

Abstract
Aims/Introduction
Under irremediable endoplasmic reticulum (ER) stress, hyperactivated inositol‐requiring enzyme 1α (IRE1α) triggers the terminal unfolded protein response (T‐UPR), causing crucial cell dysfunction and apoptosis. We hypothesized that nicotinic acetylcholine receptor (nAChR) signaling regulates IRE1α activation to protect β‐cells from the T‐UPR under ER stress.
Materials and Methods
The effects of nicotine on IRE1α activation and key T‐UPR markers, thioredoxin‐interacting protein and insulin/proinsulin, were analyzed by real‐time polymerase chain reaction and western blotting in rat INS‐1 and human EndoC‐βH1 β‐cell lines. Doxycycline‐inducible IRE1α overexpression or ER stress agents were used to induce IRE1α activation. An α7 subunit‐specific nAChR agonist (PNU‐282987) and small interfering ribonucleic acid for α7 subunit‐specific nAChR were used to modulate nAChR signaling.
Results
Nicotine inhibits the increase in thioredoxin‐interacting protein and the decrease in insulin 1/proinsulin expression levels induced by either forced IRE1α hyperactivation or ER stress agents. Nicotine attenuated X‐box‐binding protein‐1 messenger ribonucleic acid site‐specific splicing and IRE1α autophosphorylation induced by ER stress. Furthermore, PNU‐282987 attenuated T‐UPR induction by either forced IRE1α activation or ER stress agents. The effects of nicotine on attenuating thioredoxin‐interacting protein and preserving insulin 1 expression levels were attenuated by pharmacological and genetic inhibition of α7 nAChR. Finally, nicotine suppressed apoptosis induced by either forced IRE1α activation or ER stress agents.
Conclusions
Our findings suggest that nAChR signaling regulates IRE1α activation to protect β‐cells from the T‐UPR and apoptosis under ER stress partly through α7 nAChR. Targeting nAChR signaling to inhibit the T‐UPR cascade may therefore hold therapeutic promise by thwarting β‐cell death in diabetes.
Keywords: Inositol‐requiring enzyme 1α, Nicotinic acetylcholine receptor, Pancreatic β cell
Nicotinic acetylcholine receptor signaling attenuated inositol‐requiring enzyme 1α signaling and Terminal Unfolded Protein Response (T‐UPR) under irremediable endoplasmic reticulum stress in rat INS‐1 and human EndoC‐βH1 β‐cell lines. The effects of nicotine on terminal unfolded protein response were reduced by pharmacological and genetic inhibition of nicotinic acetylcholine receptor α7 subunit. Finally, nicotine suppressed apoptosis induced by either forced inositol requiring enzyme 1α activation or endoplasmic reticulum stress.

Introduction
Endoplasmic reticulum (ER)‐resident enzymatic activities, such as chaperones, play a critical role in maintaining cellular homeostasis during both constitutive secretory protein folding and assembly, and under conditions of excessive protein folding demand (i.e., ER stress)1. Under ER stress, intracellular signaling pathways termed the unfolded protein response (UPR) become activated to restore cellular homeostasis. Under remediable levels of ER stress, cytoprotective adaptive‐UPR (A‐UPR) programs become activated, thereby enhancing protein folding capacity, reducing upstream secretory protein loads and degrading ER unfolded proteins. However, under irremediably high and chronic ER stress, these adaptive systems become attenuated as alternative terminal‐UPR (T‐UPR) programs trigger apoptosis; this switch from the A‐UPR to a T‐UPR might underlie many human cell degenerative diseases, including diabetes mellitus2, 3, 4.
The UPR is triggered by the activation of three ER transmembrane sensors – protein kinase RNA‐like ER kinase (PERK), activating transcription factor 6 and inositol‐requiring enzyme 1α (IRE1α) – by changing their oligomerization state4, 5. Under ER stress, IRE1α, the most highly conserved UPR sensor, dimerizes and undergoes trans‐autophosphorylation and activation of endoribonuclease (RNase) to site‐specifically splice X‐box‐binding protein‐1 (XBP1) messenger ribonucleic acid (mRNA) to trigger A‐UPR transcriptional programs4, 6, 7, 8. However, under high (irremediable) ER stress, oligomerized and autophosphorylated IRE1α activates the RNase catalytic rate further, causing cleavage of myriad ER‐localized mRNAs, ribosomal RNAs and microRNAs, which ultimately induces apoptosis. Therefore, modulation of IRE1α activities is central to the A‐UPR to T‐UPR “switch,” and thereby to control cell fate2. This regulation of stability of multiple RNAs by IRE1α is known as regulated IRE1α‐dependent decay4, 9. Indeed, we have shown in vivo, that IRE1α‐specific inhibitors, kinase‐inhibitory RNase attenuators (KIRA), can spare pancreatic β‐cells from T‐UPR and ultimate apoptosis to reverse or ameliorate diabetes in Akita and NOD mice2, 10. Notably, the switch from A‐UPR to T‐UPR has recently been shown in NOD mice10. These findings suggest that modulating the activity of IRE1α in β‐cells might have a general therapeutic potential against type 2 and autoimmune diabetes by rescuing β‐cells from proapoptotic T‐UPR signaling.
The nicotinic acetylcholine receptor (nAChR) is a cation channel receptor that responds to acetylcholine and several ligands, including nicotine. nAChR was believed to exist only in the central and peripheral nervous system, and neuromuscular junction of muscles11. However, recent studies showed that nAChR also exists in the cells of many kinds of non‐neuronal tissues, such as the liver and muscle, and its stimulation shows functional effects12. Similarly, it was also shown that nAChR is expressed in murine islets and rodent pancreatic β‐cell lines13. Interestingly, recent reports emphasize the importance of nAChR signaling by showing that although cholinergic innervation of human islets is limited, ‐cells can substitute to secrete ACh to stimulate surrounding endocrine cells in a paracrine manner14. Supporting an important role of nAChR signaling in glucose metabolism, in vivo, it was reported that nicotine ameliorates the diabetic phenotype in db/db mice, Zucker fatty rats, streptozotocin‐induced diabetic mice and even NOD mice15, 16, 17, 18, 19. Furthermore, stimulation of nAChR by nicotine increases insulin secretion in rodent β‐cells, and decreases cytokine‐induced apoptosis in human and rodent islets at least through the α7 nAChR subunit13, 20.
Regarding the link between UPR and nAChR signaling, a limited number of studies show controversial evidence of this link in non‐β cells. Although, it is reported that pharmacological chaperoning of nAChR signaling could suppress ER stress and UPR, leading to the neuroprotection of Neuro‐2a cells21; however, another study showed the induction of ER stress by nicotine in rat placental trophoblast giant cells22. These findings might be attributed to the tissue‐specific effect of nAChR signaling on a specific UPR arm, and further questions are raised as to whether these two factors, nAChR and IRE1α signaling, are linked in β‐cells. Here, we aimed to determine whether and if so, how nAChR signaling regulates IRE1α signaling, and subsequently T‐UPR, in rat and human pancreatic β‐cell lines.
Methods
Chemicals and reagents
Nicotine, thapsigargin (Tg), doxycycline hyclate (Dox) and PNU‐282987 (PNU) were purchased from Sigma‐Aldrich (St. Louis, MO, USA). Tunicamycin (Tm) was from Merk (Darmstadt, Germany). α‐Bungarotoxin was from Tocris Bioscience (Bristol, UK).
