

Abstract
Poly(ADP-ribose) polymerase (PARP) inhibitors are a class of anticancer drugs that block the catalytic activity of PARP proteins. Optimization of our lead compound 1 ((Z)-2-benzylidene-3-oxo-2,3-dihydrobenzofuran-7-carboxamide; PARP-1 IC50 = 434 nM) led to a tetrazolyl analogue (51, IC50 = 35 nM) with improved inhibition. Isosteric replacement of the tetrazole ring with a carboxyl group (60, IC50 = 68 nM) gave a promising new lead, which was subsequently optimized to obtain analogues with potent PARP-1 IC50 values (4 nM – 200 nM). PARP enzyme profiling revealed that the majority of compounds are selective toward PARP-2 with IC50 values comparable to clinical inhibitors. X-ray crystal structures of the key inhibitors bound to PARP-1 illustrated the mode of interaction with analogue appendages extending toward the PARP-1 adenosine-binding pocket. Compound 81, an isoform-selective PARP-1/−2 (IC50 = 30 nM/2 nM) inhibitor, demonstrated selective cytotoxic effect toward BRCA1-deficient cells compared to isogenic BRCA1–proficient cells.
Graphical Abstract
INTRODUCTION
Exogenous and endogenous genotoxic injuries lead to DNA single strand breaks (SSBs) and DNA double strand breaks (DSBs), which collectively trigger the DNA damage response (DDR) in cells. In cancer cells, SSBs occur at a frequency of up to 10,000 per cell each day.1 While SSBs are repaired by base excision repair (BER),2 repair of DSBs requires a functional homologous recombination (HR) or non-homologous end joining (NHEJ) repair mechanism.3, 4 Computational analyses indicated the involvement of approximately 400 proteins in the regulation of the DDR process.5, 6 Poly(ADP-ribose) polymerase-1 (PARP-1) plays an important role in BER-mediated DNA damage repair as well as other pathways7 by binding to the damaged DNA through the coordinated action of its N-terminal zinc finger motifs. The C-terminal catalytic site of PARP-1 hydrolyzes NAD+ substrate into ADP-ribose and nicotinamide (NI). Branched and linear chains of ADP-ribose units are covalently transferred onto a wide range of target proteins such as DNA polymerases, histones, DNA ligases, p53 and topoisomerase I/II (heteromodification), and onto PARP itself (automodification).8 Thus, PARP-1 acts as a “writer” of poly (ADP-ribosylation) (PARylation).9 PARylation has been shown to play a role in cellular processes such as DNA damage repair, maintaining genomic stability, regulation of transcription, and cell death.10, 11 PARylation of PARP-1 is necessary for non-covalent recruitment of DNA repair proteins, including DNA ligase III, DNA polymerase β (pol β) and XRCC1 to the sites of DNA breaks.12–14 PARylation of PARP-1 is also thought to promote its dissociation from DNA damage sites to allow repair.15, 16 Therefore, targeting PARP-1 with small molecule inhibitors is an attractive strategy to enhance antitumor effect.17–23
Synthetic lethality is a strategy that exploits gene defects in cancer for therapeutic benefit.24 The foremost example of synthetic lethality as a targeted cancer therapy is the use of PARP inhibitors in the treatment of cancer in individuals with germline mutations in BRCA1 or BRCA2. In addition to blocking the catalytic activity of PARP proteins, some PARP inhibitors (niraparib, olaparib, rucaparib and talazoparib) act at least in part by trapping PARP on damaged DNA.23 This trapping interferes with DNA replication causing double stranded breaks that cannot be repaired in HR-defective tumor cells. PARP-1 inhibitors as single agents are, therefore, efficacious in treating tumors deficient in HR components, including BRCA1/2, but are of limited utility in tumors with normal or restored function of HR repair mechanism.25–29 Consequently, the use of FDA approved PARP-1 inhibitors such as olaparib,30 niraparib,31 rucaparib32 and talazoparib33 has mainly focused on their therapeutic role as a monotherapy to treat BRCA-deficient tumors based on the concept of synthetic lethality.23 These drugs and another PARP-1 inhibitor, veliparib,34 are also currently undergoing advanced clinical trials as combination and/or single agents in cancer therapy (Figure 1).35 Clinical PARP-1 inhibitors are also useful for the treatment of other cancers with DNA DSB repair deficiency such as those with BRCAness.36 Taking these factors into consideration, PARP-1 remains an attractive target for anticancer drug development.
Figure 1.
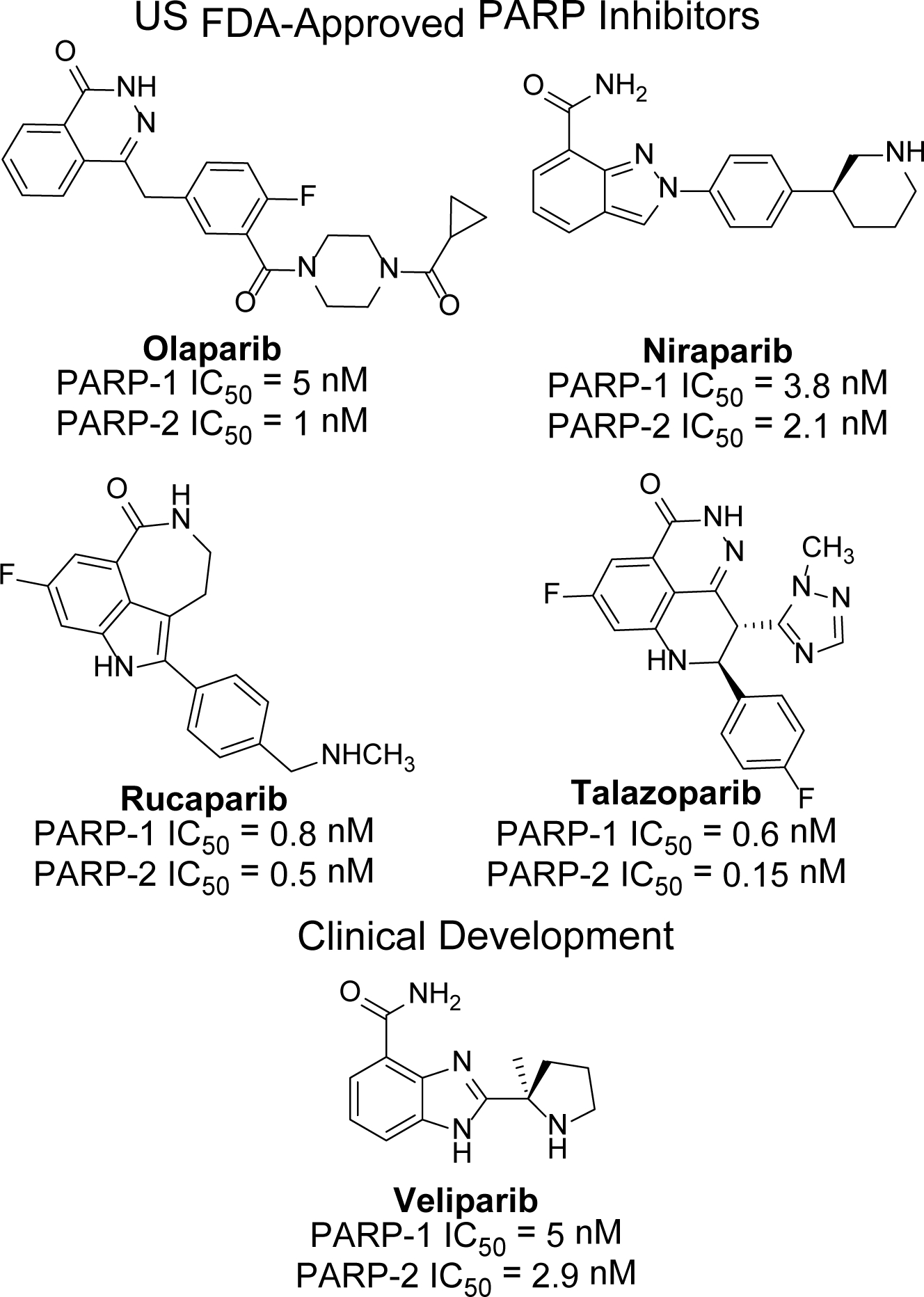
Structures and PARP-1/PARP-2 inhibitory activity of clinical compounds either US FDA approved or undergoing Phase III clinical trials.
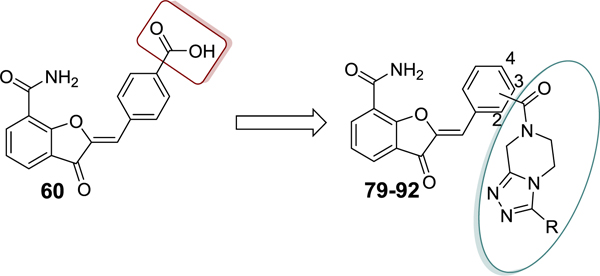
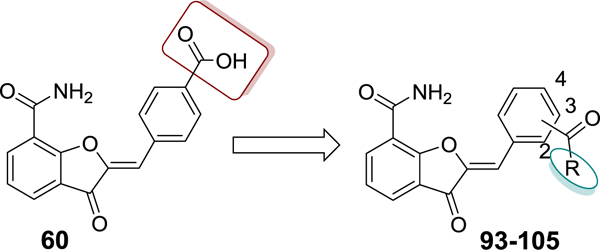
In this article, we report the design, synthesis, structure-activity relationship (SAR), and in vitro evaluation of our previously published lead compound 1,37 thereby leading to the identification of several unique PARP-1 inhibitors (Figure 2A and 2B). Compound 1 binds to the NI pocket of the PARP-1 catalytic fold. Based on previous structural data,37 we hypothesized that installation of a 4’-carboxyl group in compound 1 is an ideal vector to facilitate the incorporation of a wide range of substituents (pyridine, pyrimidine, pyrazine, 1,3,5-triazine, 1,3,4-thiadiazole, 5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine (THTP), and benzimidazole) directed toward engaging the adenosine-binding pocket (ABP) of PARP-1 with the goal of developing PARP-1 inhibitors with improved potency and a unique mode of engaging the PARP-1 active site. Indeed, we show that these new compounds act as potent inhibitors of PARP-1 and PARP-2 with desirable (low nM) IC50 values. Key target compounds showed high selectivity toward PARP-1 and PARP-2 over other catalytic PARP isoforms and specifically inhibited growth of BRCA1-mutant cells, thus providing refined leads for further optimization to produce preclinical candidates.
Figure 2.
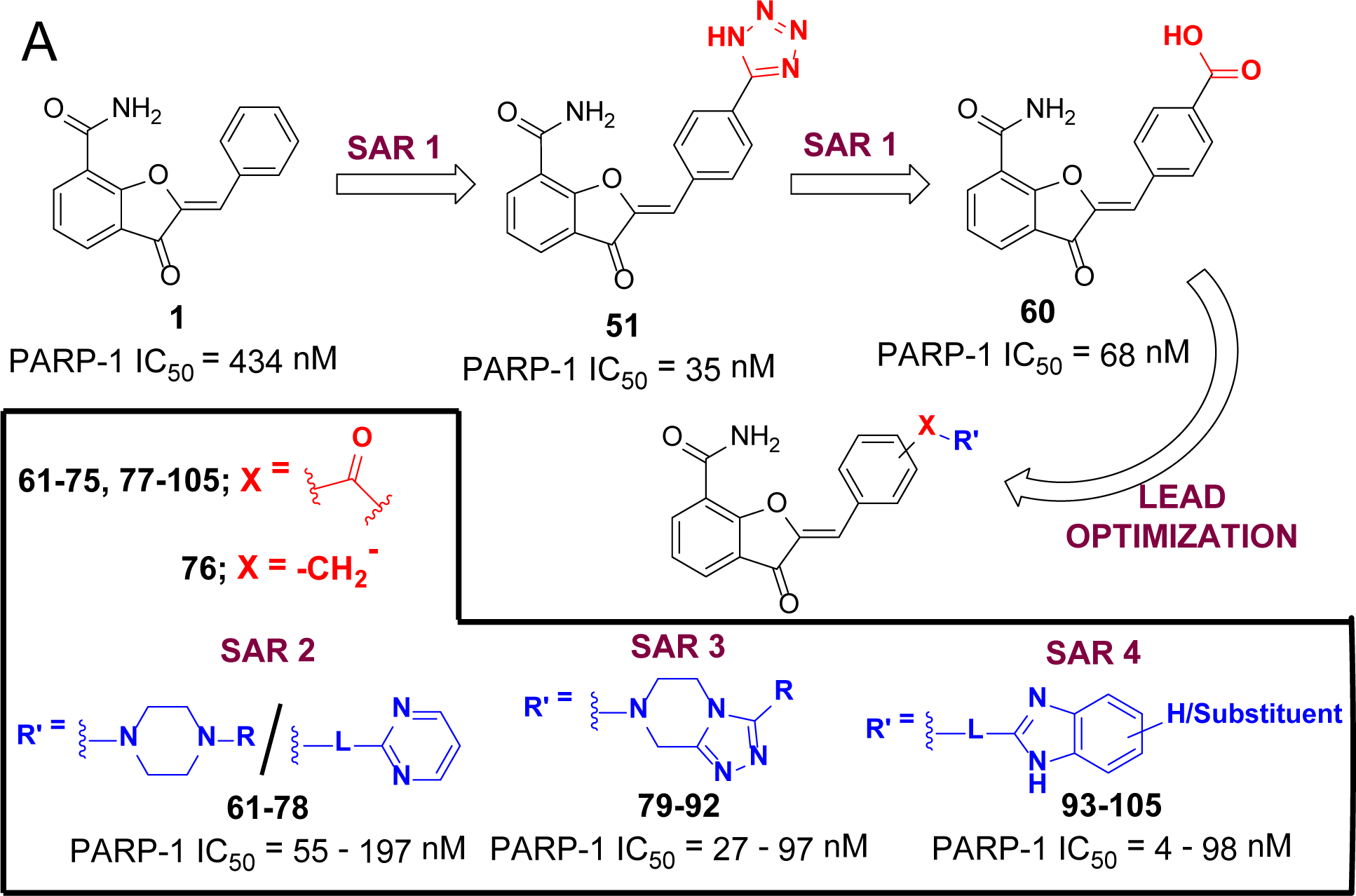
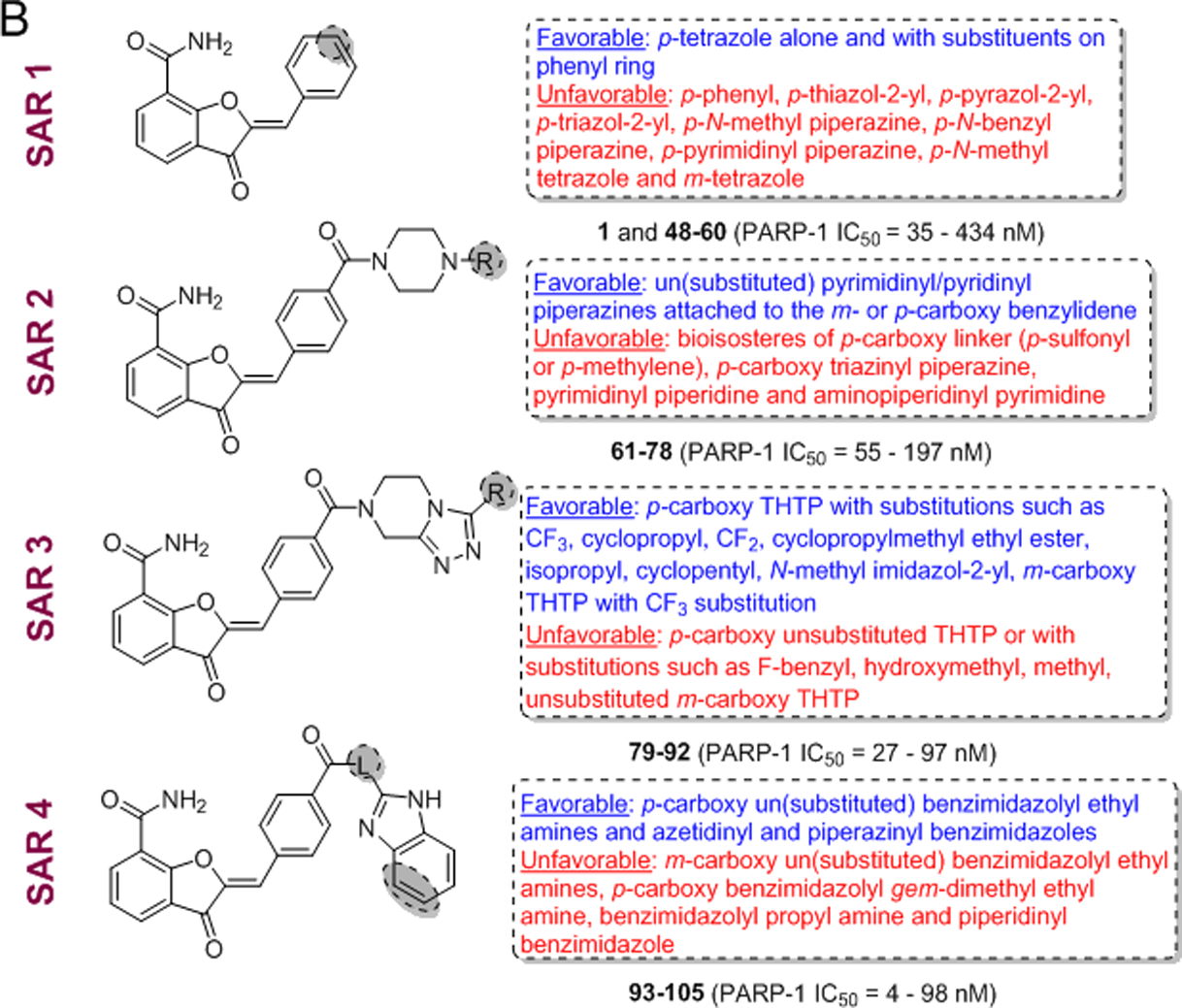
(A) Design strategy for sequential optimization of high nanomolar inhibitory lead 60. (B) Favorable and unfavorable substitutions outlined for the four phases of SAR (Linkers in the structures are abbreviated as ‘L’).
RESULTS AND DISCUSSION
Chemistry.
Benzaldehyde derivatives and the other intermediates as precursors to the synthesis of target compounds were prepared according to Schemes 1–4.
Scheme 1. Synthesis of Benzaldehyde Intermediates 2–8a.
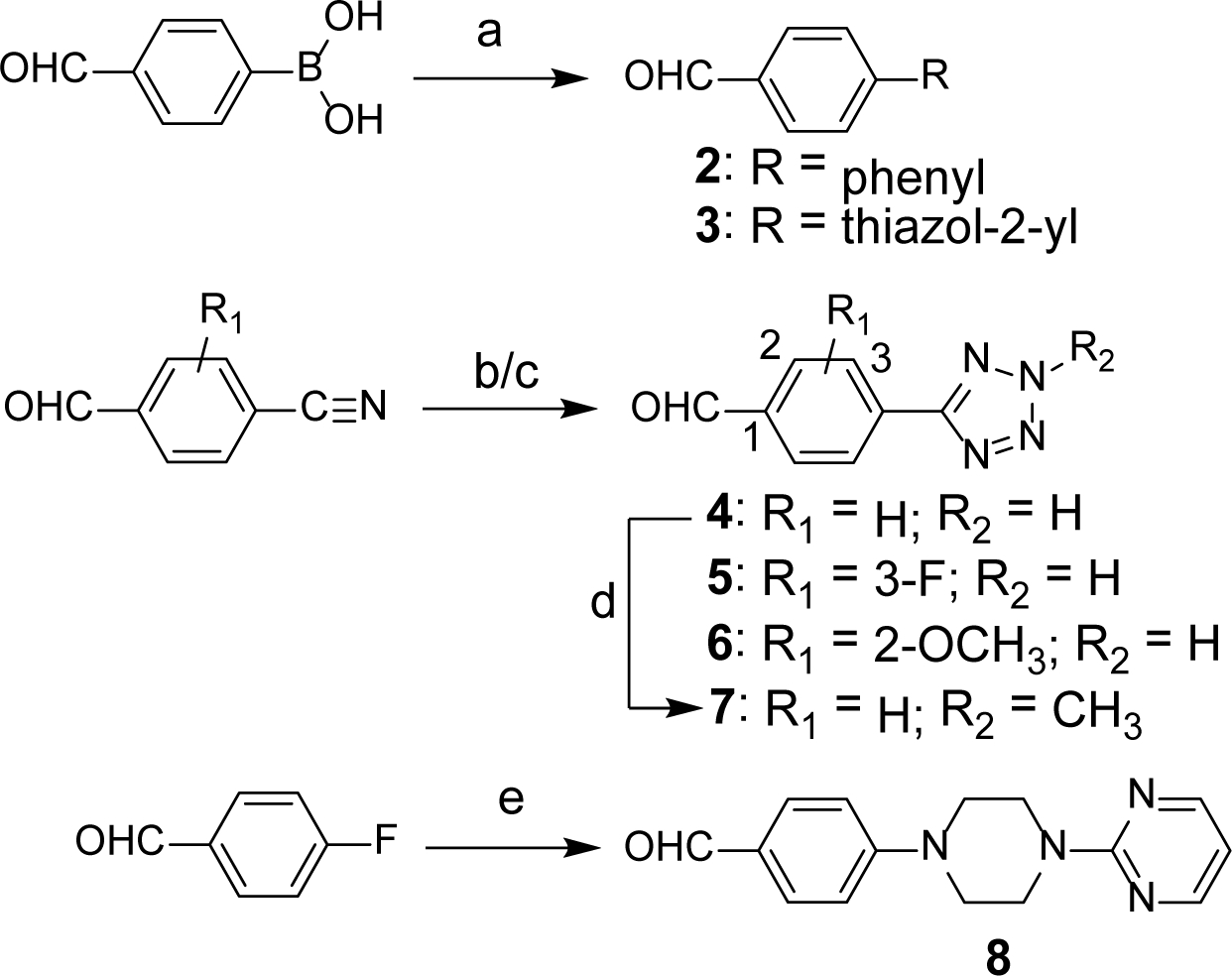
aReagents and conditions: (a) bromobenzene for 2 and 2-chlorothiazole for 3, Pd(PPh3)4, K2CO3, H2O, THF, 80°C, 12 h; (b) NaN3, Et3N, DMF, 180°C, overnight for 4; (c) NaN3, Et2NH.HCl, toluene, reflux, 24 h for 5 and 6; (d) 4, CH3I, K2CO3, DMF, rt, 4 h; (e) pyrimidin-2-yl-piperazine, K2CO3, DMF, 130°C, 24 h.
Scheme 4. Synthesis of Intermediates 43, 43a and 47a.
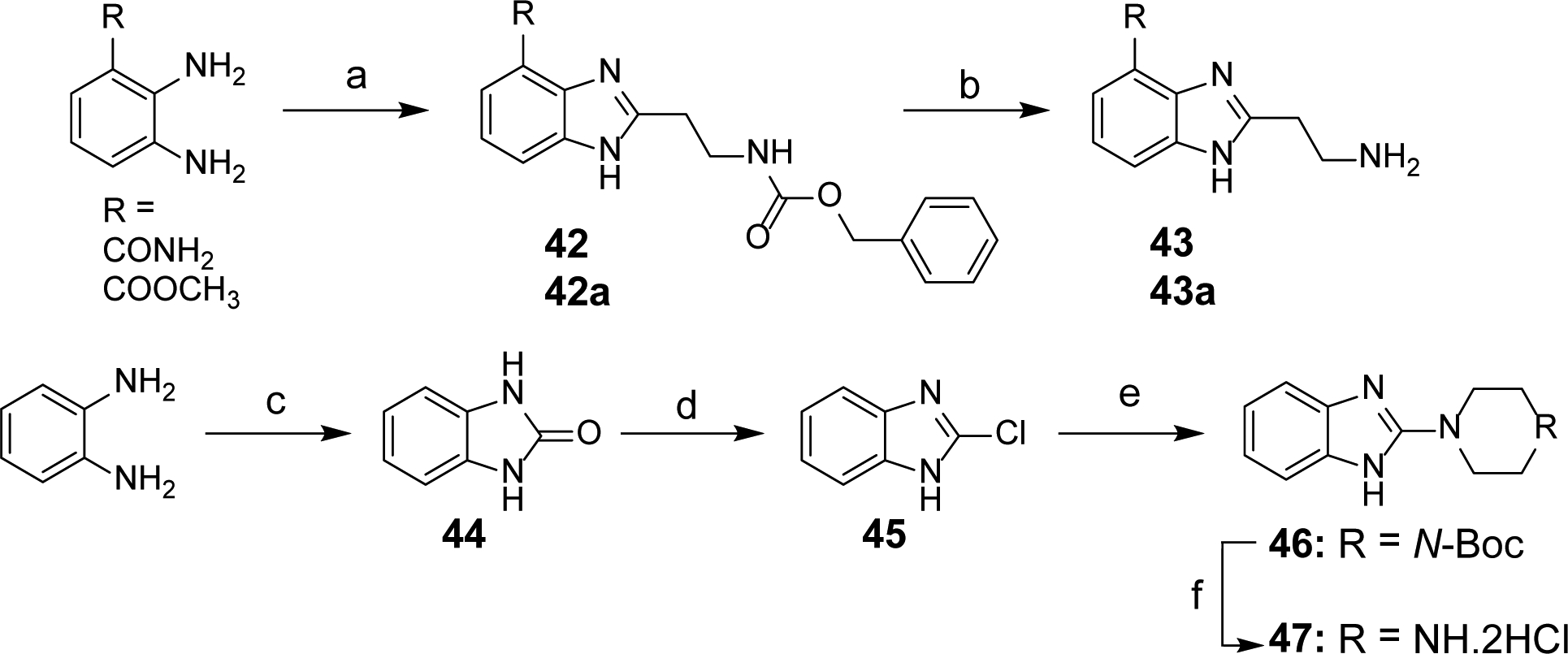
aReagents and conditions: (a) Benzyl 3-oxopropylcarbamate, NH4OAc, DMF, 80°C, 6 h (for 42); benzyl 3-oxopropylcarbamate, HCTU, EtN(i-Pr)2, DMF, rt to reflux, 10 h (for 42a); (b) H2, Pd/C, CH3OH, rt, 5 h; (c) 1,1′-carbonyldiimidazole, THF, rt, 22 h; (d) POCl3, 95°C, 16 h; (e) N-Boc piperazine, toluene, MW, 150°C, 6h; (f) 4N HCl, dioxane, rt, overnight.
Synthesis of Benzaldehyde Intermediates 2–8 (Scheme 1).
Scheme 1 represents the synthesis of substituted benzaldehydes. For the synthesis of 4-phenyl or 4-thiazol-2-yl benzaldehydes 2 and 3, reported Suzuki coupling conditions were utilized.38, 39 The [2+3] cycloaddition reaction of 4-cyanobenzaldehyde with sodium azide in the presence of triethylamine produced tetrazolyl derivative 4.40 An alternate procedure (sodium azide/diethylamine hydrochloride/toluene)41 was used for conversion of 4-cyano-3-fluorobenzaldehyde and 4-cyano-2-methoxybenzaldehyde to corresponding tetrazolyl substituted benzaldehydes 5 and 6 because the conditions used for the synthesis of 4 proved unsuccessful. N-Methyl derivative 7 was prepared from benzaldehyde 4 using iodomethane.42 Further, SNAr reaction of 4-fluorobenzaldehyde with pyrimidin-2-yl-piperazine yielded benzaldehyde derivative 8.43 These benzaldehyde intermediates (2-8) and the other commercially available benzaldehydes (see experimental) served as precursors for the synthesis of target compounds shown in Table 1.
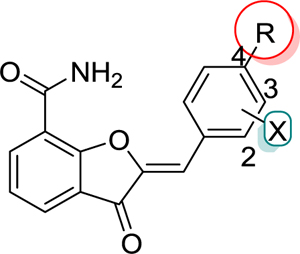
Table 1.
Initial Optimization of Lead Compound 1
 |
||||||
|---|---|---|---|---|---|---|
| Compd | X | R | PARP-1 IC50 (nM)a | pIC50 ± S.D (nM) | PARP-2 IC50 (nM)a | pIC50 ± S.D (nM) |
| 1 | H | -H | 434 | 6.41 ± 0.20 | NT | - |
| 48 | H |  |
>50 (2%b) | - | NT | - |
| 49 | H | 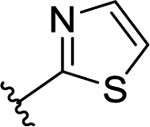 |
>50 (7%) | - | NT | - |
| 50 | H | 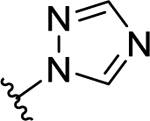 |
>50 (5%) | - | NT | - |
| 51 | H | 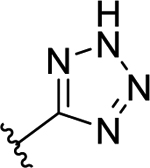 |
35 | 7.50 ± 0.20 | 2.1 | 8.69 ± 0.01 |
| 52 | H | 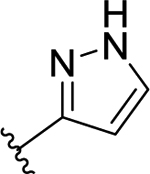 |
>50 (4%) | - | NT | - |
| 53 | 3-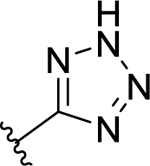
|
-H | >50 (14%) | - | <50 (76% c) | - |
| 54 | H | 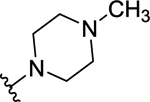 |
>50 (7%) | - | NT | - |
| 55 | H | 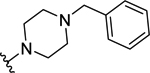 |
>50 (10%) | - | NT | - |
| 56 | H | 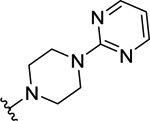 |
>50 (19%) | - | <50 (56%) | - |
| 57 | 3-F | 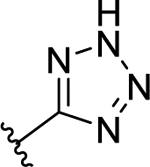 |
56 | 7.28 ± 0.14 | <50 (100%) | - |
| 58 | 2-OCH3 | 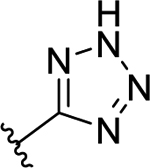 |
47 | 7.33 ± 0.07 | 1.6 | 8.80 ± 0.05 |
| 59 | H | 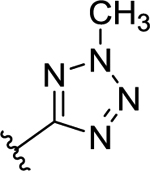 |
>50 (4%) | - | NT | - |
| 60 | H | -COOH | 68 | 7.17 ± 0.08 | NT | - |
| Olad | - | - | 1.2 | 8.93 ± 0.07 | 0.5 | 9.40 ± 0.30 |
| Vele | - | - | 1.5 | 8.83 ± 0.06 | 100% @ 10 nM | - |
Data shown are mean values obtained from two independent experiments performed in duplicates.
% inhibition screening at a single concentration (50 nM unless otherwise specified) was performed in duplicates and data shown is an average of two independent experiments;
% inhibition screening of PARP-2, was performed at 50 nM concentration, in duplicates by one experiment;
Olaparib;
Veliparib;
Not tested (NT).
Synthesis of Benzaldehyde Intermediates 9–17 and 20–28 (Scheme 2).
Scheme 2. Synthesis of Benzaldehyde Intermediates 9–17 and 20–28a.
