

Abstract
The cause and underlying mechanisms that contribute to asthma pathogenesis are not well known. Both genome- and epigenome-wide association studies have identified genes associated with asthma risk. It is unknown to what extent genes identified in these two types of studies overlap. Based on existing literature and the DisGeNET database, we extracted overlapping genes identified in genetic and epigenetic studies of childhood asthma. Through analyses of variance, we assessed whether DNA methylation (DNAm) at 5′-C-phosphate-G-3′ (CpGs) on the overlapping genes was associated with neighboring single-nucleotide polymorphisms (SNPs) within 1M base pairs (bps) and with low linkage disequilibrium (r2 < 0.2) in the childhood asthma-related genes. In total, 285 genes from genetic studies and 226 genes from epigenetic studies were shown to be associated with asthma risk, of which six overlap. Of the six genes, 79 CpGs and 8229 unique neighboring SNPs (1M bps) were included in methylation quantitative loci (methQTL) assessment analyses. We tested the association of DNAm at each of the 79 CpG sites with its neighboring SNPs. After adjusting for multiple testing by controlling the false discovery rate to 0.05 when testing methQTL for each CpG site, we found statistically significant associations in three genes with their neighboring SNPs and identified 34 unique methQTLs. The rather limited overlap in genes between genetic and epigenetic studies on asthma and the absence of methQTL in some of the overlapping genes highlight a need to jointly, rather than independently, examine genetic and epigenetic effects on asthma risk to improve our understanding of the underlying mechanisms of asthma.
Keywords: Genetic, epigenetic, DNA methylation, SNPs, methQTLs, asthma
Introduction
The Global Initiative for Asthma defined asthma as a heterogeneous disease characterized by long-term airway inflammation, with respiratory symptoms such as wheeze, shortness of breath, chest tightness, and cough. Approximately 339 million people worldwide1 suffer from asthma, with an estimated 23 million cases reported in the United States as of August 2015 including more than six million children.2 The cause of asthma is not well understood, and the underlying biological mechanisms are poorly recognized.3
Previous genome-wide association studies (GWASs) suggest a substantial genomic contribution to the cause of asthma,2 and various studies demonstrated that single-nucleotide polymorphisms (SNPs) were associated with the risk of asthma.4-6 However, as of now, genetics could only explain a fraction of variation in asthma risk.5,7-9 In addition, replication of findings failed across different populations.10,11 These limitations have caused a redirection of focus toward epigenetic studies.
Apart from genetic studies, the importance of epigenetic factors in asthma pathogenesis, such as DNA methylation (DNAm) at 5′-C-phosphate-G-3′ (CpG) sites, has been increasingly recognized. Epigenome-wide association studies have shown that DNAm in whole blood is associated with asthma.12-14 In a recent study, it was observed that individuals with asthma (from 4 years to adolescence) had consistently lower methylation at certain CpG sites. Such an association was not observed at birth indicating postnatal effects of exposure and/or disease on DNAm.15 In a meta-analysis by Reese et al13 based on findings in eight cohorts, DNAm in newborns at specific CpG sites was shown to be associated with asthma in school-aged children. Similar findings were also identified through meta-analyses based on cross-sectional studies in nine cohorts.13
Both genetic and epigenetic components play important roles in the development of asthma. Genetic studies aim to identify genetic variants associated with asthma independent of known risk factors, while epigenetic studies have the potential to assess environmental effects on the risk of asthma as epigenetics may represent a molecular intermediate between environmental exposure and disease. Both genetic and epigenetic studies have the potential to identify genes that play an important role in disease biology. However, little is known as to what extent that the findings of genetic studies agree with the findings of epigenetic studies, and whether, if the same genes are identified as containing genetic and epigenetic variation associated with the disease, the effect of the SNP on asthma risk is mediated via changes in DNAm, ie, whether SNPs in these genes are methylation quantitative loci (methQTL). The goal of this study was to examine the agreement of genes identified between genetic and epigenetic studies on asthma. At each CpG site on the overlapping genes, we assessed whether neighboring SNPs on asthma-related genes (from genetic or epigenetic studies) are associated with DNAm of that CpG site. Findings from such investigations will benefit future plans to design genetic and epigenetic asthma studies to further improve our understanding of the underlying mechanisms of asthma from a different perspective.
Methods
Asthma candidate genes detected in literature
We considered genes identified in studies of both genetic variation and DNAm on the risk of childhood asthma. We identified three GWASs of childhood-onset asthma,4-6 as well as a database, DisGeNET, which is a database of gene-trait association data from expert-curated repositories, GWAS catalogs, animal models, and scientific literature that provides gene lists for different health conditions. In epigenetic studies, a recent epigenome-wide DNAm meta-analysis showed that DNAm at specific CpG sites was associated with childhood asthma.13 To improve homogeneity between genetic and epigenetic studies, we focused on childhood-onset asthma and included findings based on data collected in European ancestry. Genes identified from both sources (genetic and DNAm studies on asthma) were included in our assessment.