Cell experiments
Cell and tissue culture
Rat insulinoma cell line, INS‐1 cells were maintained as described previously3. Briefly, the cells were cultured in RPMI 1640 medium (Invitrogen, Carlsbad, CA, USA) containing 11.1 mmol/L glucose, 10 mmol/L HEPES (Invitrogen), 10% heat‐inactivated fetal bovine serum (Sigma‐Aldrich), penicillin–streptomycin–glutamine (which contains 100 units/L penicillin, 100 µg/L streptomycin, 292 µg/mL of l‐glutamine; Invitrogen) and 1 mmol/L sodium pyruvate. EndoC‐βH1 cells (Univercell Biosolutions, Toulouse, France) were cultured in accordance with the manufacturer’s protocol23. Briefly, the cells were plated on a coat with βCOAT (Univercell Biosolutions) or MaxGel ECM coat (Sigma‐Aldrich) containing Dulbecco’s modified Eagle’s medium with 4.5 g/L glucose (Invitrogen) and cultured in OPTIβ1 complete premixed culture medium (Univercell Biosolutions). Both INS‐1 and EndoC‐βH1 cells were cultured in a humidified atmosphere of 95% O2 and 5% CO2. As all the experiments are limited to in vitro cultured cell work, the local ethics committee of Wakayama Medical University does not require approval for them.
Doxycyclin‐inducible transgenic WT IRE1α overexpressing INS‐1 cells
Transgenic WT mouse IRE1α overexpressing INS‐1 cells line in this study are tightly inducible by Dox, and maintained as described previously3.
Reverse transcription quantitative polymerase chain reaction
Total RNA was extracted from whole cells using a RNeasy Plus kit (Qiagen, Venlo, the Netherlands). For standard mRNA detection, 1 μg of total RNA was reverse transcribed to complementary deoxyribonucleic acid (cDNA) using a reverse transcription kit (Qiagen) after DNase digestion. For quatitative polymerase chain reaction (PCR), we used SYBR green (Applied Biosystems, Warrington, UK) and the StepOnePlus Real‐Time PCR System (Applied Biosystems). Gene expression levels were normalized to β‐actin expression level. The primers used for quantitative PCR were as shown in Table 1.
Table 1.
Forward and reverse sequences for the primers used for reverse transcription polymerase chain reaction gene
| Gene | Primer |
|---|---|
| Rat TXNIP | F: 5′‐CTGATGGAGGCACAGTGAGA‐3′ |
| R: 5′‐ CTCGGGTGGAGTGCTTAGAG‐3′ | |
| Rat Ins1 | F: 5′‐GTCCTCTGGGAGCC CAAG‐3′ |
| R: 5′‐ ACAGAGCCTCCACCAGG‐3′ | |
| Rat sXBP1 | F: 5′‐CTGAGTCCGAATCAGGTGCAG‐3′ |
| R: 5′‐ATCCATGGGAAGATGTTCTGG‐3′ | |
| Human sXBP1 | F: 5′‐GAGTCCGCAGCAGGTG‐3′ |
| R: 5′‐TCCTTCTGGGTA GACCTCTGGGAG‐3′ | |
| Human TXNIP | F: 5′‐CCTCTGGGAACATCCTTCAA‐ 3′ |
| R: 5′‐GGGGTATTGACATCCACCAG‐ 3′ | |
| Rat CHRNA7 | F: 5′‐TGGCCAACGACTCGCAGCCG‐3′ |
| R: 5′‐ACTCCGGGGTACTCAGACAT‐3′ | |
| Rat CHOP | F: 5′‐CCAGCAGAGGTCACAAGCAC‐3′ |
| R: 5′‐CGCACTGACCACTCTGTTTC‐3′ | |
| Rat β‐actin: | F: 5′‐GCAAATGCTTCTAGGCGGAC‐3′ |
| R: 5′‐AAGAAAGGGTGTAAAACGCAGC‐3′ | |
| Human β‐actin: | F: 5′‐AGAGCTACGAGCTGCCTGAC‐3′ |
| R: 5′‐AGCACTGTGTTGGCGTACAG‐3′ |
Measurements of XBP‐1 mRNA splicing
XBP‐1 splicing mRNA was isolated from whole cells and reverse transcribed as described above to obtain total cDNA. Then, XBP‐1 primers (F:5′‐AAACAGAGTAGCACAGACTGC‐3′, R:5′‐GGATCTCTAAGACTAGAGGCTTGGTG‐3′) were used to amplify an XBP‐1 amplicon spanning the 26 nt intron from the cDNA samples in reverse transcription PCR as previously described10. PCR fragments were digested with PstI and resolved on 3% agarose gels.
Western blot analysis
We carried out western blot as previously described10. Briefly, for protein analysis, cells were lysed in M‐PER buffer (Thermo Scientific, Rockford, IL, USA) mixed with a protease inhibitor, containing 4 µg/mL aprotinin (Wako, Osaka, Japan), 4 µg/mL leupeptin (Nacalai Tesque, Kyoto, Japan), 1 mmol/L PMSF (Wako), 1 mg/mL pepstatin (Wako) and a phosphatase inhibitor cocktail (Sigma‐Aldrich). The lysis solution was mixed and centrifuged at 15,000 g for 5 min at 4°C. The supernatant was collected and quantified by colorimetric protein assay (BioRad, Berkeley, CA, USA). Loading samples were prepared by mixing the protein extract with NuPAGE sample buffer (Invitrogen) and heated at 98°C for 5 min to denature the proteins.
Blocking, incubation with antibodies and washing were carried out in Tris‐buffered saline with 0.05% (v/v) Tween‐20 and 5% (w/v) non‐fat dry milk. Primary antibodies against target proteins were diluted in the blocking solution. Secondary antibodies were diluted in a blocking solution to detect the species‐specific portion of the primary antibodies. Antibody‐binding was detected using LAS‐3000 mini (FUJI Film, Tokyo, Japan), and quantified by densitometry using ImageJ (NIH, Bethesda, MD, USA). β‐Actin was used as the loading control.
Antibody
As primary antibodies, we used mouse anti‐insulin antibody (1:1,000; Cell Signaling Technology Inc., Danvers, MA, USA), mouse anti‐TXNIP antibody (1:1,000, JY2; MBL International, Woburn, MA, USA), rabbit anti‐IRE1α antibody (1:1,000; Cell Signaling Technology Inc.), mouse anti‐phosphorylated eIF2α (Ser51) antibody (1:1,000; Cell Signaling Technology Inc.), mouse anti‐eIF2a antibody (1:1,000; Cell Signaling Technology Inc.) and mouse anti‐β‐Actin antibody (1:5,000; Sigma‐Aldrich).
Sodium dodecyl sulfate polyacrylamide gel electrophoresis, phos‐tag gel separation
For measuring phosphorylated IRE1α, we used Supersep Phos‐tag gel (Wako), which separates phosphorylated proteins from their unphosphorylated counterparts in sodium dodecyl sulfate polyacrylamide gel electrophoresis. In accordance with the manufacturer’s instructions, the gel was soaked in 1× transfer buffer with 10 mmol/L ethylenediaminetetraacetic acid for 20 min with gentle mixing, and the buffer was exchanged three times before loading samples. Western blot was carried out as described above.
