

Abstract
Aims/Introduction
The relationship of chromogranin A (CgA) levels above the normal range with various outcomes, such as glycated hemoglobin levels, enterochromaffin‐like cell hyperplasia and autoimmune gastritis, was investigated in type 1 diabetes patients with special regard to the progression of comorbidities.
Materials and Methods
A cohort study on 153 type 1 diabetes patients was carried out with a prospective branch on clinical and laboratory data, and a retrospective branch on histological data obtained by gastroscopy.
Results
Patients with CgA levels above the upper limit of the normal range (n = 28) had significantly higher glycated hemoglobin levels (P = 0.0160) than those with CgA in the normal range (n = 125). The correlation between CgA and glycated hemoglobin was significant (P < 0.0001), but weak (R = +0.32). A slight, but steady elevation (P = 0.0410) in CgA level was observed to co‐vary with the duration of type 1 diabetes. Enterochromaffin‐like cell hyperplasia and autoimmune gastritis was significantly more frequent (P = 0.0087 for both) in the high CgA group. Detailed analyses on gastric tissue samples confirmed a progression of enterochromaffin‐like cell hyperplasia (P = 0.0192) accompanied by CgA elevation (P = 0.0316).
Conclusions
The early detection and follow up of the later progression of enterochromaffin‐like cell hyperplasia and autoimmune gastritis into gastric neuroendocrine tumors, which have ~100‐fold greater incidence in type 1 diabetes patients, can be achieved by assessment of CgA levels. Therefore, the use of CgA could be considered as a novel auxiliary biomarker in the care of these type 1 diabetes complications.
Keywords: Autoimmune gastritis, Chromogranin A, Enterochromaffin‐like cell hyperplasia
A slight, but steady elevation in chromogranin A (CgA) level was observed to co‐vary with the duration of type 1 diabetes. Approximately 20% of the patients had elevated serum CgA levels, which was associated with higher glycated hemoglobin levels. Enterochromaffin‐like cell hyperplasia and autoimmune gastritis was significantly more frequent in the high CgA group. Detailed analyses on gastric tissue samples confirmed a progression of enterochromaffin‐like cell hyperplasia accompanied by CgA elevation. Therefore, the use of CgA could be considered as a novel auxiliary biomarker in the care of these type 1 diabetes complications.

Introduction
Gastric neuroendocrine tumors (NET; or gastric carcinoid) are very rare (1:200,000) in the general population1, but in patients with type 1 diabetes, approximately 100‐fold more cases are registered2, 3, 4. Gastric NETs might develop from the hyperplastic proliferation of enterochromaffin‐like (ECL) cells5, 6 or from autoimmune gastritis (AIG)7, 8. AIG is present in 5–10% of type 1 diabetes patients9, 10 compared with approximately 2% of the non‐type 1 diabetes population10, and gastric NETs develop in approximately 4–9% of type 1 diabetes patients with AIG7. Furthermore, ECL hyperplasia, AIG and gastric NETs are observed more often in women compared with men4, 8, 11, 12.
Chromogranin A (CgA) is an acidic and hydrophilic family member of the granin glycoproteins, and primarily produced by neurons, endocrine and neuroendocrine cells, such as the intrinsic neurons of the enteric nervous system, the chromaffin cells of the adrenal medulla and the ECL cells of the stomach13. It is synthesized from 439 amino acids (molecular weight 48kDa) in humans14. CgA takes part in the peptide and protein sorting in the Golgi apparatus through its high‐capacity and low‐affinity calcium‐binding function15, 16, 17. Furthermore, various biologically active peptides are generated from CgA by the cleavage of different endoproteases during and after secretion18. Several of these cleavage products have been found in human tissues: pancreastatin, WE‐14, catestatin, cateslytin, vasostatin‐I and ‐II19. Serum CgA is one of the most prominently used markers to confirm the diagnosis, and to monitor the effectiveness of the treatment in patients with various neuroendocrine tumors20. Blood CgA level might be altered in some other medical conditions20, 21, 22, although these do not lessen its impact as a marker generally.
Recent human and animal model studies have suggested that both the CgA protein by itself and some of its cleavage products, most importantly pancreastatin and WE‐14, regulate carbohydrate metabolism and contribute to diabetes13, 23. Pancreastatin suppresses insulin signaling and inhibits insulin release24, and it has an elevated serum level in type 2 and gestational diabetes patients24, 25. WE‐14 functions as an autoantigen for the diabetogenic, pancreatic‐cell‐destroying CD4+ T‐cell populations in both non‐obese diabetic mice and patients with type 1 diabetes26, 27, 28. Some relationships between serum CgA level and type 1 diabetes are also described. High CgA levels and an increased frequency of ECL hyperplasia (n = 7) have been reported in type 1 diabetes patients with AIG and parietal cell antibody (PCA) positivity (n = 17)6; however, CgA has not been assessed independently from the PCA status, or correlated with ECL progression. Nevertheless, CgA has been identified as the only independent risk factor for ECL hyperplasia when PCA and gastrin were tested in type 1 diabetes patients6.
An observational cohort study was carried out in type 1 diabetes patients to search for relationships between serum CgA levels above the normal range and the various laboratory and clinical outcomes, such as AIG and ECL hyperplasia. Clinical and laboratory measurement data of type 1 diabetes patients were collected in a prospective manner, whereas tissue samples of the stomach were examined for AIG and ECL hyperplasia in a retrospective way. In addition to the latter, frozen samples from a tissue bank were also investigated to search for evidence for AIG and ECL hyperplasia progression.
