

Abstract
Type 2 diabetes mellitus (T2DM) is a frequent comorbidity of cancer. Hyperinsulinemia secondary to T2DM promotes cancer progression, whereas antidiabetic agents, such as metformin, have anticancer effects. However, the detailed mechanism for insulin and metformin-regulated cancer cell proliferation remains unclear. This study identified a mechanism by which insulin upregulated the expression of c-Myc, sterol regulatory element-binding protein 1 (SREBP1), and acetyl-coenzyme A (CoA) carboxylase 1 (ACC1), which are important regulators of lipogenesis and cell proliferation. Thymine DNA glycosylase (TDG), a DNA demethylase, was transactivated by c-Myc upon insulin treatment, thereby decreasing 5-carboxylcytosine (5caC) abundance in the SREBP1 promoter. On the other hand, metformin-activated AMP-activated protein kinase (AMPK) increased DNA methyltransferase 3A (DNMT3A) activity to increase 5-methylcytosine (5mC) abundance in the TDG promoter. This resulted in decreased TDG expression and enhanced 5caC abundance in the SREBP1 promoter. These findings demonstrate that c-Myc activates, whereas AMPK inhibits, TDG-mediated DNA demethylation of the SREBP1 promoter in insulin-promoted and metformin-suppressed cancer progression, respectively. This study indicates that TDG is an epigenetic-based therapeutic target for cancers associated with T2DM.
Keywords: insulin, metformin, c-Myc, AMPK, TDG, DNA demethylation, SREBP1, cancer, diabetes
Graphical Abstract
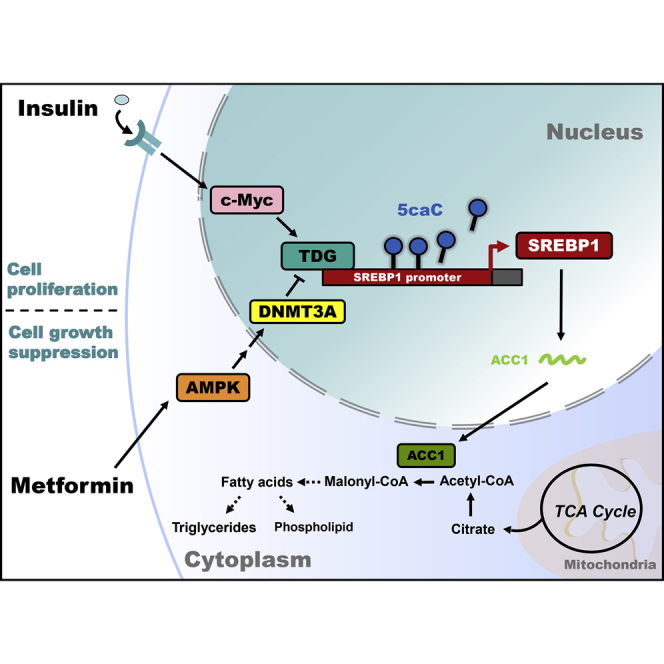
Peng and colleagues demonstrate that c-Myc activates, whereas AMP-activated protein kinase (AMPK) inhibits, thymine DNA glycosylase (TDG)-mediated DNA demethylation of the sterol regulatory element-binding protein 1 (SREBP1) promoter in insulin-promoted and metformin-suppressed cancer progression, respectively. Therefore, targeting TDG represents an epigenetic-based therapeutic strategy for cancers associated with type 2 diabetes mellitus.
Introduction
Type 2 diabetes mellitus (T2DM) is a frequent comorbidity of cancer. T2DM is associated with a higher risk of liver, breast, and colorectal cancers due to hyperinsulinemia, secondary to insulin resistance or exogenous insulin administration.1, 2, 3, 4, 5 Clinical data indicate that high serum insulin concentration is positively correlated with cancer development,6,7 whereas impairment of insulin signaling stalls cancer progression.8,9 The common antidiabetic drug, metformin, has been shown to have preventative and interventional antineoplastic effects on cancers associated with T2DM,10 in part, by activating AMP-activated protein kinase (AMPK).11 Activated AMPK directly phosphorylates proteins to modulate gene transcription of multiple processes critical to cell proliferation, such as lipid synthesis, oxidation, and lipolysis.12 Metformin-activated AMPK has been shown to promote global DNA demethylation in blood and muscle cells.13,14 Metformin is also shown to increase global DNA methylation in colon, breast, and endometrial cancer cells.15,16 However, DNA methylation on lipogenic genes regulated by metformin in cancers associated with T2DM remains unknown.
Cancer progression is associated with increased de novo lipogenesis, which is required for the biosynthesis of membranes, organelles, and signaling molecules, involved in cancer cell proliferation.17,18 Several enzymes that mediate fatty acid (FA) synthesis, such as acetyl-coenzyme A (CoA) carboxylase 1 (ACC1),19,20 are upregulated in a number of human cancers and are important for cancer cell survival and proliferation.21,22 ACC1 is regulated at the transcriptional and post-translational levels. Transcriptionally, insulin induces sterol regulatory element-binding protein 1 (SREBP1) binding to the ACC1 promoter, resulting in ACC1 transactivation.23,24 c-Myc, a well-known oncogenic transcription factor, regulates anabolic processes related to cancer progression,25, 26, 27 in part, by activating ACC1.28,29 Consistent with this, c-Myc is enhanced and stabilized by insulin, suggesting its participation in insulin-induced ACC1 transactivation.30, 31, 32, 33 Both the transcriptional suppression and inactivation of ACC1 are mediated by AMPK. Glucagon-activated AMPK phosphorylates and inhibits both SREBP1 and ACC1.34,35 Insulin, however, inhibits AMPK, which corresponds to enhanced SREBP1 and ACC1 activation.36 Ultimately, the opposing regulation of SREBP1 and ACC1 through AMPK activates or inhibits lipogenesis and cancer cell growth, respectively.37, 38, 39 Although insulin has been shown to increase lipogenic gene expression through transcriptional regulation, it is unclear whether insulin can also affect DNA methylation to regulate lipogenesis in liver and breast cancer cells.
In addition to transcriptional regulation, epigenetic modifications, such as DNA methylation and histone acetylation, alter gene expression to promote cancer initiation and progression.40 DNA methylation, catalyzed by DNA methyltransferases (DNMT1, DNMT3A, and DNMT3B), occurs on cytosines located within CpG dinucleotides to form 5-methylcytosine (5mC) and inhibit transcription.41 To restart gene expression, thymine DNA glycosylase (TDG) replaces two oxidized forms of 5mC, 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC), with unmodified cytosines.42 c-Myc has been shown to modulate gene expression by promoting TDG expression,25 suggesting the involvement of c-Myc in regulating promoter demethylation. AMPK also functions as an epigenetic regulator through modulating DNMT1- and DNMT3B-mediated DNA methylation.15,16,43 However, the roles of AMPK, c-Myc, DNA methylation, and DNA demethylation in the regulation of lipid metabolism in cancers associated with T2DM remain unclear.
In this study, we demonstrate that c-Myc and AMPK regulate SREBP1/ACC1 expression through TDG-mediated DNA demethylation. These findings provide mechanistic insights into the epigenetic regulation of insulin-promoted, metformin-suppressed lipogenesis and cancer cell proliferation that support clinical trials for lipogenesis inhibitors as a therapeutic intervention for cancer therapy44 and uncover TDG as a target for epigenetic therapies.
