

Abstract
Atherosclerosis is a major cause of coronary artery disease and stroke. A massive and new type of data has finally arrived in the field of atherosclerosis: single cell RNA sequencing (scRNAseq). Recently, scRNAseq has been successfully applied to the study of atherosclerosis to identify previously uncharacterized cell populations. scRNAseq is an effective approach to evaluate heterogeneous cell populations by measuring the transcriptomic profiles at the single cell level. Besides the studies of atherosclerosis, scRNAseq is being employed in various areas of biology, including cancer research and organ development. In order to analyze these new massive datasets, various analytic approaches have been developed. This review aims to enhance the understanding of this new technology by exploring how the single cell transcriptome has been applied to the study of atherosclerosis and further discuss potential analysis of using scRNAseq.
Keywords: Atherosclerosis, Single-cell analysis, Transcriptome
Graphical Abstract
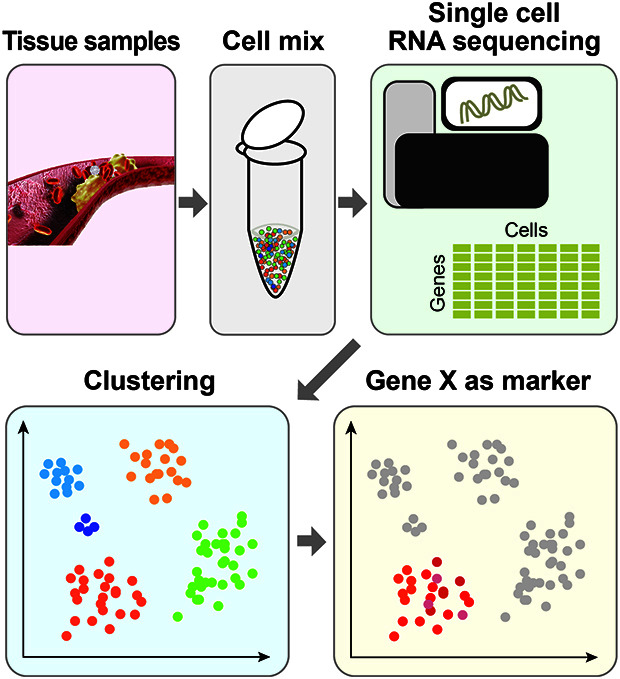
1. scRNAseq is a powerful approach to understand heterogeneous cell population.
2. scRNAseq can help reveal cell identity and their transcriptomic changes.
3. Computational algorithms have been developed to cluster scRNAseq data, identify marker genes and perform other subsequent analysis.
INTRODUCTION
Atherosclerosis is a chronic inflammatory disease driven by the interplay of many types of cells, including immune and stromal cells, with diverse phenotypic and transcriptomic changes.1,2,3,4 A major limitation to the current transcriptomic methods, such as bulk RNA-sequencing (seq) analysis, is that they only capture transcriptional changes of the whole population rather than individual cells. Gene expression is heterogeneous, even in similar cell types.5 The stochastic nature of gene expression has a functional role and can lead to cell fate decisions.6,7 By measuring transcriptomic profiles at the single cell level, single cell RNA seq (scRNAseq) is an effective approach to deal with heterogeneous cell populations. The scRNAseq can detect the transcriptome of a rare cell population8 and study the trend of gene expression across the population of cells.9 The scRNAseq has been applied to various species, tissues (human and mouse), and studies to reveal cell-to-cell gene expression variability.10 Compared with the analysis of bulk RNAseq, which is mainly focused on identifying differentially expressed genes, scRNAseq provides various angles to study heterogeneity, cell interactions and transcriptomic changes along development or upon treatment. Algorithmic development has followed the new type of data generation. Therefore, the appropriate analytical approaches must be applied to effectively handle scRNAseq data.