Detection of nAChR α7 subunit in EndoC‐βH1 cells
Total RNA was extracted from EndoC‐βH1 cells and was reverse transcribed to cDNA as described above. Human CHRNA7 primers (F: 5′‐CCGACTCTGGGTAGTGTGT‐3′, R: 5′‐ATGGTGCAGATGATGGTGAA‐3′) were used for a regular three‐step PCR, as previously described. The pcDNA3.1‐human CHRNA7 plasmid (Addgene, Watertown, MA, USA) was a gift from Sherry Leonard and Henry Lester (Addgene, #62276), and as a positive control24.
RNA interference
A negative control (Qiagen, #1022563) or two small interfering RNAs (siRNAs) against rat CHRNA7 (Qiagen, #SI00253365, #SI03045028), encoding nAChR α7 subunit, were transfected using lipofectamine RNAiMAX lipid reagent (Invitrogen) in Dox‐inducible IRE1α‐overexpressing INS‐1 cells, in accordance with the manufacturer’s protocol. A total of 24 h after the transfection, cells were treated with Dox with or without nicotine or PNU for 24 h. Then, relative TXNIP or Ins1 mRNA expression levels were determined by quantitative PCR as described above.
Apoptosis
For apoptosis assay, INS‐1 cells were treated with Tm, or Dox‐inducible transgenic WT IRE1α overexpressing INS‐1 cells were incubated with Dox. Translocation of phosphatidylserine to the external cell surface was detected by flow cytometry with annexin V‐fluorescein isothiocyanate staining fluorescein isothiocyanate‐annexin V apoptosis detection kit (BD Biosciences, San Jose, CA, USA). Flow cytometry was carried out using BD FACSVerse (BD Biosciences), and the results were analyzed using FACSuite (BD Biosciences, v.1.0.5.3841). Activation of caspase‐3 and ‐7, which are markers of the early stages of apoptosis, was detected using a Caspase‐Glo 3/7 assay kit (Promega Corporation, Madison, WI, USA) through luminescence assay in accordance with the manufacturer’s instructions.
Statistical analysis
The results of the present study are expressed as the mean ± standard error of the mean. All experiments were replicated more than three times. The significance of differences (P < 0.05) between normalized mean values was evaluated by one‐way anova followed by Tukey’s post‐hoc test or the Student’s t‐test, as appropriate. All statistical analyses were carried out using JMP® 14 (SAS Institute Inc., Cary, NC, USA).
Results
Nicotine reverses the T‐UPR induced by IRE1α hyperactivation
To elucidate whether nAChR signaling has the cell protective effect against T‐UPR induced by IRE1α signaling in pancreatic β‐cells, we first examined T‐UPR markers in Dox‐inducible FLAG‐tagged IRE1α‐overexpressing INS‐1 cells. We previously validated this cell system as a tool to determine the sufficiency of specific and focal hyperactivation of IRE1α (without the need to resort to upstream ER stress inducing agents)3. Overexpression of IRE1α increased a key T‐UPR marker, TXNIP, which activates the NLRP3 inflammasome to promote β‐cell death (Figure 1a, Figure S1)25, 26. Furthermore, it decreased insulin 1 (Ins1) mRNAs, which was identified as IRE1α RNase substrate, and consecutive proinsulin protein levels over time (Figure 1b, Figure S1)3. The effect of IRE1α overexpression on PERK arm is limited in this system, based on its substrate eIF2α phosphorylation levels and CHOP mRNA expression being unchanged (Figure S2a,b). Using this IRE1α‐overexpressing INS‐1 cell line, nicotine reversed the increase of TXNIP mRNA expression levels in a dose‐dependent manner, starting at 10 μmol/L, with complete reversal at 1 mmol/L (Figure 1a). Similarly, nicotine reversed the decrease in Ins1 mRNA expression levels dose‐dependently (Figure 1b).
Figure 1.
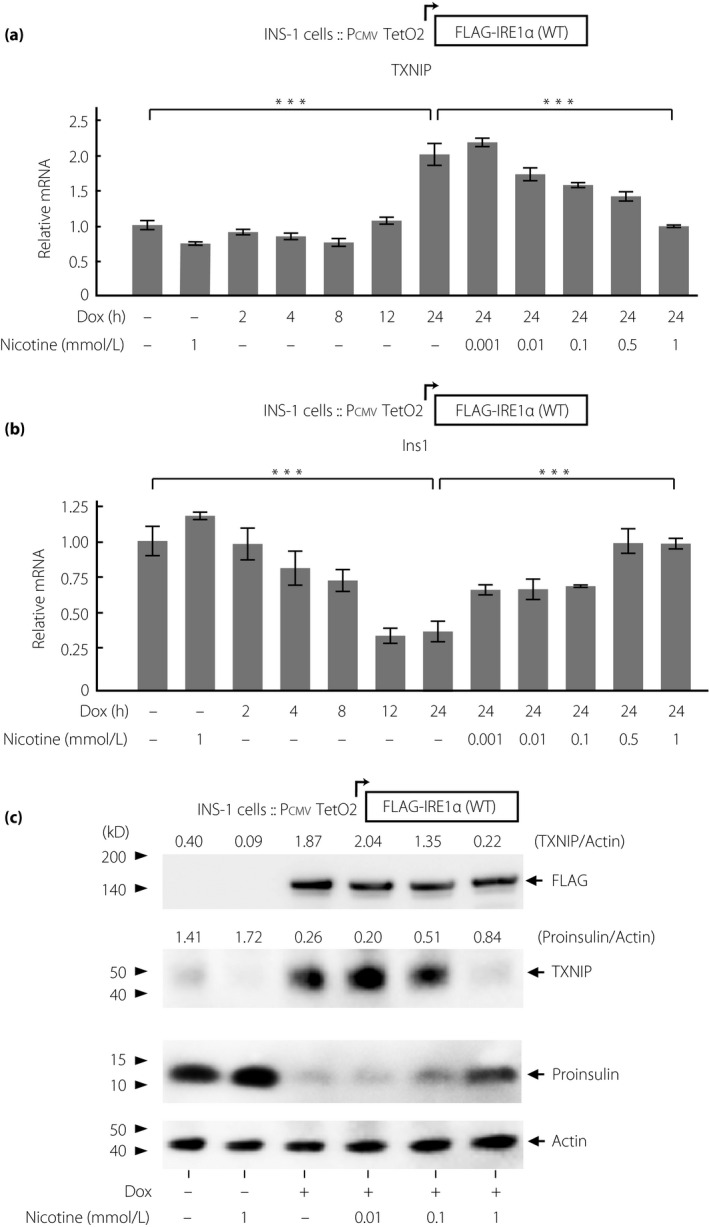
Nicotine reverses terminal unfolded protein response induced by overexpression of Flag‐tagged full‐length inositol‐requiring enzyme 1α in INS‐1 cells. (a,b) Relative messenger ribonucleic acid (mRNA) expression levels of (a) TXNIP and (b) insulin 1 (Ins1) in IRE1α overexpressing INS‐1 cells (n = 3‐8). The cells were treated with doxycycline hyclate (Dox; 1 μg/mL) with or without nicotine at indicated concentrations for indicated times. Relative mRNA levels of those in the cells treated with only nicotine for 24 h are shown in the second lane as a control. (c) Western blots of TXNIP and proinsulin in IRE1α overexpressing INS‐1 cells treated with Dox with or without 1 mmol/L nicotine for 24 h. All data are expressed as the mean ± standard error of the mean. ***P < 0.001 versus control.
