

Abstract
We identified tryptic peptides in yeast cell lysates that map to translation initiation sites downstream of the annotated start sites using the peptide-spectrum matching algorithms OMSSA and Mascot. To increase the accuracy of peptide-spectrum matching, both algorithms were run using several standardized parameter sets, and Mascot was run utilizing a, b, and y ions from collision-induced dissociation. A large fraction (22%) of the detected N-terminal peptides mapped to translation initiation downstream of the annotated initiation sites. Expression of several truncated proteins from downstream initiation in the same reading frame as the full-length protein (frame 1) was verified by western analysis. To facilitate analysis of the larger nroteome of Drosophila, we created a streamlined sequence library from which all duplicated trypsin fragments had been removed. OMSSA assessment using this “stripped” library revealed 171 peptides that map to downstream translation initiation sites, 76% of which are in the same reading frame as the full-length annotated proteins, although some are in different reading frames creating new protein sequences not in the annotated proteome. Sequences surrounding implicated downstream AUG start codons are associated with nucleotide preferences with a pronounced three-base periodicity N1^G2^A3.
Keywords: peptide mass spectrometry, OMSSA, Mascot, streamlined sequence libraries
Graphical Abstract
1. INTRODUCTION
Building our understanding of cell function requires uncovering the repertoire of proteins expressed by each mRNA and dissecting their functional relationships with other cellular components. Recent analyses of ribosome footprinting1,2 and MS/MS data3–7 suggest that proteomes are far richer and more diverse than previously realized. Ribosome footprinting experiments in yeast show signal profiles that outline open reading frames (ORFs) and reveal characteristic profiles at the annotated translation initiation sites as well as at additional upstream and downstream implicated start sites.1,2,8,9 Translation initiation sites downstream of the annotated start sites of many mRNAs have also been revealed in MS/MS analysis of yeast cell lysates, indicating that mRNAs can code for multiple proteins: N-terminally truncated proteins as well as new proteins read in different reading frames.5 Recent MS/MS analysis of human tissue and cell culture lysates enriched for N-terminal peptides revealed N-terminal truncation proteoforms, many of which were accounted for by alternative translation initiation sites or post-translational proteolytic processing.3,4,6,7 Integration of MS/MS and ribosome profiling of human cell lines has also revealed alternative translation start sites.8
Peptide MS/MS experiments output large sets of spectra that can be matched to peptide fragments through peptide-spectrum matching (PSM) algorithms (also described as sequence database search engines). The algorithms take into account that the cell lysate proteins have been digested after cell lysis with an endopeptidase, typically trypsin, which reliably digests the proteins after lysines and arginines (except when flanked by a proline). The algorithms receive as input the sequences of all annotated proteins of the organism under study (the sequence “database” or library), and the algorithms output the computed trypsin fragments that give the best peptide-spectrum matches. By incorporating into the sequence library additional sequences that one suspects might also be expressed in the cell lysate, it is possible to discover proteins not previously annotated as part of the proteome.
This protein discovery strategy relies on having high confidence in PSM by the algorithms. The algorithm stringencies can be tuned to ensure minimal false discovery rates (FDRs) by including reverse decoy sequences in the sequence library and monitoring the false decoy-matching rate.10,11 In addition, we have assessed algorithm performance using a parent-protein profiling approach,12 in which parent proteins prior to trypsin digestion are partitioned through electrophoresis into known size ranges, and the matched peptides are scored according to whether their parent proteins before trypsin digestion were in the correct size ranges. Using this approach, we have shown that different PSM algorithms, SEQUEST,13 Open Mass Spectrometry Search Algorithm14 (OMSSA), and Mascot,15 provide overlapping yet different high-confidence samplings of detected peptides in lysates. We have found12 that the confidence in OMSSA and Mascot matches can be increased by running the algorithm with multiple sets of parameter choices (parameters for mass tolerances, etc.) and only retaining matches detected with two or more of the standardized parameter sets. We have also found that confidence in Mascot matches can be increased by screening for a, b, and y ions (the ions from collision-induced dissociation (CID) of trypsin fragments) rather than screening for just b and y ions, the protocol normally applied.
Using these strategies to improve algorithm performance, we examined peptides from yeast cell lysates that map to translation initiation start sites located downstream of the standard annotated sites. We previously used the SEQUEST algorithm to examine downstream start sites within 100 nt of the annotated sites and found that 35% of the detected amino-terminal peptides mapped to the downstream sites.5 We have now expanded considerably the set of detected peptides that map to downstream start sites using high-confidence matching by the OMSSA and Mascot algorithms.
Analysis of budding yeast is simplified by its relatively small proteome and the fact that yeast have very few cases of alternative splicing. In contrast, alternative splicing in higher organisms such as Drosophila leads to a considerably bigger proteome.16 Hence the (redundant) sequence space to be interrogated by MS/MS algorithms can be much larger and lead to slower and less sensitive screening of spectra as well as potential memory overload problems. For example, taking into account splice isoforms, the annotated proteome of Drosophila has a combined length of 31.5 million amino acids as compared to 6.2 million amino acids for yeast. However, largely due to alternative splicing, many stretches of protein sequence are present multiple times in the sequence space for interrogation. To address this sequence redundancy, we tested a new alternative approach in which PSM algorithms were presented with a sequence library from which all duplicates of trypsin fragments were removed resulting in a much smaller sequence space (57% reduction for Drosophila), which we refer to as a “stripped” sequence library. We found that screening yeast spectra with the stripped library gave a largely overlapping but somewhat different set of PSMs of similar high confidence. Stripped libraries for yeast and Drosophila were used to detect peptides that map to translation initiation downstream of annotated start sites.
2. METHODS
2.1. Processing of PSM Algorithm Outputs
Spectra were analyzed from 47 PeptideAtlas data sets for yeast cell lysates (Supplementary Table 1).17 We also analyzed a data set from Fournier et al.,5 in which cell lysates had been treated with glutaraldehyde to increase detection of amino-termini of proteins.18 The OMSSA and Mascot algorithms were run with standardized parameter sets (Supplementary Table 2)12 using a 52-node (24 GB/node; 104 core) cluster (OMSSA) and a Dell XPS 8300 server (Mascot).
We used a standard sequence “database” or library of all of the currently annotated proteins in Saccharomyces cerevisiae. As previously described,5 we appended to this library all theoretical proteins that could initiate translation at an AUG within 100 nucleotides downstream of the annotated start site (down-Peptides). Appended downPeptides that were in-frame with the full-length annotated protein were truncated after the first trypsin site. We also included downPeptides from which the N-terminal methionine was cleaved if the next amino acid was A, C, S, T, G, V, or P because these methionines are commonly cleaved by amino peptidases.19 In addition, we included versions of downPeptides from which signal sequences predicted by SignalP20 were removed. The resulting sequence library had 6.2 million amino acids of annotated protein sequence and 0.9 million amino acids of downPeptide sequence.
We matched to b and y ions when running OMSSA. However, for Mascot, we included a, b, and y ions because parent protein conformance tests indicate that the small fraction of PSMs detected by b/y but not a/b/y screening have low parent protein conformance (Figure 1).12
Figure 1.