Results
Insulin Regulates SREBP1/ACC1 Expression through c-Myc/TDG-Mediated DNA Demethylation
Insulin promotes liver and breast cancer cell proliferation,1, 2, 3,45 in part, by increasing lipid synthesis.17,18 Therefore, we tested whether insulin induces the expression of genes associated with cancer cell proliferation and lipid synthesis, including c-Myc, TDG, SREBP1, and ACC1. We used 200 nM insulin to mimic the condition of diabetes, according to several papers using 200 nM insulin to establish hyperinsulinemia in vitro.46, 47, 48 Indeed, 200 nM insulin increased the expression of c-Myc, TDG, SREBP1, and ACC1 on the mRNA (Figures 1A–1C) and protein (Figures 1D–1F and S1A) in liver cancer (HepG2), breast cancer (MCF7), and normal-like breast epithelial cells (MCF10A). Next, because TDG is a CpG demethylase, and epigenetics is essential to the regulation of gene expression, we investigated whether these expression changes are due to chromatin structural changes. As TDG demethylates DNA at CpG dinucleotides, we examined the promoter region of the SREBP1 gene and identified a CpG island spanning +229/−243 base pairs (bp) flanking the transcription start site (Figure 1G). This suggested possible transcriptional regulation by TDG-mediated demethylation in the SREBP1 promoter region in response to insulin. DNA immunoprecipitation (DIP) assay revealed that insulin lowered 5caC abundance in the SREBP1 promoter region in HepG2, MCF7, and MCF10A cells (Figure 1H). Further, insulin increased TDG binding to the SREBP1 promoter in HepG2, MCF7, and MCF10A cells (Figure 1I). To provide additional evidence that TDG mediates insulin-induced SREBP1/ACC1 expression, TDG was knocked down or inhibited by gemcitabine, a DNA demethylation inhibitor.49 Illustrated in Figures 2A–2F and S1A, SREBP1/ACC1 mRNA and protein expression were attenuated when TDG was knocked down or inhibited with 200 nM gemcitabine in HepG2 and MCF7 cells regardless of insulin treatment. Further, knockdown of TDG increased 5caC abundance and moderated the lowering effect of insulin on 5caC abundance in the SREBP1 promoter in HepG2 and MCF7 cells (Figures 2G and 2H). On the other hand, overexpression of TDG in HepG2 cells increased SREBP1/ACC1 expression (Figure 2I), decreased 5caC abundance in the SREBP1 promoter (Figure 2J), and increased TDG binding to the SREBP1 promoter (Figure 2K). These findings indicate that TDG binds to the SREBP1 promoter to decrease 5caC and increase its expression. Next, we determined if these effects occur in human cancer samples. Consistent with our in vitro findings, SREBP1 mRNA expression levels were higher in breast and liver cancer tissues compared to adjacent noncancerous tissues from database analyses (Figures 2L and 2M). Further, there was an inverse correlation between SREBP1 mRNA and promoter 5caC abundance in hepatocellular carcinoma (HCC) tumor tissues compared to peri-tumor tissues (R = −0.51, p = 0.242) of 7 patients (Figures 2N, S1C, and S1D) and to normal liver tissues mixed from 3 healthy liver donors (R = −0.657, p = 0.109) (Figures 2O, S1C, and S1D). This result corroborates our in vitro findings that 5caC abundance in the SREBP1 promoter represents transcriptional repression. However, SREBP1 mRNA levels were found to be lower in HCC tumor tissues compared to peri-tumor in 4 patients (Figure S1C). It could be explained by that the SREBP1 expression may be decreased in certain cancer stages while we collected these samples. In summary, these findings indicate that insulin activates TDG, resulting in decreased 5caC in the SREBP1 promoter, leading to elevated expression.
Figure 1.

Insulin Upregulates c-Myc, TDG, SREBP1, and ACC1 Expression; Lowers 5caC Abundance in the SREBP1 Promoter; and Increases TDG Binding to the SREBP1 Promoter
(A and D) HepG2 cells, (B and E) MCF7 cells, and (C and F) MCF10A cells were treated with 200 nM insulin for 48 h (A–C) or for indicated time periods (D–F). Relative c-Myc, TDG, SREBP1, and ACC1 mRNA, determined via qRT-PCR, was shown as the mean plus SEM of a representative experiment performed in triplicates. ∗∗p < 0.01 and ∗∗∗p < 0.001 (Student’s t test). Cell lysates were analyzed by western blot with specified antibodies. flSREBP1, full-length SREBP1. (G) Schematic representation of the CpG distribution in the upstream of 2,000 bp from the transcription start site (+1) and exon 1 of the SREBP1 gene. The CpG sites are represented by vertical tick marks, and the CpG island predicted by MethPrimer is labeled. (H) Relative 5caC abundance in the SREBP1 promoter upon 200 nM insulin treatment for 48 h was detected by the DIP assay using the 5caC antibody followed by qPCR with specific primers in HepG2, MCF7, and MCF10A cells. (I) The relative amount of TDG binding to the SREBP1 promoter upon 200 nM insulin treatment for 48 h was analyzed by the ChIP assay using the TDG antibody followed by qPCR with specific primers in HepG2, MCF7, and MCF10A cells. Fold enhancement represented the abundance of enriched DNA fragments over an IgG control, and the number was shown as the mean plus SEM of a representative experiment performed in triplicates. ∗∗∗p < 0.001 (Student’s t test).
Figure 2.

Insulin Induces SREBP1/ACC1 Expression through TDG-Mediated DNA Demethylation
(A–C and E) HepG2 or MCF7 cells stably infected with nontargeting control or TDG#1 shRNA were cultured in the absence or presence of 200 nM insulin for 72 h. (D and F) HepG2 or MCF7 cells were treated with 200 nM demethylase inhibitor gemcitabine (Gem) for 30 min prior to 200 nM insulin treatment for 48 h. Relative TDG, SREBP1, and ACC1 mRNA, determined via qRT-PCR, was shown as the mean plus SEM of a representative experiment performed in triplicates. ∗∗∗p < 0.001 (Student’s t test). Cell lysates were analyzed by western blot with specified antibodies. flSREBP1, full-length SREBP1. (G and H) HepG2 (G) or MCF7 (H) cells stably infected with nontargeting control or TDG#1 shRNA were cultured in the absence or presence of 200 nM insulin for 48 h. Relative 5caC abundance in the SREBP1 promoter was detected by the DIP assay. ∗p < 0.05 and ∗∗∗p < 0.001 (ANOVA). (I–K) HepG2 cells were stably transfected with vector or pBabe-TDG. Cell lysates were analyzed by western blot with specified antibodies (I). Relative 5caC abundance in the SREBP1 promoter was detected by the DIP assay (J). The relative amount of TDG binding to the SREBP1 promoter was analyzed by the ChIP assay (K). (L and M) SREBP1 expression values were shown in breast (L; TCGA) and liver (M; GSE87630) cancer tissues compared to adjacent noncancerous tissues (normal) from database analysis. ∗∗p < 0.01 and ∗∗∗p < 0.001 (Welch’s t test). (N and O) Correlations of HCC tumor/peri-tumor tissue (N) and HCC tumor/normal liver tissue (O) mixture ratios of SREBP1 mRNA and promoter 5caC abundance on a logarithmic scale were depicted.
Since TDG was induced along with upregulated c-Myc, the importance of oncogenic c-Myc in insulin-regulated lipogenesis was also investigated. c-Myc knockdown decreased TDG, SREBP1, and ACC1 mRNA expression in HepG2 cells (Figure 3A). Moreover, insulin-induced TDG, SREBP1, and ACC1 protein expression was attenuated when c-Myc was knocked down or inhibited with 50 μM 10058-F4 in HepG2 cells (Figures 3B and 3C). On the other hand, c-Myc activation with the MycER system (pBabe-MycER expression vector25) enhanced TDG, SREBP1, and ACC1 protein expression at 24 h (Figure 3D). Inhibition or activation of c-Myc had similar effects in MCF7 cells (Figures 3E—3G and S1E) and MCF10A cells (Figures 3H and S1F). Next, to verify that insulin regulates the c-Myc/TDG/SREBP1/ACC1 axis, SREBP1 was knocked down or inhibited with betulin, with or without insulin treatment. SREBP1 and ACC1 expression was reduced by knockdown or inhibition of SREBP1 in HepG2 cells (Figures 3I and 3J), MCF7 cells (Figure 3K), and MCF10A cells (Figure 3L) regardless of insulin treatment. Furthermore, as illustrated in Figures S1G–S1I, the induction of SREBP1 and ACC1 protein expression by TDG or c-Myc was attenuated with betulin treatment. These data indicate a previously undescribed mechanism in which insulin regulates SREBP1 and ACC1 expression through the c-Myc-TDG axis (Figure 3M).
Figure 3.