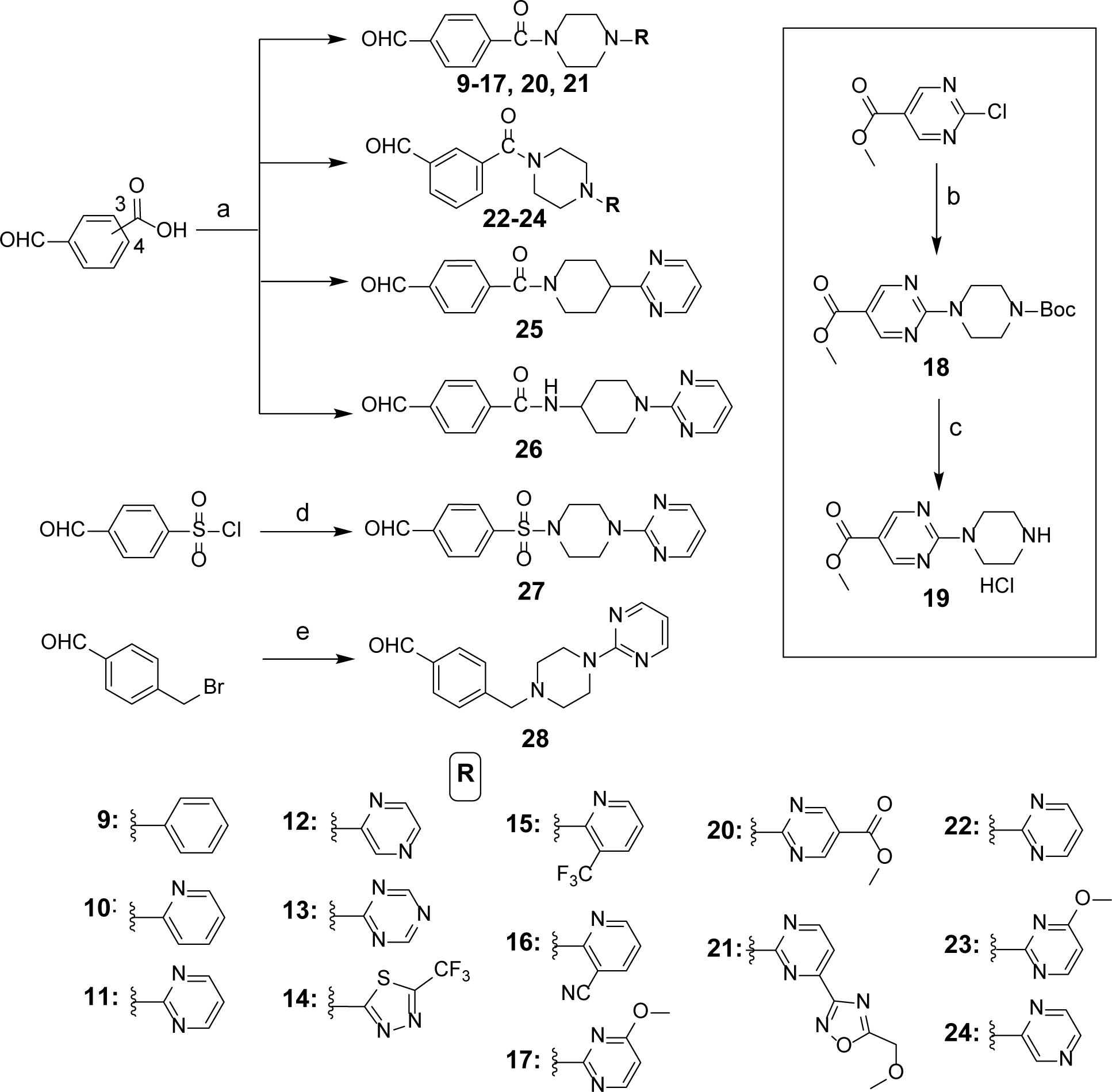
aReagents and conditions: (a) substituted commercial piperazine or piperidine or 4-aminopiperidine or synthesized piperazine 19 (see scheme in inset), HCTU, HOBt, EtN(i-Pr)2, DCM, 0°C to rt, overnight; (b) N-Boc piperazine, K2CO3, CH3CN, reflux, overnight; (c) 4N HCl, dioxane, rt, overnight; (d) pyrimidin-2-yl-piperazine, Et3N, DCM, 0°C to rt, 12 h; (e) pyrimidin-2-yl-piperazine, K2CO3, CH3CN, reflux, overnight.
Benzaldehyde intermediates 9-17 were prepared by coupling commercially available N4-substituted piperazines with commercially available 4-carboxybenzaldehyde in the presence of HCTU, HOBt and N,N-diisopropylethylamine (DIPEA).44 Ester 18 was synthesized by SNAr reaction on methyl 2-chloropyrimidinyl-5-carboxylate using N4-Boc protected piperazine as the nucleophile.37, 45 The Boc protection was then removed with 4N HCl in dioxane to obtain the hydrochloride salt 19. Piperazinyl derivative 19 was next coupled with 4-carboxybenzaldehyde in the presence of HCTU, HOBt and DIPEA to produce benzaldehyde intermediate 20. Benzaldehydes 21-24 were prepared using commercially available N4-substituted piperazines and 3- or 4-carboxybenzaldehyde in the presence of previously mentioned peptide coupling conditions. Similarly, intermediates 25 and 26 were prepared by coupling commercially available piperidine or aminopiperidine with 4-carboxybenzaldehyde. Intermediate 27 was obtained by reacting commercially available 4-formylbenzene sulfonyl chloride with pyrimidin-2-yl-piperazine in the presence of triethylamine.37 Intermediate 28 was obtained via an SN2 reaction using commercially available 4-bromomethyl benzaldehyde and pyrimidin-2-yl-piperazine.37 These benzaldehyde intermediates were used for the preparation of target compounds shown in Table 2.
Table 2.
The Effect of Substituted Piperazine/Piperidine Substituents on the Phenyl Portion of Benzylidene Moiety
 | |||||
|---|---|---|---|---|---|
| Compd | Position | R | PARP-1 IC50 (nM)a | pIC50 ± S.D (nM) | PARP-2 IC50 (nM)a |
| 51 | - | - | 35 | 7.50 ± 0.20 | 2.1 |
| 60 | - | - | 68 | 7.17 ± 0.08 | NT |
| 61 | 4- |  |
>50 (19%b) | - | NT |
| 62 | 4- |  |
66 | 7.18 ± 0.01 | NT |
| 63 | 4- |  |
55 | 7.27 ± 0.10 | <50 (89%c) |
| 64 | 4- | 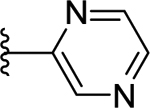 |
77 | 7.12 ± 0.04 | <50 (89%) |
| 65 | 4- | 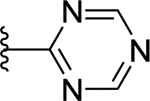 |
>50 (8%) | - | NT |
| 66 | 4- | 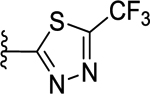 |
>50 (22%) | - | NT |
| 67 | 4- | 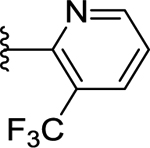 |
>50 (10%) | - | NT |
| 68 | 4- | 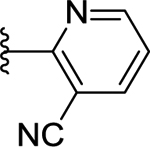 |
66 | 7.18 ± 0.04 | NT |
| 69 | 4- | 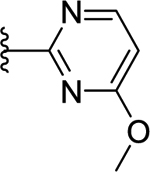 |
66 | 7.18 ± 0.05 | <50 (92%) |
| 70 | 4- | 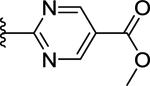 |
>50 (34%) | - | NT |
| 71 | 4- | 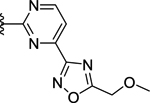 |
>50 (36%) | - | <50 (98%) |
| 72 | 3- | 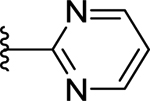 |
58 | 7.24 ± 0.06 | <50 (89%) |
| 73 | 3- | 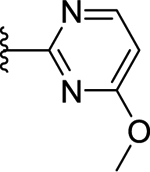 |
>50 (32%) | - | <50 (96%) |
| 74 | 3- | 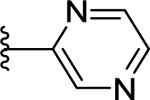 |
>50 (16%) | - | <50 (88%) |
| 75 | 4- | 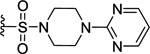 |
197 | 6.71 ± 0.08 | NT |
| 76 | 4- | 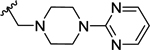 |
>50 (NA) | - | <50 (51%) |
| 77 | 4- | 112 | 6.95 ± 0.01 | NT | |
| 78 | 4- | 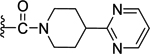 |
>50 (3%) | - | >50 (21%) |
| Olad | - | - | 1.2 | 8.93 ± 0.07 | 0.50 ± 0.30 |
| Vele | - | - | 1.5 | 8.83 ± 0.06 | 100% @ 10 nM |
Data shown are mean obtained from two independent experiments performed in duplicates.
% inhibition screening at a single concentration (50 nM unless otherwise specified) was performed in duplicates and data shown is an average of two independent experiments;
% inhibition screening of PARP-2, was performed at 50 nM concentration, in duplicates by one experiment;
Olaparib;
Veliparib;
No activity (NA);
Not tested (NT).
Synthesis of Benzaldehyde Intermediates 29–41 (Scheme 3).
Scheme 3. Synthesis of Benzaldehyde Intermediates 29–41a.
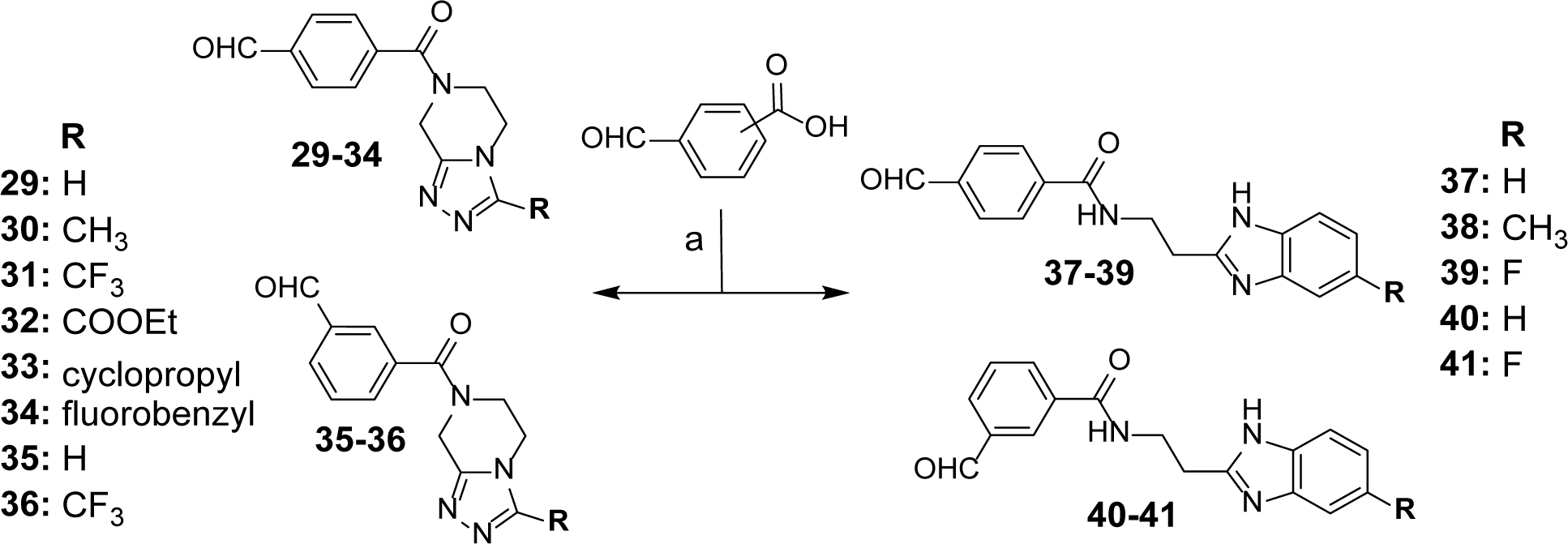
aReagents and conditions: (a) 4-formylbenzoic acid or 3-formylbenzoic acid, appropriate (un) substituted THTP or benzimidazole-2-yl-ethylamine, HCTU, HOBt, EtN(i-Pr)2, DCM, 0°C to rt, overnight.
Scheme 3 depicts the preparation of benzaldehyde intermediates 29-41, which were utilized to synthesize target compounds listed in Tables 3 and 4. The 3-carboxy or 4-carboxy benzaldehydes were coupled with commercially available (un)substituted THTPs to obtain intermediates 29-36 or (un)substituted benzimidazole-2-yl-ethylamines for intermediates 37-41 in the presence of HCTU/HOBt coupling conditions.44
Table 3.
The Effect of 1,2,4-Triazolopiperazine Amide Substituents on the Phenyl Ring of Benzylidene Moiety
 |
||||||
|---|---|---|---|---|---|---|
| Compd | Position | R | PARP-1 IC50 (nM)a | pIC50 ± S.D (nM) | PARP-2 IC50 (nM)a | pIC50 ± S.D (nM) |
| 51 | - | - | 35 | 7.50 ± 0.20 | 2.1 | 8.69 ± 0.01 |
| 60 | - | - | 68 | 7.17 ± 0.08 | NT | - |
| 79 | 4- | -H | 97 | 7.02 ± 0.10 | >10 (18%c) | - |
| 80 | 4- | -CH3 | 81 | 7.10 ± 0.09 | <10 (55%) | - |
| 81 | 4- | -CF3 | 30 | 7.53 ± 0.07 | 2 | 8.80 ± 0.01 |
| 82 | 4- | 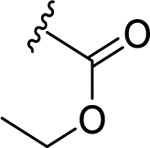 |
40 | 7.42 ± 0.12 | 3.7 | 8.44 ± 0.03 |
| 83 | 4- | 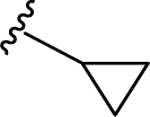 |
27 | 7.57 ± 0.05 | 1.9 | 8.72 ± 0.02 |
| 84 | 4- |  |
>50 (25%b) | - | <10 (59%) | - |
| 85 | 4- | -CHF2 | 30 | 7.52 ± 0.01 | 3 | 8.52 ± 0.03 |
| 86 | 4- | 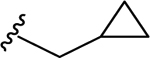 |
47 | 7.33 ± 0.06 | 3.5 | 8.46 ± 0.04 |
| 87 | 4- | 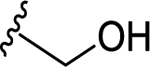 |
>50 | - | <10 (61%) | - |
| 88 | 4- | 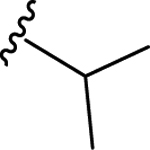 |
42 | 7.43 ± 0.21 | 4.6 | 8.34 ± 0.02 |
| 89 | 4- | 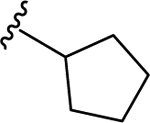 |
37 | 7.48 ± 0.22 | 3.9 | 8.42 ± 0.05 |
| 90 | 4- | 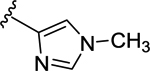 |
32 | 7.55 ± 0.22 | 3.3 | 8.48 ± 0.01 |
| 91 | 3- | -H | >50 (13%) | - | >10 (6%) | - |
| 92 | 3- | -CF3 | 42 | 7.38 ± 0.01 | >10 (47%) | - |
| Olad | - | - | 1.2 | 8.93 ± 0.07 | 0.5 | 9.40 ± 0.30 |
| Vele | - | - | 1.5 | 8.83 ± 0.06 | <10 nM | - |
Data shown are mean values obtained from two independent experiments performed in duplicates.
% inhibition screening at a single concentration (50 nM unless otherwise specified) was performed in duplicates and data shown is an average of two independent experiments;
% inhibition screening of PARP-2, was performed at 10 nM concentration, in duplicates by one experiment.
Olaparib;
Veliparib.
Not tested (NT).
Table 4.
Effect of Benzimidazolylethyl/ Benzimidazolylazetidine/ Benzimidazolylpiperazine Amide Substituents on the Phenyl Ring of Benzylidene Moiety
 |
||||||
|---|---|---|---|---|---|---|
| Compd | Position | R | PARP-1 IC50 (nM)a | pIC50 ± S.D (nM) | PARP-2 IC50 (nM)a | pIC50 ± S.D (nM) |
| 51 | - | - | 35 | 7.50 ± 0.20 | 2.1 | 8.69 ± 0.01 |
| 60 | - | - | 68 | 7.17 ± 0.08 | NT | - |
| 93 | 4- | 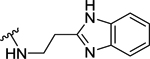 |
36 | 7.45 ± 0.10 | ~10 (50%c) | - |
| 94 | 4- | 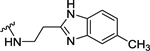 |
22 | 7.66 ± 0.02 | 5 | 8.32 ± 0.12 |
| 95 | 4- | 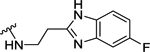 |
51 | 7.31 ± 0.14 | >10 (33%) | - |
| 96 | 3- | 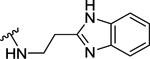 |
88 | 7.07 ± 0.10 | >10 (18%) | - |
| 97 | 3- | 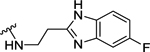 |
97 | 7.02 ± 0.07 | >10 (24%) | - |
| 98 | 4- | 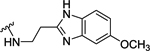 |
28 | 7.55 ± 0.02 | >10 (44%) | - |
| 99 | 4- | 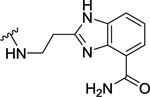 |
4 | 8.41 ± 0.11 | 0.7 | 9.19 ± 0.03 |
| 100 | 4- | 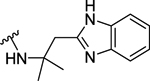 |
>50 (33%b) | - | >10 (22%) | - |
| 101 | 4- | 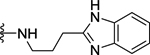 |
>50 (49%) | - | >10 (48%) | - |
| 102 | 4- | 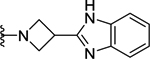 |
30 | 7.54 ± 0.10 | >10 (44%) | - |
| 103 | 4- | 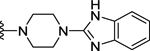 |
18 | 7.77 ± 0.15 | 4 | 8.40 ± 0.08 |
| 104 | 4- | 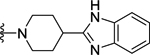 |
58 | 7.24 ± 0.05 | >10 (28%) | - |
| 105 | 4- | 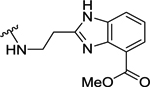 |
98 | 7.01 ± 0.02 | NT | - |
| Olad | - | - | 1.2 | 8.93 ± 0.07 | 0.5 | 9.40 ± 0.30 |
| Vele | - | - | 1.5 | 8.83 ± 0.06 | <10 nM | - |
Data shown are mean values obtained from two independent experiments performed in duplicates.
% inhibition screening at a single concentration (50 nM unless otherwise specified) was performed in duplicates and data shown is an average of two independent experiments;
% inhibition screening of PARP-2, was performed at 10 nM concentration, in duplicates by one experiment.
Olaparib;
Veliparib.
Not tested (NT).
Synthesis of Intermediate Amines 43, 43a and 47 (Scheme 4).
Scheme 4 represents the synthesis of key amine intermediates as precursors to obtain target compounds shown in Table 4. The 2,3-diaminobenzamide or methyl 2,3-diaminobenzoate were condensed with benzyl 3-oxopropylcarbamate leading to cyclized intermediates 42 or 42a, which were subjected to hydrogenolysis to obtain amines 43 or 43a. To synthesize piperazine intermediate 47, ortho-phenylenediamine was reacted with 1,1′-carbonyldiimidazole to generate a cyclic urea compound 44, followed by a chlorination reaction using neat POCl3 to obtain 2-chlorobenzimidazole 45. The chloro group in compound 45 was then replaced via microwave assisted SNAr reaction by N-Boc piperazine to obtain 46 and a subsequent Boc-deprotection using 4N HCl/dioxane mixture gave 47 as a di-hydrochloride salt.
Preparation of Target Compounds 48–105 (Scheme 5).
Scheme 5. Synthesis of Target Compounds 48–105a.
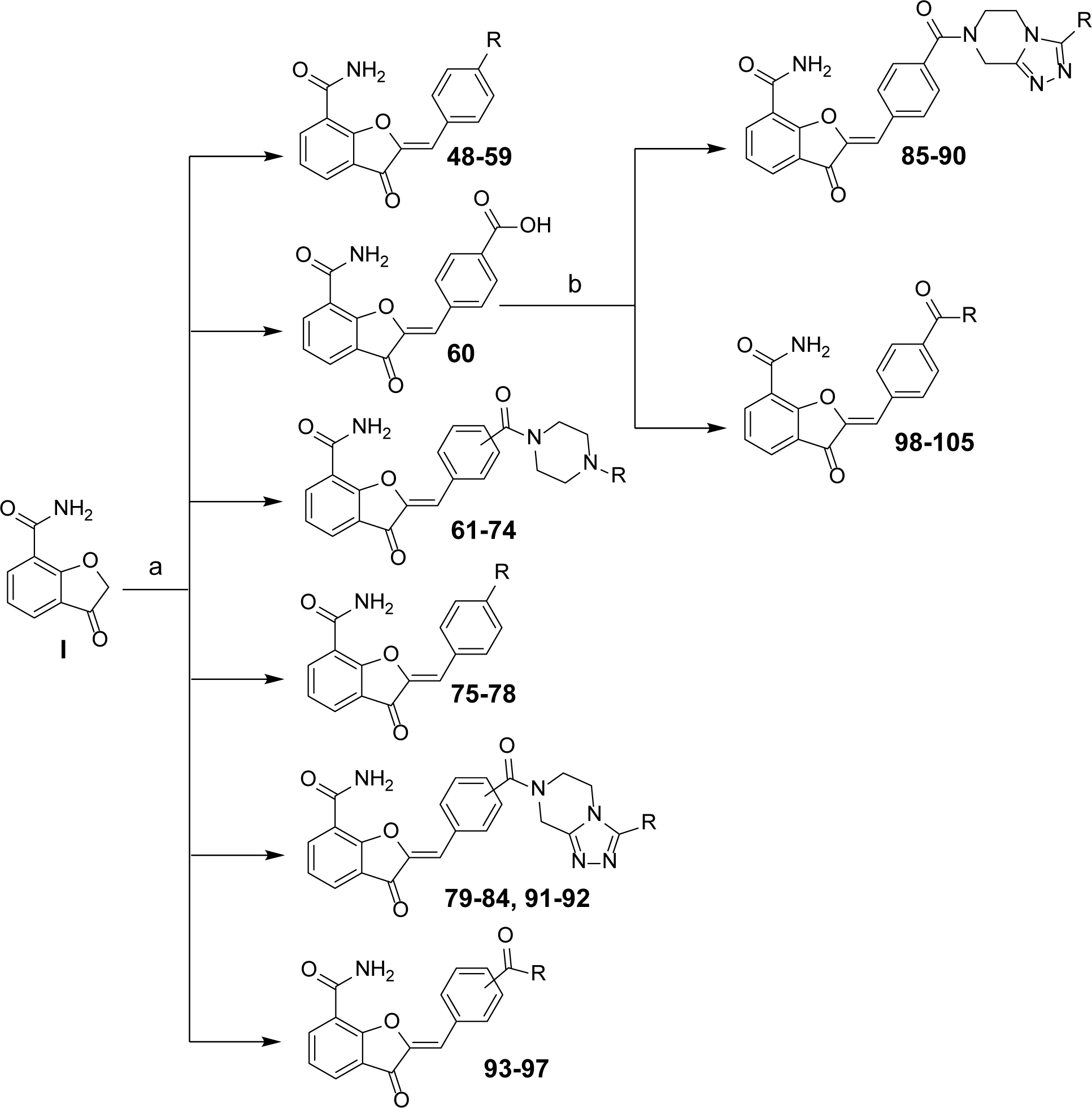
aReagents and conditions: (a) synthesized or commercial benzaldehydes, NH4OAc, toluene, reflux, 4–12 h; (b) appropriately substituted commercially obtained amines or synthesized amines 43, 47 and 43a HCTU, HOBt, EtN(i-Pr)2, DCM, 0°C to rt, overnight.
Target compounds were synthesized using the key intermediate 3-oxo-2,3-dihydrobenzofuran-7-carboxamide (I) (Supporting Information, Scheme S1 for details regarding synthesis of I). Scheme 5 depicts the synthesis of target compounds 48-84 and 91-97, using modified Knoevenagel condensation reaction of intermediate I with various synthesized or commercially obtained benzaldehydes.37 Target compounds 85-90, 98, 100-102 and 104 were prepared by coupling 60 with commercially obtained amines whereas target compounds 99, 103 and 105 were respectively prepared using synthesized amines 43, 47 and 43a in the presence of HCTU/HOBt coupling conditions.
Structure-Activity Relationship.
The present SAR study is based on our lead compound 1 ((Z)-2-benzylidene-3-oxo-2,3-dihydrobenzofuran-7-carboxamide), which showed PARP-1 inhibitory activity at a sub-micromolar concentration (IC50 = 434 nM) in PARP-1 enzyme assay. We began optimizing lead 1 to obtain a new and potent PARP-1 inhibitory series of dihydrobenzofuran-7-carboxamide (DHBF) compounds. This lead optimization program led to four innovative SAR phases as outlined in Tables 1–4. All newly synthesized compounds, along with positive controls, olaparib and veliparib, were tested using PARP-1 and PARP-2 chemiluminescence assay to obtain IC50 and pIC50 values (Supporting Information Figure S1 and S2 indicate PARP-1 and PARP-2 IC50 dose-response curves of representative compounds). Our IC50 values for the positive controls, olaparib and veliparib, were comparable to the reported values.30, 34
From each of the four SAR studies, X-ray crystal structures were determined for key target compounds in complex with the minimal ADP-ribosyltransferase (ART) fold of human PARP-1, representing a constitutively active form of PARP-1.46 ART interaction with NAD+ is primarily mediated through a NI-binding site and ABP. Attached to the ART fold is an autoinhibitory helical domain (HD) that acts to selectively block access to substrate NAD+ by interfering with the ABP (Figure 3A).46, 47 Upon binding to damaged DNA, the N-terminal regulatory domains of PARP-1 assemble in a way that leads to unfolding of the HD, thus relieving autoinhibition and allowing for binding of NAD+ to the active site.46–48 Our initial binding analysis of the newly synthesized compounds by differential scanning fluorimetry (DSF) revealed that most of these compounds were unable to bind to the catalytic domain (CAT) in the presence of a folded HD, but they successfully bound to the CAT with the deleted helical domain (CATΔHD) (Figure 3B). These compounds thus behave similar to the NAD+ analogue benzamide adenine dinucleotide (BAD), which can only bind to PARP-1 when the HD is deleted, or unfolded in the presence of DNA damage.47 This biophysical study suggests that our compounds may display increased efficacy and cancer cell specificity while showing reduced cytotoxicity in normal cells. Moreover, the binding analysis indicated that the designed extensions to compound 1 were likely to engage the ABP. Based on these data, the CATΔHD construct was used for crystallographic analysis. Five structures were determined using X-ray diffraction data extending to resolutions between 1.5 to 2.2 Å (Supporting Information Table S1 and Figure S3 and Figure 4). Each of the bound inhibitors exhibited hydrogen bonding interactions with key amino acid residues in the NI-binding site such as Ser904 and Gly863 and a water-mediated hydrogen bond with the catalytic residue Glu988. The crystal structures helped to understand the molecular basis of PARP-1 inhibition and allowed us to rationalize the observed SAR trends, as described in the sections below.
Figure 3.
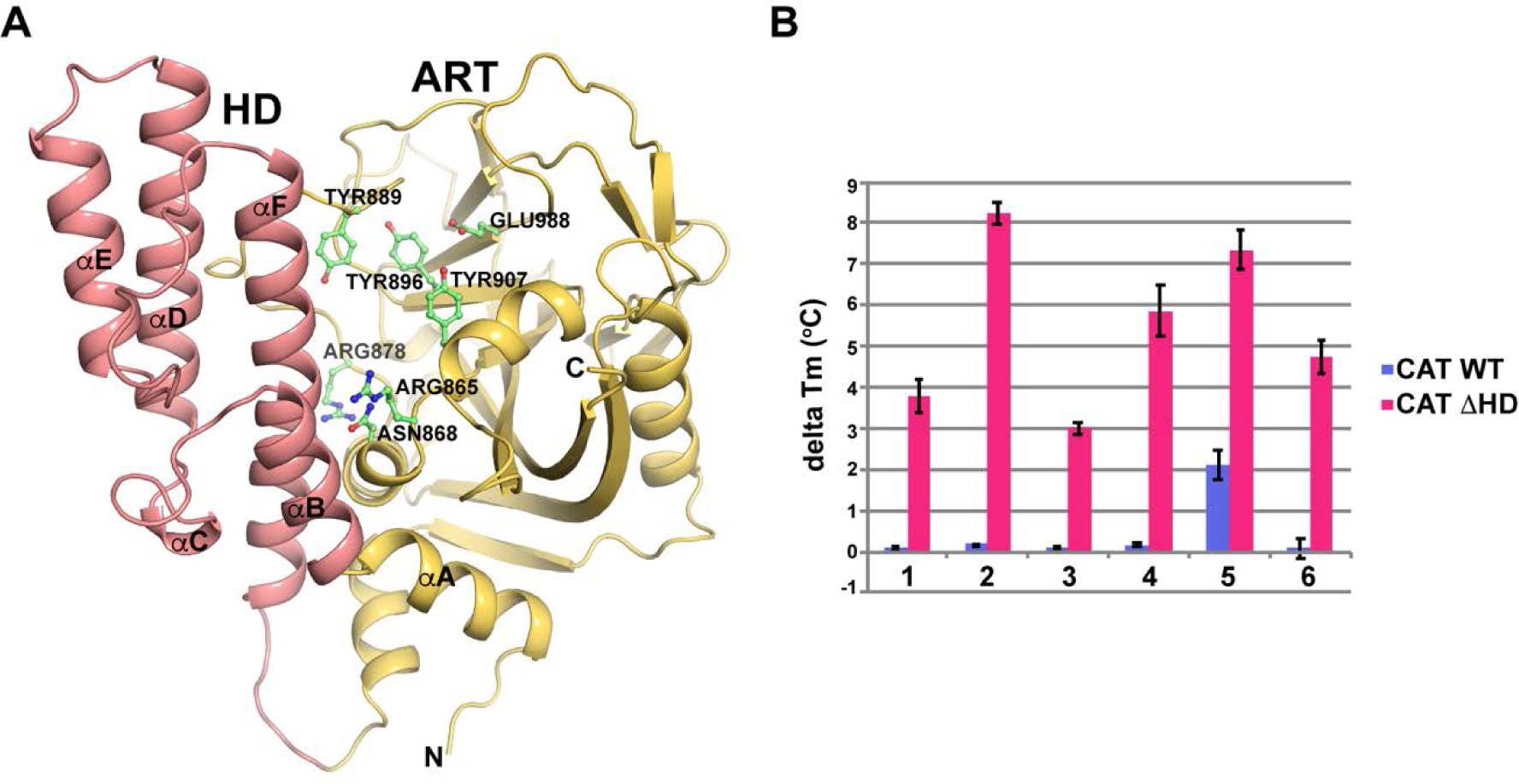
Inhibitor binding to the PARP-1 catalytic domain. (A) Ribbon representation of the PARP-1 catalytic domain (CAT) using PDB code 3gjw.71 The HD and ART domains are indicated in red and orange, respectively. Several residues are labeled and numbered, and the N-terminus (labelled as N) and C-terminus (labelled as C) are noted. (B) DSF was used to assess the capacity of inhibitors to bind to the catalytic domain of PARP-1 (CAT), or the catalytic domain with the HD deleted (CATΔHD). The change in melting temperature (delta TM) of PARP-1 CAT and CATΔHD in the presence of the indicated PARP inhibitors was measured. The delta TM was calculated by subtracting the TM values of CAT or CATΔHD alone from the values obtained in the presence of inhibitor. The averages of three experiments are shown, and the error bars represent the standard deviations. Note: (1) pyrimidinyl piperazine analogue 63; (2) tetrazolyl analogue 57; (3) benzimidazole-2-yl-ethylamine analogue 93; (4) cyclopropyl THTP analogue 83; (5) benzimidazole-2-yl-piperazine analogue 103; (6) benzamide adenine dinucleotide.
Figure 4.