Methods
Patients and study design
A total of 153 Hungarian, c‐peptide confirmed, islet‐cell‐ and/or glutamic acid decarboxylase antibody‐positive29 type 1 diabetes patients with European ancestry, who attended the Metabolic Clinic of the 2nd Department of Internal Medicine, Semmelweis University, Budapest, Hungary, from 2010 to 2018, were enrolled for the study. Written informed consent was collected from all study participants. Exclusion criteria included the use of acid reducer medications, any kidney diseases, known tumors, ulcerative colitis, Crohn’s disease, systemic rheumatoid arthritis or any conditions changing the serum concentrations of CgA21, 22. Patients with autoimmune diseases associated with type 1 diabetes, including autoimmune thyroid disease, celiac disease, pernicious anemia, alopecia areata, vitiligo, Addison’s disease and Sjögren’s syndrome were not excluded. Glycemic control of patients was determined as described in the 2019 guidelines of the American Diabetes Association30.
An observational cohort study was carried out, where cohorts were defined by the serum CgA level. On average, the patients (n = 153) had been suffering from diabetes at the time of baseline measurements for 13.47 ± 9.77 years. Follow‐up measurements (n = 94) were carried out 18.88 ± 9.80 years from the time of diagnosis. A subpopulation of patients (n = 34) who agreed to undergo gastroscopy all had the disease at the time of the procedure for nearly the same duration as the follow‐up patients (20.53 ± 7.60 years).
Clinical data and measurements
Anamnestic, bodyweight and height data were collected, and blood samples were drawn after an 8‐h fasting period. Complete blood count, glycated hemoglobin (HbA1C), total‐, high‐ and low‐density lipoprotein cholesterols, triglycerides, creatinine, high‐sensitivity C‐reactive protein, and thyroid‐stimulating hormone were measured at the Central Laboratory of Semmelweis University. The estimated glomerular filtration rate was calculated using the Chronic Kidney Disease‐Epidemiology Collaboration equations31.
Serum CgA levels were measured at the Central Endocrine and Genetic Laboratory of Semmelweis University using the CGA‐RIACT radioimmunoassay kit (CISbio International, Gif‐sur‐Yvette, France) on a RIA‐mat 280 (Laborexpert Kft., Diosd, Hungary) automated analyzer. The normal range of serum CgA (19.4–98.1 ng/mL) was obtained from the 95% confidence interval (CI) of 162 healthy individuals, which has been confirmed in previous publications32, 33.
Gastroscopy and chromogranin A‐specific immunohistochemical staining
Gastroscopy was carried out at the Gastroenterology Clinic of the 2nd Department of Internal Medicine, Semmelweis University. A total of 19 patients with serum CgA levels above the normal range, and 15 patients with normal CgA levels, whose indications were not related to type 1 diabetes, underwent the procedure. Biopsy samples for routine histological examinations and CgA‐specific immunohistochemical staining were collected from the fundus, oral and aboral parts of the antrum, and pylorus.
Biopsy samples were processed at the 1st Department of Pathology and Experimental Cancer Research, Semmelweis University. Routine histopathological procedures included hematoxylin–eosin staining34. The presence of Helicobacter pylori was investigated by modified Giemsa staining35. The updated Sydney system was used to evaluate activity, atrophy and inflammation36. CgA‐specific immunohistochemical staining was carried out with the Clone DAK‐A3 Monoclonal Mouse Anti‐Human Chromogranin A immunohistochemistry kit (M0869, dilution 1:1000; Dako Denmark A/S, Glostrup, Denmark). Immune reactions were carried out in formalin‐fixed, paraffin‐embedded tissue with 3,3'‐diaminobenzidine as chromogen with the Leica Biosystems BondTM Polymer Refine Detection kit on a Leica BondTM automated immunostainer (Leica Biosystems Newcastle Ltd., Newcastle Upon Tyne, UK). Endogenous neuroendocrine cells of the gastric mucosa served as positive controls.
Histopathological classification of the CgA‐positive, ECL cell growths were identified based on descriptions by Solcia et al.5 Briefly, cells are located randomly and individually in the case of diffuse ECL hyperplasia, cell clusters form a chain of five or more cells in linear ECL hyperplasia, while cells are orderly arranged, forming knots between 100 and 150 µm in size in micronodular ECL hyperplasia.
Statistical analysis
Statistical analyses were carried out within the R for Windows version 3.5.0 environment (R Foundation for Statistical Computing, Vienna, Austria)37. Two sample t‐tests and permutation‐based paired t‐tests were used for comparisons between groups and paired data, respectively38, 39. Fisher’s exact test was used to compare count data. Pearson’s correlation, and linear and logistic regression models were used for testing the in‐between effects of various factors. Receiver operating characteristic analysis was carried out to test the sensitivity and specificity of CgA. P < 0.05 was considered as statistically significant; P‐values were corrected with the false discovery rate method for the multiple comparisons problem.
A random intercept linear mixed effects model was used to determine the changes in CgA with the progression of type 1 diabetes. R package nlme 40 was used to run the model. The dependent variable in the final model was the natural logarithmic value of serum CgA level, the fixed effect was the duration of type 1 diabetes and patient IDs were used as the random effect.
Ethics approval
This study was carried out in concordance with the World Medical Association Declaration of Helsinki, and approved by the Regional and Institutional Committee of Science and Research Ethics of Semmelweis University. Handling of patient data was in accordance with the General Data Protection Regulation issued by the European Union.
Results
Baseline measurements
A CgA level above the upper limit of the normal range (>98.1 ng/mL) was observed in 18.30% of patients with type 1 diabetes. Patients were divided into two cohorts based on their baseline serum CgA levels. Study participants having levels within the normal range (19.4–98.1 ng/mL) were assigned to the Normal CgA group (n = 125), whereas patients with levels higher than the normal range were assigned to the High CgA group (n = 28).
To test whether other factors, such as age, sex, body mass index and/or the duration of type 1 diabetes, affects the cohorts outlined above, univariate and multivariate logistic regression models were used. None of these factors showed any effect or connection to the grouping variable. Study participants consisted of homogeneous ethnicity (only Hungarians with European ancestry); therefore, no analysis was carried out on the effects of ethnicity. No difference was found in the overall statistical analysis stratified by latent autoimmune diabetes of adults. Therefore, latent autoimmune diabetes of adults and conventional type 1 diabetes patients will not be subgrouped hereinafter.