The Isle of Wight birth cohorts
The Isle of Wight (IoW) birth cohort (IoWF1) was composed of children born between January 1, 1989, and February 28, 1990, on the IoW, United Kingdom.16 Of the 1536 children born and recruited in this period, 1456 were available for further follow-ups. Since 2010, we have recruited 503 children (IoWF2) born to the participants in IoWF1 (mother and/or father). National Research Ethics Service Committee South Central—Hampshire B (09/H0504/129) approved the IoWF2 study. In the current study, DNAm data from Guthrie cards in the IoWF2 cohort were included in the analyses. Asthma at age 10 years was defined as “ever had asthma” and “wheezing or whistling in the chest in the last 12 months” or “current treatment for asthma.”
Genome-wide SNPs
The genomic DNA was isolated from blood samples by using QIAamp DNA Blood Kits (Qiagen, Valencia, CA, USA) or the ABI PRISM 6100 Nucleic Acid PrepStation (Applied Biosystems, Foster City, CA, USA) or from saliva using Oragene DNA Self Collection Kits (DNA Genotek, Ottawa, ON, Canada). Genotyping was performed using Illumina’s OmniExpressExome BeadChip (v1.2). Genotypes were extracted from image data using GenomeStudio software (Illumina), and technical failure of genotyping assays was determined by significant deviation of genotype frequencies from expectations under Hardy-Weinberg equilibrium (P < .01, χ2 tests). In total, genome-wide SNP data of 139 participants of IoWF2 (third generation) were included in the study.
Genome-wide DNAm
Blood sample was obtained from Guthrie cards of 141 newborns (IoWF2), and DNA was extracted from these blood samples by a procedure reported by Beyan et al.17 The methylation level for each CpG was assessed using the MethylationEPIC BeadChip (Illumina, Inc., San Diego, CA, USA). DNA concentration was estimated by fluorometric quantitation. For each sample, one microgram DNA was bisulfite treated for cytosine to thymine conversion using the EZ 96-DNA methylation kit (Zymo Research, Irvine, CA, USA), following the manufacturer’s protocol. The MethylationEPIC BeadChip interrogates >850 000 CpGs associated with more than 24 000 genes. Arrays were processed using a standard protocol17 with multiple identical control samples assigned to each bisulfite conversion batch to assess assay variability. DNA methylation was measured by a ratio of intensities, denoted as β values for each CpG site. It is a ratio of methylated (M) over the sum of methylated and unmethylated (U) probes (β = M/[c + M + U]), where c is a constant to prevent from zero in the denominator.
DNA methylation preprocessing
Probes not reaching a detection P value of 10 to 16 in at least 95% of samples were excluded. CpGs on sex chromosomes were also excluded to avoid biases. DNA methylation was quantile normalized using the R package, minfi,18 and batch effects were removed using the combat function in R available in the SVA Bioconductor package. In addition, probes that contained SNPs within 10 base pairs (bps) of a targeted CpG site with minor allele frequency in at least 0.7% of subjects were excluded due to their potential influence on DNAm. After preprocessing and removing unmapped and non-CpG sites, 300 217 CpGs from 141 participants in IoWF2 were included in the study.
Statistical analysis
We first examined the overlap of genes between published findings from genetic and epigenetic studies on asthma. For genes identified from epigenetic studies, we considered two types of situations, genes that a CpG site is mapped to and genes such that DNAm at a CpG site is associated with expression of those genes. Next, for each gene identified in both types of studies, we examined the existence of methQTL. For a CpG site linked to an overlapping gene, when selecting neighboring SNPs, to assess how window size influenced the results, we considered three different windows, 500k bps (250k bps upstream and 250k bps downstream of the CpG site), 1M bps, and 2M bps, of which the 1M bp window was applied in previous studies.19 To increase statistical power, among the SNPs within the window, SNPs with genotype frequency >1% (ie, with rare SNPs excluded) and r2 < 0.2 (ie, with strongly dependent SNPs excluded) were included in the assessment. Next, we assessed whether DNAm at a given CpG site was associated with any of its neighboring SNPs using the analysis of variance (ANOVA). DNA methylation and SNP data in the IoWF2 cohort were included in this methQTL analysis. Often β values suffer from severe heteroscedasticity. In the ANOVAs, logit-transformed DNAm levels (ie, M values) were used as suggested in earlier studies.20 Multiple testing was adjusted for each CpG site by controlling a false discovery rate (FDR) of 0.05. All the analyses were performed using the R computing package.21
Results
In the study by Demenais et al,5 13 genes were shown to be associated with childhood asthma at genome-wide significance (P < 5 × 10−8) in European-ancestry populations. Based on genetic variants and expressions, the study by Pividori et al4 suggested that 155 genes are linked to childhood asthma in a white population. Finally, in the study by Ferreira et al,6 30 genes selected from previous genetic studies on childhood asthma were analyzed in individuals with European ancestry. In total, these three genetic studies identified in total 177 unique genes. Using the database DisGeNET (with the category “Disease name: Childhood asthma, cui: C0264408”), we extracted 126 genes linked to childhood asthma. However, in this database, we were not able to find genes based on race. Among these 126 genes, 18 were also on the list of the 177 genes. Altogether, we extracted 285 unique genes from genetic studies shown to be associated with childhood asthma.
