

Abstract
CD19-directed chimeric antigen receptor-T (CAR-T) cells with a 4-1BB or CD28 co-stimulatory domain have shown impressive antitumor activity against relapsed or refractory B cell acute lymphoblastic leukemia (r/r B-ALL). However, a parallel comparison of their performances in r/r B-ALL therapy has not been sufficiently reported. Here, we manufactured 4-1BB- and CD28-based CD19 CAR-T cells using the same process technology and evaluated their efficacy and safety in r/r B-ALL therapy based on pre-clinical and exploratory clinical investigations. In B-ALL-bearing mice, a similar antitumor effect and CAR-T kinetics in peripheral blood were observed at the CAR-T dose of 1 × 107/mouse. However, when the dose was decreased to 1 × 106/mouse, 4-1BB CAR-T cells were more potent in eradicating tumor cells and showed longer persistence than CD28 CAR-T cells. Retrospective analysis of an exploratory clinical study that used 4-1BB- or CD28-based CAR-T cells to treat r/r B-ALL was performed. Compared with CD28 CAR-T cells, 4-1BB CAR-T cells resulted in higher antitumor efficacy and less severe adverse events. This study demonstrated that the performance of 4-1BB CAR-T cells was superior to that of CD28 CAR-T cells in suppressing CD19+ B-ALL, at least under our manufacturing process.
Keywords: CAR-T, co-stimulatory domain, B-ALL therapy, dose
Graphical Abstract
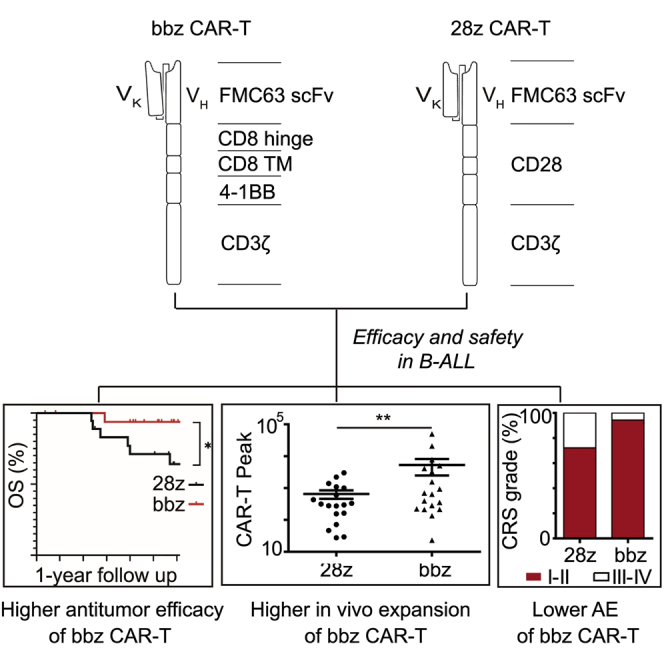
Huang, Lu, and colleagues compared the efficacy and safety of 4-1BB- and CD28-based CD19 CAR-T cells in B cell acute lymphoblastic leukemia and demonstrated that 4-1BB-based CAR-T cells showed higher antitumor efficacy and less severe adverse events.
Introduction
CD19-specific chimeric antigen receptor-T (CAR-T) cell therapy is one of the most promising therapeutic approaches to the treatment of relapsed or refractory (r/r) B cell malignancies, including acute lymphoblastic leukemia (B-ALL) and non-Hodgkin’s lymphoma.1, 2, 3, 4 Clinical investigations have shown over 70% minimal residual disease-negative (MRD−) complete remission (CR) rate in r/r B-ALL after infusion of CAR-modified autologous T cells.5, 6, 7, 8, 9, 10, 11 Two CAR-T products, axicabtagene ciloleucel (marketed as Yescarta) and tisagenlecleucel (marketed as Kymriah), have been approved to treat CD19+ r/r B-ALL in several regions.6,7,12 Many other CD19-targeted CAR-T cells are being developed worldwide. However, different initial response rate, relapse rate, CAR-T persistence, and adverse events have been reported among these CD19-targeted CAR-T cells. The composition of CAR molecules, manufacturing process of CAR-T, and disease attributes are regarded to be associated with the clinical performances.13,14
CAR molecules are composed of an antigen-recognition domain (normally single-chain variable fragment), hinge region, transmembrane region, and intracellular signaling domains.3 Currently, the majority of CARs are designed to incorporate co-stimulatory signal domains to improve the expansion, persistence, and activity of CAR-T cells.15, 16, 17 CD28 and 4-1BB are the most extensively used co-stimulatory domains, and CAR-T cells engineered with either molecule have shown strong antileukemic activities in vitro and in vivo.6,7,18, 19, 20, 21, 22 For CD28-based CAR-T cells, Martyniszyn et al.18 reported the dramatic effect of a CD20-CD19 bispecific CAR-T cell on eliminating leukemia cells in vitro and in mice at a dose of 2 × 107 cells, and An et al.19 found that anti-CD19 CAR-T cells efficiently lysed target cells in vitro and prolonged the survival time of B-ALL-bearing mice at doses of 1 × 107 and 5 × 106 cells. 4-1BB-based CD19-targeted CAR-T cells killed leukemia cells and suppressed the leukemic burden in mice by 100-fold at a dose of 2 × 107 cells.20 Furthermore, 4-1BB-based CAR-T cells (1 × 107) targeting the thymic stromal lymphopoietin receptor eradicated ALL cells in mice.21 Moreover, Li et al.22 investigated the efficacy of CD33-targeted CAR-T cells with CD28, 4-1BB, or both co-stimulatory domains in inhibiting acute myeloid leukemia. All CAR-T cells (1 × 107) decreased tumor burden and increased the survival time of mice. Meanwhile, the antileukemic activities of CAR-T cells with either CD28 or 4-1BB were similar, while the efficacy of CAR-T cells containing both co-stimulatory molecules was slightly greater.22 These studies demonstrate that CD28- and 4-1BB-based CAR-T cells exhibit similar and high inhibitory effects against leukemia in vitro and in animal models. However, they all used high doses of CAR-T (∼107), and the powerful antitumor activity may mask their different effects. Most importantly, variations in the CAR-T cell manufacturing process and the designs of these studies restrict the reliability of comparisons made between different CAR-T types.