HbA1C levels in the High CgA group were significantly higher compared with those in the Normal CgA group (P = 0.160; Table 1). A significant positive correlation was found between CgA and HbA1C (Pearson’s R = +0.32, P < 0.0001). However, as determined by a regression model, the increase in HbA1C only slightly explained the increase in CgA (adjusted R 2 = 0.0992, P < 0.0001). Based on the HbA1C values, 69.6% of the patients had poor glycemic control (HbA1C ≥7.0%, which equals to ≥53 mmol/mol) in the Normal CgA group, whereas this figure rose to 96.4% of patients in the High CgA group (P = 0.0326). The occurrence of type 1 diabetes‐specific antibodies, latent autoimmune diabetes of adults and the commonly associated comorbidities of type 1 diabetes, such as hypertension, thyroid diseases (both autoimmune and non‐autoimmune forms), celiac disease, alopecia areata, pernicious anemia, vitiligo, Addison’s disease and Sjögren’s syndrome, did not differ between the two groups (Table 1).
Table 1.
Anamnestic and laboratory measurement data
| Normal CgA (n = 125) | High CgA (n = 28) | P‐value | |
|---|---|---|---|
| Age (years) | 35.37 ± 12.27 | 36.46 ± 9.88 | 0.7486 |
| Duration of diabetes (years) | 12.89 ± 10.01 | 16.07 ± 8.29 | 0.2432 |
| C‐peptide (ng/mL) | 0.41 ± 0.76 | 0.33 ± 1.20 | 0.8554 |
| Islet cell antibody positivity | 62 | 10 | 1.0000 |
| Glutamic acid decarboxylase antibody positivity | 71 | 11 | 1.0000 |
| Chromogranin A (ng/mL) | 49.80 ± 19.58 | 291.73 ± 248.32 | 0.0006 |
| HbA1C (%) | 8.29 ± 1.96 | 9.62 ± 2.02 | 0.0160 |
| HbA1C (mmol/mol) | 67 ± 21.4 | 82 ± 22.1 | 0.0160 |
| Glycemic control of patients | |||
| Well controlled diabetes | 38 (30.4%) | 1 (3.6 %) | 0.0326 |
| Poorly controlled diabetes | 87 (69.6%) | 27 (94.4 %) | |
| White blood cell count (G/L) | 7.41 ± 2.16 | 7.94 ± 2.03 | 0.3828 |
| Red blood cell count (T/L) | 4.97 ± 0.51 | 4.64 ± 0.55 | 0.0356 |
| Hemoglobin (g/L) | 144.70 ± 15.04 | 131.71 ± 19.37 | 0.0160 |
| Hematocrit (L/L) | 0.429 ± 0.043 | 0.398 ± 0.045 | 0.0160 |
| Mean corpuscular volume (fL) | 86.30 ± 3.34 | 85.44 ± 6.76 | 0.7075 |
| Mean corpuscular hemoglobin (pg) | 29.12 ± 1.39 | 28.41 ± 3.00 | 0.3828 |
| Mean corpuscular hemoglobin concentration (g/L) | 337.54 ± 11.80 | 332.19 ± 16.75 | 0.2978 |
| Red blood cell distribution width (%) | 13.01 ± 0.80 | 13.81 ± 1.60 | 0.0620 |
| Platelets (G/L) | 273.74 ± 65.88 | 309.59 ± 69.25 | 0.0682 |
| Creatinine (µmol/L) | 72.71 ± 13.06 | 79.14 ± 26.35 | 0.3828 |
| Estimated glomerular filtration rate (mL/min/1.73 m2) | 103.94 ± 16.98 | 97.99 ± 25.07 | 0.3828 |
| Total cholesterol (mmol/L) | 5.06 ± 0.97 | 5.68 ± 1.70 | 0.2432 |
| High‐density lipoprotein cholesterol (mmol/L) | 1.66 ± 0.51 | 1.65 ± 0.48 | 0.9552 |
| Low‐density lipoprotein cholesterol (mmol/L) | 2.88 ± 0.72 | 3.38 ± 1.51 | 0.2972 |
| Triglyceride (mmol/L) | 1.18 ± 1.00 | 1.49 ± 1.24 | 0.3828 |
| High sensitivity C‐reactive protein (mg/L) | 1.80 ± 2.20 | 2.43 ± 2.27 | 0.3828 |
| Thyroid‐stimulating hormone (mIU/L) | 2.09 ± 2.35 | 1.85 ± 1.87 | 0.7369 |
| Body mass index (kg/m2) | 24.97 ± 4.95 | 24.51 ± 4.84 | 0.7924 |
| Sex (female/male) | 66/59 (52.8%/47.2%) | 16/12 (57.1%/42.9%) | 0.9907 |
| Latent autoimmune diabetes of adults | 29 (23.2%) | 4 (14.3%) | 0.7237 |
| Hypertension | 40 (32%) | 12 (42.9%) | 0.6380 |
| Autoimmune disease | 15 (12%) | 8 (28.6%) | 0.1231 |
| Vitiligo | 4 (3.2%) | 2 (7.1%) | 0.6380 |
| Alopecia areata | 5 (4%) | 0 (0%) | 0.8342 |
| Pernicious anemia | 2 (1.6%) | 4 (14.3%) | 0.1009 |
| Celiac disease | 2 (1.6%) | 1 (3.6%) | 0.7237 |
| Addison’s disease | 1 (0.8%) | 0 (0%) | 1.0000 |
| Sjögren’s syndrome | 0 (0%) | 1 (3.6%) | 0.4697 |
| Psoriasis vulgaris | 1 (0.8%) | 0 (0%) | 1.0000 |
| Thyroid disease | 39 (31.2%) | 10 (35.7%) | 0.8342 |
| Hashimoto’s thyroiditis | 31 (24.8%) | 5 (17.9%) | 0.8342 |
| Hypothyroidism | 7 (5.6%) | 3 (10.7%) | 0.7237 |
| Hyperthyroidism | 1 (0.8%) | 0 (0%) | 1.0000 |
| Thyroid nodules | 0 (0%) | 2 (7.1%) | 0.1231 |
Unit of non‐laboratory data is the number of observations. CgA, chromogranin A; HbA1C , glycated hemoglobin. P‐values of statistically different results between the two cohorts are indicated with bold text.