These effects of nicotine were confirmed at the protein expression levels of TXNIP and proinsulin (Figure 1c). Thus, nAChR signaling protects T‐UPR signaling driven by IRE1α‐specific activation in INS‐1 cells.
Nicotine modulates IRE1α signaling under ER stress
Encouraged by the protective effect of nicotine against the T‐UPR by IRE1α‐specific hyperactivation, we next examined the effect of nicotine on IRE1α signaling under global ER stress. To induce ER stress, we used two ER stress inducers: Tg and Tm. Tg is an inhibitor of the ER Ca2+ ATPase, which activates calcium‐dependent ER chaperones, leading to the accumulation of unfolded proteins27. Tm blocks glycoprotein biosynthesis in the ER causing accumulation of unfolded glycoproteins, which in turn leads to ER stress28. We first showed that nicotine minimized site‐specific XBP1 mRNA splicing induced by Tg in INS‐1 cells (Figure 2a). Furthermore, this effect of nicotine was confirmed by the quantitative measurement of the spliced form of XBP1 mRNAs in Tg‐ and Tm‐exposed INS‐1 cells (Figure 2b,c). As passing the threshold of autophosphorylation of IRE1α drives RNase activity into the hyperactivation state, we next investigated the phosphorylation levels of IRE1α using Phos‐tag gel, which can separate phosphorylated forms from non‐phosphorylated forms of protein on the basis of phosphorylation levels. Tm increased the ratio of the level of phosphorylated IRE1α to that of non‐phosphorylated IRE1α, and nicotine decreased it (Figure 2d). Then, as shown in the rodent β‐cell line, we confirmed for the first time the expression of the α7 nAChR subunit in the well‐validated human β‐cell line, EndoC‐βH1 cells (Figure 2e). In this cell line, nicotine also reduced Tg‐induced spliced XBP1 mRNA expression levels (Figure 2f). The effect of nicotine on the PERK arm was limited under ER stress, based on its substrate eIF2α phosphorylation levels being unchanged (Figure S3). Therefore, nicotine modulates kinase and RNase activation of IRE1α under ER stress in pancreatic β‐cells.
Figure 2.
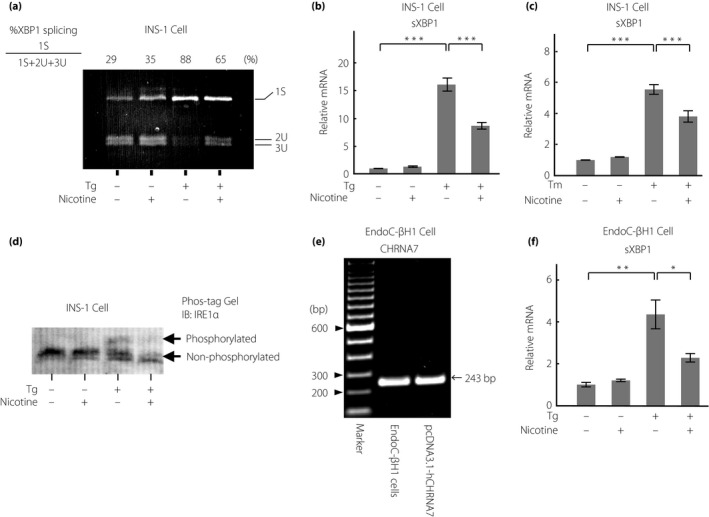
Nicotine regulates inositol‐requiring enzyme 1α (IRE1α) activation induced by endoplasmic reticulum stress in INS‐1 and EndoC‐βH1 cells. (a) EtBr‐stained agarose gel electrophoresis of Pst1‐digested X‐box‐binding protein‐1 (XBP1) amplicons obtained by polymerase chain reaction in INS‐1 cells treated with Tg (500 nmol/L, 6 h) with or without nicotine (1 mmol/L) for 3 h. (b,c) The numbers above are calculated from the ratio metric quantitation of the level of spliced XBP1 complementary deoxyribonucleic acid to that of total XBP1 complementary deoxyribonucleic acids. sXBP1 expression measured by quantitative reverse transcription polymerase chain reaction treated with (b) thapsigargin (Tg; 500 nmol/L, 6 h; n = 10) or (c) tunicamycin (Tm; 100 μg/mL, 3 h; n = 3) with or without nicotine (1 mmol/L) for 6 h in INS‐1 cells. (d) Immunoblots with anti‐IRE1α antibody for proteins separated using Phos‐tag gel for INS‐1 cells treated with Tg (500 nmol/L) with or without nicotine (1 mmol/L) for 6 h. (e) Expression of CHRNA7 messenger ribonucleic acid (mRNA) by polymerase chain reaction in human EndoC‐βH1 cells. The expression of the in pcDNA3.1‐human CHRNA7 vector is shown as the positive control. (f) Relative sXBP1 mRNA expression in EndoC‐βH1 cells treated with Tg (10 μmol/L) with or without nicotine (1 mmol/L) for 6 h (n = 3). All data are expressed as the mean ± standard error of the mean. *P < 0.05, **P < 0.01, ***P < 0.001 versus control.
Nicotine attenuates T‐UPR induced by ER stress
As nicotine showed the effect of reducing the direct IRE1α signaling under ER stress, we next examined whether nicotine also attenuates T‐UPR induced by high and chronic ER stress. ER stress inducers increased mRNA expression levels of TXNIP and decreased those of Ins1, whereas nicotine inhibited these effects of ER stress inducers (Figure 3a‐d, Figure S4). Similar results were found for TXNIP and proinsulin at the protein level (Figure 3e). The effects of attenuating the ER stress‐induced increase in TXNIP and decrease in insulin mRNA expression levels by nicotine were also confirmed in EndoC‐βH1 cells (Figure 3f,g). To evaluate the effect of nicotine under physiological ER stress, we investigated it at high glucose levels29. A high ambient glucose concentration (28 mmol/L) increased the protein expression levels of TXNIP and reduced those of proinsulin protein levels as previously reported10, and nicotine attenuated these effects (Figure 3h). These results suggest that nicotine attenuates T‐UPR induced by ER stress.
Figure 3.
