

Abstract
Trachycarpus fortunei (Hook.) is a typical dioecious plant, which has important economic value. There is currently no sex identification method for the early stages of T. fortunei growth. The aim of this study was to obtain expression and site differences between male and female T. fortunei transcriptomes. Using the Illumina sequencing platform, the transcriptomes of T. fortunei male and female plants were sequenced. By analyzing transcriptomic differences, the chromosomal helical binding protein (CHD1), serine/threonine protein kinase (STPK), cytochrome P450 716B1, and UPF0136 were found to be specifically expressed in T. fortunei males. After single nucleotide polymorphism (SNP) detection, a total of 12 male specific sites were found and the THUMP domain protein homologs were found to be male-biased expressed. Cytokinin dehydrogenase 6 (CKX6) was upregulated in male flowers and the lower concentrations of cytokinin (CTK) may be more conducive to male flower development. During new leaf growth, flavonoid and flavonol biosynthesis were initiated. Additionally, the flavonoids, 3′,5′-hydroxylase (F3′5′H), flavonoids 3′-hydroxylase, were upregulated, which may cause the pale yellow phenotype. Based on these data, it can be concluded that inter-sex differentially expressed genes (DEGs) and specific SNP loci may be associated with sex determination in T. fortunei.
Subject terms: Plant breeding, Plant genetics
Introduction
Trachycarpus fortunei (Hook.) H. Wendl. (Fam.: Trachycarpus) are commonly known as "mountain palm" or "windmill palm" evergreen trees. Its flowers are unisexual and dioecious. T. fortunei is widely planted throughout China, where its leaf sheath fiber is often used as a rope and its unopened flower buds, also known as "brown fish," are edible and consumed1. T. fortunei is an important economic and landscaping plant. Plants of different genders often have different economic values; if seeds and fruits are used as harvesting objects, a large number of female plants are needed, while greening forests dominated by vegetative organs require male plants due to their higher economic value2. The sex of mature T. fortunei plants is generally identified by the inflorescence phenotype. The male upper inflorescence has 2–3 branches and the lower part is branched, while the female inflorescence has 4–5 conical branches and 6 staminodes, and often bears residual fruit in the inflorescences from the previous year. With the exception of different inflorescences and floral organs, other morphological signs of male and female T. fortunei do not exhibit obvious sexual dimorphism. T. fortunei also has adult non-inflorescence strains. The lack of inflorescence at the seedling stage leads to the lack of markers for the identification of male and female plants in the early stage of T. fortunei. Studying T. fortunei flower inflorescence and flower body development is a key factor for understanding the evolutionary relationship between the palm family and other angiosperm families3.
Current theories on plant sex determination concentrate on dioecious species4. The emergence of high-throughput sequencing technology and generation of large-scale data from non-model species has been a turning point in uncovering potential parthenogenetic development genes5. Transcriptomes are investigated by deep sequencing technology (i.e., RNA-Seq), which is essential for interpreting the functional elements of the genome, revealing the molecular components of cells and tissues, and understanding development and disease6. Analyzing transcriptomic differences in dioecious flower buds contributes to the screening of gender-related differentially expressed genes (DEGs), such as in studies on shrub willow7, Quercus suber8, Diospyros lotus9, and Ginkgo biloba10. Currently, no studies have investigated the relevant mechanisms of T. fortunei sex determination and only the screening of improved varieties during the introduction process has been conducted. In this study, RNA-Seq was used to provide a molecular basis for revealing differences in the transcriptional levels between T. fortunei male and female flowers and leaves. The findings of this study will enhance our understanding of the differences between male and female T. fortunei, facilitate the development of gender markers, and lay a foundation for furthering our understanding of T. fortunei sex determination and flower organ development.
Results
Raw data quality assessment and composition
In order to improve the quality and integrity of the assembly, 12 cDNA libraries were combined for assembly. Through the quality control of raw reads (Table 1), the Q20 ratio of all samples was > 97.56%, Q30 was > 93.82%, GC content ranged from 47.1 to 48.55%, and the unknown N base ratio was < 0.01%. The GC content of each sample was horizontally distributed, indicating that the sequencing quality was generally good and the data was reliable. Through the Trinity read assembly, a total of 431,753 transcripts and 158,533 unigenes were obtained. The N50 of transcripts and unigenes were 2,639 and 1,867 bp, respectively, indicating that the assembly integrity was relatively high. Raw data were uploaded to the NCBI/SRA database (accession Nos. SRR10120876–SRR10120887).
Table 1.
Sample sequencing output data quality assessment form.