In recent years, we have observed a number of atherosclerosis studies using scRNAseq.11,12,13,14,15,16 In combination with various computational algorithms, the use of scRNAseq provided new information and knowledge about atherosclerosis, and it is expected that this new technology will become more popular. In this review, we aim to provide a guide about the scRNAseq technology used in recent studies of atherosclerosis. We will discuss the results from recent studies using scRNAseq in the field of atherosclerosis and introduce how the scRNAseq data were analyzed.
THE scRNAseq FOR THE STUDY OF ATHEROSCLEROSIS
The scRNAseq was first utilized in the field of atherosclerosis to investigate the driven plasticity of Forhead box P3+ T regulatory cells (Tregs) in an apolipoprotein E (ApoE) deficient mice model, a widely used system to study cardiovascular and respiratory diseases.11 Briefly, scRNAseq data were generated from 270 cells which were clustered into 3 groups: Treg, type 1 T helper (Th1)/Treg, and Th1. It has been known that atherosclerosis promotes the formation of an intermediately plastic Th1/Treg subset. Th scRNAseq data was used to confirm the results of flow cytometry about the existence of the previously uncharacterized Th1/Tregs group. The scRNAseq was further used to expand the list of genes that are differentially regulated in the Th1/Treg cell population. For instance, compared with Treg and Th1 cell populations, the Th1/Treg cell population is characterized by the downregulation of multiple Treg immunosuppressive genes, such as Tnfrsf4, Tnfrsf9, Tnfrsf18, Icos, Ctla4, and Treg-lineage transcription factors, such as Ikzf2, Ikzf4, and Foxp3.11
In 2018, Cochain et al.12 obtained transcriptome profiles of 372 control cells and 854 cells from diseased aortas. Among 13 aortic cell clusters, they identified 3 types of macrophages (resident-like, inflammatory and the previously uncharacterized triggering receptor expressed on myeloid cells 2 [TREM2+]). The scRNAseq was essential to identify TREM2+ macrophages, which do not belong to any of the previously known 2 types (M1- and M2-polarized) of macrophages. They found that the inflammatory macrophages and TREM2+ macrophages were almost exclusively observed in the cells from diseased aortas. In his study, Cochain et al.12 used the Seurat package (https://satijalab.org/seurat/)17 for clustering cells and identifying the marker genes associated with each cluster. Furthermore, they compared the number of diseased and control cells in each cluster.
In a similar study, Winkel et al.13 clustered 555 cells from control and 909 from disease cells and identified 11 leukocyte populations, including B cell subsets, using the Seurat package. Besides the cell subpopulation, they studied cell composition in the media, adventitia, and lesion and adventitia + tertiary lymphoid organs (ATLO). They calculated the composition of cells using the decomposition analysis based on bulk RNAseq and scRNAseq and reported that samples from lesions had a significantly smaller number of B cells compared to other samples.
More recently, Gu et al.14 also examined cell populations in normal and ApoE-deficient murine aortic adventitia. Besides clustering analysis to identify sub-populations, they studied cell communication between mesenchymal cells and macrophages in ApoE-deficient adventitia by evaluating the expression of ligand-receptor pairs.14 This analysis identified that chemokine (C-C motif) ligand 2, a chemokine secreted mainly by inflammatory cells and dysfunctional endothelial cells in atherosclerosis, was selectively expressed in a mesenchymal population for communication with macrophages.
Moreover, Kim et al.15 identified 11 leukocyte subpopulations, including diverse macrophage sub-clusters, in an atherosclerotic aorta using scRNAseq. These results were compared with the results from the foamy and non-foamy macrophage populations identified using flow cytometry.15 Another study investigated the features of monocyte-to-macrophage transition using scRNAseq in combination with genetic fate mapping of myeloid cells derived from CX3C chemokine receptor+ precursors during atherosclerosis progression and regression. They performed pseudo-time analysis after aligning cells along the pseudo-transition time.16
To generate scRNAseq data, Butcher et al.11 used Fluidigm C1. The C1 system enables size-based cell selection and is currently able to collect up to 800 cells. Other studies used the Chromium System (10× Genomics) in which a droplet-based microfluidic platform is used to sequence thousands of cells in parallel. A recent review paper summarized the commercially available instruments for single-cell collection.18
Because of its ability to measure the transcriptome at the single cell level, scRNAseq is increasingly used in various areas in biology. We aim to review some of the analyses made possible by scRNAseq output, which can be used for the study of atherosclerosis as well.
