genome Medicine. 2017;9:75. doi: 10.1186/s13073-017-0467-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fasterius E, Uhlén M, Szigyarto CA-K. Single-cell RNA-seq variant analysis for exploration of genetic heterogeneity in cancer. Scientific Reports. 2019;9:9524. doi: 10.1038/s41598-019-45934-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mathys, H. et al. Single-cell transcriptomic analysis of alzheimer’s disease. Nature 1 (2019). [DOI] [PMC free article] [PubMed]
- 8.Crowell, H. L. et al. On the discovery of population-specific state transitions from multi-sample multi-condition single-cell RNA sequencing data. BioRxiv 713412 (2019).
- 9.Olsen TK, Baryawno N. Introduction to single-cell RNA sequencing. Current Protocols in Molecular Biology. 2018;122:e57. doi: 10.1002/cpmb.57. [DOI] [PubMed] [Google Scholar]
- 10.Saadatpour A, Lai S, Guo G, Yuan G-C. Single-cell analysis in cancer genomics. Trends in Genetics. 2015;31:576–586. doi: 10.1016/j.tig.2015.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shalek, A. K. & Benson, M. Single-cell analyses to tailor treatments. Science Translational Medicine9 (2017). [DOI] [PMC free article] [PubMed]
- 12.Lawson DA, et al. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature. 2015;526:131. doi: 10.1038/nature15260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Andrews, T. S. & Hemberg, M. Identifying cell populations with scRNASeq. Molecular Aspects of Medicine (2017). [DOI] [PubMed]
- 14.Yuan G-C, et al. Challenges and emerging directions in single-cell analysis. Genome Biology. 2017;18:84. doi: 10.1186/s13059-017-1218-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Angerer P, et al. Single cells make big data: new challenges and opportunities in transcriptomics. Current Opinion in Systems Biology. 2017;4:85–91. [Google Scholar]
- 16.Menon V. Clustering single cells: a review of approaches on high-and low-depth single-cell rna-seq data. Briefings in Functional Genomics. 2017;17:240–245. doi: 10.1093/bfgp/elx044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu, C. & Su, Z. Identification of cell types from single-cell transcriptomes using a novel clustering method. Bioinformatics btv088 (2015). [DOI] [PMC free article] [PubMed]
- 18.Zappia L, Phipson B, Oshlack A. Exploring the single-cell RNA-seq analysis landscape with the scRNA-tools database. PLoS Computational Biology. 2018;14:e1006245. doi: 10.1371/journal.pcbi.1006245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duò, A., Robinson, M. D. & Soneson, C. A systematic performance evaluation of clustering methods for single-cell RNA-seq data. F1000Research7 (2018). [DOI] [PMC free article] [PubMed]
- 20.Ester M, Kriegel H-P, Sander J, Xu X. A density-based algorithm for discovering clusters in large spatial databases with noise. In Kdd. 1996;96:226–231. [Google Scholar]
- 21.Becht E, et al. Dimensionality reduction for visualizing single-cell data using umap. Nature Biotechnology. 2019;37:38. doi: 10.1038/nbt.4314. [DOI] [PubMed] [Google Scholar]
- 22.Pierson E, Yau C. ZIFA: Dimensionality reduction for zero-inflated single-cell gene expression analysis. Genome Biology. 2015;16:241. doi: 10.1186/s13059-015-0805-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baron M, et al. A single-cell transcriptomic map of the human and mouse pancreas reveals inter-and intra-cell population structure. Cell Systems. 2016;3:346–360. doi: 10.1016/j.cels.2016.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Campbell JN, et al. A molecular census of arcuate hypothalamus and median eminence cell types. Nature Neuroscience. 2017;20:484. doi: 10.1038/nn.4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Macosko EZ, et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 2015;161:1202–1214. doi: 10.1016/j.cell.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Segerstolpe Å, et al. Single-cell transcriptome profiling of human pancreatic islets in health and type 2 diabetes. Cell Metabolism. 2016;24:593–607. doi: 10.1016/j.cmet.2016.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guyon I, Elisseeff A. An introduction to variable and feature selection. The Journal of Machine Learning Research. 2003;3:1157–1182. [Google Scholar]
- 28.Kiselev VY, et al. SC3: consensus clustering of single-cell RNA-seq data. Nature Methods. 2017;14:483. doi: 10.1038/nmeth.4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jolliffe, I. Principal component analysis (Wiley Online Library, 2002).
- 30.Draghici, S. Statistics and Data Analysis for Microarrays using R and Bioconductor (Chapman and Hall/CRC Press, 2011).