c-Myc and SREBP1 Are Required for Insulin-Induced ACC1 Expression
(A and B) HepG2 cells stably infected with nontargeting control or c-Myc#1 shRNA were cultured in the absence or presence of 200 nM insulin for 72 h. (C) HepG2 cells were treated with 50 μM c-Myc inhibitor 10058-F4 for 30 min prior to 200 nM insulin treatment for 48 h. (D) HepG2-MycER cells were treated with 500 nM 4-OHT for indicated time periods. (E) MCF7 cells were stably infected with nontargeting control or Myc#1 shRNA. (F) MCF7 cells and (H) MCF10A cells were treated with 50 μM c-Myc inhibitor 10058-F4 for 30 min prior to 200 nM insulin treatment for 48 h. (G) MCF7 cells were stably transfected with vector or pBabe-Myc. (I) HepG2 cells, stably infected with nontargeting control, SREBP1#1 shRNA, or SREBP1#2 shRNA, were cultured in the absence or presence of 200 nM insulin for 72 h. (J) HepG2 cells and (K) MCF7 cells were treated with 50 μM SREBP1 inhibitor betulin for 30 min prior to 200 nM insulin treatment for 48 h. (L) MCF10A cells were treated with 25 μM SREBP1 inhibitor betulin for 30 min prior to 200 nM insulin treatment for 48 h. Relative c-Myc, TDG, SREBP1, and ACC1 mRNA, determined via qRT-PCR, were shown as the mean plus SEM of a representative experiment performed in triplicates. ∗∗∗p < 0.001 (Student’s t test). Cell lysates were analyzed by western blot with specified antibodies. flSREBP1, full-length SREBP1. (M) A diagram illustrating that insulin-activated c-Myc-TDG signaling downregulates 5caC abundance in the SREBP1 promoter to increase SREBP1 expression.
Insulin Promotes Cell Proliferation through c-Myc/TDG/SREBP1 Signaling
Next, the biological consequences of inhibiting c-Myc, TDG, and SREBP1 expression were determined. Cell growth was attenuated by 50 μM 10058-F4, 200 nM gemcitabine, and 50 or 25 μM betulin (Figures 4A–4E), with or without insulin cotreatment. Moreover, c-Myc or TDG knockdown decreased cell proliferation in MCF7 cells (Figures S2A and S2B). On the other hand, overexpression of c-Myc or TDG enhanced cell growth (Figures 4F and 4G), and proliferation was further enhanced in cells overexpressing c-Myc and treated with insulin (Figure 4F). In summary, these results demonstrate that c-Myc/TDG/SREBP1 signaling is vital and required for insulin-induced lipogenesis and cell growth.
Figure 4.

Insulin Promotes Cell Proliferation through c-Myc/TDG/SREBP1 Signaling
(A) HepG2 cells and (B) MCF7 cells were pretreated with 50 μM 10058-F4, 200 nM gemcitabine (Gem), and 50 μM betulin for 30 min and then treated with 200 nM insulin for 10 days. Colony formation ability was measured by crystal violet staining, and colony numbers were counted. Relative colony formation was shown as the mean plus SEM of 3 independent experiments. ∗p < 0.05 and ∗∗∗p < 0.001 (Student’s t test). (C–E) MCF10A cells were pretreated with 50 μM 10058-F4 (C), 200 nM Gem (D), or 25 μM betulin (E) for 30 min and then treated with 200 nM insulin for indicated time periods. (F and G) MCF10A cells, stably transfected with vector, pBabe-Myc (F), or pBabe-TDG (G), were cultured in the absence or presence of 200 nM insulin for indicated time periods. Cell growth was measured by MTT assay and shown as the mean plus SEM of a representative experiment performed in triplicates. Comparisons of growth curves were analyzed by the mixed regression model; ##p < 0.01 and ###p < 0.001 were considered to be statistically significant as compared with control or vector-transfected cells. ∗p < 0.05 (Student’s t test at 72 h as compared with control cells).
Activation of AMPK Attenuates Cancer Cell Proliferation by Impairing TDG, SREBP1, and ACC1
The importance of AMPK signaling in regulating lipogenic gene expression and cell growth was assessed using AMPK activators metformin and 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR). Metformin reduced TDG, SREBP1, and ACC1 mRNA (Figure 5A) and protein (Figure 5B) abundance in HepG2 cells. Further, metformin reduced TDG binding and raised 5caC abundance in the SREBP1 promoter (Figures 5C and 5D). Likewise, metformin decreased TDG, SREBP1, and ACC1 mRNA (Figure 5E) and protein (Figure 5F) abundance. Further, metformin attenuated TDG binding and enhanced 5caC abundance in the SREBP1 promoter (Figures 5G and 5H) in MCF7 cells. AICAR also decreased the expression of TDG, SREBP1, and ACC1 mRNA (Figure S2C) and protein (Figure S2D) abundance, attenuated TDG binding, and increased 5caC abundance in the SREBP1 promoter (Figures S2E and S2F). To provide additional evidence that metformin acts through AMPK to reduce TDG and lipogenic gene expression, cells were treated with metformin in the presence or absence of compound C, an AMPK inhibitor. Following treatment, compound C attenuated the effect of metformin on the protein abundance of TDG, SREBP1, and ACC1 (Figures 5I and 5J). Consistent with previous findings,50,51 these data show that activation of AMPK increases 5caC accumulation in the SREBP1 promoter to downregulate SREBP1 expression. Moreover, metformin and AICAR reduced cell proliferation (Figures 5K and 5L). In summary, these results demonstrate that the AMPK-TDG/SREBP1/ACC1 pathway impairs cell proliferation.
Figure 5.

Activation of AMPK by Metformin Reduces TDG, SREBP1, and ACC1 Expression; Lowers TDG Binding to the SREBP1 Promoter; and Increases 5caC Abundance in the SREBP1 Promoter
(A–D) HepG2 cells were treated with 10 mM metformin for 48 h (A, C, and D) or indicated time periods (B). (E–H) MCF7 cells were treated with 10 mM metformin for 48 h (E, G, and H) or indicated time periods (F). Relative TDG, SREBP1, and ACC1 mRNA, determined via qRT-PCR, were shown as the mean plus SEM of a representative experiment performed in triplicates. Cell lysates were analyzed by western blot with specified antibodies. flSREBP1, full-length SREBP1. The relative amount of TDG binding to the SREBP1 promoter was analyzed by the ChIP assay. Relative 5caC abundance in the SREBP1 promoter was detected by the DIP assay. ∗∗p < 0.01 and ∗∗∗p < 0.001 (Student’s t test). (I and J) HepG2 (I) or MCF7 (J) cells were treated with 5 μM or 1 μM AMPK inhibitor compound C for 30 min prior to 10 mM metformin treatment for 48 h, respectively. Cell lysates were analyzed by western blot with specified antibodies. (K and L) HepG2 (K) or MCF7 (L) cells were treated with 10 mM metformin or 1 mM AICAR for indicated time periods. Cell growth was measured by MTT assay and shown as the mean plus SEM of a representative experiment performed in triplicates. Comparisons of growth curves were analyzed by the mixed regression model, and ##p < 0.01 and ###p < 0.001 were considered to be statistically significant.
AMPK Increases DNA Methylation in the TDG Promoter through DNMT3A
Notably, the TDG promoter contains a CpG island that might be regulated through methylation (Figure 6A). Treatment with decitabine, a DNMT inhibitor, enhanced TDG, SREBP1, and ACC1 protein expression in HepG2 (Figure 6B) and MCF7 cells (Figure 6C). Furthermore, metformin and AICAR increased DNMT activity (Figures 6D and 6G) and elevated abundance of 5mC in the TDG promoter region (Figures 6E, 6H, S2G, and S2H). To determine which DNMT is involved in the regulation of methylation in the TDG promoter, we initially examined the expression of DNMT1, DNMT3A, and DNMT3B upon metformin and AICAR treatment. As illustrated in Figures S3A and S3B, the activation of AMPK downregulated DNMT1 and DNMT3B but sustained DNMT3A protein expression, indicating that DNMT3A is involved in regulating TDG, SREBP1, and ACC1 expression. Indeed, DNMT3A knockdown in HepG2 and MCF7 cells upregulated TDG, SREBP1, and ACC1 protein expression (Figure S3C), whereas gemcitabine blocked its effect (Figure S3D). Further, metformin and AICAR enhanced DNMT3A binding to the TDG promoter (Figures 6F and 6I). Metformin- and AICAR-reduced TDG, SREBP1, and ACC1 expressions were abrogated by DNMT3A knockdown (Figures 6J and S2E). These data demonstrated that TDG expression is modulated by the AMPK-DNMT3A axis, thereby impeding SREBP1 expression in response to metformin and AICAR (Figure 6K).
Figure 6.