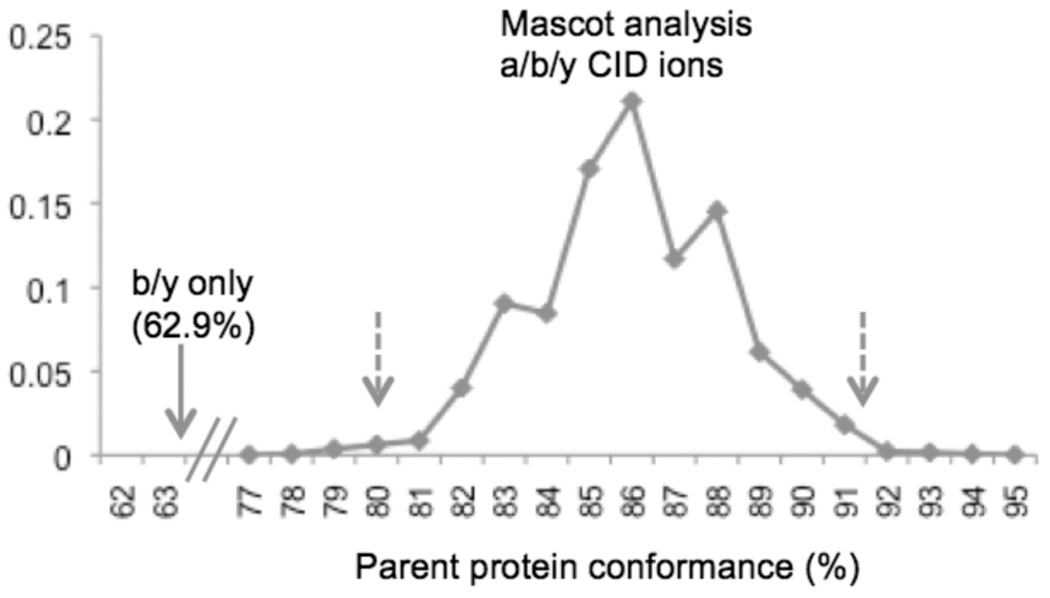
Mascot is significantly more accurate when screening a, b, and y ions. In assessment of conformance to parent proteins before trypsin digestion,12 all 4560 peptides detected by Mascot screening for a, b, and y CID ions were also detected when screening for only b and y ions. However, the 264 additional peptides detected only in the b/y screen had very poor conformance to parent protein MWs (62.9%; arrow). Bootstrap analysis with 2000 samples of 264 peptides (with replacement) from the a/b/y screen revealed 99% confidence limits between 79.5 and 90.9% (broken arrows). This confirms that the poor b/y conformance of 62.9% is significantly lower than the a/b/y conformance.
Peptide-spectrum match data sets from OMSSA and Mascot were uploaded into an MS SQL database and processed as follows:
Remove peptides with internal trypsin sites; the algorithms were run allowing one internal trypsin site, but these were subsequently filtered out. This step ensures that spectra that match well to a peptide with an internal trypsin site are not incorrectly matched to other peptides with much poorer scores.
Compute 1 and 5% FDR threshold score (where FDR = no. reverse decoy peptides/no. forward peptides)10,11 for each standardized parameter set of each PeptideAtlas (PAe) experimental series, counting each peptide no more than once per MS/MS experiment.
Remove PSMs with scores below the 1 or 5% FDR threshold from reverse decoy analysis;10,11 because of the high default stringency of the OMSSA algorithm, the actual decoy FDR was 1.2% when 5% FDR thresholds were applied.
Only retain peptides unique to one gene.
Only retain PSMs that were detected with two or more standardized parameter sets.
The Mascot algorithm reports the ranks of all matches to the same spectrum, and we only retained the highest ranking (rank-1) matches and only rank-1 matches were used to compute the FDR thresholds. Although the OMSSA algorithm does not report ranks of matches, for our computation of FDR thresholds and assessment of matches to peptides, we excluded all peptide-spectrum matches where another peptide matched the spectrum with a better score.
High-confidence subsets of the presented PSMs are notated (in Supplementary Data Files 2, 3, and 5) as conforming to 1% FDR thresholds computed using rank-1 PSMs from OMSSA for all peptides with no internal trypsin sites (internal and N- or C-terminal trypsin fragments). We also illustrate even higher stringency data subsets with scores >10-fold better than the 1% FDR thresholds. The equivalent scoring thresholds were also computed for the Mascot data.
Annotated spectra from Mascot outputs were obtained using Mascot Daemon2.4.0.15 Selected experiments from PeptideAtlas data sets were rerun using SearchGUI2.2.221 and Peptide-Shaker1.2.2 to obtain annotated spectra from OMSSA.22 Samples of annotated spectra are presented according to the same FDR threshold stringencies discussed above (Supplementary Data Files 6, 7, and 8).
2.2. Creating a Stripped Library
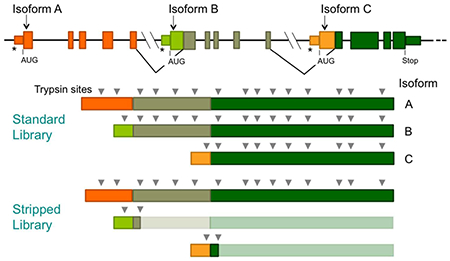
To reduce the size of the sequence library to be screened by PSM algorithms, we created a “stripped” library from which all duplicated trypsin fragments had been removed (see Results and Discussion and Figure 3; Supplementary Files 9 and 10). A Python script screened all theoretical trypsin fragments of the library and removed duplicates concatenating the sequences on either side of the removed fragment. Although concatenation created artificial sequences, these sequences could not be present in the final set of spectrum-matched peptides because peptides with an internal trypsin site were filtered out during the processing of the algorithm outputs.
Figure 3.
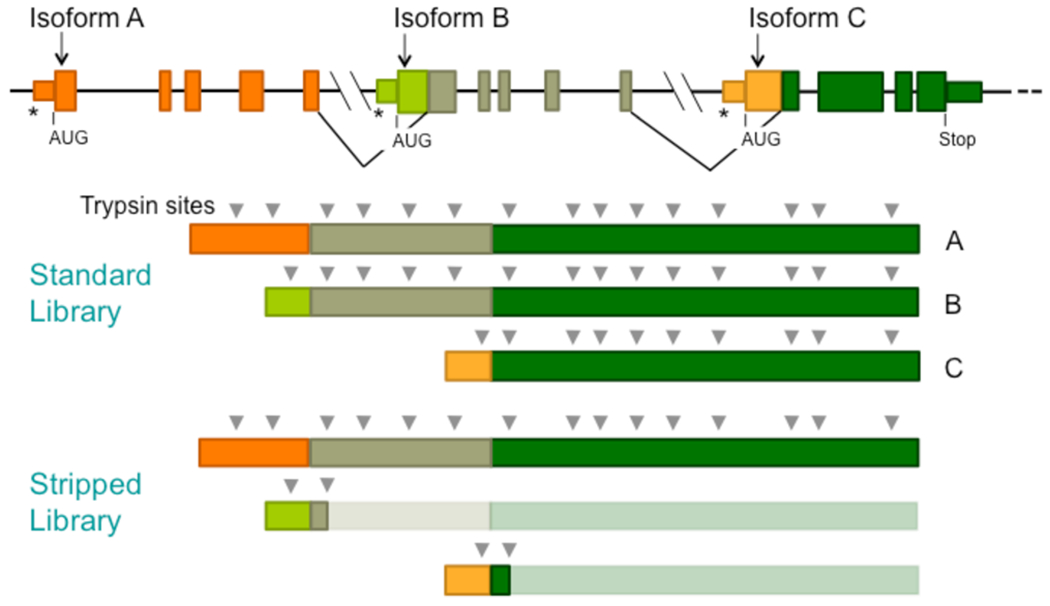
Stripped sequence libraries facilitate MS/MS analysis of large proteomes. Sequence libraries stripped of duplicated trypsin fragments give much smaller search spaces for PSM algorithms. The mRNAs from alternative splicing produce proteins with high sequence redundancy, most of which is removed when duplicated trypsin fragments are removed from the proteins of the sequence library. Alternative transcription start sites are shown (*).
