

Abstract

Poly(propylene carbonate) (PPC) from CO2 and propylene oxide (PO) has wide potential applications as a degradable “plastic”. However, the thermal stability and mechanical properties of PPC cannot meet most of the application requirements. Herein, we focus on improving these properties. A (maleic anhydride/cis-1,2,3,6-tetrahydrophthalic anhydride) (MA/THPA) oligomer containing several cyclocarboxylic anhydride groups, which can copolymerize with PO, has been readily synthesized and used as the third comonomer to prepare PPC with cross-linked networks. The gel contents increase from 16 to 42% with increasing MA/THPA oligomer feed contents from 0.5 to 4 wt % of PO. The formation of cross-linked networks in PPC greatly improves the thermal, mechanical, and dimensional properties. The 5% weight-loss degradation temperature increases from 217 °C to nearly 290 °C before and after cross-linking, which ensures that PPC does not decompose in melt processing. The tensile strength of the copolymer is in the range of 22.2–44.3 MPa with elongation at break of 11–312%. The maximum tensile strength is improved by 143% compared to that of PPC. When the MA/THPA oligomer feed is above 3 wt % of PO, the hot-set elongation of the copolymer at 65 °C decreases more than 10 times when compared with that of PPC, and the permanent deformation is close to 0, while it is 145% for PPC. The dimensional stability is improved sharply. It can overcome the cold flow phenomenon of PPC. The improvement of the above comprehensive properties is of great significance to the practical application of PPC in various fields.
Introduction
As a “two-way” environmental protection material, poly(propylene carbonate) (PPC) can not only consume CO2 greenhouse gas in the production process but also be degraded by microorganisms in nature, which has received great attention of the world.1,2 PPC has potential applications in adhesives,3 mulch film,4 packaging,5 polymer electrolyte,6,7 toughening agent,8 biomedical materials,9 and other fields. This makes it become a possible alternative to traditional nonbiodegradable polymers. However, for large-scale applications, besides the need of improving the catalytic activity of catalysts for CO2/PO copolymerization, another important demand is to improve the properties of PPC. First, the low onset decomposition temperature in the range of 150–180 °C makes it unable to be processed smoothly. If PPC processing is performed at a low temperature, there may be substantial stress in the sample. This stress will relax at room temperature and deform the sample.10 A thermally more resilient PPC is thus highly desirable. Second, PPC’s low glass transition temperature (Tg) (<42 °C) and amorphism leads to weak mechanical strength and poor dimensional stability. Therefore, improving the thermal, mechanical, and dimensional properties is of great significance for the practical application of PPC.
For PPC modification, many attempts have been made, including physical and chemical methods, such as blending with various inorganic powders, fibers, organics, or other degradable or nondegradable polymers, end-capping the terminal hydroxy of PPC with other groups, chain extension, cross-linking, and ternary polymerization with other comonomers, some of which have achieved good results in some properties.10−17 In fact, the difficulty of PPC modification lies in how to comprehensively improve the thermal, mechanical, and dimensional properties on the premise of introducing a small amount of other components. For example, end-capping is very effective for increasing decomposition temperature, but it cannot enhance the mechanical performance. In addition, if more amounts of other components are introduced, the CO2 utilization will be reduced, which is the significance of continuous research on this topic in the world for decades. Perhaps the most fundamental way to modify PPC is to crystallize PPC without introducing other components. Like crystal polyethylene and polypropylene, although their Tgs are less than 0 °C, their melting points are above 100 °C. They can be used as structural materials. However, PPC is difficult to crystallize even if various isotactic PPCs have been prepared18,19 and other crystalline CO2-based polycarbonates have been synthesized.20,21 In other various modifications, cross-linking is an effective method to meet the above requirements. For blending modification, as long as other components such as graphene oxide (GO),22 microfibrillated cellulose,23 SiC/GO nanosheets,24 or hyperbranched polyester amide25 are uniformly dispersed in the PPC matrix and physical cross-linking is formed through noncovalent bonds such as hydrogen bonds, the properties of PPC can be fully improved using a small amount of other components with the contents of less than 5 wt % of PPC, even when the Tg is low. However, the incorporated fills should have good compatibility with PPC in this method. Chemical cross-linking is relatively more effective. For instance, the chemically cross-linked PPCs have an improved 5% weight-loss degradation temperature (Td,–5%) of over 260 °C and an enhanced mechanical strength of about 40 MPa.26−28 The permanent deformation of cross-linked PPC in the hot elongation test at 65 °C is close to 0,29 while that of PPC is more than 10%, that is, PPC cannot be recovered after being stretched.
For chemical cross-linking of PPC, initially, it was conducted through the reaction between PPC with various cross-linking agents such as triallyl isocyanurate (TAIC), diisopropyl peroxide (DCP),30 polyvinyl polyphenyl isocyanate (PAPI),31 trimethylopropane triacrylate, pentaerythritol triacrylate, poly(ethylene glycol dimethyl methacrylate), and poly(ethylene glycol dimethyl methacrylate)32 by heating or radiation. Nevertheless, this method is not universally applicable because PPC itself has neither double bonds nor other cross-linkabe pendant groups. Therefore, people try to introduce double bonds into PPC chains, and then add the initiator to realize cross-linking by heating or by ultraviolet irradiation. For example, PPC bearing C=C groups was prepared by terpolymerization of maleic anhydride (MA),26 vinyl oxide,33 or allyl glycidyl ether29 with CO2 and PO, respectively. Then, it was cross-linked by DCP, ethylene glycol bis(3-mercaptoproionate), pentaerythritol tetrakis(mercaptoacetate), or by ultraviolet irradiation. Besides, organosilylated PPC was synthesized by CO2/PO/γ-glycidopropyltrimethoxysilane terpolymerization, and the cross-linked structure was formed after hydrolysis.34 The cross-linking effect by the above methods is excellent, but the process is complicated. Later, it was found that cross-linked PPC can be achieved by one-pot terpolymerization of a multifunctional monomer with CO2 and PO. The multifunctional monomer always contains several epoxides, isocyanates, or cyclic carboxylic anhydrides, such as vinylcyclohexene dioxide,35 diphenylmethane diisocyanate,36 1,2,7,8-diepoxyoctane,37 1,2,9,10-diepoxydecane,37 pyromellitic dianhydride,27 triglycidyl isocyanurate,38 and bicyclo(2,2,2)oct-7-ene-2,3,5,6-tetracarboxylic dianhydride.28 Itaconic anhydride that contains both C=C bond and cyclic anhydride was also used as a third monomer to prepare cross-linked PPC.39 In addition, our group prepared PPC with networks by CO2/PO/MA/furfuryl glycidyl ether copolymerization,40 in which the functional groups that can react with each other were introduced into the PPC pendants.