Metformin and AICAR Downregulate TDG Expression through DNMT3A-Mediated Methylation in the TDG Promoter
(A) Schematic representation of the CpG distribution in the upstream of 2,000 bp from the transcription start site (+1) and exon 1 of the TDG gene. The CpG sites are represented by vertical tick marks, and the CpG island predicted by MethPrimer is labeled. (B and C) HepG2 (B) or MCF7 (C) cells were treated with the indicated concentration of decitabine for 24 h. (D–F) HepG2 cells and (G–I) MCF7 cells were treated with 10 mM metformin or 1 mM AICAR for 24 h. DNMT activity was determined and shown as the mean plus SEM of 3 independent experiments (D and G). Relative 5mC abundance in the TDG promoter was detected by the DIP assay (E and H). The relative amount of DNMT3A binding to the TDG promoter was analyzed by the ChIP assay (F and I). ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 (Student’s t test). (J) HepG2 or MCF7 cells stably infected with nontargeting control or DNMT3A#1 shRNA were cultured in the absence or presence of 10 mM metformin for 48 h. Cell lysates were analyzed by western blot with indicated antibodies. flSREBP1, full-length SREBP1. (K) A diagram illustrating that metformin and AICAR-activated AMPK-DNMT3A signaling upregulates 5mC abundance in the TDG promoter and 5caC abundance in the SREBP1 promoter to suppress TDG and SREBP1 expression, respectively.
Discussion
This study demonstrates that insulin regulates SREBP1 and ACC1 expression through c-Myc-TDG-promoted 5caC removal in the SREBP1 promoter. Since insulin-activated phosphatidylinositol 3-kinase (PI3K)-Akt signaling pathways have been shown to upregulate the expression and processing of SREBP1,23,52, 53, 54 and c-Myc expression is shown to be upregulated by Akt signaling,55 we examined whether c-Myc/TDG/SREBP1 signaling is downstream of Akt or independent of Akt in response to insulin. Our results showed that inhibition of Akt by Akt inhibitor (Akti) did not abolish the effects of insulin on SREBP1 expression in MCF7 cells at 24 h, indicating insulin-induced c-Myc/TDG/SREBP1 is Akt independent (Figure S3F). However, Akti decreased SREBP1 at 48 h and had an effect on ACC1 expression. These results suggest that insulin-induced ACC1 is resulted from temporal actions of c-Myc/TDG/SREBP1 and Akt pathways. Ample evidence indicates that highly proliferating cancer cells require enhanced lipid production for membrane biosynthesis and resilience to oxidative stress.56,57 In line with this evidence, the targeting of SREBP1 and ACC1 as a strategy for cancer treatment has been suggested.44,58, 59, 60 Our results further indicate the targeting of TDG as a strategy to treat hyperinsulinemia-promoted cancer progression in diabetic cancer patients. The current epigenetic therapy primarily involves inhibitors of DNMTs, histone methyltransferases, and histone deacetylation.41,61,62 Identification of specific inhibitors of TDG and combinations with other therapies, such as chemotherapy, targeted therapies, and immunotherapy, may be further investigated to broaden therapeutic efficacy.63
In addition to insulin signaling, this study also finds that metformin-activated AMPK reduces SREBP1/ACC1 expression and cancer cell proliferation through modulating DNMT3A and TDG. AMPK activity is downregulated in most cancers,64 and reduced AMPK phosphorylation in cancer cells is correlated with aggressive progression and poor diagnosis of patients with HCC.65 Our data provide a detailed mechanism for AMPK-mediated DNMT3A-TDG signaling that promotes the suppression of lipogenesis and cancer growth, which is consistent with the expected anticancer role of AMPK. Activated AMPK has been shown to phosphorylate and inhibit DNMT1 activity directly in endothelial cells.43 However, our data demonstrate that metformin-activated AMPK stimulates DNMT3A activity to increase the abundance of 5mC in the TDG promoter, which is consistent with the finding that metformin modulates global DNA methylation through AMPK-promoted DNMT3B activity.15,16 Notably, TDG has been shown to interact with DNMT3A and inhibit its activity.66 Nevertheless, this study finds that AMPK-activated DNMT3A downregulates TDG expression, suggesting a reciprocal inhibition of TDG by DNMT3A.
TDG plays a central role in DNA repair, DNA demethylation, and transcriptional regulation.67 Active DNA demethylation starts from oxidation of 5mC to generate 5-hydroxymethylcytosine (5hmC) or be further oxidized to 5fC and 5caC by the ten-eleven translocation (TET) family of enzymes, followed by TDG-mediated 5fC and 5caC replacements.68 Metformin-activated AMPK is shown to phosphorylate TET2, thereby stabilizing TET2 to increase 5hmC abundance and global DNA demethylation in blood cells.13 Another study shows that AMPK increases the production of α-ketoglutaric acid to promote TET-facilitated DNA demethylation in the PR domain-containing 16 (PRDM16) promoter and brown adipogenesis.69 However, this study demonstrates that metformin-activated AMPK decreases TDG expression, therefore inhibiting the final step of 5caC removal in the SREBP1 promoter. Thus, AMPK activation may preferentially promote or inhibit DNA demethylation in a context-dependent manner. In addition, AMPK-inhibited TDG may only result in hypermethylation of particular genes, which is supported by a recent report that only a small fraction of the genome undergoes hypermethylation after TDG inactivation.70 Moreover, the roles of 5mC and 5hmC in epigenetic regulation of gene expression are well established, and the functional effects of 5caC on transcription have been characterized recently.51 RNA polymerase II elongation efficiencies in DNA regions consisting of 5caC are significantly lower than regions containing unmodified cytosine.50 Further, transcriptional repressors are recruited by 5caC, which also contributes to transcriptional repression.71 Our in vitro and in vivo studies further validate that 5caC attenuates SREBP1 expression to explore the functional roles of 5caC (Figures 2 and 5).
Metformin has been shown to have antineoplastic effects on cancers associated with T2DM.10 Metformin-activated AMPK reduces mammalian target of rapamycin complex 1 (mTORC1) signaling, which leads to the inhibition of protein synthesis and cell proliferation in cancer cells.72 Nevertheless, there is no study directly showing the metformin effect in cancer patients with T2DM containing high insulin levels or cultured cancer cells under hyperinsulinemia regarding the epigenetic regulation and lipogenesis in cancers. In order to investigate the fact that insulin coexisted with metformin in cancer cells, HepG2 and MCF7 cells were cotreated with 200 nM insulin and 10 mM metformin to inspect the c-Myc/TDG/SREBP1 signaling. Our results showed that metformin overrode the insulin-promoting effect on lipogenesis and decreased c-Myc, TDG, SREBP1, and ACC1 expression in HepG2 and MCF7 cells (Figure S3G). These results demonstrate and support that metformin treatment in cancer patients with T2DM containing high insulin levels still inhibits lipogenesis and growth in cancer cells.10 In this study, we discovered the insulin and metformin effects on TDG/SREBP1/ACC1 signaling and 5caC changes in the SREBP1 promoter in cultured liver and breast cancer cells. We also used human cancer samples to validate our in vitro findings. SREBP1 mRNA expression levels were higher in breast and liver cancer tissues compared to adjacent noncancerous tissues from database analyses (Figures 2L and 2M). Although only 7 patient samples were used, the results revealed that the 5caC abundance in the SREBP1 promoter represented transcriptional repression among these HCC patients (Figures 2N and 2O). We are extending our study by expanding the sample size of the patients, which allows us to divide the samples into diabetic and nondiabetic HCC groups among large patient populations and to determine the differences of 5caC abundance in the SREBP1 promoter and SREBP1 expression between them.
In conclusion, this study demonstrates that c-Myc and AMPK can modulate TDG expression to affect 5caC abundance in the SREBP1 promoter, therefore amending cellular lipogenesis and proliferation upon insulin and metformin treatment. This study indicates that TDG is a viable therapeutic target, and inhibitors of TDG can be used for treatment of cancers associated with T2DM.
Materials and Methods
Antibodies and Reagents
Antibodies used were as follows: ACC (#3676; Cell Signaling Technology), phospho-AMPKα Thr172 (#2531; Cell Signaling Technology), AMPKα1 (#2795; Cell Signaling Technology), AMPKα2 (#2757; Cell Signaling Technology), AKT serine/threonine kinase (Akt; #9272; Cell Signaling Technology), phospho-Akt S473 (#9271; Cell Signaling Technology), normal rabbit immunoglobulin G (IgG; #2729; Cell Signaling Technology), SREBP1 (sc-13551; Santa Cruz Biotechnology), DNMT1 (sc-271729; Santa Cruz Biotechnology), TDG (ABE1440; Millipore), β-actin (MAB1501; Millipore), glyceraldehyde 3-phosphate dehydrogenase (GAPDH; MAB374; Millipore), DNMT3A (GTX129125; GeneTex), DNMT3B (GTX129127; GeneTex), 5caC (GTX60801; GeneTex), c-Myc (#21034; SAB Signalway), 5mC (A3001-200; Zymo Research), and normal mouse IgG (#31903; Thermo Fisher Scientific). 4-hydroxytestosterone (4-hydroxytamoxifen [4-OHT]; H7904; 500 nM) was purchased from Sigma-Aldrich. Insulin (Humulin R; 200 nM) was purchased from Eli Lilly. 10058-F4 (HY-12702) (50 μM) was purchased from MCE. Betulin (sc-234016; 50 μM or 25 μM) was purchased from Santa Cruz Biotechnology. Metformin (#13118; 10 mM) was purchased from Cayman Chemical. AICAR (S1802; 1 mM), Akti (S7776; 10 μM), and decitabine (S1200; 1–20 μM) were purchased from Selleckchem. Compound C (#171260; 5 μM or 1 μM) and gemcitabine (#504094; 200 nM) were purchased from Millipore. The short hairpin RNA (shRNA) targeting c-Myc, TDG, DNMT3A, SREBP1, and β-galactosidase (LacZ) control and nontargeting control shRNA were purchased from Sigma-Aldrich or National RNAi Core Facility, Academia Sinica.