| Sample ID | Read sum | Base sum | Q20 | Q30 | GC (%) | N (%) |
|---|---|---|---|---|---|---|
| Pt-ff1 | 44,913,516 | 6,737,027,400 | 97.67 | 94.04 | 47.56 | 0 |
| Pt-ff2 | 48,017,968 | 7,202,695,200 | 97.63 | 93.96 | 48.55 | 0 |
| Pt-ff3 | 48,645,406 | 7,296,810,900 | 97.67 | 94.06 | 47.26 | 0 |
| Pt-fl1 | 48,517,790 | 7,277,668,500 | 97.8 | 94.31 | 47.22 | 0 |
| Pt-fl2 | 48,059,210 | 7,208,881,500 | 97.7 | 94.12 | 47.5 | 0 |
| Pt-fl3 | 47,985,230 | 7,197,784,500 | 97.73 | 94.16 | 47.4 | 0 |
| Pt-mf1 | 50,344,970 | 7,551,745,500 | 97.67 | 94.06 | 47.42 | 0 |
| Pt-mf2 | 48,395,086 | 7,259,262,900 | 97.62 | 93.91 | 47.8 | 0 |
| Pt-mf3 | 49,530,474 | 7,429,571,100 | 97.64 | 94.14 | 47.5 | 0.01 |
| Pt-ml1 | 44,413,774 | 6,662,066,100 | 97.72 | 94.15 | 47.1 | 0 |
| Pt-ml2 | 46,870,966 | 7,030,644,900 | 97.56 | 93.82 | 47.59 | 0 |
| Pt-ml3 | 48,786,860 | 7,318,029,000 | 97.67 | 94.04 | 47.26 | 0 |
Functional comment
Unigenes were compared with the major databases (Table 2). A total of 69,074 (43.57%) unigenes were obtained from the aforementioned databases. The maximum number of annotated unigenes was obtained from the NCBI NR database (66,355 unigenes, 41.86%), and the minimum number was obtained from the GO database (13,511 unigenes, 8.52%).
Table 2.
Unigene comment statistics.
| Anno_Database | Annotated_Number | 300 ≤ length < 1,000 | Length ≥ 1,000 |
|---|---|---|---|
| COG_Annotation | 16,197 | 8,640 | 7,557 |
| GO_Annotation | 13,511 | 5,364 | 8,147 |
| KEGG_Annotation | 19,581 | 8,181 | 11,400 |
| KOG_Annotation | 35,348 | 18,853 | 16,495 |
| Pfam_Annotation | 24,446 | 12,406 | 12,040 |
| Swissprot_Annotation | 32,535 | 14,572 | 17,963 |
| eggNOG_Annotation | 52,992 | 27,298 | 25,694 |
| nr_Annotation | 66,355 | 37,197 | 29,158 |
Gene expression analysis
Bowtie compared the sequenced reads with the unigene library by using the expected number of Fragments Per Kilobase of transcript sequence per Millions (FPKM) base pairs sequenced as the expression abundance of the corresponding unigenes. Pt-ff and Pt-mf were grouped together, Pt-ml and Pt-fl were grouped together, and flowers and leaves were distinguishable (Fig. 1a), wherein the value of PCA1 was 57.8% and the value of PCA2 was 9% (Fig. 1b).
Figure 1.

(a) Heatmaps of the expression levels between two samples; (b) PCA; (c) Comparison of Venn diagrams; (d) Heatmaps of DEGs in Pt-ff/Pt-mf and Pt-fl/Pt-ml.
Functional enrichment analysis of DEGs
There were 127 DEGs between Pt-ff and Pt-mf (Fig. 1c), which consisted of 76 upregulated and 51 downregulated genes. Between Pt-fl and Pt-ml, 32 genes were upregulated and eight were downregulated. There were 17 DEGs in Pt-ff/Pt-mf and Pt-fl/Pt-ml (Fig. 1d). In Pt-ff/Pt-mf and Pt-fl/Pt-ml, 5 DEGs (i.e., TRINITY_DN56254_c0_g1, TRINITY_DN82812_c2_g2, TRINITY_DN70437_c2_g4, TRINITY_DN61762_c0_g1, and TRINITY_DN81704_c4_g4) were expressed in male plants. Additionally, 1 DEG, β-carotene 3-hydroxylase 2 (TRINITY_DN74609_c4_g1), was expressed in four combinations.
The GO enrichment analysis revealed that the Pt-ff/Pt-mf DEGs grouped into 56 categories, including transcriptional regulation, DNA-templated (GO: 0006355), sequence-specific DNA binding (GO: 0043565), and nucleus (GO: 0005634). Pt-fl/Pt-ml DEGs grouped into 19 categories, including cell wall (GO: 0005618), cell wall modification (GO: 0045545), and pectinesterase activity (GO: 0030599). The Pt-ff/Pt-ml DEGs grouped into 637 categories, including protein phosphorylation (GO: 0006468), regulation of transcription, DNA-templated (GO: 0006355), microtubule-based movement (GO: 0007018) and so on. The Pt-mf/Pt-fl DEGs grouped into 670 categories, including regulation of transcription, DNA-templated (GO: 0006355), protein phosphorylation (GO: 0006468), microtubule-based movement (GO: 0007018) and so on.
In Pt-ff/Pt-mf, the KEGG enrichment analysis and mapping of the DEGs (Fig. 2a) revealed that plant hormone signal transduction (ko04075) had the lowest q-value, indicating an obvious difference fold. Zeatin biosynthesis (ko00908) also had a large abscissa enrichment factor. In Pt-fl/Pt-ml (Fig. 2b), alpha-linolenic acid metabolism (ko00592), ribosome biogenesis in eukaryotes (ko03008), and starch and sucrose metabolism (ko00500), among other terms, had q-values that sequentially decreased. In Pt-ff/Pt-fl and Pt-mf/Pt-ml (Fig. 2c,d), the enrichment factors of flavonoid and flavonol biosynthesis (ko00944) were > 1.
Figure 2.

(a) Pt-ff/Pt-mf enrichment pathway factor map; (b) Pt-fl/Pt-ml enrichment pathway factor map; (c) Pt-ff/Pt-fl enrichment pathway factor map; (d) Pt-Mf/Pt-ml enrichment pathway factor map.