It should be noted that sometimes, certain types of multifunctional third monomers, such as diepoxides41,42 and polyisocyanates,43 cannot form cross-linked networks in copolymerizing them with CO2 and PO. This may be related to either the reactivity of monomers or the catalysts used. Nevertheless, cyclic dianhydrides could do without exception. Anhydride groups are vulnerable to react with PO. For example, various monofunctional cyclic anhydrides could copolymerize with epoxides in the presence/absence of CO2 under various types of catalysts, including double-metal cyanide, salen–metal complex, biphenol-linked dinuclear Al(III) complex, and zinc dicarboxylate,44−50 in which the linear polymers were formed. Our group has used several multifunctional third comonomers to prepare PPC with cross-linked networks.27,28 It is found that adding a small amount of third monomers can improve the thermal and mechanical properties. In particular, for elevating the thermal decomposition temperature, the required amount of third monomers is smaller. Another preliminary discovery is that in addition to cross-linking, the rigidity or flexibility of the third monomer molecule will also affect the mechanical strength and toughness of the final copolymers. If the third monomer is too flexible, the tensile strength would be significantly reduced, even the cross-linked networks are contained.40 However, it is difficult to further verify the above finding because there are not many multifunctional small molecules, commercially, that can participate in CO2/PO copolymerization, and their custom synthesis is not economical. We have tried to use a maleic anhydride oligomer as the third monomer to prepare cross-linked PPC.51 We speculate that the mechanical strength and toughness of the copolymer can be tailored by the co-oligomer from different rigid or flexible monomers and MA. In this work, we select a relatively rigid monomer, cis-1,2,3,6-tetrahydrophthalic anhydride (THPA), to prepare a MA/THPA oligomer, and use it to copolymerize with PO and CO2 to synthesize PPC with cross-linked networks in one pot. The effect on the thermal stability, mechanical properties, and dimensional stability were fully investigated. After ring opening copolymerization, the MA unit is linear and flexible, and the THPA unit contains a rigid ring. Based on this, we speculate that the copolymer will have strong mechanical strength and certain toughness. Moreover, compared with the case where the MA oligomer is used as the third monomer, the mechanical strength will be stronger and the toughness slightly worse.
Results and Discussion
Synthesis
In the synthysis of the MA/THPA oligomer, according to the integral area of each peak of the 1H NMR spectroscopy (Figure S1), the proportion of MA and THPA units in the oligomer is 1:1. It is well known that MA does not easily homopolymerize but readily copolymerizes with other monomers. Combined with the GPC result of the MA/THPA oligomer that the Mn is 894 g·mol–1 (Figure S2), it is inferred that each molecule contains three MA and three THPA units on average, that is, it contains six cyclic anhydride functional groups. As mentioned in the Introduction section, a cyclic anhydride can copolymerize with epoxides to prepare polyesters under various types of catalysts, including ZnGA. After the addition of the MA/THPA oligomer in the CO2/PO copolymerization catalyzed by ZnGA, the terminal alkoxy anion of the CO2/PO propagation chain can nucleophilically attack the cyclic anhydride group in a MA/THPA oligomer molecule, causing it to ring-open and transform into a terminal carboxylate anion propagation chain. The terminal carboxylate anion attacks PO and is converted again into a terminal alkoxide anion propagation chain, which can then react with CO2 or cyclic anhydride. Because the amount of the MA/THPA oligomer in the system does not exceed 4 wt % of PO, that is, PO and CO2 are much more than the MA/THPA oligomer, therefore, each ring-opened cyclic anhydride can lead to two PPC chains. In theory, one molecule of the MA/THPA oligomer that contains six cyclic anhydrides can link twelve PPC chains on average, so the MA/THPA oligomer acts as an effective cross-linker (Scheme 1). The formation of the cross-linked networks is confirmed by the presence of gel in the copolymers. Here, we define the copolymers as PPCx, where x represents the MA/THPA oligomer feed content (x wt %) of PO. The gel contents increased from 16.1 to 42.2% with increasing the MA/THPA oligomer feed contents from 0.5 to 4 wt % of PO. Figure 1 shows the typical photos of the polymers before and after purification. It is seen that PPC looks soft because of the presence of more unreacted PO. While the copolymers look like a block, and they feel more elastic when pulled with tweezers. In fact, the yields of copolymers are 2–3 times that of PPC (Table 1). In combination with previous studies on the CO2/PO/third monomer copolymerization catalyzed by ZnGA,27,28,52,53 it is shown that the polymer yield increased when the third monomer was cyclic anhydrides but decreased when it was epoxides, when compared with the CO2/PO copolymerization. This related to the higher activity of cyclic anhydrides/PO copolymerization than epoxides/CO2. The typical 1H NMR of PPC4 is similar to that of PPC (Figure S3) after purification. However, the 1H NMR spectroscopy only represents the soluble fraction and does not display the signal peaks from the MA/THPA oligomer units because the MA/THPA oligomer is a multifunctional monomer that formed a gel and is insoluble in chloroform-d. In order to know whether the MA/THPA oligomer is completely involved in the copolymerization and the effect of the MA/THPA oligomer on the copolymerization, we examined the 1H NMR of the reaction mixture after copolymerization (Figure S4) and the GPC of the soluble fraction of the polymers (Figure S5). In Figure S4, no signal peaks of the MA/THPA oligomer like those in Figure S1 are found. The reaction mixture mainly contains PPC, cyclic propylene carbonate (PC), and PO. It indicates that the MA/THPA oligomer can completely participate in the CO2/PO copolymerization to form cross-linked networks, but it is confined to the gel. In addition, after adding the MA/THPA oligomer, the selectivity of the polymer over PC increases slightly and the carbonate linkage contents over ether linkage in polymer slightly decrease (Table 1). What is more, the molecular weights of the copolymers declined with increasing MA/THPA oligomer feed (Table 1), and the molecular weight distribution (Mw/Mn) is higher than that of PPC. When the MA/THPA oligomer feed is less than 2 wt % of PO, the molecular weights of the copolymers are greater than that of PPC; when it is more than 2 wt %, the molecular weights are slightly lower than that of PPC. This relates to the following two effects: the copolymer chains are slightly branched because of the multifunctionality of the MA/THPA oligomer, which is favorable for increasing the molecular weights upon adding the MA/THPA oligomer first. When the MA/THPA oligomer feed further increases, the movement of propagation chains becomes less active because of the increase of the cross-linked networks, which in turn is not conducive to increasing the molecular weight. Note that the Mn, selectivity for the polymer, and carbonate linkage content of the copolymers only represents the soluble fraction, excluding the gel.
Scheme 1. Formation of PPC with Networks in CO2/Propylene Oxide Copolymerization in the Presence of the (Maleic Anhydride/cis-1,2,3,6-Tetrahydrophthalic Anhydride) (MA/THPA) Oligomer.

Figure 1.

Photographs of PPC and PPC with networks before and after purification, as well as the corresponding dumbbell specimens. PPC0.5–4 presents the copolymer with a 0.5–4 wt % (maleic anhydride/cis-1,2,3,6-tetrahydrophthalic anhydride) (MA/THPA) oligomer feed of propylene oxide in the copolymerization, respectively.
Table 1. Results of Copolymerizationa.
| sample | MA/THPA oligomer feedb | polymer yieldc | gel (%) | Mnd | Mw/Mne | selectivity for polymerf | carbonate linkagesg |
|---|---|---|---|---|---|---|---|
| PPC | 0 | 25 | 0 | 70 | 3.0 | 96 | 98 |
| PPC0.5 | 0.5 | 55 | 16 ± 1.3 | 91 | 4.5 | 98 | 97 |
| PPC1 | 1 | 60 | 23 ± 1.5 | 82 | 4.7 | 98 | 97 |
| PPC2 | 2 | 66 | 29 ± 1.7 | 68 | 4.4 | 98 | 97 |
| PPC3 | 3 | 70 | 37 ± 2.2 | 58 | 4.6 | 99 | 97 |
| PPC4 | 4 | 74 | 42 ± 2.5 | 58 | 4.8 | 99 | 97 |
Polymerization conditions: zinc glutarate (ZnGA, 0.15 g), propylene oxide (PO, 45 mL), CO2 pressure 5.0 MPa, 70 °C, 30 h.
