

Abstract
Aims/Introduction
The incidence of type 2 diabetes mellitus is increasing worldwide, and it might partly cause metabolic disorder and type 2 diabetes mellitus susceptibility in patients’ offspring through epigenetic modification. However, the underlying mechanisms remain largely unclear. Recent studies have shown a potential link between deoxyribonucleic acid methylation in paternal sperm and susceptibility to type 2 diabetes mellitus in offspring, so this article focuses on whether the whole‐genome methylation profiles of spermatozoa in type 2 diabetes mellitus patients have changed.
Materials and Methods
We investigated the genome‐wide deoxyribonucleic acid methylation profiles in spermatozoa by comparing eight individuals with type 2 diabetes mellitus and nine non‐diabetic controls using whole‐genome bisulfite sequencing method.
Results
First, we found that the proportion of methylated cytosine in the whole genome of the type 2 diabetes mellitus group was slightly lower than that of the control group. Interestingly, the proportion of methylated cytosines in the CG context decreased, and the proportion of methylated cytosines in the CHG context (H = A, T or C) increased in the type 2 diabetes mellitus group, but the proportion of methylated cytosines in the CHH context (H = A, T or C) barely changed. The methylated cytosines in the CG context were mainly distributed at the high methylated level, whereas methylated cytosines in the CHG context and methylated cytosines in the CHH context were mainly distributed at the low and middle methylated level in both groups. Second, functional enrichment analysis showed that differentially methylated genes played a significant role in nervous system development and cell metabolism. Finally, we identified 10 top type 2 diabetes mellitus‐related differentially methylated genes, including IRS1, PRKCE, FTO, PPARGC1A, KCNQ1, ATP10A, GHR, CREB1, PRKAR1A and HNF1B.
Conclusions
Our study provides the first evidence for deoxyribonucleic acid methylation reprogramming in spermatozoa of type 2 diabetes mellitus patients, and provides a new basis for explaining the complex mechanism of type 2 diabetes mellitus susceptibility in offspring.
Keywords: Sperm deoxyribonucleic acid methylation, Type 2 diabetes mellitus, Whole‐genome bisulfite sequencing
Type 2 diabetes mellitus has an increasing global prevalence, and partly contributes to the susceptibility to metabolic dysregulation and type 2 diabetes mellitus in offspring through epigenetic modifications. However, the underlying mechanism remains largely obscure. Recent work has shown a potential link between paternal sperm deoxyribonucleic acid methylation and susceptibility to type 2 diabetes mellitus in offspring.

Introduction
The global incidence of type 2 diabetes mellitus is increasing in modern societies1. According to several human epidemiological studies, the susceptibility to diabetes and related chronic diseases is increasing in offspring among type 2 diabetes mellitus patients2, 3. However, the molecular mechanisms of paternal type 2 diabetes mellitus that mediate these impacts remain largely unclear, particularly the role of epigenetics factors, such as deoxyribonucleic acid (DNA) methylation.
DNA methylation is a common pattern of epigenetic modification that mainly occurs at the CpG sites, adding a methyl to the fifth position of cytosine. DNA methylation plays an important role in regulating gene activities through modulating DNA transcription4, 5, and protecting genome integrity by restricting transposal element activity in the germ line and perhaps in somatic cells6, 7.
To our knowledge, fathers contribute to the developing offspring mostly just by sperm, and studies have found that sperm DNA methylation is crucial in transmitting epigenetic information from father to offspring. Furthermore, although sperm DNA methylation is erased and established anew during germ cell and pre‐implantation development, the DNA methyl group can be inherited in approximately 20% of each generation8, 9, 10.
Several independent research groups have shown that abnormal sperm DNA methylation usually increases the offspring’s susceptibility to disorders and affects their phenotypes11, 12, 13, 14, 15, 16. For example, a study of C57B/6J mice showed that offspring of fathers who received 12 weeks of long exercise training were more likely to be adversely affected by a high‐fat diet, characterized by weight and fat gain, impaired glucose tolerance, and elevated insulin levels. Interestingly, several differentially methylated metabolic genes are also expressed in the skeletal muscle of offspring, including Oga, Ogt, H19, Pdk4, Glut4 and Ptpn1 13. In another study, the connection of abnormal DNA methylation in fathers’ sperm with the risk of autism in offspring was tested, and suggested that several differentially methylated regions (DMRs) in fathers’ sperm were associated with autism risk in offspring14. In addition, sperm DNA methylation was altered with age, and possibly increased susceptibility to disease in the next generation15.
Although accumulating evidence shows that many paternal factors might contribute to differential DNA methylation in sperm and influence the health of the next generation, there are very limited studies focused on the relationship between sperm DNA methylation in type 2 diabetes mellitus fathers and the susceptibility to type 2 diabetes mellitus in their offspring. Wei et al. found that male mice with prediabetes altered overall DNA methylation patterns in sperm that matched changed methylation in their pancreatic islets in offspring, and increased their susceptibility to diabetes17. However, whether there are observed differences in sperm DNA methylation in humans is still unknown.