Cell Lines and Culture
HepG2 and MCF7 cells were cultured in the Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum (FBS), 100 units/mL penicillin, and 100 μg/mL streptomycin. Nontumorigenic epithelial MCF10A cells were maintained in the DMEM and F-12 medium supplemented with 5% horse serum, 100 units/mL penicillin, 100 μg/mL streptomycin, 20 ng/mL epidermal growth factor (EGF), 0.5 μg/mL hydrocortisone, and 0.1 μg/mL cholera toxin. The c-Myc-, c-MycER-, or TDG-expressing cells were established by transfection with retroviral infection of the pBabe-Myc, pBabe-MycER, or pBabe-TDG construct and selected with 1 μg/mL puromycin as a stable cell pool. The c-Myc-, TDG-, SREBP1-, and DNMT3A-silencing cells were established by transfection with lentiviral infection of the c-Myc, TDG, SREBP1, or DNMT3A shRNA and selected with 1 μg/mL puromycin for stable cell pools.
Cell Growth Assay
Cell growth rate was measured by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay. Briefly, HepG2 or MCF7 cells were seeded at 104 cells per well, and MCF10A cells were seeded at 5 × 103 cells per well in 24-well culture plates for 24 h and then treated with various treatments. Cells were washed with phosphate buffer saline (PBS), followed by staining with the 1-mg/mL MTT solution at 37°C for 4–6 h. After removal of the MTT solution, formazan crystals were dissolved in dimethyl sulfoxide (DMSO), and the absorbance was measured at 570 nm with a reference wavelength of 630 nm. Cell growth was also measured by colony formation assay. Briefly, 104 cells were seeded in 100 mm dishes for 24 h and then chronically treated with various treatments for 10 days. Cells were fixed with the 4% formaldehyde overnight, followed by staining with the 1% crystal violet. After washing with sterile water, the plates were air dried, and the colonies were scanned. The number of colonies was counted using GeneTools software (Syngene).
Western Blotting
After indicated treatments, cells were harvested and lysed with a lysis buffer containing 10 mM Tris-HCl (pH 7.4), 100 mM NaCl, 10% glycerol, 1 mM EDTA, 0.1% SDS, 1 mM NaF, 0.5% sodium deoxycholate, 1% Triton X-100, 20 mM Na4P2O7, and 2 mM Na3VO4, supplemented with protease and phosphatase inhibitors and phenylmethylsulfonyl fluoride (PMSF). After the protein quantification, an equal amount of total protein was resolved in 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to a polyvinylidene fluoride (PVDF) membrane. The samples were immunoblotted with primary antibodies, as indicated, followed by secondary antibodies conjugated with the horseradish peroxidase. The recognized bands were visualized by using the enhanced chemiluminescence (ECL) detection kit (Millipore; WBKLS0500) or the Trident Femto-ECL (GeneTex; GTX14698).
Quantitative Reverse Transcription PCR (qRT-PCR)
Total RNA was isolated with the TRIzol Reagent (Invitrogen). Reverse transcription was performed using 3 mg of total RNA with the high capacity cDNA Reverse Transcription Kit (Applied Biosystems). The synthesized cDNA was used for real-time quantitative PCR (qPCR) with the Fast SYBR Green Master Mix (Applied Biosystems) on the StepOnePlus (Applied Biosystems). Primers are shown in Table S1. The relative abundance of specific mRNAs was normalized to human GAPDH or β-actin mRNA as the invariant control.
DNA Methyltransferase Activity Assay
Nuclear proteins were isolated using the EpiQuik Nuclear Extraction Kit (OP-0002). DNMT activity measurements and calculations were performed according to the manufacturer’s protocols and formulas. DNMT activities were measured using 10 μg of nuclear protein extracts by the EpiQuik DNMT activity assay kits (P-3009; EpiGentek, Farmingdale, NY, USA).
CpG Islands Prediction Analysis
The CpG islands were predicted by MethPrimer (http://www.urogene.org/methprimer/). Upstream of 2,000 bp from the transcription start site (+1) and exon 1 of the SREBP1 or TDG genes was analyzed. Criteria for CpG islands prediction is obtained by calculating parameters in a window-size 100 bp, GC content of island > 50%, and observed/expected (Obs/Exp) > 0.6.
Chromatin Immunoprecipitation (ChIP) Assay
ChIP assays were performed, according to standard protocols available at Abcam. Proteins were cross-linked to DNA with the addition of 1% formaldehyde for 10 min and then quenched by 125 mM glycine. Cells were washed 2 times with PBS and then scraped into PBS with protease inhibitors. Cells were collected after centrifugation, and lysed in the ChIP lysis buffer (50 mM HEPES-KOH, pH 7.5, 140 mM NaCl, 1 mM EDTA, pH 8.0, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, and protease inhibitors). Resulting cell lysates were sonicated with the sonicator (Bioruptor UCD-200). Target proteins were immunoprecipitated overnight at 4°C with the Dynabeads Protein G (Invitrogen; #10004D) in the presence of TDG, DNMT3A, or normal rabbit IgG antibodies in the dilution buffer (1% Triton X-100, 2 mM EDTA, pH 8.0, 150 mM NaCl, 20 mM Tris-HCl, pH 8.0, and protease inhibitors). The beads were washed 3 times with the wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, pH 8.0, 150 mM NaCl, and 20 mM Tris-HCl, pH 8.0) and 1 time with the final wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, pH 8.0, 500 mM NaCl, and 20 mM Tris-HCl, pH 8.0) and finally eluted in the elution buffer (1% SDS, 100 mM NaHCO3, and proteinase K), followed by shaking at 55°C overnight and inactivating proteinase K at 95°C for 10 min. All DNA samples were purified using the DNA cleanup kit (Zymo Research; D4034) before qPCR analysis. Primers for ChIP are shown in Table S1.
DIP Assay
Genomic DNA was isolated from cells using a lysis buffer containing 100 mM Tris-HCl (pH 8.0), 5 mM EDTA, 0.2% SDS, 200 mM NaCl, as well as 100 μg/mL proteinase K, precipitated by isopropanol and washed with 75% ethanol. Genomic DNA was digested with RNase A for 1 h at 37°C and sonicated with the sonicator (Bioruptor UCD-200). DNA fragments were subjected to immunoprecipitation with Dynabeads Protein G (Invitrogen; #10004D) in the presence of anti-5mC, anti-5caC, or normal IgG antibodies in the DIP buffer (10 mM sodium phosphate, pH 7.0, 0.14 M NaCl, 0.05% Triton X-100, and protease inhibitors) at 4°C overnight. The beads were washed 3 times with the DIP buffer and eluted in the digestion buffer (50 mM Tris-HCl, pH 8.0, 10 mM EDTA, 0.5% SDS, and proteinase K), followed by shaking at 55°C overnight and inactivating proteinase K at 95°C for 10 min. All DNA samples were purified using the DNA cleanup kit (Zymo Research; D4034) before qPCR analysis. Primers for DIP are shown in Table S1.
McrBC-qPCR
McrBC-qPCR was performed, as described previously.73 Briefly, genomic DNA digested with the McrBC enzyme (New England Biolabs), which cuts methylated DNA, was followed by qPCR (McrBC-qPCR) with specific primers. McrBC digestion was carried out using 100 ng of genomic DNA. qPCR was performed on equal amounts (20 ng) of digested and undigested DNA samples with the Fast SYBR Green Master Mix (Applied Biosystems) on the StepOnePlus (Applied Biosystems). Primers are listed in Table S1. The relative levels of qPCR products of digested DNA samples were normalized to undigested DNA samples. Methylated DNA can be digested by McrBC; thus, higher qPCR levels indicate lower 5mC abundance, and lower qPCR levels indicate higher 5mC abundance.