Despite the great potency of both CD28 and 4-1BB in the antileukemic activity of CAR-T cells, the different effects of these two co-stimulatory molecules on the activation and proliferation of CAR-T cells in vitro have been reported,23 which may influence the efficacy and safety of CAR-T cells. Salter et al.24 compared the antitumor effects of CD28- and 4-1BB-based CD19 CAR-T cells in lymphoma-bearing mice and demonstrated that adoptive transfer of both CAR-T cells at a dose of 3 × 106 cells mediated complete tumor regression; however, infusion of fewer CAR-T cells (8 × 105 cells) led to lower antitumor activity in CD28 CAR-T cells.24 This suggests that CD28 and 4-1BB have different contributions to CAR-T cell function and that the infusion dose is important in comparing the two CAR-T cell types. Although the study by Salter et al.24 utilized a low dose of CAR-T (∼105), the authors investigated the effects of CAR-T cells against lymphoma rather than B-ALL, and CAR-T cell durability was not addressed.
Besides pre-clinical studies, previous case series have revealed that B-ALL patients receiving 4-1BB-based CD19 CAR-T cells achieve 83%–93% CR, while the CR of patients treated with CD28 CAR-T cells is lower (70%–88%).6,7,11,25,26 It seems that 4-1BB is more applicable as a component of CAR compared with CD28 after reviewing these studies. However, the two CAR-T types that were reported in previous studies were not manufactured under the same production process, which restricted the reliability of comparing the performances of CD28 and 4-1BB.
To address the lack of studies comparing the performance of CD28- and 4-1BB-based CAR-T cells, we manufactured CD19-directed CAR-T cells with either of these co-stimulatory molecules using identical techniques and evaluated their antitumor activities, durability, and adverse effects through pre-clinical investigations and retrospective analysis of an exploratory clinical study.
Results
Comparison of the In Vitro Activation and Killing Efficiency of CD28- and 4-1BB-Based CAR-T Cells against CD19+ Leukemia Cells
To compare the contribution of different co-stimulatory domains to the potency of CAR-T cells, 4-1BB- and CD28-based CAR molecules targeting CD19 (Figure S1) were generated under identical manufacturing processes, and the activation and killing effects of both CAR-T types against CD19+ cells were evaluated. The levels of interleukin 6 (IL-6), IL-10, tumor necrosis factor (TNF), and interferon gamma (IFN-γ) secreted by both CAR-T types increased dramatically after co-culture with Daudi cells for 12 h (Figure S2A). Furthermore, higher cytokine secretion was observed in 4-1BB CAR-T cells (Figure S2A), indicating stronger activation of 4-1BB- than CD28-based CAR-T cells. Furthermore, both CAR-T cells specifically eradicated CD19+ cells including Daudi, NALM6, and Raji cells, instead of CD19− cells (K562) with similar killing efficiency (Figure S2B).
Comparison of the Antileukemic Activity and Safety of CD28- and 4-1BB-Based CAR-T Cells in B-ALL-Bearing Mice
After confirming the specificity and efficiency of both CAR-T types in vitro, we compared the antitumor effect of 4-1BB and CD28 CAR-T cells in B-ALL-bearing mice. When infused at a dose of 1 × 107 cells/mouse, these CAR-T cells eradicated tumor cells (Figure 1A). The peripheral blood levels of human IL-6, IL-10, TNF, and IFN-γ increased after CAR-T cell infusion (Figure 1B). However, no significant differences were observed in cytokine levels in the 4-1BB and CD28 CAR-T cell groups (Figure 1B).
Figure 1.

Comparison of the Anti-leukemic Efficacy and Adverse Effects of CD28- and 4-1BB-Based Chimeric Antigen Receptor-T (CAR-T) Cells in Tumor-Bearing Mice
(A) Bioluminescence images of tumor-bearing mice treated with saline, CD28 CAR-T cells, or 4-1BB CAR-T cells at a dose of 1 × 107 cells/mouse. (B) Cytokine expression level in the sera of mice after CAR-T cell infusion at a dose of 1 × 107 cells/mouse. (C) Bioluminescence images of tumor-bearing mice treated with saline, CD28 CAR-T cells, or 4-1BB CAR-T cells at a dose of 1 × 106 cells/mouse. (D) The survival curve of mice treated with a lower dose of CAR-T cells. (E) Serum cytokine levels after CAR-T cell injection at low dose. ∗∗p < 0.01, and the error bars represent the standard derivation.
The similar efficacy and safety of 4-1BB and CD28 CAR-T cells in the animal model may be attributed to the high CAR-T dose (∼107). Thus, we further investigated the antitumor effect of 4-1BB CAR-T cells and CD28 CAR-T cells infused at a low dose (1 × 106 cells/mouse). Bioluminescence imaging showed that both types of CAR-T cells inhibited tumor progression within a week; however, severe tumor recurrence appeared in CD28 CAR-T cell-treated mice after a week (Figure 1C; Table S1). Furthermore, higher survival rates were observed in 4-1BB CAR-T-treated mice (Figure 1D). The median survival time of mice in saline, CD28-CAR-T, and 4-1BB CAR-T was 20, 29, and 58 days. And higher levels of IL-6, IL-10, TNF, and IFN-γ were detected in 4-1BB CAR-T cell-treated mouse sera (Figure 1E).
We further analyzed the persistence of CD28- and 4-1BB-based CAR-T cells in B-ALL-bearing mice. Both CAR-T cell types were detectable in peripheral blood after infusion of 1 × 107 cells, and the amount decreased along with time (Figure 2A). However, when injected at a dose of 1 × 106 cells, the persistence of 4-1BB CAR-T cells was significantly greater than that of CD28 CAR-T cells (Figure 2B). These results demonstrate that co-stimulation with 4-1BB conferred superior antitumor activity and longer persistence to CD19 CAR-T cells than that with CD28 in B-ALL-bearing mice.
Figure 2.

Persistence of CD28- and 4-1BB-Based CAR-T Cells in Tumor-Bearing Mice
(A) Number of CAR-T cells in the peripheral blood of tumor-bearing mice infused with CD28- or 4-1BB-based CAR-T cells at high dose for different time intervals. (B) Number of CAR-T cells in peripheral blood after CAR-T cell injection at low dose. ∗∗p < 0.01, and the error bars represent the standard derivation.