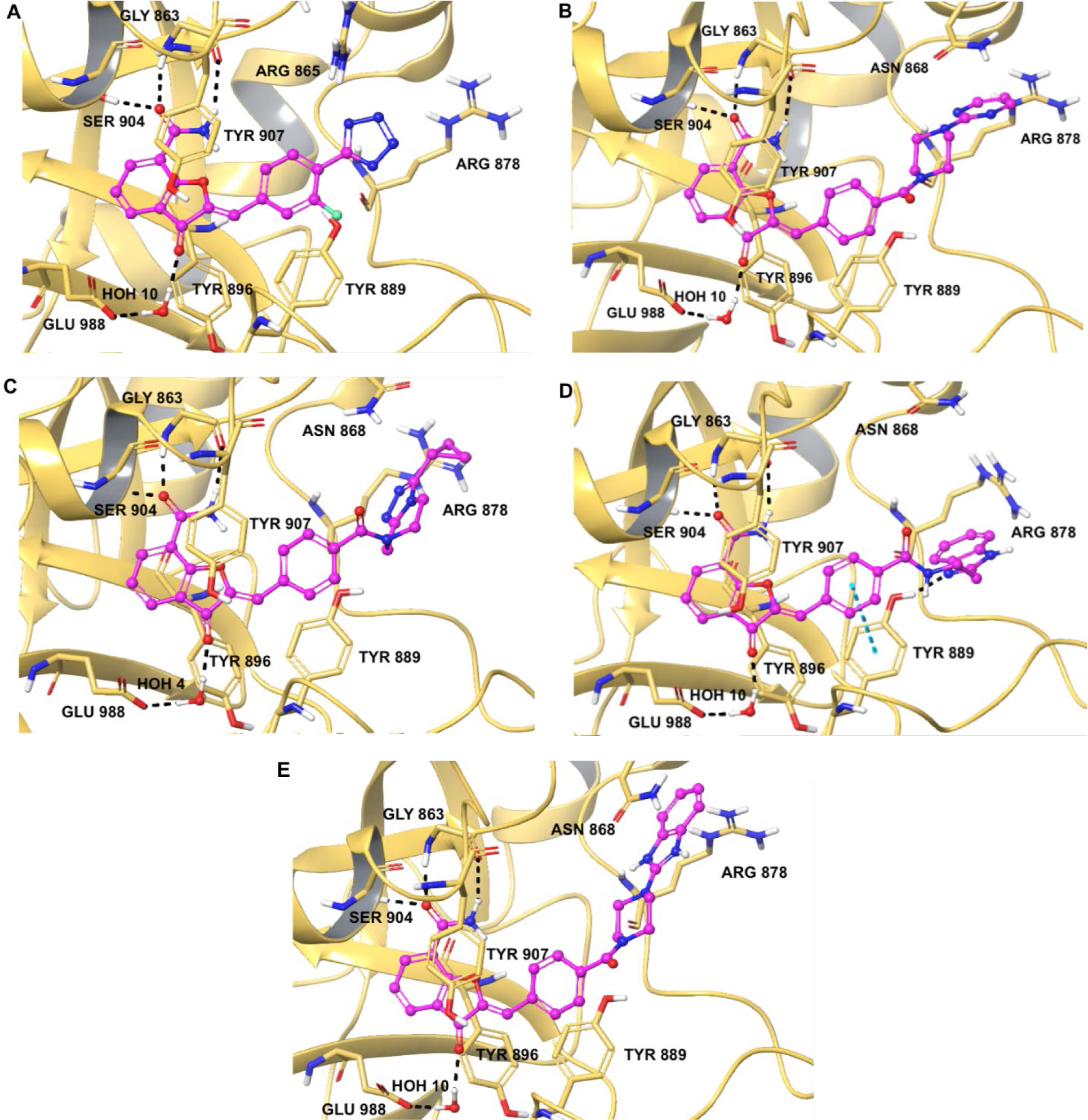
X-ray crystal structures of selected PARP inhibitors bound to CATΔHD of PARP-1. Structures in case of inhibitors are represented in the form of ball and stick model and in case of amino acid residues as tube model. Nitrogen and oxygen atoms are represented in blue and red colors respectively. Carbons in case of inhibitors are represented in magenta color whereas amino acid residues are shown in faded orange color. Hydrogen bond interactions are shown as broken black lines and pi-pi stacking interactions are shown as blue broken lines. (A) Representation of tetrazolyl analogue 57 bound to PARP-1; (B) representation of pyrimidin-2-yl-piperazine analogue 63 bound to PARP-1; (C) representation of cyclopropyl THTP analogue 83 bound to PARP-1; (D) representation of benzimidazole-2-yl-ethylamine analogue 93 bound to PARP-1 and (E) representation of benzimidazole-2-yl-piperazine analogue 103 bound to PARP-1.
Exploration of para-/meta-Aryl/Heteroaryl Substituted Benzylidene Analogues of Lead 1 (SAR 1).
Table 1 represents various substituents at C2-position of DHBF scaffold (I, see Scheme 5). The initial optimization efforts were mainly focused on exploring simplified substitution of various aryl/heteroaryl ring systems at the para-position of the benzylidene moiety present in lead 1. A biphenyl analogue 48 failed to show any PARP-1 inhibition at the tested concentration of 50 nM. Therefore, we replaced the para-phenyl ring with various saturated and unsaturated heterocyclic rings with the intention of capturing hydrogen bonding and/or ionic interactions within the ABP of PARP-1. Amongst the unsaturated heterocycles (49-52) such as thiazole, 1,2,4-triazole, 2H-tetrazole, and 1H-pyrazole, only the tetrazolyl analogue, 51, gave a promising enzyme inhibition with an IC50 value of 35 nM. Encouragingly, 51 showed ~12-fold improvement in inhibition as compared to lead 1. The 4-position was optimal for the tetrazole ring as moving it to the meta-position of the benzylidene moiety (53) proved detrimental to the activity. Next, the distal aromatic/heteroaromatic ring in the above-mentioned analogues was replaced with basic saturated heterocycle such as substituted piperazine. The N-methylpiperazine analogue 54, showed loss of activity as compared to 51. Replacing the N-methylpiperazine of 54 with bulky and hydrophobic N-benzylpiperazine and pyrimidin-2-yl-piperazine yielded compounds 55 and 56; however, both proved inferior to 51. Having established 51 as the best inhibitor from this series, we conducted a limited SAR on 51 by introducing electron-withdrawing 3-fluoro (57, IC50 = 56 nM) and electron-donating 2-methoxy (58, IC50 = 47 nM) substituents, both of which gave a degree of PARP-1 inhibition comparable to that of 51. X-ray crystallographic analysis revealed favorable localization of the tetrazole moiety of a representative analogue 57 in the vicinity of active site residue Arg865 as shown in Figure 4A. This observation was also corroborated by the detrimental result obtained by replacing the acidic tetrazole proton with a methyl group as observed in 59.
Since the tetrazole ring is not amenable for further chemical optimization, we sought to determine if the tetrazole ring can be isosterically replaced with a carboxyl group, because the carboxyl derivative could further be efficiently derivatized to improve potency similar to what has been done during the development of olaparib.30 Toward this goal, we prepared a 4-carboxybenzylidene analogue (60, IC50 = 68 nM) and noted appreciable inhibitory potency, suggesting this compound would serve as a refined lead for subsequent SAR studies. To further confirm the role of acidic group at the para-position of a benzylidene moiety, we replaced the carboxyl group with a cyano substituent and as expected, it showed decreased activity as compared to 60 (data not shown). We found that replacement of a carboxyl group in 60 with a primary carboxamide group resulted in the retention of activity (data not shown), which prompted us to explore further SAR using various alicyclic amines such as N4-heteroaryl substituted piperazines, un(substituted) THTPs and un(substituted) benzimidazoles with various linkers.
Exploration of the Vector Addressing ABP of PARP-1 with Heteroaryl Piperazine/Piperidine Motifs (SAR 2).
Compounds displayed in Table 2 were designed to extend carboxyl group of the newly identified lead 60 to capture additional interactions within the ABP of PARP-1. The activities of analogues in this series were also compared to compound 51, which was the best compound from SAR 1. To obtain more potent compounds, first we coupled the pendant carboxyl group in 60 with an N-phenylpiperazine moiety to obtain 61, which showed a detrimental effect on activity compared to 60. We next replaced the hydrophobic phenyl ring with polar isosteric heteroaromatic rings to obtain potent inhibitors. For example, pyridin-2-yl (62), pyrimidin-2-yl (63), and pyrazin-2-yl (64) derivatives showed tolerance for heteroaryl-piperazine extensions with IC50s ranging from 55 nM to 77 nM. X-ray crystal structure of 63 bound to CATΔHD PARP-1 demonstrated extension of pyrimidine moiety in the vicinity of Asn868 and the ABP residue Arg878 as shown in Figure 4B. The 1,3,5-triazin-2-yl (65) and 5-trifluoromethyl-1,3,4-thiadiazol-2-yl (66) analogues were found inferior compared to 63. Because pyridin-2-yl (62) and pyrimidin-2-yl (63) analogues gave appreciable enzyme inhibition, we decided to determine the influence of electron-withdrawing groups (67, 68, 70, and 71) or electron-donating group (69) on pyridine or pyrimidine rings. While 3-trifluoromethylpyridin-2-yl analogue (67) was inferior, the 3-cyanopyridin-2-yl analogue (68, IC50 = 66 nM) was as active as unsubstituted analogue 62. Amongst small to large substituents, 4-methoxy substitution on pyrimidine ring (IC50 = 66 nM) showed appreciable inhibition as evidenced from analogue 69, whereas, analogues 70 and 71 with an ester and methoxymethyl oxadiazolyl substitutions led to a moderate inhibition. Having established a favorable role of heteroaryl substituted piperazine motifs at the para-position of the benzylidene, we next determined whether these motifs could be tolerated at the meta-position. Toward this objective, we made meta-counterparts of 63, 69, and 64 to obtain 72-74, which were not well tolerated except for analogue 72 (IC50 = 58 nM). To investigate the significance of a carbonyl group in the disposition of a pyrimidin-2-yl-piperazine moiety in 63, compounds 75 and 76 were prepared. Compound 75, a non-classical rigid sulfone isostere, showed ~3.5-fold decreased enzyme inhibition as compared to 63. Similarly, compound 76, a non-classical flexible methylene isostere, also led to decreased activity. These results underscore the contribution of a carbonyl group in directing the pyrimidin-2-yl-piperazine moiety toward the amino acid residues located within ABP (Figure 4B). Next, we sought to explore the role of the piperazine ring in 63 by replacing it with a 4-aminopiperidine ring (77, IC50 = 112 nM) and found that this substitution resulted in two-fold loss of activity. We replaced the piperazine linker in 63 with a piperidine linker to obtain 78, which also showed a detrimental effect on potency as compared to 63 indicating the requirement of terminal piperazine ring nitrogen for an improved inhibition. Because substitutions on pyrimidin-2-yl- or pyridin-2-yl-piperazines failed to give us better enzyme inhibition than analogue 51, we decided to generate a new series with a fused bicyclic ring system containing sp2 N atoms with the expectation of obtaining potent inhibitors.
Exploration of THTP Amides Linked to the meta- or para-Position of Benzylidene Moiety (SAR 3).
SAR 3 optimization involved extension of carboxyl group toward ABP of PARP-1 by coupling with THTP as a rigid isostere of pyrimidinylpiperazine moiety as shown in Table 3. Unsubstituted THTP analogue (79) gave appreciable enzyme inhibition (IC50 = 97 nM). To enhance productive interactions with residues from ABP of PARP-1, we inserted functional groups at the 3-position of THTP ring with varying molecular size and electronic properties such as electron neutral (methyl, isopropyl, cyclopropylmethyl, cyclopentyl and -CH2OH), electron-withdrawing (-CHF2, -CF3, -COOEt, 3-flurobenzyl, and N-methyl imidazole) and pi-electron donor (cyclopropyl). These efforts led to a series of target compounds (80-90, and 92) with improved inhibitory profile as compared to SAR 2 analogues. The C3-methyl substitution on THTP (80) resulted in a slight improvement in enzyme inhibition as compared to 79. Similarly, C3-trifluoromethyl analogue (81) gave a 3-fold improvement in the activity as compared to unsubstituted analogue 79 and methyl analogue 80. The C3-ethyl ester analogue (82, IC50 = 40 nM) was also well tolerated, which indicates the favorable contribution of moderately sized electron-withdrawing groups at C3- position of THTP. Based on the recent review on versatile role of a cyclopropyl ring in medicinal chemistry,49 next we added a cyclopropyl group at C3-position to obtain analogue 83 (IC50 = 27 nM) with the best enzyme inhibition of the THTP series. X-ray structure of 83 bound to CATΔHD PARP-1 revealed localization of a cyclopropyl ring in the vicinity of Asn868 and ABP residue Arg878 (Figure 4C). Increasing the steric bulk and hydrophobicity at C3-position of THTP with meta-fluorobenzyl substituent yielded 84 with unfavorable enzyme inhibition. Replacing the trifluoromethyl group at C3-position of THTP in 81 by a difluoromethyl group (85, IC50 = 30 nM) was well tolerated. Insertion of a methylene bridge between the C3 of a THTP ring and a cyclopropyl ring in 83 gave 86 (IC50 = 47 nM) with considerable retention of the activity of 83. A polar C3-hydroxymethyl substituent (87) proved to be a weak inhibitor. Substitution of an isopropyl group, noncyclic isostere of a cyclopropyl ring, at C3-position of THTP (88, IC50 = 42 nM) was well tolerated. Replacement of a cyclopropyl ring in 83 with cyclopentyl (89, IC50 = 37 nM) or 1-methylimidazol-4-yl (90, IC50 = 32 nM) also showed comparable potency to that observed for 83. Based on the inhibition profile of various substitutions at C3-position of THTP, it is evident that smaller substituents with electronegative property improve inhibition by interacting with polar residues in ABP of PARP-1.
The evaluation of the effect of moving THTP from para- to the meta-position led to two representative analogues. For example, meta-version of 79 led to a loss of activity as exemplified by 91. However, meta-version of 81 produced 92 (IC50 = 42 nM) with retention of inhibitory activity. In summary, this SAR study revealed favorable impact of THTP scaffold on PARP-1 inhibition as compared to the lead compounds 51 and 60.
Exploration of Benzimidazoles with Various Linkers as ABP Motifs, Coupled to the meta- or para-Position of Benzylidene Moiety (SAR 4).
Table 4 shows the extension of a carboxyl group of 60 for ligand occupancy within the ABP of PARP-1. Carboxyl group of 60 was subjected to coupling with various (un)substituted benzimidazolyl ethylamines as novel ABP-motifs. An initial lead from this series, compound 93 (IC50 = 36 nM) with no substituent on the benzimidazole ring, had set the stage for obtaining potent PARP-1 inhibitors. Further SAR work on lead 93 involved exploration of different substituents on the benzimidazole ring with electronic properties such as electron neutral (CH3) and electron withdrawing (F) groups as exemplified by 94 (IC50 = 22 nM) and 95 (IC50 = 51 nM), respectively. As anticipated, moving benzimidazole ethylamine moiety in 93 and 95 to the meta-position produced 96 (IC50 = 88 nM) and 97 (IC50 = 97 nM) with a 2-fold decreased potency. Introducing electron donor methoxy group at 5-position of the benzimidazole moiety led to 98 (IC50 = 28 nM) with comparable activity to analogue 93. These results conclude that 4-position of the benzylidene moiety is the ideal vector to access and produce productive interactions within ABP of PARP-1. Replacement of the benzimidazole ring in 93 with benzimidazole-4-carboxamide led to 99 (IC50 = 4 nM) with 9-fold improvement in potency compared to 93. Further SAR involved modification of the ethyl linker and, toward this goal, we synthesized analogues with a gem-dimethyl50 substitution (100) which led to a slight decrease in activity. Replacement of the ethyl linker in 93 with the propyl linker produced 101 with marginally decreased activity compared to 93.
Finally, we explored the impact of replacing flexible ethylamine linker with azetidine, piperazine or piperidine linkers to limit the conformational flexibility and allow for entropically favorable binding within the ABP of PARP-1. These efforts led to the synthesis of azetidine analogue 102 (IC50 = 30 nM), piperazine analogue 103 (IC50 = 18 nM) and piperidine analogue 104 (IC50 = 58 nM) amongst which 103 exhibited the best inhibition. Since benzimidazole-4-carboxamide in 99 serves as an excellent NI mimic, we decided to elucidate whether DHBF-7-carboxamide or benzimidazole-4-carboxamide binds to the NI site. Toward this objective, we synthesized a methyl ester analogue 105 (PARP-1 IC50 = 98 nM) and observed a 25-fold decrease in activity as compared to 99, and thus, validated the switched positioning of benzimidazole-4-carboxamide and DHBF-7-carboxamide, respectively, within NI and ABP of PARP-1 active site. Further biophysical characterization of 99 and 105 will confirm above-mentioned observation. In summary, SAR 4 revealed potent analogues such as 99 and 103 with 108- and 24-fold increase in activity, respectively, as compared to lead 1.
X-ray crystal structures of 93 and 103 in complex with CATΔHD PARP-1 revealed that the benzimidazole portion was directed toward Arg878 of ABP (Figure 4D and 4E). Additionally, compound 93 exhibited pi-pi stacking and hydrogen bonding interactions with the side chain of Tyr889.
Investigation of PARP-2 Enzyme Inhibition by Selected Target Compounds.
PARP-2, via physical interaction or PARylation of various target proteins, plays an important role in a wide range of cellular processes that are dysregulated in tumorigenesis.51 PARP-2−/− mice are highly sensitive to alkylating agents as well as ionizing radiation.52, 53 Further, both PARP-1 and PARP-2 are required for efficient BER as evidenced from global decrease in PARP activity upon PARP-2 depletion.54 Because the C-terminal catalytic domains of PARP-1 and PARP-2 exhibit ~69% homology,55 it is not surprising that clinically utilized PARP inhibitors potently inhibit both PARP-1 and PARP-2 (Figure 1). We, therefore, conducted a screen of highly active PARP-1 inhibitors against PARP-2 as shown in Tables 1–4. The tetrazolyl and piperazinyl analogues 51, 53, and 56-58 from SAR 1 (Table 1) demonstrated potent PARP-2 inhibition (IC50 = 2.1 nM, 76%, 56%, 100% at 50 nM and IC50 = 1.6 nM, respectively). Compound 58 showed a 29-fold greater potency against PARP-2 as compared to PARP-1 and its PARP-2 IC50, was comparable to that observed for olaparib (PARP-2 IC50 = 0.5 nM). PARP-2 inhibition profiles of representative compounds 63, 64, 69, 71-74 and 76 from SAR 2 (Table 2) also showed greater selectivity toward PARP-2 compared to PARP-1 as evidenced by 51–98% inhibition of PARP-2 at 50 nM concentration. We further investigated the inhibition profile of THTP analogues, from SAR 3 (Table 3), in PARP-2 enzyme assay at 10 nM concentration. Compound 51 displayed potent PARP-2 inhibition (IC50 = 2.1 nM) and it was used for comparison in Tables 3 and 4. Compounds 81 (IC50 = 2 nM) and 83 (IC50 = 1.9 nM) both displayed high potency against PARP-2 with IC50s comparable to that of olaparib and with a 15- and 14-fold selectivity, respectively, as compared to PARP-1 inhibition. Compounds 80, 84 and 87 inhibited PARP-2 by 55–61% at 10 nM concentration and thus demonstrate higher potency toward PARP-2 as compared to PARP-1. Analogues 82, 85, 86, and 88-90 also exhibited potent inhibition of PARP-2 with IC50 values ranging from 3 – 4.6 nM. PARP-2 screening of compounds 93, 98, and 101 from Table 4 at 10 nM concentration showed 44–50% inhibition. Compounds 94 (IC50 = 5 nM), 99 (IC50 = 0.7 nM) and 103 (IC50 = 4 nM) also exhibited potent PARP-2 inhibition and moderate selectivity toward PARP-2 over PARP-1. Modest PARP-2 inhibitory activity was observed for the meta-analogues 96 and 97. Overall, most of these compounds exhibited selectivity toward PARP-2, which is a common trend for the FDA approved drugs olaparib, niraparib, rucaparib and talazoparib. However, the extent of the selectivity for the compounds toward PARP-2, in the current study, is greater than that observed for the clinically used PARP inhibitors.
PARP-Isoform Profiling for Selected Target Compounds.
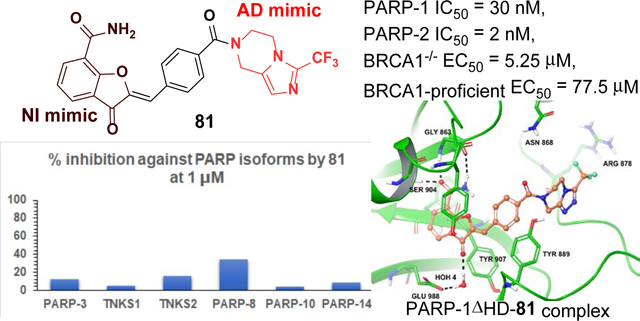
Discovery of isoform-selective inhibitors is at the forefront of medicinal chemistry and chemical biology research.56 Because the majority of clinically validated PARP-1 inhibitors show a wide spectrum of inhibitory activity toward catalytically active PARP-isoforms,57, 58 we obtained selectivity profiles of our four best PARP-1 inhibitors (81, 83, 99 and 103) at 500 nM concentration against a panel of six catalytic PARP-isoforms (Figure 5). Based on this data, compounds 81 and 83 will serve as high affinity chemical probes for PARP-1 and PARP-2 without interfering with other catalytic PARP-isoforms (PARP-3, TNKS1, TNKS2, PARP-8, PARP-10 and PARP-14). Further evaluation of compound 81 at 1 μM concentration against above mentioned catalytic PARP-isoforms led to minimal inhibition (12%, 5%, 16%, 34%, 4% and 9%, respectively). Thus, compound 81 demonstrated >33 and 500-fold selectivity toward PARP-1 and PARP-2 compared to the other catalytic PARP-isoforms.
Figure 5.
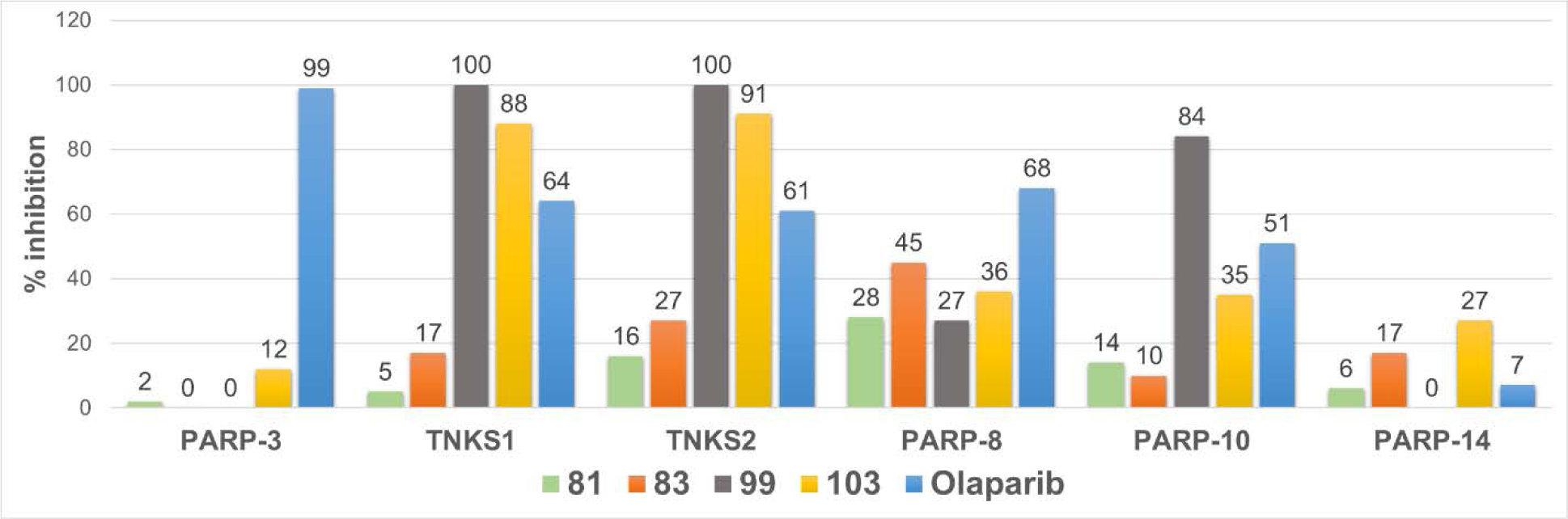
Inhibitory profile of selected PARP inhibitors against several PARP isoforms at 500 nM concentration. Screening was performed in duplicates and % inhibition was represented as the average of obtained values.
Since compounds 99 and 103 showed significant inhibition of anticancer targets TNKS1 and TNKS2 at 500 nM concentration, we obtained their IC50 values (Table 5). Compound 99 inhibited TNKS1 and TNKS2 with IC50 values of 6.3 nM and 8.8 nM, respectively, which was comparable to TNKS-selective analogue XAV939.59 Compound 99 thus inhibits clinically significant isoforms of PARP (PARP-1, PARP-2, TNKS1 and TNKS2) with low nM IC50 values (Table 5). Compound 103, however, moderately inhibited TNKS1 and TNKS2 with 131 nM and 198 nM IC50 values, respectively (Supporting Information, dose-response curves of 99 and 103 toward TNKS1 and TNKS2). It may be concluded that a bicyclic ring system attached to a flexible linker is important for the inhibition of TNKS1 and TNKS2 as evidenced from flexible analogue 99 and rigid analogues 81 and 103. Inhibition data of 81 against PARP-1, PARP-2, TNKS1 and TNKS2 is also shown in Table 5 for comparison.
Table 5.
Inhibition Data for Compounds 81, 99 and 103 against PARP-Isoforms
| Compd | PARP-1 IC50 (nM) a |
PARP-2 IC50 (nM) a |
TNKS1 IC50 (nM) a |
TNKS2 IC50 (nM) a |
|---|---|---|---|---|
| 81 | 30 | 2 | >1000b | >1000b |
| 99 | 4 | 0.7 | 6.3 | 8.8 |
| 103 | 18 | 4 | 131 | 198 |
| Olaparib | 1.2 | 0.5 | NT | NT |
| XAV939 | NT | NT | 4.2 | 2.1 |
Data shown are mean values obtained from two independent experiments performed in duplicates.
% inhibition of 81 was 5% and 16% at 1000 nM for TNKS1 and TNKS2, respectively.
Investigation of the Cellular Activity of PARP Inhibitors in BRCA1-mutant Cells.
Extensive preclinical and clinical data have established that loss of BRCA1 or BRCA2 is associated with increased sensitivity to small molecule inhibitors targeting PARP-1/−2 (PARPi).23 To determine whether compounds 81 and 83, the most selective PARP-1/−2 inhibitors of the series, demonstrate specific cytotoxicity, we tested them in a pair of isogenic BRCA1-deficient and -proficient SUM149 breast cancer cell lines.60 We found that BRCA1-mutant cells are >10-fold more sensitive to both compounds 81 and 83 when compared to BRCA1-proficient cells (Figure 6A, 6B and Supporting Information Figure S4). This differential sensitivity was similar to talazoparib or olaparib treatment (Figure 6C and 6D). This finding suggests that compounds 81 and 83 have PARP-specific cytotoxicity in the context of BRCA1 loss.
Figure 6.