In the present study, our aim was to assess the distribution of genome‐wide DNA methylation in sperm isolated from type 2 diabetes mellitus patients and non‐diabetic individuals through whole‐genome bisulfite sequencing (WGBS) technology. Here, we found that sperm DNA methylation profiles are altered in type 2 diabetes mellitus patients, which is of great significance to explain the susceptibility of type 2 diabetes mellitus and the overall health in offspring.
Methods
Study cohorts
The study protocol was approved by the ethics committee of Fujian Province Hospital (K2016‐09‐014). Semen samples were collected from age‐matched volunteers aged 20–45 years. There were eight type 2 diabetes mellitus patients (fasting plasma glucose >6.1 mmol/L), and nine control volunteers without diabetes (fasting plasma glucose 3.9–6.1 mmol/L). More detailed information was provided in Table S1.
Isolation of motile spermatozoa
The routine operation of sperm treatment was mainly referred to the 5th edition of the World Health Organization laboratory manual for human semen examination and processing18. Motile spermatozoa were isolated and purified by the pellet swim‐up procedure19. The corresponding volume of preheated HTF solution was added to a round‐bottom tube containing 1–5 mL of liquefied semen, then incubated at a 45° angle for 30 min in a 37°C incubator. The upper components were collected and placed in centrifuge tubes and centrifuged at 340 g for 15 min. The supernatant was discarded, and 1~2 mL of HTF solution was slowly added for suspension. The spermatozoa were then counted by microscopy and the presence of somatic cells was checked. The purified spermatozoa were frozen and stored in the refrigerator at −80°C for subsequent DNA methylation analysis.
Library construction and WGBS
DNA from semen samples was extracted by tissue/cell genome DNA isolation kit (Aidlab Biotechnologies, Beijing, China). The WGBS library was constructed according to the manufacturer’s instructions. Genomic DNA was fragmented to an average size of approximately 250 bp by sonication using a bioruptor (Diagenode, Liege, Belgium). The fragments were then followed by end‐repaired, dA addition to 3'‐end and adaptor ligation. According to the manufacturer’s instructions, ligated DNA was bisulfite converted using the EZ DNA Methylation‐Gold Kit (Zymo Research, Irvine, CA, USA). Then, 200–250 bp fragments were excised from the same lane of a 2% TAE agarose gel electrophoresis. The selected fragments were purified by using a QIAquick Gel Extraction Kit (Qiagen, Hilden, Germany) and amplified by polymerase chain reaction. At last, the qualified WGBS library was sequenced using the Illumina HiSeq 2000 (BGI, Shenzhen, China).
WGBS data procession and analysis pipeline
The reads from WGBS were aligned to hg19 reference genome converted by bisulfite using Bismark v0.19.0 software20. We used the Bismark methylation extractor script in Bismark to extract the methylated status. Only CpGs covered by at least three reads were used for downstream analysis. Differentially methylated CpGs (DMCs) were identified by methylKit v1.10.021 and RRBSeeqer22. DMRs were selected as regions that contain at least five DMCs in the 250 bp window. Promoters were defined as the regions containing 2 kb upstream and downstream of the transcription start site of the reference genome. Promoter methylation for each gene was calculated based on the mean of methylation levels from all CpGs covered in the promoter. We only selected DMRs with >10% methylation change and with a Q‐value <0.01, pval_perm_1000 ≤ 0.01 were included for further analysis.
Functional enrichment analysis of differentially methylated genes (DMGs) was realized through the OmicsBean proteomics kit (www.omicsbean.com:88). Gene Ontology (GO) analysis was carried out by using Fisher’s exact test and the GO database (http://www.ebi.ac.uk/QuickGO/) 23. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis was carried out by KEGG Mapper v2.8 software (http://www.kegg.jp/kegg/mapper.html) 24. Protein–protein interaction analysis mainly used Cytoscape Web to generate a protein–protein interaction network model25.
Results
DNA methylation mapping
To identify the characteristics of the methylome in human spermatozoa, we collected WGBS data from nine controls and eight type 2 diabetes mellitus donors. After filtering low‐quality reads, N reads and adapter sequences, each sample generated an average of 93.036 GB clean bases. Clean reads were then mapped to the human reference genome (hg19), with genome mapping rates ranging from 87.33% to 90.70% across all genomic CpG sites. Approximately 6.56% and 6.71% of all genomic C loci were methylated in the type 2 diabetes mellitus group and control group, respectively (Table 1).
Table 1.