Antitumor Efficacy of 4-1BB- and CD28-Based CD19 CAR-T Cells in Clinical Studies
To assess the performance of 4-1BB and CD28 CAR-T cells, we retrospectively analyzed an exploratory clinical study that was conducted to evaluate the efficacy and safety of 4-1BB- and CD28-based CD19 CAR-T cells at the dose of 1 × 105 cells/kg against r/r B-ALL. Thirty-six patients (22 men and 14 women) were analyzed (18 patients were enrolled for each treatment). Patients in both treatment groups showed the same gender ratio, age, and percentage (5%) of refractory or relapsed patients. Karyotypes, BCR-ABL fusion genes, and tumor burden differed slightly between the two groups (Table S2). Clinically applicable CAR-T cells for all patients were manufactured via the same process and the median manufacturing time was 9 days (Figure 3A). The cell proliferation rate, CAR expression level, and differentiation subtypes were characterized. The mean proliferation rate of CD28 CAR-T and 4-1BB CAR-T from days 2 to 7 was 17 times and 16 times, respectively (Figure 3B). The transduction efficiency of CD28 CAR and 4-1BB CAR was 39.44% ± 3.46% and 48.01% ± 5.33%, respectively (Figure 3C). The average CD4:CD8 composition ratio of CD28 CAR-T cells was higher than that of 4-1BB CAR-T cells (Figure 3D). The ratio of naive T cells, central memory T cells, and effector memory T cells were similar between CD28 CAR-T and 4-1BB CAR-T cells (Figure 3E; Figure S3).
Figure 3.

Manufacturing Parameters and Characteristics of CD28- and 4-1BB-Based CAR-T Cells
(A) Manufacturing time. (B) Proliferation rate from days 2 to 7. (C) CAR expression ratio. (D) CD4:CD8 composition ratio. (E) Percentage of naive T cells (CD45RA+ CD62L+), central memory T cell (CD45RA− CD62L+), and effector memory T cell (CD45RA− CD62L−) in both CAR-T types. ∗p < 0.05, and the error bars represent the standard derivation; ns, not significant.
The therapeutic efficacy was evaluated 15–30 days post-T-cell infusion and followed up thereafter. Ten subjects (56%) from the CD28 CAR-T cell group and 12 from the 4-1BB CAR-T cell group (67%) were bridged to allo-HSCT (Hematopoietic stem cell transplantation) following CAR-T cell treatment. According to multiparameter flow cytometry analysis, the initial MRD− CR rates were 89% (Figure 4A) and 100% (Figure 4B) in the CD28 and 4-1BB CAR-T cell groups, respectively. Two patients showed no response to CD28 CAR-T cell therapy (Figure 4A), one of which died from rapid disease progression. The median duration of remission time was 6.8 months for the CD28-based group and 7.25 months for the 4-1BB-based group. Disease relapse occurred in six patients (33%) showing initial response to CD28 CAR-T cells (Figure 4A), and four patients (22%) treated with 4-1BB CAR-T cells (Figure 4B).
Figure 4.

Therapeutic Efficacy of CD28- and 4-1BB-Based CAR-T Cells
(A and B) Response rate and disease progression status of patients after treatment with CD28-based CAR-T cells (A) or 4-1BB-based CAR-T cells (B). (C) Kaplan-Meier curves of relapse-free survival rate after infusion with CD28 CAR-T or 4-1BB CAR-T. (D) Kaplan-Meier curves of overall survival rate after infusion with different CAR-T types. ∗p < 0.05.
Patients’ survival was monitored up to 12 months post-CAR-T infusion. The relapse-free survival rate of patients in the 4-1BB or CD28 CAR-T group was 69% versus 44% within 1-year follow-up (hazards ratio [95% confidence interval] = 3.003 [0.9024–9.9927], p = 0.073; Figure 4C). Six patients died from disease progression due to resistance or relapse after CD28 CAR-T infusion, and only one death occurred in patients infused with 4-1BB CAR-T cells (Figures 4A and 4B). Patients in the 4-1BB CAR-T group showed much higher overall survival rate than those in CD28 CAR-T group (hazards ratio [95% confidence interval] = 6.762 [1.691–27.04], p = 0.038; Figure 4D). Among the patients who were bridged to allo-HSCT, seven patients in the CD28 CAR-T group and 11 patients in the 4-1BB CAR-T group showed complete response during follow-up within 12 months (Figures 4A and 4B).
To address the different therapeutic efficacy between the two CAR-T cell types, their persistence in each patient after infusion was analyzed. All subjects exhibited CAR-T cell expansion in peripheral blood (Figure 5), and the variation trend was similar between the two groups (Figures 5A and 5B). The median time to reach peak number in peripheral blood was 11.56 and 11.11 days for CD28 and 4-1BB CAR-T cells, respectively (Figures 5A and 5B). However, the peak number of 4-1BB CAR-T cells was significantly higher than that of the CD28 CAR-T cells (5.3 × 108 cells/L versus 6.5 × 107 cells/L, respectively; Figure 5C). Peripheral blood CAR-T cell peak number did not correlate with the infusion dose or tumor burden for either group. We also detected the level of CD19+ B cells in the peripheral blood of each patient. An extremely low level of B cells was detected in patients showing responses to CD28 CAR-T or 4-1BB CAR-T since day 4–10 after CAR-T infusion (Table S3).
Figure 5.

Persistence of CD28- and 4-1BB-Based CAR-T Cells after Infusion for Different Time Intervals
(A and B) Variation trends of engrafted CD28 (A) and 4-1BB CAR-T cells (B) over different time periods determined by flow cytometry analysis of CAR-T cells in peripheral blood. (C) Peak numbers of CD28- and 4-1BB-based CAR-T cells. ∗∗p < 0.01, and the error bars represent the standard derivation.