The deleted trypsin fragments were entered into a database lookup table that recorded the identities of each protein in which they were present and the trypsin fragment coordinates in these proteins. The lookup table was used in subsequent processing to determine if matched peptides were present in more than one protein isoform from a single gene or multiple genes.
The OMSSA algorithm was run exactly as previously described except using the stripped library and newly computed 1 and 2% FDR decoy thresholds. DownPeptides were appended to the stripped library as previously described. Initial testing of the yeast stripped library was performed using parent-protein conformance tests with the spectrum data set of Lin et al.12 As expected, OMSSA run times were shorter with the smaller stripped library. Run times were typically 25% shorter with the stripped yeast library, which is 17% smaller than the standard library. Apparent memory issues prevented us from running large-scale tests of the standard (nonstripped) library of Drosophila. Testing of individual PeptideAtlas experiments using SearchGUI2.2.2 showed that run times were 80% longer using the standard library.
3. RESULTS AND DISCUSSION
3.1. Detection of DownPeptides
The PSM algorithms used in MS/MS analysis are based on techniques such as cross-correlation (e.g., SEQUEST13) or model-based approaches using statistical significance (OMSSA,14 Mascot15). Although the different algorithms detect many of the same peptide-spectrum matches, each algorithm also outputs matches not detected by the others. Indeed, even different settings of algorithm parameters give rise to partially overlapping sets of matches. Using decoy10,11 and parent-protein12 analyses, we have developed protocols for applying OMSSA and Mascot that result in sets of high-confidence PSMs. Specifically, both algorithms were run using multiple sets of standardized parameters (Supplementary Table 2),12 and we retained only those PSMs detected by two or more parameter sets. In addition, for Mascot, we screened for a, b, and y CID ions, a departure from the standard screening of just b and y ions. Peptides matched by the b/y but not the a/b/y screen had significantly lower conformance to their parent proteins before trypsin digestion12 (Figure 1). We used these approaches to screen for translation initiation events downstream of the annotated translation start codons. We analyzed 48 data sets of spectra (PeptideAtlas17 and Fournier et al. 2012;5 Supplementary Table 1A), expanding significantly on our previous analysis of 21 data sets using just the SEQUEST algorithm.5
Translation initiation at downstream start sites gives rise to diagnostic N-terminus-derived trypsin fragments (downPeptides) that are not created when the longer annotated protein is digested with trypsin.5 We screened for downPeptides that map to AUG start sites within 100 nucleotides downstream of the annotated start codon. We detected 138 downPeptides and 478 annotated start peptides (annPeptides) using 1% FDR thresholds; 226 downPeptides and 631 annPeptides were detected with slightly less stringent 5% FDR thresholds (Supplementary Files 1, 2, and 3). It is striking that the downPeptides represented 22 to 26% of all the detected N-terminal peptides, suggesting that downPeptide expression is quite common in yeast, as previously discussed.5 31 of the downPeptides detected by OMSSA or Mascot (5% FDR) were also detected previously using the SEQUEST algorithm. 42 annPeptides and 7 downPeptides were detected by both OMSSA and Mascot.
Confidence in peptide-spectrum matches is based on false-detection rates calculated for each PeptideAtlas data set. Because these are average rates for each data set, the peptide-spectrum matches closer to the FDR thresholds have lower than the average confidence, and those further from the threshold have higher confidence.12 The distance measures defined and presented in Supplementary File 1 show the distance from the 5% FDR threshold for the best-scoring PSM of each down-Peptide.
As previously observed in our SEQUEST screen,5 although the downPeptide ORFs (downORFs) were detected in all three reading frames, for all three algorithms there is a tendency for the downORFs to be in the same reading frame as the gene’s annotated protein (frame 1), resulting in N-terminal truncated proteins (Table 1). However, many of the detected downPeptide ORFs (52%) are in frames 2 or 3, which translate into different amino acid sequences from the annotated frame-1 proteins. The frames 2 or 3 ORFs are significantly longer than would be expected from randomly chosen downORFs (Figure 4). Indeed the median length of ORFs for downPeptides detected by OMSSA or Mascot was 66, suggesting selection against stop codons.
Table 1.
DownPeptide Reading Frames
| yeast | Frame 1 | Frame 2 | Frame 3 |
|---|---|---|---|
| detected downPeptides (OMSSA) | 0.52a | 0.40 | 0.08 |
| detected downPeptides (Mascot) | 0.46a | 0.50 | 0.04 |
| all downAUGs within 100 nt of annAUG | 0.34 | 0.54 | 0.12 |
| Drosophila | Frame 1 | Frame 2 | Frame 3 |
| detected downPeptides | 0.73a | 0.20 | 0.06 |
| all downAUGs within 100 nt of annAUG | 0.44 | 0.43 | 0.13 |
Chi-square tests indicated that frame-1 downPeptide frequencies are significantly elevated compared to all downAUGs within 100 nt of the annAUG and with downORF > 12 nt (yeast Mascot: p = 0.014, 5% FDR; yeast OMSSA: p = 3.10 × 10−5, 5% FDR; Drosophila: p = 2.15 × 10−25, 2% FDR).
Figure 4.
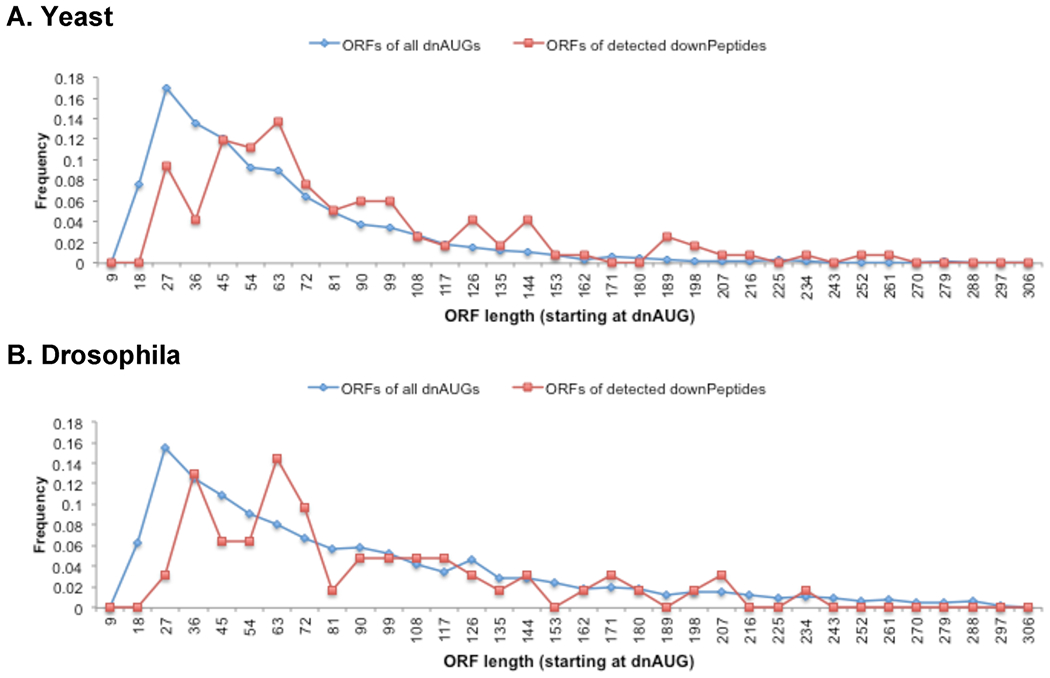
Lengths of detected frame-2 and -3 downORFs compared with all frame-2 and −3 ORFs initiated within 100 nucleotides downstream of the annAUG (and ≥15 nucleotides long). The ORFs for downPeptides detected in yeast (A) are longer than expected by random selection (chi-square goodness of fit p < 0.01). Although generally longer, the Drosophila downORFs (B) are not significantly longer by chi-square test. Illustrated are ORF lengths for downPeptides detected by OMSSA or Mascot with the standard yeast library (A; 5% FDR) and OMSSA with the stripped Drosophila library (B; 2% FDR).