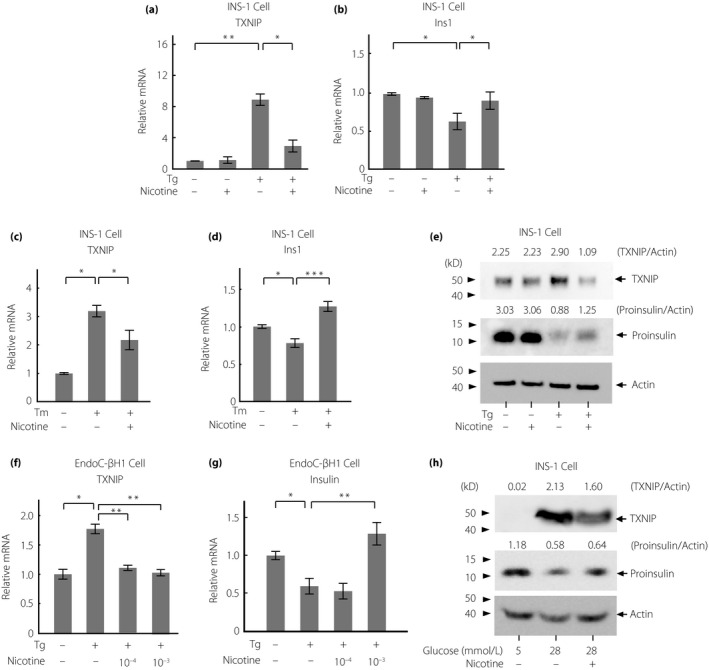
Nicotine attenuates terminal unfolded protein response induced by endoplasmic reticulum stress in INS‐1 and EndoC‐βH1 cells. (a–d) Relative messenger ribonucleic acid (mRNA) levels of (a,c) TXNIP and (b,d) Ins1 by quantitative polymerase chain reaction in INS‐1 cells treated with thapsigargin (Tg; 500 nmol/L, 6 h) or tunicamycin (Tm; 200 nmol/L, 24 h) with or without nicotine (1 mmol/L; n = 6‐9). (e,h) Western blots of TXNIP and proinsulin in INS‐1 cells incubated with (e) Tg (500 nmol/L, 6 h; n = 3) or with (h) high medium glucose (28 mmol/L, 24 h) with or without nicotine (1 mmol/L). (f,g) Relative mRNA levels of (f) TXNIP and (g) insulin in EndoC‐βH1 cells treated with Tg (10 μmol/L) with or without nicotine (100 μmol/L) for 6 h. All data are expressed as the mean ± standard error of the mean. *P < 0.05, **P < 0.01, ***P < 0.001 versus control.
nAChR signaling regulates IRE1α signaling through the α7 subunit
To investigate nAChR signaling in detail, we focused on signaling through the α7 subunit for the following reasons. First, it is the most commonly expressed subunit among the nAChR subunits in rodent β‐cells11, 30. Second, the mechanisms underlying the recently revealed anti‐apoptotic effects of a complete and highly specific nAChR α7 agonist, PNU, in β‐cells are still unknown. PNU dose‐dependently attenuated IRE1α‐induced T‐UPR markers similarly to nicotine (Figure 4a‐c). Next, PNU attenuated the increased mRNA expression levels of XBP1 spliced form induced by ER stress in INS‐1 and EndoC‐βH1 cells (Figure 4d,e). Furthermore, PNU inhibited the increase of TXNIP and decrease of Ins1/proinsulin expression at mRNA and protein levels (Figure 4f‐h). The effect of nicotine was canceled by the α7‐specific antagonist, α‐bungarotoxin (Figure 5a). Finally, the protective effects of nicotine and PNU on T‐UPR signaling were canceled by two specific small interfering RNAs targeting α7 nAChR with approximately 60% reduction of CHRNA7 mRNA expression levels under overexpression of IRE1α in INS‐1 cells (Figure 5b,c, Figure S5). Taken together, the effects of nAChR signaling on T‐UPR were, at least in part, through the α7 nAChR in pancreatic β‐cells.
Figure 4.
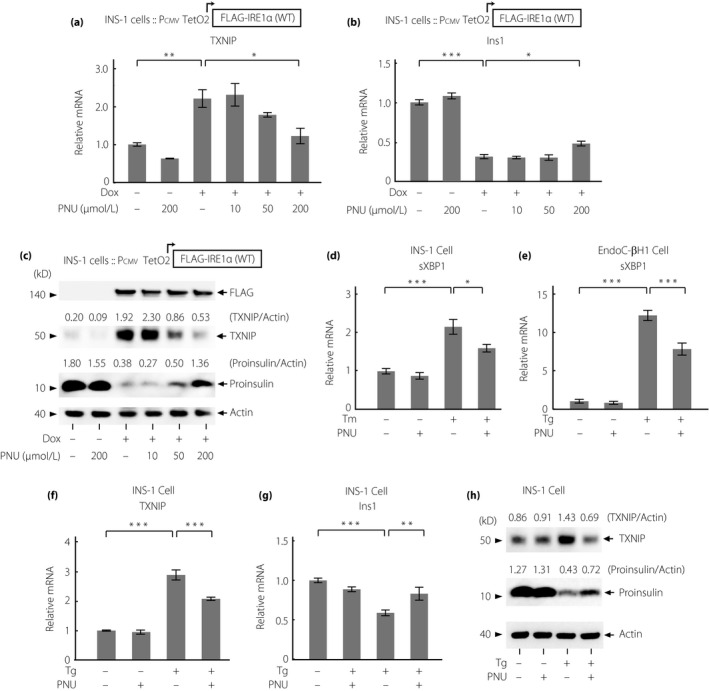
The nicotinic acetylcholine receptor (nAChR) α7 subunit‐specific agonist, PNU‐282987, regulates terminal unfolded protein response induced by inositol‐requiring enzyme 1α (IRE1α) overexpression and endoplasmic reticulum stress in INS‐1 and EndoC‐βH1 cells. (a,b) Relative messenger ribonucleic acid (mRNA) expression levels of (a) TXNIP and (b) Ins1 in IRE1α‐overexpressing INS‐1 cells treated with doxycycline hyclate (Dox; 1 μg/mL) with or without PNU‐282987 (PNU), the agonist of nAChR α7 at indicated doses for 24 h (n = 4). (c) Western blots of TXNIP and proinsulin in IRE1α‐overexpressing INS‐1 cells treated with Dox (1 μg/mL) with or without PNU at indicated doses for 24 h. (d,e) Relative mRNA expression levels of splice X‐box‐binding protein‐1 (sXBP1) in (d) INS‐1 cells treated with Tm (200 nmol/L, 24 h) or in (e) EndoC‐βH1 cells with thapsigargin (Tg; 10 μmol/L, 6 h) with or without PNU (10 μmol/L) (n = 3–4). (f,g) Relative mRNA levels of (f) TXNIP (n = 3) and (g) Ins1 (n = 6) in INS‐1 cells treated with Tg (500 nmol/L) with or without PNU (10 μmol/L) for 6 h. (h) Western blots of TXNIP and proinsulin in INS‐1 cells treated with Tg (500 nmol/L) with or without PNU (10 μmol/L) for 6 h. All data are expressed as the mean ± standard error of the mean. *P < 0.05, **P < 0.01, ***P < 0.001 versus control.
Figure 5.
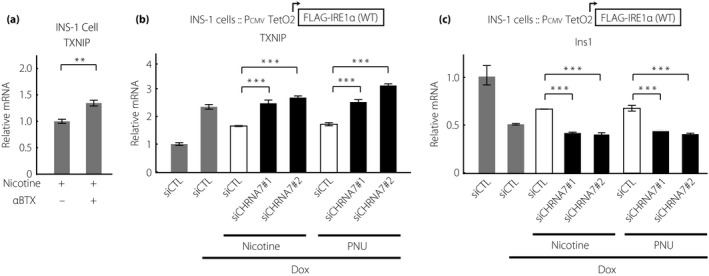
Nicotinic acetylcholine receptor (nAChR) signaling regulates inositol‐requiring enzyme 1α (IRE1α) signaling through the α7 subunits in INS‐1 cells. (a) Relative messenger ribonucleic acid (mRNA) levels of TXNIP in INS‐1 cells co‐treated with tunicamycin (Tm; 200 nmol/L) and nicotine (1 mmol/L) for 24 h, pretreated with or without α‐bungarotoxin (αBTX, 1 μmol/L), an α7‐specific nAChR inhibitor, for 3 h (n = 6). (b,c) IRE1α‐overexpressing INS‐1 cells were transfected with a non‐specific control small interfering ribonucleic acid (siCTL) or two independent small interfering ribonucleic acids targeting CHRNA7 (siCHRNA7 #1 and #2). After 24 h, the cells were treated with doxycycline hyclate (Dox; 1 μg/mL) with or without nicotine (1 mmol/L) or PNU (20z0 μmol/L) for 24 h. Relative (b) TXNIP and(c) Ins1 mRNA levels were determined by quantitative polymerase chain reaction (n = 3‐5). All data are expressed as the mean ± standard error of the mean. **P < 0.01, ***P < 0.001 versus control.