Changes in serum chromogranin A levels with the duration of type 1 diabetes
To determine whether the CgA level changes with respect to the duration of type 1 diabetes, we chose two prospective approaches. First, the patients were recalled for a follow‐up measurement of CgA. We were able to reach 94 patients from the original 153 (call‐back rate 61.4%). The mean duration between the baseline and follow‐up measurements was 4.73 ± 2.27 years. The elevation of serum CgA levels in the Normal CgA group was significant (n = 77, P = 0.0277), whereas no significant change was observed in the High CgA group (n = 17, P = 0.2202), or in both groups combined (P = 0.0571; Table 2). The CgA levels of five (6.5%) Normal CgA group patients were above the normal range at the follow‐up measurement.
Table 2.
Baseline and follow‐up chromogranin A measurements of the study participants
| Baseline CgA (ng/mL) | Follow‐up CgA (ng/mL) | P‐value | |
|---|---|---|---|
| Normal CgA group (n = 77) | 47.53 ± 19.51 (35.10–56.40) | 53.25 ± 27.85 (32.00–63.20) | 0.0277 |
| High CgA group (n = 17) | 365.65 ± 316.83 (151.20–410.40) | 466.16 ± 625.03 (168.20–572.10) | 0.2202 |
| Groups combined (n = 94) | 105.05 ± 180.93 (36.60–81.55) | 127.93 ± 305.57 (36.33–95.08) | 0.0571 |
Duration between the two measurements was 4.73 ± 2.27 years. Mean ± Standard Deviation (Interquartile range). CgA, chromogranin A. P‐values of statistically different results between the two cohorts are indicated with bold text.
Second, a random intercept linear mixed effect model was constructed, where not only the paired, but all the baseline and further repeated measurements from all of the 153 study participants were used. A total of 364 CgA measurements were collected. Based on the estimations given by the statistical model, a significant 0.40–1.82% average increase in the serum CgA concentrations can be expected annually (P = 0.0410).
Gastroscopy results
Gastroscopy was carried out in 15 patients of the Normal CgA group, and 19 patients of the High CgA group. Frequencies of gastroesophageal reflux disease, Helicobacter pylori positivity and/or chronic gastritis were statistically not different between these groups (Table 3). The serum gastrin level was also investigated in patients with gastroscopy, and gastrin levels in the High CgA group were significantly higher (48.12 ± 50.97 pg/mL vs 351.05 ± 252.66 pg/mL, P < 0.0001). A significant positive correlation was found between CgA and gastrin (Pearson’s R = +0.59, P = 0.0009), and the increase in gastrin moderately explained the increase in CgA (adjusted R 2 = 0.3286, P = 0.0004).
Table 3.
Histological and clinical results of gastroscopies carried out
| Normal CgA (n = 15) | High CgA (n = 19) | P‐value | |
|---|---|---|---|
| Chromogranin A‐specific immunohistochemistry | |||
| Negative | 8 (53%) | 3 (15.8%) | 0.0087 |
| Diffuse ECL hyperplasia | 7 (47%) | 4 (26.3%) | |
| Linear ECL hyperplasia | 0 (0%) | 7 (36.8%) | |
| Micronodular ECL hyperplasia | 0 (0%) | 4 (21%) | |
| Autoimmune gastritis | 0 (0%) | 9 (47.4%) | 0.0087 |
| Chronic gastritis | 9 (60%) | 16 (84.2%) | 0.1949 |
| Helicobacter pylori positivity | 1 (6.7%) | 2 (10.5%) | 1.0000 |
| Gastroesophageal reflux disease | 10 (66.7%) | 12 (63.2%) | 1.0000 |
Unit of data is the number of observations. P‐values were the same after false discovery rate correction of multiple comparisons. CgA, chromogranin A; ECL, enterochromaffin‐like. P‐values of statistically different results between the two cohorts are indicated with bold text.
ECL hyperplasia positivity (ECL+) occurs significantly more often in female patients (85% of 20 women vs 42.9% of 14 men, P = 0.0383) in accordance with previous findings12. ECL+ was present in 16 of the 19 High CgA patients (odds ratio 5.74, 95% CI 1.0001–44.1645) compared with seven of the 15 Normal CgA patients (P = 0.0087). The specificity and sensitivity of CgA were analyzed to predict ECL+; CgA had 90.9% specificity and 45.5% sensitivity, with an optimal CgA cut‐off point of 183.35 ng/mL (Figure 1a). The distribution of hyperplasia types was significantly different (P = 0.0087, P‐values were the same after FDR correction of multiple comparisons) between the two groups. Diffuse ECL hyperplasia occurred in both the Normal CgA group and the High CgA group, but more advanced forms of hyperplasia were found only in the patients of the High CgA group (Table 3).
Figure 1.