THE scRNAseq DATA ANALYSIS
1. Pre-processing
Raw data generated by a sequencing machine are processed to have read counts or number of molecules. The count information can be represented in a matrix which shows the gene expression across cells (Fig. 1A). To exclude low-quality information from scRNAseq, a series of quality controls (QC) are generally required. QC is employed to ensure that the quality of scRNAseq data is sufficient for subsequent downstream analysis. Common practices include removing cells with a low count depth or few detected genes. Some transcriptomic information can be from multiple cells (doublets). These unwanted doublets can be removed using doublet detection tools including Scrublet19 and DoubletFinder.20
Fig. 1. The scRNAseq data processing for atherosclerosis. (A) scRNA procedure. The scRNAseq data are collected from each sample which are represented in a table. PCA and/or tSNE is applied to reduce the dimension for the sake of clustering. Genes expressed in each cluster are examined. (B) Clustering results using scRNAseq data from the study by Lin et al.16.

scRNAseq, single cell RNA sequencing; PCA, principal components analysis; tSNE, t-distributed stochastic neighbor embedding; Ear2, V-erbA-related protein 2; Irf7, interferon regulatory factor 7.
Once the matrix is obtained, normalization can be considered to compensate for cells with different numbers of barcodes or read depth. Normalization scales count data to a relative expression abundance. In addition, the batch effect should be removed when considering scRNAseq data from several sources. The batch effect is a common source of technical variation that can arise from various sources, including disparate cell dissociation protocol, library preparation, and sequencing platforms. Comprehensive benchmarking tests were applied for normalization and batch effect correction for the dataset for droplet-based scRNAseq data21 showing that pooling-based size factor estimation by Scran22 is one of the best working normalization approaches. The Seurat package is equipped with batch effect correction,17 but trying diverse approaches after considering the underlying biological processes is also recommended for batch effect correction.
2. Clustering
Clustering is a useful approach to identify various types of cells from scRNAseq. Hierarchical clustering has been widely used for bulk-cell RNAseq analysis. Butcher et al.11 used hierarchical clustering for 270 cells and identified 3 groups of cells. As the number of cells increases, the need for new approaches to handle high-dimensional scRNAseq data arises. Various algorithms for processing multi-dimensional scRNAseq data have been developed. Generally, these clustering methods rely on dimension reduction algorithms to avoid unnecessary noise (features). Dimensionality reduction approaches, such as principal components analysis (PCA), have been widely used to analyze both bulk RNA-seq data as well as scRNAseq data.17,23 For instance, PCA allows the conversion of a higher dimensional dataset into a lower, often 2 or 3, dimensional dataset with more important and uncorrelated variables (dimensions) called principal components. Subsequently, cells can be clustered and envisioned in 2- or 3-dimensional (2D or 3D) space. Similarly, t-distributed stochastic neighbor embedding (tSNE) has also been successfully used to visualize cells in a reduced space.17 Moreover, tSNE is used to visualize cells in 2D or 3D space while reflecting true distances in the original space as far as possible so that cells of a particular cell type tend to be located nearby in 2D space. Finally, clustering is performed on cells by grouping them in the reduced space. Fig. 1B shows the tSNE-based analysis using the dataset by Lin et al.16 to understand macrophage heterogeneity during atherosclerosis progression and repression. From the tSNE plot, we can identify sub-clusters with V-erbA-related protein 2 (Ear2) expression (corresponds to RentnlahiEar2hi macrophage) and interferon (IFN) regulatory factor 7 (type 1 IFN signature) (Fig. 1B). In a similar manner, cell types are assigned based on a priori known marker genes. Cochain et al.12 identified cells, such as B cells (using Cd79a, Cd79b, Ly6d, and Mzb1), C-X-C chemokine receptor type (CXCR)6+ T cells (using CXCR6, Icos, Cd3g, and Il7R) and natural killer cells (using Klrb1c, Ncr1, Klra8, and Klrc1), based on the associated marker genes.