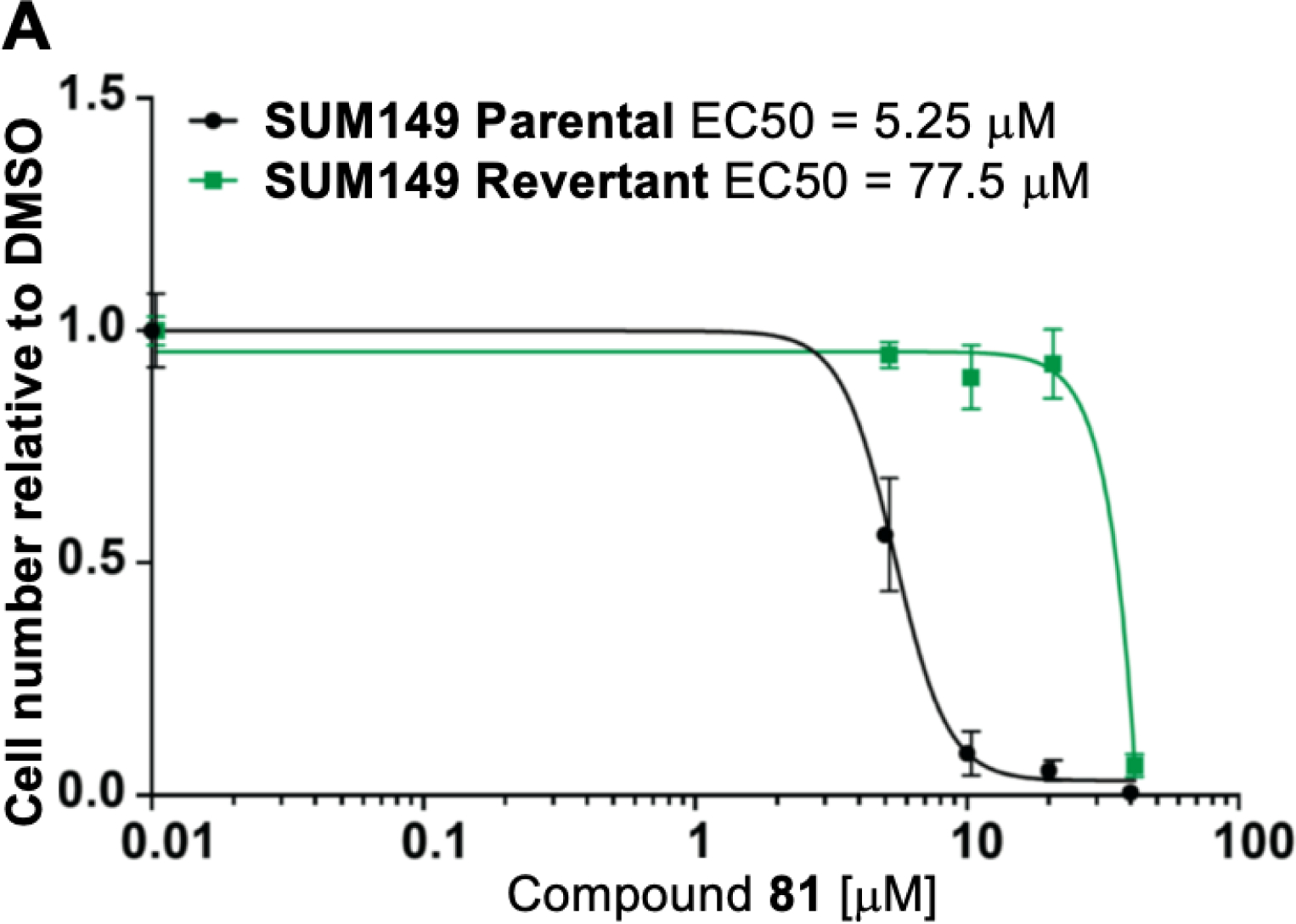
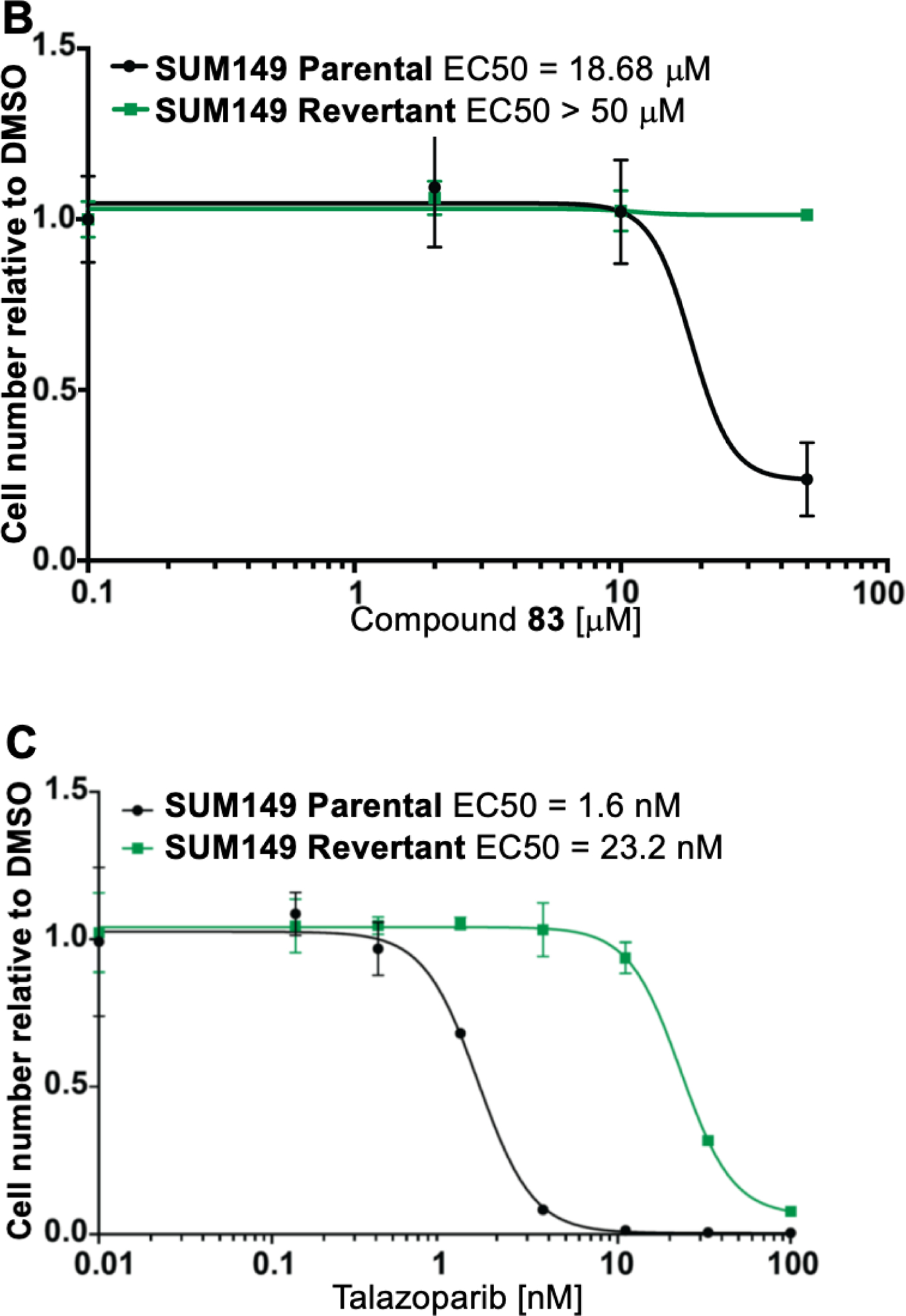
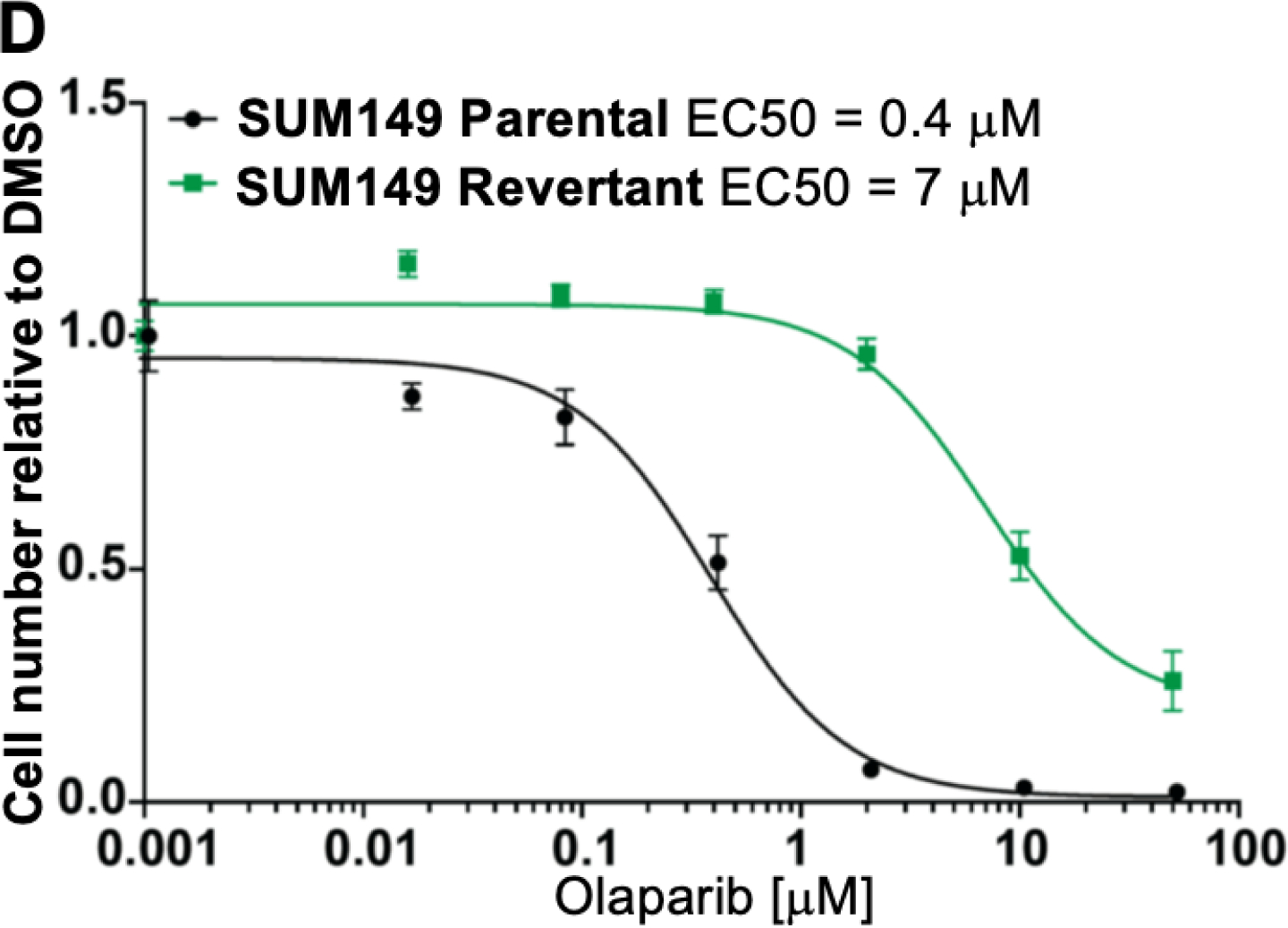
Dose-response survival curves for SUM149 parental (BRCA1−/−, black line) and SUM149 corrected/revertant (BRCA1-proficient, green line) cells treated with (A) compound 81, (B) compound 83, (C) Talazoparib and (D) Olaparib at the indicated concentrations. Data were normalized to vehicle treated cells and error bars indicate standard deviation derived from technical replicates (n=3).
CONCLUSIONS
A series of dihydrobenzofuran-7-carboxamides was designed, starting from the X-ray crystal structure of moderately active lead 1 (Z-2-benzylidene-3-oxo-2,3-dihydrobenzofuran-7-carboxamide, PARP-1 IC50 = 434 nM) in complex with a full length multi-domain PARP-1.37 In this study, four different SARs were explored at the meta- or para-position of the benzylidene portion of lead 1 to identify effective adenosine-binding motifs. For example, the 4-tetrazole motif yielded analogues with PARP-1 IC50 values of 35 nM - 56 nM. The pyridinyl/pyrimidinyl piperazine motifs displayed IC50 values ranging from 55 nM - 197 nM. Modifications on THTP motif demonstrated IC50 values of 27 nM - 97 nM and benzimidazolyl ethylamine/piperazine /azetidine/piperidine motifs also gave desirable IC50s in the range of 4 nM - 98 nM. Additionally, most of the compounds in the series were PARP-2 selective and their IC50s were similar to clinically utilized PARP inhibitors as exemplified by compounds with <5 nM IC50s against PARP-2. Differential scanning fluorimetry (DSF) of selected compounds from each of the SARs revealed that these compounds are unable to bind to the CAT domain in the presence of a folded helical domain; however, they efficiently bound to the CAT with the helical domain deleted (CATΔHD). Therefore, we propose that these compounds can bind to PARP-1 either with HD deleted or unfolded potentially as a result of DNA damage. X-ray crystal structures of selected compounds from four different SARs in complex with CATΔHD PARP-1 provided insights into the binding mechanism and will form the basis for optimization efforts in the future. Compounds 81 and 83 showed selective inhibition of PARP-1 and PARP-2 over other catalytic PARP-isoforms such as PARP-3, TNKS1, TNKS2, PARP-8, PARP-10, and PARP-14. Compound 99 exhibited single digit nM IC50 values against clinically significant PARP-isoforms (PARP-1, PARP-2, TNKS1 and TNKS2). PARP-isoform selective compounds 81 and 83 demonstrated BRCA1-dependent cytotoxic effect in the SUM149 cell line suggesting that they are promising lead compounds for further optimization.
EXPERIMENTAL
Chemical Synthesis.
Materials and Instrumentation.
All chemicals were procured from Accela Chembio (San Diego, CA), Aldrich Chemical Co. (Milwaukee, WI), Alfa Aesar (Ward Hill, MA), Arkpharm, Inc. (Arlington Heights, IL), Chem-Impex Int. Inc. (Wood Dale, IL), Combi-Blocks Inc. (San Diego, CA), Enamine LLC (Monmouth Jct., NJ), Oakwood Products (West Columbia, SC), Oxchem Corporation (Wood Dale, IL), Synthonix (Wake Forest, NC) and were used without additional purification. Qualitative analysis of reactions was performed by thin layer chromatography (TLC) with silica gel G as the adsorbent (250 microns) on aluminum backed plates (Agela Technologies) and Ultraviolet (UV) light at 254 nm or 365 nm for visualization purposes. 1H NMR experiments were performed using a Bruker 400 Ultrashield™ spectrometer (1H at 400 MHz and 13C at 100 MHz) equipped with a z-axis gradient probe. 1H NMR chemical shifts were reported downfield from tetramethylsilane (TMS as an internal standard) in parts per million (δ ppm) for majority of the intermediates and all the target compounds. The 1H NMR data are depicted as: chemical shift (multiplicity s (singlet), bs (broad singlet), d (doublet), t (triplet), dd (doublet of doublets), ddd (doublet of doublets of doublets), dq (doublet of quartets), dt (doublet of triplets), tt (triplet of triplets), td (triplet of doublets), h (hextate), m (multiplet), qd (quartet of doublets), number of protons and coupling constant). Column chromatography purifications were performed using silica gel (40–63 μm) purchased from Silicycle Inc. (Quebec City, CANADA) and flash chromatography was conducted using Reveleris® X2 flash chromatography system (BUCHI Corporation, New Castle, DE). Preparative TLC was performed using Silica Gel GF 1000 μm 20×20 cm glass backed plates procured from Analtech (Miles Scientific, Newark, DE). Purity analysis for target compounds 48-78 and mass analysis of all the target compounds was performed on an Agilent 1260 infinity series liquid chromatography (LC) system connected with Agilent 6120 quadrupole mass spectrometer (MS) (Agilent, Santa Clara, CA). Purity analysis of compounds 79-105 were carried out using Agilent 1260 infinity series HPLC system (Agilent, Santa Clara, CA). Purity and mass analysis was performed using Agilent Eclipse plus C18, 3.5 μm, 4.6 mm × 100 mm column and the runs were monitored at 254 nm. All target compounds were analyzed to be ≥ 95% pure (based on major peak area/total area of combined peaks). Acetonitrile (ACN) and water (0.1% formic acid) mixtures were used as mobile phase for purity analysis of compounds 48-78. For analogues 48 and 52, a 12 min gradient run was performed with 30 – 70% ACN in water. For analogue 50, a gradient run was performed with 60 – 40% ACN in water over 8 min. For analogues 51, 56, 57, 60, 61, 63 – 75, 77 and 78, a gradient run was performed with 40 – 60% ACN in water over 8 min. For analogue 54, an 8 min isocratic run was performed with 60% ACN in water. The flow rate was 0.5 mL/min for analysis of all the above-mentioned target compounds. For mass analysis of compounds 79-92, an 8 min gradient run of 70–90% ACN in water was used with a flow rate of 0.5 mL/min and for compounds 93-105, the flow rate was increased to 1 mL/min. For the purity analysis of compounds 49, 53, 58, 59, 62, 76, 79-105, ACN (0.1% DEA) and water (0.1% DEA) combination was used as the mobile phase. A gradient run with 10% ACN to 90% ACN in water over 8 min (flow rate of 1 mL/min) was used as the mobile phase. The elemental analyses (C, H, and N) were carried out by Atlantic Microlabs, Inc., (Norcross, GA), and the observed values were within ± 0.4% of the calculated values.
Synthesis.
Procedures for synthesizing the key intermediate I and conditions for Knoevenagel condensation to obtain the target compounds were adapted from our previously reported work.37 Target compounds obtained via Knoevenagel condensation were either washed with methanol and water, thereby resulting in pure compounds or were purified using chromatographic techniques such as preparative TLC or flash chromatography.
General Procedure for Suzuki Coupling Reaction (A).
Reactions were performed using conditions reported in previously published studies.38, 39
[1,1’-Biphenyl]-4-carbaldehyde (2).
Intermediate 2 was obtained by Suzuki coupling (procedure A) of 4-formylphenylboronic acid (500 mg, 3.34 mmol) with bromobenzene (0.35 mL, 3.34 mmol), as a pale yellow solid (485 mg, 80% yield).61 1H NMR (400 MHz, CDCl3; TMS) δ 9.93 (s, 1H), 7.82 (d, J = 8.2 Hz, 2H), 7.61 (d, J = 8.2 Hz, 2H), 7.54 – 7.48 (m, 5H).
4-(Thiazol-2-yl)benzaldehyde (3).
Intermediate 3 was synthesized using procedure A and 4-formylphenylboronic acid (500 mg, 3.34 mmol) and 2-chlorothiazole (0.29 mL, 3.34 mmol), as a pale brown solid (424 mg, 67% yield); 1H NMR (400 MHz, DMSO-d6; TMS) δ 10.11 (s, 1H), 8.13 (d, J = 7.8 Hz, 2H), 8.01 (d, J = 3.1 Hz, 1H), 7.97 (d, J = 7.6 Hz, 2H), 7.90 (d, J = 3.2 Hz, 1H).
4-(2H-Tetrazol-5-yl)benzaldehyde (4).
To a solution of 4-formylbenzonitrile (1 g, 7.63 mmol) dissolved in N,N-dimethylformamide (DMF), triethylamine (2.13 mL, 15.25 mmol) was added with subsequent addition of sodium azide (1.49 g, 22.88 mmol) and ensuing reaction mixture was heated to 180°C for overnight. The reaction mixture was then vacuum dried on a rotary evaporator to remove majority of DMF. The resulting crude mixture was then partitioned between 1N aqueous HCl and ethyl acetate and the organic layer was collected and further extracted 3X with brine to remove the residual DMF from the organic layer. Later, ethyl acetate layer was dried over MgSO4, filtered and evaporated to obtain brown solid, which was suspended in ethyl acetate and washed with ethyl acetate to yield pure compound 4 as a cream colored solid (897 mg, 67% yield). 1H NMR (400 MHz, DMSO-d6) δ 10.11 (s, 1H), 8.31 (d, J = 7.3 Hz, 2H), 8.13 (d, J = 7.3 Hz, 2H).
General Procedure for Preparation of Substituted 2H-tetrazol-5-yl benzaldehyde Intermediates (B).
These intermediates were prepared by slightly modifying reported procedure,41 wherein sodium azide and diethylamine hydrochloride were added to a solution of the appropriate benzonitrile in toluene. The reaction mixture was then allowed to reflux in an inert condition for a period of 24 h. Thereafter, toluene was evaporated and subsequently extracted with 1X 1N aqueous HCl and ethyl acetate. Ethyl acetate layer was then dried over MgSO4 and evaporated to obtain the crude benzaldehyde derivative that was purified by flash chromatography.
3-Fluoro-4-(2H-tetrazol-5-yl)benzaldehyde (5).
Intermediate 5 was obtained using the general procedure B, by reacting 2-fluoro-4-formylbenzonitrile (500 mg, 3.35 mmol) with sodium azide (371 mg, 5.7 mmol) and diethylamine hydrochloride (625mg, 5.7 mmol), as a white solid (173 mg, 27% yield). 1H NMR (400 MHz, DMSO-d6) δ 10.09 (s, 1H), 8.32 (t, J = 7.4 Hz, 1H), 8.03 – 7.95 (m, 2H).
2-Methoxy-4-(2H-tetrazol-5-yl)benzaldehyde (6).
Intermediate 6 was prepared using the general procedure B, by reacting 3-methoxy-4-formylbenzonitrile (500 mg, 3.10 mmol), sodium azide (343 mg, 5.27 mmol) and diethylamine hydrochloride (578 mg, 5.27 mmol) as a white solid (520 mg, 82% yield); 1H NMR (400 MHz, DMSO-d6) δ 10.40 (s, 1H), 7.93 – 7.83 (m, 2H), 7.75 (dt, J = 8.0, 1.1 Hz, 1H), 4.04 (s, 3H).
4-(2-Methyl-2H-tetrazol-5-yl)benzaldehyde (7).
Intermediate 7 was obtained by reacting tetrazole intermediate 4 (250 mg, 0.57 mmol), as described in a reported procedure,42 as a yellow solid (196 mg, 73% yield). 1H NMR (400 MHz, DMSO-d6; TMS) δ 10.10 (s, 1H), 8.29 (d, J = 8.3 Hz, 2H), 8.10 (d, J = 8.0 Hz, 2H), 4.47 (s, 3H).
4-(4-(Pyrimidin-2-yl)piperazin-1-yl)benzaldehyde (8).
Intermediate 8 was obtained using a reported procedure43 by reacting 4-fluorobenzaldehyde (1 g, 8.06 mmol) with pyrimidin-2-yl-piperazine (2.16 g, 8.06 mmol) and potassium carbonate (2.23 g, 16.11 mmol), as a white solid (1.575 g, 73% yield). 1H NMR (400 MHz, CDCl3; TMS) δ 10.05 (s, 1H), 8.32 (d, J = 4.8 Hz, 2H), 7.95 (d, J = 8.2 Hz, 2H), 7.59 (d, J = 8.0 Hz, 2H), 6.55 (t, J = 4.7 Hz, 1H), 4.00 – 3.75 (m, 6H), 3.53 – 3.39 (m, 2H)
General Procedure for Peptide Coupling Reactions (C).
To a suspension of appropriate carboxylic acid [(3-carboxy or 4-carboxy benzaldehyde or 60) 1 eq] in dichloromethane, HCTU (1.5 eq) and HOBt (1.5 eq) were added and the temperature was brought down to 0°C while the reaction was stirring. To this mixture, DIPEA was added (2 eq) and the resultant mixture was left stirring at 0°C for 15 min. Subsequently, the amine (1.1 eq) was added as such or by dissolving in a minimum volume of dichloromethane (for amines which were liquids at rt) to the reaction mixture and the reaction was stirred at rt for overnight. The reaction was then diluted with DCM and washed 3X with small portions of water. Resultant organic phase was dried over MgSO4, filtered and evaporated to yield crude coupled products, which were either used as obtained or purified by flash chromatography using gradient DCM and methanol combinations as the mobile phase, wherein the concentration of methanol in dichloromethane was varied from 1–8% based on the nature of product to be purified.
4-(4-Phenylpiperazine-1-carbonyl)benzaldehyde (9).
Intermediate 9 was prepared using the general procedure C, where 4-formylbenzoic acid (500 mg, 3.33 mmol) was reacted with 1-phenylpiperazine (594 mg, 3.66 mmol), as a brown solid (724 mg, 74% yield). 1H NMR (400 MHz, CDCl3; TMS) δ 10.01 (s, 1H), 7.92 (d, J = 7.9 Hz, 2H), 7.57 (d, J = 7.8 Hz, 2H), 7.27 (t, J = 7.8 Hz, 2H), 6.96 – 6.81 (m, 3H), 4.01 – 3.86 (m, 2H), 3.60 – 3.46 (m, 2H), 3.31 – 3.18 (m, 2H), 3.18 – 3.03 (m, 2H).
4-(4-(Pyridin-2-yl)piperazine-1-carbonyl)benzaldehyde (10).
Intermediate 10 was synthesized using general procedure C, by reacting 4-formylbenzoic acid (500 mg, 3.33 mmol) with pyridin-2-yl-piperazine (598 mg, 3.66 mmol), as a brown oil (638 mg, 65% yield) that was used as such in the next step; 1H NMR (400 MHz, CDCl3; TMS) δ 10.02 (s, 1H), 8.20 – 8.13 (m, 1H), 7.92 (dd, J = 8.2, 2.1 Hz, 2H), 7.57 (dd, J = 8.3, 2.0 Hz, 2H), 7.49 (ddd, J = 10.6, 6.5, 2.0 Hz, 1H), 6.65 (dd, J = 7.5, 4.8 Hz, 2H), 4.02 – 3.82 (m, 2H), 3.66 – 3.43 (m, 6H).
4-(4-(Pyrimidin-2-yl)piperazine-1-carbonyl)benzaldehyde (11).
Intermediate 11 was obtained by using general procedure C, where 4-formylbenzoic acid (500 mg, 3.33 mmol) was treated with pyrimidin-2-yl-piperazine (602 mg, 3.66 mmol), as an off-white solid after flash purification (838 mg, 85% yield); 1H NMR (400 MHz, DMSO-d6; TMS) δ 10.08 (s, 1H), 8.39 (d, J = 4.8 Hz, 2H), 8.00 (d, J = 8.3 Hz, 2H), 7.67 (d, J = 7.9 Hz, 2H), 6.68 (t, J = 4.7 Hz, 1H), 3.96 – 3.64 (m, 6H), 3.45 – 3.30 (m, 2H).
4-(4-(Pyrazin-2-yl)piperazine-1-carbonyl)benzaldehyde (12).
12 was synthesized by using 4-formylbenzoic acid (500 mg, 3.33 mmol), pyrazin-2-yl-piperazine (602 mg, 3.66 mmol) and the general procedure C, as a brown oil that was used as such without any purification for subsequent synthesis; 1H NMR (400 MHz, CDCl3; TMS) δ 10.04 (s, 1H), 8.20 – 8.14 (m, 1H), 8.13 – 8.05 (m, 1H), 7.96 (d, J = 7.9 Hz, 2H), 7.87 (d, J = 2.7 Hz, 1H), 7.61 (d, J = 7.9 Hz, 2H), 3.98 – 3.83 (m, 2H), 3.78 – 3.66 (m, 2H), 3.65 – 3.49 (m, 4H).
4-(4-(1,3,5-Triazin-2-yl)piperazine-1-carbonyl)benzaldehyde (13).
Aldehyde 13 was prepared using the general procedure C, where 4-formylbenzoic acid (500 mg, 3.33 mmol) was reacted with triazin-2-yl-piperazine (605 mg, 3.66 mmol), as a brown oil (738 mg, 75% yield) that was as such subjected to the next step; 1H NMR (400 MHz, CDCl3; TMS) δ 10.04 (s, 1H), 8.52 (s, 2H), 7.98 (d, J = 7.9 Hz, 2H), 7.63 (d, J = 7.9 Hz, 2H), 4.05 – 3.94 (m, 2H), 3.92 – 3.80 (m, 4H), 3.55 – 3.44 (m, 2H).
4-(4-(5-(Trifluoromethyl)-1,3,4-thiadiazol-2-yl)piperazine-1-carbonyl)benzaldehyde (14).
Aldehyde 14 was prepared by using general procedure C, where 4-formylbenzoic acid (500 mg, 3.33 mmol) was treated with 2-(piperazin-1-yl)-5-(trifluoromethyl)-1,3,4-thiadiazole (503 mg, 3.66 mmol), as a pale yellow solid (838 mg, 85% yield), after evaporating the organic layer and washing the crude solid with ethyl acetate; 1H NMR (400 MHz, DMSO-d6; TMS) δ 10.08 (s, 1H), 8.02 (d, J = 8.2 Hz, 2H), 7.68 (d, J = 8.1 Hz, 2H), 3.89 – 3.41 (m, 8H).
4-(4-(3-(Trifluoromethyl)pyridin-2-yl)piperazine-1-carbonyl)benzaldehyde (15).
Intermediate 15 was prepared via general procedure C by using 4-formylbenzoic acid (500 mg, 3.33 mmol) and 1-(3-(trifluoromethyl)pyridin-2-yl)piperazine (847 mg, 3.66 mmol) as a dark brown oil (757 mg, 63% yield), which was directly used as obtained for subsequent synthesis; 1H NMR (400 MHz, CDCl3; TMS) δ 10.06 (s, 1H), 8.48 (dd, J = 5.0, 1.9 Hz, 1H), 8.00 – 7.96 (m, 2H), 7.93 (dd, J = 7.8, 1.9 Hz, 1H), 7.65 – 7.58 (m, 2H), 7.18 – 7.10 (m, 1H), 3.99 – 3.90 (m, 2H), 3.60 – 3.52 (m, 2H), 3.39 – 3.31 (m, 2H), 3.26 – 3.21 (m, 2H).
2-(4-(4-Formylbenzoyl)piperazin-1-yl)nicotinonitrile (16).
Aldehyde 16 was prepared using the general procedure C, where 4-formylbenzoic acid (500 mg, 3.33 mmol) was allowed to react with 2-(piperazin-1-yl)nicotinonitrile (689 mg, 3.66 mmol) as a brown oil (638 mg, 60% yield) that was directly used as obtained for subsequent synthesis; 1H NMR (400 MHz, CDCl3; TMS) δ 10.06 (s, 1H), 8.38 (dd, J = 4.9, 2.0 Hz, 1H), 7.97 (d, J = 7.8 Hz, 2H), 7.84 (dd, J = 7.7, 2.1 Hz, 1H), 7.60 (d, J = 7.8 Hz, 2H), 6.89 (dd, J = 7.6, 4.9 Hz, 1H), 4.01 – 3.92 (m, 2H), 3.85 – 3.77 (m, 2H), 3.68 – 3.62 (m, 2H), 3.62 – 3.52 (m, 2H).
4-(4-(4-Methoxypyrimidin-2-yl)piperazine-1-carbonyl)benzaldehyde (17).
Aldehyde 17 was synthesized by using 4-formylbenzoic acid (250 mg, 1.67 mmol) and 4-methoxy-2-(piperazin-1-yl)pyrimidine (356 mg, 1.83 mmol) as per the general procedure C as a dark yellow oil, which was used as obtained for subsequent synthesis; 1H NMR (400 MHz, CDCl3; TMS) δ 10.04 (s, 1H), 8.00 (dd, J = 23.9, 6.7 Hz, 3H), 7.63 (d, J = 7.8 Hz, 2H), 6.03 (d, J = 5.7 Hz, 1H), 3.99 – 3.90 (m, 2H), 3.90 – 3.77 (m, 7H), 3.52 – 3.43 (m, 2H).
Methyl 2-(4-(tert-butoxycarbonyl)piperazin-1-yl)pyrimidine-5-carboxylate (18).
Intermediate 18 was prepared according to reported procedures.37, 45 To a solution of N-Boc piperazine (270 mg, 1.45 mmol) in acetonitrile, potassium carbonate (400 mg, 2.9 mmol) and methyl 2-chloropyrimidine-5-carboxylate (250 mg, 1.45 mmol) were added and the suspension was allowed to reflux for overnight. Subsequently, the solvent was evaporated and the residue was subjected to extraction with ethyl acetate and water. The organic phase was then dried over MgSO4 and was further vacuum dried to obtain the N-Boc intermediate 18 as an off-white solid (413 mg, 88% yield). 1H NMR (400 MHz, CDCl3; TMS) δ 8.86 (s, 2H), 3.99 – 3.91 (m, 4H), 3.90 (s, 3H), 3.59 – 3.46 (m, 4H), 1.51 (s, 9H).
Methyl 2-(piperazin-1-yl)pyrimidine-5-carboxylate hydrochloride (19).
Intermediate 19 was prepared by using the N-Boc piperazine 18 (413 mg, 1.28 mmol) and dissolving it in dioxane, followed by lowering the temperature of the reaction to 0°C, using ice. Further, 4N aqueous solution of HCl (4.7 mL, 12.81 mmol) was added drop wise and the reaction mixture was stirred at rt for overnight. The solvent was then evaporated and the resultant semi-solid mass was triturated with a small amount of methanol to obtain a white suspension, which was filtered and dried to obtain 19 (278 mg, 88% yield) as a hydrochloride salt. 1H NMR (400 MHz, D2O) δ 8.70 (s, 2H), 4.05 – 3.98 (m, 4H), 3.77 (s, 3H), 3.30 – 3.24 (m, 4H).
Methyl 2-(4-(4-formylbenzoyl)piperazin-1-yl)pyrimidine-5-carboxylate (20).
Intermediate 20 was synthesized using general procedure C and by reacting 4-formylbenzoic acid (125 mg, 0.83 mmol) with intermediate 19 (237 mg, 0.92 mmol) as a white solid (185 mg, 63% yield); 1H NMR (400 MHz, DMSO-d6; TMS) δ 10.08 (s, 1H), 8.82 (s, 2H), 8.01 (d, J = 7.9 Hz, 2H), 7.68 (d, J = 7.9 Hz, 2H), 4.10 – 3.68 (m, 9H), 3.51 – 3.37 (m, 2H).
4-(4-(4-(5-(Methoxymethyl)-1,2,4-oxadiazol-3-yl)pyrimidin-2-yl)piperazine-1-carbonyl)benzaldehyde (21).
Aldehyde 21 was prepared using the general procedure C, where 4-formylbenzoic acid (130 mg, 0.87 mmol) was allowed to react with 5-(methoxymethyl)-3-(2-(piperazin-1-yl)pyrimidin-4-yl)-1,2,4-oxadiazole (250 mg, 0.95 mmol) as a cream colored solid (189 mg, 53% yield), by washing the crude solid with ethyl acetate; 1H NMR (400 MHz, CDCl3; TMS) δ 10.07 (s, 1H), 7.98 (d, J = 8.1 Hz, 1H), 7.61 (d, J = 8.1 Hz, 1H), 7.54 (d, J = 8.4 Hz, 2H), 7.46 (d, J = 8.4 Hz, 2H), 4.79 (s, 2H), 4.12 – 3.97 (m, 2H), 3.97 – 3.79 (m, 4H), 3.60 – 3.47 (m, 5H).
3-(4-(Pyrimidin-2-yl)piperazine-1-carbonyl)benzaldehyde (22).