Although we screened for downPeptides that map to within 100 nucleotides of the annAUG, some of the frame-1 downAUGs were sufficiently downstream for their truncated protein to be detectable in western analysis as distinct from the full-length protein. Figure 2 shows examples of downPeptide genes that express both the truncated and full-length proteins, either simultaneously or under different growth conditions. One of our downPeptide matches, DOT1, has been described independently as a likely case of leaky translation initiation.23
Figure 2.
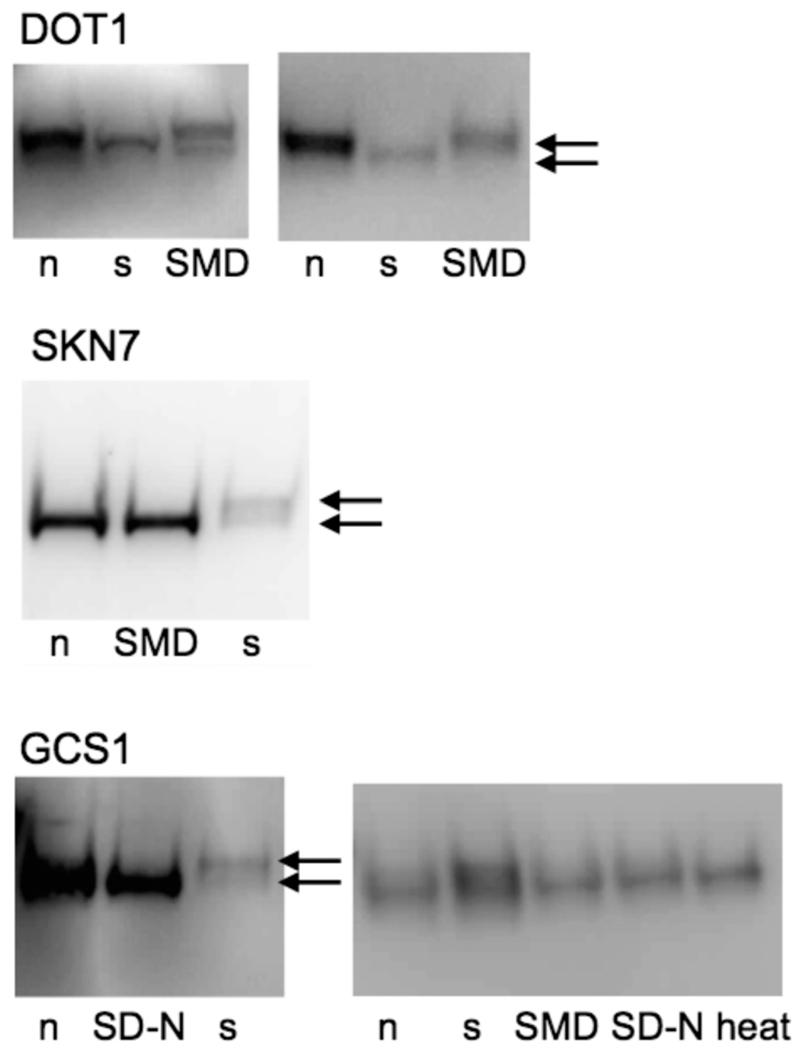
Western analysis of frame-1 truncated proteins. DownPeptide genes with predicted frame-1 truncated proteins were tested in TAP-epitope-tag expression lines (DOT1, SKN7, GCS1). Cells were grown under various conditions: normal (n), stationary (s), synthetic minimal medium with glucose (SMD), starved in synthetic minimal medium lacking nitrogen (SD-N), or heat-treated (37 °C, 15 min). Five experiments are illustrated where truncated, and full-length proteins (arrows) were detected under different conditions. A truncated protein of DOT1 was also previously reported.23 Note that both the annPeptide and downPeptide were detected by MS/MS for DOT1, but only the downPeptide was detected by MS/MS for SKN7. The downPeptide for GCS1 was only detected with one OMSSA parameter set. The detected truncated and full-length proteins ran appropriately according to MW size markers (not shown).
3.2. Optimizing the Sequence Library
Having detected high frequencies of downPeptides in budding yeast, we were interested to assess whether this is also a property of higher organisms such as Drosophila. However, this is a challenge given that higher organisms have more complex proteomes that result from larger genomes and alternative splicing, and this can increase drastically the sequence space to be searched by PSM algorithms. This problem has been addressed previously by creating sequence libraries that contain a small number of diagnostic peptides representing a large portion of the proteome, an approach that significantly reduces the search space and allows confident identification of proteins but does not provide comprehensive peptide coverage.24 We wished to develop an alternative approach that would provide fuller coverage of the proteome and reduce the chances that a matched spectrum actually has a better peptide-spectrum match to an annotated protein’s peptide that had been removed from the sequence library. Other approaches have been taken to speed up searches using indexed peptide libraries,25,26 which can reduce the sizes of the searched sequence libraries.
In this study, we created a yeast sequence library (stripped sequence library; Figure 3) in which blocks of one or more trypsin fragments present more than once in the proteome were deleted from all but one of the protein sequences. Hence, duplicate trypsin fragments were only represented once in the library and were also entered into a database lookup table that recorded the identities of each protein in which they were present and the trypsin fragment coordinates. This reduced the number of amino acids in the yeast library of annotated proteins by 17%. The stripped library of Drosophila showed an even more dramatic reduction of 57%. A set of downPeptides was appended to each stripped library as described later.
We compared the performance of the OMSSA algorithm with the yeast stripped and standard sequence libraries using spectrum data for which parent proteins had been separated into different size ranges before trypsin digestion.12 When the algorithm outputs were filtered using 5% FDR thresholds based on decoy analysis, the stripped library detected more peptides (1973) than the standard library (1840) (Table 2A). Although filtered at 5% FDR, the actual FDRs were lower (0.5 to 1.7% for the standard library and 0.9 to 1.7% for the stripped library) due to the high stringency of the default implementation of the OMSSA algorithm. Consistent with this, conformance to parent proteins was slightly lower for the stripped library (86.2%) compared with 87.1% for the standard library (Table 2A). 1578 of the 1840 peptides detected with the standard library were also detected with the stripped library (Table 2A). This suggests that the stripped and standard libraries provide different but largely overlapping samplings of high-confidence peptide matches. Our analyses below of yeast and Drosophila stripped libraries were performed using 1 and 2% FDR filters.
Table 2.
Peptides Detected by Standard and Stripped Libraries
| A. Yeast | |||
|---|---|---|---|
| FDR threshold | standard library (conformance) | stripped library (conformance) overlap | stripped library (conformance) overlap |
| 5%a | 1840 (87.1%) | 1973 (86.2%) | 1578 |
| 1% | 1838 (87.1%) | 1920 (86.3%)b | 1574 |
| B. Yeast | |||
| N-terminal AnnPeptides (FDR) | standard (5%) | stripped (2%) | |
| total distinct | 464 | 509 | |
| uniquec | 16 | 51 | |
| overlapd | 448 | ||
| N-terminal DownPeptides (FDR) | standard (5%) | stripped (2%) | |
| total distinct | 139 | 149 | |
| uniquec | 27 | 37 | |
| overlapd | 112 | ||
| C. Yeast and Drosophila | |||
| species library (FDR) | yeast stripped (2%) | Drosophila stripped (2%) | |
| N-terminal AnnPeptides | 509 | 504 | |
| N-terminal DownPeptides | 149 | 178 | |
Because of the default stringency of the OMSSA algorithm, the 5% threshold data had actual decoy FDRs of 0.5 to 1.7% (standard library) and 0.9 to 1.7% (stripped library).