A number of algorithms have also been exclusively proposed for scRNAseq data analysis, including SC324 and SIMLR.25 CellBIC was designed to identify small cell subpopulations without losing information by dimension reduction.26 GiniClust has also been proposed to identify rare cell population8. Recent advances allow for ultra-fast clustering of more than 1 million cells.27
3. Cell composition comparison
One of the downstream applications of scRNAseq analysis is the comparison of cell compositions. For instance, Cochain et al.12 compared the number of cells from the control and the diseased aortas for each of the 3 clusters of macrophages and found that TREM2+ macrophages were almost exclusively observed in the cells from diseased aortas. In addition, the same quantitation could also provide an estimation of cell composition of bulk-cell RNAseq. This approach may be particularly useful when samples are collected from a different section of tissue. If scRNAseq is provided for a section (so that cell subpopulations are obtained), the cell composition of another section can be estimated from the bulk RNAseq using computational deconvolution based on scRNAseq28 (Fig. 2). Winkel et al.13 used CIBERSORT29 to perform deconvolution of cells using bulk-RNA-seq from the media, adventitia, lesion and adventitia + ATLO.
Fig. 2. Cell decomposition using scRNAseq. When bulk RNAseq and scRNAseq are available, cell decomposition may be used to obtain the cell composition.

scRNAseq, single cell RNA sequencing; RNAseq, RNA sequencing.
4. Pseudo-time analysis
When cells are represented in a lower dimensional space, those with similar transcriptomes will be located nearby on a plot, e.g. using tSNE. When cells are collected in different time stamps during differentiation, mature cells will be located far from progenitors, and cells being differentiated will be located in the middle. The path that links the cells can be regarded as a “pseudo” time9 (Fig. 3). This allows for longitudinal analysis of gene expression (e.g. development). Pseudo-time can be used to model transcriptomic changes during the development of atherosclerosis. Gene expressions can be analyzed along pseudo-time. For instance, the expression level of elastin deceases during direct cardiomyocyte conversion, while the expression level of troponin I1, slow skeletal type increases (Fig. 3). Furthermore, Lin et al.16 used the pseudo-time analysis along the fate-mapping during atherosclerosis progression and regression. This analysis found 53 genes significantly correlated with pseudo-time score, including CXCR4 and Ctsd. Monocle has been used for pseudo-time analysis.9 TSCAN combines clustering with pseudo-time analysis.30 Partition-based graph abstraction could be useful when complex trajectories are expected.31
Fig. 3. Pseudo-time analysis using scRNAseq. The scRNAseq are obtained from cells during direct conversion to cardiomyocytes49 and reprocessed. Fibroblast cells are located on the left side and cardiomyocyte cells on the top right. Cells can be aligned in between based on their transcriptomic similarities. When aligned, pseudo-time analysis is applied. The expression level of Eln, a fibroblast marker, decreases along the pseudo-time.

scRNAseq, single cell RNA sequencing; Eln, elastin; Dlk1, delta like non-canonical notch ligand 1; Tnni1, troponin I1, slow skeletal type; Tnni3, troponin I3, cardiac type.
5. Reconstruction of gene regulatory networks
Reverse engineering reconstructs gene regulatory networks from gene expression information.32 It usually requires a large amount of expression data. By providing transcriptomic information for each single cell, scRNAseq can be a good resource for reconstructing the regulatory networks. Pseudo-time has also been used to identify potential downstream target genes9 (Fig. 3). Software tools such as SCODE were developed to reconstruct gene regulatory networks from scRNAseq data.