- 31.Tracy CA, Widom H. Level-spacing distributions and the airy kernel. Communications in Mathematical Physics. 1994;159:151–174. [Google Scholar]
- 32.Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genetics. 2006;2:e190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kiselev, V. Y., Andrews, T. S. & Hemberg, M. Challenges in unsupervised clustering of single-cell RNA-seq data. Nature Reviews Genetics 1 (2019). [DOI] [PubMed]
- 34.Maaten LVD, Hinton G. Visualizing data using t-SNE. Journal of Machine Learning Research. 2008;9:2579–2605. [Google Scholar]
- 35.Grün D, et al. Single-cell messenger rna sequencing reveals rare intestinal cell types. Nature. 2015;525:251–255. doi: 10.1038/nature14966. [DOI] [PubMed] [Google Scholar]
- 36.Lin J-T, et al. A new electron bridge channel 1T-DRAM employing underlap region charge storage. IEEE Journal of the Electron Devices Society. 2017;5:59–63. [Google Scholar]
- 37.Li H, et al. Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nature Genetics. 2017;49:708. doi: 10.1038/ng.3818. [DOI] [PubMed] [Google Scholar]
- 38.Guo M, Wang H, Potter SS, Whitsett JA, Xu Y. SINCERA: a pipeline for single-cell RNA-seq profiling analysis. PLoS Computational Biology. 2015;11:e1004575. doi: 10.1371/journal.pcbi.1004575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Monti S, Tamayo P, Mesirov J, Golub T. Consensus clustering: a resampling-based method for class discovery and visualization of gene expression microarray data. Machine Learning. 2003;52:91–118. [Google Scholar]
- 40.Wilkerson MD, Hayes DN. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics. 2010;26:1572–1573. doi: 10.1093/bioinformatics/btq170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tseng GC, Wong WH. Tight clustering: a resampling-based approach for identifying stable and tight patterns in data. Biometrics. 2005;61:10–16. doi: 10.1111/j.0006-341X.2005.031032.x. [DOI] [PubMed] [Google Scholar]
- 42.Ward J., Jr. Hierarchical grouping to optimize an objective function. Journal of the American Statistical Association. 1963;58:236–244. [Google Scholar]
- 43.Joost S, et al. Single-cell transcriptomics reveals that differentiation and spatial signatures shape epidermal and hair follicle heterogeneity. Cell Systems. 2016;3:221–237. doi: 10.1016/j.cels.2016.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Draghici, S. & Nguyen, T. C. PINS: A Perturbation Clustering Approach for Data Integration and Disease Subtyping US Patent App. 15/068,048 (2016).
- 45.Hennig C. Cluster-wise assessment of cluster stability. Computational Statistics & Data Analysis. 2007;52:258–271. [Google Scholar]
- 46.Hennig C. Dissolution point and isolation robustness: robustness criteria for general cluster analysis methods. Journal of Multivariate Analysis. 2008;99:1154–1176. [Google Scholar]
- 47.Hubert L, Arabie P. Comparing partitions. Journal of Classification. 1985;2:193–218. [Google Scholar]
- 48.Vinh, N. X., Epps, J. & Bailey, J. Information theoretic measures for clusterings comparison: is a correction for chance necessary? In Proceedings of the 26th annual international conference on machine learning, 1073–1080 (2009).
- 49.Vinh NX, Epps J, Bailey J. Information theoretic measures for clusterings comparison: Variants, properties, normalization and correction for chance. Journal of Machine Learning Research. 2010;11:2837–2854. [Google Scholar]
- 50.Rosenberg, A. & Hirschberg, J. V-measure: A conditional entropy-based external cluster evaluation measure. In Proceedings of the 2007 joint conference on empirical methods in natural language processing and computational natural language learning (EMNLP-CoNLL), 410–420 (2007).
- 51.Klein AM, et al. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell. 2015;161:1187–1201. doi: 10.1016/j.cell.2015.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Patel AP, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396–1401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Treutlein B, et al. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature. 2014;509:371. doi: 10.1038/nature13173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kiselev VY, Yiu A, Hemberg M. scmap: projection of single-cell RNA-seq data across data sets. Nature Methods. 2018;15:359. doi: 10.1038/nmeth.4644. [DOI] [PubMed] [Google Scholar]
- 55.Lun, A., Risso, D. & Korthauer, K. SingleCellExperiment: S4 classes for single cell data. R package version1 (2018).
- 56.McCarthy, D., Campbell, K., Lun, A. & Wills, Q. Scater: pre-processing, quality control, normalisation and visualisation of single-cell RNA-seq data in r. bioRxiv, 10.1101/069633 (2016). [DOI] [PMC free article] [PubMed]
- 57.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biology. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Amir E-aD, et al. viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nature Biotechnology. 2013;31:545. doi: 10.1038/nbt.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jaccard P. Étude comparative de la distribution florale dans une portion des Alpes et des jura. Bull Soc Vaudoise Sci Nat. 1901;37:547–579. [Google Scholar]
- 60.Shannon CE. A mathematical theory of communication. ACM SIGMOBILE Mobile Computing and Communications Review. 2001;5:3–55. [Google Scholar]
- 61.Hennig, C. fpc: Flexible procedures for clustering, http://CRAN.R-project.org/package=fpc. R package version 2.1-7. (2014).
- 62.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nature Biotechnology. 2018;36:411. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.