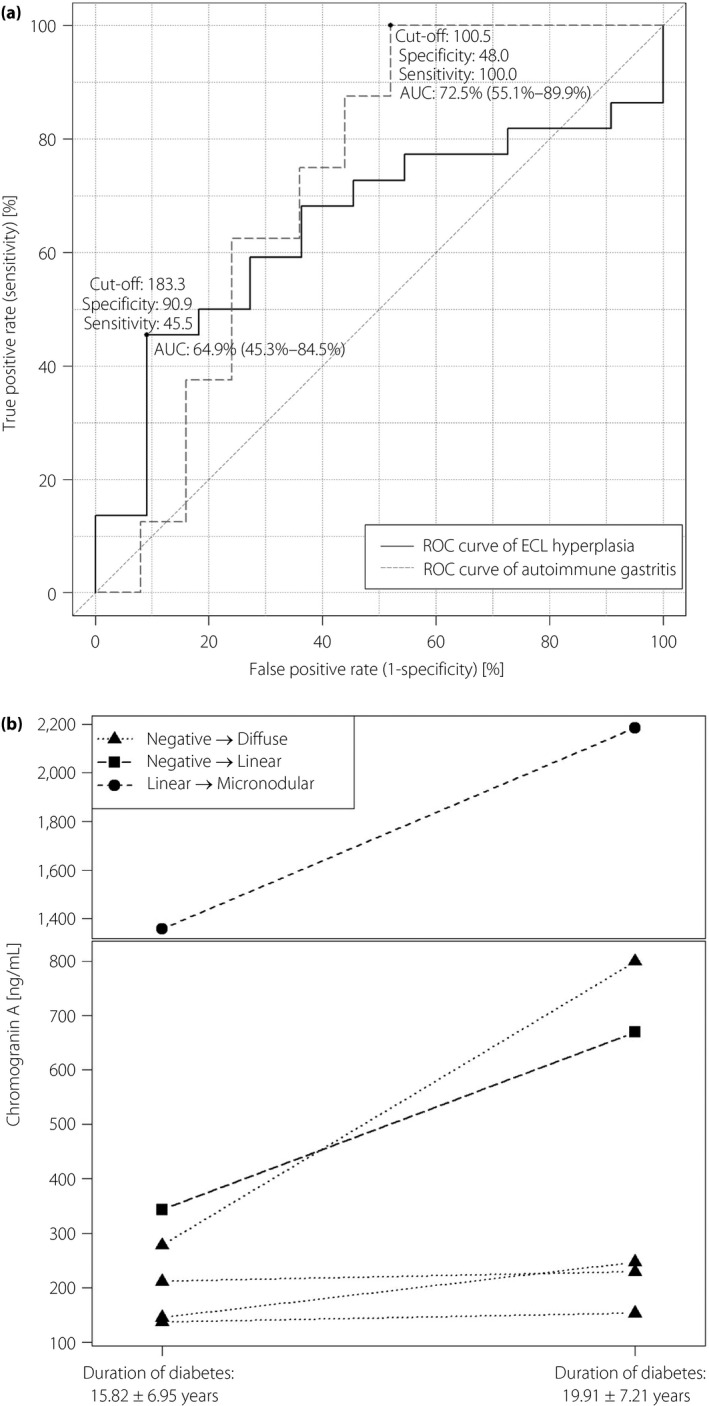
(a) Receiver operating characteristic (ROC) curve of chromogranin A to detect enterochromaffin‐like neuroendocrine hyperplasia and autoimmune gastritis (n = 34). The two curves do not differ significantly (P = 0.5615). (b) Changes in serum chromogranin A levels of the patients who had progression in their neuroendocrine enterochromaffin‐like (ECL) hyperplasia types between the two gastroscopies (P = 0.0316). AUC, area under the curve.
AIG was also more frequent in women (P = 0.0023), as described before11. Histologically‐confirmed AIG was significantly more frequent (P = 0.0087) in the High CgA group (9 AIG cases vs 0 cases, odds ratio ∞ due to division by zero, CI 95% 2.23–∞). To detect AIG, CgA has a 48% specificity and 100% sensitivity, with an optimal CgA cut‐off point of 100.50 ng/mL (Figure 1a). Red blood cell counts (P = 0.0336), hemoglobin (P = 0.0167) and hematocrit (P = 0.0167) levels in the High CgA group were significantly lower (Table 1), in accordance with the described consequences of AIG7, 41.
Progression of ECL hyperplasia in patients with continuously high serum CgA levels
The examination of archive biopsy samples could be carried out in 11 of the 19 patients in the High CgA group. Archived biopsy samples were collected 3.94 ± 1.96 years before the gastroscopy. A significantly (P = 0.0192) higher number of more advanced hyperplasia stages were observed at the recent gastroscopic examination compared with the archive samples (Table 4). Just four of the patients had no changes at all, and a progression was observed in seven patients.
Table 4.
Distributional changes of neuroendocrine enterochromaffin‐like hyperplasia types and serum chromogranin A levels at the two gastroscopy examinations
| High CgA (n = 11) | Archived gastroscopy samples | Recent gastroscopy | P‐value |
|---|---|---|---|
| Chromogranin A‐specific immunohistochemistry | |||
| Negative | 8 (72.7%) | 2 (18.2%) | 0.0192 |
| Diffuse ECL hyperplasia | 0 (0%) | 5 (45.4%) | |
| Linear ECL hyperplasia | 2 (18.2%) | 2 (18.2%) | |
| Micronodular ECL hyperplasia | 1 (9.1%) | 2 (18.2%) | |
| Serum chromogranin A (ng/mL) | |||
| Patients with histological change (n = 6)† | 411.75 ± 469.88 (161.00–327.05) | 713.53 ± 766.97 (233.05–767.15) | 0.0316 |
| Patients without histological change (n = 4) | 197.53 ± 131.31 (120.83–219.9) | 164.68 ± 67.82 (128.43–178.85) | 0.3752 |
Unit of enterochromaffin‐like (ECL) hyperplasia data is the number of observations. Mean ± standard deviation (interquartile range). †One patient from the group had no serum chromogranin A (CgA) measurement around the time of archive gastroscopy results. P‐values of statistically different results between the two cohorts are indicated with bold text. [Correction added on 18 May 2020, after first online publication: A missing number of observation has been inserted.]
The serum CgA levels of patients with and without progression were compared with a one‐sided paired t‐test. Serum CgA levels were significantly higher (P = 0.0316) at recent gastroscopy in patients with ECL hyperplasia progression (Figure 1b), whereas they did not differ in patients without histological progressions (P = 0.3752).