Nicotine inhibits apoptosis induced by IRE1α activation and ER stress
Finally, if indeed nicotine attenuates T‐UPR, we hypothesized that nicotine could regulate ultimate apoptosis induced by IRE1α activation or ER stress. To evaluate apoptosis, we measured two markers, caspase‐3 and ‐7 activity and annexin V. Caspase‐3 and ‐7 is an apoptotic marker, and its activation is specific to cell apoptosis. Caspase‐3 and ‐7 are normally present in cells as inactive precursors. When apoptosis occurs, these caspases are proteolysed and activated, initiating a caspase cascade that leads to the degradation of proteins essential for cell survival31. Annexin V is a marker of apoptotic cells based on its ability to bind to phosphatidylserine32. The translocation of phosphatidylserine to the surface of the cell membrane occurs downstream in the apoptotic process33. Not only nicotine, but also PNU suppressed caspase‐3/7 activity caused by IRE1α overexpression (Figure 6a,b). Notably, the concentration of PNU was lower than that of nicotine, suggesting that the effect of the α7 subunit specifically attenuates IRE1α‐induced apoptosis. Furthermore, nicotine attenuated the increased caspase‐3/7 activation induced by ER stress (Figure 6c). Finally, nicotine suppressed the number of Annexin V‐positive cells induced by ER stress (Figure 6d, Figure S6). Taken together, nAChR signaling suppresses ultimate apoptosis induced by ER stress and IRE1α activation.
Figure 6.
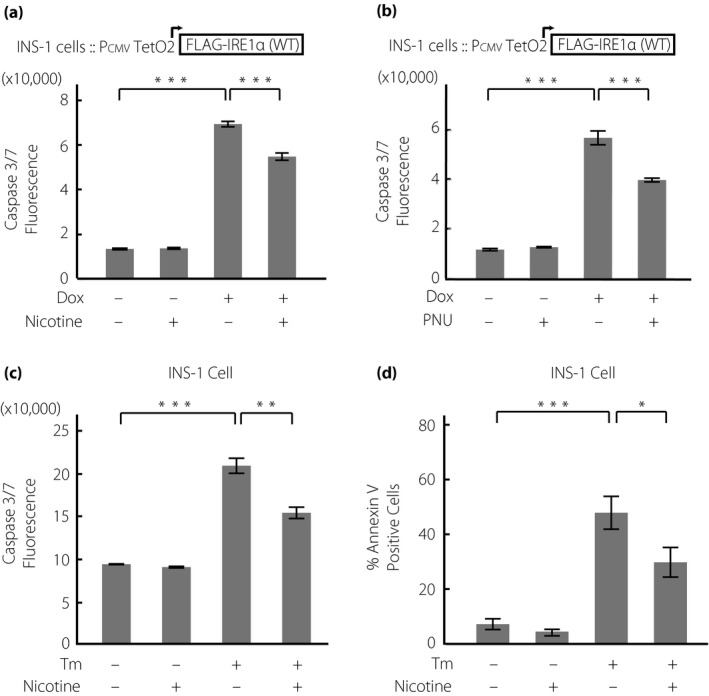
Nicotinic acetylcholine receptor (nAChR) signaling inhibits apoptosis induced by inositol‐requiring enzyme 1α (IRE1α) activation and endoplasmic reticulum stress in INS‐1 cells. (a,b) Caspase‐3/7 fluorescence in IRE1α overexpressing INS‐1 cells treated with (a) doxycycline hyclate (Dox; 1 μg/mL) with or without nicotine (1 mmol/L) or (b) PNU (10 nmol/L) for 72 h (n = 4). (c) Caspase‐3/7 fluorescence (n = 4) or (d) percentage of annexin V‐positive cells (n = 6) in INS‐1 cells treated with tunicamycin (Tm; 100 ng/mL) with or without nicotine (1 mmol/L) for 48 h. All data are expressed as the mean ± standard error of the mean. *P < 0.05, **P < 0.01, ***P < 0.001 versus control.
Discussion
We previously showed the critical role of IRE1α signaling in diabetes by demonstrating that its endoribonuclease activities trigger cell death and crucial dysfunction in β‐cells3. Furthermore, we showed that the monoselective IRE1α inhibitor KIRA8 rescues β‐cells from T‐UPR and ultimate apoptosis to ameliorate or reverse diabetes in Akita or NOD mice, as shown by the observation that KIRA spares cell survival A‐UPR, but shuts down T‐UPR, showing the essential role of tuning IRE1α output for glucose metabolism in vivo 2, 10. In contrast, recent reports have emphasized the protective role of global and α7 subunit‐specific nAChR signaling against apoptosis in rodent β‐cells, but the underlying mechanisms are still unknown34. These backgrounds encouraged us to investigate the link between nAChR signaling and IRE1α signaling. In the present study, we showed that the modulation of global and α7 subunit‐specific nAChR signaling attenuated IRE1α activation, which protected rat and human β‐cells from T‐UPR and ultimate apoptosis under ER stress.
The vagus nerve system was previously shown to have a cytoprotective effect by balancing and attenuating the pro‐inflammatory activation in islets35. It has also been shown that nicotine or the systemic increase in ACh levels ameliorates the diabetic phenotypes by stimulating the Th1 to Th2 cytokine switch or pancreatic T‐cell differentiation in NOD mice and multiple low‐dose streptozotocin mice18, 36. In addition, the specific stimulation of nAChR α7 has recently been shown to activate anti‐inflammatory and prosurvival pathways in cytokine‐treated INS‐1 cells and murine islets34. In contrast, we and other groups have recently shown that UPR markers are induced in β‐cells from type 1 diabetes patients and NOD mice, and that established diabetes in NOD mice could be reversed by KIRA10, 37. In addition, we previously showed that IRE1α activation induces TXNIP to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress by reducing the expression levels of a TXNIP mRNA destabilizing microRNA, miR‐1710, 25. The findings of the present study might support the anti‐inflammatory role of nAChR signaling in the immune/UPR‐mediated pathogenic state of pancreatic β‐cells by showing its novel pathway of regulation of proapoptotic TXNIP expression induced by IRE1α activation and ER stress.