Adverse Events of CD28- and 4-1BB-Based CAR-T Cells in Clinical Studies
There were no toxicity-related deaths from either CAR-T at the infused dose of 1 × 105 cells/kg. Cytometric bead array analysis of IL-6, IL-10, and IFN-γ in the peripheral blood revealed that CD28 CAR-T cell infusion led to higher peak levels of these cytokines (median peak level of IL-6, 98.49 pg/mL for CD28 CAR-T versus 43.20 pg/mL for 4-1BB CAR-T; median peak level of IL-10, 75.05 pg/mL for CD28 CAR-T versus 31.68 pg/mL for 4-1BB CAR-T; median peak level of IFN-γ, 37.59 pg/mL for CD28 CAR-T versus 28.78 pg/mL for 4-1BB CAR-T; Figures 6A and 6B). Cytokine release syndrome (CRS) occurred in almost all patients despite different severities. A higher percentage of patients receiving CD28 CAR-T cells experienced grade III–IV CRS (28% for CD28 CAR-T cells versus 6% for 4-1BB CAR-T cells; Figure 6C). The average starting time (Figure 6D) and duration (Figure 6E) of CRS were similar for both groups (starting time, day 4.0 ± 2.6 for CD28 CAR-T versus day 5.0 ± 2.5 for 4-1BB CAR-T; duration time, day 9.8 ± 8.7 for CD28 CAR-T versus day 8.1 ± 8.0 for 4-1BB CAR-T). Regarding immune effector cell-associated neurotoxicity syndrome (ICANS), four patients (22%) in the CD28 group and one patient (5.6%) in the 4-1BB group experienced grade I–II neurotoxicity (Figure 6F). Severe ICANS occurred in three patients after infusion of CD28 CAR-T cells and did not occur in the 4-1BB group (Figure 6F).
Figure 6.

Adverse Effects of CD28- and 4-1BB-Based CAR-T Cells
(A and B) Analysis of cytokine levels in the peripheral blood of patients after infusion of CD28- (A) and 4-1BB-based CAR-T cells (B). (C) Ratio of different cytokine release syndrome (CRS) grades induced by both CAR-T types. (D) Comparison of the CRS emergence time between the two treatment groups. (E) Comparison of the CRS duration time between the two treatment groups. (F) Number of patients with neurotoxicity after treatment with both CAR-T types. The error bars represent the standard derivation; ns, not significant.
Discussion
In this study, we systematically compared the effects of CD28 and 4-1BB on the therapeutic efficacy and adverse effects of CD19-specific CAR-T cells for the treatment of r/r B-ALL through pre-clinical and clinical investigations. Our results showed that 4-1BB CAR-T cells exhibited higher B-ALL inhibitory activity, longer persistence, and less severe adverse events than CD28 CAR-T cells.
The performances of 4-1BB and CD28 CAR-T cells was compared using an in vitro experiment, tumor-bearing mice, and clinical investigation. Both CAR-T cell types specifically killed CD19+ leukemia cells within 6 h in vitro. Their tumor-inhibitory efficiencies were similar at a dose of 1 × 107 cells in B-ALL-bearing mice (Figure 1A; Figure S2B). Previous studies also showed similar and high antileukemic efficacies for both CAR-T cell types in mouse models with similar infusion doses (from 5 × 106 to 1 × 107 cells/mouse).18, 19, 20, 21, 22 However, retrospective analyses of our prospective clinical studies revealed that 4-1BB CAR-T cells showed higher therapeutic efficacy than CD28 CAR-T cells with regard to MRD− remission rate, relapse ratio, and overall survival rate (Figure 4). In-depth analyses of these results suggested that the controversy was likely due to the different CAR-T infusion doses, since the treatment dose in mice was approximately 5–10 times of the clinically relevant dosage. Thus, we further evaluated the antitumor effects of both CAR-T cells at a low dose (1 × 106 cells/mouse). We observed that 4-1BB CAR-T cells exhibited stronger antitumor activity than CD28 CAR-T cells via the elimination of leukemia cells, suppression of tumor recurrence, and survival rate improvement. These results correlate with a previous report showing the superior antitumor effect of 4-1BB CAR-T cells at a dose of 8 × 105 cells in lymphoma-bearing mice.24 Thus, the treatment dose is important for the comparison of 4-1BB- and CD28-based CAR-T cells in animal models. According to a review of previous case series conducted by Novartis, Juno, and the National Institutes of Health (NIH), B-ALL patients receiving 4-1BB-based CD19 CAR-T cell therapy achieved 83%–93% CR, compared to 70%–88% in CD28 CAR-T cell-treated patients.4,7,9,25,26 However, the CAR-T types in these independent studies were not manufactured via the same process, and the design principles of these clinical trials varied, which restricted the reliability of the comparison between CD28 and 4-1BB. Li et al.27 have evaluated the effect of CD28 and 4-1BB on CD19 CAR-T treatment in 10 r/r B-ALL patients and reported that both types of CAR-T cells induced similar response rates and adverse events, although the response pattern differed. The contradiction between this study and our results probably lies in the differences in manufacturing process of CAR-T cells, evaluated patient number, and most importantly, the infusion dose. In this report, most patients were infused with CAR-T cells at the dose of 3 × 105 to 9 × 106/kg that was 3–90 times as high as our dose.
Our pre-clinical and clinical results consistently demonstrate that 4-1BB CAR-T cells are more effective in suppressing B-ALL than CD28 CAR-T cells at a low infusion dose that is usually correlated with less severe adverse events. The variation in the antileukemic efficacy of the two CAR-T types is likely attributable to the different signaling pathways of 4-1BB and CD28.28 CD28 belongs to the immunoglobulin superfamily and leads to activation of phosphatidylinositol 3-kinase (PI3K)-Akt pathway and Ras-mitogen-activated protein kinase (MAPK) pathway, endowing CAR-T cells with the features of robust activation, differentiation into effector memory phenotype, and glycolytic metabolism.23,24,29 In contrast, 4-1BB, a TNF receptor superfamily member, elicits progressive activation of CAR-T cells by inducing recruitment of TRAF (Tumor necrosis factor [TNF] receptor associated factor) adaptors and subsequent activation of the nuclear factor of activated T cells (NFAT) and nuclear factor κB (NF-κB) pathways. CAR-T cells with 4-1BB co-receptor showed long-term survival, high ratio of central memory phenotype, great capacity of oxidative metabolism, increased fatty acid oxidation, and mitochondrial biogenesis.23,24,29 The high infusion dose (1 × 107 cells) probably led to high levels of activated cells in both CAR-T types and confounded the differences between CD28 and 4-1BB signaling, resulting in obvious tumor elimination in both treatment groups.