(Maleic anhydride/cis-1,2,3,6-tetrahydrophthalic anhydride) (MA/THPA) oligomer feed of PO in the copolymerization (wt %).
g Polymer/g ZnGA.
Mn = Number average molecular weight (Kg·mol–1), determined by GPC.
Molecular weight distribution: Mw = weight average molecular weight (Kg·mol–1).
Selectivity for polymer over cyclic carbonate in % units, which was determined by the 1H NMR spectroscopy of the reaction mixture after copolymerization.
Carbonate linkages over ether linkages in % units, which were determined by the 1H NMR spectroscopy of the soluble fraction of polymers after purification. The Mn, polymer selectivity and carbonate linkages do not include the gel, because the gel is insoluble.
Thermal Properties
Figures 2 and S6 show that the TGA and the corresponding differential thermogravimetry (DTG) curves of PPC, copolymers as well as their soluble fraction, respectively. Table 2 lists the data. The result shows that the decomposition temperature of the copolymers increases significantly when the MA/THPA oligomer is introduced, even if a small amount of the MA/THPA oligomer is used, such as 0.5 wt % of PO. It was reported that PPC begins to decompose at 150–180 °C,10,54 depending on the existence of the catalyst or water, molecular weight, and the heating rate in the test. Here, PPC begins to lose weight at 197 °C under the experimental conditions, which is higher compared with those reported. This is related to the faster heating rate in the TGA test.55 The copolymers begin to lose weight at the temperature range of 256–275 °C, and the 5% and maximum weight-loss decomposition temperature (Td,max) reaches up to about 290 and 300 °C, respectively. These values exceed 60–70 °C, higher than the corresponding ones of PPC. This great improvement in thermal stability can ensure that the copolymers do not decompose during melt processing.
Figure 2.

Thermogravimetric analysis curves of PPC and PPC with networks. PPC0.5-S–PPC4-S refers the soluble fraction of the corresponding copolymer.
Table 2. Thermal Properties of PPC and PPC with Networks.
| sample | temperature for onset of weight loss (°C) | temperature for 5% weight loss (°C) | temperature for maximum weight loss (°C) | glass transition temperature (°C) |
|---|---|---|---|---|
| PPC | 197 | 217 | 231/251c | 51,e 35f |
| PPC0.5 | 256,a 256b | 288,a 277b | 298,a 293b | 47, 31 |
| PPC1 | 264, 258 | 288, 281 | 297, 294 | 50, 33 |
| PPC2 | 273, 264 | 289, 282 | 297, 293 | 50, 34 |
| PPC3 | 272, 263 | 292, 282 | 302, 295 | 53, 38 |
| PPC4 | 275, 266 | 291, 282 | 312, 288/299d | 51, 37 |
The copolymers.
The soluble fraction of the copolymers.
PPC has two main maximum weight-loss peaks at 231 and 251 °C.
The soluble fraction of the PPC4 has two maximum weight-loss peaks at 288 and 299 °C.
Measured from tan δ curves in a DMA test.
Measured from DSC curves.
It is also found that two apparent maximum weight-loss peaks at 231 and 251 °C appear in the DTG curve of PPC (Figure S6) which derive from chain scission and unzipping degradation, respectively.56 It should be noted that when PPC was heated at 200 °C, the unzipping degradation product, PC, was detected.54,56 The DTG curve shows the maximum weight loss peak around 250 °C because the boiling point of PC is 242 °C and the weight loss is not obvious before this temperature. However, each copolymer has only one maximum weight loss peak, which is far away from that of PPC at high temperatures. This indicates that the formed cross-linked networks in the PPC matrix by introducing the MA/THPA oligomer play a significant role in restraining various degradation reactions. In order to further understand the thermal properties, we also conducted the TGA test for the soluble fraction of the copolymers (the scatter curves in Figure 2). Interestingly, it is quite different from that of PPC. Several key weight-loss temperatures, such as the onset weight-loss temperature, Td,–5% and Td,max, are still much higher than the corresponding ones of PPC. They are only a few degrees lower than those of the copolymers. It further implies that the soluble fraction of the copolymers is not just the neat PPC but the slightly branched PPC with a very low fraction of the MA/THPA oligomer incorporated in the PPC backbone. First, the molecular weight is not the cause for elevation of the decomposition temperature because compared with PPC, PPC0.5, and PPC1 has a higher Mn, while PPC2, PPC3, and PPC4 has a lower Mn, but their decomposition temperatures are similar to each other and much higher than that of PPC. What is more, within a certain range, the molecular weight of PPC has little effect on the thermal decomposition temperature. For example, it was reported that the Td,maxs of the PPCs with Mns of 26.9, 56, and 144.6 Kg·mol–1 are 255, 255, and 251 °C, respectively.56 Second, although the increasing ether linkage helps elevate the thermal decomposition temperature, it is not enough to greatly improve the decomposition temperature with an elevation of more 60 °C because each of the ether linkage contents of the copolymers is only a little more than that of PPC. The low content of carbonate linkages in the copolymers listed in Table 1 means that there is a high content of ether linkages. In the properties report of PPC that was also prepared using ZnGA, the Td,–5% of the PPC with 96.6% of carbonate linkage content was only about 10 °C higher than that of the PPC with 98.2% carbonate linkage content, and their Mns were 93 and 104 Kg·mol–1, respectively.57 A similar phenomenon was also reported by Tang and coworkers.58 Here, the carbonate linkage content of the PPC and the copolymers is almost equal; therefore, we infer that the elevation of 60 °C of Td,–5% is not because of the slightly increased ether linkage content, but a small amount of MA/THPA units that are incorporated into the soluble fraction of the copolymers.
The tanδ versus temperature curves from the DMA test (Figure 3) and DSC (Figure S7) curves show that Tgs slightly increases and then decreases with increasing MA/THPA oligomer feed under the experimental conditions. This phenomenon is related to two factors that have opposite effects on Tg: cross-linking and the flexibility of MA/THPA units. Table 2 lists the data. A maximum Tg of 53 °C is reached when the MA/THPA oligomer feed content is 3 wt % of PO. The restriction of the polymer movement by the cross-linked networks is clear. Note that the Tgs of copolymers are lower than that of PPC when the MA/THPA oligomer feeds are less than 2 wt % of PO, especially when 0.5 wt %. The gel content at this time is low with only 16% (Table 1). Therefore, if the cross-linking effect is excluded, introducing the MA/THPA oligomer will increase the flexibility of the polymer chains and result in low Tg. This effect becomes the main factor and reduces Tg when the MA/THPA oligomer feed increases up to 4 wt % of PO. It was reported that introducing more flexible MA units into the PPC backbone severely reduced Tg,59 in which, no cross-linked networks formed because MA is not a multifunctional comonomer.
Figure 3.