6. Adding spatial information to scRNAseq
Another major limitation of current transcriptomic analysis workflow is that once the cells are isolated from tissue for scRNAseq, the cell location and orientation information is lost. To restore approximate location information, tissues can be mechanically sampled from different spots. For instance, Winkel et al.,13 used the spatial information by comparing cells from whole-atherosclerotic aortas versus aortic leukocytes. Another strategy has also been introduced in which barcoding the native tissue location has been proposed.33 This approach dissects the histological section with a grid, and each spot is barcoded to provide position information. Currently, a grid contains 10–30 cells depending on the tissue. The RNAs along with the position information are sequenced together. Spatial transcriptomics is now part of 10× Genomics (https://spatialtranscriptomics.com/). Another approach called MERFISH uses fluorescence in situ hybridization to provide spatial information in order to map the identity and location of specific cell types.34 Statistical test can be applied to identify genes whose spatial patterns are significantly different.35
7. Studying cell communications
The expression of ligand-receptor pairs can be utilized to study cell communication.36 Specifically, Gu et al.14 investigated the ligand-receptor interaction between cell subtypes by considering the transcriptomic levels of ligands and their corresponding receptors. In cells from the adventitia, this computational prediction showed the importance of mesenchyme populations for maintaining adventitial homeostasis. For instance, cells expressing Cd34 and Cav1 interacted with Sell and Icam1 expressed by inflammatory macrophages, respectively, which may potentially modulate leukocyte influx to the adventitia. This ligand-receptor pair analysis predicted the manner in which resident mesenchyme cells interacted with and attracted immune cells in vivo.
CONCLUSION
By providing transcriptomic profiles at single cell resolution, scRNAseq is a powerful tool to study heterogeneity and dynamic changes in cell populations.9,37,38,39,40,41 However, scRNAseq has limitations, such as biases in transcript coverage and low capture efficiency.42,43 We discussed how to handle some of these limitations in the pre-processing section.
In this review, we discussed certain scRNAseq studies about atherosclerosis which tried to identify heterogeneous cell populations by scRNAseq assay. Focusing on the data analysis aspect, we discussed some of the computational approaches that may be applied to the study of atherosclerosis. Clustering has been applied to all studies and has contributed to the identification of previously uncharacterized populations.11,12,13,15,16,17 Additionally, clustering various computational algorithms has provided new angles in studying atherosclerosis. For instance, pseudo-time analysis revealed key genes associated with atherosclerosis development.16 Cell-to-cell communications was investigated by studying the co-expression of ligand-receptor pairs.14 We also discussed spatial transcriptomic analysis which attempts to provide information about the locations of cells in the cell transcriptome. This can be used to identify tissue-morphology-specific gene expression.
There have been attempts to provide additional data collected from the same set of cells. G&T-seq44 provides information on DNA methylation as well as transcriptome from the same set of cells at the same time. TARGET-seq detects genetic mutations which together with transcriptome from a single cell.45 However, TARGET-seq only detects a limited number of known mutations. In comparison, SIDR46 enables parallel sequencing of the entire genomic DNA and messenger RNA of a cell. CITE-seq47 and REAP-seq48 quantify transcriptome with protein level using antibodies conjugated to a tripartite DNA sequence that contains a primer for amplification and sequencing (polymerase chain reaction handle), a unique oligonucleotide that acts as an antibody barcode, and an oligo (dA). These technologies can provide new insights into scRNAseq analysis. Overall, single-cell-resolution data will enhance our understanding about the development of morbid metabolic disorders such as atherosclerosis.
Footnotes
Funding: The Novo Nordisk Foundation Center for Stem Cell Biology is supported by a Novo Nordisk Foundation grant No. NNF17CC0027852.
Conflict of Interest: The authors have no conflicts of interest to declare.
- Funding acquisition: Won KJ.
- Supervision: Won KJ.
- Writing - original draft: Hajkarim MC, Won KJ.