RT-qPCR verification
Quantitative fluorescence analysis and RNA expression quantification were followed by linear regression analysis (Fig. 3a). Results revealed that the expression levels of STPK, UPF, and P450, which are specifically expressed in male plants, were significantly different compared with female plants; genes that were specifically expressed are presented (Fig. 3b). For example, the expression level of STPK in Pt-ml and Pt-mf was 23.76, which was 10.52 times that of Pt-ff.
Figure 3.
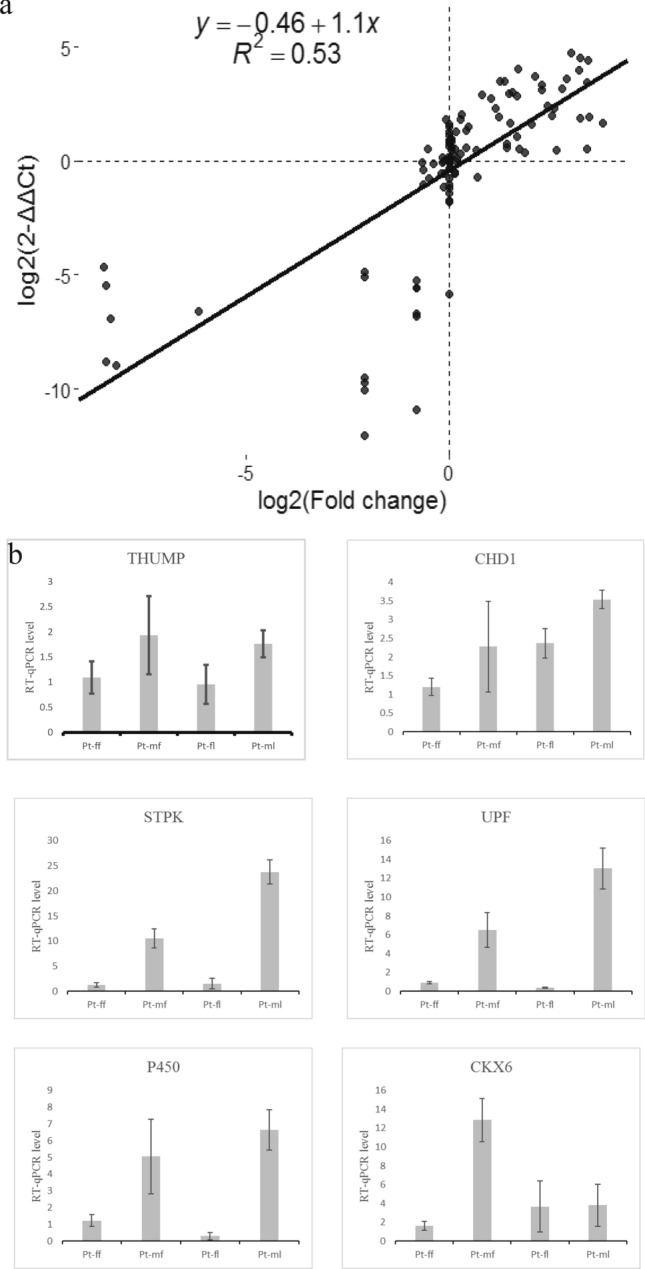
(a) The relative quantification of fluorescence (log2(2−ΔΔCt)) correlated with log2(fold change) linear regression. (b) Fluorescence relative quantification results (2−ΔΔCt) were calculated from the mean Pt-ff control (Ct).
SNP full transcriptome detection
By searching transcriptomic single nucleotide polymorphisms (SNPs), 12 SNP loci in the male and female strains were identified with base substitutions and homozygous homology from the same sample (Table 3); these sites may be gender markers that distinguish the male sex in T. fortunei. It is worth noting that TRINITY_DN73064_c0_g1 is 1 DEG in Pt-ff/Pt-mf and Pt-fl/Pt-ml that was upregulated in male plants at the 1,066 site (5′–3′ locus). There is a base-switching event where the G nucleotide is fixed in T. fortunei females and A in T. fortunei males. SSR detection found that there was a double-base repeat event inside the TRINITY_DN73064_c0_g1 gene and the repeating unit was 34 times. Male and female plants have C ↔ A, G ↔ A, T ↔ C, and C ↔ T, among other base conversion events, at the same base sites of the remaining seven genes.
Table 3.
Hypothetical sex-related genes in T. fortunei.