The epigenetic study by Reese et al identified 188 CpGs individually associated with childhood asthma and 71 differentially methylated regions (DMRs) related to asthma. These CpGs and DMRs were mapped to 226 genes.13 Among these 226 genes, only 6 genes were among the 285 genes from genetic studies. That is, in total, 505 genes were identified to be associated with the risk of childhood asthma based on genetic and epigenetic studies. Among these six overlapping genes, CpG sites on gene AP5B1 were not available in our data and, hence, were excluded from subsequent analysis. For the remaining five genes, DNAm at birth on 79 CpG sites were available in 141 newborns in the IoWF2 cohort.
For each of the 79 CpG sites, we identified its neighboring SNPs within 1M bps (500 kb upstream and downstream) and with minor allele frequency (MAF) > 1% and r2 < 0.2 (Figure 1). Following this criterion, we identified in total 8229 unique neighboring SNPs within 1M bps of their corresponding CpGs. The number of neighboring SNPs ranged from 847 to 2250 across 79 CpGs (Table 1). Next, for each CpG site, we tested the association of genotypes of each neighboring SNP with DNAm to detect possible methQTLs.
Figure 1.
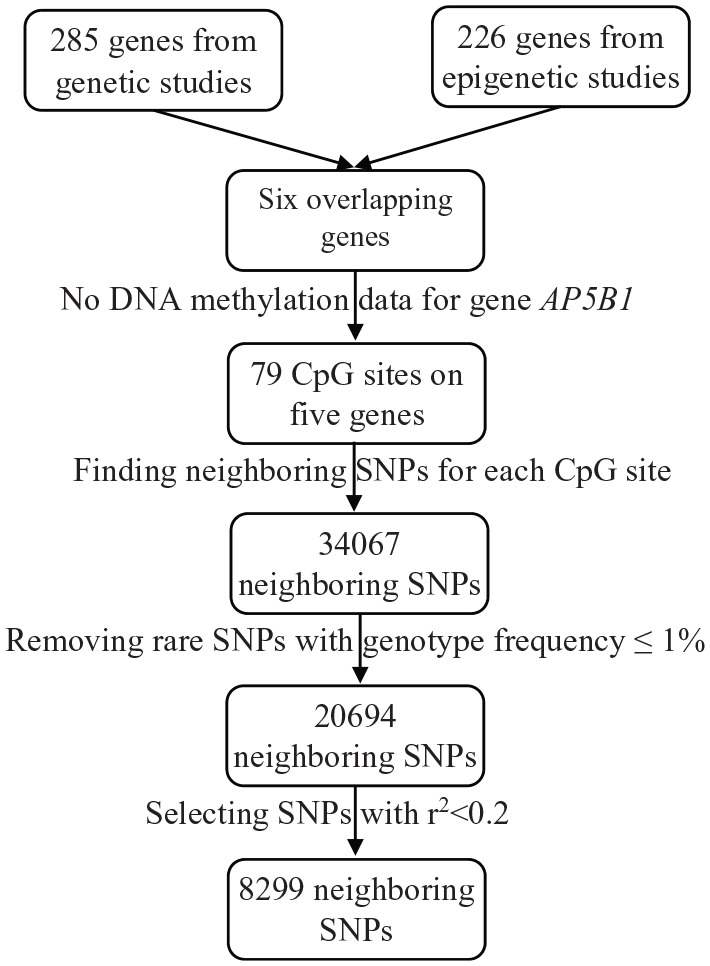
The flowchart of selecting neighboring SNPs for methQTL analyses based on window size of 1M bps.
bps indicates base pairs; CpG, 5′-C-phosphate-G-3′; methQTL, methylation quantitative loci; SNP, single-nucleotide polymorphism.
Table 1.
Summary of the number of CpGs on each gene and their neighboring SNPs (based on a window size of 1M bps), and the number of ANOVA models fitted for each gene, along with the number of significant results after controlling FDR.
| Gene | Number of CpGs | Number of unique SNPs | Number of models | Number of significant resultsa |
|---|---|---|---|---|
| DUSP22 | 24 | 1290 | 29 521 | 14 |
| IKZF3 | 7 | 2481 | 15 736 | 9 |
| IL4 | 4 | 850 | 3390 | 0 |
| RUNX1 | 7 | 2243 | 67 843 | 12 |
| TLE4 | 37 | 1365 | 9529 | 0 |
Abbreviations: ANOVA, analysis of variance; bps, base pairs; FDR, false discovery rate; methQTLs, methylation quantitative loci; SNP, single-nucleotide polymorphism.