To investigate the mechanisms underlying the superior therapeutic effects of 4-1BB CAR-T cells, we analyzed the number of CAR-T cells in peripheral blood after infusion for different time intervals. In B-ALL-bearing mice, we observed similar variation trends for both types of CAR-T cells infused at high dose (Figure 2A) and a more gradual decrease in 4-1BB CAR-T cell numbers at a clinically equivalent dose (Figure 2B). The peak number of 4-1BB CAR-T cells was > 8 times that of CD28 CAR-T cells (Figure 5C), although their variation trends at different time points were similar. These clinical data together with our animal study indicated the longer persistence of 4-1BB CAR-T cells in comparison to that of CD28 CAR-T cells, which was likely due to the progressive activation and memory phenotype differentiation of 4-1BB-based CAR-T cells.23 The superior persistence of 4-1BB CAR-T cells may account for greater antileukemic efficacy. In our in vitro studies, both types of CAR-T cells were activated and evaluated within a short period, which may mask the differences in their persistence capacity. Further studies on the evaluation of long-term in vitro CAR-T survival and killing efficiency are required and are likely to show the different performance of both CAR-T types.
Besides favorable antileukemic activity, we also observed less severe adverse events induced by 4-1BB CAR-T cells. Grade III–IV CRS occurred in 28% of CD28 CAR-T-cell-treated subjects compared to only 6% of 4-1BB CAR-T-cell-treated patients (Figure 6C). Regarding ICANS, another typical adverse effect of CAR-T cell therapy,30,31 only one patient experienced mild neurotoxicity post-4-1BB CAR-T cell infusion (Figure 6F). Seven CD28 CAR-T-cell-treated patients experienced neurotoxicity, among whom three showed grade III–IV toxicity. The different safety profile is likely attributed to the different activation intensity and downstream signaling pathway as described above.
One limitation of this study is that the hinge and transmembrane domains of CD28 CAR and 4-1BB CAR were different. Previous studies have shown that the length and composition of hinge and transmembrane regions can affect the activity of CAR-T cells.32,33 We chose similar numbers of amino acids in the hinge and transmembrane domains of both CAR molecules, although one contained the domains from CD28 and another contained the domains from CD8α. Alabanza et al.33 reported that CD19 CAR-T cells with CD8α hinge and transmembrane domains expressed lower levels of cytokines than those with CD28 domains, but both types of CAR-T cells showed similar antitumor activity. It seems that this phenomenon is inconsistent with our experimental data. We observed higher expression levels of cytokines as well as better antitumor efficacy in 4-1BB CAR-T that used CD8α hinge and transmembrane domains. One possible explanation is that the effect of the co-stimulatory domain is more powerful than that of the hinge and transmembrane domains on the functional activities of CAR-T cells. Thus, we can deduce the different contributions of CD28 and 4-1BB to the efficacy and safety of CD19 CAR-T cells in this study. Another concern is that the effect of CD28 or 4-1BB co-stimulatory domains on CAR-T cells may be associated with the specificity of target tumor antigen and the binding affinity of single-chain variable fragment (scFv) fragment to the tumor antigen. A recent study reported that 4-1BB and CD28 co-stimulation could be combined to enhance the efficacy of CAR-T cells when scFv fragment with low binding affinity to target antigen was used.34
In conclusion, CAR-T cells engineered with the 4-1BB co-stimulatory domain hold great potential in the treatment of CD19-specific r/r B-ALL under our manufacturing technology. This is the first study to provide evidence comparing the effects of CD28 and 4-1BB on the functions and toxicities of CD19 CAR-T cells in suppressing B-ALL at a low infusion dose. Nevertheless, the efficacy and safety of CAR-T cell therapy are associated with many other factors that call for further investigations.
Materials and Methods
Vector Construction and Lentivirus Production
The CD19-targeted CAR contained an FMC63-derived CD19-specific scFv, either a CD28 or a 4-1BB co-stimulatory domain, and a CD3ζ signaling domain. The PCR products of both CAR molecules were ligated to the third-generation EF1α promoter-based lentiviral transfer plasmid pLenti6.3/V5 (Thermo Fisher, Waltham, MA, USA). Lentivirus stock was prepared by transient transfection of transfer plasmid, packaging plasmids (pLP1 and pLP2; Thermo Fisher, Waltham, MA, USA) and envelope plasmid (pLP/VSVG; Thermo Fisher, Waltham, MA, USA) to 293T cells using polyethyleneimine, collection of the culture medium 48 and 72 h after transfection, ultrafiltration of the culture medium, and subsequent purification of the lentiviral particles using Core 700 chromatography (GE Healthcare, USA).
Manufacturing Process of CAR-T Cell Product
CAR-T cells were produced using the GMP facilities at Immunochina Pharmaceuticals (Beijing, China). Peripheral blood mononuclear cells were collected from patients via apheresis, and CD3+ T cells were separated and stimulated with CD3/CD28 Dynabeads (Thermo Fisher, Waltham, MA, USA) at a T cell:bead ratio of 1:1.5. CD3+ T cells were cultured in X-VIVO 15 medium (Lonza Group, Basel, Switzerland) supplemented with 100 U/mL of IL-2. The T cells were then transduced with CAR lentivirus within 48 h, forming CD28 CAR-T cells or 4-1BB CAR-T cells. Transduction efficiency and cell viability were examined 5–7 days after CAR lentivirus transduction. CAR-T cells were harvested and cryopreserved once they had reached sufficient levels for testing and patient infusion. The viability, potency, copy number, replication-competent lentivirus, sterility, mycoplasma, and endotoxin of CAR-T cells were analyzed for quality control purposes.
Evaluation of In Vitro Tumor-Cell-Killing Efficiency
Tumor cells, including Daudi, NALM6, Raji, and K562, were collected, labeled with calcein-acetoxymethyl ester (AM) (5 μg/1 × 107 cells; Thermo Fisher Scientific), suspended in X-VIVO 15 medium containing 100 U/mL of IL-2 (1 × 107 cells/mL), and added to a 48-well plate (1 × 105 cells/well). CAR-T cells and T cells were subsequently collected, suspended in X-VIVO 15 medium containing 100 U/mL IL-2, and added to the 48-well plate at a CAR-T number:tumor cell number ratio of 5:1. After 6 h, the plate was centrifuged and the supernatants from each well were transferred to an ELISA plate. The fluorescence intensity (FL) of released calcein-AM in each well was detected in triplicate using a Microplate Reader (Varioscan Lux, Thermo Fisher Scientific; excitation, 495 nm; emission, 515 nm). Additionally, three calcein-AM-labeled cell samples were lysed using 2% Triton X-100/saline (positive control [PC]). The FL of the supernatant from calcein-AM-labeled cells that were not treated with CAR-T or T cells was used as the negative control (NC). The tumor-cell-killing efficiency was calculated as ([FL-NC]/[PC-NC]) × 100%.