Whole‐genome deoxyribonucleic acid bisulfite sequencing data
| Groups | Sample ID | Clean reads | Mapping rate (%) | Bisulfite conversion rate (%) | Total mC (%) |
|---|---|---|---|---|---|
| T2DM | T2DM 1 | 633333338 | 88.62 | 99.53 | 7.04 |
| T2DM 2 | 609888844 | 88.61 | 99.53 | 6.83 | |
| T2DM 3 | 633333336 | 90.60 | 99.55 | 6.77 | |
| T2DM 4 | 564932690 | 89.18 | 99.58 | 7.12 | |
| T2DM 5 | 633333336 | 90.66 | 99.47 | 6.27 | |
| T2DM 6 | 633333342 | 90.70 | 99.53 | 5.98 | |
| T2DM 7 | 633333342 | 89.84 | 99.49 | 6.17 | |
| T2DM 8 | 608786878 | 90.12 | 99.56 | 6.30 | |
| Control | Control 1 | 633333342 | 90.16 | 99.53 | 6.86 |
| Control 2 | 633333338 | 89.37 | 99.56 | 6.96 | |
| Control 3 | 633333346 | 90.49 | 99.52 | 6.76 | |
| Control 4 | 633333338 | 90.18 | 99.53 | 6.70 | |
| Control 5 | 587013322 | 89.35 | 99.55 | 6.86 | |
| Control 6 | 633333336 | 88.00 | 99.54 | 7.04 | |
| Control 7 | 627205050 | 89.31 | 99.51 | 6.02 | |
| Control 8 | 633333338 | 87.33 | 99.47 | 6.92 | |
| Control 9 | 579568080 | 88.04 | 99.52 | 6.26 |
We randomly selected 633333336‐46 reads when the sequenced data were more than 95G. mC, methylated cytosines; T2DM, type 2 diabetes mellitus.
General characteristics of methylated cytosines in the CG context, methylated cytosines in the CHG context and methylated cytosines in the CHH context
The methylated cytosine in DNA sequences can be divided into three types: CG, CHG and CHH (H = A, G or T), and these types showed a similar proportion in both groups. We also found that the whole‐genome methylated cytosine levels in the type 2 diabetes mellitus group were 89.20% CGs, 2.35% CHGs and 8.45% CHHs, whereas those in the control group were 89.97% CGs, 2.13% CHGs and 8.45% CHHs (Figure 1a). Furthermore, compared with the control group, the proportion of methylated cytosines in the CG context (mCG) decreased slightly, whereas methylated cytosines in the CHG context (mCHG) showed an upward trend in the type 2 diabetes mellitus group. By comparing the methylation levels of different types of cytosine, we found that the methylated cytosines in the CPC context (mCpG) sites were highly methylated, whereas mCHG and mCHH sites were at low levels of methylation with wide sections (Figure 1b).
Figure 1.
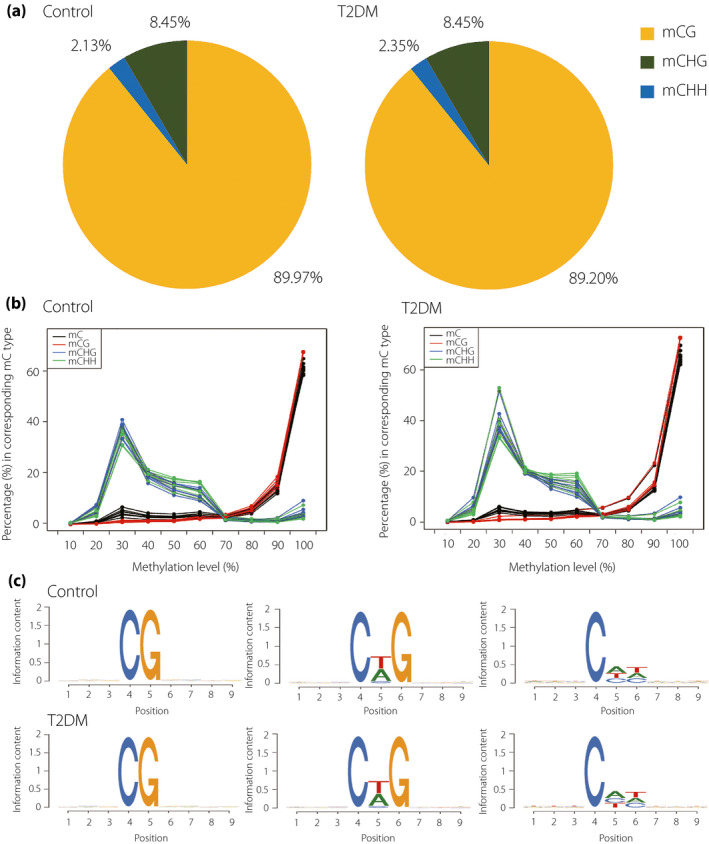
(a) The average ratio of deoxyribonucleic acid methylation types in the whole genome in type 2 diabetes mellitus (T2DM) patients and control groups (H = A, C or T), the methylated cytosines in the CG context (mCG), methylated cytosines in the CHG context (mCHG) and methylated cytosines in the CHH context (mCHH) are denoted by yellow, green and blue colors respectively. (b) Distribution of methylation level of methylated cytosines (mC) in different sequence contexts. The x‐axis is defined as the percentage of mC readings shown at the reference cytosine site, and the y‐axis represents the score of the total mC calculated in 10% of the bins. (c) Sequence preferences for methylation in CG, CHG and CHH contexts. The horizontal axis represents the base position and methylated cytosine is in the fourth position, whereas the vertical axis showed the entropy of the base.