Bootstrap analysis of 1000 samplings with replacement of the 1920 peptides showed 95% confidence limits between 85.6 and 87.1%.
Unique: distinct peptides in either standard or stripped library.
Overlap: distinct peptides detected with both standard and stripped library.
When applied to the 48 data sets (from PeptideAtlas17 and Fournier et al.5), the stripped library (containing annotated proteins and downPeptides) detected sets of annPeptides and downPeptides that overlapped with the sets detected by the standard library (Table 2B). Hence, utilizing the stripped library expanded the set of detected downPeptides, which are presented in Supplementary File 1.
For both the standard and stripped libraries, we selected rank-1 peptides with no internal trypsin sites. The selected peptides had to win against other peptides with 0 or 1 internal trypsin sites, including the artificial junction fragments created during stripping. Because the competing peptides did not include many peptides that could be present in the cell lysates (e.g., from unscreened post-translation processing) and the competing sets in the stripped and standard libraries differed, the confidence in detected peptide-spectrum matches relied on the decoy analysis and in many cases visual inspection of individual annotated spectra (Supplementary Files 6 and 7).
3.3. DownPeptide Detection in Drosophila melanogaster
The stripped library for annotated Drosophila proteins contained 13.6 million amino acids and was considerably smaller than the standard library of 31.5 million amino acids. We appended to the stripped library downPeptides for all AUG codons within 100 nucleotides downstream of the annotated AUG start codons. This increased the stripped library size by 890 512 amino acids. Only the first trypsin fragment (>4 amino acids) was included for each protein, initiating at a downAUG in the same reading frame (frame 1) as the main protein product.
The stripped library with downPeptides was used to screen with OMSSA 53 Drosophila data sets deposited at Peptide Atlas (Supplementary Table 1B). We detected 494 annPeptides using 1% FDR thresholds (546 peptides with 2% FDR), of which a large fraction (70%, 1% FDR; 66% 2% FDR) were missing their N-terminal methionine presumably due to aminopeptidase cleavage of methionine, which often occurs after A, C, S, T, G, V, or P.19 We detected 171 downPeptides using 1% FDR (230 downPeptides, 2% FDR), which corresponds to 26% (30%, 2% FDR) of the detected N-terminal peptides (Table 2C; Supplementary Files 4, 5, and 8), suggesting that, like yeast, translation initiation at downAUGs is quite common in Drosophila. Moreover, 76% (73% for 2% FDR) of the detected downPeptide ORFs were in frame-1, significantly higher than would be predicted by random sampling of the screened theoretical downPeptides (Table 1), although unlike yeast, the frame 2 and 3 downPeptides did not show significant selection for long downORFs (Figure 4). Translation initiation at downAUGs may result from leaky ribosome scanning past the annAUG. In addition to leaky scanning, translation initiation at downAUGs may also be a consequence of alternative transcription initiation or splicing that gives rise to unannotated transcripts that lack the annAUG.
We investigated whether the observed preference for frame-1 initiation at downAUGs could be related to 3-nucleotide periodicities encountered while scanning the full-length protein’s ORF. It has been suggested previously that the three-base periodicity observed in ORFs may help stabilize secondary structures of mRNAs27 as well as contribute to mRNA base pairing with bacterial rRNA sequences.28 Examination of sequences upstream and downstream of aligned frame-1 downAUGs revealed depression of G and A at positions 2 and 3 of codons, respectively. This G2A3 depression was significantly more pronounced downstream of the implicated downAUGs of frame-1 downPeptide genes (Figure 5A,C) compared with randomly selected downAUGs. Depression of G2 and A3 was also significantly more pronounced upstream of the downAUGs. This depression of G and A at positions 2 and 3 of codons suggests that ribosomes have a preference for alternative nucleotides at these positions (N1^G2^A3) and that this may influence which downAUGs initiate translation. Moreover, we found that detected frame-2 downPeptide genes had strong preferences for depression of G at position 2 of the frame-2 codons immediately downstream of the aligned downAUGs (N1^G2N3; Figure 5B,C). Bootstrap analysis confirmed that this depression in G was significantly higher than in samples of randomly selected frame-2 downAUG regions. Position 2 in frame-2 codons corresponds to the wobble third position in frame 1 and may therefore be more free to evolve away from G. In contrast, neither of the codon positions 2 or 3 of frame-3 downPeptide ORFs correspond to the frame-1 wobble position, and this could account in part for the underrepresentation of frame-3 downPeptides compared with frames 1 and 2.
Figure 5.
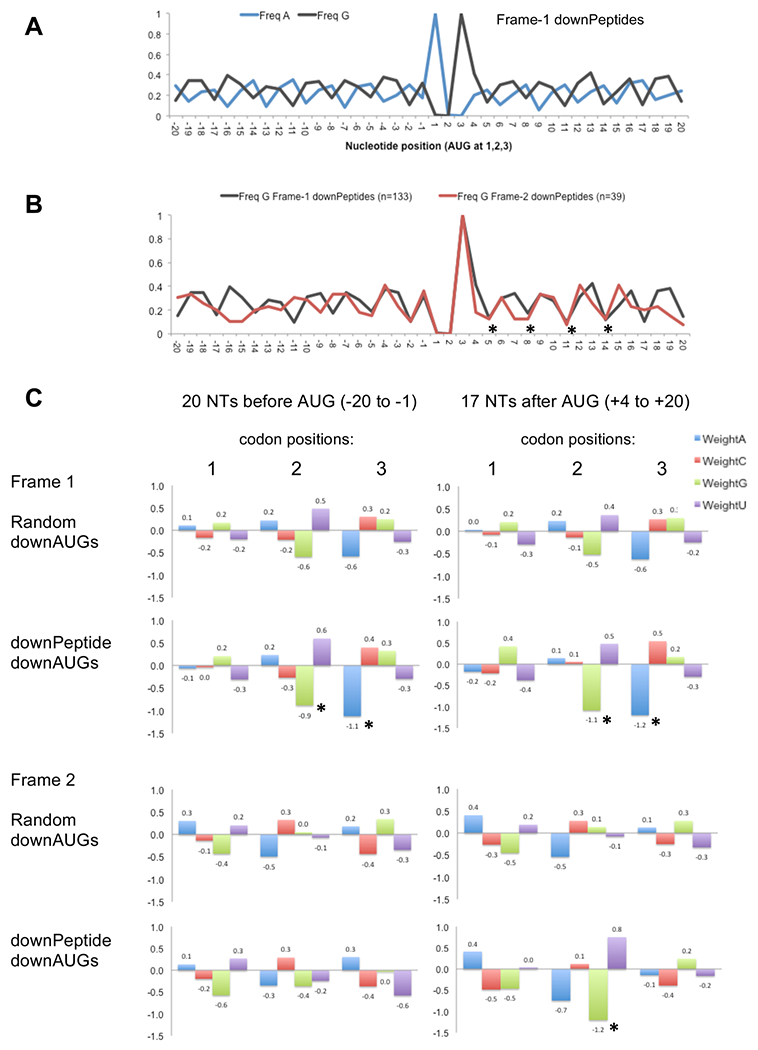
(A) Sequences flanking the implicated frame-1 downAUGs of Drosophila downPeptide genes have a pronounced 3-nucleotide periodicity with depression of G at position 2 and A at position 3 of the codons. Average nucleotide frequencies of aligned sequences are shown relative to the downAUG at positions 1, 2, and 3. (B) Frequencies of G are depressed at the second nucleotide of codons downstream of the AUG of frame-1 and frame-2 downPeptide ORFs (*) despite the frame-2 ORF being out of frame with the overlapping frame-1 ORF. (C) Average deviations from background, measured as log2(freqobs/freqbackground), are illustrated for codon positions 1, 2, and 3 for windows upstream (positions −20 to −1) and downstream (positions +4 to +20) of the start codons of frame-1 and frame-2 downPeptide ORFs (based on background nucleotide frequencies in ORFs; fA: 25.6%, fC: 27.1%, fG: 26.8%, fU: 20.5%). Compared with random samples of 1000 downAUGs, frame-1 downPeptides show G2 and A3 depression at positions 2 and 3 of codons upstream and downstream of the start codon (*). Frame-2 downPeptides show G2 depression (*) downstream of the start codon at positions corresponding to the wobble position of frame-1. The G2 and A3 depressions (*) are significant by bootstrap analysis (p < 0.01). Only downPeptides (2% FDR) with downAUGs > 20 nucleotides downstream of the annAUG were used in this analysis.