References
- 1.Vengrenyuk Y, Nishi H, Long X, Ouimet M, Savji N, Martinez FO, et al. Cholesterol loading reprograms the microRNA-143/145-myocardin axis to convert aortic smooth muscle cells to a dysfunctional macrophage-like phenotype. Arterioscler Thromb Vasc Biol. 2015;35:535–546. doi: 10.1161/ATVBAHA.114.304029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rong JX, Shapiro M, Trogan E, Fisher EA. Transdifferentiation of mouse aortic smooth muscle cells to a macrophage-like state after cholesterol loading. Proc Natl Acad Sci U S A. 2003;100:13531–13536. doi: 10.1073/pnas.1735526100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Feil S, Fehrenbacher B, Lukowski R, Essmann F, Schulze-Osthoff K, Schaller M, et al. Transdifferentiation of vascular smooth muscle cells to macrophage-like cells during atherogenesis. Circ Res. 2014;115:662–667. doi: 10.1161/CIRCRESAHA.115.304634. [DOI] [PubMed] [Google Scholar]
- 4.Li Y, Lui KO, Zhou B. Reassessing endothelial-to-mesenchymal transition in cardiovascular diseases. Nat Rev Cardiol. 2018;15:445–456. doi: 10.1038/s41569-018-0023-y. [DOI] [PubMed] [Google Scholar]
- 5.Huang S. Non-genetic heterogeneity of cells in development: more than just noise. Development. 2009;136:3853–3862. doi: 10.1242/dev.035139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maamar H, Raj A, Dubnau D. Noise in gene expression determines cell fate in Bacillus subtilis. Science. 2007;317:526–529. doi: 10.1126/science.1140818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eldar A, Elowitz MB. Functional roles for noise in genetic circuits. Nature. 2010;467:167–173. doi: 10.1038/nature09326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiang L, Chen H, Pinello L, Yuan GC. GiniClust: detecting rare cell types from single-cell gene expression data with Gini index. Genome Biol. 2016;17:144. doi: 10.1186/s13059-016-1010-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol. 2014;32:381–386. doi: 10.1038/nbt.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen G, Ning B, Shi T. Single-cell RNA-seq technologies and related computational data analysis. Front Genet. 2019;10:317. doi: 10.3389/fgene.2019.00317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Butcher MJ, Filipowicz AR, Waseem TC, McGary CM, Crow KJ, Magilnick N, et al. Atherosclerosis-driven Treg plasticity results in formation of a dysfunctional subset of plastic IFNγ+ Th1/Tregs. Circ Res. 2016;119:1190–1203. doi: 10.1161/CIRCRESAHA.116.309764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cochain C, Vafadarnejad E, Arampatzi P, Pelisek J, Winkels H, Ley K, et al. Single-cell RNA-seq reveals the transcriptional landscape and heterogeneity of aortic macrophages in murine atherosclerosis. Circ Res. 2018;122:1661–1674. doi: 10.1161/CIRCRESAHA.117.312509. [DOI] [PubMed] [Google Scholar]
- 13.Winkels H, Ehinger E, Vassallo M, Buscher K, Dinh HQ, Kobiyama K, et al. Atlas of the immune cell repertoire in mouse atherosclerosis defined by single-cell RNA-sequencing and mass cytometry. Circ Res. 2018;122:1675–1688. doi: 10.1161/CIRCRESAHA.117.312513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gu W, Ni Z, Tan YQ, Deng J, Zhang SJ, Lv ZC, et al. Adventitial cell atlas of wt (wild type) and ApoE (apolipoprotein E)-deficient mice defined by single-cell RNA sequencing. Arterioscler Thromb Vasc Biol. 2019;39:1055–1071. doi: 10.1161/ATVBAHA.119.312399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim K, Shim D, Lee JS, Zaitsev K, Williams JW, Kim KW, et al. Transcriptome analysis reveals nonfoamy rather than foamy plaque macrophages are proinflammatory in atherosclerotic murine models. Circ Res. 2018;123:1127–1142. doi: 10.1161/CIRCRESAHA.118.312804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin JD, Nishi H, Poles J, Niu X, Mccauley C, Rahman K, et al. Single-cell analysis of fate-mapped macrophages reveals heterogeneity, including stem-like properties, during atherosclerosis progression and regression. JCI Insight. 