Discussion
Newly‐diagnosed type 1 diabetes patients have shown elevated WE‐14‐induced CD4+ 26, CgA10‐19‐ and CgA43‐52‐fragment‐related, CD8+ β‐cell‐specific diabetogenic T‐cell antigenicity27, suggesting that these CgA cleavage products contribute to the pathogenesis of type 1 diabetes13, 23, 26, 27, 28, 42, 43. However, there is little information about the relationship between CgA and the latter stage of type 1 diabetes long after the onset of the disease. The present study showed that approximately 20% of the type 1 diabetes patients had CgA levels above the upper limit of the normal range. There are no pancreatic β‐cells in the patients long after the first onset44; therefore, our observation is not connected to the CgA product‐sensitive, β cell‐specific T cells in all probability.
ECL hyperplasia and AIG occurred more frequently in our type 1 diabetes patients with high CgA levels, similar to the finding that ECL hyperplasia and AIG more often occur in type 1 diabetes patients with PCA positivity6. Approximately 50% of our patients with high CgA levels and ECL+ also had AIG, in accordance with the known connection between ECL+ and AIG6, 7, 11, 45. Significantly lower hemoglobin and hematocrit values, and red blood cell counts, were detected in the High CgA group, which can be explained by the higher presence of AIG within the group, accompanied by various forms of anemia7, 41. In addition, ECL hyperplasia and AIG occur more often in women8, 11, 12, which we also observed.
The prospective branch of the current study showed a slow, but steady, rise in CgA level over time, and the patients with high CgA levels had more progressed ECL hyperplasia stages, indicating the progression of ECL hyperplasia. Although the low number of archived gastroscopy samples did not enable us to carry out a full prospective investigation on tissue samples, the comparison of biopsy samples from recent gastroscopy to archived biopsy samples showed that the ECL hyperplasia stages in type 1 diabetes patients of the High CgA group progressed, which was also accompanied by a rise in serum CgA levels. CgA levels did not change significantly in the patients of the High CgA group who had a lack of ECL hyperplasia progression. The observed connection between the serum and histological results strongly suggest that the histological changes in the gastric mucosa cause the elevation of CgA levels in type 1 diabetes patients. This is also supported by the higher serum gastrin levels in the High CgA group, which indicates the gastric origin of CgA46, although CgA is an independent risk factor from PCA and gastrin for ECL hyperplasia in type 1 diabetes patients6. Unfortunately, the present findings do not allow us to determine whether ECL hyperplasia or AIG develops first, and what is the exact pathomechanism. As AIG was only observed together with linear or micronodular ECL hyperplasia stages and high CgA levels, and ECL hyperplasia was also present in patients within normal CgA levels and without AIG, it might be considered as a sign that ECL hyperplasia is the one that develops first. Nevertheless, ECL cell hyperplasia5, 6 and AIG7, 8 can both be a basis of developing gastric NETs1, which has 100‐fold increased prevalence in type 1 diabetes patients2, 3, 4. The significance of CgA levels in the diagnosis of type 1 diabetes was shown by one of our cases47; high CgA levels helped to detect a silent gastric NET on the basis of ECL hyperplasia in a female type 1 diabetes patient. After tumor removal, her serum CgA level returned to the normal range47.
Serum CgA is used as a marker of various neuroendocrine tumors20 and it has been proven to be a good marker to track the progression of both ECL hyperplasia48 and gastric NETs20. CgA levels far above the normal range clearly indicated the presence of ECL hyperplasia in patients with type 1 diabetes according to the data of receiver operating characteristic curves, but the optimal cut‐off point was higher than the upper limit of the normal range, resulting in a high number of unidentified ECL hyperplasia with CgA values between these two points. The value of the optimal CgA cut‐off point for AIG was practically the same as the upper limit of the normal range, and there were no unidentified AIG cases based on these CgA levels. Last but not least, CgA levels far above the normal range characterized well the progression of ECL hyperplasia.
In all probability, elevated serum CgA levels in patients far from first onset of type 1 diabetes were caused by gastric ECL hyperplasia and AIG. AIG is a known comorbidity of type 1 diabetes, appearing during the progression of type 1 diabetes; therefore, it is not surprising that the CgA level showed a weak, but significant correlation with HbA1C, and was also associated with the glycemic control of patients. During the present study, we made a preliminary, but clinically interesting, observation that glycemic control could not have been efficiently normalized in most of our patients with high CgA, despite their good compliance. We do not know the potential cause of the bad glycemic control in patients with high CgA levels and without compliance issues, but we cannot rule out that elevated CgA or one of its cleavage products (for example, pancreastatin) due to ECL hyperplasia‐AIG exert an influence on glycemic control.
There is no consensus regarding the usefulness of CgA as a marker in diabetes at the present time. It has been previously suggested that further testing is necessary to evaluate the usefulness of CgA in diabetes23, whereas others have suggested that testing the combination of serum CgA, gastrin and PCA might be equivalent to histological examinations to detect premalignant lesions of gastric NETs, and would be recommended6. G cells producing gastrin, ECL cells producing CgA and parietal cells covered by the antigens of PCA have a complex, functional relationship in gastric glands49. PCA is applied to indicate AIG8, 45, 50. However, PCA cannot be used to follow up the progression of the AIG8, 50 due to the loss of gastric parietal cells, nor can it be used to predict and follow up premalignant histological changes and gastric NETs8, contrary to CgA7. The gastrin serum level gives only indirect information on ECL cell progression, because it is produced by gastric G cells49, whereas CgA is a direct product of gastric ECL cells13. Based on the observations of the current study, the routine measurement of CgA in patients with type 1 diabetes would indicate ECL+ and AIG reasonably well. Progressively increasing CgA levels would be a good indicator of the need for histological examination by gastroscopy. It would be possible to follow up the progression of ECL hyperplasia toward gastric neuroendocrine tumors by using CgA measurements in type 1 diabetes patients who already have CgA levels above the normal range and ECL hyperplasia‐AIG confirmed by biopsy. Therefore, CgA should be considered as an auxiliary marker for the early detection and follow up of ECL hyperplasia and AIG in diabetes care.