| Gene ID | POS | Pt-ff1|Depth; Pt-ff2|Depth; Pt-ff3|Depth | Pt-fl1|Depth; Pt-fl2|Depth; Pt-fl3|Depth | Pt-mf1|Depth; Pt-mf2|Depth; Pt-mf3|Depth | Pt-ml1|Depth; Pt-ml2|Depth; Pt-ml3|Depth |
|---|---|---|---|---|---|
| TRINITY_DN69373_c0_g2 | 89 | C|7; C|7; C|7 | C|7; C|6; C|7 | A|7; A|4; A|6 | A|2; A|12; A|6 |
| 143 | G|20; G|7; G|20 | G|7; G|7; G|10 | A|9; A|5; A|7 | A|3; A|11; A|7 | |
| TRINITY_DN70551_c5_g1 | 1762 | T|24; T|22; T|23 | T|26; T|47; T|20 | C|76; C|45; C|69 | C|69; C|70; C|69 |
| TRINITY_DN73064_c0_g1 | 1,066 | G|8; G|2; G|4 | G|4; G|4; G|4 | A|22; A|9; A|19 | A|9; A|15; A|19 |
| TRINITY_DN73101_c1_g3 | 728 | C|28; C|15; C|24 | C|5; C|3; C|5 | T|24; T|27; T|6 | T|4; T|2; T|6 |
| 1,276 | A|22; A|15; A|26 | A|7; A|4; A|8 | C|20; C|14; C|3 | C|2; C|2; C|3 | |
| TRINITY_DN77827_c1_g1 | 1,395 | C|23; C|11; C|12 | C|9; C|19; C|7 | T|28; T|17; T|4 | T|3; T|6; T|4 |
| TRINITY_DN82283_c0_g1 | 176 | A|5; A|4; A|6 | A|31; A|11; A|22 | C|7; C|7; C|22 | C|25; C|8; C|22 |
| TRINITY_DN85795_c1_g1 | 1693 | T|7; T|3; T|2 | T|6; T|9; T|2 | C|10; C3; C|23 | C|20; C|2; C|23 |
| 1805 | A|7; A|3; A|2 | A|10; A|10; A|6 | G|11; G|5; G|23 | G|20; G|7; G|23 | |
| 1901 | A|5; A|3; A|4 | A|6; A|4; A|5 | C|11; C|3; C|11 | C|9; C|2; C|11 | |
| TRINITY_DN87151_c2_g1 | 885 | G|83; G|96; G|67 | G|27; G|20; G|23 | A|160; A|206; A|20 | A|18; A|19; A|20 |
POS is the site of the unigene where the sequence-specific site was located (5′–3′).
Discussion
There are 15,600 dioecious angiosperms in 987 genera and 175 families that account for 5–6% of the total species11. Early ascertaining the sex of seedlings can accelerate the artificial selection in breeding programs12. Sex determination is a major shift in the evolutionary history of angiosperms, as dioecious, sex-determining genes are usually located in the non-recombinant regions of sex chromosomes13. Next generation sequencing (NGS) technology is a widely used to study the promotion of sex determination in flowering plants. De novo RNA‐Seq transcriptome assembly and expression analysis can inform the investigation of gender determination in dioecious specie, exploring genome-wide sex-biased expression patterns in different species revealed a broad variation in the percentage of sex-biased genes (ranging from 2% of transcripts in Littorina saxatilis to 90% in Drosophila melanogaster), a study show that significantly more genes exhibited male-biased than female-biased expression in Asparagus officinalis14. Sex-biased expression was pervasive in floral tissue in Populus balsamifera, but nearly absent in leaf tissue15. Sex-specifically expressed genes may be derived from silencing the inhibition of related genes or the deletion of homologous genes in corresponding sex tissues. There are several scenarios for the origin of sex-biased genes, including single-locus antagonism, sexual antagonism plus gene duplication and duplication of sex-biased genes16. In this study, transcripts were constructed and assembled from T. fortunei female and male flowers and leaves to identify genes with sex-biased expression. Our study showed that more genes exhibited male-biased than female-biased expression in T. fortunei. Genes with sex-biased expression, often contribute largely to the expression of sexually dimorphic traits, SNPs calling from segregated populations of dioecious plant can help to identify the sex-associated SNPs and corresponding loci, five DEGs were polymorphic with common SNPs in each sex type in Eucommia ulmoides17. By searching transcriptomic SNPs, 12 SNP loci in the male and female strains were identified with base substitutions and homozygous homology from the same sample. S4U (4-thiouridine), a modified nucleoside, is located in the receptor arm and D arm of eubacterial and archaeal tRNA. S4U8 is synthesized by 4-thiouridine synthase (Thil) to stabilize the folding of tRNA and acts as a sensitive trigger for UV irradiation response mechanisms18,19. TRINITY_DN73064_c0_g1, the THUMP domain-containing protein 1 homolog, is a homologue with a THUMP domain and is one of the Thil constituent domains; it has a double base (CT) 34-unit repeat and a male-specific site. It is differentially expressed in both T. fortunei male and female plants; such polymorphic biased genes may be linked together in the non-recombinant region of sex determination regions (SDR)20.
CHD1 (TRINITY_DN61762_c0_g1) is highly expressed in T. fortunei male plants. In a previous study, rice CHD1 was highly expressed in plant leaves and the number of cells in the leaves and stems of CHD1 mutant plants decreased, the surface epidermis increased, and the chlorophyll a/b content of leaves decreased21. CHD1 has little difference in terms of exon and intron changes, and variations are mainly concentrated in the introns. Interestingly, it has been used as a marker for early sex identification in birds and poultry22–24. Protein kinases are a class of enzymes that use ATP to phosphorylate other proteins and play important roles in controlling many aspects of cellular life, and are divided into three major subclasses: receptor tyrosine kinase (RTK), serine/su protein kinase (STPK), and histidine kinase25. TRINITY_DN81704_c4_g4 acts as a STPK that is synergistic with cyclins and serves as an important cellular regulatory factor. In Arabidopsis, the gene encoding the STPK, OXI1, is induced in response to extensive H2O2 stimulation and serves as an important part of the signal transduction pathway that links several oxidative signals with downstream responses26. STPK are highly expressed in T. fortunei males compared to female flowers and leaves, suggesting that there may be differences in the stress transmission of oxidative signals between male and female plants. UPF0136 (TRINITY_DN70437_c2_g4) and cytochrome P450 716B1 (cytochrome P450 716B1-like) (TRINITY_DN82812_c2_g2) were also highly expressed in male plants, however, their specific functions have yet to be explored.