Assessment of Gene Ontology terms (Supplementary Table 3; Supplementary Figure 1) indicates that the Drosophila frame-1 downPeptide genes, but not frame-2/3 genes, show enrichment for some Gene Ontology (GO) terms above background levels. Although Drosophila downPeptides were detected in multiple tissue types (Supplementary Table 1B), the overrepresented cellular function GO terms (Supplementary Figure 1) included terms related to retinal cell-programmed cell death. We examined 13 downPeptide genes associated with eye development (Table 3). Several encode transcription factors or GTPases and some are involved in cellular processes including vesicle formation and endocytosis. The predicted truncated regions of these proteins overlap or are close to conserved or functional domains (Table 3). For example, the truncated form of the Irregular chiasm C-roughest (rst-RA) protein would have its signal peptide domain deleted and not be imported into the ER. Similarly, the predicted amino truncations ofEchinus splice form 3 (ec-RA), Ras-related protein Ral-a (Rala-RB), and ADP-ribosylation factor 1 (Arf79f-RA) partially overlap the most amino-terminal portions of their IG-like, ras, and Arf domains, respectively. In contrast with the frame-1 downPeptides, the Drosophila frame-2/3 downPeptides genes did not have enrichment for any GO terms. Whether the newly identified proteins from downAUG initiation contribute to the same or new functions of genes awaits future studies.
Table 3.
Truncated Drosophila Proteins Involved in Eye Development
| domainb |
||||||
|---|---|---|---|---|---|---|
| gene (mRNA) | truncation length (aa) | sourcea | domain | start (aa) | end (aa) | protein class |
| ec-RA | 29 | Prosite | IG-like | 21 | 120 | ubiquitin protein ligase |
| bun-RD | 21 | transcription factor | ||||
| Mitf-RA | 26 | basic helix–loop–helix transcription factor | ||||
| Rala-RB | 31 | Pfam | Ras | 13 | 174 | small GTPase |
| futsch-RC | 25 | microtubule associated protein | ||||
| exd-RA | 14 | homeodomain transcription factor | ||||
| Arf79F-RA | 21 | Pfam | Arf | 4 | 177 | small GTPase involved in vesicle formation |
| grh-RM | 24 | transcription factor | ||||
| rin-RA | 2 | Pfam | NTF2 | 16 | 129 | signaling molecule and RNA binding protein |
| shi-RB | 21 | Pfam | Dynamin N | 29 | 202 | dynamin protein essential for vesicle formation in endocytosis |
| rst-RA | 31 | sig_p | signal peptide | 1 | 19 | transmembrane adhesion protein |
| Hsp83-RA | 17 | Pfam | HATPase_c | 27 | 181 | Hsp90 family chaperone |
| InterPro | HATPase_C | 6 | 214 | |||
| arm-RE | 10 | β-catenin adhesion and signaling protein | ||||
Database sources for domain prediction: Prosite (http://prosite.expasy.org),29 Pfam (http://pfam.xfam.org),30 SignalP (sig_p; http://www.cbs.dtu.dk/services/SignalP-4.0),31 and InterProScan (InterPro; http://www.ebi.ac.uk/interpro).32
Protein domains that overlap or are within 15 amino acids of the region deleted in truncated proteins from translation initiation at downAUGs.
3.4. Conclusions
We conclude that translation at downstream AUGs is common in both yeast and Drosophila. Using a stripped library, it has been possible to screen mass spectra from Drosophila, which has a large annotated proteome due in part to alternative splicing. This screen revealed amino-terminal trypsin fragments that map to translation initiation downstream of the annotated start sites, suggesting that downstream initiation may be a property common to higher organisms and that proteomes are more complex than generally assumed. This conclusion is supported by recent MS/MS analyses of several human cell lines and tissues.3,4,6–9 A three-nucleotide periodicity in the region following the downAUG, with especially pronounced depression of G occurrences in the second codon position, may facilitate downstream initiation and favor the predominance of translation from frame-1 downAUGs.
Supplementary Material
Supplementary Table 1A. Analyzed yeast datasets from PeptideAtlas. Supplementary Table 1B. Analyzed Drosophila datasets from PeptideAtlas. Supplementary Table 2. Standardized parameter settings for three algorithms. Supplementary Table 3. Enriched GO terms associated with Drosophila Frame-1 downPeptide genes. Supplementary Figure 1. Scatterplot showing Gene Ontology terms that are enriched above background for Drosophila frame-1 downPeptide genes. (PDF)
Supplementary File 1. Summary of yeast N-terminal annPeptides and downPeptides detected with Mascot, OMSSA, and SEQUEST PSM algorithms. OMSSA was used with standard and stripped sequence libraries. Numerical entries for each algorithm indicate a peptide’s maximum PSM confidence (d) where d = −log10(PSM score/FDR threshold), which measures the relative distance between an algorithm PSM score and the decoy FDR confidence threshold (using the following PSM scores: SEQUEST probability score, OMSSA e-value score, Mascot peptide score × 0.1; Lin et al.12). A confidence value of 2, for example, indicates the peptide’s best PSM score is 100 times better than the poorest acceptable threshold score computed from decoy analysis. A 5% FDR threshold was used for all three algorithms except OMSSA_stripped, where FDR = 2%. The reference entries show yeast gene id, the start and stop coordinate of the downORF (where the annAUG is at 1001), the frame (1, 2, or 3), and optionally whether the N-terminal methionine is cleaved (trimmed) or whether a signal sequence was cleaved (cleavedNTerm at the ith amino acid). (XLSX)
Supplementary File 2. Yeast peptide-spectrum matches by Mascot using the standard sequence library at 5% FDR. Higher stringency subsets of downPeptide matches are illustrated that conform to 1% FDR. Also illustrated is the subset of PSMs with peptide-matching scores 10 times better than the 1% FDR threshold. Included are the Peptide-Atlas experiment (PAe), spectrum (dta), number of algorithm parameter sets (runcount). Matched peaks of spectra for downPeptides are listed. Supplementary Files 3 and 5 also show this information. (XLSX)
Supplementary File 3. Yeast peptide-spectrum matches by OMSSA using the standard sequence library at 5% FDR. Subsets of downPeptides are shown for 1% FDR and are 10 times better than these thresholds. (XLSX)
Supplementary File 4. Summary of Drosophila N-terminal annPeptides and downPeptides detected by OMSSA. The reference entries show transcript and protein names followed by the downAUG location (measured in nucleotides after the annAUG), the frame (1, 2 or 3), and optionally whether the N-terminal methionine is cleaved (trimmed). (XLSX)
Supplementary File 5. Drosophila peptide-spectrum matches by OMSSA using the stripped sequence library at 2% FDR. Subsets of downPeptides are shown for 1% FDR and are 10 times better than these thresholds. (XLSX)
Supplementary File 6a. Samples of annotated spectra of yeast downPeptides detected by OMSSA. Subsets of spectra are presented conforming to different stringencies: 5% FDRbut not 1% FDR. Supplementary Files 7 and 8 are similarly presented. (ZIP)