2019;4:124574. doi: 10.1172/jci.insight.124574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Valihrach L, Androvic P, Kubista M. Platforms for single-cell collection and analysis. Int J Mol Sci. 2018;19:E807. doi: 10.3390/ijms19030807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wolock SL, Lopez R, Klein AM. Scrublet: computational identification of cell doublets in single-cell transcriptomic data. Cell Syst. 2019;8:281–291.e9. doi: 10.1016/j.cels.2018.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McGinnis CS, Murrow LM, Gartner ZJ. DoubletFinder: doublet detection in single-cell RNA sequencing data using artificial nearest neighbors. Cell Syst. 2019;8:329–337.e4. doi: 10.1016/j.cels.2019.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Büttner M, Miao Z, Wolf FA, Teichmann SA, Theis FJ. A test metric for assessing single-cell RNA-seq batch correction. Nat Methods. 2019;16:43–49. doi: 10.1038/s41592-018-0254-1. [DOI] [PubMed] [Google Scholar]
- 22.Lun AT, Bach K, Marioni JC. Pooling across cells to normalize single-cell RNA sequencing data with many zero counts. Genome Biol. 2016;17:75. doi: 10.1186/s13059-016-0947-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lall S, Sinha D, Bandyopadhyay S, Sengupta D. Structure-aware principal component analysis for single-cell RNA-seq data. J Comput Biol. 2018;25:1365–1373. doi: 10.1089/cmb.2018.0027. [DOI] [PubMed] [Google Scholar]
- 24.Kiselev VY, Kirschner K, Schaub MT, Andrews T, Yiu A, Chandra T, et al. SC3: consensus clustering of single-cell RNA-seq data. Nat Methods. 2017;14:483–486. doi: 10.1038/nmeth.4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang B, Ramazzotti D, De Sano L, Zhu J, Pierson E, Batzoglou S. SIMLR: a tool for large-scale genomic analyses by multi-kernel learning. Proteomics. 2018;18:1700232. doi: 10.1002/pmic.201700232. [DOI] [PubMed] [Google Scholar]
- 26.Kim J, Stanescu DE, Won KJ. CellBIC: bimodality-based top-down clustering of single-cell RNA sequencing data reveals hierarchical structure of the cell type. Nucleic Acids Res. 2018;46:e124. doi: 10.1093/nar/gky698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wan SJ, Kim J, Won KJ. SHARP: single-cell RNA-seq hyper-fast and accurate processing via ensemble random projection. bioRxivorg. 2018 doi: 10.1101/gr.254557.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shen-Orr SS, Gaujoux R. Computational deconvolution: extracting cell type-specific information from heterogeneous samples. Curr Opin Immunol. 2013;25:571–578. doi: 10.1016/j.coi.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12:453–457. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ji Z, Ji H. TSCAN: pseudo-time reconstruction and evaluation in single-cell RNA-seq analysis. Nucleic Acids Res. 2016;44:e117. doi: 10.1093/nar/gkw430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wolf FA, Hamey FK, Plass M, Solana J, Dahlin JS, Göttgens B, et al. PAGA: graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells. Genome Biol. 2019;20:59. doi: 10.1186/s13059-019-1663-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tegner J, Yeung MK, Hasty J, Collins JJ. Reverse engineering gene networks: integrating genetic perturbations with dynamical modeling. Proc Natl Acad Sci U S A. 2003;100:5944–5949. doi: 10.1073/pnas.0933416100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ståhl PL, Salmén F, Vickovic S, Lundmark A, Navarro JF, Magnusson J, et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science. 2016;353:78–82. doi: 10.1126/science.aaf2403. [DOI] [PubMed] [Google Scholar]