KEGG enrichment of the DEGs in Pt-ff/Pt-mf revealed that zeatin biosynthesis (ko00908) was enriched and cytokinin dehydrogenase 6 (TRINITY_DN64789_c0_g1) was upregulated 6.98 times higher in male flowers. Cytokinin oxidase/dehydrogenase (CKXs) catalyze the irreversible degradation of cytokinins27. During the development of male and female T. fortunei flowers, the IAA, Abscisic acid (ABA) and zeatin riboside (ZR) contents of male flowers are lower than female flowers in the corresponding period28. Transgenic overexpression of 6 CKX members in Arabidopsis transgenic plants increased the breakdown of cytokinin with a content equivalent to 30–45% of wild-type CTK content; the existence of CTK is essential for survival and a lack of CTK leads to reduced plant apical meristem and leaf primordium activities29. Most Brassica napus BnCKXs are highly expressed in reproductive organs, such as buds, flowers, or siliques30. TRINITY_DN64789_c0_g1 was highly expressed in male flowers, indicating that CKXs were upregulated in male flowers and that lower CTK concentrations may be more favorable for male flower development. This spatial expression pattern may be related to the different CTK functions.
In Pt-ff/Pt-fl and Pt-mf/Pt-ml, flavonoid and flavonol biosynthesis (ko00944) were both > 1. Flavonoid 3′,5′-hydroxylase (F3′5′H) is a member of the cell pigment P450 family and is the key enzyme for the synthesis of 3′,5′-hydroxylhydrochemical pigmentation31. Flavonoid 3′-hydroxylase (F3′H) is involved in flavonoid biosynthesis. The F3′H gene is expressed in different tissues such as roots, stems, leaves, flowers, etc., and can change the color of plant flowers or seed coats32. Flavonoids, carotenoids, and beetroot are the main flower pigments33. In flavonoids, orange and charone are yellow pigments; thus, flavonoids and flavonols are colorless or very light yellow34. The upward expression of F3′5′H and F3′H in T. fortunei new leaf growth indicates that these components may cause the light yellow leaf color phenotype.
Conclusion
By analyzing the transcriptomic differences between T. fortunei female and male flowers and leaves, CHD1, STPK, cytochrome P450 716B1, UPF0136 and THUMP domain-containing protein 1 homolog were found to be highly expressed in T. fortunei males. Through SNP site detection, a total of 12 male and female specific sites were found. CKX6 exhibited increased expression in male flowers. Lower CTK concentrations may be more conducive to the development of male flowers. In the early stages of leaf growth, flavonoid and flavonol biosynthesis were initiated and the up-regulated expression of F3′5′H and F3′H may cause the pale yellow phenotyper of the leaf.
Materials and methods
Materials
Materials were collected from an artificially planted T. fortunei forest located in Guiding County, Guizhou Province, China. The test site belongs to the mid-subtropical monsoon humid climate. The soil in the forest is yellow soil with an annual rainfall of 1,143 mm and annual average temperature of 15 °C. Three male and female T. fortunei were selected for sampling. Male and female flowers in the unexpanded flower buds were collected and labeled as Pt-ff (female flower) or Pt-mf (male flower). The middle part of unexpanded leaves at the top of the treetop was collected (light yellow) and labeled as Pt-fl (female leaves) or Pt-ml (male leaves), and then promptly placed in liquid nitrogen. There are three biological repeats in each group.
RNA extraction and library preparation
The isolation of total RNA from above samples were isolated according to the instruction manual of the Trizol Reagent (Invitrogen, Carlsbad, CA, USA). Nandrop 2000 (Thermo Fisher Scientific, Waltham, Massachusetts, USA) was used to detect the purity of RNA. Qubit was used to accurately quantify RNA concentration. Agilent 2100 (Agilent Technologies, Santa Clara, California, USA) accurately detected RNA integrity. After passing these quality checks, magnetic beads with Oligo (dT) were enriched for eukaryotic mRNA. Subsequently, fragmentation buffer was used to break the mRNA into short fragments, which was used as a template to synthesize a strand of cDNA with a 6-base random primer. Then, double-stranded cDNA was synthesized, purified, and subjected to end repair, poly-(A) tail synthesis, and ligation to the sequencing link. Fragment size selection was conducted using AMPure XP beads. The second strand of the cDNA containing U was degraded using USER enzymes and the strand orientation of the mRNA was retained. After the prepared library was tested, sequencing was performed on Illumina HiSeq 2500 system machine. There are a total of 12 samples, and each sample is constructed separately for library sequencing.
Evaluation, assembly, and annotation of raw data quality
The raw sequencing data was quality-controlled; low-quality reads and unknown bases > 1% were removed using the Trimmomatic tool35. Trinity software36 was used for clean reads assembly. Unigene sequences (longest transcript) were aligned with the NCBI NR37, Swiss-Prot38, Gene Ontology (GO)39, COG40, EuKaryotic Orthologous Groups (KOG)41, eggNOG42, and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases43.