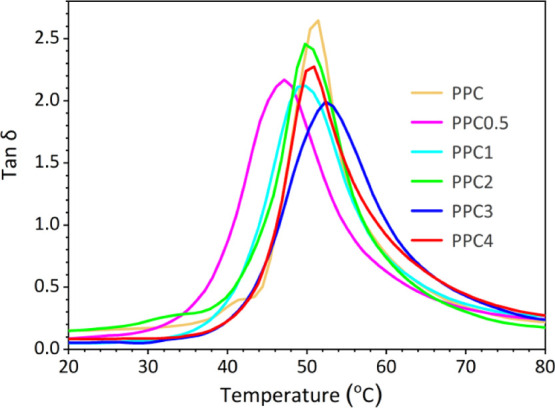
Tan δ vs temperature curves of PPC and PPC with networks.
Tensile Strength
The typical mechanical properties of PPC are brittle at low temperatures and weak at high temperatures.25 Cross-linking of PPC is an effective method to improve and balance the above two properties. Figure 4 shows the strain–stress curves in tensile tests and Table S1 lists the data. It shows that the mechanical strength can be significantly improved by introducing the MA/THPA oligomer. The tensile strength increases up to 44.3 MPa, that is, above twice that of PPC when the MA/THPA oligomer feed content is 4 wt % of PO. Accordingly, the elongation at the break decreases gradually. It is still 11% of PPC4, retaining a certain toughness. As mentioned above, introducing the MA/THPA oligomer into the PPC chain makes it flexible and leads to low Tg. From this point, it is disadvantageous for improving mechanical properties. Therefore, the significant increase in mechanical strength, here, comes from the fact that the PPC matrix contains cross-linked networks, which limits the PPC chain movement. The more gel content (Table 1), the higher the degree of cross-linking, so the tensile strength gradually increases. Similarly, after introducing MA into PPC chains, the tensile strength decreased sharply before cross-linking treatment59 and increased significantly after cross-linking.26 The PPC chain mainly contains carbonate linkages and a small amount of ether linkages without polar groups. The weak intermolecular interactions make PPC slip easily, so the mechanical strength is weak. When the cross-linked networks are contained in the PPC matrix, the PPC chain movement is restricted, thereby increasing the mechanical strength. In addition, compared with the result of using the MA oligomer as the third monomer,51 introducing the rigid THPA in the co-oligomer, here, does make the prepared copolymers have greater tensile strength and smaller elongation at break. This further confirms our previous find that the rigidity or flexibility of the multifunctional third comonomer has corresponding influence on the tensile strength and toughness of the prepared cross-linked copolymers.
Figure 4.

Strain–stress curves of PPC and PPC with networks.
It is noted that the reported values of mechanical properties of PPC varied greatly. The tensile strength ranged from a few MPa to more than 40 MPa, and the elongation at the break ranged from a few percent to more than 1000 percent.10,22,24,31,55,59−62 The most important factors are the content of ether linkages in PPC chains, the plasticization of residual small molecules such as PC, and test conditions such as the temperature and crosshead speed. In addition, the molecular weight and distribution of PPC also have some influence. In Figure 4, the PPC specimens were cut after pressing at 140 °C, and the copolymer specimens were prepared at 190 °C. At first, in order to make the conditions consistent, PPC was also prepared at 190 °C, under which the tensile strength is only 13.2 MPa and the elongation at break is 520%. It is suspected that PPC may decompose to produce small molecular substances when it was pressed at high temperatures. Therefore, the 1H NMR of PPC and copolymers that were pressed at 190 °C were detected. The result shows that the signal peak of PC does appear in PPC (Figure S8), but the 1H NMR of the copolymers does not change. This also exhibits that the copolymers have highly enhanced thermal stability. The presence of PC plays a plasticizing role and leads to a decrease in tensile strength and an increase in elongation at break of the PPC specimens that were pressed at 190 °C.
Dimensional Stability
In addition to the weak mechanical strength of PPC, another knotty problem is that PPC will soften and deform when heated, even if it is held in hand for a short time. Therefore, in order to use PPC as a real biodegradable material, it is necessary to improve its dimensional stability. The hot-set tests were conducted at 65 °C to characterize the dimensional stability of the prepared polymers according to a literature.29 As shown in Figure 5 and Table S1, the hot-set elongation decreases by 90% from 237 to 23%, and the permanent deformation also rapidly decreases from 145 to 43.4, 2.1, and 0.3%, until 0 when the MA/THPA oligomer feed increases from 0 to 0.5, 1, 2, 3, and 4 wt % of PO, respectively. Note that when PPC specimens were prepared at 190 °C, the hot-set elongation and the permanent deformation are larger with 513 and 325%, respectively, because of the presence of PC from the unzipping decomposition (Figure S8). The dimensional stability of PPC is greatly improved by introducing the MA/THPA oligomer. Except PPC3, each Tg of copolymers is lower than that of PPC (Table 2) but their hot-set elongation and permanent deformation are much lower than that of PPC. This also indirectly proves that the copolymers contain cross-linked networks, which plays a very positive role in improving the dimensional stability. Figure S9 shows the photos of dumbbell specimens before and after the hot-set test. Figure 6 shows the storage modulus (E′) versus temperature curves of PPC and the copolymers from a DMA test. The E′s of PPC, PPC0.5–4 are 0.040, 0.38, 0.79, 0.95, 1.1, and 1.5 GPa at 40 °C under a frequency of 1 Hz, respectively. The higher E′s of the copolymers indicates that they are more rigid and more deformation resistant than PPC, which is consistent with the mechanical strength results. In addition, the state change of the polymers when they were strongly heated was also observed through combustion experiments. As shown in Video S1, during the PPC burning, the deformation is very serious and even dripping occurs. While the shape retention of the copolymers becomes better and better with increasing the MA/THPA oligomer feed. PPC0.5 and PPC1 still drips, but not as serious as PPC (Video S2). PPC2–4 has no dripping and the shape is well maintained, but the volume gradually shrinks as the combustion progresses (Video S3). The above characteristics demonstrate that the copolymers have more rigid and stronger resistance to strain and deformation than PPC at higher temperatures, where the formed cross-linked networks in the PPC matrix by introducing the MA/THPA oligomer play an important role in restricting the PPC chain movement. Compared with the case of the MA oligomer as the third monomer,51 where the dimensional stability did not reach 0 when the amount of the MA oligomer is 3.74 wt % of PO, the permanent deformation rate reaches 0 when the MA/THPA feed is 3 wt % of PO, and the test temperature is 5 °C higher than the former. Therefore, when a rigid monomer THPA is introduced into the MA co-oligomer, it is more beneficial to improve the dimensional stability.
Figure 5.

Hot-set elongation and permanent deformation of PPC and PPC with networks.
Figure 6.

Storage modulus (E′) vs temperature curves of PPC and PPC with networks.
Conclusions
PPC with cross-linked networks was synthesized in one pot by incorporating a multifunctional MA/THPA oligomer in the CO2/PO copolymerization. The thermal stability, mechanical properties, and dimensional stability of the obtained copolymers can be tuned by changing the amount of the MA/THPA oligomer feed. The cross-linked networks in the PPC matrix play a positive role in improving the above performance. The flexible MA units make the copolymer retain certain toughness. In addition, compared with the MA oligomer as the third monomer, the copolymers have stronger tensile strength, smaller elongation at break, and more dimensional stability because THPA units that contain rings are more rigid than MA units. This confirms our speculation mentioned in the Introduction section. The synthesis of co-oligomers of MA with other monomers is convenient and economical, and there are many kinds of other monomers to choose. It will allow conveniently and economically tailoring the tensile strength and toughness of PPC for various applications.