These 35 statistically significant results represented 34 unique methQTLs. Single-nucleotide polymorphism rs11706690 was associated with DNAm at two CpG sites on DUSP22 (details are in Table 2).
Among the five overlapping genes, we found a statistically significant association for three genes: DUSP22, IKZF3, and RUNX1 (Table 1), and 34 unique methQTLs were identified, after adjusting for multiple testing among the neighboring SNPs for each CpG site by controlling FDR of 0.05. When we used windows of 500k and 2M bps for the definition of neighboring SNPs, methQTLs also showed on these three genes only (Supplemental Tables 1 and 2).
Among these three genes, DUSP22, IKZF3, and RUNX1, most methQTLs were observed on gene DUSP22, after adjusting for multiple testing. In gene DUSP22, after fitting in total 29 521 ANOVA models to the 24 CpGs and 1290 SNPs, 13 SNPs showed statistically significant associations with five CpGs (Table 2). Nine methQTLs were identified in gene IKZF3 contributed by two CpG sites after fitting 15 736 ANOVA models (Table 3). For gene RUNX1, in total, 67 843 ANOVA models were fitted to the seven CpGs and 2243 SNPs, leading to 12 methQTLs (Table 4).
Table 2.
Detected methQTLs based on 1 MB neighboring SNPs of CpGs in gene DUSP22 between genetic and epigenetic association studies with asthma.
| Gene | CpG | SNPs | Gtypea | DNAm mean | DNAm SD | Effects (95% CIb) | P value | PFDR c |
|---|---|---|---|---|---|---|---|---|
| DUSP22 | cg01516881 | rs10418525 | AA | −6.2 | 0.056 | — | 2.25 × 10−5 | 2.77 × 10−2 |
| AG | −1.1 | 1.36 | 5.1; (3.2, 7.0) | |||||
| GG | −1.8 | 2.91 | 4.3; (2.5, 6.2) | |||||
| cg01516881 | rs10423498 | AA | −2.68 | 4.45 | — | 5.61 × 10−5 | 2.77 × 10−2 | |
| AG | −0.97 | 0.49 | 1.7; (0.61, 2.80) | |||||
| GG | −4.77 | 7.73 | −2.1; (−3.87, −0.31) | |||||
| cg01516881 | rs11706690 | AA | −1.7 | 3.8 | — | 6.50 × 10−5 | 2.77 × 10−2 | |
| AC | −1.2 | 1.1 | 0.51; (−0.75, 1.77) | |||||
| CC | −4.5 | 5.1 | −2.79; (−4.39, −1.19) | |||||
| cg03624871 | rs10118798 | AA | 0.72 | 0.14 | — | 4.78 × 10−5 | 2.92 × 10−2 | |
| AG | 1.80 | 0.27 | 1.08; (0.61, 1.56) | |||||
| GG | 1.68 | NA | 0.97; (0.17, 1.76) | |||||
| cg03624871 | rs1410968 | AA | 0.72 | 0.14 | — | 4.78 × 10−5 | 2.92 × 10−2 | |
| AG | 1.80 | 0.27 | 1.08; (0.61, 1.56) | |||||
| GG | 1.68 | NA | 0.97; (0.17, 1.76) | |||||
| cg11235426 | rs11706690 | AA | −1.5 | 0.95 | — | 1.74 × 10−5 | 2.23 × 10−2 | |
| AC | −1.4 | 0.43 | 0.097; (−0.55, 0.75) | |||||
| CC | −3.3 | 0.93 | −1.723; (−2.55, −0.90) | |||||
| cg22949951 | rs7840893 | AG | 1.98 | NA | — | 2.04 × 10−5 | 1.32 × 10−2 | |
| GG | 0.55 | 0.084 | −1.4; (−2.0, −0.84) | |||||
| cg22949951 | exm2216266 | AA | 0.55 | 0.084 | — | 2.04 × 10−5 | 1.32 × 10−2 | |
| GG | 1.98 | NA | 1.43; (0.84, 2.03) | |||||
| cg24325581 | rs4797173 | AA | 4.1 | NA | — | 3.95 × 10−5 | 4.65 × 10−2 | |
| AG | 3.1 | 0.14 | −1.0; (−1.6, −0.43) | |||||
| GG | 2.8 | 0.039 | −1.3; (−1.9, −0.72) | |||||
| cg24325581 | rs12970203 | AA | 2.8 | 0.046 | — | 1.03 × 10−4 | 4.91 × 10−2 | |
| AC | 3.1 | 0.219 | 0.29; (0.066, 0.52) | |||||
| CC | 4.1 | NA | 1.24; (0.661, 1.82) | |||||
| cg24325581 | rs34383812 | AA | 3.6 | 0.614 | — | 1.51 × 10−4 | 4.91 × 10−2 | |
| AG | 2.9 | 0.051 | −0.70; (−1.1, −0.35) | |||||
| GG | 2.7 | 0.044 | −0.91; (−1.3, −0.52) | |||||
| cg24325581 | rs910858 | AA | 2.8 | 0.051 | — | 2.27 × 10−4 | 4.91 × 10−2 | |
| AG | 2.9 | 0.071 | 0.017; (−0.18, 0.22) | |||||
| GG | 3.6 | 0.500 | 0.806; (0.45, 1.17) | |||||
| cg24325581 | rs10409912 | AA | 2.8 | 0.05 | — | 2.36 × 10−4 | 4.91 × 10−2 | |
| AG | 3.1 | 0.18 | 0.24; (0.025, 0.46) | |||||
| GG | 4.1 | NA | 1.24; (0.641, 1.83) | |||||
| cg24325581 | rs7236111 | AA | 2.8 | 0.047 | — | 2.50 × 10−4 | 4.91 × 10−2 | |
| AG | 3.0 | 0.127 | 0.21; (0.012, 0.4) | |||||
| GG | 3.7 | 0.237 | 0.93; (0.501, 1.4) |
Abbreviations: ANOVA, analysis of variance; CpG, 5′-C-phosphate-G-3′; DNAm, DNA methylation; FDR, false discovery rate; methQTLs, methylation quantitative loci; NA, not applicable; SNPs, single-nucleotide polymorphisms.