To show the relationship between sequence context and human methylation preference, we calculated the methylation percentage of all cytosine 9‐mer sequences in which methylated cytosine located in the fourth position. We found that there was no significant difference in methylation preference in both groups. Furthermore, the sequence motifs of mCHG and mCHH have almost the same frequency in both groups (Figure 1c).
DMRs analysis
DMRs were defined as regions containing at least five DMCs (CG, CHH or CHG) within a 250bp window. A total of 9,025 DMRs were significantly differentially methylated between diabetes patients and controls (methylation difference ≥10%, Q < 0.01, p_permutation_1000 ≤ 0.01). Of these, 5,756 DMRs were hypomethylated and 3269 DMRs were hypermethylated (Table S2).
Functional annotation of DMGs
DMGs were defined as a class of genes whose promoter, intron or exon regions overlap with DMRs. Apart from the overlap, we found approximately 2,019 DMGs in all (Table S3–S5). To probe gene functions of DMGs in human sperm exposed to type 2 diabetes mellitus, GO and KEGG pathway analyses were carried out to characterize the DMGs (Table S6–S7). The GO analysis showed that DMGs were significantly enriched in multiple biological processes, such as multicellular organism development, system development, cell differentiation, neuron system development, neurogenesis, regulation of cell communication, signal transduction and so on (Figure 2a). The KEGG analysis showed that DMGs were significantly enriched in the phosphoinositide‐3‐kinase–protein kinase B signaling pathway, cyclic adenosine monophosphate signaling pathway, calcium signaling pathway, insulin signaling pathway and so on (Figure 2b). More importantly, we found that some DMGs were involved in the endocrine and digestive system, including insulin secretion, thyroid hormone synthesis and pancreatic secretion. These results suggested that these DMGs could affect insulin secretion and energy metabolism, subsequently contributing to metabolism dysregulation and the pathogenesis of type 2 diabetes mellitus in offspring.
Figure 2.
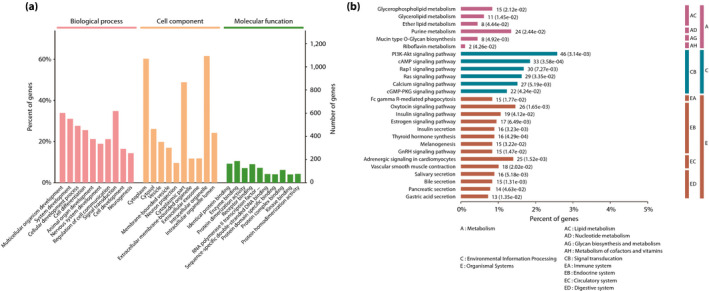
(a) Gene Ontology analysis of differentially methylated genes. A total of 10 significantly enriched terms of biological processes, cell components and molecular functions are shown, respectively. The P‐value was set to 0.05, and terms of the same category were sorted by P‐value. (b) Kyoto Encyclopedia of Genes and Genomes pathway analysis of differentially methylated genes. Kyoto Encyclopedia of Genes and Genomes pathways were divided into the following subcategories, containing metabolism, environmental information processing and organismal systems, and the P‐value was set to 0.05.
Identification of the type 2 diabetes mellitus‐related DMGs
To further explore the relationship between aberrant sperm DNA methylation and type 2 diabetes mellitus, we searched for type 2 diabetes mellitus‐related differential DNA methylation genes. First, a protein–protein interaction network model of DMGs was built by choosing type 2 diabetes mellitus‐related biological processes and pathways, such as insulin receptor signaling pathway (P‐value = 2.17E‐07), insulin signaling pathway (P‐value = 0.0412), insulin secretion (P‐value = 2.63E‐10), calcium signaling pathway (P‐value = 0.00519) and so on (Figure 3a). The genes‐matched confidence score (>400) was shown in the network. Second, we compared 2,019 DMGs and 832 top relevant score genes (>8) searched by the term “diabetes” in the GeneCards website, and found that 77 DMGs overlap with type 2 diabetes mellitus candidate genes identified by the GeneCards (Figure 3b). Finally, we identified 10 top type 2 diabetes mellitus‐related DMGs, including IRS1, PRKCE, FTO, PPARGC1A, KCNQ1, ATP10A, GHR, CREB1, PRKAR1A and HNF1B (Figure 3c).