Experimental Section
Materials
Most of the materials are of analytical purity, purchased from Shanghai Aladdin Company, and used as received unless otherwise specified. CO2 (99.99%) was purchased from Shenzhen Shente Industrial Gas Co. PO was distilled after reflux over calcium hydride for 8 h under a high pure nitrogen flow and then stored with 0.4 nm molecular sieves prior to use. High pure nitrogen was obtained from Zhanjiang Oxygen Plant. A zinc glutarate (ZnGA) catalyst was synthesized following the literature procedure.51 Toluene, chloroform, and methanol were dried over 0.4 nm molecular sieves for more than 24 h before use. Benzoyl peroxide (BPO) was recrystallized from a mixed solvent of chloroform and methanol before use.
Synthesis of the MA/THPA Oligomer
The MA/THPA oligomer was synthesized according to the literature63 and modified slightly. Briefly, MA (10 g, 102 mmol) and THPA (14.6 g, 96 mmol) were melted together and stirred under nitrogen at 90 °C. BPO (2.16 g, 8.9 mmol) that was plasticized with 1.5 g of dimethyl phthalate was added in three equal portions at half hour intervals with stirring under nitrogen. The reaction was allowed to proceed for a total of 4 h. Finally, the reactant became purple and viscous, and then it was poured into 600 mL of toluene to give a granular precipitate. The precipitate was filtered off, dissolved in acetone, and reprecipitated in 600 mL of toluene. The fine precipitate was filtered off and dried under vacuum at 80 °C. The final product was characterized by the 1H NMR spectrum (Figure S1). 1H NMR (400 MHz, DMSO-d6): δ 2.54 (s, 4H), 2.29 (s, 2H), 2.03 (d, 2H, J = 38.9 Hz), 1.20 (d, 2H, J = 24.0 Hz). The number molecular weight (Mn) of the MA/THPA oligomer was 894 g·mol–1 with a polymer dispersity index of 1.17, which was determined by gel permeation chromatography (GPC) (Figure S2).
General Copolymerization Procedure
Typically, the pre-dried 0.15 g of ZnGA and a certain proportion of the MA/THPA oligomer were put into a 100 mL autoclave with a magnetic stirrer and dried in vacuum at 100 °C for 8 h. Then, the autoclave was cooled to below 15 °C, cleaned carefully with nitrogen, and then evacuated 3 times alternately. Afterward, 45 mL of PO was injected into the autoclave and CO2 was filled to 2 MPa. When the temperature was raised to 70 °C, CO2 was pressured to 5.0 MPa. After stirring at 70 °C for 30 h, the autoclave was cooled to room temperature and released the pressure. The hard copolymer was dissolved in enough acetone containing 5% hydrochloric acid solution, stirred to decompose ZnGA, and then precipitated in the distilled water under vigorous stirring. Such dissolution and precipitation was repeated to completely purify the copolymer. The final acetone solution of the copolymer was precipitated in ethanol and washed three times to reduce the water content of the copolymer and facilitate drying. Then, the copolymer was dried to constant weight in vacuum at 80 °C.
PPC was also synthesized in a similar procedure to that of copolymers, except that the MA/THPA oligomer was not added into the autoclave.
Analytical Methods
1H Nuclear magnetic resonance (1H NMR) spectra were detected on a 400 MHz NMR instrument of DRX-400, with dimethylsulfoxide-d6 (DMSO-d6), or chloroform-d (CDCl3) as a solvent. A differential scanning calorimetry (DSC) analysis was performed to measure the Tgs of polymers on a Q100 TA instrument using a heat ramp of 10 K·min–1 to reach 200 °C under 40 mL·min–1 nitrogen flow. A thermogravimetric analysis (TGA) was conducted on a STA 6000 simultaneous thermal analyzer using a 20 K·min–1 rate for heating up to 400 °C under 40 mL·min–1 nitrogen flow. The average molecular weight of the MA/THPA oligomer was determined on a Waters GPC system equipped with a differential refraction detector with tetrahydrofuran as an eluent using a polystyrene standard. A dynamic mechanical analysis (DMA) was performed on a NETZSCH DMA 242E in tension mode at a constant heating rate of 3 K·min–1 from −30 to 100 °C at a frequency of 1 Hz. The dimensions of the specimens were 20 × 4 × 2 mm3.
Gel Content
The ASTM D2765 method was used for determination. The sample was refluxed in boiling chloroform for 24 h. The insoluble fraction was dried to constant weight at 80 °C in vacuum. Gel content refers to the weight percentage of insoluble matter in the sample. The data was recorded as the average of three parallel measurements.
Tensile Measurement
It was conducted on an Instron 3360 electronic tensile tester at a crosshead speed of 50 mm·min–1 at 23 °C and 45–55% humidity in accordance with ASTM d368. The data was recorded as the average value of five parallel measurements. The copolymer specimens were prepared by hot pressing at 190 °C, and then cut to 75 × 4 × 2 mm3 by a dumbbell cutter. PPC specimens were prepared by hot pressing at 140 and at 190 °C, respectively.
Hot-Set Test
The dumbbell specimen that was premarked as the reference length with L0 (L0 = 20 mm) was loaded with 0.14 MPa and placed in an oven at 65 °C. After 10 min, the length between the marks was detected and recorded as L1. The load was then released, relaxed at 65 °C for 5 min, and the specimen was allowed to relax at room temperature until it no longer shortened. The length between the marks was recorded as L2. The hot-set elongation and the permanent deformation were calculated by (L1 – L0)/L0 × 100% and (L2 – L0)/L0 × 100%, respectively. The data were recorded as the average of three parallel measurements.
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (nos. 51403183, 51003092), the Natural Science Foundation of Guangdong Province (no. 2018A030307068), Research Group of Rare Earth Resource Exploiting and Luminescent Materials (no. 2017KCXTD022), Characteristic Innovation Project of Innovation and Strengthening of Higher Education in Guangdong (no. 2016KTSCX080), and Key Laboratory of Tropical Crop Products Processing of Ministry of Agriculture and Rural Affairs , P.R. China (no. KFKT201905).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c02608.
1H NMR spectrum of (maleic anhydride/cis-1,2,3,6-tetrahydrophthalic anhydride) oligomer; GPC curve of (maleic anhydride/cis-1,2,3,6-tetrahydrophthalic anhydride) oligomer; 1H NMR spectra of poly(propylene carbonate) (PPC) (upper) and the soluble fraction of PPC with networks with 4 wt % of (maleic anhydride/cis-1,2,3,6-tetrahydrophthalic anhydride) oligomer feed of propylene oxide (below) after purification; 1H NMR spectrum of the reaction mixture after terpolymerization of CO2, propylene oxide (PO), and (maleic anhydride/cis-1,2,3,6-tetrahydrophthalic anhydride) oligomer (4 wt % of PO); GPC curves of PPC and PPC with networks (soluble fraction); PPC0.5–PPC4 presents the copolymer with 0.5–4 wt % (maleic anhydride/cis-1,2,3,6-tetrahydrophthalic anhydride) (MA/THPA) oligomer feed of PO in the copolymerization, respectively; thermogravimetric analysis curves of PPC and PPC with networks; PPC0.5-S–PPC4-S presents the soluble fraction of the corresponding copolymer; differential scanning calorimetry curves of PPC and PPC with networks; tensile and hot-set results of PPC and PPC with networks; 1H NMR of PPC specimen prepared by pressing at 190 °C; photographs of dumbbell specimens before (upper) and after (below) hot-set test; photograph below is the permanent deformation; and PPC and PPC4 specimens were prepared by hot pressing at 140 °C and at 190 °C, respectively (PDF)
PPC burning (MP4)
PPC0.5–1 burning (MP4)
PPC2–4 burning (MP4)
The authors declare no competing financial interest.