Disclosure
The authors declare no conflict of interest.
Acknowledgments
The authors thank all members of staff who worked on laboratory measurements at Semmelweis University. We are grateful to Mark Eyre for English proofreading. ZH was supported by the “Wörwag Research Prize for PhD students”, announced by Wörwag Pharma Kft. MD was supported by Janos Bolyai Research Scholarship from the Hungarian Academy of Sciences, and the UNKP‐19‐4 New National Excellence Program of the Ministry for Innovation and Technology. Research was supported by a grant from the National Research, Development and Innovation Office (K‐116128), and by a Research Grant from the Hungarian Diabetes Association.
J Diabetes Investig 2020; 11: 865–873
References
- 1. Gluckman CR, Metz DC. Gastric neuroendocrine tumors (carcinoids). Curr Gastroenterol Rep 2019; 21: 13–20. [DOI] [PubMed] [Google Scholar]
- 2. Tseng CH. Diabetes conveys a higher risk of gastric cancer mortality despite an age‐standardised decreasing trend in the general population in Taiwan. Gut 2011; 60: 774–779. [DOI] [PubMed] [Google Scholar]
- 3. Shu X, Ji J, Li X, et al Cancer risk among patients hospitalized for type 1 diabetes mellitus: a population‐based cohort study in Sweden. Diabet Med 2010; 27: 791–797. [DOI] [PubMed] [Google Scholar]
- 4. Harding JL, Shaw JE, Peeters A, et al Cancer risk among people with type 1 and type 2 diabetes: disentangling true associations, detection bias, and reverse causation. Diabetes Care 2015; 38: 264–270. [DOI] [PubMed] [Google Scholar]
- 5. Solcia E, Bordi C, Creutzfeldt W, et al Histopathological classification of nonantral gastric endocrine growths in man. Digestion 1988; 41: 185–200. [DOI] [PubMed] [Google Scholar]
- 6. De Block CE, Colpin G, Thielemans K, et al Neuroendocrine tumor markers and enterochromaffin‐like cell hyper/dysplasia in type 1 diabetes. Diabetes Care 2004; 27: 1387–1393. [DOI] [PubMed] [Google Scholar]
- 7. De Block CE, De Leeuw IH, Van Gaal LF. Autoimmune gastritis in type 1 diabetes: a clinically oriented review. J Clin Endocrinol Metab 2008; 93: 363–371. [DOI] [PubMed] [Google Scholar]
- 8. Bizzaro N, Antico A, Villalta D. Autoimmunity and gastric cancer. Int J Mol Sci 2018; 19: 377–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Krzewska A, Ben‐Skowronek I. Effect of associated autoimmune diseases on type 1 diabetes mellitus incidence and metabolic control in children and adolescents. Biomed Res Int 2016; 2016: Article ID 6219730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hansen MP, Matheis N, Kahaly GJ. Type 1 diabetes and polyglandular autoimmune syndrome: A review. World J Diabetes 2015; 6: 67–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kalkan C, Karakaya F, Soykan I. Factors associated with elevated serum chromogranin A levels in patients with autoimmune gastritis. Turk J Gastroenterol 2016; 27: 515–520. [DOI] [PubMed] [Google Scholar]
- 12. Plockinger U. Diagnosis and treatment of gastric neuroendocrine tumours. Wien Klin Wochen 2007; 119: 570–572. [DOI] [PubMed] [Google Scholar]
- 13. Herold Z, Doleschall M, Kovesdi A, et al Chromogranin A and its role in the pathogenesis of diabetes mellitus. Endokrynol Pol 2018; 69: 598–610. [DOI] [PubMed] [Google Scholar]
- 14. Konecki DS, Benedum UM, Gerdes HH, et al The primary structure of human chromogranin A and pancreastatin. J Biol Chem 1987; 262: 17026–17030. [PubMed] [Google Scholar]
- 15. Yoo SH, Lewis MS. Effects of pH and Ca2+ on monomer‐dimer and monomer‐tetramer equilibria of chromogranin A. J Biol Chem 1992; 267: 11236–11241. [PubMed] [Google Scholar]
- 16. Gorr SU, Dean WL, Radley TL, et al Calcium‐binding and aggregation properties of parathyroid secretory protein‐I (chromogranin A). Bone Miner 1988; 4: 17–25. [PubMed] [Google Scholar]
- 17. Dirkx R Jr, Solimena M. Cholesterol‐enriched membrane rafts and insulin secretion. J Diabetes Investig 2012; 3: 339–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Eskeland NL, Zhou A, Dinh TQ, et al Chromogranin A processing and secretion: Specific role of endogenous and exogenous prohormone convertases in the regulated secretory pathway. J Clin Invest 1996; 98: 148–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Helle KB, Metz‐Boutigue MH, Cerra MC, et al Chromogranins: from discovery to current times. Pflugers Arch 2018; 470: 143–154. [DOI] [PubMed] [Google Scholar]
- 20. Vinik AI, Silva MP, Woltering EA, et al Biochemical testing for neuroendocrine tumors. Pancreas 2009; 38: 876–889. [DOI] [PubMed] [Google Scholar]
- 21. Glinicki P, Jeske W. Chromogranin A (CgA) ‐ the influence of various factors in vivo and in vitro, and existing disorders on it's concentration in blood. Endokrynol Pol 2010; 61: 384–387. [PubMed] [Google Scholar]
- 22. Pregun I, Herszenyi L, Juhasz M, et al Effect of proton‐pump inhibitor therapy on serum chromogranin a level. Digestion 2011; 84: 22–28. [DOI] [PubMed] [Google Scholar]
- 23. Broedbaek K, Hilsted L. Chromogranin A as biomarker in diabetes. Biomark Med 2016; 10: 1181–1189. [DOI] [PubMed] [Google Scholar]
- 24. Troger J, Theurl M, Kirchmair R, et al Granin‐derived peptides. Prog Neurogibol 2017; 154: 37–61. [DOI] [PubMed] [Google Scholar]