Database Analysis
Bioinformatics analyses of SREBP1 expression in breast cancer were performed with R and Python coding languages, with support from The Cancer Genome Atlas (TCGA) Workflow Data, TCGAbiolinks, ggplot2, data.table, Comprehensive R Archive Network (CRAN), and Bioconductor packages. For SREBP1 expression in liver cancer, a public dataset containing normal and liver cancer specimens (GEO: GSE87630) was analyzed from GEO datasets (https://www.ncbi.nlm.nih.gov/gds/) via analysis platform of GEO2R.
Patients’ Characteristics at National Cheng Kung University Hospital
A total of 7 patients, who underwent liver resection for HCC from May 2012 to July 2014, and normal liver mixed from 3 healthy liver donors were enrolled in the present study. Informed consent regarding the use of specimens for this research was obtained from all patients, and all protocols were reviewed and approved through the National Cheng Kung University Hospital Institutional Review Board (B-ER-108-132). The patients included 6 men and 1 woman, ranging in age from 50 to 72 years (mean age 59.7 years). The average follow-up time was 30.7 months (range, 3.3 to 110.6 months). At the end of the follow-up, 2 patients had died of disease.
Statistical Analyses
Unless indicated, results are expressed as mean ± SEM of a representative experiment from 3 independent experiments performed in triplicates. Experiments comparing 2 groups were analyzed with the Student’s t test or Welch’s t test. Differences among multiple groups were initially evaluated by ANOVA, followed by the Dunnett or Bonferroni post hoc test with GraphPad Prism 5 for Windows (GraphPad Software, San Diego, CA). Pearson’s correlation (with a two-tailed test of significance) was performed for correlation analysis using SPSS 17.0 software (SPSS; Chicago, IL, USA) on a logarithmic scale. The longitudinal data analyses were performed to assess the growth curves. The analyses were mainly carried out using PROC MIXED in the SAS 9.4 (SAS Institute, Cary, NC). The statistical significance level was 0.05. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 (Student’s t test, Welch’s t test, or ANOVA) and #p < 0.05, ##p < 0.01, and ###p < 0.001 (the mixed regression model) were considered to be statistically significant.
Author Contributions
J.-B.Y., C.-C.L., and I.-C.P. conceived the ideas, designed the experiments, and wrote the first draft of the manuscript. J.-B.Y., C.-C.L., J.-W.J., H.-Y.C., C.-J.Y., and I.-C.P. performed the experiments and analyzed the data. B.G., T.L.M., and S.-C.L. performed the database analysis. L.-Y.W. performed statistical analyses with the mixed regression model. I.-C.P., B.G., and T.L.M. reviewed and edited the manuscript. All authors read and commented on the manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
We thank Drs. Wei-Xing Zong, Etienne Lefei, Michael Hsiao, Hung-Jiun Liaw, Hao-Ven Wang, Wen-Tai Chiu, Yau-Sheng Tsai, Horng-Yih Ou, Yun-Wen Chen, and Shun-Fen Tzeng for sharing plasmids, cell lines, and reagents. We thank Drs. Wei-Xing Zong and Richard Lin for reading and discussion. We also thank Miss Huan-Tzu Hou for drawing diagrams. This work was supported by grants from the Ministry of Science and Technology, Taiwan (MOST 104-2320-B-006-006, MOST 104-2320-B-006-031, and MOST 105-2628-B-006-005-MY3 to I.-C.P.), and the Headquarters of University Advancement at the National Cheng Kung University, sponsored by the Ministry of Education, Taiwan, to I.-C.P.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omto.2020.06.010.
Supplemental Information
References
- 1.Guo Y., Zhu S.L., Wu Y.K., He Z., Chen Y.Q. Omega-3 free fatty acids attenuate insulin-promoted breast cancer cell proliferation. Nutr. Res. 2017;42:43–50. doi: 10.1016/j.nutres.2017.04.008. [DOI] [PubMed] [Google Scholar]
- 2.Pivovarova O., von Loeffelholz C., Ilkavets I., Sticht C., Zhuk S., Murahovschi V., Lukowski S., Döcke S., Kriebel J., de las Heras Gala T. Modulation of insulin degrading enzyme activity and liver cell proliferation. Cell Cycle. 2015;14:2293–2300. doi: 10.1080/15384101.2015.1046647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wairagu P.M., Phan A.N., Kim M.-K., Han J., Kim H.-W., Choi J.-W., Kim K.W., Cha S.K., Park K.H., Jeong Y. Insulin priming effect on estradiol-induced breast cancer metabolism and growth. Cancer Biol. Ther. 2015;16:484–492. doi: 10.1080/15384047.2015.1016660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Giovannucci E., Harlan D.M., Archer M.C., Bergenstal R.M., Gapstur S.M., Habel L.A., Pollak M., Regensteiner J.G., Yee D. Diabetes and cancer: a consensus report. Diabetes Care. 2010;33:1674–1685. doi: 10.2337/dc10-0666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vigneri P., Frasca F., Sciacca L., Pandini G., Vigneri R. Diabetes and cancer. Endocr. Relat. Cancer. 2009;16:1103–1123. doi: 10.1677/ERC-09-0087. [DOI] [PubMed] [Google Scholar]
- 6.Kabat G.C., Kim M., Caan B.J., Chlebowski R.T., Gunter M.J., Ho G.Y., Rodriguez B.L., Shikany J.M., Strickler H.D., Vitolins M.Z., Rohan T.E. Repeated measures of serum glucose and insulin in relation to postmenopausal breast cancer. Int. J. Cancer. 2009;125:2704–2710. doi: 10.1002/ijc.24609. [DOI] [PubMed] [Google Scholar]
- 7.Bowers L.W., Rossi E.L., O’Flanagan C.H., deGraffenried L.A., Hursting S.D. The Role of the Insulin/IGF System in Cancer: Lessons Learned from Clinical Trials and the Energy Balance-Cancer Link. Front. Endocrinol. (Lausanne) 2015;6:77. doi: 10.3389/fendo.2015.00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang H., Fagan D.H., Zeng X., Freeman K.T., Sachdev D., Yee D. Inhibition of cancer cell proliferation and metastasis by insulin receptor downregulation. Oncogene. 2010;29:2517–2527. doi: 10.1038/onc.2010.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Djiogue S., Nwabo Kamdje A.H., Vecchio L., Kipanyula M.J., Farahna M., Aldebasi Y., Seke Etet P.F. Insulin resistance and cancer: the role of insulin and IGFs. Endocr. Relat. Cancer. 2013;20:R1–R17. doi: 10.1530/ERC-12-0324. [DOI] [PubMed] [Google Scholar]
- 10.Kheirandish M., Mahboobi H., Yazdanparast M., Kamal W., Kamal M.A. Anti-cancer Effects of Metformin: Recent Evidences for its Role in Prevention and Treatment of Cancer. Curr. Drug Metab. 2018;19:793–797. doi: 10.2174/1389200219666180416161846. [DOI] [PubMed] [Google Scholar]
- 11.Zi F., Zi H., Li Y., He J., Shi Q., Cai Z. Metformin and cancer: An existing drug for cancer prevention and therapy. Oncol. Lett. 2018;15:683–690. doi: 10.3892/ol.2017.7412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Q., Liu S., Zhai A., Zhang B., Tian G. AMPK-Mediated Regulation of Lipid Metabolism by Phosphorylation. Biol. Pharm. Bull. 2018;41:985–993. doi: 10.1248/bpb.b17-00724. [DOI] [PubMed] [Google Scholar]
- 13.Wu D., Hu D., Chen H., Shi G., Fetahu I.S., Wu F., Rabidou K., Fang R., Tan L., Xu S. Glucose-regulated phosphorylation of TET2 by AMPK reveals a pathway linking diabetes to cancer. Nature. 2018;559:637–641. doi: 10.1038/s41586-018-0350-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang T., Guan X., Choi U.L., Dong Q., Lam M.M.T., Zeng J., Xiong J., Wang X., Poon T.C.W., Zhang H. Phosphorylation of TET2 by AMPK is indispensable in myogenic differentiation. Epigenetics Chromatin. 2019;12:32. doi: 10.1186/s13072-019-0281-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhong T., Men Y., Lu L., Geng T., Zhou J., Mitsuhashi A., Shozu M., Maihle N.J., Carmichael G.G., Taylor H.S., Huang Y. Metformin alters DNA methylation genome-wide via the H19/SAHH axis. Oncogene. 2017;36:2345–2354. doi: 10.1038/onc.2016.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cuyàs E., Fernández-Arroyo S., Verdura S., García R.Á.-F., Stursa J., Werner L., Blanco-González E., Montes-Bayón M., Joven J., Viollet B. Metformin regulates global DNA methylation via mitochondrial one-carbon metabolism. Oncogene. 2018;37:963–970. doi: 10.1038/onc.2017.367. [DOI] [PubMed] [Google Scholar]