Gtype: genotype; the first genotype for each SNP is the reference in the ANOVA test.
95% CI: 95% confidence interval of the effects. Also, “—” indicates the reference group.
PFDR: P values after adjusting for multiple testing by controlling the FDR of 0.05 based on the P values from ANOVA tests for each gene.
Table 3.
Detected methQTLs based on 1 MB neighboring SNPs of CpGs in gene IKZF3 between genetic and epigenetic association studies with asthma.
| Gene | CpG | SNPs | Gtypea | DNAmean | DNAm SD | Effects (95% CIb) | P value | PFDR c |
|---|---|---|---|---|---|---|---|---|
| IKZF3 | cg03293732 | rs4795405 | AA | 0.64 | 0.054 | — | 4.04 × 10−5 | 3.63 × 10−2 |
| AG | 0.92 | 0.053 | 0.28; (0.11, 0.45) | |||||
| GG | 1.22 | 0.033 | 0.58; (0.35, 0.81) | |||||
| cg03293732 | rs10445308 | AA | 0.64 | 0.060 | — | 5.26 × 10−5 | 3.63 × 10−2 | |
| AG | 0.92 | 0.051 | 0.28; (0.11, 0.45) | |||||
| GG | 1.26 | 0.031 | 0.62; (0.37, 0.88) | |||||
| cg03293732 | rs12603332 | AA | 0.68 | 0.049 | — | 7.16 × 10−5 | 3.63 × 10−2 | |
| AG | 0.94 | 0.059 | 0.25; (0.091, 0.42) | |||||
| GG | 1.22 | 0.033 | 0.54; (0.315, 0.77) | |||||
| cg03293732 | rs4378650 | AA | 0.68 | 0.049 | — | 7.16 × 10−5 | 3.63 × 10−2 | |
| AG | 0.94 | 0.059 | 0.25; (0.091, 0.42) | |||||
| GG | 1.22 | 0.033 | 0.54; (0.315, 0.77) | |||||
| cg03293732 | rs12950743 | AA | 1.26 | 0.031 | — | 9.68 × 10−5 | 3.63 × 10−2 | |
| AG | 0.93 | 0.057 | −0.33; (−0.57, −0.10) | |||||
| GG | 0.68 | 0.053 | −0.58; (−0.82, −0.33) | |||||
| cg03293732 | rs907091 | AA | 1.26 | 0.031 | — | 9.68 × 10−5 | 3.63 × 10−2 | |
| AG | 0.93 | 0.057 | −0.33; (−0.57, −0.10) | |||||
| GG | 0.68 | 0.053 | −0.58; (−0.82, −0.33) | |||||
| cg20709984 | rs2598068 | AA | 2.1 | 0.194 | — | 1.21 × 10−5 | 2.14 × 10−2 | |
| AG | 2.6 | 0.118 | 0.50; (0.24, 0.76) | |||||
| GG | 1.4 | 0.081 | −0.71; (−1.20, −0.22) | |||||
| cg20709984 | exm614782 | AA | 1.4 | 0.081 | — | 2.86 × 10−5 | 2.14 × 10−2 | |
| AG | 2.6 | 0.124 | 1.22; (0.71, 1.7) | |||||
| GG | 2.2 | 0.196 | 0.75; (0.25, 1.3) | |||||
| cg20709984 | rs2722371 | AA | 1.4 | 0.081 | — | 2.86 × 10−5 | 2.14 × 10−2 | |
| AG | 2.6 | 0.124 | 1.22; (0.71, 1.7) | |||||
| GG | 2.2 | 0.196 | 0.75; (0.25, 1.3) |
Abbreviations: ANOVA, analysis of variance; CpG, 5′-C-phosphate-G-3′; DNAm, DNA methylation; FDR, false discovery rate; methQTLs, methylation quantitative loci; SNPs, single-nucleotide polymorphisms.