Supplementary Material
References
- Beckman E. J. Making Polymers from Carbon Dioxide. Science 1999, 283, 946–947. 10.1126/science.283.5404.946. [DOI] [Google Scholar]
- Grignard B.; Gennen S.; Jérôme C.; Kleij A. W.; Detrembleur C. Advances in the use of CO2 as a renewable feedstock for the synthesis of polymers. Chem. Soc. Rev. 2019, 48, 4466–4514. 10.1039/c9cs00047j. [DOI] [PubMed] [Google Scholar]
- Fritz N.; Dao H.; Allen S. A. B.; Kohl P. A. Polycarbonates as temporary adhesives. Int. J. Adhes. Adhes. 2012, 38, 45–49. 10.1016/j.ijadhadh.2012.04.001. [DOI] [Google Scholar]
- Touchaleaume F.; Angellier-Coussy H.; César G.; Raffard G.; Gontard N.; Gastaldi E. How performance and fate of biodegradable mulch films are impacted by field ageing. J. Polym. Environ. 2018, 26, 2588–2600. 10.1007/s10924-017-1154-7. [DOI] [Google Scholar]
- Seo J.; Jeon G.; Jang E. S.; Bahadar Khan S.; Han H. Preparation and properties of poly(propylene carbonate) and nanosized ZnO composite films for packaging applications. J. Appl. Polym. Sci. 2011, 122, 1101–1108. 10.1002/app.34248. [DOI] [Google Scholar]
- Zhang J.; Zang X.; Wen H.; Dong T.; Chai J.; Li Y.; Chen B.; Zhao J.; Dong S.; Ma J.; Yue L.; Liu Z.; Guo X.; Cui G.; Chen L. High-voltage and free-standing poly(propylene carbonate)/Li6.75La3Zr1.75Ta0.25O12 composite solid electrolyte for wide temperature range and flexible solid lithium ion battery. J. Mater. Chem. A 2017, 5, 4940–4948. 10.1039/c6ta10066j. [DOI] [Google Scholar]
- Yang H.; Zhang Y.; Tennenbaum M. J.; Althouse Z.; Ma Y.; He Y.; Wu Y.; Wu T.-H.; Mathur A.; Chen P.; Huang Y.; Fernandez-Nieves A.; Kohl P. A.; Liu N. Polypropylene carbonate-based adaptive buffer layer for stable interfaces of solid polymer lithium metal batteries. ACS Appl. Mater. Interfaces 2019, 11, 27906–27912. 10.1021/acsami.9b08285. [DOI] [PubMed] [Google Scholar]
- Chen S.; Chen B.; Fan J.; Feng J. Exploring the application of sustainable poly(propylene carbonate) copolymer in toughening epoxy thermosets. ACS Sustainable Chem. Eng. 2015, 3, 2077–2083. 10.1021/acssuschemeng.5b00343. [DOI] [Google Scholar]
- Zheng Y.; Li Y.; Hu X.; Shen J.; Guo S. Biocompatible shape memory blend for self-expandable stents with potential biomedical applications. ACS Appl. Mater. Interfaces 2017, 9, 13988–13998. 10.1021/acsami.7b04808. [DOI] [PubMed] [Google Scholar]
- Luinstra G. A.; Borchardt E.. Material properties of poly(propylene carbonates). In Synthetic Biodegradable Polymers; Rieger B.; Kunkel A.; Coates G. W.; Reichardt R.; Dinjus E.; Zevaco T. A., Eds.; Springer: Heidelberg, 2012; pp 29–48. [Google Scholar]
- Manavitehrani I.; Fathi A.; Wang Y.; Maitz P. K.; Dehghani F. Reinforced poly(propylene carbonate) composite with enhanced and tunable characteristics, an alternative for poly(lactic acid). ACS Appl. Mater. Interfaces 2015, 7, 22421–22430. 10.1021/acsami.5b06407. [DOI] [PubMed] [Google Scholar]
- Qin Y.; Sheng X.; Liu S.; Ren G.; Wang X.; Wang F. Recent advances in carbon dioxide based copolymers. J. CO2 Util. 2015, 11, 3–9. 10.1016/j.jcou.2014.10.003. [DOI] [Google Scholar]
- Xu Y.; Lin L.; Xiao M.; Wang S.; Smith A. T.; Sun L.; Meng Y. Synthesis and properties of CO2-based plastics: Environmentally-friendly, energy-saving and biomedical polymeric materials. Prog. Polym. Sci. 2018, 80, 163–182. 10.1016/j.progpolymsci.2018.01.006. [DOI] [Google Scholar]
- Muthuraj R.; Mekonnen T. Recent progress in carbon dioxide (CO2) as feedstock for sustainable materials development: Co-polymers and polymer blends. Polymer 2018, 145, 348–373. 10.1016/j.polymer.2018.04.078. [DOI] [Google Scholar]
- Subhani M. A.; Köhler B.; Gürtler C.; Leitner W.; Müller T. E. Light-mediated curing of CO2-based unsaturated polyethercarbonates via thiol-ene click chemistry. Polym. Chem. 2016, 7, 4121–4126. 10.1039/c6py00458j. [DOI] [Google Scholar]
- Liu L.; Wang Y.; Hu Q.; Li T.; Ma P.; Zhang H.; Du M.; Chen M.; Dong W. Core–shell starch nanoparticles improve the mechanical and thermal properties of poly(propylene carbonate). ACS Sustainable Chem. Eng. 2019, 7, 13081–13088. 10.1021/acssuschemeng.9b02256. [DOI] [Google Scholar]
- An J.; Ke Y.; Cao X.; Ma Y.; Wang F. A novel method to improve the thermal stability of poly(propylene carbonate). Polym. Chem. 2014, 5, 4245–4250. 10.1039/c4py00013g. [DOI] [Google Scholar]
- Li B.; Wu G.-P.; Ren W.-M.; Wang Y.-M.; Rao D.-Y.; Lu X.-B. Asymmetric, regio- and stereo-selective alternating copolymerization of CO2 and propylene oxide catalyzed by chiral chromium Salan complexes. J. Polym. Sci., Part A: Polym. Chem. 2008, 46, 6102–6113. 10.1002/pola.22922. [DOI] [Google Scholar]
- Nakano K.; Hashimoto S.; Nakamura M.; Kamada T.; Nozaki K. Stereocomplex of poly(propylene carbonate): Synthesis of stereogradient poly(propylene carbonate) by regio- and enantioselective copolymerization of propylene oxide with carbon dioxide. Angew. Chem., Int. Ed. 2011, 50, 4868–4871. 10.1002/anie.201007958. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Ren W.-M.; Zhang W.-P.; Zhao R.-R.; Lu X.-B. Crystalline CO2-based polycarbonates prepared from racemic catalyst through intramolecularly interlocked assembly. Nat. Commun. 2015, 6, 8594. 10.1038/ncomms9594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Ren W.-M.; He K.-K.; Lu X.-B. Crystalline-gradient polycarbonates prepared from enantioselective terpolymerization of meso-epoxides with CO2. Nat. Commun. 2014, 5, 5687. 10.1038/ncomms6687. [DOI] [PubMed] [Google Scholar]