Evaluation of Antitumor Efficacy in Tumor-Bearing Mice
NCG mice (female, 6–8 weeks old) were purchased from the National Resource Center of Model Mice (Nanjing, Jiangsu, China). All animal studies were approved by the Tsinghua University Animal Care and Use Committee (Beijing, China). To establish the B-ALL animal model, NCG mice were injected with 1 million NALM6-GFP-FLuc cells, which stably expressed luciferase via the tail vein. Tumor burden was evaluated via in vivo bioluminescent imaging using the Xenogen IVIS imaging system (Xenogen, Alameda, CA, USA). One week later, mice were intravenously injected with 10 million (high dose) or 1 million (low dose) 4-1BB or CD28 CAR-T cells in saline. Mice treated with saline only served as controls. Tumor burden was monitored via bioluminescence imaging, and data were acquired by the Living Image software (Xenogen, Alameda, CA, USA).
Patient Eligibility Criteria for the Exploratory Clinical Trial
Patients suffering from CD19+ r/r B-ALL were enrolled according to the following inclusion and exclusion criteria: primary refractory or relapse after chemotherapy or allo-HSCT, and performance status of Eastern Cooperative Oncology Group score ≤2. Patients with intracranial hypertension, unconsciousness, respiratory failure, disseminated intravascular coagulation, hematosepsis, uncontrolled active infection, uncontrolled diabetes, confusion, pregnancy, breastfeeding, alanine aminotransferase/aspartate aminotransferase >3 times the upper limit of normal, creatinine >1.5 times the upper limit of normal, bilirubin > 2 times the upper limit of normal, World Health Organization score >3, previous treatment with gene therapy product, or any other uncontrolled medical disorders that investigators considered would rather exclude them from the clinical trial.
Clinical Study Design
The exploratory clinical trial (NCT03173417) was launched for r/r CD19+ B-ALL patients who showed primary resistance or recurrence after conventional chemotherapy or allo-HSCT. The study was carried out in accordance with the Declaration of Helsinki and approved by the Lu Daopei Hospital Ethics Committee. All participants in this trial signed informed consent after a discussion of the possible risks and adverse effects.
Patients received intravenous injections of fludarabine (25 mg/m2/day) and cyclophosphamide (250 mg/m2/day) for 3 consecutive days to deplete endogenous lymphocytes before CAR-T cell infusion. Pre-treatment ended 48 h before CAR-T cell infusion. After infusion with 4-1BB CAR-T or CD28 CAR-T cells, the patients’ response rate, peripheral CAR-T cell number, adverse events including CRS and ICANS, routine blood analysis, and blood biochemistry were monitored.
Flow Cytometry Analysis of CAR-T Cells
Flow cytometry was used to detect CAR expression ratio, CD4:CD8 ratio, differentiation status of the manufactured CAR-T cells, and CAR-T cell levels in peripheral blood. In brief, CAR-T cells (1 × 106) were suspended in 100 μL of Dulbecco’s phosphate-buffered saline (DPBS; Thermo Fisher Scientific) and incubated with fluorescent molecule-labeled antibodies for 30 min at 25°C. The cells were analyzed using a flow cytometer (NOVOCYTE 2060R, ACEA Biosciences, San Diego, CA, USA) after washing DPBS twice. Allophycocyanin-anti-CD3 (BD Biosciences, Franklin Lakes, NJ, USA) and fluorescein isothiocyanate (FITC)-anti-CAR (Immunochina, Beijing, China) that recognized the scFv fragment were used to detect CAR-T cells. The specificity of FITC-anti-CAR was detected and was shown in Figure S4. Phycoerythrin (PE)-anti-CD4 and FITC-anti-CD8 (BD Biosciences) were used to determine the CD4:CD8 ratio. Allophycocyanin-anti-CD45RA and PE-anti-CD62L (BioLegend, San Diego, CA, USA) were used to evaluate the differentiation status.
Response Rate Assessment
The primary efficacy endpoint was the evaluation of the overall MRD− CR through bone marrow aspiration 1, 2, and 3 months after cell infusion. Therapeutic responses were identified according to standard ALL criteria. Secondary endpoints included the overall response rate at 6 months after cell infusion, duration of response, and overall survival rate.
Assessment and Management of Adverse Events
Adverse events were evaluated according to the Common Terminology Criteria for Adverse Events (CTCAE) version 5.0. The severity of CRS and ICANS was assessed according to the reported gradings.35,36 Antipyretics were used to control low-grade CRS (grade I–II). Supportive care, tocilizumab, dexamethasone, or other steroids were employed to treat severe CRS.
Cytometric Bead Array Assay of Cytokines
Cytokines in the peripheral blood of patients were measured using the cytometric bead array human Th1/Th2/Th17 kit (BD Biosciences, San Jose, CA, USA). The lyophilized cytokines in the kit were resolved and were serially diluted for preparation of cytokine standards. Patients’ serum samples or cytokine standards were incubated with the antibody-coated capture beads and PE-labeled secondary antibody in the dark for 30 min, washed, and analyzed using flow cytometry (NOVOCYTE 2060R, ACEA Biosciences, San Diego, CA, USA). Cytokine standards were used to plot the standard curve between cytokine concentration and mean FL.
MRD Analysis
Patients’ bone marrow samples were suspended using DPBS and prepared as a single-cell suspension. Cells were incubated in the dark with a panel of antibodies including FITC-anti-CD38, PE-anti-CD10, PerCP-anti-CD34, PECy7-anti-CD19, allophycocyanin (APC)-anti-CD13, APC-anti-CD33, V500-anti-CD45, and APC Cy7-anti-CD20, or antibodies including FITC-anti-cTdT, PE-anti-CD81, PerCP-anti-CD34, PECy7-anti-CD19, APC-anti-CD10, and V500-anti-CD45. Then cells were detected using FACS Canto II (BD Biosciences, San Jose, CA, USA). Cell size, morphology, and the FL of above markers were analyzed for evaluation of MRD.
Statistical Analysis
Two-sided Wilcoxon-Mann-Whitney tests were performed to assess differences. Kaplan-Meier curves were calculated for overall survival and relapse-free survival, and log-rank tests were performed to determine significant differences between groups. Analyses were performed using SPSS and GraphPad Prism 5 software. p <0.05 was considered statistically significant for all analyses.