Gene expression quantification, differential analysis, and functional enrichment
The reads obtained from sequencing were compared with the unigene library using Bowtie software44. Expression levels were estimated according to the comparison results and RSEM45. Pearson's correlation coefficient was used as an indicator for evaluating biological repetitive correlations46. R software (https://www.r-project.org/) was used to calculate the correlation coefficients, plot the correlation heatmap, and conducted a principal component analysis (PCA). Use DESeq47 software to standardize the number of Unigene counts in each sample (basemean value is used to estimate expression), calculate the difference multiple, and use NB (negative binomial distribution test) to test the difference reads significance, and finally screen differentially expressed gene (DEG) according to the difference multiple and the difference significance test results. DEG screening threshold was set to |fold change| ≥ 2 and FDR < 0.01. Perform hierarchical clustering (hcluster, hierarchical clustering) analysis on DEG, annotate the function of the database with DEG, and perform functional enrichment analysis on DEG using software topGO48 and KOBAS49.
RT-qPCR verification
Real-time quantitative PCR (RT-qPCR) amplification was conducted using an SYBR Premix Ex TaqTM II kit. Twelve genes were selected; the actin gene was used as the reference gene, Primer Primer5 software was used to design primers (refer to Table 4 for the primer list). All samples reported in the transcriptome were quantitatively verified; each sample had three biological and three technical replicates. The first strand of the cDNA fragment was synthesized from total RNA. The RT-qPCR reaction conditions were as follows: preheating at 95 °C for 30 s, 40 cycles at 95 °C for 5 s, and annealing at 60 °C for 34 s. Relative expression levels were calculated using the 2−ΔΔCt method50.
Table 4.
List of primers used in this study.
| No. | Gene symbol | GeneBank | Forward primer | Reverse primer | Product length(bp) | Ta (℃) |
|---|---|---|---|---|---|---|
| 1 | CHD1 | TRINITY_DN61762_c0_g1 | GAGTGAAGAGGAGCCATTTG | GGCATTCCCACCATAAGTATC | 61 | 60 |
| 2 | STPK | TRINITY_DN81704_c4_g4 | AGTACCATCACCCATGACC | CGAAGATGAGTGCTGTTGT | 83 | 60 |
| 3 | P450 716B1 | TRINITY_DN82812_c2_g2 | CCAGTTCTACTTGGCTAGTC | CATTGATAGTGTGAGCAAACC | 64 | 60 |
| 4 | UPF0136 | TRINITY_DN70437_c2_g4 | TCTATGTATGCACTGGGCTTC | ATCAACAGCCACATTCTCG | 90 | 60 |
| 5 | Cytokinin dehydrogenase 6 | TRINITY_DN64789_c0_g1 | AATGGAGGATGGGTATTCACA | GGGACATGATAGAGCACCGA | 89 | 60 |
| 6 | Thump | TRINITY_DN73064_c0_g1 | ATATGGGCAATTATTAGCTGGT | AGTGAGTAGGATTCTAGTGCTT | 60 | 60 |
| 7 | FT | TRINITY_DN75953_c2_g6 | GCACATGAAGACATGAACCT | GTAACTGCTTGGGATGATGAC | 74 | 60 |
| 8 | PI | TRINITY_DN70832_c3_g1 | TTACCGGAAACTCAGCAGTC | TATCCAAGCTCCAGCTCCCTTA | 116 | 60 |
| 9 | AG | TRINITY_DN69681_c4_g1 | AGTACAGCAAGTAGGTATTGTG | ATGGTAATAGTTTCGGGAATCG | 79 | 60 |
| 10 | AG9 | TRINITY_DN81743_c0_g1 | ATGGAAAGTAATGGAAACCAGT | GGATTATGATGTGTGAAGCTCT | 132 | 60 |
| 11 | Sterile apetala | TRINITY_DN72937_c1_g3 | CTCGCCACAGTGCTAAAC | CAGGGCATTGGTGGATTC | 126 | 60 |
| 12 | FLC | TRINITY_DN71780_c5_g1 | AGGCTAAGGAACTGTCGAT | CAGTGGGCGAGAACATGA | 63 | 60 |
| 13 | Actin | TRINITY_DN66424_c1_g2 | TGAATCTGGTCCATCCATTGTC | AGAACATACCATAACCAAGCTC | 60 | 60 |
Acknowledgements
The work was supported by Major Science and Technology Projects in Guizhou Province (Ministry of Science and Technology Major Project [2014] 6024).