Figure 3.
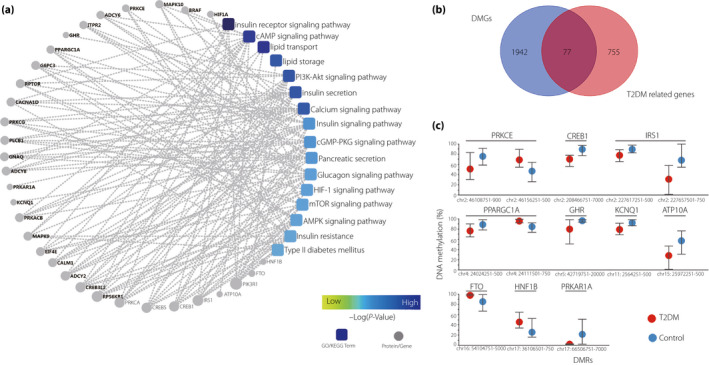
(a) A protein–protein interaction network model was generated using the Cytoscape Web application. The circle nodes represent genes/proteins, and the rectangle represents the Kyoto Encyclopedia of Genes and Genomes pathway or biological process. The pathway color changes from yellow to blue, yellow represents a smaller P‐value, and blue represents a larger P‐value. (b) Venn diagram comparing the 2019 differentially methylated genes (DMGs) and 832 top relevance score (RS >8) genes that were searched by the term “diabetes”. (c) Differentially methylated regions (DMRs) of the 10 top type 2 diabetes mellitus (T2DM)‐related DMGs are shown. The circles represent the median values of methylation levels, and the whiskers represent the minimum and maximum levels of methylation.
Discussion
Does type 2 diabetes mellitus change DNA methylation profiles in human spermatozoa? To answer this question, we carried out the present study and uniquely showed DNA methylome alterations in the spermatozoa of type 2 diabetes mellitus patients. As the main type of epigenetic regulation mechanism, DNA methylation plays a key role in regulating gene expression and genomic imprinting. Previous studies have shown that DNA methylation was closely related to the pathogenesis of type 2 diabetes mellitus26, 27, 28, and an abnormal DNA methylation spectrum from type 2 diabetes mellitus patients was found in different somatic cells, such as pancreatic islets29, 30, 31, 32, liver33, 34, skeletal muscle35, 36 and adipose tissue37, 38, 39. However, there is no report about the DNA methylation pattern in sperm from type 2 diabetes mellitus patients at the genome‐wide level. In the mouse model, Wei et al. first found that overall DNA methylation profiles of spermatozoa in male mice with prediabetes were changed, with most of DMGs overlapping with that of pancreatic islets in offspring, and increasing the susceptibility to diabetes in offspring17. To extend this previous study, we explored the epigenetic changes of DNA methylation in type 2 diabetes mellitus spermatozoa by WGBS, which allowed us to comprehensively study the DNA methylation of the whole genome at single‐nucleotide resolution40.
In the present study, we found that the proportion of methylated cytosine in the whole genome in the type 2 diabetes mellitus group was slightly lower than that in the control group, similar to the previous study41. Interestingly, the proportion of mCG decreased, whereas mCHG showed an upward trend in the type 2 diabetes mellitus group. Unlike mCHH, there is a certain symmetry in mCG and mCHG, especially as mCG methylation occurs predominantly at the symmetrical dinucleotide CpG, and as a result of that, DNA methyltransferase 1 prefers a hemimethylated substrate to maintain specific patterns of methylation in the genome42. This provided a means to explain how differentially methylated patterns could be maintained by populations of cells. Furthermore, mCGs were mainly distributed at the high methylated level, whereas the mCHGs and mCHHs were at the low and middle methylated level in both groups. In addition, sequence preferences in mCGs, mCHGs and mCHHs were obtained in the present study, and CTG and CAG were the most common sequence motifs in mCHG sites, and the sequence motif between the two groups had no obvious difference.
We also compared the whole‐genome methylation sites between type 2 diabetes mellitus and control groups. Overall, we identified 9,025 DMRs. Recent studies have shown that both promoter and gene body regions could regulate gene expression, and DNA methylation in the gene body regions was related to the regulation of alternative splicing4, 43.
The GO analysis showed that DMGs were significantly enriched in terms related to nervous system development and cell metabolism. Previous epigenetic studies have shown that genes associated with energy metabolism and neurological disorders escaped demethylation in human primordial germ cells, suggesting that certain genomic regions, especially those located between the central nervous system and metabolic regulation, are hot spots of epigenetic variation of gametes44, 45. Interestingly, DMGs were significantly enriched in the KEGG signaling pathway “insulin secretion” (P = 0.00323), which was closely associated with the occurrence and development of type 2 diabetes mellitus. Furthermore, many pathways involved in energy metabolism were also enriched, including calcium signaling pathway, adenosine monophosphate protein kinase signaling pathway, cyclic guanosine monophosphate‐dependent protein kinase signaling pathway, cyclic adenosine monophosphate signaling pathway, glycerophospholipid metabolism, lipid metabolism and glycerolipid metabolism. These results suggested that aberrant DNA methylation in spermatozoa might affect cell metabolic function and increase the risk of diabetes in offspring.