- 34.Moffitt JR, Bambah-Mukku D, Eichhorn SW, Vaughn E, Shekhar K, Perez JD, et al. Molecular, spatial, and functional single-cell profiling of the hypothalamic preoptic region. Science. 2018;362:eaau5324. doi: 10.1126/science.aau5324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Svensson V, Teichmann SA, Stegle O. SpatialDE: identification of spatially variable genes. Nat Methods. 2018;15:343–346. doi: 10.1038/nmeth.4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ramilowski JA, Goldberg T, Harshbarger J, Kloppmann E, Lizio M, Satagopam VP, et al. A draft network of ligand-receptor-mediated multicellular signalling in human. Nat Commun. 2015;6:7866. doi: 10.1038/ncomms8866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grün D, Lyubimova A, Kester L, Wiebrands K, Basak O, Sasaki N, et al. Single-cell messenger RNA sequencing reveals rare intestinal cell types. Nature. 2015;525:251–255. doi: 10.1038/nature14966. [DOI] [PubMed] [Google Scholar]
- 38.Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396–1401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shalek AK, Satija R, Shuga J, Trombetta JJ, Gennert D, Lu D, et al. Single-cell RNA-seq reveals dynamic paracrine control of cellular variation. Nature. 2014;510:363–369. doi: 10.1038/nature13437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pollen AA, Nowakowski TJ, Shuga J, Wang X, Leyrat AA, Lui JH, et al. Low-coverage single-cell mRNA sequencing reveals cellular heterogeneity and activated signaling pathways in developing cerebral cortex. Nat Biotechnol. 2014;32:1053–1058. doi: 10.1038/nbt.2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Proserpio V, Piccolo A, Haim-Vilmovsky L, Kar G, Lönnberg T, Svensson V, et al. Single-cell analysis of CD4+ T-cell differentiation reveals three major cell states and progressive acceleration of proliferation. Genome Biol. 2016;17:103. doi: 10.1186/s13059-016-0957-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kolodziejczyk AA, Kim JK, Svensson V, Marioni JC, Teichmann SA. The technology and biology of single-cell RNA sequencing. Mol Cell. 2015;58:610–620. doi: 10.1016/j.molcel.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 43.Haque A, Engel J, Teichmann SA, Lönnberg T. A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications. Genome Med. 2017;9:75. doi: 10.1186/s13073-017-0467-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Macaulay IC, Haerty W, Kumar P, Li YI, Hu TX, Teng MJ, et al. G&T-seq: parallel sequencing of single-cell genomes and transcriptomes. Nat Methods. 2015;12:519–522. doi: 10.1038/nmeth.3370. [DOI] [PubMed] [Google Scholar]
- 45.Rodriguez-Meira A, Buck G, Clark SA, Povinelli BJ, Alcolea V, Louka E, et al. Unravelling intratumoral heterogeneity through high-sensitivity single-cell mutational analysis and parallel RNA sequencing. Mol Cell. 2019;73:1292–1305.e8. doi: 10.1016/j.molcel.2019.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Han KY, Kim KT, Joung JG, Son DS, Kim YJ, Jo A, et al. SIDR: simultaneous isolation and parallel sequencing of genomic DNA and total RNA from single cells. Genome Res. 2018;28:75–87. doi: 10.1101/gr.223263.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, et al. Simultaneous epitope and transcriptome measurement in single cells. Nat Methods. 2017;14:865–868. doi: 10.1038/nmeth.4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Peterson VM, Zhang KX, Kumar N, Wong J, Li L, Wilson DC, et al. Multiplexed quantification of proteins and transcripts in single cells. Nat Biotechnol. 2017;35:936–939. doi: 10.1038/nbt.3973. [DOI] [PubMed] [Google Scholar]
- 49.Liu Z, Wang L, Welch JD, Ma H, Zhou Y, Vaseghi HR, et al. Single-cell transcriptomics reconstructs fate conversion from fibroblast to cardiomyocyte. Nature. 2017;551:100–104. doi: 10.1038/nature24454. [DOI] [PMC free article] [PubMed] [Google Scholar]