Author Contributions
X. Zhao, X.-A.L., P.L., J.Y., F.Q., T.H., X. Huang, and F.D. conceived and designed the study; X. Hu conducted the in vitro and pre-clinical experiments; J.Y., X. Zhang., J.Z., X. Zhou, and P.L. performed clinical examinations; X. Zhao, X.-A.L., J.Y., X. Zhang, F.Q., T.H., Y.D., and P.L. analyzed and interpreted the data; F.Q., X.-A.L., and T.H. designed the CAR and prepared the CAR-T cell product; Y.D., X. Huang, T.H., P.L., X.L., and J.Y. wrote the manuscript; all authors read and approved the final manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
We thank all patients who participated in our exploratory clinical studies. This work was supported by grants from the National Key Research and Development Program of China (no. 2017YFA0104500); the National Natural Science Foundation of China (nos. 81670166, 81530046, and 81870140); the Innovative Research Groups of the National Natural Science Foundation of China (no. 81621001); the Beijing Municipal Science & Technology Commission (no. Z171100001017098); the Project of Health Collaborative Innovation of Guangzhou city (no. 201704020214); the Peking University Clinical Scientist Program (no. BMU2019LCKXJ003); and the Scientific Research Foundation for Capital Medicine Development (no. 2018-2-4084).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omto.2020.06.016.
Contributor Information
Peihua Lu, Email: peihua_lu@126.com.
Xiaojun Huang, Email: huangxiaojun@bjmu.edu.cn.
Supplemental Information
References
- 1.Kochenderfer J.N., Rosenberg S.A. Treating B-cell cancer with T cells expressing anti-CD19 chimeric antigen receptors. Nat. Rev. Clin. Oncol. 2013;10:267–276. doi: 10.1038/nrclinonc.2013.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Frey N.V., Porter D.L. CAR T-cells merge into the fast lane of cancer care. Am. J. Hematol. 2016;91:146–150. doi: 10.1002/ajh.24238. [DOI] [PubMed] [Google Scholar]
- 3.June C.H., O’Connor R.S., Kawalekar O.U., Ghassemi S., Milone M.C. CAR T cell immunotherapy for human cancer. Science. 2018;359:1361–1365. doi: 10.1126/science.aar6711. [DOI] [PubMed] [Google Scholar]
- 4.June C.H., Sadelain M. Chimeric antigen receptor therapy. N. Engl. J. Med. 2018;379:64–73. doi: 10.1056/NEJMra1706169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maude S.L., Frey N., Shaw P.A., Aplenc R., Barrett D.M., Bunin N.J., Chew A., Gonzalez V.E., Zheng Z., Lacey S.F. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014;371:1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Park J.H., Rivière I., Gonen M., Wang X., Sénéchal B., Curran K.J., Sauter C., Wang Y., Santomasso B., Mead E. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N. Engl. J. Med. 2018;378:449–459. doi: 10.1056/NEJMoa1709919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maude S.L., Laetsch T.W., Buechner J., Rives S., Boyer M., Bittencourt H., Bader P., Verneris M.R., Stefanski H.E., Myers G.D. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med. 2018;378:439–448. doi: 10.1056/NEJMoa1709866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haso W., Lee D.W., Shah N.N., Stetler-Stevenson M., Yuan C.M., Pastan I.H., Dimitrov D.S., Morgan R.A., FitzGerald D.J., Barrett D.M. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood. 2013;121:1165–1174. doi: 10.1182/blood-2012-06-438002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davila M.L., Riviere I., Wang X., Bartido S., Park J., Curran K., Chung S.S., Stefanski J., Borquez-Ojeda O., Olszewska M. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci. Transl. Med. 2014;6:224ra25. doi: 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu Y., Wu Z., Luo Y., Shi J., Yu J., Pu C., Liang Z., Wei G., Cui Q., Sun J. Potent anti-leukemia activities of chimeric antigen receptor-modified T cells against CD19 in Chinese patients with relapsed/refractory acute lymphocytic leukemia. Clin. Cancer Res. 2017;23:3297–3306. doi: 10.1158/1078-0432.CCR-16-1799. [DOI] [PubMed] [Google Scholar]
- 11.Gardner R.A., Finney O., Annesley C., Brakke H., Summers C., Leger K., Bleakley M., Brown C., Mgebroff S., Kelly-Spratt K.S. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood. 2017;129:3322–3331. doi: 10.1182/blood-2017-02-769208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Salmikangas P., Kinsella N., Chamberlain P. Chimeric antigen receptor T-cells (CAR T-cells) for cancer immunotherapy - moving target for industry? Pharm. Res. 2018;35:152–159. doi: 10.1007/s11095-018-2436-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Finney O.C., Brakke H.M., Rawlings-Rhea S., Hicks R., Doolittle D., Lopez M., Futrell R.B., Orentas R.J., Li D., Gardner R.A., Jensen M.C. CD19 CAR T cell product and disease attributes predict leukemia remission durability. J. Clin. Invest. 2019;129:2123–2132. doi: 10.1172/JCI125423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oldham R.A.A., Medin J.A. Practical considerations for chimeric antigen receptor design and delivery. Expert Opin. Biol. Ther. 2017;17:961–978. doi: 10.1080/14712598.2017.1339687. [DOI] [PubMed] [Google Scholar]
- 15.van der Stegen S.J., Hamieh M., Sadelain M. The pharmacology of second-generation chimeric antigen receptors. Nat. Rev. Drug Discov. 2015;14:499–509. doi: 10.1038/nrd4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang J., Jensen M., Lin Y., Sui X., Chen E., Lindgren C.G., Till B., Raubitschek A., Forman S.J., Qian X. Optimizing adoptive polyclonal T cell immunotherapy of lymphomas, using a chimeric T cell receptor possessing CD28 and CD137 costimulatory domains. Hum. Gene Ther. 2007;18:712–725. doi: 10.1089/hum.2007.028. [DOI] [PubMed] [Google Scholar]