- 17.Luo X., Cheng C., Tan Z., Li N., Tang M., Yang L., Cao Y. Emerging roles of lipid metabolism in cancer metastasis. Mol. Cancer. 2017;16:76. doi: 10.1186/s12943-017-0646-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li J., Cheng J.X. Direct visualization of de novo lipogenesis in single living cells. Sci. Rep. 2014;4:6807. doi: 10.1038/srep06807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stoiber K., Nagło O., Pernpeintner C., Zhang S., Koeberle A., Ulrich M., Werz O., Müller R., Zahler S., Lohmüller T. Targeting de novo lipogenesis as a novel approach in anti-cancer therapy. Br. J. Cancer. 2018;118:43–51. doi: 10.1038/bjc.2017.374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mounier C., Bouraoui L., Rassart E. Lipogenesis in cancer progression (review) Int. J. Oncol. 2014;45:485–492. doi: 10.3892/ijo.2014.2441. [DOI] [PubMed] [Google Scholar]
- 21.Chajès V., Cambot M., Moreau K., Lenoir G.M., Joulin V. Acetyl-CoA carboxylase α is essential to breast cancer cell survival. Cancer Res. 2006;66:5287–5294. doi: 10.1158/0008-5472.CAN-05-1489. [DOI] [PubMed] [Google Scholar]
- 22.Jones J.E., Esler W.P., Patel R., Lanba A., Vera N.B., Pfefferkorn J.A., Vernochet C. Inhibition of Acetyl-CoA Carboxylase 1 (ACC1) and 2 (ACC2) Reduces Proliferation and De Novo Lipogenesis of EGFRvIII Human Glioblastoma Cells. PLoS ONE. 2017;12:e0169566. doi: 10.1371/journal.pone.0169566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Owen J.L., Zhang Y., Bae S.-H., Farooqi M.S., Liang G., Hammer R.E., Goldstein J.L., Brown M.S. Insulin stimulation of SREBP-1c processing in transgenic rat hepatocytes requires p70 S6-kinase. Proc. Natl. Acad. Sci. USA. 2012;109:16184–16189. doi: 10.1073/pnas.1213343109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Osborne T.F. Sterol regulatory element-binding proteins (SREBPs): key regulators of nutritional homeostasis and insulin action. J. Biol. Chem. 2000;275:32379–32382. doi: 10.1074/jbc.R000017200. [DOI] [PubMed] [Google Scholar]
- 25.Bott A.J., Peng I.-C., Fan Y., Faubert B., Zhao L., Li J., Neidler S., Sun Y., Jaber N., Krokowski D. Oncogenic Myc induces expression of glutamine synthetase through promoter demethylation. Cell Metab. 2015;22:1068–1077. doi: 10.1016/j.cmet.2015.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dang C.V. MYC on the path to cancer. Cell. 2012;149:22–35. doi: 10.1016/j.cell.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hsieh A.L., Walton Z.E., Altman B.J., Stine Z.E., Dang C.V. MYC and metabolism on the path to cancer. Semin. Cell Dev. Biol. 2015;43:11–21. doi: 10.1016/j.semcdb.2015.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin C.Y., Lovén J., Rahl P.B., Paranal R.M., Burge C.B., Bradner J.E., Lee T.I., Young R.A. Transcriptional amplification in tumor cells with elevated c-Myc. Cell. 2012;151:56–67. doi: 10.1016/j.cell.2012.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zeller K.I., Zhao X., Lee C.W., Chiu K.P., Yao F., Yustein J.T., Ooi H.S., Orlov Y.L., Shahab A., Yong H.C. Global mapping of c-Myc binding sites and target gene networks in human B cells. Proc. Natl. Acad. Sci. USA. 2006;103:17834–17839. doi: 10.1073/pnas.0604129103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kerkhoff E., Houben R., Löffler S., Troppmair J., Lee J.-E., Rapp U.R. Regulation of c-myc expression by Ras/Raf signalling. Oncogene. 1998;16:211–216. doi: 10.1038/sj.onc.1201520. [DOI] [PubMed] [Google Scholar]
- 31.Sears R., Nuckolls F., Haura E., Taya Y., Tamai K., Nevins J.R. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 2000;14:2501–2514. doi: 10.1101/gad.836800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fernandes J.C., Rodrigues Alves A.P.N., Machado-Neto J.A., Scopim-Ribeiro R., Fenerich B.A., da Silva F.B., Simões B.P., Rego E.M., Traina F. IRS1/β-Catenin Axis Is Activated and Induces MYC Expression in Acute Lymphoblastic Leukemia Cells. J. Cell. Biochem. 2017;118:1774–1781. doi: 10.1002/jcb.25845. [DOI] [PubMed] [Google Scholar]
- 33.Masui K., Tanaka K., Akhavan D., Babic I., Gini B., Matsutani T., Iwanami A., Liu F., Villa G.R., Gu Y. mTOR complex 2 controls glycolytic metabolism in glioblastoma through FoxO acetylation and upregulation of c-Myc. Cell Metab. 2013;18:726–739. doi: 10.1016/j.cmet.2013.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li Y., Xu S., Mihaylova M.M., Zheng B., Hou X., Jiang B., Park O., Luo Z., Lefai E., Shyy J.Y. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011;13:376–388. doi: 10.1016/j.cmet.2011.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peng I.-C., Chen Z., Sun W., Li Y.-S., Marin T.L., Hsu P.-H., Su M.I., Cui X., Pan S., Lytle C.Y. Glucagon regulates ACC activity in adipocytes through the CAMKKβ/AMPK pathway. Am. J. Physiol. Endocrinol. Metab. 2012;302:E1560–E1568. doi: 10.1152/ajpendo.00504.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Deng H.P., Chai J.K., Shen C.A., Zhang X.B., Ma L., Sun T.J., Hu Q.G., Chi Y.F., Dong N. Insulin down-regulates the expression of ubiquitin E3 ligases partially by inhibiting the activity and expression of AMP-activated protein kinase in L6 myotubes. Biosci. Rep. 2015;35:e00242. doi: 10.1042/BSR20150017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guo D., Hildebrandt I.J., Prins R.M., Soto H., Mazzotta M.M., Dang J., Czernin J., Shyy J.Y., Watson A.D., Phelps M. The AMPK agonist AICAR inhibits the growth of EGFRvIII-expressing glioblastomas by inhibiting lipogenesis. Proc. Natl. Acad. Sci. USA. 2009;106:12932–12937. doi: 10.1073/pnas.0906606106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wen Y.-A., Xiong X., Zaytseva Y.Y., Napier D.L., Vallee E., Li A.T., Wang C., Weiss H.L., Evers B.M., Gao T. Downregulation of SREBP inhibits tumor growth and initiation by altering cellular metabolism in colon cancer. Cell Death Dis. 2018;9:265. doi: 10.1038/s41419-018-0330-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Luo Z., Zang M., Guo W. AMPK as a metabolic tumor suppressor: control of metabolism and cell growth. Future Oncol. 2010;6:457–470. doi: 10.2217/fon.09.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jones P.A., Baylin S.B. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 2002;3:415–428. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- 41.Schübeler D. Function and information content of DNA methylation. Nature. 2015;517:321–326. doi: 10.1038/nature14192. [DOI] [PubMed] [Google Scholar]
- 42.Kohli R.M., Zhang Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature. 2013;502:472–479. doi: 10.1038/nature12750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marin T.L., Gongol B., Zhang F., Martin M., Johnson D.A., Xiao H., Wang Y., Subramaniam S., Chien S., Shyy J.Y.-J. AMPK promotes mitochondrial biogenesis and function by phosphorylating the epigenetic factors DNMT1, RBBP7, and HAT1. Sci. Signal. 2017;10:eaaf7478. doi: 10.1126/scisignal.aaf7478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Beloribi-Djefaflia S., Vasseur S., Guillaumond F. Lipid metabolic reprogramming in cancer cells. Oncogenesis. 2016;5:e189. doi: 10.1038/oncsis.2015.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Orgel E., Mittelman S.D. The links between insulin resistance, diabetes, and cancer. Curr. Diab. Rep. 2013;13:213–222. doi: 10.1007/s11892-012-0356-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Misra U.K., Pizzo S.V. Receptor-recognized α2-macroglobulin binds to cell surface-associated GRP78 and activates mTORC1 and mTORC2 signaling in prostate cancer cells. PLoS ONE. 2012;7:e51735. doi: 10.1371/journal.pone.0051735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cen H.H., Botezelli J.D., Wang S., Noursadeghi N., Johnson J.D. Downregulation of muscle cell Insr and insulin receptor signaling by hyperinsulinemia in vitro and in vivo. bioRxiv. 2020 doi: 10.1101/556571. [DOI] [Google Scholar]