The effect of nAChR signaling on the IRE1α arm has not been previously investigated. Regarding the effect of nAChR signaling on UPR in non‐β‐cells, one study showed that nicotine inhibits the translocation of endogenously expressed activating transcription factor 6 and the phosphorylation of eIF2α in Neuro‐2a cells21; however, another study controversially showed that nicotine dose‐dependently induces the phosphorylation of PERK and eIF2α, and its antagonist blocks the phosphorylation of PERK in rat placental trophoblast cells22. One of the reasons for the conflicting observations of the effect of nicotine on UPR might be related to the tissue specificity of the effect of nicotine. In β‐cells, previous studies showed the expressions of the α7 subunit in rat islet β‐cells13, and the α2, α3, α4, α7 and β2 subunits in INS‐1 cells34. We also preliminarily confirmed the expression of the α2, α5 and β2 subunits, as well as the α7 subunit (Figure S7; Table S1). The α7 subunit is the most common subunit in the previous islet studies, and its specific agonist has recently been shown to have a protective effect against cytokine‐induced β‐cell apoptosis and eventual diabetes in vivo 19, 34. On the basis of these findings, we investigated the α7 subunit‐specific effect on T‐UPR and apoptosis under ER stress. In addition to previously reported protective mechanisms of modulation of nAChR α7 signaling against β‐cell apoptosis, which were attributed to attenuated JAK2–STAT3 signaling34, the results of our present study provide a novel protective role of nAChR α7 signaling by linking to the regulation of IRE1α signaling under ER stress.
One of the limitations of the present study is that, although we showed that the modulation of global and α7‐specific nAChR signaling could regulate IRE1α signaling, its detailed pathway(s) is still widely unknown. Initially, UPR signaling was viewed as a direct and linear transduction of ER stress levels4. However, recent findings have shown that UPR sensors are tightly regulated through their binding to cofactors and post‐translational modifications4, 38. For example, it has recently been reported that many cytosolic proteins characterized as components of other signaling pathways interact with UPR sensors to regulate their outputs, and their complexes are conceptually termed ‘‘UPRosome’’38. As for IRE1α, we and other groups showed that ABL kinase, as well as protein‐tyrosine phosphatase 1B, BAX inhibitor‐1, ASK1 and RACK1, could tune IRE1α outputs to affect cell fate10, 39, 40, 41, 42, 43. As nAChR is a type of ligand‐gated ion channel, it could be speculated that a change in intracellular calcium flux through nAChR might widely affect myriad molecules, including UPRosome‐related proteins, or the post‐translational modulation of IRE1α to attenuate the amplification of the T‐UPR cascade. Further studies are required to elucidate the detailed mechanism(s) of cross‐talk of nAChR‐IRE1α signaling. Another limitation is that all the experiments were in vitro studies. Thus, as we could speculate the effect of vagus systems on nAChR signaling in β‐cells or the paracrine effect of other islet component cells on this signaling in in vivo β‐cells, the present findings regarding the roles of nAChR‐IRE1α signaling should be carefully interpreted. Further studies using animal models are required35, 44.
In conclusion, we showed that nAChR signaling regulates IRE1α signaling to protect against T‐UPR and eventual apoptosis induced by ER stress in both rat and human pancreatic β‐cells. Furthermore, it regulates IRE1α activation, at least in part, through the α7 nAChR subunit. We elucidated the novel role of nAChR signaling as a protective factor for β‐cells under ER stress. nAChR signaling could be one of the candidates of a therapeutic target to prevent or reverse β‐cell loss and dysfunction in diabetes.
Disclosure
The authors declare no conflict of interest.
Supporting information
Fig. S1 | Time courses of FLAG, TXNIP and proinsulin expressions in doxycycline hyclate (Dox)‐inducible inositol‐requiring enzyme 1α (IRE1α)‐overexpressing INS‐1 cells.
Fig. S 2 | Overexpression of inositol‐requiring enzyme 1α (IRE1α) does not affect phosphorylation of eIF2α nor CHOP messenger ribonucleic acid expression in INS‐1 cells.
Fig. S 3 | Effects of nicotine on phosphorylation of eIF2α is limited under endoplasmic reticulum (ER) stress in INS‐1 cells.
Fig. S 4 | Nicotine does not affect basal TXNIP expression in INS‐1 cells.
Fig. S 5 | Knock down of rat α7 nicotinic acetylcholine receptor (CHRNA7) by specific small interfering ribonucleic acids in INS‐1 cells.
Fig. S 6 | Representative histograms of fluorescein isothiocyanate annexin V‐stained INS‐1 cells.
Fig. S 7 | Messenger ribonucleic acid expression levels of nicotinic acetylcholine receptor subunits in INS‐1 cells.
Table S1 | Primers used for Figure S7.
Acknowledgments
We thank Dr Asako Doi for technical assistance, and Dr Yoshito Ihara for helpful suggestions and comments. We acknowledge proofreading and editing by Benjamin Phillis at the Clinical Study Support Center, Wakayama Medical University. This work was supported by SRF (TA, HA), JSPS KAKENHI #JP17H07033 (SM), #JP18K16242 (SM) and Japan Diabetes Foundation (SM).
J Diabetes Investig 2020; 11: 801–813
References
- 1. Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 2007; 8: 519–529. [DOI] [PubMed] [Google Scholar]
- 2. Ghosh R, Wang L, Wang ES, et al Allosteric inhibition of the IRE1alpha RNase preserves cell viability and function during endoplasmic reticulum stress. Cell 2014; 158: 534–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Han D, Lerner AG, Vande Walle L, et al IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell 2009; 138: 562–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hetz C, Papa FR. The unfolded protein response and cell fate control. Mol Cell 2018; 69: 169–181. [DOI] [PubMed] [Google Scholar]
- 5. Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science 2011; 334: 1081–1086. [DOI] [PubMed] [Google Scholar]
- 6. Calfon M, Zeng H, Urano F, et al IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP‐1 mRNA. Nature 2002; 415: 92–96. [DOI] [PubMed] [Google Scholar]
- 7. Shen X, Ellis RE, Lee K, et al Complementary signaling pathways regulate the unfolded protein response and are required for C. elegans development. Cell 2001; 107: 893–903. [DOI] [PubMed] [Google Scholar]
- 8. Yoshida H, Matsui T, Yamamoto A, et al XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001; 107: 881–891. [DOI] [PubMed] [Google Scholar]
- 9. Hollien J, Lin JH, Li H, et al Regulated Ire1‐dependent decay of messenger RNAs in mammalian cells. J Cell Biol 2009; 186: 323–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Morita S, Villalta SA, Feldman HC, et al Targeting ABL‐IRE1alpha signaling spares ER‐stressed pancreatic beta cells to reverse autoimmune diabetes. Cell Metab 2017; 25, 883–897. e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Somm E, Guerardel A, Maouche K, et al Concomitant alpha7 and beta2 nicotinic AChR subunit deficiency leads to impaired energy homeostasis and increased physical activity in mice. Mol Genet Metab 2014; 112: 64–72. [DOI] [PubMed] [Google Scholar]
- 12. Vu CU, Siddiqui JA, Wadensweiler P, et al Nicotinic acetylcholine receptors in glucose homeostasis: the acute hyperglycemic and chronic insulin‐sensitive effects of nicotine suggest dual opposing roles of the receptors in male mice. Endocrinology 2014; 155: 3793–3805. [DOI] [PubMed] [Google Scholar]