Abnormal insulin secretion and insulin resistance are the main characteristics of type 2 diabetes mellitus. In the DMGs list, we found 16 genes associated with insulin secretion and 14 genes associated with insulin resistance. To find the key genes associated with type 2 diabetes mellitus, the 10 top DMGs were identified by the Genecards database and protein–protein interaction analysis, including IRS1, PRKCE, FTO, PPARGC1A, KCNQ1, ATP10A, GHR, CREB1, PRKAR1A and HNF1B, and some of which have been validated in previous studies. The IRS1 and FTO genes were reported to have significant differences in CpG sites in human pancreatic islets from individuals with type 2 diabetes versus non‐diabetic donors46, 47. Another genome‐wide methylome study of the liver found that the activating transcription factor‐motif of the PRKCE gene showed decreased DNA methylation from obese men with type 2 diabetes mellitus compared with non‐obese controls34. Type 2 diabetes mellitus is associated with differential DNA methylation in human tissues. A total of 23 DMGs have been reported in the liver, adipose tissue and islets26. Importantly, nine DMGs were also found in the present study, including IRS1, ATP10A, PPARGC1A, KCNQ1, FTO, PRKCE, PDE7B, GRB10 and MOGAT1. Interestingly, we also found 10 paternal methylation imprinting sites, including KCNQ1, CALCR, RNU5D‐1, INPP5F, PHACTR2, RB1, GRB10, DSCAM, DLGAP2 and SNHG14. The KCNQ1 and CALCR genes are closely related to the occurrence and development of type 2 diabetes mellitus. These results suggested that sperm DNA methylation profiles associated with type 2 diabetes mellitus might be established and passed on to offspring at the early stage of development, and participate in insulin secretion or insulin resistance in offspring.
In conclusion, the present study uniquely showed that global cytosine methylation profiles were altered in spermatozoa from men with type 2 diabetes mellitus. Although we identified several candidate DMGs in type 2 diabetes mellitus men that might affect islet β‐cell function and insulin resistance, the extent to which these DMGs influence energy metabolism of their offspring is not clear. Thus, we will validate those DMGs in different tissues of the offspring from type 2 diabetes mellitus patients in further studies by flow sorting technology or single‐cell sequencing technology. We believe this work not only provides some new clues for deciphering the epigenetic mechanisms of the susceptibility to diabetes in offspring, but also inspires new ideas for the diagnosis and treatment of type 2 diabetes mellitus susceptibility.
Disclosure
The authors declare no conflict of interest.
Supporting information
Table S1 | Clinical information on sperm donors.
Table S2 | Differentially methylated regions were significantly differentially methylated between type 2 diabetes mellitus patients and controls (methylation difference ≥10%, Q < 0.01, pval_perm_1000 ≤ 0.01).
Table S3 | Differentially methylated genes associated with promoters were significantly differentially methylated between type 2 diabetes mellitus patients and controls (methylation difference ≥10%, Q < 0.01, pval_perm_1000 ≤ 0.01).
Table S4 | Differentially methylated genes associated with exons were significantly differentially methylated between type 2 diabetes mellitus patients and controls (methylation difference ≥10%, Q < 0.01, pval_perm_1000 ≤ 0.01).
Table S5 | Differentially methylated genes associated with introns were significantly differentially methylated between type 2 diabetes mellitus patients and controls (methylation difference ≥10%, Q < 0.001, pval_perm_1000 ≤ 0.01).
Table S6 | Gene Ontology analysis of differentially methylated genes.
Table S7 | Kyoto Encyclopedia of Genes and Genomes pathway analysis of differentially methylated genes.
Acknowledgments
This study was supported by Joint Funds for the Innovation of Science and Technology, Fujian province (grant number: 2016Y9007). WGBS sequencing data have been archived in a publicly accessible database at Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo) with accession number GEO: GSE138598.
J Diabetes Investig 2020; 11: 856–864
The first three authors contributed equally to this work.
Contributor Information
Deli Liu, Email: del2017@med.cornell.edu.
Gang Chen, Email: chengangfj@163.com.