Intermediate 22 was prepared by using general procedure C, where 3-formylbenzoic acid (500 mg, 3.33 mmol) was treated with pyrimidin-2-yl-piperazine (602 mg, 3.66 mmol) as a dark yellow oil (812 mg, 82% yield) that was directly used for subsequent synthesis without additional purification; 1H NMR (400 MHz, CDCl3; TMS) δ 10.05 (s, 1H), 8.33 (d, J = 4.7 Hz, 2H), 8.03 – 7.92 (m, 2H), 7.74 (d, J = 7.6 Hz, 1H), 7.65 (t, J = 7.8 Hz, 1H), 6.58 (t, J = 4.7 Hz, 1H), 4.02 – 3.76 (m, 6H), 3.61 – 3.46 (m, 2H).
3-(4-(4-Methoxypyrimidin-2-yl)piperazine-1-carbonyl)benzaldehyde (23).
Aldehyde 23 was synthesized by using 4-formylbenzoic acid (250 mg, 1.67 mmol) and 4-methoxy-2-(piperazin-1-yl)pyrimidine (356 mg, 1.83 mmol) and as per the general procedure C as a brown oil (338 mg, 62% yield) that was directly used in the subsequent step without additional purification; 1H NMR (400 MHz, CDCl3; TMS) δ 10.03 (s, 1H), 8.03 (d, J = 5.7 Hz, 1H), 7.97 – 7.92 (m, 2H), 7.70 (dt, J = 7.6, 1.5 Hz, 1H), 7.61 (t, J = 7.9 Hz, 1H), 6.02 (d, J = 5.6 Hz, 1H), 3.97 – 3.74 (m, 9H), 3.57 – 3.41 (m, 2H).
3-(4-(Pyrazin-2-yl)piperazine-1-carbonyl)benzaldehyde (24).
Intermediate 24 was prepared using the general procedure C, where 3-formylbenzoic acid (500 mg, 3.33 mmol) was allowed to react with pyrazin-2-yl-piperazine (602 mg, 3.66 mmol) as a brown oil (753 mg, 76% yield), which was used as obtained in the subsequent step; 1H NMR (400 MHz, CDCl3; TMS) δ 10.03 (d, J = 1.3 Hz, 1H), 8.15 (d, J = 1.6 Hz, 1H), 8.07 (dt, J = 3.2, 1.6 Hz, 1H), 7.98 – 7.92 (m, 2H), 7.89 (dd, J = 2.8, 1.3 Hz, 1H), 7.71 (dq, J = 7.7, 1.6 Hz, 1H), 7.62 (t, J = 7.4 Hz, 1H), 3.99 – 3.81 (m, 2H), 3.76 – 3.48 (m, 6H).
4-(4-(Pyrimidin-2-yl)piperidine-1-carbonyl)benzaldehyde (25).
Aldehyde 25 was prepared by using general procedure C, where 4-formylbenzoic acid (250 mg, 1.67 mmol) was treated with 2-(piperidin-4-yl)pyrimidine (299 mg, 1.83 mmol) as a yellow oil (364 mg, 74% yield), which was used in the subsequent synthesis without additional purification; 1H NMR (400 MHz, CDCl3; TMS) δ 10.05 (s, 1H), 8.72 (d, J = 4.9 Hz, 2H), 7.95 (d, J = 7.7 Hz, 2H), 7.58 (d, J = 7.9 Hz, 2H), 7.25 (t, J = 5.0 Hz, 1H), 4.85 – 4.71 (m, 1H), 3.82 – 3.63 (m, 4H), 3.27 – 3.16 (m, 4H).
4-Formyl-N-(1-(pyrimidin-2-yl)piperidin-4-yl)benzamide (26).
Intermediate 26 was synthesized by using 4-formylbenzoic acid (500 mg, 3.33 mmol) and 1-(pyrimidin-2-yl)piperidin-4-amine (653 mg, 3.66 mmol) as per general procedure C as a pale yellow solid (666 mg, 64% yield); 1H NMR (400 MHz, CDCl3; TMS) δ 10.02 (s, 1H), 8.27 (d, J = 4.8 Hz, 2H), 7.95 (d, J = 8.3 Hz, 2H), 7.88 (d, J = 8.3 Hz, 2H), 7.04 (d, J = 7.9 Hz, 1H), 6.46 (t, J = 4.8 Hz, 1H), 4.78 – 4.66 (m, 2H), 4.31 – 4.19 (m, 1H), 3.98 – 3.68 (m, 2H), 3.10 – 2.99 (m, 2H), 2.11 – 2.02 (m, 2H).
4-((4-(Pyrimidin-2-yl)piperazin-1-yl)sulfonyl)benzaldehyde (27).
Intermediate 27 was prepared using procedure from our previously published work,37 where pyrimidin-2-yl-piperazine (201 mg, 1.22 mmol) was reacted with triethylamine (0.34 mL, 2.44 mmol) in dichloromethane, followed by drop wise addition of 4-formylphenyl sulfonyl chloride (dissolved in dichloromethane) under 0°C and brought to rt after which it was left stirring for a period of 12 h. The solvent was later evaporated, and the mixture was purified by flash chromatography to yield 27 as a white solid (286 mg, 70% yield). 1H NMR (400 MHz, DMSO-d6) δ 10.11 (s, 1H), 8.34 (d, J = 4.7 Hz, 2H), 8.14 (d, J = 7.9 Hz, 2H), 7.97 (d, J = 7.9 Hz, 2H), 6.64 (t, J = 4.7 Hz, 1H), 3.88 – 3.78 (m, 4H), 3.04 – 2.96 (m, 4H).
4-((4-(Pyrimidin-2-yl)piperazin-1-yl)methyl)benzaldehyde (28).
Intermediate 28 was prepared according to our previous report.37 To a solution of pyrimidin-2-yl-piperazine (454 mg, 2.76 mmol) in acetonitrile, potassium carbonate (694 mg, 5.02 mmol) and 4-bromomethyl benzaldehyde (500 mg, 2.51 mmol) were added and the suspension was allowed to reflux for overnight. Subsequently, the solvent was evaporated followed by extraction of the reaction mass with ethyl acetate and water. Ethyl acetate layer was then dried over MgSO4 and was further purified using flash chromatography to obtain 28 as a yellow solid (536 mg, 76% yield). 1H NMR (400 MHz, CDCl3; TMS) δ 10.00 (s, 1H), 8.30 (dd, J = 4.8, 0.7 Hz, 2H), 7.86 (d, J = 8.0 Hz, 2H), 7.55 (d, J = 8.0 Hz, 2H), 6.48 (td, J = 4.7, 0.7 Hz, 1H), 3.88 – 3.79 (m, 4H), 3.62 (s, 2H), 2.56 – 2.45 (m, 4H).
4-(5,6,7,8-Tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine-7-carbonyl)benzaldehyde (29).
Intermediate 29 was synthesized using general procedure C, by reacting 4-formylbenzoic acid (275 mg, 1.83 mmol) with 5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine (250 mg, 2.01 mmol) as a white solid (213 mg, 45% yield), after purification using flash chromatography with DCM and methanol as the mobile phase; 1H NMR (400 MHz, CDCl3; TMS) δ 10.08 (s, 1H), 8.25 (s, 1H), 7.99 (d, J = 8.0 Hz, 2H), 7.64 (d, J = 7.8 Hz, 2H), 5.01 (bs, 2H), 4.34 – 4.03 (m, 4H).
4-(3-Methyl-5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine-7-carbonyl)benzaldehyde (30).
Intermediate 30 was synthesized using general procedure C and by reacting 4-formylbenzoic acid (247 mg, 1.65 mmol) with 3-methyl-5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine (250 mg, 1.81 mmol) as a white solid (236 mg, 53% yield), after purification using flash chromatography with DCM and methanol as the mobile phase; 1H NMR (400 MHz, CDCl3; TMS) δ 10.07 (s, 1H), 7.98 (d, J = 8.3 Hz, 2H), 7.63 (d, J = 8.0 Hz, 2H), 4.84 (bs, 2H), 4.36 – 3.78 (m, 4H), 2.43 (s, 3H).
4-(3-(Trifluoromethyl)-5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine-7-carbonyl)benzaldehyde (31).
Intermediate 31 was synthesized using general procedure C and by reacting 4-formylbenzoic acid (178 mg, 1.19 mmol) with 3-(trifluoromethyl)-5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine (251 mg, 1.3 mmol) as a pale yellow solid (259 mg, 67% yield) after purification using flash chromatography with DCM and methanol as the mobile phase; 1H NMR (400 MHz, CDCl3; TMS) δ 10.05 (s, 1H), 7.97 (d, J = 7.8 Hz, 2H), 7.66 (d, J = 7.7 Hz, 2H), 5.01 (bs, 2H), 4.42 – 3.91 (m, 4H).
Ethyl 7-(4-formylbenzoyl)-5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine-3-carboxylate (32).
Intermediate 32 was synthesized using general procedure C and by reacting 4-formylbenzoic acid (174 mg, 1.16 mmol) with ethyl 5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine-3-carboxylate (250 mg, 1.27 mmol) as off-white solid (234 mg, 62% yield) after purification using flash chromatography with DCM and methanol as the mobile phase; 1H NMR (400 MHz, CDCl3; TMS) δ 10.06 (s, 1H), 7.99 (d, J = 7.9 Hz, 2H), 7.68 (d, J = 7.8 Hz, 2H), 5.03 (bs, 2H), 4.48 – 4.39 (m, 4H), 4.03 – 3.78 (m, 2H), 1.43 – 1.38 (m, 3H).
4-(3-Cyclopropyl-5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine-7-carbonyl)benzaldehyde (33).
Intermediate 33 was synthesized using general procedure C and by reacting 4-formylbenzoic acid (208 mg, 1.39 mmol) with 3-cyclopropyl-5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine (250 mg, 1.52 mmol) as a white solid (209 mg, 51% yield) after purification using flash chromatography with DCM and methanol as the mobile phase; 1H NMR (400 MHz, DMSO-d6) δ 10.09 (s, 1H), 8.08 – 7.97 (m, 2H), 7.74 (d, J = 7.9 Hz, 2H), 4.77 (bs, 2H), 4.20 – 3.98 (m, 3H), 3.83 – 3.65 (m, 1H), 2.00 – 1.82 (m, 1H), 1.02 – 0.81 (m, 4H).
4-(3-(3-Fluorobenzyl)-5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine-7-carbonyl)benzaldehyde (34).
Intermediate 34 was synthesized using general procedure C and by reacting 4-formylbenzoic acid (147 mg, 0.98 mmol) with 3-(3-fluorobenzyl)-5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine (250 mg, 1.08 mmol) as a white solid (178 mg, 50% yield) after purification using flash chromatography with DCM and methanol as the mobile phase; 1H NMR (400 MHz, CDCl3; TMS) δ 10.03 (s, 1H), 7.98 – 7.90 (m, 2H), 7.60 (d, J = 7.8 Hz, 2H), 7.29 (td, J = 7.9, 5.9 Hz, 1H), 7.05 – 6.87 (m, 3H), 4.91 (bs, 2H), 4.16 (s, 2H), 3.98 – 3.62 (m, 3H), 3.31 – 3.01 (m, 1H).
3-(5,6,7,8-Tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine-7-carbonyl)benzaldehyde (35).
Intermediate 35 was synthesized using general procedure C and by reacting 3-formylbenzoic acid (275 mg, 1.83 mmol) with 5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine (250 mg, 2.01 mmol) as a white solid (268 mg, 57% yield) after purification using flash chromatography with DCM and methanol as the mobile phase; 1H NMR (400 MHz, CDCl3; TMS) δ 10.05 (s, 1H), 8.04 – 7.97 (m, 2H), 7.92 (s, 1H), 7.82 – 7.73 (m, 1H), 7.67 (t, J = 7.4 Hz, 1H), 4.93 (bs, 2H), 4.44 – 3.97 (m, 4H).
3-(3-(Trifluoromethyl)-5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine-7-carbonyl)benzaldehyde (36).
Intermediate 36 was synthesized using general procedure C and by reacting 3-formylbenzoic acid (178 mg, 1.19 mmol) with 3-(trifluoromethyl)-5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine (251 mg, 1.3 mmol) as a colorless oil (242 mg, 63% yield) after purification using flash chromatography with DCM and methanol as the mobile phase; 1H NMR (400 MHz, CDCl3; TMS) δ 10.03 (s, 1H), 7.81 – 7.74 (m, 2H), 7.60 – 7.52 (m, 1H), 7.44 (t, J = 7.9 Hz, 1H), 4.81 (s, 2H), 4.16 – 3.73 (m, 4H).
N-(2-(1H-Benzo[d]imidazol-2-yl)ethyl)-4-formylbenzamide (37).
Intermediate 37 was synthesized by reaction of 4-formylbenzoic acid (212 mg, 1.41 mmol) with 2-(1H-benzo[d]imidazol-2-yl)ethan-1-amine (250 mg, 1.55 mmol) using general procedure C, where the ethyl acetate extract was subjected to evaporation and the crude residue was washed further with ethyl acetate to obtain 37 as an off-white solid (257 mg, 62% yield); 1H NMR (400 MHz, DMSO-d6) δ 12.33 (bs, 1H), 10.08 (s, 1H), 8.94 (t, J = 5.6 Hz, 1H), 8.06 – 7.95 (m, 4H), 7.54 – 7.42 (m, 3H), 7.17 – 7.09 (m, 3H), 3.75 (q, J = 7.0 Hz, 2H), 3.12 (t, J = 7.3 Hz, 2H).
4-Formyl-N-(2-(5-methyl-1H-benzo[d]imidazol-2-yl)ethyl)benzamide (38).
Aldehyde 38 was synthesized by reaction of 4-formylbenzoic acid (195 mg, 1.30 mmol) with 2-(5-methyl-1H-benzo[d]imidazol-2-yl)ethan-1-amine (250 mg, 1.43 mmol) using general procedure C, where the ethyl acetate extract was subjected to evaporation and the crude residue was washed further with ethyl acetate to obtain 38 as a pale yellow solid (283 mg, 71% yield); 1H NMR (400 MHz, DMSO-d6; TMS) δ 12.16 (bs, 1H), 10.08 (s, 1H), 8.92 (s, 1H), 8.09 – 7.90 (m, 4H), 7.48 – 7.12 (m, 2H), 6.95 (t, J = 9.1 Hz, 1H), 3.73 (q, J = 6.8 Hz, 2H), 3.08 (t, J = 7.4 Hz, 2H), 2.39 (s, 3H).
N-(2-(5-Fluoro-1H-benzo[d]imidazol-2-yl)ethyl)-4-formylbenzamide (39).
Intermediate 39 was synthesized by reaction of 4-formylbenzoic acid (190 mg, 1.27 mmol) with 2-(5-fluoro-1H-benzo[d]imidazol-2-yl)ethan-1-amine (249 mg, 1.39 mmol) using general procedure C, where the ethyl acetate extract was subjected to evaporation and the crude residue was washed further with ethyl acetate to obtain 39 as an off-white solid (271 mg, 69% yield); 1H NMR (400 MHz, DMSO-d6) δ 12.44 (bs, 1H), 10.08 (s, 1H), 8.91 (d, J = 7.6 Hz, 1H), 8.06 – 7.97 (m, 4H), 7.57 – 7.39 (m, 1H), 7.30 (ddd, J = 36.5, 9.6, 2.6 Hz, 1H), 7.05 – 6.91 (m, 1H), 3.73 (q, J = 6.8 Hz, 2H), 3.10 (td, J = 7.3, 3.6 Hz, 2H).
N-(2-(1H-Benzo[d]imidazol-2-yl)ethyl)-3-formylbenzamide (40).
Aldehyde 40 was synthesized by reaction of 3-formylbenzoic acid (212 mg, 1.41 mmol) with 2-(1H-benzo[d]imidazol-2-yl)ethan-1-amine (250 mg, 1.55 mmol) using general procedure C, where the ethyl acetate extract was subjected to evaporation and the crude residue was washed further with ethyl acetate to obtain 40 as a brown solid (209 mg, 51% yield); 1H NMR (400 MHz, DMSO-d6) δ 12.31 (bs, 1H), 10.08 (s, 1H), 8.95 (t, J = 5.6 Hz, 1H), 8.39 (t, J = 1.8 Hz, 1H), 8.16 (dt, J = 7.8, 1.5 Hz, 1H), 8.07 (dt, J = 7.7, 1.3 Hz, 1H), 7.71 (t, J = 7.7 Hz, 1H), 7.61 – 7.36 (m, 2H), 7.13 (dd, J = 6.2, 3.0 Hz, 2H), 3.75 (q, J = 6.8 Hz, 2H), 3.12 (t, J = 7.3 Hz, 2H).
N-(2-(5-Fluoro-1H-benzo[d]imidazol-2-yl)ethyl)-3-formylbenzamide (41).
Intermediate 41 was synthesized by reaction of 3-formylbenzoic acid (190 mg, 1.27 mmol) with 2-(5-fluoro-1H-benzo[d]imidazol-2-yl)ethan-1-amine (249 mg, 1.39 mmol) using general procedure C, where the ethyl acetate extract was evaporated and the crude residue was further purified by preparative TLC with DCM and 7N ammonia in methanol solution as the solvent system to obtain 41 as a brown solid (229 mg, 58% yield); 1H NMR (400 MHz, CDCl3; TMS) δ 9.91 (s, 1H), 8.62 (t, J = 5.7 Hz, 1H), 8.30 (s, 1H), 8.08 (d, J = 7.6 Hz, 1H), 7.92 (d, J = 7.7 Hz, 1H), 7.50 (t, J = 7.7 Hz, 1H), 7.45 – 7.34 (m, 1H), 7.14 (ddd, J = 8.9, 4.1, 2.2 Hz, 1H), 6.95 – 6.80 (m, 1H), 4.03 – 3.92 (m, 2H), 3.28 (t, J = 6.4 Hz, 2H).
Tert-butyl (2-(4-carbamoyl-1H-benzo[d]imidazol-2-yl)ethyl)carbamate (42).
To a solution of 500 mg of 2,3-diaminobenzamide (3.30 mmol) in DMF, benzyl 3-oxopropylcarbamate (754 mg, 3.64 mmol) and ammonium acetate (382 mg, 4.96 mmol) were added, followed by heating the mixture at 60°C for a period of 6 h. The resulting mixture was then dissolved in ethyl acetate and extracted 3X with saturated NaHCO3 and 3X with brine solution. The resultant organic layer is dehydrated using MgSO4 and concentrated under vacuum to obtain 42 as an orange colored oil (457 mg, 45% yield), which was used as obtained in the next step. 1H NMR (400 MHz, DMSO-d6; TMS) δ 12.79 (s, 1H), 9.30 (s, 1H), 7.85 – 7.60 (m, 3H), 7.50 (s, 1H), 7.38 – 7.22 (m, 6H), 5.02 (s, 2H), 3.56 – 3.45 (m, 2H), 3.07 (t, J = 7.1 Hz, 2H).
Methyl 2-(2-(((benzyloxy)carbonyl)amino)ethyl)-1H-benzo[d]imidazole-7-carboxylate (42a).
Intermediate 42a was prepared according to a reported procedure62 by dissolving methyl 2,3-diaminobenzoate (500 mg, 3.01 mmol) in DMF, followed by adding HCTU (2490 mg, 6.02 mmol) and DIPEA (0.79 mL, 4.52 mmol) to the mixture. The reaction was then allowed to stir for 4 h at room temperature. Subsequently, the mixture was subjected to reflux conditions for a period of 6 h. Reaction was then subjected to evaporation under vacuum and purified using a preparative TLC using 3% of 2.33 M NH3 containing methanol in DCM to obtain 42a as a brown solid (212 mg, 20% yield). 1H NMR (400 MHz, DMSO-d6) δ 12.29 (s, 1H), 7.84 (d, J = 7.8 Hz, 1H), 7.77 (d, J = 7.5 Hz, 1H), 7.47 – 7.41 (m, 1H), 7.38 – 7.29 (m, 5H), 7.26 (t, J = 7.8 Hz, 1H), 5.02 (s, 2H), 3.94 (s, 3H), 3.50 (q, J = 6.7 Hz, 2H), 3.08 (t, J = 7.1 Hz, 2H).
Methyl 2-(2-aminoethyl)-1H-benzo[d]imidazole-7-carboxylate (43a).
Intermediate 43a was prepared from 42a (200 mg, 0.57 mmol) by following similar protocol used for preparation of 43 as a white solid (56 mg, 45% yield). 1H NMR (400 MHz, DMSO-d6) δ 7.94 – 7.75 (m, 4H), 7.30 (t, J = 7.8 Hz, 1H), 3.95 (s, 3H), 3.42 – 3.33 (m, 2H), 3.24 (t, J = 6.7 Hz, 2H).
2-(2-Aminoethyl)-1H-benzo[d]imidazole-4-carboxamide (43).
Intermediate 42 (300 mg, 0.99 mmol) was dissolved in methanol and catalytic amounts of palladium over carbon was added to the solution followed by transferring the mixture into a Parr-hydrogenation apparatus. The apparatus was purged (3X) with nitrogen and then evacuated followed by introduction of hydrogen into the vessel to attain a pressure of 60 psi. The reaction was monitored for the consumption of hydrogen and approximately after 5 h, the reaction was stopped, filtered on celite bed to remove palladium. This was followed by subjecting the reaction mixture to column chromatography using DCM and 2.33M ammonia in methanol mixture to obtain amide 43 as a white solid (120 mg, 60% yield). 1H NMR (400 MHz, DMSO-d6; TMS) δ 9.35 (s, 1H), 8.06 – 7.95 (m, 4H), 7.84 (d, J = 7.3 Hz, 1H), 7.68 (s, 1H), 7.28 (t, J = 7.9 Hz, 1H), 3.19 – 3.10 (m, 2H), 2.98 (t, J = 5.7 Hz, 2H).
1,3-Dihydro-2H-benzo[d]imidazol-2-one (44).
Intermediate 44 was synthesized using a reported procedure63 by reacting ortho-phenylelediamine (1000 mg, 9.25 mmol) with CDI (3006 mg, 18.5 mmol) in DMF. The reaction mixture thus obtained was concentrated and washed with ethyl acetate to obtain the cyclic urea intermediate 44 as a white solid (1190 mg, 96% yield). 1H NMR (400 MHz, DMSO-d6; TMS) δ 10.60 (s, 2H), 7.05 – 6.81 (m, 4H).
2-Chloro-1H-benzo[d]imidazole (45).
Intermediate 45 was prepared using a reported protocol.63 Intermediate 44 (2000 mg, 14.91 mmol) was allowed to react with neat POCl3. The resulting reaction mixture was carefully treated with ethyl acetate and NaHCO3 to quench unreacted POCl3. Organic layer was collected and subsequently concentrated after drying with MgSO4 and the resulting solid was washed with minimal amount of ethyl acetate to obtain 45 as a white solid (1810 mg, 80% yield). 1H NMR (400 MHz, DMSO-d6; TMS) δ 13.23 (s, 1H), 7.64 – 7.39 (m, 2H), 7.34 – 7.11 (m, 2H).
Tert-butyl 4-(1H-benzo[d]imidazol-2-yl)piperazine-1-carboxylate (46).
Intermediate 46 was obtained as a white solid using a reported protocol.64 Intermediate 45 (1000 mg, 6.55 mmol) was transferred to a 20 mL microwave vial and N-Boc-piperazine (2441 mg, 13.1 mmol) and toluene were added to the same vial and the mixture was subjected to microwave irradiation for 6 h at 150 °C. Subsequently, the reaction mixture was purified using reverse phase (C18) flash chromatography to obtain 47 as a white solid (1200 mg, 61% yield). 1H NMR (400 MHz, CDCl3; TMS) δ 7.38 – 7.29 (m, 2H), 7.14 – 7.04 (m, 2H), 3.65 – 3.42 (m, 8H), 1.50 (s, 9H).
2-(Piperazin-1-yl)-1H-benzo[d]imidazole dihydrochloride (47).
Intermediate 47 (1000 mg, 3.31 mmol) was prepared by using the conditions mentioned for the synthesis of 19 in quantitative yields as a white solid. 1H NMR (400 MHz, DMSO-d6; TMS) δ 13.92 (s, 2H), 9.79 (s, 2H), 7.56 – 7.38 (m, 2H), 7.36 – 7.20 (m, 2H), 4.14 – 3.98 (m, 4H), 3.38 – 3.26 (m, 4H).
General Procedure for Knoevenagel Condensation (D).
Reaction was performed using the procedure mentioned in our previous report.37 To a suspension of I in toluene, appropriate aldehyde [1.1 eq with respect to I (synthesized or commercially obtained)] was added along with ammonium acetate (1.5 eq for 60, 2 eq for 48-59, 61-78, 93-97 and 5 eq for 79-84, 91 and 92, with respect to I, optimized for respective class of compounds based on yields obtained) and allowed to reflux for a period for 4–12 h based on the compounds to be synthesized. The reaction was then removed and solvent was subjected to evaporation under vacuum and the resultant mass was either stirred, filtered and washed with methanol and water to obtain solid with the desired purity, or purified by using preparative TLC or flash chromatography.
(Z)-2-([1,1’-Biphenyl]-4-ylmethylene)-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (48).
Target compound 48 was obtained by reacting amide I (75 mg, 0.23 mmol) with aldehyde 2 (85 mg, 0.47 mmol), as a fluorescent yellow solid (39 mg, 27% yield), by treatment with methanol and water as mentioned in the general procedure D. 1H NMR (400 MHz, DMSO-d6) δ 8.16 (d, J = 8.1 Hz, 2H), 8.07 (d, J = 7.7 Hz, 1H), 8.03 – 7.91 (m, 2H), 7.91 – 7.74 (m, 5H), 7.52 (t, J = 7.6 Hz, 2H), 7.42 (q, J = 6.9 Hz, 2H), 7.12 (s, 1H); ESI-MS: m/z 342.1 (C22H15NO3 requires 342.11, [M + H]+). HPLC Purity: 95% (tR = 11.54 min).
(Z)-3-Oxo-2-(4-(thiazol-2-yl)benzylidene)-2,3-dihydrobenzofuran-7-carboxamide (49).
Target compound 49 was obtained by reacting amide I (75 mg, 0.23 mmol) with aldehyde 3 (88 mg, 0.47 mmol), using general procedure D, as a fluorescent yellow solid (43 mg, 29% yield), by treatment with methanol and water as mentioned in the general procedure D; mp 287–288 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 8.18 (d, J = 8.2 Hz, 2H), 8.13 – 8.04 (m, 3H), 8.01 (d, J = 3.2 Hz, 1H), 7.96 (d, J = 6.5 Hz, 2H), 7.92 – 7.85 (m, 2H), 7.42 (t, J = 7.5 Hz, 1H), 7.11 (s, 1H); ESI-MS: m/z 349.1 (C19H12N2O3S requires 349.06, [M + H]+). HPLC Purity: 96% (tR = 7.21 min).
(Z)-2-(4-(1H-1,2,4-Triazol-1-yl)benzylidene)-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (50).
Target compound 50 was obtained by reacting amide I (50 mg, 0.28 mmol) with commercially obtained 4-(1H-1,2,4-triazol-1-yl)benzaldehyde (54 mg, 0.31 mmol) as per general procedure D, as a fluorescent yellow solid (32 mg, 34% yield) and by treatment with methanol and water as mentioned in the general procedure D; mp 302–303 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 9.46 (s, 1H), 8.31 (s, 1H), 8.25 (d, J = 8.3 Hz, 2H), 8.11 – 7.93 (m, 5H), 7.86 (s, 1H), 7.41 (t, J = 7.6 Hz, 1H), 7.13 (s, 1H); ESI-MS: m/z 333.1 (C18H12N4O3 requires 333.09, [M + H]+); HPLC Purity: 97% (tR = 3.67 min).
(Z)-2-(4-(2H-Tetrazol-5-yl)benzylidene)-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (51).
Target compound 51 was obtained by reacting amide I (75 mg, 0.23 mmol) with aldehyde 4 (81 mg, 0.47 mmol) as per general procedure D, as a fluorescent yellow solid (76 mg, 54% yield) and by treatment with methanol and water as mentioned in the general procedure D; mp 285–287 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 8.25 (d, J = 8.5 Hz, 2H), 8.15 (d, J = 8.3 Hz, 2H), 8.09 (dd, J = 7.6, 1.4 Hz, 1H), 8.01 – 7.88 (m, 3H), 7.42 (t, J = 7.6 Hz, 1H), 7.12 (s, 1H); ESI-MS: m/z 334.1 (C17H11N5O3 requires 334.09, [M + H]+); HPLC Purity: 98% (tR = 2.81 min); Anal. Calcd for C17H11N5O3.0.25 H2O: C, 60.44; H, 3.43; N, 20.73; Found: C, 60.50; H, 3.59; N, 20.58.
(Z)-2-(4-(1H-Pyrazol-3-yl)benzylidene)-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (52).
Target compound 52 was obtained by reacting amide I (75 mg, 0.23 mmol) with commercially obtained 4-(1H-pyrazol-3-yl)benzaldehyde (80 mg, 0.47 mmol), using general procedure D, as a fluorescent yellow solid (66 mg, 47% yield) and by treatment with methanol and water as mentioned in the general procedure D; mp >310 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 13.07 (s, 1H), 8.09 (dd, J = 15.7, 7.8 Hz, 3H), 8.01 – 7.92 (m, 4H), 7.87 (d, J = 17.5 Hz, 2H), 7.41 (t, J = 7.6 Hz, 1H), 7.08 (s, 1H), 6.88 (d, J = 2.2 Hz, 1H); ESI-MS: m/z 332.1 (C19H13N3O3 requires 332.10, [M + H]+); HPLC Purity: 98% (tR = 3.12 min).
(Z)-2-(3-(2H-Tetrazol-5-yl)benzylidene)-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (53).
Target compound 53 was obtained by reacting amide I (100 mg, 0.56 mmol) with commercially obtained 3-(1H-tetrazol-5-yl)benzaldehyde (108 mg, 0.62 mmol), using general procedure D, as an off-white solid (67 mg, 36% yield) and by treatment with methanol and water as mentioned in the general procedure D; mp 287–288 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 8.62 (t, J = 1.8 Hz, 1H), 8.32 (dt, J = 7.9, 1.4 Hz, 1H), 8.13 – 8.05 (m, 2H), 7.97 (dd, J = 7.6, 1.5 Hz, 1H), 7.90 (d, J = 14.0 Hz, 2H), 7.75 (t, J = 7.8 Hz, 1H), 7.42 (t, J = 7.6 Hz, 1H), 7.14 (s, 1H); ESI-MS: m/z 334.1 (C17H11N5O3 requires 334.09, [M + H]+); HPLC Purity: 95% (tR = 3.78 min).
(Z)-2-(4-(4-Methylpiperazin-1-yl)benzylidene)-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (54).
Target compound 54 was obtained by reacting amide I (100 mg, 0.56 mmol) with commercially obtained 4-(4-methylpiperazin-1-yl)benzaldehyde (127 mg, 0.62 mmol) as per general procedure D, as a red solid (45 mg, 22% yield) and by purification using preparative TLC; mp 283–285 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 8.02 (dd, J = 7.6, 1.5 Hz, 1H), 7.95 – 7.87 (m, 4H), 7.82 (s, 1H), 7.37 (t, J = 7.5 Hz, 1H), 7.04 (d, J = 8.8 Hz, 2H), 6.99 (s, 1H), 3.40 – 3.32 (m, 4H), 2.48 – 2.41 (m, 4H), 2.23 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 182.49, 165.37, 162.09, 152.41, 144.57, 136.40, 134.06, 126.94, 123.79, 122.93, 121.68, 121.30, 115.25, 114.66, 54.76, 46.97, 46.19; ESI-MS: m/z 364.2 (C21H21N3O3 requires 364.16, [M + H]+); HPLC Purity: >99% (tR = 2.78 min).