- Gao J.; Chen F.; Wang K.; Deng H.; Zhang Q.; Bai H.; Fu Q. A promising alternative to conventional polyethylene with poly(propylene carbonate) reinforced by graphene oxide nanosheets. J. Mater. Chem. 2011, 21, 17627–17630. 10.1039/c1jm14300j. [DOI] [Google Scholar]
- Qi X.; Yang G.; Jing M.; Fu Q.; Chiu F.-C. Microfibrillated cellulose-reinforced bio-based poly(propylene carbonate) with dual shape memory and self-healing properties. J. Mater. Chem. A 2014, 2, 20393–20401. 10.1039/c4ta04954c. [DOI] [Google Scholar]
- Qu H.; Wang Y.; Ye Y. S.; Zhou W.; Bai S. P.; Zhou X. P.; Peng H. Y.; Xie X. L.; Mai Y.-W. A promising nanohybrid of silicon carbide nanowires scrolled by graphene oxide sheets with a synergistic effect for poly(propylene carbonate) nanocomposites. J. Mater. Chem. A 2017, 5, 22361–22371. 10.1039/c7ta06080g. [DOI] [Google Scholar]
- Chen L.; Qin Y.; Wang X.; Li Y.; Zhao X.; Wang F. Toughening of poly(propylene carbonate) by hyperbranched poly(ester-amide) via hydrogen bonding interaction. Polym. Int. 2011, 60, 1697–1704. 10.1002/pi.3132. [DOI] [Google Scholar]
- Song P.-f.; Wang S.-j.; Xiao M.; Du F.-g.; Gan L.-q.; Liu G.-q.; Meng Y.-z. Cross-linkable and thermally stable aliphatic polycarbonates derived from CO2, propylene oxide and maleic anhydride. J. Polym. Res. 2009, 16, 91–97. 10.1007/s10965-008-9206-2. [DOI] [Google Scholar]
- Gao L.; Feng J. A one-step strategy for thermally and mechanically reinforced pseudo-interpenetrating poly(propylene carbonate) networks by terpolymerization of CO2, propylene oxide and pyromellitic dianhydride. J. Mater. Chem. A 2013, 1, 3556–3560. 10.1039/c3ta01177a. [DOI] [Google Scholar]
- Chen X.; Wang L.; Feng J.; Huang X.; Guo X.; Chen J.; Xiao Z.; Liang X.; Gao L. Enhanced poly(propylene carbonate) with thermoplastic networks: A one-pot synthesis from carbon dioxide, propylene oxide, and a carboxylic dianhydride. Polymers 2018, 10, 552. 10.3390/polym10050552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Y.; Wang X.; Zhao X.; Li J.; Wang F. Crosslinkable poly(propylene carbonate): High-yield synthesis and performance improvement. J. Polym. Sci., Part A: Polym. Chem. 2006, 44, 5329–5336. 10.1002/pola.21595. [DOI] [Google Scholar]
- Wang X. L.; Meng Y. Z.; Li R. K. Y. Crosslinking of poly(propylene carbonate) by peroxide crosslinking agent dicumyl peroxide (DCP). Acta Sci. Nat. Univ. Sunyatseni 2007, 46, 4–6. [Google Scholar]
- Hao Y.; Ge H.; Han L.; Liang H.; Zhang H.; Dong L. Thermal, mechanical, and rheological properties of poly(propylene carbonate) cross-linked with polyaryl polymethylene isocyanate. Polym. Bull. 2013, 70, 1991–2003. 10.1007/s00289-013-0912-5. [DOI] [Google Scholar]
- Qin Y. S.; MA Q. W.; Wang X. H.; Sun J. Z.; Zhao X. J.; Wang F. S. Electron-beam irradiation on poly(propylene carbonate) in the presence of polyfunctional monomers. Polym. Degrad. Stab. 2007, 92, 1942–1947. 10.1016/j.polymdegradstab.2007.06.007. [DOI] [Google Scholar]
- Darensbourg D. J.; Wang Y. Terpolymerization of propylene oxide and vinyl oxides with CO2: copolymer cross-linking and surface modification via thiol-ene click chemistry. Polym. Chem. 2015, 6, 1768–1776. 10.1039/c4py01612b. [DOI] [Google Scholar]
- Xia L.; Chen L.-b. Silicone modified poly(propylene carbonate). Polym. Mater. Sci. Eng. 2003, 19, 202–204. [Google Scholar]
- Cyriac A.; Lee S. H.; Lee B. Y. Connection of polymer chains using diepoxide in CO2/propylene oxide copolymerizations. Polym. Chem. 2011, 2, 950–956. 10.1039/c0py00365d. [DOI] [Google Scholar]
- Wu J.; Xiao M.; He H.; Wang S.; Han D.; Meng Y. Copolymerization of propylene oxide and carbon dioxide in the presence of diphenylmethane diisoyanate. J. Polym. Res. 2011, 18, 1479–1486. 10.1007/s10965-010-9553-7. [DOI] [Google Scholar]
- Okada A.; Kikuchi S.; Yamada T. Alternating copolymerization of propylene oxide/alkylene oxide and carbon dioxide: Tuning thermal properties of polycarbonates. Chem. Lett. 2011, 40, 209–211. 10.1246/cl.2011.209. [DOI] [Google Scholar]
- Feng J.-Y.; Gao L.-J.; Chen B.; Wu X.-J.; Luo Q.-L.; Wu C.-Y.; Zheng C.-X.; Lin L.-Z.; Deng S.-L.; Huang X.-M. A one-step strategy for reinforced poly(propylene carbonate) with partial crosslinking via terpolymerization of CO2 and propylene oxide using triglycidyl isocyanurate. Chem. Lett. 2013, 42, 714–716. 10.1246/cl.130165. [DOI] [Google Scholar]
- Song P.; Mao X.; Zhang X.; Zhu X.; Wang R. A one-step strategy for cross-linkable aliphatic polycarbonates with high degradability derived from CO2, propylene oxide and itaconic anhydride. RSC Adv. 2014, 4, 15602–15605. 10.1039/c4ra01514b. [DOI] [Google Scholar]
- Gao L.; Chen X.; Liang X.; Guo X.; Huang X.; Chen C.; Wan X.; Deng R.; Wu Q.; Wang L.; Feng J. A novel one-pot synthesis of poly(propylene carbonate) containing cross-linked networks by copolymerization of carbon dioxide, propylene oxide, maleic anhydride, and furfuryl glycidyl ether. Polymers 2019, 11, 881. 10.3390/polym11050881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Y.; Wang X.; Zhao X.; Li J.; Wang F. Double propagation based on diepoxide, a facile route to high molecular weight poly(propylene carbonate). Polymer 2006, 47, 7368–7373. 10.1016/j.polymer.2006.08.035. [DOI] [Google Scholar]