- 17.Hombach A.A., Rappl G., Abken H. Arming cytokine-induced killer cells with chimeric antigen receptors: CD28 outperforms combined CD28-OX40 “super-stimulation”. Mol. Ther. 2013;21:2268–2277. doi: 10.1038/mt.2013.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martyniszyn A., Krahl A.C., André M.C., Hombach A.A., Abken H. CD20-CD19 bispecific CAR T cells for the treatment of B-cell malignancies. Hum. Gene Ther. 2017;28:1147–1157. doi: 10.1089/hum.2017.126. [DOI] [PubMed] [Google Scholar]
- 19.An N., Tao Z., Li S., Xing H., Tang K., Tian Z., Rao Q., Wang M., Wang J. Construction of a new anti-CD19 chimeric antigen receptor and the anti-leukemia function study of the transduced T cells. Oncotarget. 2016;7:10638–10649. doi: 10.18632/oncotarget.7079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Foster J.B., Choudhari N., Perazzelli J., Storm J., Hofmann T.J., Jain P., Storm P.B., Pardi N., Weissman D., Waanders A.J. Purification of mRNA encoding chimeric antigen receptor is critical for generation of a robust T-cell response. Hum. Gene Ther. 2019;30:168–178. doi: 10.1089/hum.2018.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qin H., Cho M., Haso W., Zhang L., Tasian S.K., Oo H.Z., Negri G.L., Lin Y., Zou J., Mallon B.S. Eradication of B-ALL using chimeric antigen receptor-expressing T cells targeting the TSLPR oncoprotein. Blood. 2015;126:629–639. doi: 10.1182/blood-2014-11-612903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li S., Tao Z., Xu Y., Liu J., An N., Wang Y., Xing H., Tian Z., Tang K., Liao X. CD33-specific chimeric antigen receptor T cells with different co-stimulators showed potent anti-leukemia efficacy and different phenotype. Hum. Gene Ther. 2018;29:626–639. doi: 10.1089/hum.2017.241. [DOI] [PubMed] [Google Scholar]
- 23.Kawalekar O.U., O’Connor R.S., Fraietta J.A., Guo L., McGettigan S.E., Posey A.D., Jr., Patel P.R., Guedan S., Scholler J., Keith B. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity. 2016;44:380–390. doi: 10.1016/j.immuni.2016.01.021. [DOI] [PubMed] [Google Scholar]
- 24.Salter A.I., Ivey R.G., Kennedy J.J., Voillet V., Rajan A., Alderman E.J., Voytovich U.J., Lin C., Sommermeyer D., Liu L. Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Sci. Signal. 2018;11:eaat6753. doi: 10.1126/scisignal.aat6753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee D.W., Kochenderfer J.N., Stetler-Stevenson M., Cui Y.K., Delbrook C., Feldman S.A., Fry T.J., Orentas R., Sabatino M., Shah N.N. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385:517–528. doi: 10.1016/S0140-6736(14)61403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Turtle C.J., Hanafi L.A., Berger C., Gooley T.A., Cherian S., Hudecek M., Sommermeyer D., Melville K., Pender B., Budiarto T.M. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Invest. 2016;126:2123–2138. doi: 10.1172/JCI85309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li S., Zhang J., Wang M., Fu G., Li Y., Pei L., Xiong Z., Qin D., Zhang R., Tian X. Treatment of acute lymphoblastic leukaemia with the second generation of CD19 CAR-T containing either CD28 or 4-1BB. Br. J. Haematol. 2018;181:360–371. doi: 10.1111/bjh.15195. [DOI] [PubMed] [Google Scholar]
- 28.Zhong Q., Zhu Y.M., Zheng L.L., Shen H.J., Ou R.M., Liu Z., She Y.L., Chen R., Li C., Huang J. Chimeric Antigen Receptor-T Cells with 4-1BB Co-Stimulatory Domain Present a Superior Treatment Outcome than Those with CD28 Domain Based on Bioinformatics. Acta Haematol. 2018;140:131–140. doi: 10.1159/000492146. [DOI] [PubMed] [Google Scholar]
- 29.Chen L., Flies D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013;13:227–242. doi: 10.1038/nri3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu Y., Sun J., Wu Z., Yu J., Cui Q., Pu C., Liang B., Luo Y., Shi J., Jin A. Predominant cerebral cytokine release syndrome in CD19-directed chimeric antigen receptor-modified T cell therapy. J. Hematol. Oncol. 2016;9:70. doi: 10.1186/s13045-016-0299-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kroschinsky F., Stölzel F., von Bonin S., Beutel G., Kochanek M., Kiehl M., Schellongowski P., Intensive Care in Hematological and Oncological Patients (iCHOP) Collaborative Group New drugs, new toxicities: severe side effects of modern targeted and immunotherapy of cancer and their management. Crit. Care. 2017;21:89. doi: 10.1186/s13054-017-1678-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hudecek M., Sommermeyer D., Kosasih P.L., Silva-Benedict A., Liu L., Rader C., Jensen M.C., Riddell S.R. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol. Res. 2015;3:125–135. doi: 10.1158/2326-6066.CIR-14-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alabanza L., Pegues M., Geldres C., Shi V., Wiltzius J.J.W., Sievers S.A., Yang S., Kochenderfer J.N. Function of Novel Anti-CD19 Chimeric Antigen Receptors with Human Variable Regions Is Affected by Hinge and Transmembrane Domains. Mol. Ther. 2017;25:2452–2465. doi: 10.1016/j.ymthe.2017.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Drent E., Poels R., Ruiter R., van de Donk N.W.C.J., Zweegman S., Yuan H., de Bruijn J., Sadelain M., Lokhorst H.M., Groen R.W.J. Combined CD28 and 4-1BB Costimulation Potentiates Affinity-tuned Chimeric Antigen Receptor-engineered T Cells. Clin. Cancer Res. 2019;25:4014–4025. doi: 10.1158/1078-0432.CCR-18-2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Neelapu S.S., Tummala S., Kebriaei P., Wierda W., Gutierrez C., Locke F.L., Komanduri K.V., Lin Y., Jain N., Daver N. Chimeric antigen receptor T-cell therapy - assessment and management of toxicities. Nat. Rev. Clin. Oncol. 2018;15:47–62. doi: 10.1038/nrclinonc.2017.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee D.W., Santomasso B.D., Locke F.L., Ghobadi A., Turtle C.J., Brudno J.N., Maus M.V., Park J.H., Mead E., Pavletic S. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol. Blood Marrow Transplant. 2019;25:625–638. doi: 10.1016/j.bbmt.2018.12.758. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.