- 13. Yoshikawa H, Hellstrom‐Lindahl E, Grill V. Evidence for functional nicotinic receptors on pancreatic beta cells. Metabolism 2005; 54: 247–254. [DOI] [PubMed] [Google Scholar]
- 14. Egleton RD, Brown KC, Dasgupta P. Nicotinic acetylcholine receptors in cancer: multiple roles in proliferation and inhibition of apoptosis. Trends Pharmacol Sci 2008; 29: 151–158. [DOI] [PubMed] [Google Scholar]
- 15. Marrero MB, Lucas R, Salet C, et al An alpha7 nicotinic acetylcholine receptor‐selective agonist reduces weight gain and metabolic changes in a mouse model of diabetes. J Pharmacol Exp Ther 2010; 332: 173–180. [DOI] [PubMed] [Google Scholar]
- 16. Liu RH, Kurose T, Matsukura S. Oral nicotine administration decreases tumor necrosis factor‐alpha expression in fat tissues in obese rats. Metabolism 2001; 50: 79–85. [DOI] [PubMed] [Google Scholar]
- 17. Wang H, Yu M, Ochani M, et al Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 2003; 421: 384–388. [DOI] [PubMed] [Google Scholar]
- 18. Mabley JG, Pacher P, Southan GJ, et al Nicotine reduces the incidence of type I diabetes in mice. J Pharmacol Exp Ther 2002; 300: 876–881. [DOI] [PubMed] [Google Scholar]
- 19. Somm E. Nicotinic cholinergic signaling in adipose tissue and pancreatic islets biology: revisited function and therapeutic perspectives. Arch Immunol Ther Exp 2014; 62: 87–101. [DOI] [PubMed] [Google Scholar]
- 20. Delbro DS. Expression of the non‐neuronal cholinergic system in rat beta‐cells. Auton Neurosci 2012; 167: 75–77. [DOI] [PubMed] [Google Scholar]
- 21. Srinivasan R, Richards CI, Xiao C, et al Pharmacological chaperoning of nicotinic acetylcholine receptors reduces the endoplasmic reticulum stress response. Mol Pharmacol 2012; 81: 759–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wong MK, Holloway AC, Hardy DB. Nicotine directly induces endoplasmic reticulum stress response in rat placental trophoblast giant cells. Toxicol Sci 2016; 151: 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ravassard P, Hazhouz Y, Pechberty S, et al A genetically engineered human pancreatic beta cell line exhibiting glucose‐inducible insulin secretion. J Clin Investig 2011; 121: 3589–3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang Y, Xiao C, Indersmitten T, et al The duplicated alpha7 subunits assemble and form functional nicotinic receptors with the full‐length alpha7. J Biol Chem 2014; 289: 26451–26463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lerner AG, Upton JP, Praveen PV, et al IRE1alpha induces thioredoxin‐interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab 2012; 16: 250–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Oslowski CM, Hara T, O'Sullivan‐Murphy B, et al Thioredoxin‐interacting protein mediates ER stress‐induced beta cell death through initiation of the inflammasome. Cell Metab 2012; 16: 265–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Keestra‐Gounder AM, Byndloss MX, Seyffert N, et al NOD1 and NOD2 signalling links ER stress with inflammation. Nature 2016; 532: 394–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Oslowski CM, Urano F. Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Method Enzymol 2011; 490: 71–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lipson KL, Fonseca SG, Ishigaki S, et al Regulation of insulin biosynthesis in pancreatic beta cells by an endoplasmic reticulum‐resident protein kinase IRE1. Cell Metab 2006; 4: 245–254. [DOI] [PubMed] [Google Scholar]
- 30. Klee P, Bosco D, Guerardel A, et al Activation of nicotinic acetylcholine receptors decreases apoptosis in human and female murine pancreatic islets. Endocrinology 2016; 157: 3800–3808. [DOI] [PubMed] [Google Scholar]
- 31. Fischer U, Janicke RU, Schulze‐Osthoff K. Many cuts to ruin: a comprehensive update of caspase substrates. Cell Death Differ 2003; 10: 76–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Concha NO, Head JF, Kaetzel MA, et al Annexin V forms calcium‐dependent trimeric units on phospholipid vesicles. FEBS Lett 1992; 314: 159–162. [DOI] [PubMed] [Google Scholar]
- 33. Koopman G, Reutelingsperger CP, Kuijten GA, et al Annexin V for flow cytometric detection of phosphatidylserine expression on B cells undergoing apoptosis. Blood 1994; 84: 1415–1420. [PubMed] [Google Scholar]
- 34. Gupta D, Lacayo AA, Greene SM, et al beta‐Cell mass restoration by alpha7 nicotinic acetylcholine receptor activation. J Biol Chem 2018; 293: 20295–20306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pavlov VA, Tracey KJ. The vagus nerve and the inflammatory reflex–linking immunity and metabolism. Nat Rev Endocrinol 2012; 8: 743–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. George JA, Bashir G, Qureshi MM, et al Cholinergic stimulation prevents the development of autoimmune diabetes: evidence for the modulation of Th17 effector Cells via an IFNgamma‐dependent mechanism. Front Immunol 2016; 7: 419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tersey SA, Nishiki Y, Templin AT, et al Islet beta‐cell endoplasmic reticulum stress precedes the onset of type 1 diabetes in the nonobese diabetic mouse model. Diabetes 2012; 61: 818–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sepulveda D, Rojas‐Rivera D, Rodriguez DA, et al Interactome screening identifies the ER luminal chaperone Hsp47 as a regulator of the unfolded protein response transducer IRE1alpha. Mol Cell 2018; 69: 238–252. e7. [DOI] [PubMed] [Google Scholar]
- 39. Gu F, Nguyen DT, Stuible M, et al Protein‐tyrosine phosphatase 1B potentiates IRE1 signaling during endoplasmic reticulum stress. The J Biol Chem 2004; 279: 49689–49693. [DOI] [PubMed] [Google Scholar]
- 40. He Y, Beatty A, Han X, et al Nonmuscle myosin IIB links cytoskeleton to IRE1alpha signaling during ER stress. Dev Cell 2012; 23: 1141–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lisbona F, Rojas‐Rivera D, Thielen P, et al BAX inhibitor‐1 is a negative regulator of the ER stress sensor IRE1alpha. Mol Cell 2009; 33: 679–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nishitoh H, Matsuzawa A, Tobiume K, et al ASK1 is essential for endoplasmic reticulum stress‐induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev 2002; 16: 1345–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Qiu Y, Mao T, Zhang Y, et al A crucial role for RACK1 in the regulation of glucose‐stimulated IRE1alpha activation in pancreatic beta cells. Sci Signal 2010; 3: ra7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rodriguez‐Diaz R, Dando R, Jacques‐Silva MC, et al Alpha cells secrete acetylcholine as a non‐neuronal paracrine signal priming beta cell function in humans. Nat Med 2011; 17: 888–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 | Time courses of FLAG, TXNIP and proinsulin expressions in doxycycline hyclate (Dox)‐inducible inositol‐requiring enzyme 1α (IRE1α)‐overexpressing INS‐1 cells.
Fig. S 2 | Overexpression of inositol‐requiring enzyme 1α (IRE1α) does not affect phosphorylation of eIF2α nor CHOP messenger ribonucleic acid expression in INS‐1 cells.
Fig. S 3 | Effects of nicotine on phosphorylation of eIF2α is limited under endoplasmic reticulum (ER) stress in INS‐1 cells.
Fig. S 4 | Nicotine does not affect basal TXNIP expression in INS‐1 cells.
Fig. S 5 | Knock down of rat α7 nicotinic acetylcholine receptor (CHRNA7) by specific small interfering ribonucleic acids in INS‐1 cells.
Fig. S 6 | Representative histograms of fluorescein isothiocyanate annexin V‐stained INS‐1 cells.
Fig. S 7 | Messenger ribonucleic acid expression levels of nicotinic acetylcholine receptor subunits in INS‐1 cells.
Table S1 | Primers used for Figure S7.