Gtype: genotype; the first genotype for each SNP is the reference in the ANOVA test.
95% CI: 95% confidence interval of the effects. Also, “—” indicates the reference group.
PFDR: P values after adjusting for multiple testing by controlling the FDR of 0.05 based on the P values from ANOVA tests for each gene.
Table 4.
Detected methQTLs based on 1 MB neighboring SNPs of CpGs in gene RUNX1 between genetic and epigenetic association studies with asthma.
| Gene | CpG | SNPs | Gtypesa | DNAm mean | DNAm SD | Effects (95% CIb) | P value | PFDR c |
|---|---|---|---|---|---|---|---|---|
| RUNX1 | cg04357830 | rs2231304 | – | −3.1 | 0.214 | — | 4.60 × 10−8 | 8.13 × 10−5 |
| AA | −4.0 | 0.069 | −0.9; (−1.2, −0.64) | |||||
| cg04357830 | rs9648428 | AG | −3.4 | 0.321 | — | 1.19 × 10−5 | 1.06 × 10−2 | |
| GG | −4.0 | 0.063 | −0.66; (−0.93, −0.4) | |||||
| cg05973398 | rs16960097 | AA | −4.5 | 0.040 | — | 3.33 × 10−5 | 2.95 × 10−2 | |
| AG | −5.0 | 0.011 | −0.48; (−0.69, −0.28) | |||||
| cg05973398 | rs9950297 | AA | −5.0 | 0.011 | — | 3.33 × 10−5 | 2.95 × 10−2 | |
| AC | −4.5 | 0.040 | 0.48; (0.28, 0.69) | |||||
| cg06758350 | rs9544614 | AA | −3.5 | NA | — | 5.38 × 10−5 | 4.41 × 10−2 | |
| AC | −1.6 | 0.088 | 1.9; (1.12, 2.6) | |||||
| CC | −2.0 | 0.130 | 1.5; (0.77, 2.2) | |||||
| cg06758350 | rs975284 | AA | −2.0 | 0.130 | — | 5.38 × 10−5 | 4.41 × 10−2 | |
| AC | −1.6 | 0.088 | 0.38; (0.083, 0.68) | |||||
| CC | −3.5 | NA | −1.50; (−2.22, −0.77) | |||||
| cg06758350 | rs753921 | AA | −3.5 | NA | — | 7.22 × 10−5 | 4.41 × 10−2 | |
| AG | −1.8 | 0.14 | 1.7; (0.99, 2.5) | |||||
| GG | −2.1 | 0.12 | 1.4; (0.71, 2.2) | |||||
| cg06758350 | rs700164 | AA | −3.5 | NA | — | 1.05 × 10−4 | 4.83 × 10−2 | |
| AG | −2.1 | 0.189 | 1.4; (0.65, 2.2) | |||||
| GG | −1.8 | 0.089 | 1.7; (0.93, 2.4) | |||||
| cg08433011 | rs4678516 | AA | −3.4 | 0.078 | — | 1.20 × 10−5 | 2.14 × 10−2 | |
| AG | −3.1 | 0.035 | 0.32; (0.14, 0.505) | |||||
| GG | −3.6 | 0.081 | −0.18; (−0.41, 0.053) | |||||
| cg08433011 | rs10510676 | AA | −3.6 | 0.095 | — | 6.44 × 10−5 | 3.83 × 10−2 | |
| AG | −3.1 | 0.036 | 0.52; (0.2921,0.74) | |||||
| GG | −3.4 | 0.079 | 0.24; (−0.0028, 0.48) | |||||
| cg08433011 | rs10865872 | AA | −3.6 | 0.095 | — | 6.44 × 10−5 | 3.83 × 10−2 | |
| AG | −3.1 | 0.036 | 0.52; (0.2921,0.74) | |||||
| GG | −3.4 | 0.079 | 0.24; (−0.0028, 0.48) | |||||
| cg13030790 | rs4670546 | AA | −5.3 | 0.12 | — | 6.40 × 10−6 | 1.20 × 10−2 | |
| AG | −5.6 | 0.11 | −0.25; (−0.48, −0.016) | |||||
| GG | −4.8 | 0.04 | 0.54; (0.26, 0.823) |
Abbreviations: ANOVA, analysis of variance; CpG, 5′-C-phosphate-G-3′; DNAm, DNA methylation; FDR, false discovery rate; methQTLs, methylation quantitative loci; NA, not applicable; SNPs, single-nucleotide polymorphisms.
Gtype: genotype; the first genotype for each SNP is the reference in the ANOVA test.
95% CI: 95% confidence interval of the effects. Also, “—” indicates the reference group.
PFDR: P values after adjusting for multiple testing by controlling the FDR of 0.05 based on the P values from ANOVA tests for each gene.