Author contributions
X.F. and Z.Y. designed and executed the experiment; W.X.R. and W.Y. conducted additional analyses; X.F. wrote the manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Yunfa D. The many uses and exploitation of Trachycarpus fortunei. Chin. Wild Plant Resour. 2005;24:25–27. [Google Scholar]
- 2.Li R, Lu L, Gao W, et al. Advances in sex identification of dioecious plants. Guihaia. 2006;26:387–391. [Google Scholar]
- 3.Adam H, Jouannic S, Escoute J, Duval Y, Verdeil JL, Tregear JW. Reproductive developmental complexity in the African oil palm (Elaeis guineensis, Arecaceae) Am. J. Bot. 2005;92:1836–1852. doi: 10.3732/ajb.92.11.1836. [DOI] [PubMed] [Google Scholar]
- 4.Pannell JR. Plant sex determination. Curr. Biol. 2017;27:R191–R197. doi: 10.1016/j.cub.2017.01.052. [DOI] [PubMed] [Google Scholar]
- 5.Sobral R, Silva HG, Morais-Cecílio L, Costa MM. The quest for molecular regulation underlying unisexual flower development. Front/ Plant Sci. 2016;7:160. doi: 10.3389/fpls.2016.00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Z, Gerstein M, Snyder M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009;10:57. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu J, Yin T, Ye N, Chen Y, Yin T, Liu M, Hassani D. Transcriptome analysis of the differentially expressed genes in the male and female shrub willows (Salix suchowensis) PLoS ONE. 2013;8:e60181. doi: 10.1371/journal.pone.0060181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rocheta M, Sobral R, Magalhães J, Amorim MI, Ribeiro T, Pinheiro M, et al. Comparative transcriptomic analysis of male and female flowers of monoecious Quercus suber. Front. Plant Sci. 2014;5:599. doi: 10.3389/fpls.2014.00599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Akagi T, Henry IM, Tao R, Comai L. Plant genetics. A Y-chromosome-encoded small RNA acts as a sex determinant in persimmons. Science (New York, N.Y.) 2014;346:646–650. doi: 10.1126/science.1257225. [DOI] [PubMed] [Google Scholar]
- 10.Tang, H., Du, S., Xing, S., Sang, Y., Li, J., & Liu, X., et al. Screening of sex determination related genes in ginkgo biloba. Sci. Silvae Sin.53, 76–82 https://kns.cnki.net/KXReader/Detail?TIMESTAMP=637140104015600000&DBCODE=CJFQ&TABLEName=CJFDLAST2017&FileName=LYKE201702009&RESULT=1&SIGN=SaSFe%2fwtBU47RmMTYoDsRf0DHtU%3d (2017).
- 11.Renner SS. The relative and absolute frequencies of angiosperm sexual systems: Dioecy, monoecy, gynodioecy, and an updated online database. Am. J. Bot. 2014;101:1588–1596. doi: 10.3732/ajb.1400196. [DOI] [PubMed] [Google Scholar]
- 12.Wang, W., Yang, G., Deng, X., Shao, F., Li, Y., Guo, W., & Zhang, X. et al. Molecular sex identification in the hardy rubber tree (Eucommia ulmoides Oliver) via ddRAD markers. Int. J. Genom.2020, 2420976 (2020). [DOI] [PMC free article] [PubMed]
- 13.Zhang J, Boualem A, Bendahmane A, Ming R. Genomics of sex determination. Curr. Opin. Plant Biol. 2014;18:110–116. doi: 10.1016/j.pbi.2014.02.012. [DOI] [PubMed] [Google Scholar]
- 14.Harkess A, Mercati F, Shan HY, Sunseri F, Falavigna A, Leebens-Mack J. Sex-biased gene expression in dioecious garden asparagus (Asparagus officinalis) New Phytol. 2015;207:883–892. doi: 10.1111/nph.13389. [DOI] [PubMed] [Google Scholar]
- 15.Sanderson BJ, Wang L, Tiffin P, Wu Z, Olson MS. Sex-biased gene expression in flowers, but not leaves, reveals secondary sexual dimorphism in Populus balsamifera. New Phytol. 2019;221:527–539. doi: 10.1111/nph.15421. [DOI] [PubMed] [Google Scholar]
- 16.Ellegren H, Parsch J. The evolution of sex-biased genes and sex-biased gene expression. Nat. Rev. Genet. 2007;8:689–698. doi: 10.1038/nrg2167. [DOI] [PubMed] [Google Scholar]
- 17.Wang W, Zhang X. Identification of the sex-biased gene expression and putative sex-associated genes in Eucommia ulmoides Oliver using comparative transcriptome analyses. Molecules. 2017;22:2255. doi: 10.3390/molecules22122255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Waterman DG, Ortiz-Lombardía M, Fogg MJ, Koonin EV, Antson AA. Crystal structure of Bacillus anthracis ThiI, a tRNA-modifying enzyme containing the predicted RNA-binding THUMP domain. J. Mol. Biol. 2006;356:97–110. doi: 10.1016/j.jmb.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 19.Neumann P, Lakomek K, Naumann PT, Erwin WM, Lauhon CT, Ficner R. Crystal structure of a 4-thiouridine synthetase—RNA complex reveals specificity of tRNA U8 modification. Nucleic Acids Res. 2014;42:6673–6685. doi: 10.1093/nar/gku249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu H, Fu J, Du H, Hu J, Wuyun TD. novo sequencing of Eucommia ulmoides flower bud transcriptomes for identification of genes related to floral development. Genom. Data. 2016;9:105–110. doi: 10.1016/j.gdata.2016.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yongfeng, H. Fuctional analysis of chromatin regulators in rice (D). Huazhong Agricultural University. https://kns.cnki.net/KCMS/detail/detail.aspx?dbcode=CDFD&dbname=CDFD0911&filename=2010271724.nh&v=MDQxNDExMjZIckcvSDliT3E1RWJQSVI4ZVgxTHV4WVM3RGgxVDNxVHJXTTFGckNVUjdxZlp1Wm9GeS9nVTc3TFY= (2009).
- 22.Zhiming S, Qi X, Lu X, Guyin N, Jinmei W, Zoo JW. Gender identification of white-naped crane and red crowned crane based on genetic polymorphism of ee0.6 gene and chd gene. Chin. J. Wildlife. 2018;39:151–153. [Google Scholar]
- 23.Ndlovu, S. Standardization of a PCR-HRM assay for DNA sexing of birds (Doctoral dissertation, North-West University, 2018).