References
- 1. Xu G, Liu B, Sun Y, et al Prevalence of diagnosed type 1 and type 2 diabetes among US adults in 2016 and 2017: population based study. BMJ 2018; 362: k1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Penesova A, Bunt JC, Bogardus C, et al Effect of paternal diabetes on prediabetic phenotypes in adult offspring. Diabetes Care 2010; 33: 1823–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Volkov P, Bacos K, Ofori JK, et al Whole‐genome bisulfite sequencing of human pancreatic islets reveals novel differentially methylated regions in Type 2 diabetes pathogenesis. Diabetes 2017; 66: 1074–1085. [DOI] [PubMed] [Google Scholar]
- 4. Borgel J, Guibert S, Li Y, et al Targets and dynamics of promoter DNA methylation during early mouse development. Nat Genet 2010; 42: 1093–1100. [DOI] [PubMed] [Google Scholar]
- 5. Bartke T, Vermeulen M, Xhemalce B, et al Nucleosome‐interacting proteins regulated by DNA and histone methylation. Cell 2010; 143: 470–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Levin HL, Moran JV. Dynamic interactions between transposable elements and their hosts. Nat Rev Genet 2011; 12: 615–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Guibert S, Weber M. Functions of DNA methylation and hydroxymethylation in mammalian development. Curr Top Dev Biol 2013; 104: 47–83. [DOI] [PubMed] [Google Scholar]
- 8. Wang L, Zhang J, Duan J, et al Programming and inheritance of parental DNA methylomes in mammals. Cell 2014; 157: 979–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Heard E, Martienssen RA. Transgenerational epigenetic inheritance: myths and mechanisms. Cell 2014; 157: 95–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Van DJ, Nivard MG, Willemsen G, et al Genetic and environmental influences interact with age and sex in shaping the human methylome. Nat Commun 2016; 7: 111–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Anway MD, Cupp AS, Uzumcu M, et al Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science 2005; 308: 1466–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Radford EJ, Ito M, Shi H, et al In utero effects: in utero undernourishment perturbs the adult sperm methylome and intergenerational metabolism. Science 2014; 345: 1255903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Murashov AK, Pak ES, Koury M, et al Paternal long‐term exercise programs offspring for low energy expenditure and increased risk for obesity in mice. FASEB J 2016; 30: 775–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Feinberg JI, Bakulski KM, Jaffe AE, et al Paternal sperm DNA methylation associated with early signs of autism risk in an autism‐enriched cohort. Int J Epidemiol 2015; 44: 1199–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jenkins TG, Aston KI, Pflueger C, et al Age‐associated sperm DNA methylation alterations: possible implications in offspring disease susceptibility. PLoS Genet 2014; 10: 1004458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Skaar DA, Li Y, Bernal AJ, et al The human imprintome: regulatory mechanisms, methods of ascertainment, and roles in disease susceptibility. ILAR J 2012; 53: 341–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wei Y, Yang CR, Wei YP, et al Paternally induced transgenerational inheritance of susceptibility to diabetes in mammals. Proc Natl Acad Sci USA 2014; 111: 1873–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. World Health Organization . WHO laboratory manual for the examination and processing of human semen, 5th edn WHO Press, Geneva, 2010. [Google Scholar]
- 19. Jayaraman V, Upadhya D, Narayan PK, et al Sperm processing by swim‐up and density gradient effective in elimination of sperm with DNA damage. J Assist Reprod Genet 2012; 29: 557–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite‐Seq applications. Bioinformatics 2011; 27: 1571–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Akalin A, Kormaksson M, Li S, et al methylKit: a comprehensive R package for the analysis of genome‐wide DNA methylation profiles. Genome Biol 2012; 13: R87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pan H, Jiang Y, Boi M, et al Epigenomic evolution in diffuse large B‐cell lymphomas. Nat Commun 2015; 6: 6921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Binns D, Dimmer E, Huntley R, et al QuickGO: a web‐based tool for gene ontology searching. Bioinformatics 2009; 25: 3045–3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kanehisa M, Sato Y, Kawashima M, et al KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res 2016; 44: 457–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Szklarczyk D, Franceschini A, Wyder S, et al STRING v10: protein‐protein interaction networks, integrated over the tree of life. Nucleic Acids Res 2015; 43: 447–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ling C, Rönn T. Epigenetics in human obesity and type 2 diabetes. Cell Metab 2019; 29: 1028–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dabelea D, Crume T. Maternal environment and the transgenerational cycle of obesity and diabetes. Diabetes 2011; 60: 1849–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Simmons R. Epigenetics and maternal nutrition: nature v. nurture. Proc Nutr Soc 2011; 70: 73–81. [DOI] [PubMed] [Google Scholar]