(Z)-2-(4-(4-Benzylpiperazin-1-yl)benzylidene)-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (55).
Target compound 55 was obtained by reacting amide I (100 mg, 0.56 mmol) with commercially obtained 4-(4-benzylpiperazin-1-yl)benzaldehyde (174 mg, 0.62 mmol), using general procedure D, as an orange solid (56 mg, 23% yield) and by purification using preparative TLC; mp 251–253 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 8.02 (dd, J = 7.6, 1.4 Hz, 1H), 7.98 – 7.87 (m, 4H), 7.82 (s, 1H), 7.43 – 7.31 (m, 5H), 7.31 – 7.21 (m, 1H), 7.04 – 7.00 (m, 2H), 6.98 (s, 1H), 3.53 (s, 2H), 3.40 – 3.34 (m, 6H), 2.50 – 2.45 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ 182.48, 165.35, 162.10, 144.57, 136.40, 134.05, 129.42, 128.71, 127.51, 122.93, 121.32, 115.24, 114.66, 62.45, 52.72, 47.11; ESI-MS: m/z 440.2 (C27H25N3O3 requires 440.19, [M + H]+); HPLC Purity: >99% (tR = 2.78 min); Anal. Calcd for C27H25N3O3.0.4H2O: C, 72.60; H, 5.82; N, 9.41; Found: C, 72.70; H, 5.84; N, 9.18.
(Z)-3-Oxo-2-(4-(4-(pyrimidin-2-yl)piperazin-1-yl)benzylidene)-2,3-dihydrobenzofuran-7-carboxamide (56).
Target compound 56 was obtained by reacting amide I (100 mg, 0.56 mmol) with aldehyde 8 (167 mg, 0.62 mmol) as per general procedure D, as a red solid (57 mg, 24% yield) and by treatment with methanol and water as mentioned in the general procedure D; mp 248–249 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 8.40 (d, J = 4.7 Hz, 2H), 8.03 (dd, J = 7.6, 1.4 Hz, 1H), 7.98 – 7.87 (m, 4H), 7.84 (s, 1H), 7.37 (t, J = 7.6 Hz, 1H), 7.07 (d, J = 8.7 Hz, 2H), 7.00 (s, 1H), 6.67 (t, J = 4.7 Hz, 1H), 3.92 – 3.85 (m, 4H), 3.51 – 3.44 (m, 4H). ESI-MS: m/z 428.2 (C24H21N5O3 requires 428.16, [M + H]+); HPLC Purity: >99% (tR = 4.68 min); Anal. Calcd for C24H21N5O3.0.3CH3COCH3: C, 67.22; H, 5.17; N, 15.74; Found: C, 67.47; H, 5.09; N, 15.53.
(Z)-2-(3-Fluoro-4-(2H-tetrazol-5-yl)benzylidene)-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (57).
Target compound 57 was obtained by reacting amide I (75 mg, 0.23 mmol) with aldehyde 5 (89 mg, 0.47 mmol) as per general procedure D, as a fluorescent yellow solid (78 mg, 52% yield) and by treatment with methanol and water as mentioned in the general procedure D; mp >310 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 8.10 – 7.90 (m, 6H), 7.85 (d, J = 8.0 Hz, 1H), 7.39 (t, J = 7.6 Hz, 1H), 7.02 (s, 1H); ESI-MS: m/z 352.1 (C17H10FN5O3 requires 352.08, [M + H]+); HPLC Purity: 97% (tR = 2.84 min).
(Z)-2-(2-Methoxy-4-(2H-tetrazol-5-yl)benzylidene)-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (58).
Target compound 58 was obtained by reacting amide I (100 mg, 0.56 mmol) with aldehyde 6 (127 mg, 0.62 mmol), using general procedure D, as a fluorescent yellow solid (103 mg, 50% yield) and by treatment with methanol and water as mentioned in the general procedure D; mp 284–285 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 8.44 (d, J = 8.1 Hz, 1H), 8.08 (dd, J = 7.6, 1.4 Hz, 1H), 7.98 – 7.90 (m, 3H), 7.80 – 7.69 (m, 2H), 7.40 (t, J = 7.6 Hz, 1H), 7.23 (s, 1H), 4.04 (s, 3H); ESI-MS: m/z 364.1 (C18H13N5O4 requires 364.10, [M + H]+); HPLC Purity: 96% (tR = 3.99 min).
(Z)-2-(4-(2-Methyl-2H-tetrazol-5-yl)benzylidene)-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (59).
Target compound 59 was obtained by reacting amide I (100 mg, 0.56 mmol) with aldehyde 7 (116 mg, 0.62 mmol) as per general procedure D, as a pale brown solid (55 mg, 30% yield) and by treatment with methanol and water as mentioned in the general procedure D; mp 188–191 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.24 (d, J = 8.5 Hz, 2H), 8.16 (d, J = 8.4 Hz, 2H), 8.09 (dd, J = 7.6, 1.4 Hz, 1H), 8.04 – 7.93 (m, 2H), 7.90 (s, 1H), 7.42 (t, J = 7.6 Hz, 1H), 7.13 (s, 1H), 4.47 (s, 3H); ESI-MS: m/z 348.1 (C18H13N5O3 requires 348.10, [M + H]+); HPLC Purity: 95% (tR = 6.21 min).
(Z)-4-((7-Carbamoyl-3-oxobenzofuran-2(3H)-ylidene)methyl)benzoic acid (60).
Target compound 60 was obtained by reacting amide I (1000 mg, 5.64 mmol) with commercially obtained 4-formylbenzoic acid (932 mg, 6.21 mmol) as per general procedure D, as a pale yellow solid (1200 mg, 69% yield) and by treatment with methanol and water; mp 302–303 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 13.24 (bs, 1H), 8.19 – 8.12 (m, 2H), 8.08 (dd, J = 7.6, 1.4 Hz, 1H), 8.05 – 7.99 (m, 2H), 7.99 – 7.92 (m, 2H), 7.89 (s, 1H), 7.42 (t, J = 7.6 Hz, 1H), 7.11 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 183.70, 167.28, 164.99, 163.05, 147.20, 137.53, 136.36, 132.23, 131.89, 130.08, 127.44, 124.45, 122.03, 121.74, 111.91; ESI-MS: m/z 310.1 (C17H11NO5 requires 310.06, [M + H]+); HPLC Purity: 96% (tR = 2.95 min); Anal. Calcd for C17H11NO5.0.1H2O: C, 60.44; H, 3.43; N, 20.73; Found: C, 60.50; H, 3.59; N, 20.58.
(Z)-3-Oxo-2-(4-(4-phenylpiperazine-1-carbonyl)benzylidene)-2,3-dihydrobenzofuran-7-carboxamide (61).
Target compound 61 was obtained by reacting amide I (100 mg, 0.56 mmol) with aldehyde 9 (183 mg, 0.62 mmol), using general procedure D, as a dark yellow solid (63 mg, 25% yield) and by treatment with methanol and water as mentioned in the general procedure D; mp 250–252 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 8.14 (d, J = 8.3 Hz, 2H), 8.07 (dd, J = 7.6, 1.5 Hz, 1H), 8.00 – 7.93 (m, 2H), 7.87 (s, 1H), 7.60 – 7.53 (m, 2H), 7.42 (t, J = 7.6 Hz, 1H), 7.28 – 7.19 (m, 2H), 7.11 (s, 1H), 7.01 – 6.94 (m, 2H), 6.87 – 6.78 (m, 1H), 3.89 – 3.66 (m, 2H), 3.60 – 3.38 (m, 2H), 3.30 – 3.03 (m, 4H); ESI-MS: m/z 454.2 (C27H23N3O4 requires 454.17, [M + H]+); HPLC Purity: 99% (tR = 3.65 min).
(Z)-3-Oxo-2-(4-(4-(pyridin-2-yl)piperazine-1-carbonyl)benzylidene)-2,3-dihydrobenzofuran-7-carboxamide (62).
Target compound 62 was obtained by reacting amide I (100 mg, 0.56 mmol) with aldehyde 10 (184 mg, 0.62 mmol), using general procedure D, as a fluffy yellow solid (72 mg, 28% yield) and by treatment with methanol and water as mentioned in the general procedure D; mp 250–251 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 8.18 – 8.11 (m, 3H), 8.08 (dd, J = 7.6, 1.4 Hz, 1H), 7.97 (dt, J = 7.6, 2.0 Hz, 2H), 7.87 (s, 1H), 7.61 – 7.52 (m, 3H), 7.42 (t, J = 7.6 Hz, 1H), 7.11 (s, 1H), 6.86 (d, J = 8.6 Hz, 1H), 6.68 (dd, J = 7.1, 4.9 Hz, 1H), 3.83 – 3.40 (m, 8H); ESI-MS: m/z 455.2 (C26H22N4O4 requires 455.16, [M + H]+); HPLC Purity: 96% (tR = 6.07 min); Anal. Calcd for C26H22N4O4.0.65H2O: C, 66.99; H, 5.04; N, 12.02; Found: C, 66.92; H, 4.98; N, 12.18.
(Z)-3-Oxo-2-(4-(4-(pyrimidin-2-yl)piperazine-1-carbonyl)benzylidene)-2,3-dihydrobenzofuran-7-carboxamide (63).
Target compound 63 was obtained by reacting amide I (100 mg, 0.56 mmol) with aldehyde 11 (185 mg, 0.62 mmol), using general procedure D, as a yellow solid (65 mg, 25% yield) and by flash purification as mentioned in the general procedure D; mp 268–270 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 8.39 (d, J = 4.8 Hz, 2H), 8.14 (d, J = 8.0 Hz, 2H), 8.07 (dd, J = 7.6, 1.4 Hz, 1H), 7.99 – 7.93 (m, 2H), 7.87 (s, 1H), 7.57 (d, J = 8.0 Hz, 2H), 7.41 (t, J = 7.6 Hz, 1H), 7.11 (s, 1H), 6.68 (t, J = 4.8 Hz, 1H), 3.88 – 3.83 (m, 4H), 3.47 – 3.42 (m, H); 13C NMR (101 MHz, DMSO-d6) δ 183.71, 169.00, 165.15, 163.04, 161.55, 158.49, 146.84, 137.60, 137.35, 133.45, 132.01, 128.10, 127.36, 124.36, 122.15, 121.85, 112.44, 110.99; ESI-MS: m/z 456.2 (C25H21N5O4 requires 456.16, [M + H]+); HPLC Purity: 96% (tR = 3.21 min); Anal. Calcd for C25H21N5O4.0.6H2O: C, 64.40; H, 4.80; N, 15.02; Found: C, 64.27; H, 4.81; N, 15.20.
(Z)-3-Oxo-2-(4-(4-(pyrazin-2-yl)piperazine-1-carbonyl)benzylidene)-2,3-dihydrobenzofuran-7-carboxamide (64).
Target compound 64 was obtained by reacting amide I (100 mg, 0.56 mmol) with aldehyde 12 (230 mg, 0.62 mmol), using general procedure D, as a yellow solid (76 mg, 30% yield) and by treatment with methanol and water as mentioned in the general procedure D; mp 230–232 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 8.35 (d, J = 1.5 Hz, 1H), 8.18 – 8.10 (m, 3H), 8.07 (dd, J = 7.6, 1.5 Hz, 1H), 8.00 – 7.93 (m, 2H), 7.91 – 7.83 (m, 2H), 7.61 – 7.54 (m, 2H), 7.42 (t, J = 7.6 Hz, 1H), 7.11 (s, 1H), 3.85 – 3.43 (m, 8H); ESI-MS: m/z 456.2 (C25H21N5O4 requires 456.16, [M + H]+); HPLC Purity: 98% (tR = 2.87 min); Anal. Calcd for C25H21N5O4.0.75H2O: C, 64.03; H, 4.84; N, 14.93; Found: C, 64.00; H, 4.79; N, 14.90.
(Z)-2-(4-(4-(1,3,5-Triazin-2-yl)piperazine-1-carbonyl)benzylidene)-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (65).
Target compound 65 was obtained by reacting amide I (100 mg, 0.56 mmol) with aldehyde 13 (226 mg, 0.62 mmol) as per general procedure D as a pale yellow solid (45 mg, 17% yield) and by treatment with methanol and water; mp 245–247 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 8.62 (s, 2H), 8.15 (d, J = 8.0 Hz, 2H), 8.07 (dd, J = 7.5, 1.5 Hz, 1H), 8.03 – 7.92 (m, 2H), 7.87 (s, 1H), 7.58 (d, J = 7.9 Hz, 2H), 7.42 (t, J = 7.6 Hz, 1H), 7.11 (s, 1H), 4.00 – 3.68 (m, 6H), 3.55 – 3.40 (m, 2H); ESI-MS: m/z 457.2 (C24H20N6O4 requires 457.15, [M + H]+); HPLC Purity: 95% (tR = 2.65 min); Anal. Calcd for C24H20N6O4.0.85H2O: C, 61.10; H, 4.64; N, 17.81; Found: C, 60.99; H, 4.43; N, 17.65.
(Z)-3-Oxo-2-(4-(4-(5-(trifluoromethyl)-1,3,4-thiadiazol-2-yl)piperazine-1-carbonyl)benzylidene)-2,3-dihydrobenzofuran-7-carboxamide (66).
Target compound 66 was obtained by reacting amide I (100 mg, 0.56 mmol) with aldehyde 14 (199 mg, 0.62 mmol) as per general procedure D, as a pale brown solid (114 mg, 38% yield) and by treatment with methanol and water as mentioned in the general procedure D; mp 256–257 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 8.18 – 8.12 (m, 2H), 8.08 (dd, J = 7.5, 1.4 Hz, 1H), 7.96 (dd, J = 7.6, 1.4 Hz, 2H), 7.87 (s, 1H), 7.62 – 7.55 (m, 2H), 7.42 (t, J = 7.6 Hz, 1H), 7.11 (s, 1H), 3.92 – 3.49 (m, 8H); ESI-MS: m/z 530.1 (C24H18F3N5O4S requires 530.10, [M + H]+); HPLC Purity: >99% (tR = 4.26 min); Anal. Calcd for C24H18F3N5O4S.0.35H2O: C, 53.80; H, 3.52; N, 13.07; Found: C, 53.94; H, 3.56; N, 12.94.
(Z)-3-Oxo-2-(4-(4-(3-(trifluoromethyl)pyridin-2-yl)piperazine-1-carbonyl)benzylidene)-2,3-dihydrobenzofuran-7-carboxamide (67).
Target compound 67 was obtained by reacting amide I (100 mg, 0.56 mmol) with aldehyde 15 (203 mg, 0.62 mmol) as per general procedure D as a pale yellow solid (57 mg, 19% yield) and by treatment with methanol and water as mentioned in the general procedure D; mp 217–219 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 8.56 (dd, J = 4.8, 1.8 Hz, 1H), 8.15 (s, 1H), 8.12 (d, J = 7.5 Hz, 2H), 8.07 (dd, J = 7.5, 1.4 Hz, 1H), 7.99 – 7.93 (m, 2H), 7.87 (s, 1H), 7.57 (d, J = 8.3 Hz, 2H), 7.41 (t, J = 7.6 Hz, 1H), 7.26 (dd, J = 7.8, 4.9 Hz, 1H), 7.11 (s, 1H), 3.86 – 3.71 (m, 2H), 3.60 – 3.44 (m, 2H), 3.31 – 3.12 (m, 4H); ESI-MS: m/z 523.2 (C27H21F3N4O4 requires 523.15, [M + H]+); HPLC Purity: 96% (tR = 5.59 min); Anal. Calcd for C27H21F3N4O4.0.55H2O: C, 60.91; H, 4.18; N, 10.52; Found: C, 60.95; H, 4.13; N, 10.43.
(Z)-2-(4-(4-(3-Cyanopyridin-2-yl)piperazine-1-carbonyl)benzylidene)-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (68).
Target compound 68 was obtained by reacting amide I (100 mg, 0.56 mmol) with aldehyde 16 (220 mg, 0.62 mmol), using general procedure D, as a yellow solid (68 mg, 25% yield) and by treatment with methanol and water as mentioned in the general procedure D; mp 224–226 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 8.44 (dd, J = 4.8, 1.9 Hz, 1H), 8.17 – 8.10 (m, 3H), 8.07 (dd, J = 7.6, 1.5 Hz, 1H), 8.00 – 7.93 (m, 2H), 7.88 (s, 1H), 7.62 – 7.55 (m, 2H), 7.42 (t, J = 7.6 Hz, 1H), 7.11 (s, 1H), 6.98 (dd, J = 7.7, 4.8 Hz, 1H), 3.86 – 3.47 (m, 8H); ESI-MS: m/z 480.2 (C27H21N5O4 requires 480.16, [M + H]+); HPLC Purity: 96% (tR = 3.73 min); Anal. Calcd for C27H21N5O4.1.55 H2O: C, 53.80; H, 3.52; N, 13.07; Found: C, 53.94; H, 3.56; N, 12.94.
(Z)-2-(4-(4-(4-Methoxypyrimidin-2-yl)piperazine-1-carbonyl)benzylidene)-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (69).
Target compound 69 was obtained by reacting amide I (75 mg, 0.23 mmol) with aldehyde 17 (184 mg, 0.47 mmol), using general procedure D, as a dark yellow solid (83 mg, 40% yield) and by treatment with methanol and water as mentioned in the general procedure D; mp 266–268 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 8.17 – 8.10 (m, 3H), 8.07 (dd, J = 7.6, 1.5 Hz, 1H), 7.99 – 7.93 (m, 2H), 7.87 (s, 1H), 7.60 – 7.54 (m, 2H), 7.42 (t, J = 7.6 Hz, 1H), 7.11 (s, 1H), 6.12 (d, J = 5.6 Hz, 1H), 3.92 – 3.66 (m, 9H), 3.51 – 3.38 (m, 2H); ESI-MS: m/z 486.2 (C26H23N5O5 requires 486.17, [M + H]+); HPLC Purity: 96% (tR = 3.68 min).
Methyl (Z)-2-(4-(4-((7-carbamoyl-3-oxobenzofuran-2(3H)-ylidene)methyl)benzoyl)piperazin-1-yl)pyrimidine-5-carboxylate (70).
Target compound 70 was obtained by reacting amide I (75 mg, 0.23 mmol) with aldehyde 20 (138 mg, 0.47 mmol) as per general procedure D, as a pale yellow solid (37 mg, 17% yield) and by using preparative TLC for purification; mp 294–295 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 8.83 (s, 2H), 8.14 (d, J = 8.1 Hz, 2H), 8.07 (dd, J = 7.6, 1.4 Hz, 1H), 7.99 – 7.93 (m, 2H), 7.87 (s, 1H), 7.58 (d, J = 8.2 Hz, 2H), 7.41 (t, J = 7.6 Hz, 1H), 7.11 (s, 1H), 4.06 – 3.84 (m, 4H), 3.81 (s, 3H), 3.79 – 3.58 (m, 2H), 3.58 – 3.41 (m, 2H); ESI-MS: m/z 514.2 (C27H23N5O6 requires 514.16, [M + H]+); HPLC Purity: 95% (tR = 3.83 min); Anal. Calcd for C27H23N5O6.0.75H2O: C, 61.53; H, 4.69; N, 13.29; Found: C, 61.25; H, 4.68; N, 13.58.
(Z)-2-(4-(4-(4-(5-(Methoxymethyl)-1,2,4-oxadiazol-3-yl)pyrimidin-2-yl)piperazine-1-carbonyl)benzylidene)-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (71).
Target compound 71 was obtained by reacting amide I (75 mg, 0.23 mmol) with aldehyde 21 (152 mg, 0.47 mmol), using general procedure D, as a fluffy yellow solid (56 mg, 23% yield) and by treatment with methanol and water as mentioned in the general procedure D; mp 268–270 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 8.66 (d, J = 4.9 Hz, 1H), 8.15 (d, J = 8.3 Hz, 2H), 8.07 (dd, J = 7.6, 1.5 Hz, 1H), 8.00 – 7.93 (m, 2H), 7.88 (s, 1H), 7.59 (d, J = 8.2 Hz, 2H), 7.42 (t, J = 7.6 Hz, 1H), 7.27 (d, J = 4.9 Hz, 1H), 7.12 (s, 1H), 4.86 (s, 2H), 4.05 – 3.68 (m, 6H), 3.60 – 3.46 (m, 2H), 3.43 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 183.67, 177.93, 169.04, 167.42, 165.11, 163.04, 161.68, 160.82, 153.78, 146.84, 137.51, 137.36, 133.49, 131.99, 128.15, 127.36, 124.35, 122.15, 121.82, 112.42, 108.83, 64.93, 59.31; ESI-MS: m/z 568.2 (C29H25N7O6 requires 568.19, [M + H]+); HPLC Purity: 98% (tR = 3.86 min); Anal. Calcd for C29H25N7O6: C, 61.37; H, 4.44; N, 17.28; Found: C, 61.16; H, 4.55; N, 17.12.
(Z)-3-Oxo-2-(3-(4-(pyrimidin-2-yl)piperazine-1-carbonyl)benzylidene)-2,3-dihydrobenzofuran-7-carboxamide (72).
Target compound 72 was obtained by reacting amide I (100 mg, 0.56 mmol) with aldehyde 22 (184 mg, 0.62 mmol) as per general procedure D, as a pale yellow solid (47 mg, 18% yield) and by treatment with methanol and water as mentioned in the general procedure D; mp 282–283 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 8.39 (d, J = 4.7 Hz, 2H), 8.16 (dt, J = 7.8, 1.5 Hz, 1H), 8.12 (t, J = 1.7 Hz, 1H), 8.07 (dd, J = 7.6, 1.5 Hz, 1H), 8.03 – 7.92 (m, 2H), 7.88 (s, 1H), 7.61 (t, J = 7.7 Hz, 1H), 7.55 (dt, J = 7.7, 1.4 Hz, 1H), 7.41 (t, J = 7.6 Hz, 1H), 7.12 (s, 1H), 6.67 (t, J = 4.7 Hz, 1H), 3.96 – 3.66 (m, 6H), 3.54 – 3.40 (m, 2H); ESI-MS: m/z 456.2 (C25H21N5O4 requires 456.16, [M + H]+). HPLC Purity: 99% (tR = 2.65 min); Anal. Calcd for C25H21N5O4.0.35H2O: C, 65.03; H, 4.74; N, 15.17; Found: C, 64.78; H, 4.69; N, 15.39.
(Z)-2-(3-(4-(4-Methoxypyrimidin-2-yl)piperazine-1-carbonyl)benzylidene)-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (73).
Target compound 73 was obtained by reacting amide I (100 mg, 0.56 mmol) with aldehyde 23 (203 mg, 0.62 mmol), using general procedure D, as a dark yellow solid (58 mg, 21% yield) and by treatment with methanol and water as mentioned in the general procedure D; mp 266–268 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 8.16 (d, J = 7.8 Hz, 1H), 8.14 – 8.09 (m, 2H), 8.07 (dd, J = 7.6, 1.4 Hz, 1H), 7.99 – 7.92 (m, 2H), 7.91 – 7.86 (m, 1H), 7.61 (t, J = 7.6 Hz, 1H), 7.58 – 7.52 (m, 1H), 7.41 (t, J = 7.6 Hz, 1H), 7.13 (s, 1H), 6.11 (d, J = 5.6 Hz, 1H), 3.96 – 3.68 (m, 9H), 3.53 – 3.40 (m, 2H); ESI-MS: m/z 486.2 (C26H23N5O5 requires 486.17, [M + H]+); HPLC Purity: >99% (tR = 2.78 min); Anal. Calcd for C26H23N5O5.0.5H2O: C, 63.15; H, 4.89; N, 14.16; Found: C, 62.99; H, 4.70; N, 14.43.
(Z)-3-Oxo-2-(3-(4-(pyrazin-2-yl)piperazine-1-carbonyl)benzylidene)-2,3-dihydrobenzofuran-7-carboxamide (74).
Target compound 74 was obtained by reacting amide I (100 mg, 0.56 mmol) with aldehyde 24 (184 mg, 0.62 mmol) as per general procedure D, as a brown solid (52 mg, 20% yield) and by treatment with methanol and water as mentioned in the general procedure D; mp 270–271 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 8.35 (d, J = 1.5 Hz, 1H), 8.16 (dt, J = 7.9, 1.5 Hz, 1H), 8.14 – 8.10 (m, 2H), 8.07 (dd, J = 7.6, 1.4 Hz, 1H), 7.96 (dd, J = 7.6, 1.4 Hz, 2H), 7.88 (d, J = 2.7 Hz, 2H), 7.62 (t, J = 7.7 Hz, 1H), 7.55 (dt, J = 7.7, 1.5 Hz, 1H), 7.41 (t, J = 7.6 Hz, 1H), 7.13 (s, 1H), 3.90 – 3.42 (m, 8H); ESI-MS: m/z 456.2 (C25H21N5O4 requires 456.16, [M + H]+); HPLC Purity: 98% (tR = 2.96 min).
(Z)-3-Oxo-2-(4-((4-(pyrimidin-2-yl)piperazin-1-yl)sulfonyl)benzylidene)-2,3-dihydrobenzofuran-7-carboxamide (75).
Target compound 75 was obtained by reacting amide I (75 mg, 0.23 mmol) with aldehyde 27 (155 mg, 0.47 mmol), using general procedure D, as a dark yellow solid (50 mg, 24% yield) and by treatment with methanol and water as mentioned in the general procedure D; mp 290–291 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 8.33 (d, J = 4.7 Hz, 2H), 8.28 (d, J = 8.2 Hz, 2H), 8.08 (dd, J = 7.6, 1.4 Hz, 1H), 7.99 – 7.93 (m, 2H), 7.84 (d, J = 7.8 Hz, 3H), 7.42 (t, J = 7.5 Hz, 1H), 7.13 (s, 1H), 6.63 (t, J = 4.8 Hz, 1H), 3.87 – 3.80 (m, 4H), 3.05 – 2.98 (m, 4H); ESI-MS: m/z 492.1 (C24H21N5O5S requires 492.13, [M + H]+); HPLC Purity: 97% (tR = 6.67 min).
(Z)-3-Oxo-2-(4-((4-(pyrimidin-2-yl)piperazin-1-yl)methyl)benzylidene)-2,3-dihydrobenzofuran-7-carboxamide (76).
Target compound 76 was obtained by reacting amide I (100 mg, 0.56 mmol) with aldehyde 28 (175 mg, 0.62 mmol), using general procedure D, as a dark yellow solid (49 mg, 20% yield) and by treatment with methanol and water as mentioned in the general procedure D; mp 268–269 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.40 – 8.30 (m, 2H), 8.10 – 7.99 (m, 3H), 7.99 – 7.83 (m, 3H), 7.48 (d, J = 7.8 Hz, 2H), 7.40 (t, J = 7.7 Hz, 1H), 7.06 (s, 1H), 6.66 – 6.59 (m, 1H), 3.80 – 3.68 (m, 4H), 3.60 (s, 2H), 2.48 – 2.37 (m, 4H); ESI-MS: m/z 442.2 (C25H23N5O3 requires 442.18, [M + H]+); HPLC Purity: 98% (tR = 6.43 min); Anal. Calcd for C25H23N5O3.0.45H2O: C, 66.79; H, 5.36; N, 15.58; Found: C, 66.67; H, 5.24; N, 15.58.
(Z)-3-Oxo-2-(4-((1-(pyrimidin-2-yl)piperidin-4-yl)carbamoyl)benzylidene)-2,3-dihydrobenzofuran-7-carboxamide (77).
Target compound 77 was obtained by reacting amide I (100 mg, 0.56 mmol) with aldehyde 25 (193 mg, 0.62 mmol) as per general procedure D, as a pale yellow solid (84 mg, 32% yield) and by treatment with methanol and water as mentioned in the general procedure D; mp >310 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 8.44 (d, J = 7.8 Hz, 1H), 8.38 (d, J = 4.7 Hz, 2H), 8.13 (d, J = 8.2 Hz, 2H), 8.08 (dd, J = 7.6, 1.4 Hz, 1H), 8.00 – 7.92 (m, 4H), 7.88 (s, 1H), 7.41 (t, J = 7.6 Hz, 1H), 7.09 (s, 1H), 6.62 (t, J = 4.7 Hz, 1H), 4.66 (d, J = 13.3 Hz, 2H), 4.18 – 4.08 (m, 1H), 3.04 (t, J = 12.6 Hz, 2H), 1.89 (dd, J = 13.5, 4.1 Hz, 2H), 1.50 (qd, J = 12.2, 4.1 Hz, 2H); 13C NMR (100 MHz, DMSO-d6) δ 183.68, 168.79, 165.13, 163.04, 157.64, 146.82, 138.13, 137.36, 133.28, 132.04, 127.74, 127.36, 124.36, 122.17, 121.85, 112.48, 36.10; ESI-MS: m/z 470.2 (C26H23N5O4 requires 470.18, [M + H]+); HPLC Purity: >99% (tR = 3.245 min); Anal. Calcd for C26H23N5O4.0.25H2O: C, 65.88; H, 5.00; N, 14.77; Found: C, 66.07; H, 5.00; N, 14.58.
(Z)-3-Oxo-2-(4-(4-(pyrimidin-2-yl)piperidine-1-carbonyl)benzylidene)-2,3-dihydrobenzofuran-7-carboxamide (78).
Target compound 78 was obtained by reacting amide I (100 mg, 0.56 mmol) with aldehyde 26 (183 mg, 0.62 mmol), using general procedure D, as an off-white solid (61 mg, 24% yield) and by treatment with methanol and water as mentioned in the general procedure D; mp 263–265 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 8.78 (d, J = 4.9 Hz, 2H), 8.16 – 8.10 (m, 2H), 8.07 (dd, J = 7.6, 1.4 Hz, 1H), 7.99 – 7.92 (m, 2H), 7.87 (s, 1H), 7.56 – 7.49 (m, 2H), 7.45 – 7.34 (m, 2H), 7.10 (s, 1H), 4.70 – 4.39 (m, 1H), 3.81 – 3.56 (m, 1H), 3.30 – 2.92 (m, 3H), 2.17 – 1.87 (m, 2H), 1.85 – 1.64 (m, 2H); ESI-MS: m/z 455.2 (C26H22N4O4 requires 455.16, [M + H]+); HPLC Purity: 99% (tR = 2.8 min).
(Z)-3-Oxo-2-(4-(5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine-7-carbonyl)benzylidene)-2,3-dihydrobenzofuran-7-carboxamide (79).
Target compound 79 was obtained by reacting amide I (100 mg, 0.56 mmol) with aldehyde 29 (159 mg, 0.62 mmol) as per general procedure D, as a pale yellow solid (43 mg, 18% yield) and by treatment with methanol and water as mentioned in the general procedure D; mp 275–277 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 8.16 (d, J = 8.0 Hz, 2H), 8.07 (dd, J = 7.6, 1.4 Hz, 1H), 8.03 – 7.93 (m, 3H), 7.87 (s, 1H), 7.65 (d, J = 7.9 Hz, 2H), 7.42 (t, J = 7.6 Hz, 1H), 7.12 (s, 1H), 4.90 (bs, 2H), 4.34 – 3.75 (m, 4H); 13C NMR (100 MHz, DMSO-d6) δ 183.71, 165.11, 163.07, 151.41, 148.88, 146.95, 137.39, 136.61, 133.96, 132.04, 128.21, 127.36, 124.38, 122.14, 121.86, 112.27; ESI-MS: m/z 416.1 (C22H17N5O4 requires 416.13, [M + H]+); HPLC Purity: >99% (tR = 4.58 min).