- Han B.; Zhang L.; Zhang H.; Ding H.; Liu B.; Wang X. One-pot synthesis and postpolymerization functionalization of cyclic carbonate/epoxide-difunctional polycarbonates prepared by regioselective diepoxide/CO2 copolymerization. Polym. Chem. 2016, 7, 4453–4457. 10.1039/c6py00563b. [DOI] [Google Scholar]
- Lee S. H.; Cyriac A.; Jeon J. Y.; Lee B. Y. Preparation of thermoplastic polyurethanes using in situ generated poly(propylene carbonate)-diols. Polym. Chem. 2012, 3, 1215–1220. 10.1039/c2py00010e. [DOI] [Google Scholar]
- Darensbourg D. J. Making plastics from carbon dioxide: Salen metal complexes as catalysts for the production of polycarbonates from epoxides and CO2. Chem. Rev. 2007, 107, 2388–2410. 10.1021/cr068363q. [DOI] [PubMed] [Google Scholar]
- DiCiccio A. M.; Coates G. W. Ring-opening copolymerization of maleic anhydride with epoxides: A chain-growth approach to unsaturated polyesters. J. Am. Chem. Soc. 2011, 133, 10724–10727. 10.1021/ja203520p. [DOI] [PubMed] [Google Scholar]
- Liu J.; Bao Y.-Y.; Liu Y.; Ren W.-M.; Lu X.-B. Binuclear chromium-salan complex catalyzed alternating copolymerization of epoxides and cyclic anhydrides. Polym. Chem. 2013, 4, 1439–1444. 10.1039/c2py20842c. [DOI] [Google Scholar]
- Hua Z.; Qi G.; Chen S. Ring-opening copolymerization of maleic anhydride with propylene oxide by double-metal cyanide. J. Appl. Polym. Sci. 2004, 93, 1788–1792. 10.1002/app.20635. [DOI] [Google Scholar]
- Liu Y.; Xiao M.; Wang S.; Xia L.; Hang D.; Cui G.; Meng Y. Mechanism studies of terpolymerization of phthalic anhydride, propylene epoxide, and carbon dioxide catalyzed by ZnGA. RSC Adv. 2014, 4, 9503–9508. 10.1039/c3ra46343e. [DOI] [Google Scholar]
- Van Zee N. J.; Coates G. W. Alternating copolymerization of propylene oxide with biorenewable terpene-based cyclic anhydrides: A sustainable route to aliphatic polyesters with high glass transition temperatures. Angew. Chem., Int. Ed. 2015, 54, 2665–2668. 10.1002/anie.201410641. [DOI] [PubMed] [Google Scholar]
- Li J.; Liu Y.; Ren W.-M.; Lu X.-B. Asymmetric alternating copolymerization of meso-epoxides and cyclic anhydrides: Efficient access to enantiopure polyesters. J. Am. Chem. Soc. 2016, 138, 11493–11496. 10.1021/jacs.6b07520. [DOI] [PubMed] [Google Scholar]
- Gao L.; Huang M.; Wu Q.; Wan X.; Chen X.; Wei X.; Yang W.; Deng R.; Wang L.; Feng J. Enhanced poly(propylene carbonate) with thermoplastic networks: A cross-linking role of maleic anhydride oligomer in CO2/PO copolymerization. Polymers 2019, 11, 1467. 10.3390/polym11091467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L. J.; Xiao M.; Wang S. J.; Meng Y. Z. Thermally stable poly(propylene carbonate) synthesized by copolymerizing with bulky naphthalene containing monomer. J. Appl. Polym. Sci. 2008, 108, 1037–1043. 10.1002/app.27271. [DOI] [Google Scholar]
- Gao L. J.; Du F. G.; Xiao M.; Wang S. J.; Meng Y. Z. Thermally stable aliphatic polycarbonate containing bulky carbazole pendants. J. Appl. Polym. Sci. 2008, 108, 3626–3631. 10.1002/app.27994. [DOI] [Google Scholar]
- Varghese J. K.; Na S. J.; Park J. H.; Woo D.; Yang I.; Lee B. Y. Thermal and weathering degradation of poly(propylene carbonate). Polym. Degrad. Stab. 2010, 95, 1039–1044. 10.1016/j.polymdegradstab.2010.03.006. [DOI] [Google Scholar]
- Lu X. L.; Zhu Q.; Meng Y. Z. Kinetic analysis of thermal decomposition of poly(propylene carbonate). Polym. Degrad. Stab. 2005, 89, 282–288. 10.1016/j.polymdegradstab.2004.12.025. [DOI] [Google Scholar]
- Li X. H.; Meng Y. Z.; Zhu Q.; Tjong S. C. Thermal decomposition characteristics of poly(propylene carbonate) using TG/IR and PY-GC/MS techniques. Polym. Degrad. Stab. 2003, 81, 157–165. 10.1016/s0141-3910(03)00085-5. [DOI] [Google Scholar]
- Jintang D.; Jiajun W.; Lianfang F.; Long W.; Xueping G. Pressure dependence of the CO2/propylene oxide copolymerization catalyzed by zinc glutarate. J. Appl. Polym. Sci. 2010, 118, 366–371. 10.1002/app.32399. [DOI] [Google Scholar]
- Tang L.; Xiao M.; Xu Y.; Wang S.; Meng Y. Zinc adipate/tertiary amine catalytic system: efficient synthesis of high molecular weight poly(propylene carbonate). J. Polym. Res. 2013, 20, 190. 10.1007/s10965-013-0190-9. [DOI] [Google Scholar]
- Song P. F.; Xiao M.; Du F. G.; Wang S. J.; Gan L. Q.; Liu G. Q.; Meng Y. Z. Synthesis and properties of aliphatic polycarbonates derived from carbon dioxide, propylene oxide and maleic anhydride. J. Appl. Polym. Sci. 2008, 109, 4121–4129. 10.1002/app.28449. [DOI] [Google Scholar]
- Zhang H.-l.; Sun X.-h.; Chen Q.-y.; Ren M.-q.; Zhang Z.-h.; Zhang H.-f.; Mo Z.-s. Miscibility, crystallization and mechanical properties of PPC/PBS blends. Chin. J. Polym. Sci. 2007, 25, 589–597. 10.1142/s0256767907002503. [DOI] [Google Scholar]
- Li Y.; Yu Y.-C.; Han C.-Y.; Wang X.-H.; Huang D.-X. Sustainable blends of poly(propylene carbonate) and stereocomplex polylactide with enhanced rheological properties and heat resistance. Chin. J. Polym. Sci. 2020, 10.1007/s10118-020-2408-8. [DOI] [Google Scholar]
- Li Y.-h.; Zhou M.; Geng C.-z.; Chen F.; Fu Q. Simultaneous improvements of thermal stability and mechanical properties of poly(propylene carbonate) via incorporation of environmental-friendly polydopamine. Chin. J. Polym. Sci. 2014, 32, 1724–1736. 10.1007/s10118-014-1518-6. [DOI] [Google Scholar]
- Smallman R. V.Maleic anhydride copolymer. U.S. Patent 3,838,113 A, 24 Sept. 1974.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.