- 24.Huang J, Mao H, Xu H, Tang J, Ouyang J, Wu Z, et al. SEX-typing of blue-crowned laughingthrush (Garrluax courtoisi) using chd1-based polymerase chain reaction. Appl. Ecol. Environ. Res. 2018;16:6341–6347. [Google Scholar]
- 25.Azad I, Alemzadeh A. Bioinformatic and empirical analysis of a gene encoding serine/threonine protein kinase regulated in response to chemical and biological fertilizers in two maize (Zea mays L.) cultivars. Mol. Biol. Res. Commun. 2017;6:65. [PMC free article] [PubMed] [Google Scholar]
- 26.Rentel MC, Lecourieux D, Ouaked F, Usher SL, Petersen L, Okamoto H, et al. OXI1 kinase is necessary for oxidative burst-mediated signalling in Arabidopsis. Nature. 2004;427:858. doi: 10.1038/nature02353. [DOI] [PubMed] [Google Scholar]
- 27.Galuszka P, Frébort I, Šebela M, Peč P. Degradation of cytokinins by cytokinin oxidases in plants. Plant Growth Regul. 2000;32:315–327. [Google Scholar]
- 28.Ying, W., Yang, Z., & Jie, R. (2018). Variation of endogenous hormone in the development of female and male flowers of palm. Seed03, 7–11 (2018).
- 29.Werner T, Motyka V, Laucou V, Smets R, Van Onckelen H, Schmülling T. Cytokinin-deficient transgenic Arabidopsis plants show multiple developmental alterations indicating opposite functions of cytokinins in the regulation of shoot and root meristem activity. Plant Cell. 2003;15:2532–2550. doi: 10.1105/tpc.014928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu P, Zhang C, Ma JQ, Zhang LY, Yang B, Tang XY, et al. Genome-wide identification and expression profiling of cytokinin oxidase/dehydrogenase (CKX) genes reveal likely roles in pod development and stress responses in oilseed rape (Brassica napus L.) Genes. 2018;9:168. doi: 10.3390/genes9030168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shimada Y, Nakano-Shimada R, Ohbayashi M, Okinaka Y, Kiyokawa S, Kikuchi Y. Expression of chimeric P450 genes encoding flavonoid-3′,5′-hydroxylase in transgenic tobacco and petunia plants 1. Febs Lett. 1999;461:241–245. doi: 10.1016/s0014-5793(99)01425-8. [DOI] [PubMed] [Google Scholar]
- 32.Castellarin SD, Di Gaspero G, Marconi R, et al. Colour variation in red grapevines (Vitis vinifera L.): Genomic organisation, expression of flavonoid 3'-hydroxylase, flavonoid 3',5'-hydroxylase genes and related metabolite profiling of red cyanidin-/blue delphinidin-based anthocyanins in berry skin. BMC Genom. 2006;7:12. doi: 10.1186/1471-2164-7-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tanaka Y, Sasaki N, Ohmiya A. Biosynthesis of plant pigments: Anthocyanins, betalains and carotenoids. Plant J. 2008;54:733–749. doi: 10.1111/j.1365-313X.2008.03447.x. [DOI] [PubMed] [Google Scholar]
- 34.Tanaka Y, Brugliera F, Chandler S. Recent progress of flower colour modification by biotechnology. Int. J. Mol. Sci. 2009;10:5350–5369. doi: 10.3390/ijms10125350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bolger AM, Lohse M, Usadel B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grabherr MG, Haas BJ, Yassour M, et al. Trinity: Reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 2011;29:644. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deng YY, Li JQ, Wu SF, et al. Integrated nr database in protein annotation system and its localization. Comput. Eng. 2006;32:71–74. [Google Scholar]
- 38.Apweiler R, Bairoch A, Wu CH, Barker WC, Boeckmann B, Ferro S, et al. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2004;32(suppl_1):D115–D119. doi: 10.1093/nar/gkh131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tatusov RL, Galperin MY, Natale DA, Koonin EV. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28:33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koonin EV, Fedorova ND, Jackson JD, Jacobs AR, Krylov DM, Makarova KS, et al. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genom. Biol. 2004;5:R7. doi: 10.1186/gb-2004-5-2-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huerta-Cepas J, Szklarczyk D, Forslund K, Cook H, Heller D, Walter MC, et al. eggNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic acids Res. 2015;44:D286–D293. doi: 10.1093/nar/gkv1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M. The KEGG resource for deciphering the genome. Nucleic acids Res. 2004;32(suppl_1):D277–D280. doi: 10.1093/nar/gkh063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li B, Dewey CN. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schulze SK, Kanwar R, Gölzenleuchter M, Therneau TM, Beutler AS. SERE: Single-parameter quality control and sample comparison for RNA-Seq. BMC Genom. 2012;13:524. doi: 10.1186/1471-2164-13-524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alexa, A., & Rahnenführer, J. Gene set enrichment analysis with topGO. Bioconductor Improv27 (2009).
- 49.Mao X, Cai T, Olyarchuk JG, Wei L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics. 2005;21:3787–3793. doi: 10.1093/bioinformatics/bti430. [DOI] [PubMed] [Google Scholar]
- 50.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C T method. Nat. Protoc. 2008;3:1101. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]