Supplementary File 6b. Samples of annotated spectra of yeast downPeptides detected by OMSSA: 1% FDR but not 10 times better. (ZIP)
Supplementary File 6c. Samples of annotated spectra of yeast downPeptides detected by OMSSA: at least 10-fold better scores than 1% FDR thresholds. (ZIP)
Supplementary File 7a. Annotated spectra of yeast downPeptides detected by Mascot: 5% FDR but not 1% FDR. (ZIP)
Supplementary File 7b. Annotated spectra of yeast downPeptides detected by Mascot: 1% FDR but not 10 times better. (ZIP)
Supplementary File 7c. Annotated spectra of yeast downPeptides detected by Mascot: at least 10-fold better scores than 1% FDR thresholds. (ZIP)
Supplementary File 8a. Samples of annotated spectra of Drosophila downPeptides detected by OMSSA showing the standard 2% FDR. (ZIP)
Supplementary File 8b. Samples of annotated spectra of Drosophila downPeptides detected by OMSSA showing higher stringencies: 1% FDR but not 10 times better. (ZIP)
Supplementary File 8c. Samples of annotated spectra of Drosophila downPeptides detected by OMSSA showing higher stringencies: at least 10-fold better scores than 1% FDR thresholds. (ZIP)
Fasta sequence library with full yeast proteome plus all downPeptides within 100 nucleotides of annotated start sites. (ZIP)
Fasta sequence library with full Drosophila proteome plus all downPeptides within 100 nucleotides ofannotated start sites. (ZIP)
ACKNOWLEDGMENTS
We thank Scott Holmes, Amy MacQueen, Ruth Johnson, and Mary Vallo for discussions. This work was supported in part by funds from the Howard Hughes Medical Institute to support undergraduate initiatives in the life sciences, NSF grant CNS-0959856, and NIH grant 1R15GM096228.
ABBREVIATIONS:
- FDR
false discovery rate
- MS/MS
tandem mass spectrometry
- CID
collision-induced dissociation
- OMSSA
open mass spectrometry search algorithm
- PSM
peptide spectrum match
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jproteome.5b00996.
The authors declare no competing financial interest.
REFERENCES
- (1).Ingolia NT; Ghaemmaghami S; Newman JR; Weissman JS Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 2009, 324 (5924), 218–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Ingolia NT; Lareau LF; Weissman JS Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell 2011, 147 (4), 789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Eckhard U; Marino G; Abbey SR; Tharmarajah G; Matthew I; Overall CM The Human Dental Pulp Proteome and N-Terminome: Levering the Unexplored Potential ofSemitryptic Peptides Enriched by TAILS to Identify Missing Proteins in the Human Proteome Project in Underexplored Tissues. J. Proteome Res 2015, 14 (9), 3568–82. [DOI] [PubMed] [Google Scholar]
- (4).Fortelny N; Pavlidis P; Overall CM The path of no return–Truncated protein N-termini and current ignorance of their genesis. Proteomics 2015, 15 (14), 2547–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Fournier CT; Cherny JJ; Truncali K; Robbins-Pianka A; Lin MS; Krizanc D; Weir MP Amino termini of many yeast proteins map to downstream start codons. J. Proteome Res 2012, 11 (12), 5712–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Lange PF; Huesgen PF; Nguyen K; Overall CM Annotating N termini for the human proteome project: N termini and Nalpha-acetylation status differentiate stable cleaved protein species from degradation remnants in the human erythrocyte proteome. J. Proteome Res 2014, 13 (4), 2028–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Prudova A; Serrano K; Eckhard U; Fortelny N; Devine DV; Overall CM TAILS N-terminomics of human platelets reveals pervasive metalloproteinase-dependent proteolytic processing in storage. Blood 2014, 124 (26), e49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Koch A; Gawron D; Steyaert S; Ndah E; Crappe J; De Keulenaer S; De Meester E; Ma M; Shen B; Gevaert K; Van Criekinge W; Van Damme P; Menschaert G A proteogenomics approach integrating proteomics and ribosome profiling increases the efficiency of protein identification and enables the discovery of alternative translation start sites. Proteomics 2014, 14 (23–24), 2688–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Lee S; Liu B; Lee S; Huang SX; Shen B; Qian SB Global mapping of translation initiation sites in mammalian cells at single-nucleotide resolution. Proc. Natl. Acad. Sci. U. S. A 2012, 109 (37), E2424–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Elias JE; Gygi SP Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 2007, 4 (3), 207–14. [DOI] [PubMed] [Google Scholar]
- (11).Fitzgibbon M; Li Q; McIntosh M Modes of inference for evaluating the confidence of peptide identifications. J. Proteome Res 2008, 7 (1), 35–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Lin MS; Cherny JJ; Fournier CT; Roth SJ; Krizanc D; Weir MP Assessment of MS/MS Search Algorithms with Parent-Protein Profiling. J. Proteome Res 2014, 13 (4), 1823–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Eng J; McCormack AL; Yates JR 3rd An Approach to Correlate Tandem Mass Spectral Data of Peptides with Amino Acid Sequences in a Protein Database. J. Am. Soc. Mass Spectrom 1994, 5, 976–989. [DOI] [PubMed] [Google Scholar]
- (14).Geer LY; Markey SP; Kowalak JA; Wagner L; Xu M; Maynard DM; Yang X; Shi W; Bryant SH Open mass spectrometry search algorithm. J. Proteome Res 2004, 3 (5), 958–64. [DOI] [PubMed] [Google Scholar]
- (15).Perkins DN; Pappin DJ; Creasy DM; Cottrell JS Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 1999, 20 (18), 3551–67. [DOI] [PubMed] [Google Scholar]
- (16).Celotto AM; Graveley BR Alternative splicing of the Drosophila Dscam pre-mRNA is both temporally and spatially regulated. Genetics 2001, 159 (2), 599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Desiere F; Deutsch EW; King NL; Nesvizhskii AI; Mallick P; Eng J; Chen S; Eddes J; Loevenich SN; Aebersold R The PeptideAtlas project. Nucleic Acids Res 2006, 34 (Database issue), D655–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Russo A; Chandramouli N; Zhang L; Deng H Reductive glutaraldehydation of amine groups for identification of protein N-termini. J. Proteome Res 2008, 7 (9), 4178–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Chen S; Vetro JA; Chang YH The specificity in vivo of two distinct methionine aminopeptidases in Saccharomyces cerevisiae. Arch. Biochem. Biophys 2002, 398 (1), 87–93. [DOI] [PubMed] [Google Scholar]
- (20).Bendtsen JD; Nielsen H; von Heijne G; Brunak S Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol 2004, 340 (4), 783–95. [DOI] [PubMed] [Google Scholar]
- (21).Vaudel M; Barsnes H; Berven FS; Sickmann A; Martens L SearchGUI: An open-source graphical user interface for simultaneous OMSSA and X!Tandem searches. Proteomics 2011, 11 (5), 996–9. [DOI] [PubMed] [Google Scholar]
- (22).Vaudel M; Burkhart JM; Zahedi RP; Oveland E; Berven FS; Sickmann A; Martens L; Barsnes H PeptideShaker enables reanalysis of MS-derived proteomics data sets. Nat. Biotechnol 2015, 33 (1), 22–4. [DOI] [PubMed] [Google Scholar]