(Z)-2-(4-(3-Methyl-5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine-7-carbonyl)benzylidene)-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (80).
Target compound 80 was obtained by reacting amide I (100 mg, 0.56 mmol) with aldehyde 30 (168 mg, 0.62 mmol), using general procedure D, as a bright yellow solid (56 mg, 23% yield) and by treatment with methanol and water as mentioned in the general procedure D; mp 287–288 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 8.16 (d, J = 8.1 Hz, 2H), 8.07 (dd, J = 7.6, 1.4 Hz, 1H), 8.04 – 7.93 (m, 2H), 7.87 (s, 1H), 7.62 (d, J = 7.9 Hz, 2H), 7.42 (t, J = 7.6 Hz, 1H), 7.12 (s, 1H), 4.89 (bs, 2H), 4.16 – 3.67 (m, 4H), 2.32 (s, 3H); ESI-MS: m/z 430.1 (C23H19N5O4 requires 430.14, [M + H]+); HPLC Purity: 98% (tR = 4.37 min).
(Z)-3-Oxo-2-(4-(3-(trifluoromethyl)-5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine-7-carbonyl)benzylidene)-2,3-dihydrobenzofuran-7-carboxamide (81).
Target compound 81 was obtained by reacting amide I (100 mg, 0.56 mmol) with aldehyde 31 (201 mg, 0.62 mmol) as per general procedure D, as a pale yellow solid (72 mg, 26% yield) and by treatment with methanol and water as mentioned in the general procedure D; mp 291–293 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 8.20 – 8.13 (m, 2H), 8.07 (dd, J = 7.6, 1.5 Hz, 1H), 7.96 (dd, J = 7.6, 1.5 Hz, 2H), 7.87 (s, 1H), 7.65 (d, J = 8.2 Hz, 2H), 7.42 (t, J = 7.6 Hz, 1H), 7.12 (s, 1H), 4.95 (bs, 2H), 4.33 – 3.73 (m, 4H); ESI-MS: m/z 484.1 (C23H16F3N5O4 requires 484.12, [M + H]+); HPLC Purity: 95% (tR = 5.4 min); Anal. Calcd for C23H16F3N5O4.0.4H2O.0.25CH3COOC2H5: C, 56.23; H, 3.70; N, 13.66; Found: C, 56.21; H, 3.31; N, 13.27.
Ethyl (Z)-7-(4-((7-carbamoyl-3-oxobenzofuran-2(3H)-ylidene)methyl)benzoyl)-5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine-3-carboxylate (82).
Target compound 82 was obtained by reacting amide I (100 mg, 0.56 mmol) with aldehyde 32 (204 mg, 0.62 mmol) as per general procedure D, as a pale yellow solid (84 mg, 31% yield) and by treatment with methanol and water as mentioned in the general procedure D; mp 244–245 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 8.17 (d, J = 8.3 Hz, 2H), 8.08 (dd, J = 7.6, 1.4 Hz, 1H), 7.96 (dd, J = 7.6, 1.4 Hz, 2H), 7.87 (s, 1H), 7.64 (d, J = 8.1 Hz, 2H), 7.42 (t, J = 7.6 Hz, 1H), 7.12 (s, 1H), 5.00 (bs, 2H), 4.47 – 4.29 (m, 4H), 4.16 – 3.70 (m, 2H), 1.34 (t, J = 7.1 Hz, 3H); ESI-MS: m/z 488.2 (C25H21N5O6 requires 488.15, [M + H]+); HPLC Purity: 96% (tR = 5.06 min); Anal. Calcd for C25H21N5O6.0.2H2O: C, 61.15; H, 4.39; N, 14.26; Found: C, 61.18; H, 4.38; N, 14.32.
(Z)-2-(4-(3-Cyclopropyl-5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine-7-carbonyl)benzylidene)-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (83).
Target compound 83 was obtained by reacting amide I (100 mg, 0.56 mmol) with aldehyde 33 (184 mg, 0.62 mmol) as per general procedure D, as a pale yellow solid (56 mg, 22% yield) and by treatment with methanol and water as mentioned in the general procedure D. 1H NMR (400 MHz, DMSO-d6; TMS) δ 8.16 (d, J = 7.9 Hz, 2H), 8.07 (d, J = 7.5 Hz, 1H), 8.00 – 7.93 (m, 2H), 7.87 (s, 1H), 7.63 (d, J = 7.8 Hz, 2H), 7.42 (t, J = 7.5 Hz, 1H), 7.12 (s, 1H), 4.87 (bs, 2H), 4.19 – 3.69 (m, 4H), 1.92 (h, J = 5.5 Hz, 1H), 1.02 – 0.93 (m, 2H), 0.93 – 0.85 (m, 2H); ESI-MS: m/z 456.2 (C25H21N5O4 requires 456.16, [M + H]+); HPLC Purity: 96% (tR = 4.69 min).
(Z)-2-(4-(3-(3-Fluorobenzyl)-5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine-7-carbonyl)benzylidene)-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (84).
Target compound 84 was obtained by reacting amide I (100 mg, 0.56 mmol) with aldehyde 34 (226 mg, 0.62 mmol) as per general procedure D, as a pale yellow solid (47 mg, 16% yield) and by treatment with methanol and water as mentioned in the general procedure D; mp 273–274 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.23 – 7.76 (m, 6H), 7.73 – 7.52 (m, 2H), 7.52 – 7.27 (m, 2H), 7.19 – 7.03 (m, 4H), 4.88 (s, 2H), 4.17 (s, 2H), 4.10 – 3.65 (m, 4H); ESI-MS: m/z 524.2 (C29H22FN5O4 requires 524.17, [M + H]+); HPLC Purity: 97% (tR = 5.49 min).
(Z)-2-(4-(3-(Difluoromethyl)-5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine-7-carbonyl)benzylidene)-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (85).
Target compound 85 was prepared by reacting acid 60 (100 mg, 0.32 mmol) with 3-(difluoromethyl)-5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine (62 mg, 0.36 mmol) as per general procedure C, as a pale yellow solid (46 mg, 31% yield) and by extraction followed by purification using flash chromatography as mentioned in the general procedure C; mp 273–274 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 8.16 (d, J = 8.1 Hz, 2H), 8.07 (dd, J = 7.5, 1.4 Hz, 1H), 8.00 – 7.91 (m, 2H), 7.87 (s, 1H), 7.65 (d, J = 8.0 Hz, 2H), 7.57 – 7.24 (m, 2H), 7.12 (s, 1H), 4.99 (bs, 2H), 4.35 – 3.71 (m, 4H); ESI-MS: m/z 466.1 (C23H17F2N5O4 requires 466.12, [M + H]+); HPLC Purity: 96% (tR = 4.97 min); Anal. Calcd for C23H17F2N5O4.0.4H2O: C, 58.45; H, 3.80; N, 14.82; Found: C, 58.51; H, 3.93; N, 14.75.
(Z)-2-(4-(3-(Cyclopropylmethyl)-5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine-7-carbonyl)benzylidene)-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (86).
Target compound 86 was obtained by reacting acid 60 (100 mg, 0.32 mmol) with 3-(cyclopropylmethyl)-5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine (63 mg, 0.36 mmol) as per general procedure C, as a pale yellow solid (55 mg, 36% yield) upon extraction followed by purification using flash chromatography as mentioned in the general procedure C; mp 245–247 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 8.16 (d, J = 8.1 Hz, 2H), 8.07 (dd, J = 7.5, 1.4 Hz, 1H), 8.00 – 7.93 (m, 2H), 7.86 (s, 1H), 7.63 (d, J = 7.9 Hz, 2H), 7.42 (t, J = 7.6 Hz, 1H), 7.13 (s, 1H), 4.90 (bs, 2H), 4.17 – 3.65 (m, 4H), 2.64 (d, J = 6.8 Hz, 2H), 1.15 – 0.99 (m, 1H), 0.55 – 0.46 (m, 2H), 0.27 – 0.19 (m, 2H); ESI-MS: m/z 470.2 (C26H23N5O4 requires 470.18, [M + H]+); HPLC Purity: 96% (tR = 4.96 min); Anal. Calcd for C26H23N5O4.0.6H2O: C, 65.02; H, 5.08; N, 14.58; Found: C, 64.97; H, 4.95; N, 14.55.
(Z)-2-(4-(3-(Hydroxymethyl)-5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine-7-carbonyl)benzylidene)-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (87).
Target compound 87 was prepared by reacting acid 60 (100 mg, 0.32 mmol) with (5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazin-3-yl)methanol (55 mg, 0.36 mmol) as per general procedure C, as a pale yellow solid (86 mg, 60% yield), upon extraction followed by purification using flash chromatography as mentioned in the general procedure C; mp 289–290 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 8.16 (d, J = 7.9 Hz, 2H), 8.07 (d, J = 7.5 Hz, 1H), 7.96 (d, J = 8.2 Hz, 2H), 7.87 (s, 1H), 7.63 (d, J = 8.0 Hz, 2H), 7.42 (t, J = 7.6 Hz, 1H), 7.12 (s, 1H), 5.55 (s, 1H), 4.91 (bs, 2H), 4.59 (s, 2H), 4.29 – 3.63 (m, 4H); 13C NMR (100 MHz, DMSO-d6) δ 183.71, 165.13, 163.08, 153.32, 146.95, 137.39, 133.99, 132.07, 128.18, 127.37, 124.39, 122.14, 121.87, 112.26, 53.92; ESI-MS: m/z 446.1 (C23H19N5O5 requires 446.14, [M + H]+); HPLC Purity: 97% (tR = 4.81 min); Anal. Calcd for C23H19N5O5.0.45H2O.0.5CH3OH: C, 60.11; H, 4.70; N, 14.91; Found: C, 60.34; H, 4.35; N, 14.58.
(Z)-2-(4-(3-Isopropyl-5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine-7-carbonyl)benzylidene)-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (88).
Target compound 88 was obtained by reacting acid 60 (100 mg, 0.32 mmol) with 3-isopropyl-5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine (60 mg, 0.36 mmol), using general procedure C, as a pale yellow solid (45 mg, 30% yield), upon extraction followed by purification using flash chromatography as mentioned in the general procedure C; mp 292–294 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 8.16 (d, J = 8.3 Hz, 2H), 8.07 (dd, J = 7.6, 1.4 Hz, 1H), 7.96 (dd, J = 7.6, 1.4 Hz, 2H), 7.86 (s, 1H), 7.64 (d, J = 7.8 Hz, 2H), 7.42 (t, J = 7.6 Hz, 1H), 7.12 (s, 1H), 4.89 (s, 2H), 4.15 – 3.61 (m, 4H), 3.10 – 2.95 (m, 1H), 1.27 (d, J = 6.9 Hz, 6H); ESI-MS: m/z 458.2 (C25H23N5O4 requires 458.18, [M + H]+); HPLC Purity: 97% (tR = 4.81 min); Anal. Calcd for C25H23N5O4.0.55H2O: C, 64.24; H, 5.20; N, 14.98; Found: C, 64.19; H, 5.20; N, 14.96.
(Z)-2-(4-(3-Cyclopentyl-5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine-7-carbonyl)benzylidene)-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (89).
Target compound 89 was obtained by reacting acid 60 (100 mg, 0.32 mmol) with 3-cyclopentyl-5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine (69 mg, 0.36 mmol) as per general procedure C, as a pale yellow solid (73 mg, 47% yield), upon extraction followed by purification using flash chromatography as mentioned in the general procedure C; mp 281–283 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 8.16 (d, J = 8.3 Hz, 2H), 8.07 (dd, J = 7.6, 1.4 Hz, 1H), 7.96 (dd, J = 7.6, 1.4 Hz, 2H), 7.86 (s, 1H), 7.64 (d, J = 7.9 Hz, 2H), 7.42 (t, J = 7.6 Hz, 1H), 7.12 (s, 1H), 4.89 (s, 2H), 4.15 – 3.67 (m, J = 107.5 Hz, 4H), 3.15 (p, J = 8.0 Hz, 1H), 2.05 – 1.93 (m, 2H), 1.92 – 1.78 (m, 2H), 1.78 – 1.68 (m, 2H), 1.68 – 1.56 (m, 2H); ESI-MS: m/z 484.2 (C27H25N5O4 requires 484.19, [M + H]+); HPLC Purity: 96% (tR = 5.24 min); Anal. Calcd for C27H25N5O4.0.3H2O: C, 66.33; H, 5.28; N, 14.32; Found: C, 66.52; H, 5.28; N, 14.05.
(Z)-2-(4-(3-(1-Methyl-1H-imidazol-4-yl)-5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine-7-carbonyl)benzylidene)-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (90).
Target compound 90 was obtained by reacting acid 60 (100 mg, 0.32 mmol) with 3-(1-methyl-1H-imidazol-4-yl)-5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine (74 mg, 0.36 mmol) as per general procedure C, as a pale yellow solid (110 mg, 69% yield), upon extraction followed by purification using flash chromatography as mentioned in the general procedure C; mp 208–210 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 8.16 (d, J = 8.3 Hz, 2H), 8.07 (dd, J = 7.6, 1.3 Hz, 1H), 7.96 (dd, J = 7.5, 1.3 Hz, 2H), 7.87 (s, 1H), 7.83 – 7.73 (m, 2H), 7.65 (d, J = 8.2 Hz, 2H), 7.42 (t, J = 7.6 Hz, 1H), 7.12 (s, 1H), 4.95 (s, 2H), 4.43 (t, J = 5.3 Hz, 2H), 3.91 – 3.61 (m, 5H); 13C NMR (100 MHz, DMSO-d6) δ 183.72, 165.12, 163.06, 148.41, 146.94, 139.30, 137.38, 133.91, 132.05, 130.07, 128.22, 127.36, 124.39, 122.14, 121.88, 121.24, 112.32, 33.70; ESI-MS: m/z 495.2 (C26H21N7O4 requires 495.17, [M + H]+); HPLC Purity: 99% (tR = 4.47 min); Anal. Calcd for C26H21N7O4.2H2O: C, 58.75; H, 4.74; N, 18.45; Found: C, 58.50; H, 4.69; N, 18.56.
(Z)-3-Oxo-2-(3-(5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine-7-carbonyl)benzylidene)-2,3-dihydrobenzofuran-7-carboxamide (91).
Target compound 91 was obtained by reacting amide I (100 mg, 0.56 mmol) with aldehyde 35 (159 mg, 0.62 mmol) as per general procedure D, as a pale yellow solid (40 mg, 16% yield) and by treatment with methanol and water as mentioned in the general procedure D; mp 257–259 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 8.20 (s, 2H), 8.12 – 7.85 (m, 5H), 7.66 – 7.59 (m, 2H), 7.41 (t, J = 7.6 Hz, 1H), 7.15 (s, 1H), 4.91 (s, 2H), 4.35 – 3.81 (m, 4H); ESI-MS: m/z 416.1 (C22H17N5O4 requires 416.13, [M + H]+); HPLC Purity: 98% (tR = 4.58 min).
(Z)-3-Oxo-2-(3-(3-(trifluoromethyl)-5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine-7-carbonyl)benzylidene)-2,3-dihydrobenzofuran-7-carboxamide (92).
Target compound 92 was obtained by reacting amide I (100 mg, 0.56 mmol) with aldehyde 36 (201 mg, 0.62 mmol) as per general procedure D, as a pale yellow solid (46 mg, 17% yield) and by treatment with methanol and water as mentioned in the general procedure D; mp 268–269 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 8.16 (d, J = 8.0 Hz, 2H), 8.07 (dd, J = 7.6, 1.5 Hz, 1H), 8.00 – 7.93 (m, 2H), 7.87 (s, 1H), 7.65 (d, J = 7.9 Hz, 2H), 7.42 (t, J = 7.6 Hz, 1H), 7.12 (s, 1H), 4.95 (bs, 2H), 4.31 – 4.23 (m, 2H), 3.99 – 3.69 (m, 2H); ESI-MS: m/z 484.1 (C23H16F3N5O4 requires 484.12, [M + H]+); HPLC Purity: 97% (tR = 5.44 min).
(Z)-2-(4-((2-(1H-Benzo[d]imidazol-2-yl)ethyl)carbamoyl)benzylidene)-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (93).
Target compound 93 was obtained by reacting amide I (100 mg, 0.56 mmol) with aldehyde 37 (182 mg, 0.62 mmol), using general procedure D, as a yellow solid (66 mg, 26% yield) and by treatment with methanol and water as mentioned in the general procedure D; mp 255–257 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 12.34 (bs, 1H), 8.86 (t, J = 5.5 Hz, 1H), 8.13 (d, J = 8.2 Hz, 2H), 8.10 – 8.05 (m, 1H), 7.96 (d, J = 8.0 Hz, 4H), 7.89 (s, 1H), 7.60 – 7.35 (m, 3H), 7.18 – 7.06 (m, 3H), 3.75 (q, J = 6.8 Hz, 2H), 3.12 (t, J = 7.3 Hz, 2H); ESI-MS: m/z 453.2 (C26H20N4O4 requires 453.15, [M + H]+); HPLC Purity: 96% (tR = 5.43 min).
(Z)-2-(4-((2-(5-Methyl-1H-benzo[d]imidazol-2-yl)ethyl)carbamoyl)benzylidene)-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (94).
Target compound 94 was obtained by reacting amide I (100 mg, 0.56 mmol) with aldehyde 38 (191 mg, 0.62 mmol), using general procedure D, as a yellow solid (75 mg, 28% yield) and by treatment with methanol and water as mentioned in the general procedure D; mp 289–291 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 12.22 (bs, 1H), 8.86 (s, 1H), 8.18 – 8.05 (m, 3H), 8.02 – 7.82 (m, 5H), 7.45 – 7.34 (m, 2H), 7.27 (s, 1H), 7.09 (s, 1H), 6.95 (d, J = 8.2 Hz, 1H), 3.77 – 3.70 (m, 2H), 3.09 (t, J = 7.4 Hz, 2H), 2.39 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 183.68, 166.06, 165.12, 163.02, 152.92, 146.98, 137.44, 135.75, 134.88, 131.77, 130.80, 128.18, 127.39, 124.40, 123.14, 122.11, 121.81, 112.23, 38.64, 29.26, 21.75; ESI-MS: m/z 467.2 (C27H22N4O4 requires 467.16, [M + H]+); HPLC Purity: 97% (tR = 4.37 min).
(Z)-2-(4-((2-(5-Fluoro-1H-benzo[d]imidazol-2-yl)ethyl)carbamoyl)benzylidene)-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (95).
Target compound 95 was obtained by reacting amide I (100 mg, 0.56 mmol) with aldehyde 39 (193 mg, 0.62 mmol), using general procedure D, as a yellow solid (78 mg, 29% yield) and by treatment with methanol and water as mentioned in the general procedure D; mp 292–293 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 12.46 (bs, 1H), 8.85 (t, J = 5.7 Hz, 1H), 8.13 (d, J = 8.2 Hz, 2H), 8.08 (dd, J = 7.6, 1.4 Hz, 1H), 7.98 – 7.92 (m, 4H), 7.89 (s, 1H), 7.62 – 7.19 (m, 4H), 7.09 (s, 1H), 6.98 (t, J = 8.6 Hz, 1H), 3.74 (q, J = 6.8 Hz, 2H), 3.11 (t, J = 7.3 Hz, 2H); ESI-MS: m/z 471.1 (C26H19FN4O4 requires 471.14, [M + H]+); HPLC Purity: 95% (tR = 5.63 min).
(Z)-2-(3-((2-(1H-Benzo[d]imidazol-2-yl)ethyl)carbamoyl)benzylidene)-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (96).
Target compound 96 was obtained by reacting amide I (100 mg, 0.56 mmol) with aldehyde 40 (182 mg, 0.62 mmol), using general procedure D, as a yellow solid (49 mg, 19% yield) and by treatment with methanol and water; mp 276–277 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 12.35 (bs, 1H), 8.73 (t, J = 5.6 Hz, 1H), 8.48 (s, 1H), 8.21 (d, J = 7.8 Hz, 1H), 8.12 (d, J = 7.7 Hz, 1H), 8.06 (s, 1H), 8.00 – 7.86 (m, 3H), 7.61 (t, J = 7.8 Hz, 1H), 7.54 – 7.46 (m, 2H), 7.42 (t, J = 7.6 Hz, 1H), 7.17 – 7.10 (m, 2H), 7.08 (s, 1H), 3.79 (q, J = 6.8 Hz, 2H), 3.15 (t, J = 7.2 Hz, 2H); ESI-MS: m/z 453.2 (C26H20N4O4 requires 453.15, [M + H]+); HPLC Purity: 98% (tR = 5.6 min).
(Z)-2-(3-((2-(5-Fluoro-1H-benzo[d]imidazol-2-yl)ethyl)carbamoyl)benzylidene)-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (97).
Target compound 97 was obtained by reacting amide I (100 mg, 0.56 mmol) with aldehyde 41 (193 mg, 0.62 mmol), using general procedure D, as a yellow solid (56 mg, 21% yield) and by treatment with methanol and water as mentioned in the general procedure D; mp 268–269 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 12.46 (bs, 1H), 8.78 (t, J = 5.4 Hz, 1H), 8.53 (s, 1H), 8.40 (d, J = 7.8 Hz, 1H), 8.17 (dd, J = 7.6, 1.4 Hz, 1H), 8.04 – 7.87 (m, 3H), 7.74 (s, 1H), 7.63 – 7.47 (m, 2H), 7.47 – 7.22 (m, 3H), 7.05 – 6.91 (m, 1H), 3.74 (q, J = 6.8 Hz, 2H), 3.11 (t, J = 7.3 Hz, 2H); ESI-MS: m/z 471.1 (C26H19FN4O4 requires 471.14, [M + H]+); HPLC Purity: 97% (tR = 5.86 min).
(Z)-2-(4-((2-(5-Methoxy-1H-benzo[d]imidazol-2-yl)ethyl)carbamoyl)benzylidene)-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (98).
Target compound 98 was obtained by reacting acid 60 (100 mg, 0.32 mmol) with 2-(5-methoxy-1H-benzo[d]imidazol-2-yl)ethan-1-amine (68 mg, 0.36 mmol) as per general procedure C, as a pale yellow solid (55 mg, 20% yield), upon extraction followed by purification using flash chromatography as mentioned in the general procedure C; mp 290–291 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 12.15 (bs, 1H), 8.85 (t, J = 5.5 Hz, 1H), 8.19 – 8.04 (m, 3H), 8.04 – 7.83 (m, 5H), 7.48 – 7.22 (m, 2H), 7.09 (s, 1H), 6.98 – 6.68 (m, 2H), 3.82 – 3.67 (m, 5H), 3.08 (t, J = 7.3 Hz, 2H); ESI-MS: m/z 483.2 (C27H22N4O5 requires 483.16, [M + H]+); HPLC Purity: 97% (tR = 5.46 min); Anal. Calcd for C27H22N4O5.0.8H2O: C, 65.26; H, 4.79; N, 11.28; Found: C, 65.20; H, 4.71; N, 11.27.
(Z)-2-(2-(4-((7-Carbamoyl-3-oxobenzofuran-2(3H)-ylidene)methyl)benzamido)ethyl)-1H-benzo[d]imidazole-4-carboxamide (99).
Target compound 99 was obtained by reacting acid 60 (100 mg, 0.32 mmol) with amine 43 (74 mg, 0.36 mmol) as per general procedure C, as a pale yellow solid (66 mg, 41% yield), upon extraction followed by purification using reverse phase flash chromatography as mentioned in the general procedure C; mp 269–271 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 12.33 (s, 1H), 8.91 – 8.82 (m, 1H), 8.13 (d, J = 8.3 Hz, 2H), 8.08 (d, J = 6.8 Hz, 1H), 7.96 (d, J = 7.9 Hz, 4H), 7.89 (s, 1H), 7.59 – 7.37 (m, 3H), 7.20 – 7.07 (m, 3H), 3.75 (q, J = 6.3 Hz, 2H), 3.12 (t, J = 7.2 Hz, 2H); ESI-MS: m/z 496.2 (C27H21N5O5 requires 496.15, [M + H]+); HPLC Purity: 96% (tR = 4.91 min); Anal. Calcd for C27H21N5O5.1.8H2O: C, 61.43; H, 4.70; N, 13.27; Found: C, 61.67; H, 4.62; N, 13.00.
(Z)-2-(4-((1-(1H-Benzo[d]imidazol-2-yl)-2-methylpropan-2-yl)carbamoyl)benzylidene)-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (100).
Target compound 100 was obtained by reacting acid 60 (100 mg, 0.32 mmol) with 1-(1H-benzo[d]imidazol-2-yl)-2-methylpropan-2-amine (68 mg, 0.36 mmol) as per general procedure C, as a pale yellow solid (108 mg, 70% yield), upon extraction followed by purification using reverse phase flash chromatography; mp 255–257 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 12.26 (s, 1H), 8.81 (t, J = 5.3 Hz, 1H), 8.14 (d, J = 8.5 Hz, 2H), 8.08 (dd, J = 7.6, 1.4 Hz, 1H), 8.02 – 7.92 (m, 4H), 7.89 (s, 1H), 7.52 – 7.37 (m, 3H), 7.18 – 7.04 (m, 3H), 3.41 (q, J = 6.6 Hz, 2H), 2.90 (t, J = 7.5 Hz, 2H), 2.16 – 2.00 (m, 2H); ESI-MS: m/z 481.2 (C28H24N4O4 requires 481.18, [M + H]+); HPLC Purity: 98% (tR = 6.05 min); Anal. Calcd for C28H24N4O4.0.3H2O: C, 69.21; H, 5.10; N, 11.53; Found: C, 69.50; H, 5.12; N, 11.21.
(Z)-2-(4-((3-(1H-Benzo[d]imidazol-2-yl)propyl)carbamoyl)benzylidene)-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (101).
Target compound 101 was obtained by reacting acid 60 (100 mg, 0.32 mmol) with 3-(1H-benzo[d]imidazol-2-yl)propan-1-amine (63 mg, 0.36 mmol) as per general procedure C, as a pale yellow solid (76 mg, 50% yield), upon extraction followed by purification using reverse phase flash chromatography as mentioned in the general procedure C; mp 298–299 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.26 (s, 1H), 8.81 (t, J = 5.6 Hz, 1H), 8.17 – 8.11 (m, 2H), 8.08 (dd, J = 7.6, 1.4 Hz, 1H), 8.00 – 7.93 (m, 4H), 7.89 (s, 1H), 7.52 – 7.44 (m, 2H), 7.42 (t, J = 7.6 Hz, 1H), 7.13 (dd, J = 6.0, 3.2 Hz, 2H), 7.10 (s, 1H), 3.45 – 3.37 (m, 2H), 2.90 (t, J = 7.5 Hz, 2H), 2.12 – 2.02 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ 183.68, 166.01, 165.14, 163.03, 155.24, 146.97, 137.43, 135.89, 134.81, 131.78, 128.16, 127.39, 124.39, 122.12, 121.83, 121.68, 112.28, 112.21, 27.79, 26.70; ESI-MS: m/z 467.2 (C27H22N4O4 requires 467.16, [M + H]+); HPLC Purity: >99% (tR = 5.56 min); Anal. Calcd for C27H22N4O4.0.5H2O: C, 68.20; H, 4.88; N, 11.78; Found: C, 68.27; H, 4.75; N, 11.65.
(Z)-2-(4-(3-(1H-Benzo[d]imidazol-2-yl)azetidine-1-carbonyl)benzylidene)-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (102).
Target compound 102 was obtained by reacting acid 60 (100 mg, 0.32 mmol) with 2-(azetidin-3-yl)-1H-benzo[d]imidazole (62 mg, 0.36 mmol) as per general procedure C, as a pale yellow solid (102 mg, 68% yield), upon extraction followed by purification using flash chromatography as mentioned in the general procedure C; mp 287–289 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 12.52 (s, 1H), 8.18 – 8.12 (m, 2H), 8.08 (dd, J = 7.6, 1.4 Hz, 1H), 8.02 – 7.93 (m, 2H), 7.86 (s, 1H), 7.83 – 7.77 (m, 2H), 7.60 (d, J = 7.4 Hz, 1H), 7.47 (d, J = 7.3 Hz, 1H), 7.41 (t, J = 7.6 Hz, 1H), 7.22 – 7.13 (m, 2H), 7.12 (s, 1H), 4.80 (t, J = 8.7 Hz, 1H), 4.66 (t, J = 7.3 Hz, 1H), 4.52 (t, J = 9.4 Hz, 1H), 4.34 (dd, J = 9.8, 6.0 Hz, 1H), 4.17 (tt, J = 9.0, 6.0 Hz, 1H); ESI-MS: m/z 465.2 (C27H20N4O4 requires 465.15, [M + H]+); HPLC Purity: 98% (tR = 5.45 min); Anal. Calcd for C27H20N4O4.1.25H2O: C, 66.59; H, 4.66; N, 11.50; Found: C, 66.42; H, 4.37; N, 11.54.
(Z)-2-(4-(4-(1H-Benzo[d]imidazol-2-yl)piperazine-1-carbonyl)benzylidene)-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (103).
Target compound 103 was obtained by reacting acid 60 (100 mg, 0.32 mmol) with intermediate 47 (72 mg, 0.36 mmol), using general procedure C, as an orange solid (48 mg, 30% yield), upon extraction followed by purification using reverse phase flash chromatography; mp 238–239 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 11.51 (s, 1H), 8.15 (d, J = 8.2 Hz, 2H), 8.08 (dd, J = 7.5, 1.4 Hz, 1H), 7.97 (dd, J = 7.6, 1.4 Hz, 2H), 7.87 (s, 1H), 7.58 (d, J = 8.2 Hz, 2H), 7.42 (t, J = 7.6 Hz, 1H), 7.22 (dd, J = 18.4, 7.5 Hz, 2H), 7.12 (s, 1H), 6.94 (dt, J = 20.8, 7.2 Hz, 2H), 3.87 – 3.70 (m, 2H), 3.68 – 3.46 (m, 6H); ESI-MS: m/z 494.2 (C28H23N5O4 requires 494.18, [M + H]+); HPLC Purity: 99% (tR = 5.45 min); Anal. Calcd for C28H23N5O4.1.6H2O: C, 64.38; H, 5.06; N, 13.41; Found: C, 64.48; H, 5.16; N, 13.24.
(Z)-2-(4-(4-(1H-Benzo[d]imidazol-2-yl)piperidine-1-carbonyl)benzylidene)-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (104).
Target compound 104 was obtained by reacting acid 60 (100 mg, 0.32 mmol) with 2-(piperidin-4-yl)-1H-benzo[d]imidazole (73 mg, 0.36 mmol) as per general procedure C, as a pale yellow solid (104 mg, 65% yield), upon extraction followed by purification using reverse phase flash chromatography; mp 227–229 °C; 1H NMR (400 MHz, DMSO-d6; TMS) δ 12.32 (bs, 1H), 8.14 (d, J = 8.3 Hz, 2H), 8.07 (dd, J = 7.6, 1.3 Hz, 1H), 7.96 (dd, J = 7.5, 1.3 Hz, 2H), 7.86 (s, 1H), 7.58 – 7.46 (m, 4H), 7.41 (t, J = 7.6 Hz, 1H), 7.20 – 7.06 (m, 3H), 4.60 – 4.43 (m, 1H), 3.80 – 3.60 (m, 1H), 3.29 – 2.97 (m, 3H), 2.20 – 1.94 (m, 2H), 1.92 – 1.75 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ 183.68, 168.79, 165.13, 163.04, 157.64, 146.82, 138.13, 137.36, 133.28, 132.04, 127.74, 127.36, 124.36, 122.17, 121.85, 112.48, 36.10. ESI-MS: m/z 493.2 (C29H24N4O4 requires 493.18, [M + H]+); HPLC Purity: 98% (tR = 5.52 min); Anal. Calcd for C29H24N4O4.1.65H2O: C, 66.69; H, 5.27; N, 10.73; Found: C, 66.70; H, 5.20; N, 10.73.