Discussion
In this study, based on findings in large-scale genetic and epigenetic studies on childhood asthma, as well as related genes extracted from a database, we identified overlapping genes and examined whether the overlap was related to methQTLs. The number of genes overlapping between the two types of studies is small (six genes). After excluding one gene due to missingness of DNAm in the IoW cohort, 34 SNPs on three genes (DUSP22, IKZF3, and RUNX1) were shown to be methQTLs. This finding indicates that the observed epigenetic effects of CpGs on these three genes were likely to be genetic effects, although the effects of methQTLs on DNAm may change with other factors such as exposures.
Regardless of window sizes when defining neighboring SNPs, in overlapping genes IL4 and TLE4, methQTLs were not identified. The observation that the number of genes showing methQTLs is irrelevant to the selection of window sizes strengthens the potential of absent methQTLs on genes IL4 and TLE4. Thus, among the small number of genes identical between genetic and epigenetic findings on asthma, the overlapping is likely to be only partially due to methQTLs. The rather small number of overlapping genes and results from methQTL assessment might suggest distinct mechanisms for genetic and epigenetic impact on childhood asthma. We postulate an alternative explanation of this disparity that focusing on genetics only or epigenetics only provides an incomplete rather than comprehensive understanding of genes contributing to the development of asthma. Furthermore, the lack of overlapping genes emphasizes the importance and need to combine both genetic and epigenetic information through their joint effects or interactions, to further advance our understanding of the underlying mechanisms of asthma.
Previous epidemiological studies have shown that genetic variants influence DNAm which can act as effect modifiers for phenotypes of asthma. Recent studies have shown that SNPs and DNAm may interact to affect the risk of asthma.2,3,22-24 Soto-Ramirez et al demonstrated the role of genetic and epigenetic factors within the genomic region of IL4R gene of asthma risk. They found the risk of asthma is moderated by the CpG site located far away from SNPs after multiple testing adjustment. These factors (CpG site and SNP) collectively contribute to gene expression or alternative splicing.24 In addition, previous research has also shown that DNAm may act as a mediator for the association of genetic variants (SNPs) with gene expression.25 All these indicate that genetics and epigenetics may work jointly to influence the risk of asthma. Accompanied by transcriptional regulatory properties, the independent as well as joint functionality of genetic and epigenetic factors on asthma incidence can be further investigated, leading to improved understanding of the complex disease mechanisms underlying asthma. On the other hand, it is worth noting that genetic factors of asthma are sensitive to ethnicity, and careful study designs are essential when jointly assessing genetic and epigenetic effects on asthma to reduce the occurrence of false discoveries.
The small sample size is a limitation of our study. It potentially affected the power of detecting methQTLs. In addition, with small sample sizes, the probability to have rare genetic variants in our study samples was expected to be low, which also affected the power of methQTL detection. Although we have managed to improve the homogeneity of our detected genes to a certain extent concerning the age of patients with asthma and region of study populations, we acknowledge that population stratification might still exist in the findings. Nevertheless, the small fraction of asthma risk explained by genetic variants and the lack of overlap in genes between genetic and epigenetic associations suggest an opportunity to examine the combined effects of genetic variants and epigenetics. We postulate that this effort will enhance our understanding of the underlying genetic and epigenetic mechanisms of asthma. By examining their combined effects through interactions or networks, we are likely to discover novel and critical biomarkers of asthma.
Supplemental Material
Supplemental material, Supplemental_Tables_xyz29536d7d85c79 for Interweaving Between Genetic and Epigenetic Studies on Childhood Asthma by Aniruddha Rathod, Jiasong Duan, Hongmei Zhang, John W Holloway, Susan Ewart, S Hasan Arshad and Wilfried Karmaus in Epigenetics Insights
Acknowledgments
The authors are thankful to the nurses and staff at the David Hide Asthma and Allergy Research Center, Isle of Wight, UK, for their help in recruitment and sample collections and are thankful to all the cohort participants. Our special thanks also go to the High-Performance Computing facility provided by the University of Memphis.
Footnotes
Funding:The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Institutes of Health (grant numbers R01 AI121226, R01 HL132321).
Declaration of conflicting interests:The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions: HZ and WK conceived the study, designed the analytical plans, and provided guidance on the analytical and statistical aspects. SHA and SE provided data. JD did data management and performed all the statistical analyses. JWH provided guidance on epigenetics aspects. AR and HZ drafted the manuscript. All authors reviewed and approved the final manuscript.
ORCID iD: Aniruddha Rathod  https://orcid.org/0000-0002-4921-0952
https://orcid.org/0000-0002-4921-0952
Supplemental material: Supplemental material for this article is available online.