- (23).Frederiks F; Heynen GJ; van Deventer SJ; Janssen H; van Leeuwen F Two Dot1 isoforms in Saccharomyces cerevisiae as a result of leaky scanning by the ribosome. Nucleic Acids Res. 2009, 37 (21), 7047–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Brunner E; Ahrens CH; Mohanty S; Baetschmann H; Loevenich S; Potthast F; Deutsch EW; Panse C; de Lichtenberg U; Rinner O; Lee H; Pedrioli PG; Malmstrom J; Koehler K; Schrimpf S; Krijgsveld J; Kregenow F; Heck AJ; Hafen E; Schlapbach R; Aebersold R A high-quality catalog of the Drosophila melanogaster proteome. Nat. Biotechnol 2007, 25 (5), 576–83. [DOI] [PubMed] [Google Scholar]
- (25).Diament BJ; Noble WS Faster SEQUEST searching for peptide identification from tandem mass spectra. J. Proteome Res 2011, 10 (9), 3871–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Park CY; Klammer AA; Kall L; MacCoss MJ; Noble WS Rapid and accurate peptide identification from tandem mass spectra. J. Proteome Res 2008, 7 (7), 3022–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Shabalina SA; Ogurtsov AY; Spiridonov NA A periodic pattern of mRNA secondary structure created by the genetic code. Nucleic Acids Res. 2006, 34 (8), 2428–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Lagunez-Otero J; Trifonov EN mRNA periodical infrastructure complementary to the proof-reading site in the ribosome. J. Biomol Struct. Dyn 1992, 10 (3), 455–64. [DOI] [PubMed] [Google Scholar]
- (29).Sigrist CJ; de Castro E; Cerutti L; Cuche BA; Hulo N; Bridge A; Bougueleret L; Xenarios I New and continuing developments at PROSITE. Nucleic Acids Res. 2013, 41 (Database issue), D344–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Finn RD; Bateman A; Clements J; Coggill P; Eberhardt RY; Eddy SR; Heger A; Hetherington K; Holm L; Mistry J; Sonnhammer EL; Tate J; Punta M Pfam: the protein families database. Nucleic Acids Res. 2014, 42 (Database issue), D222–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Petersen TN; Brunak S; von Heijne G; Nielsen H SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8 (10), 785–6. [DOI] [PubMed] [Google Scholar]
- (32).Jones P; Binns D; Chang HY; Fraser M; Li W; McAnulla C; McWilliam H; Maslen J; Mitchell A; Nuka G; Pesseat S; Quinn AF; Sangrador-Vegas A; Scheremetjew M; Yong SY; Lopez R; Hunter S InterProScan 5: genome-scale protein function classification. Bioinformatics 2014, 30 (9), 1236–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1A. Analyzed yeast datasets from PeptideAtlas. Supplementary Table 1B. Analyzed Drosophila datasets from PeptideAtlas. Supplementary Table 2. Standardized parameter settings for three algorithms. Supplementary Table 3. Enriched GO terms associated with Drosophila Frame-1 downPeptide genes. Supplementary Figure 1. Scatterplot showing Gene Ontology terms that are enriched above background for Drosophila frame-1 downPeptide genes. (PDF)
Supplementary File 1. Summary of yeast N-terminal annPeptides and downPeptides detected with Mascot, OMSSA, and SEQUEST PSM algorithms. OMSSA was used with standard and stripped sequence libraries. Numerical entries for each algorithm indicate a peptide’s maximum PSM confidence (d) where d = −log10(PSM score/FDR threshold), which measures the relative distance between an algorithm PSM score and the decoy FDR confidence threshold (using the following PSM scores: SEQUEST probability score, OMSSA e-value score, Mascot peptide score × 0.1; Lin et al.12). A confidence value of 2, for example, indicates the peptide’s best PSM score is 100 times better than the poorest acceptable threshold score computed from decoy analysis. A 5% FDR threshold was used for all three algorithms except OMSSA_stripped, where FDR = 2%. The reference entries show yeast gene id, the start and stop coordinate of the downORF (where the annAUG is at 1001), the frame (1, 2, or 3), and optionally whether the N-terminal methionine is cleaved (trimmed) or whether a signal sequence was cleaved (cleavedNTerm at the ith amino acid). (XLSX)
Supplementary File 2. Yeast peptide-spectrum matches by Mascot using the standard sequence library at 5% FDR. Higher stringency subsets of downPeptide matches are illustrated that conform to 1% FDR. Also illustrated is the subset of PSMs with peptide-matching scores 10 times better than the 1% FDR threshold. Included are the Peptide-Atlas experiment (PAe), spectrum (dta), number of algorithm parameter sets (runcount). Matched peaks of spectra for downPeptides are listed. Supplementary Files 3 and 5 also show this information. (XLSX)
Supplementary File 3. Yeast peptide-spectrum matches by OMSSA using the standard sequence library at 5% FDR. Subsets of downPeptides are shown for 1% FDR and are 10 times better than these thresholds. (XLSX)
Supplementary File 4. Summary of Drosophila N-terminal annPeptides and downPeptides detected by OMSSA. The reference entries show transcript and protein names followed by the downAUG location (measured in nucleotides after the annAUG), the frame (1, 2 or 3), and optionally whether the N-terminal methionine is cleaved (trimmed). (XLSX)
Supplementary File 5. Drosophila peptide-spectrum matches by OMSSA using the stripped sequence library at 2% FDR. Subsets of downPeptides are shown for 1% FDR and are 10 times better than these thresholds. (XLSX)
Supplementary File 6a. Samples of annotated spectra of yeast downPeptides detected by OMSSA. Subsets of spectra are presented conforming to different stringencies: 5% FDRbut not 1% FDR. Supplementary Files 7 and 8 are similarly presented. (ZIP)
Supplementary File 6b. Samples of annotated spectra of yeast downPeptides detected by OMSSA: 1% FDR but not 10 times better. (ZIP)
Supplementary File 6c. Samples of annotated spectra of yeast downPeptides detected by OMSSA: at least 10-fold better scores than 1% FDR thresholds. (ZIP)
Supplementary File 7a. Annotated spectra of yeast downPeptides detected by Mascot: 5% FDR but not 1% FDR. (ZIP)
Supplementary File 7b. Annotated spectra of yeast downPeptides detected by Mascot: 1% FDR but not 10 times better. (ZIP)
Supplementary File 7c. Annotated spectra of yeast downPeptides detected by Mascot: at least 10-fold better scores than 1% FDR thresholds. (ZIP)
Supplementary File 8a. Samples of annotated spectra of Drosophila downPeptides detected by OMSSA showing the standard 2% FDR. (ZIP)
Supplementary File 8b. Samples of annotated spectra of Drosophila downPeptides detected by OMSSA showing higher stringencies: 1% FDR but not 10 times better. (ZIP)
Supplementary File 8c. Samples of annotated spectra of Drosophila downPeptides detected by OMSSA showing higher stringencies: at least 10-fold better scores than 1% FDR thresholds. (ZIP)
Fasta sequence library with full yeast proteome plus all downPeptides within 100 nucleotides of annotated start sites. (ZIP)
Fasta sequence library with full Drosophila proteome plus all downPeptides within 100 nucleotides ofannotated start sites. (ZIP)