- 48.Shen W.H., Boyle D.W., Wisniowski P., Bade A., Liechty E.A. Insulin and IGF-I stimulate the formation of the eukaryotic initiation factor 4F complex and protein synthesis in C2C12 myotubes independent of availability of external amino acids. J. Endocrinol. 2005;185:275–289. doi: 10.1677/joe.1.06080. [DOI] [PubMed] [Google Scholar]
- 49.Schäfer A., Schomacher L., Barreto G., Döderlein G., Niehrs C. Gemcitabine functions epigenetically by inhibiting repair mediated DNA demethylation. PLoS ONE. 2010;5:e14060. doi: 10.1371/journal.pone.0014060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kellinger M.W., Song C.-X., Chong J., Lu X.-Y., He C., Wang D. 5-formylcytosine and 5-carboxylcytosine reduce the rate and substrate specificity of RNA polymerase II transcription. Nat. Struct. Mol. Biol. 2012;19:831–833. doi: 10.1038/nsmb.2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kitsera N., Allgayer J., Parsa E., Geier N., Rossa M., Carell T., Khobta A. Functional impacts of 5-hydroxymethylcytosine, 5-formylcytosine, and 5-carboxycytosine at a single hemi-modified CpG dinucleotide in a gene promoter. Nucleic Acids Res. 2017;45:11033–11042. doi: 10.1093/nar/gkx718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Düvel K., Yecies J.L., Menon S., Raman P., Lipovsky A.I., Souza A.L., Triantafellow E., Ma Q., Gorski R., Cleaver S. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell. 2010;39:171–183. doi: 10.1016/j.molcel.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Porstmann T., Santos C.R., Griffiths B., Cully M., Wu M., Leevers S., Griffiths J.R., Chung Y.L., Schulze A. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008;8:224–236. doi: 10.1016/j.cmet.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yecies J.L., Zhang H.H., Menon S., Liu S., Yecies D., Lipovsky A.I., Gorgun C., Kwiatkowski D.J., Hotamisligil G.S., Lee C.H., Manning B.D. Akt stimulates hepatic SREBP1c and lipogenesis through parallel mTORC1-dependent and independent pathways. Cell Metab. 2011;14:21–32. doi: 10.1016/j.cmet.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rodrik V., Zheng Y., Harrow F., Chen Y., Foster D.A. Survival signals generated by estrogen and phospholipase D in MCF-7 breast cancer cells are dependent on Myc. Mol. Cell. Biol. 2005;25:7917–7925. doi: 10.1128/MCB.25.17.7917-7925.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pavlova N.N., Thompson C.B. The emerging hallmarks of cancer metabolism. Cell Metab. 2016;23:27–47. doi: 10.1016/j.cmet.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rysman E., Brusselmans K., Scheys K., Timmermans L., Derua R., Munck S., Van Veldhoven P.P., Waltregny D., Daniëls V.W., Machiels J. De novo lipogenesis protects cancer cells from free radicals and chemotherapeutics by promoting membrane lipid saturation. Cancer Res. 2010;70:8117–8126. doi: 10.1158/0008-5472.CAN-09-3871. [DOI] [PubMed] [Google Scholar]
- 58.Svensson R.U., Parker S.J., Eichner L.J., Kolar M.J., Wallace M., Brun S.N., Lombardo P.S., Van Nostrand J.L., Hutchins A., Vera L. Inhibition of acetyl-CoA carboxylase suppresses fatty acid synthesis and tumor growth of non-small-cell lung cancer in preclinical models. Nat. Med. 2016;22:1108–1119. doi: 10.1038/nm.4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mashima T., Seimiya H., Tsuruo T. De novo fatty-acid synthesis and related pathways as molecular targets for cancer therapy. Br. J. Cancer. 2009;100:1369–1372. doi: 10.1038/sj.bjc.6605007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Guo D., Bell E.H., Mischel P., Chakravarti A. Targeting SREBP-1-driven lipid metabolism to treat cancer. Curr. Pharm. Des. 2014;20:2619–2626. doi: 10.2174/13816128113199990486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ahuja N., Sharma A.R., Baylin S.B. Epigenetic therapeutics: a new weapon in the war against cancer. Annu. Rev. Med. 2016;67:73–89. doi: 10.1146/annurev-med-111314-035900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Michalak E.M., Burr M.L., Bannister A.J., Dawson M.A. The roles of DNA, RNA and histone methylation in ageing and cancer. Nat. Rev. Mol. Cell Biol. 2019;20:573–589. doi: 10.1038/s41580-019-0143-1. [DOI] [PubMed] [Google Scholar]
- 63.Morel D., Jeffery D., Aspeslagh S., Almouzni G., Postel-Vinay S. Combining epigenetic drugs with other therapies for solid tumours - past lessons and future promise. Nat. Rev. Clin. Oncol. 2020;17:91–107. doi: 10.1038/s41571-019-0267-4. [DOI] [PubMed] [Google Scholar]
- 64.He X., Li C., Ke R., Luo L., Huang D. Down-regulation of adenosine monophosphate-activated protein kinase activity: A driver of cancer. Tumour Biol. 2017;39 doi: 10.1177/1010428317697576. 1010428317697576. [DOI] [PubMed] [Google Scholar]
- 65.Zheng L., Yang W., Wu F., Wang C., Yu L., Tang L., Qiu B., Li Y., Guo L., Wu M. Prognostic significance of AMPK activation and therapeutic effects of metformin in hepatocellular carcinoma. Clin. Cancer Res. 2013;19:5372–5380. doi: 10.1158/1078-0432.CCR-13-0203. [DOI] [PubMed] [Google Scholar]
- 66.Li Y.-Q., Zhou P.-Z., Zheng X.-D., Walsh C.P., Xu G.-L. Association of Dnmt3a and thymine DNA glycosylase links DNA methylation with base-excision repair. Nucleic Acids Res. 2007;35:390–400. doi: 10.1093/nar/gkl1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xu X., Watt D.S., Liu C. Multifaceted roles for thymine DNA glycosylase in embryonic development and human carcinogenesis. Acta Biochim. Biophys. Sin. (Shanghai) 2016;48:82–89. doi: 10.1093/abbs/gmv083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bhutani N., Burns D.M., Blau H.M. DNA demethylation dynamics. Cell. 2011;146:866–872. doi: 10.1016/j.cell.2011.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yang Q., Liang X., Sun X., Zhang L., Fu X., Rogers C.J., Berim A., Zhang S., Wang S., Wang B. AMPK/α-ketoglutarate axis dynamically mediates DNA demethylation in the Prdm16 promoter and brown adipogenesis. Cell Metab. 2016;24:542–554. doi: 10.1016/j.cmet.2016.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mancuso P., Tricarico R., Bhattacharjee V., Cosentino L., Kadariya Y., Jelinek J., Nicolas E., Einarson M., Beeharry N., Devarajan K. Thymine DNA glycosylase as a novel target for melanoma. Oncogene. 2019;38:3710–3728. doi: 10.1038/s41388-018-0640-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hashimoto H., Olanrewaju Y.O., Zheng Y., Wilson G.G., Zhang X., Cheng X. Wilms tumor protein recognizes 5-carboxylcytosine within a specific DNA sequence. Genes Dev. 2014;28:2304–2313. doi: 10.1101/gad.250746.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Malek M., Aghili R., Emami Z., Khamseh M.E. Risk of cancer in diabetes: the effect of metformin. ISRN Endocrinol. 2013;2013:636927. doi: 10.1155/2013/636927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Teixeira F.K., Heredia F., Sarazin A., Roudier F., Boccara M., Ciaudo C., Cruaud C., Poulain J., Berdasco M., Fraga M.F. A role for RNAi in the selective correction of DNA methylation defects. Science. 2009;323:1600–1604. doi: 10.1126/science.1165313. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.