Methyl (Z)-2-(2-(4-((7-carbamoyl-3-oxobenzofuran-2(3H)-ylidene)methyl)benzamido)ethyl)-1H-benzo[d]imidazole-7-carboxylate (105).
Target compound 105 was obtained by reacting acid 60 (200 mg, 0.32 mmol) with 43a (156 mg, 0.35 mmol) as per general procedure C, as a pale yellow solid (100 mg, 30% yield), upon extraction followed by purification using reverse phase flash chromatography; mp 221–223 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.32 (s, 1H), 8.87 – 8.77 (m, 1H), 8.13 (d, J = 8.4 Hz, 2H), 8.08 (d, J = 6.5 Hz, 1H), 7.98 – 7.92 (m, 4H), 7.86 (d, J = 8.3 Hz, 2H), 7.78 (d, J = 7.5 Hz, 1H), 7.42 (t, J = 7.6 Hz, 1H), 7.27 (t, J = 7.8 Hz, 1H), 7.09 (s, 1H), 3.96 (s, 3H), 3.82 – 3.74 (m, 2H), 3.23 (t, J = 7.1 Hz, 2H); 13C NMR (100 MHz, DMSO-d6) δ 183.67, 166.25, 166.11, 165.12, 163.02, 155.61, 146.97, 137.43, 135.79, 134.87, 131.76, 128.18, 127.39, 124.38, 124.03, 122.11, 121.79, 121.22, 112.20, 52.47, 38.56, 29.00; ESI-MS: m/z 511.2 (C28H22N4O6 requires 511.15, [M + H]+); HPLC Purity: 96% (tR = 5.75 min).
PARP Enzymatic Inhibition Assay.
PARP inhibitor screening and IC50 determination were conducted by BPS Bioscience (San Diego, CA) using chemiluminescence assay protocol. In general, all assays were done by following the BPS PARP or TNKS assay kit protocols. The enzymatic reactions were conducted in duplicate at room temperature for 1 h in a 96 well plate coated with histone substrate. 50 μL of reaction buffer (Tris.HCl, pH 8.0) contains NAD+, biotinylated NAD+, activated DNA, a PARP enzyme and the test compound. After enzymatic reactions, 50 μL of Streptavidin-horseradish peroxidase was added to each well and the plate was incubated at room temperature for an additional 30 min. 100 μL of developer reagents were added to wells and luminescence was measured using a BioTek SynergyTM 2 microplate reader.
Data analysis was performed by PARP activity assays performed in duplicates. The luminescence data were analyzed using the computer software, Graphpad Prism. In the absence of the compound, the luminescence (Lt) in each data set was defined as 100% activity. In the absence of the PARP, the luminescence (Lb) in each data set was defined as 0% activity. The percent activity in the presence of each compound was calculated according to the following equation: % activity = [(L- Lb)/(Lt - Lb)]×100, where L= the luminescence in the presence of the compound, Lb = the luminescence in the absence of the PARP, and Lt = the luminescence in the absence of the compound. The percent inhibition was calculated according to the following equation: % inhibition = 100 - % activity.
The values of % activity versus a series of compound concentrations were then plotted using non-linear regression analysis of Sigmoidal dose-response curve generated with the equation Y = B + (T-B)/1+10((LogEC50-X)×Hill Slope), where Y = percent activity, B = minimum percent activity, T = maximum percent activity, X = logarithm of compound concentration and Hill Slope = slope factor or Hill coefficient. The IC50 value was determined by the concentration causing a half-maximal percent activity.
PARP-Isoform Screening of Representative Set of Target Compounds.
Selected PARP-1 and PARP-2 inhibitors were screened against other catalytic PARPs (PARP-3, TNKS1, TNKS2, PARP-8, PARP-10, and PARP-14) at BPS Bioscience (San Diego, CA) using chemiluminescence assay protocol. The protocol employed is similar to that used in PARP-1 enzyme assay.
Protein Expression Vectors.
PARP-1 CAT WT (residues 661–1014) was produced from a pET28 vector. The PARP-1 CATΔHD construct used for crystallization and binding analysis replaces HD residues 678–787 with an 8-residue linker (GSGSGSGG) in the pET28 construct coding for PARP-1 residues 661–1011.47
Protein Expression and Purification.
PARP-1 CAT WT and CATΔHD were expressed and purified as described.48 Note that for CATΔHD 10 mM benzamide was added to the Escherichia coli media to reduce cellular toxicity of the PARP-1 protein.
Differential Scanning Fluorimetry.
Differential scanning fluorimetry experiments were performed as described16, 48 using 5 μM protein and 250 μM of PARP-1 inhibitor. Experiments were performed on a Roche LightCycler 480 RT-PCR in the following buffer: 25 mM Hepes pH 8.0, 150 mM NaCl, 0.1 mM TCEP and 1 mM EDTA. ΔTM values were calculated by subtracting the TM determined for the protein in the absence of inhibitor from the TM determined in the presence of inhibitor. Experiments were performed in triplicate and a Boltzmann sigmoid was fit to the data to determine the TM values (KaleidaGraph).
Protein Crystallization and Structure Determination.
PARP-1 CATΔHD (30 mg/ml) was crystallized in the presence of 1.1 mM PARP-1 inhibitors (compounds 57, 63 and 93) in 19 to 24% PEG 3350, 0.2 M ammonium sulfate, 0.1 M Hepes pH 7.5 in sitting drop vapor diffusion trays at room temperature. Crystals were cryo-protected in 23% PEG 3350, 0.2 M ammonium sulfate, 0.1 M Hepes pH 7.5, 1.7 mM PARP-1 inhibitor, and 20% sucrose prior to flash-cooling in liquid nitrogen. Compounds 83 and 103 (1.1 mM) in complex with PARP-1 CATΔHD (30 mg/ml) were crystallized in 17 to 22% PEG 3350, 0.2 M sodium citrate and cryoprotected in 18–19% PEG 3350, 0.2 M sodium citrate, 1.7–1.8 mM compound, and 20% sucrose. X-ray diffraction data were collected at the Canadian Light Source and processed using XDS65 (Table S1). The structures were determined by molecular replacement using PHASER66 as implemented in the Phenix suite67 and PDB code 5ds346 as a search model. Model building was performed using COOT68 and refinement was performed using Phenix67 and REFMAC569, 70. Structure images were made using PYMOL Molecular Graphics System (Schrödinger, LLC).
Cell-Based Assays
Cell Lines.
SUM149 parental cells (BRCA1–/–) and SUM149 revertant (BRCA1 corrected) cells have been previously described.60 These cells were infected with NucLight-RFP red nuclear tag (Essen Bioscience, Ann Arbor), according to manufacturer’s protocol. All cells were cultured following the supplier’s instructions.
Small Molecule Inhibitors.
Test compounds 81, 83, olaparib and talazoparib (Selleck Chemicals) were prepared in DMSO following manufacturer protocols and stored in aliquots at −80°C.
Cell-Based Drug Exposure Assay.
Cells were seeded into 48-well or 96-well plates at a concentration of 5,000 or 500 cells per well, respectively. After 24 h, cells were exposed to increasing concentrations of each inhibitor such that final DMSO concentrations were ≤0.8% (v/v). Cell growth was monitored for 6 days using time-lapse microscopy (IncuCyte, Essen Bioscience, Ann Arbor) and survival curves were calculated by normalizing cell counts to cell numbers in vehicle-treated wells and plotted using a four-parameter logistic regression curve fit (Prism, Graphpad).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Department of Pharmaceutical Sciences and seed grant program (579-1110-6709) of St. John’s University, and through grant support from the Canadian Institute of Health Research (BMA342854 to J.M.P). AA thanks the BRCA Foundation, Susan G Komen for the Cure and the Breast Cancer Research Foundation for funding. The authors thank Dr. Vijay M. Gokhale (Arizona University) for his helpful comments.
TTT and JMP are co-founders of Hysplex, LLC, with interests in PARP-inhibitor development. AA holds patents on the use of PARP inhibitors held jointly with AstraZeneca from which he has benefitted financially (and may do so in the future) through the ICR Rewards to Inventors Scheme. AA is also co-founder of Tango Therapeutics, a consultant for TopoRx and receives grant funding from AstraZeneca. The other authors declare no competing financial interest.
ABBREVIATIONS USED
- ADP
adenosine 5’-diphosphate
- ABCB1
ATP-binding cassette family B1 transporter
- ABCG2
ATP-binding cassette family G2 transporter
- ABP
adenine binding pocket
- ART
ADP-ribosyltransferase
- BAD
benzamide adenine dinucleotide
- BER
base excision repair
- 53BP1
p53 binding protein 1
- BRCA1
breast cancer gene 1
- BRCA2
breast cancer gene 2
- DHBF
dihydrobenzofuran-7-carboxamide
- CAT
Catalytic
- DDR
DNA damage response
- DEA
diethylamine
- DIPEA
N,N-diisopropylethylamine
- DSBs
double strand breaks
- DSF
differential scanning fluorimetry
- HCTU
O-(1H-6-Chlorobenzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate
- HD
helical domain
- HOBt
1-hydroxybenzotriazole
- HR
homologous recombination
- NHEJ
non-homologous end joining
- PAR
poly(ADP)ribose
- PARP
poly(ADP-ribose) polymerases
- SSBs
single strand breaks
- THTP
5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazines
- TNBC
triple negative breast cancer
- TNKS
tankyrase
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI:
Synthesis of intermediate I. The document also contains analytical data of target compounds (such as 1H NMR, 13C NMR, and HPLC chromatograms), concentration-response curves for selected compounds against PARP-1, PARP-2, TNKS1, and TNKS2, crystallographic data and refinement statistics, X-ray crystal structures of PARP-1 CATΔHD bound to inhibitors, and dose-response survival curves for SUM149 parental (BRCA1−/−, black line) and SUM149 revertant (BRCA1 corrected, green line) cells treated with 81 (PDF)
Molecular formula strings (CSV)
Accession Codes
Coordinates and structure factors are deposited at the Protein Data Bank with codes 6NRG, 6NRH, 6NRI, 6NRJ, and 6NRF. Authors will release the atomic coordinates and experimental data upon article publication.
REFERENCES
- 1.Caldecott KW Protein ADP-ribosylation and the cellular response to DNA strand breaks. DNA Repair (Amst) 2014, 19, 108–113. [DOI] [PubMed] [Google Scholar]
- 2.Khanna A DNA damage in cancer therapeutics: a boon or a curse? Cancer Res 2015, 75, 2133–2138. [DOI] [PubMed] [Google Scholar]
- 3.Hoeijmakers JH Genome maintenance mechanisms for preventing cancer. Nature 2001, 411, 366–374. [DOI] [PubMed] [Google Scholar]
- 4.Friedberg EC; Aguilera A; Gellert M; Hanawalt PC; Hays JB; Lehmann AR; Lindahl T; Lowndes N; Sarasin A; Wood RD DNA repair: from molecular mechanism to human disease. DNA Repair (Amst) 2006, 5, 986–996. [DOI] [PubMed] [Google Scholar]
- 5.Polo SE; Jackson SP Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev 2011, 25, 409–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pearl LH; Schierz AC; Ward SE; Al-Lazikani B; Pearl FM Therapeutic opportunities within the DNA damage response. Nat. Rev. Cancer 2015, 15, 166–180. [DOI] [PubMed] [Google Scholar]
- 7.Ray Chaudhuri A; Nussenzweig A The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol 2017, 18, 610–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tan ES; Krukenberg KA; Mitchison TJ Large-scale preparation and characterization of poly(ADP-ribose) and defined length polymers. Anal. Biochem 2012, 428, 126–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barkauskaite E; Jankevicius G; Ahel I Structures and mechanisms of enzymes employed in the synthesis and degradation of PARP-dependent protein ADP-ribosylation. Mol. Cell 2015, 58, 935–946. [DOI] [PubMed] [Google Scholar]
- 10.Hottiger MO; Hassa PO; Luscher B; Schuler H; Koch-Nolte F Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem. Sci 2010, 35, 208–219. [DOI] [PubMed] [Google Scholar]
- 11.Rouleau M; Patel A; Hendzel MJ; Kaufmann SH; Poirier GG PARP inhibition: PARP1 and beyond. Nat. Rev. Cancer 2010, 10, 293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beneke S Regulation of chromatin structure by poly(ADP-ribosyl)ation. Front. Genet 2012, 3, 169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Caldecott KW XRCC1 and DNA strand break repair. DNA Repair (Amst) 2003, 2, 955–969. [DOI] [PubMed] [Google Scholar]
- 14.Audebert M; Salles B; Calsou P Involvement of poly(ADP-ribose) polymerase-1 and XRCC1/DNA ligase III in an alternative route for DNA double-strand breaks rejoining. J. Biol. Chem 2004, 279, 55117–55126. [DOI] [PubMed] [Google Scholar]
- 15.Kedar PS; Stefanick DF; Horton JK; Wilson SH Increased PARP-1 association with DNA in alkylation damaged, PARP-inhibited mouse fibroblasts. Mol. Cancer Res 2012, 10, 360–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Langelier MF; Riccio AA; Pascal JM PARP-2 and PARP-3 are selectively activated by 5’ phosphorylated DNA breaks through an allosteric regulatory mechanism shared with PARP-1. Nucleic Acids Res 2014, 42, 7762–7775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kummar S; Ji J; Morgan R; Lenz HJ; Puhalla SL; Belani CP; Gandara DR; Allen D; Kiesel B; Beumer JH; Newman EM; Rubinstein L; Chen A; Zhang Y; Wang L; Kinders RJ; Parchment RE; Tomaszewski JE; Doroshow JH A phase I study of veliparib in combination with metronomic cyclophosphamide in adults with refractory solid tumors and lymphomas. Clin. Cancer Res 2012, 18, 1726–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barazzuol L; Jena R; Burnet NG; Meira LB; Jeynes JC; Kirkby KJ; Kirkby NF Evaluation of poly (ADP-ribose) polymerase inhibitor ABT-888 combined with radiotherapy and temozolomide in glioblastoma. Radiat. Oncol 2013, 8, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Plummer R; Jones C; Middleton M; Wilson R; Evans J; Olsen A; Curtin N; Boddy A; McHugh P; Newell D; Harris A; Johnson P; Steinfeldt H; Dewji R; Wang D; Robson L; Calvert H Phase I study of the poly(ADP-ribose) polymerase inhibitor, AG014699, in combination with temozolomide in patients with advanced solid tumors. Clin. Cancer Res 2008, 14, 7917–7923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Su JM; Thompson P; Adesina A; Li XN; Kilburn L; Onar-Thomas A; Kocak M; Chyla B; McKeegan E; Warren KE; Goldman S; Pollack IF; Fouladi M; Chen A; Giranda V; Boyett J; Kun L; Blaney SM A phase I trial of veliparib (ABT-888) and temozolomide in children with recurrent CNS tumors: a pediatric brain tumor consortium report. Neuro. Oncol 2014, 16, 1661–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rajan A; Carter CA; Kelly RJ; Gutierrez M; Kummar S; Szabo E; Yancey MA; Ji J; Mannargudi B; Woo S; Spencer S; Figg WD; Giaccone G A phase I combination study of olaparib with cisplatin and gemcitabine in adults with solid tumors. Clin. Cancer Res 2012, 18, 2344–2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Samol J; Ranson M; Scott E; Macpherson E; Carmichael J; Thomas A; Cassidy J Safety and tolerability of the poly(ADP-ribose) polymerase (PARP) inhibitor, olaparib (AZD2281) in combination with topotecan for the treatment of patients with advanced solid tumors: a phase I study. Invest. New Drugs 2012, 30, 1493–1500. [DOI] [PubMed] [Google Scholar]
- 23.Lord CJ; Ashworth A PARP inhibitors: synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ashworth A; Lord CJ; Reis-Filho JS Genetic interactions in cancer progression and treatment. Cell 2011, 145, 30–38. [DOI] [PubMed] [Google Scholar]
- 25.Tutt A; Robson M; Garber JE; Domchek SM; Audeh MW; Weitzel JN; Friedlander M; Arun B; Loman N; Schmutzler RK; Wardley A; Mitchell G; Earl H; Wickens M; Carmichael J Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet 2010, 376, 235–244. [DOI] [PubMed] [Google Scholar]
- 26.Farmer H; McCabe N; Lord CJ; Tutt AN; Johnson DA; Richardson TB; Santarosa M; Dillon KJ; Hickson I; Knights C; Martin NM; Jackson SP; Smith GC; Ashworth A Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [DOI] [PubMed] [Google Scholar]
- 27.Bryant HE; Schultz N; Thomas HD; Parker KM; Flower D; Lopez E; Kyle S; Meuth M; Curtin NJ; Helleday T Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [DOI] [PubMed] [Google Scholar]
- 28.Audeh MW; Carmichael J; Penson RT; Friedlander M; Powell B; Bell-McGuinn KM; Scott C; Weitzel JN; Oaknin A; Loman N; Lu K; Schmutzler RK; Matulonis U; Wickens M; Tutt A Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet 2010, 376, 245–251. [DOI] [PubMed] [Google Scholar]
- 29.Saleh-Gohari N; Bryant HE; Schultz N; Parker KM; Cassel TN; Helleday T Spontaneous homologous recombination is induced by collapsed replication forks that are caused by endogenous DNA single-strand breaks. Mol. Cell. Biol 2005, 25, 7158–7169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Menear KA; Adcock C; Boulter R; Cockcroft XL; Copsey L; Cranston A; Dillon KJ; Drzewiecki J; Garman S; Gomez S; Javaid H; Kerrigan F; Knights C; Lau A; Loh VM Jr.; Matthews IT; Moore S; O’Connor MJ; Smith GC; Martin NM 4-[3-(4-Cyclopropanecarbonylpiperazine-1-carbonyl)-4-fluorobenzyl]-2H-phthalazin- 1-one: a novel bioavailable inhibitor of poly(ADP-ribose) polymerase-1. J. Med. Chem 2008, 51, 6581–6591. [DOI] [PubMed] [Google Scholar]
- 31.Jones P; Altamura S; Boueres J; Ferrigno F; Fonsi M; Giomini C; Lamartina S; Monteagudo E; Ontoria JM; Orsale MV; Palumbi MC; Pesci S; Roscilli G; Scarpelli R; Schultz-Fademrecht C; Toniatti C; Rowley M Discovery of 2-{4-[(3S)-piperidin-3-yl]phenyl}−2H-indazole-7-carboxamide (MK-4827): a novel oral poly(ADP-ribose)polymerase (PARP) inhibitor efficacious in BRCA-1 and −2 mutant tumors. J. Med. Chem 2009, 52, 7170–7185. [DOI] [PubMed] [Google Scholar]
- 32.Thomas HD; Calabrese CR; Batey MA; Canan S; Hostomsky Z; Kyle S; Maegley KA; Newell DR; Skalitzky D; Wang LZ; Webber SE; Curtin NJ Preclinical selection of a novel poly(ADP-ribose) polymerase inhibitor for clinical trial. Mol. Cancer Ther 2007, 6, 945–956. [DOI] [PubMed] [Google Scholar]
- 33.Wang B; Chu D; Feng Y; Shen Y; Aoyagi-Scharber M; Post LE Discovery and characterization of (8S,9R)-5-fluoro-8-(4-fluorophenyl)-9-(1-methyl-1H-1,2,4-triazol-5-yl)-2,7,8,9-te trahydro-3H-pyrido[4,3,2-de]phthalazin-3-one (BMN 673, Talazoparib), a novel, highly potent, and orally efficacious poly(ADP-ribose) polymerase-1/2 inhibitor, as an anticancer agent. J. Med. Chem 2016, 59, 335–357. [DOI] [PubMed] [Google Scholar]
- 34.Donawho CK; Luo Y; Luo Y; Penning TD; Bauch JL; Bouska JJ; Bontcheva-Diaz VD; Cox BF; DeWeese TL; Dillehay LE; Ferguson DC; Ghoreishi-Haack NS; Grimm DR; Guan R; Han EK; Holley-Shanks RR; Hristov B; Idler KB; Jarvis K; Johnson EF; Kleinberg LR; Klinghofer V; Lasko LM; Liu X; Marsh KC; McGonigal TP; Meulbroek JA; Olson AM; Palma JP; Rodriguez LE; Shi Y; Stavropoulos JA; Tsurutani AC; Zhu GD; Rosenberg SH; Giranda VL; Frost DJ ABT-888, an orally active poly(ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin. Cancer Res 2007, 13, 2728–2737. [DOI] [PubMed] [Google Scholar]
- 35.Litton JK; Rugo HS; Ettl J; Hurvitz SA; Goncalves A; Lee KH; Fehrenbacher L; Yerushalmi R; Mina LA; Martin M; Roche H; Im YH; Quek RGW; Markova D; Tudor IC; Hannah AL; Eiermann W; Blum JL Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. N. Engl. J. Med 2018, 379, 753–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chuang HC; Kapuriya N; Kulp SK; Chen CS; Shapiro CL Differential anti-proliferative activities of poly(ADP-ribose) polymerase (PARP) inhibitors in triple-negative breast cancer cells. Breast Cancer Res. Treat 2012, 134, 649–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Patel MR; Bhatt A; Steffen JD; Chergui A; Murai J; Pommier Y; Pascal JM; Trombetta LD; Fronczek FR; Talele TT Discovery and structure-activity relationship of novel 2,3-dihydrobenzofuran-7-carboxamide and 2,3-dihydrobenzofuran-3(2H)-one-7-carboxamide derivatives as poly(ADP-ribose)polymerase-1 inhibitors. J. Med. Chem 2014, 57, 5579–5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Patel BA; Krishnan R; Khadtare N; Gurukumar KR; Basu A; Arora P; Bhatt A; Patel MR; Dana D; Kumar S; Kaushik-Basu N; Talele TT Design and synthesis of L- and D-phenylalanine derived rhodanines with novel C5-arylidenes as inhibitors of HCV NS5B polymerase. Bioorg. Med. Chem 2013, 21, 3262–3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lu F; Chi SW; Kim DH; Han KH; Kuntz ID; Guy RK Proteomimetic libraries: design, synthesis, and evaluation of p53-MDM2 interaction inhibitors. J. Comb. Chem 2006, 8, 315–325. [DOI] [PubMed] [Google Scholar]
- 40.Khadtare N; Stephani R; Korlipara V Design, synthesis and evaluation of 1,3,6-trisubstituted-4-oxo-1,4-dihydroquinoline-2-carboxylic acid derivatives as ETA receptor selective antagonists using FRET assay. Bioorg. Med. Chem. Lett 2017, 27, 2281–2285. [DOI] [PubMed] [Google Scholar]
- 41.Osyanin VO; Nakushnov DV; Zemtsova M; Klimochkin Y Alkylation of 5-aryltetrazoles with 2- and 4-hydroxybenzyl alcohols. Chem. Het. Compounds 2015, 51, 984–990. [Google Scholar]
- 42.Young MB; Barrow JC; Glass KL; Lundell GF; Newton CL; Pellicore JM; Rittle KE; Selnick HG; Stauffer KJ; Vacca JP; Williams PD; Bohn D; Clayton FC; Cook JJ; Krueger JA; Kuo LC; Lewis SD; Lucas BJ; McMasters DR; Miller-Stein C; Pietrak BL; Wallace AA; White RB; Wong B; Yan Y; Nantermet PG Discovery and evaluation of potent P1 aryl heterocycle-based thrombin inhibitors. J. Med. Chem 2004, 47, 2995–3008. [DOI] [PubMed] [Google Scholar]
- 43.Mishra CB; Kumari S; Manral A; Prakash A; Saini V; Lynn AM; Tiwari M Design, synthesis, in-silico and biological evaluation of novel donepezil derivatives as multi-target-directed ligands for the treatment of Alzheimer’s disease. Eur. J. Med. Chem 2017, 125, 736–750. [DOI] [PubMed] [Google Scholar]
- 44.Patel BA; Abel B; Barbuti AM; Velagapudi UK; Chen ZS; Ambudkar SV; Talele TT Comprehensive synthesis of amino acid-derived thiazole peptidomimetic analogues to understand the enigmatic drug/substrate-binding site of P-glycoprotein. J. Med. Chem 2018, 61, 834–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Larsen MA; Hartwig JF Iridium-catalyzed C-H borylation of heteroarenes: scope, regioselectivity, application to late-stage functionalization, and mechanism. J. Am. Chem. Soc 2014, 136, 4287–4299. [DOI] [PubMed] [Google Scholar]
- 46.Dawicki-McKenna JM; Langelier MF; DeNizio JE; Riccio AA; Cao CD; Karch KR; McCauley M; Steffen JD; Black BE; Pascal JM PARP-1 activation requires local unfolding of an autoinhibitory domain. Mol. Cell 2015, 60, 755–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Langelier MF; Zandarashvili L; Aguiar PM; Black BE; Pascal JM NAD(+) analog reveals PARP-1 substrate-blocking mechanism and allosteric communication from catalytic center to DNA-binding domains. Nat. Commun 2018, 9, 844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Langelier MF; Planck JL; Roy S; Pascal JM Structural basis for DNA damage-dependent poly(ADP-ribosyl)ation by human PARP-1. Science 2012, 336, 728–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Talele TT The “cyclopropyl fragment” is a versatile player that frequently appears in preclinical/clinical drug molecules. J. Med. Chem 2016, 59, 8712–8756. [DOI] [PubMed] [Google Scholar]
- 50.Talele TT Natural-products-inspired use of the gem-dimethyl group in medicinal chemistry. J. Med. Chem 2018, 61, 2166–2210. [DOI] [PubMed] [Google Scholar]
- 51.Ali SO; Khan FA; Galindo-Campos MA; Yelamos J Understanding specific functions of PARP-2: new lessons for cancer therapy. Am. J. Cancer Res 2016, 6, 1842–1863. [PMC free article] [PubMed] [Google Scholar]
- 52.Farres J; Martin-Caballero J; Martinez C; Lozano JJ; Llacuna L; Ampurdanes C; Ruiz-Herguido C; Dantzer F; Schreiber V; Villunger A; Bigas A; Yelamos J PARP-2 is required to maintain hematopoiesis following sublethal gamma-irradiation in mice. Blood 2013, 122, 44–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Menissier de Murcia J; Ricoul M; Tartier L; Niedergang C; Huber A; Dantzer F; Schreiber V; Ame JC; Dierich A; LeMeur M; Sabatier L; Chambon P; de Murcia G Functional interaction between PARP-1 and PARP-2 in chromosome stability and embryonic development in mouse. EMBO J 2003, 22, 2255–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schreiber V; Ame JC; Dolle P; Schultz I; Rinaldi B; Fraulob V; Menissier-de Murcia J; de Murcia G Poly(ADP-ribose) polymerase-2 (PARP-2) is required for efficient base excision DNA repair in association with PARP-1 and XRCC1. J. Biol. Chem 2002, 277, 23028–23036. [DOI] [PubMed] [Google Scholar]
- 55.Ame JC; Spenlehauer C; de Murcia G The PARP superfamily. Bioessays 2004, 26, 882–893. [DOI] [PubMed] [Google Scholar]
- 56.Huggins DJ; Sherman W; Tidor B Rational approaches to improving selectivity in drug design. J. Med. Chem 2012, 55, 1424–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sonnenblick A; de Azambuja E; Azim HA Jr.; Piccart M An update on PARP inhibitors--moving to the adjuvant setting. Nat. Rev. Clin. Oncol 2015, 12, 27–41. [DOI] [PubMed] [Google Scholar]
- 58.O’Connor MJ Targeting the DNA damage response in cancer. Mol. Cell 2015, 60, 547–560. [DOI] [PubMed] [Google Scholar]
- 59.Huang SM; Mishina YM; Liu S; Cheung A; Stegmeier F; Michaud GA; Charlat O; Wiellette E; Zhang Y; Wiessner S; Hild M; Shi X; Wilson CJ; Mickanin C; Myer V; Fazal A; Tomlinson R; Serluca F; Shao W; Cheng H; Shultz M; Rau C; Schirle M; Schlegl J; Ghidelli S; Fawell S; Lu C; Curtis D; Kirschner MW; Lengauer C; Finan PM; Tallarico JA; Bouwmeester T; Porter JA; Bauer A; Cong F Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 2009, 461, 614–620. [DOI] [PubMed] [Google Scholar]
- 60.Drean A; Williamson CT; Brough R; Brandsma I; Menon M; Konde A; Garcia-Murillas I; Pemberton HN; Frankum J; Rafiq R; Badham N; Campbell J; Gulati A; Turner NC; Pettitt SJ; Ashworth A; Lord CJ Modeling therapy resistance in BRCA1/2-mutant cancers. Mol. Cancer Ther 2017, 16, 2022–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ogiwara Y; Sakurai Y; Hattori H; Sakai N Palladium-catalyzed reductive conversion of acyl fluorides via ligand-controlled decarbonylation. Org. Lett 2018, 20, 4204–4208. [DOI] [PubMed] [Google Scholar]
- 62.Barasa L; Yoganathan S An efficient one-pot conversion of carboxylic acids into benzimidazoles via an HBTU-promoted methodology. RSC Adv 2018, 8, 35824–35830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ognyanov VI; Balan C; Bannon AW; Bo Y; Dominguez C; Fotsch C; Gore VK; Klionsky L; Ma VV; Qian YX; Tamir R; Wang X; Xi N; Xu S; Zhu D; Gavva NR; Treanor JJ; Norman MH Design of potent, orally available antagonists of the transient receptor potential vanilloid 1. Structure-activity relationships of 2-piperazin-1-yl-1H-benzimidazoles. J. Med. Chem 2006, 49, 3719–3742. [DOI] [PubMed] [Google Scholar]
- 64.Devine WG; Diaz-Gonzalez R; Ceballos-Perez G; Rojas D; Satoh T; Tear W; Ranade RM; Barros-Alvarez X; Hol WG; Buckner FS; Navarro M; Pollastri MP From cells to mice to target: Characterization of NEU-1053 (SB-443342) and its analogues for treatment of human African Trypanosomiasis. ACS Infect. Dis 2017, 3, 225–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kabsch W XDS. Acta Crystallogr. D Biol. Crystallogr 2010, 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ Phaser crystallographic software. J. Appl. Crystallogr 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Adams PD; Afonine PV; Bunkoczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung LW; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Emsley P; Lohkamp B; Scott WG; Cowtan K Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Murshudov GN; Skubak P; Lebedev AA; Pannu NS; Steiner RA; Nicholls RA; Winn MD; Long F; Vagin AA REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr 2011, 67, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Winn MD; Ballard CC; Cowtan KD; Dodson EJ; Emsley P; Evans PR; Keegan RM; Krissinel EB; Leslie AG; McCoy A; McNicholas SJ; Murshudov GN; Pannu NS; Potterton EA; Powell HR; Read RJ; Vagin A; Wilson KS Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr 2011, 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Miyashiro J; Woods KW; Park CH; Liu X; Shi Y; Johnson EF; Bouska JJ; Olson AM; Luo Y; Fry EH; Giranda VL; Penning TD Synthesis and SAR of novel tricyclic quinoxalinone inhibitors of poly(ADP-ribose)polymerase-1 (PARP-1). Bioorg. Med. Chem. Lett 2009, 19, 4050–4054. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.