- 25. Sanchez‐Margalet V, Lobon JA, Gonzalez A, et al Increased plasma pancreastatin‐like levels in gestational diabetes: correlation with catecholamine levels. Diabet Care 1998; 21: 1951–1954. [DOI] [PubMed] [Google Scholar]
- 26. Gottlieb PA, Delong T, Baker RL, et al Chromogranin A is a T cell antigen in human type 1 diabetes. J Autoimmun 2014; 50: 38–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li Y, Zhou L, Li Y, et al Identification of autoreactive CD8+ T cell responses targeting chromogranin A in humanized NOD mice and type 1 diabetes patients. Clin Immunol 2015; 159: 63–71. [DOI] [PubMed] [Google Scholar]
- 28. Stadinski BD, Delong T, Reisdorph N, et al Chromogranin A is an autoantigen in type 1 diabetes. Nat Immunol 2010; 11: 225–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Decochez K, Tits J, Coolens JL, et al High frequency of persisting or increasing islet‐specific autoantibody levels after diagnosis of type 1 diabetes presenting before 40 years of age. The Belgian Diabetes Registry. Diabet Care 2000; 23: 838–844. [DOI] [PubMed] [Google Scholar]
- 30. American Diabetes Association . 6. Glycemic targets: Standards of medical care in diabetes‐2019. Diabetes Care 2019; 42: S61–S70. [DOI] [PubMed] [Google Scholar]
- 31. Schwandt A, Denkinger M, Fasching P, et al Comparison of MDRD, CKD‐EPI, and Cockcroft‐Gault equation in relation to measured glomerular filtration rate among a large cohort with diabetes. J Diabet Complicat 2017; 31: 1376–1383. [DOI] [PubMed] [Google Scholar]
- 32. Nolting S, Kuttner A, Lauseker M, et al Chromogranin a as serum marker for gastroenteropancreatic neuroendocrine tumors: a single center experience and literature review. Cancers 2012; 4: 141–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stridsberg M, Eriksson B, Oberg K, et al A comparison between three commercial kits for chromogranin A measurements. J Endocrinol 2003; 177: 337–341. [DOI] [PubMed] [Google Scholar]
- 34. Fischer AH, Jacobson KA, Rose J, et al Hematoxylin and eosin staining of tissue and cell sections. CSH Protocols 2008; 2008: pdb.prot4986–pdb.prot4986. [DOI] [PubMed] [Google Scholar]
- 35. Gray SF, Wyatt JI, Rathbone BJ. Simplified techniques for identifying Campylobacter pyloridis. J Clin Pathol 1986; 39: 1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Stolte M, Meining A. The updated Sydney system: classification and grading of gastritis as the basis of diagnosis and treatment. Can J Gastroenterol 2001; 15: 591–598. [DOI] [PubMed] [Google Scholar]
- 37. R Core Team . R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing, 2019. [Google Scholar]
- 38. Johnson DH. Statistical sirens: the allure of nonparametrics. Ecology 1995; 76: 1998–2000. [Google Scholar]
- 39. Rietveld T, van Hout R. The paired t test and beyond: Recommendations for testing the central tendencies of two paired samples in research on speech, language and hearing pathology. J Commun Disord 2017; 69: 44–57. [DOI] [PubMed] [Google Scholar]
- 40. Pinheiro J, Bates D, DebRoy S, et al {nlme}: Linear and nonlinear mixed effects models.2019.
- 41. Miceli E, Lenti MV, Padula D, et al Common features of patients with autoimmune atrophic gastritis. Clin Gastroenterol Hepatol 2012; 10: 812–814. [DOI] [PubMed] [Google Scholar]
- 42. Delong T, Baker RL, He J, et al Diabetogenic T‐cell clones recognize an altered peptide of chromogranin A. Diabetes 2012; 61: 3239–3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Baker RL, Bradley B, Wiles TA, et al Cutting edge: Nonobese diabetic mice deficient in chromogranin A are protected from autoimmune diabetes. J Immunol 2016; 196: 39–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Katsarou A, Gudbjornsdottir S, Rawshani A, et al Type 1 diabetes mellitus. Nat Rev Dis Primers 2017; 3: 17016. [DOI] [PubMed] [Google Scholar]
- 45. De Block CE, De Leeuw IH, Bogers JJ, et al Autoimmune gastropathy in type 1 diabetic patients with parietal cell antibodies: histological and clinical findings. Diabetes Care 2003; 26: 82–88. [DOI] [PubMed] [Google Scholar]
- 46. Peracchi M, Gebbia C, Basilisco G, et al Plasma chromogranin A in patients with autoimmune chronic atrophic gastritis, enterochromaffin‐like cell lesions and gastric carcinoids. Eur J Endocrinol 2005; 152: 443–448. [DOI] [PubMed] [Google Scholar]
- 47. Somogyi A, Ruzicska E, Varga T, et al Development of silent gastric carcinoid in a type 1 diabetic patient with primer hypothyreosis. Orv Hetil 2007; 148: 1667–1671 (Hungarian). [DOI] [PubMed] [Google Scholar]
- 48. Zhao CM, Chen D. The ECL cell: relay station for gastric integrity. Curr Med Chem 2012; 19: 98–108. [DOI] [PubMed] [Google Scholar]
- 49. Malfertheiner P, Kandulski A, Venerito M. Proton‐pump inhibitors: understanding the complications and risks. Nat Rev Gastroenterol Hepatol 2017; 14: 697–710. [DOI] [PubMed] [Google Scholar]
- 50. Antico A, Tampoia M, Villalta D, et al Clinical usefulness of the serological gastric biopsy for the diagnosis of chronic autoimmune gastritis. Clin Dev Immunol 2012; 2012: 520970. [DOI] [PMC free article] [PubMed] [Google Scholar]