References
- 1. Global Asthma Network. The Global Asthma Report 2018. Auckland, New Zealand: Global Asthma Network; 2018. [Google Scholar]
- 2. Kogan V, Millstein J, London SJ, et al. Genetic-epigenetic interactions in asthma revealed by a genome-wide gene-centric search. Hum Hered. 2018;83:130-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhang H, Tong X, Holloway JW, et al. The interplay of DNA methylation over time with Th2 pathway genetic variants on asthma risk and temporal asthma transition. Clin Epigenetics. 2014;6:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pividori M, Schoettler N, Nicolae DL, Ober C, Im HK. Shared and distinct genetic risk factors for childhood-onset and adult-onset asthma: genome-wide and transcriptome-wide studies. Lancet Respir Med. 2019;7:509-522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Demenais F, Margaritte-Jeannin P, Barnes KC, et al. Multiancestry association study identifies new asthma risk loci that colocalize with immune cell enhancer marks. Nat Genet. 2018;50:42-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ferreira MA, Vonk JM, Baurecht H, et al. Shared genetic origin of asthma, hay fever and eczema elucidates allergic disease biology. Nat Genet. 2017;49:1752-1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yan Q, Brehm J, Pino-Yanes M, et al. A meta-analysis of genome-wide association studies of asthma in Puerto Ricans. Eur Respir J. 2017;49:1601505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Galanter JM, Gignoux CR, Torgerson DG, et al. Genome-wide association study and admixture mapping identify different asthma-associated loci in Latinos: the Genes-environments & Admixture in Latino Americans study. J Allergy Clin Immunol. 2014;134:295-305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Acevedo N, Reinius LE, Greco D, et al. Risk of childhood asthma is associated with CpG-site polymorphisms, regional DNA methylation and mRNA levels at the GSDMB/ORMDL3 locus. Hum Mol Genet. 2015;24:875-890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lee JU, Kim JD, Park CS. Gene-environment interactions in asthma: genetic and epigenetic effects. Yonsei Med J. 2015;56:877-886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Best LG, Azure C, Segarra A, et al. Genetic variants and risk of asthma in an American Indian population. Ann Allergy Asthma Immunol. 2017;119:31.e1-36.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. DeVries A, Vercelli D. Early predictors of asthma and allergy in children: the role of epigenetics. Curr Opin Allergy Clin Immunol. 2015;15:435-439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Reese SE, Xu CJ, Den Dekker HT, et al. Epigenome-wide meta-analysis of DNA methylation and childhood asthma. J Allergy Clin Immunol. 2018;143:2062-2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yang IV, Pedersen BS, Liu A, et al. DNA methylation and childhood asthma in the inner city. J Allergy Clin Immunol. 2015;136:69-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Xu CJ, Soderhall C, Bustamante M, et al. DNA methylation in childhood asthma: an epigenome-wide meta-analysis. Lancet Respir Med. 2018;6:379-388. [DOI] [PubMed] [Google Scholar]
- 16. Arshad SH, Holloway JW, Karmaus W, et al. Cohort profile: the Isle of Wight whole population birth cohort (IOWBC). Int J Epidemiol. 2018;47:1043i-1044i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Beyan H, Down TA, Ramagopalan SV, et al. Guthrie card methylomics identifies temporally stable epialleles that are present at birth in humans. Genome Res. 2012;22:2138-2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Aryee MJ, Jaffe AE, Corrada-Bravo H, et al. Minfi: a flexible and comprehensive bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30:1363-1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Taylor DL, Jackson AU, Narisu N, et al. Integrative analysis of gene expression, DNA methylation, physiological traits, and genetic variation in human skeletal muscle. Proc Natl Acad Sci U S A. 2019;116:10883-10888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Du P, Zhang X, Huang CC, et al. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinform. 2010;11:587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. R Development Core Team. R: A Language and Environment for Statistical Computing [computer program]. Vienna, Austria: R Foundation for Statistical Computing; 2019. [Google Scholar]
- 22. Guthikonda K, Zhang H, Nolan VG, et al. Oral contraceptives modify the effect of GATA3 polymorphisms on the risk of asthma at the age of 18 years via DNA methylation. Clin Epigenetics. 2014;6:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Karmaus W, Ziyab AH, Everson T, Holloway JW. Epigenetic mechanisms and models in the origins of asthma. Curr Opin Allergy Clin Immunol. 2013;13:63-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Soto-Ramirez N, Arshad SH, Holloway JW, et al. The interaction of genetic variants and DNA methylation of the interleukin-4 receptor gene increase the risk of asthma at age 18 years. Clin Epigenetics. 2013;5:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kim S, Forno E, Yan Q, et al. SNPs identified by GWAS affect asthma risk through DNA methylation and expression of cis-genes in airway epithelium. Eur Respir J. 2020;55:1902079. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, Supplemental_Tables_xyz29536d7d85c79 for Interweaving Between Genetic and Epigenetic Studies on Childhood Asthma by Aniruddha Rathod, Jiasong Duan, Hongmei Zhang, John W Holloway, Susan Ewart, S Hasan Arshad and Wilfried Karmaus in Epigenetics Insights