- 29. Zhu Z, Chen X, Xiao Y, et al Gestational diabetes mellitus alters DNA methylation profiles in pancreas of the offspring mice. J. Diabetes Complicat 2019; 33: 15–22. [DOI] [PubMed] [Google Scholar]
- 30. Hall E, Dekker Nitert M, Volkov P, et al The effects of high glucose exposure on global gene expression and DNA methylation in human pancreatic islets. Mol. Cell. Endocrinol 2018; 472: 57–67. [DOI] [PubMed] [Google Scholar]
- 31. Yang BT, Dayeh TA, Kirkpatrick CL, et al Insulin promoter DNA methylation correlates negatively with insulin gene expression and positively with HbA(1c) levels in human pancreatic islets. Diabetologia 2011; 54: 360–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Volkmar M, Dedeurwaerder S, Cunha DA, et al DNA methylation profiling identifies epigenetic dysregulation in pancreatic islets from Type 2 diabetic patients. EMBO J 2012; 31: 1405–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bysani M, Perfilyev A, de Mello VD, et al Epigenetic alterations in blood mirror age‐associated DNA methylation and gene expression changes in human liver. Epigenomics 2017; 9: 105–122. [DOI] [PubMed] [Google Scholar]
- 34. Kirchner H, Sinha I, Gao H, et al Altered DNA methylation of glycolytic and lipogenic genes in liver from obese and type 2 diabetic patients. Mol Metab 2016; 5: 171–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nitert MD, Dayeh T, Volkov P, et al Impact of an exercise intervention on DNA methylation in skeletal muscle from first‐degree relatives of patients with Type 2 diabetes. Diabetes 2012; 61: 3322–3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Barrès R, Yan J, Egan B, et al Acute exercise remodels promoter methylation in human skeletal muscle. Cell Metab 2012; 15: 405–411. [DOI] [PubMed] [Google Scholar]
- 37. Nilsson E, Jansson PA, Perfilyev A, et al Altered DNA methylation and differential expression of genes influencing metabolism and inflammation in adipose tissue from subjects with type 2 diabetes. Diabetes 2014; 63: 2962–2976. [DOI] [PubMed] [Google Scholar]
- 38. Grundberg E, Meduri E, Sandling JK, et al Global analysis of DNA methylation variation in adipose tissue from twins reveals links to disease‐associated variants in distal regulatory elements. Am. J. Hum. Genet 2013; 93: 876–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ziller MJ, Hansen KD, Meissner A, et al Coverage recommendations for methylation analysis by whole‐genome bisulfite sequencing. Nat. Methods 2015; 12: 230–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dayeh T, Volkov P, Salo S, et al Genome‐wide DNA methylation analysis of human pancreatic islets from Type 2 diabetic and non‐diabetic donors identifies candidate genes that influence insulin secretion. PLoS Genet 2014; 10: e1004160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Feng S, Jacobsen SE, Reik W. Epigenetic reprogramming in plant and animal development. Science 2010; 330: 622–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Aran D, Toperoff G, Rosenberg M, et al Replication timing related and gene body‐specific methylation of active human genes. Hum Mol Genet 2011; 20: 670–680. [DOI] [PubMed] [Google Scholar]
- 43. Tang WW, Dietmann S, Irie N, et al A unique gene regulatory network resets the human germline epigenome for development. Cell 2015; 161: 1453–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Donkin I, Versteyhe S, Ingerslev LR. Obesity and bariatric surgery drive epigenetic variation of spermatozoa in humans. Cell Metab 2016; 23: 369–378. [DOI] [PubMed] [Google Scholar]
- 45. Dayeh T, Volkov P, Salo S, et al Genome‐wide DNA methylation analysis of human pancreatic islets from type 2 diabetic and nondiabetic donors identifies candidate genes that influence insulin secretion. PLoS Genet. 2014; 10: e1004160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hall E, Dayeh T, Kirkpatrick CL, et al DNA methylation of the glucagon‐like peptide 1 receptor (GLP1R) in human pancreatic islets. BMC Med Genet 2013; 14: 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kirchner H, Sinha I, Gao H. Altered DNA methylation of glycolytic and lipogenic genes in liver from obese and type 2 diabetic patients. Mol Metab 2016; 5: 171–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 | Clinical information on sperm donors.
Table S2 | Differentially methylated regions were significantly differentially methylated between type 2 diabetes mellitus patients and controls (methylation difference ≥10%, Q < 0.01, pval_perm_1000 ≤ 0.01).
Table S3 | Differentially methylated genes associated with promoters were significantly differentially methylated between type 2 diabetes mellitus patients and controls (methylation difference ≥10%, Q < 0.01, pval_perm_1000 ≤ 0.01).
Table S4 | Differentially methylated genes associated with exons were significantly differentially methylated between type 2 diabetes mellitus patients and controls (methylation difference ≥10%, Q < 0.01, pval_perm_1000 ≤ 0.01).
Table S5 | Differentially methylated genes associated with introns were significantly differentially methylated between type 2 diabetes mellitus patients and controls (methylation difference ≥10%, Q < 0.001, pval_perm_1000 ≤ 0.01).
Table S6 | Gene Ontology analysis of differentially methylated genes.
Table S7 | Kyoto Encyclopedia of Genes and Genomes pathway analysis of differentially methylated genes.