

Summary
The antiviral activity of antibodies reflects the bifunctional properties of these molecules. While the Fab domains mediate highly specific antigenic recognition to block virus entry, the Fc domain interacts with diverse types of Fcγ receptors (FcγRs) expressed on the surface of effector leukocytes to induce the activation of distinct immunomodulatory pathways. Fc-FcγR interactions are tightly-regulated to control IgG-mediated inflammation and immunity and are largely determined by the structural heterogeneity of the IgG Fc domain, stemming from differences in the primary amino acid sequence of the various subclasses, as well as the structure and composition of the Fc-associated N-linked glycan. Engagement of specific FcγR types on effector leukocytes has diverse consequences that affect several aspects of innate and adaptive immunity. In this review, we provide an overview of the complexity of FcγR-mediated pathways, discussing their role in the in vivo protective activity of anti-HIV-1 antibodies. We focus on recent studies on broadly neutralizing anti-HIV-1 antibodies that revealed that Fc-FcγR interactions are required to achieve full therapeutic activity through clearance of IgG-opsonized virions and elimination of HIV-infected cells. Manipulation of Fc-FcγR interactions to specifically activate distinct FcγR-mediated pathways has the potential to affect downstream effector responses, influencing thereby the in vivo protective activity of anti-HIV-1 antibodies; a strategy that has already been successfully applied to other IgG-based therapeutics, substantially improving their clinical efficacy.
Keywords: Fc receptors, Antibodies, Inflammation, AIDS, Immunotherapies, Cytotoxicity
Introduction
For many years, antibody-based therapeutics for the prevention or treatment of infectious diseases had been considered an ineffective therapeutic approach, with limited clinical benefit and no significant advantages over conventional antimicrobial pharmacologic molecules. Given the highly-specific nature of antibody-antigen interactions, antibodies against infectious diseases were thought to provide only specific protection against a very narrow spectrum of microbial sub-species, presenting thereby limited breadth. Indeed, with the exception of immunoprophylaxis against anthrax and RSV infection, antibody-based therapies have currently limited clinical use and antimicrobial drugs represent the first and in most cases the only choice for the prevention or treatment of viral, bacterial, and fungal infections. However, for many infections, current pharmacologic mediators have limited therapeutic activity, or their use is associated with systemic toxicity. In addition, the emergence of multi-drug resistant strains poses a viable threat to public health that necessitates the development of alternative therapeutic strategies against infectious diseases that cannot be adequately controlled by currently available drugs or exhibit evidence for multi-drug resistance (1).
The successful development and clinical use of numerous anti-tumor antibodies with proven safety and therapeutic efficacy has sparked tremendous interest in the isolation and characterization of monoclonal antibodies against infectious diseases. Indeed, over the past decade, the systematic study of antibodies targeting primarily highly conserved epitopes on infectious agents have led to the development of several antibody-based therapeutics with broad and potent activity. Indeed, palivizumab and raxibacumab represent two examples of FDA-approved antibodies for the prevention and treatment of RSV and anthrax infection, respectively (2, 3). Additionally, ZMApp, a monoclonal antibody cocktail against Ebola surface glycoprotein (GP) was employed as a therapeutic strategy during the 2014 Ebola outbreak (4). Currently, over a dozen of antibodies are in clinical trials to evaluate their activity against several infectious agents, including HIV, influenza, Ebola, rabies, and C.difficile (2, 3, 5, 6).
Recently described antibodies against the HIV-1 envelope protein (Env) exhibit remarkable potency and breadth against several diverse viral strains. In early phase I/IIa trials, passive administration of broadly neutralizing anti-Env antibodies in chronically infected HIV-1 patients resulted in a significant reduction in plasma viremia, lasting for several days post-antibody infusion (7–9). The clinical characterization of the protective activity of these antibodies reflect the intense efforts over the past decade to systematically isolate and study the role and function of several anti-Env antibodies with broad and potent activity (reviewed in (10). Thanks to recent advances in hybridoma and B cell cloning technologies, these antibodies have been isolated from a small fraction of patients, termed as “elite neutralizers”, who exhibit sustained control of virus replication with no evidence for plasma viremia for several years post-infection (11). Serologic analyses of these patients revealed that these individuals develop highly mutated, affinity matured antibody responses with broad activity against diverse, cross-clade virus isolates (11). In pre-clinical animal disease models, passive administration of these broadly neutralizing anti-Env antibodies confers sterilizing immunity against SHIV challenge in macaques and HIV-1 infection in humanized mouse models (12–15). Likewise, these antibodies have the capacity to suppress plasma viremia in humanized mice and non-human primates with established infection, indicative of their activity to effectively control virus replication and confer therapeutic potency (16–19).
Compared to pharmacologic compounds, whose activity is typically the result of directly blocking the function of a receptor or enzyme, antibodies are bifunctional molecules with the capacity to simultaneously engage a diverse array of effector functions to confer full therapeutic activity in vivo. While the Fab domain of IgG antibodies mediates highly specific interactions with the antigen, potent IgG activity is the result of the synergistic function of the Fab and Fc domains. Indeed, the presence of the Fc domain in the IgG structure is imperative to maintain IgG at a conformation permissive for bivalent binding of the two Fab domains to the antigen, increasing thereby the avidity of the Fab-antigen interactions. Additionally, interactions of the Fc domain with specialized recycling receptors (FcRn) account for the unusually long serum half-life of IgG (up to 3 weeks in humans). This highly-conserved system represents a homeostatic mechanism that effectively regulates the circulating levels of IgG, along with other proteins, like albumin. Loss or deletion of the Fc domain readily impacts IgG avidity for the antigen and dramatically reduces in vivo stability and half-life.
Additionally, the Fc domain of IgG has the capacity to interact with diverse types of receptors (FcγRs) expressed on the surface of effector leukocytes. Engagement of each of these receptors by the Fc domain of IgG stimulates distinct immunomodulatory signaling pathways that regulate the activity of effector functions with significant consequences for immunity and inflammation. In this article, we will provide an overview of the FcγR structural and functional characteristics, highlighting the role of Fc-FcγRs in driving effector responses and mediating in vivo protection. We will present recent evidence on the role of Fc effector activity of broadly neutralizing anti-Env antibodies and discuss these data in relation to clinical data from the use of other IgG-based therapeutic molecules.
FcγR types and activity
FcγRs are broadly divided into main types: type I and II that are distinguished based on their structural characteristics and their capacity to interact with the two main conformational states of the IgG Fc domain (Figure 1).
Figure 1: Overview of the human type I and type II FcγR family.
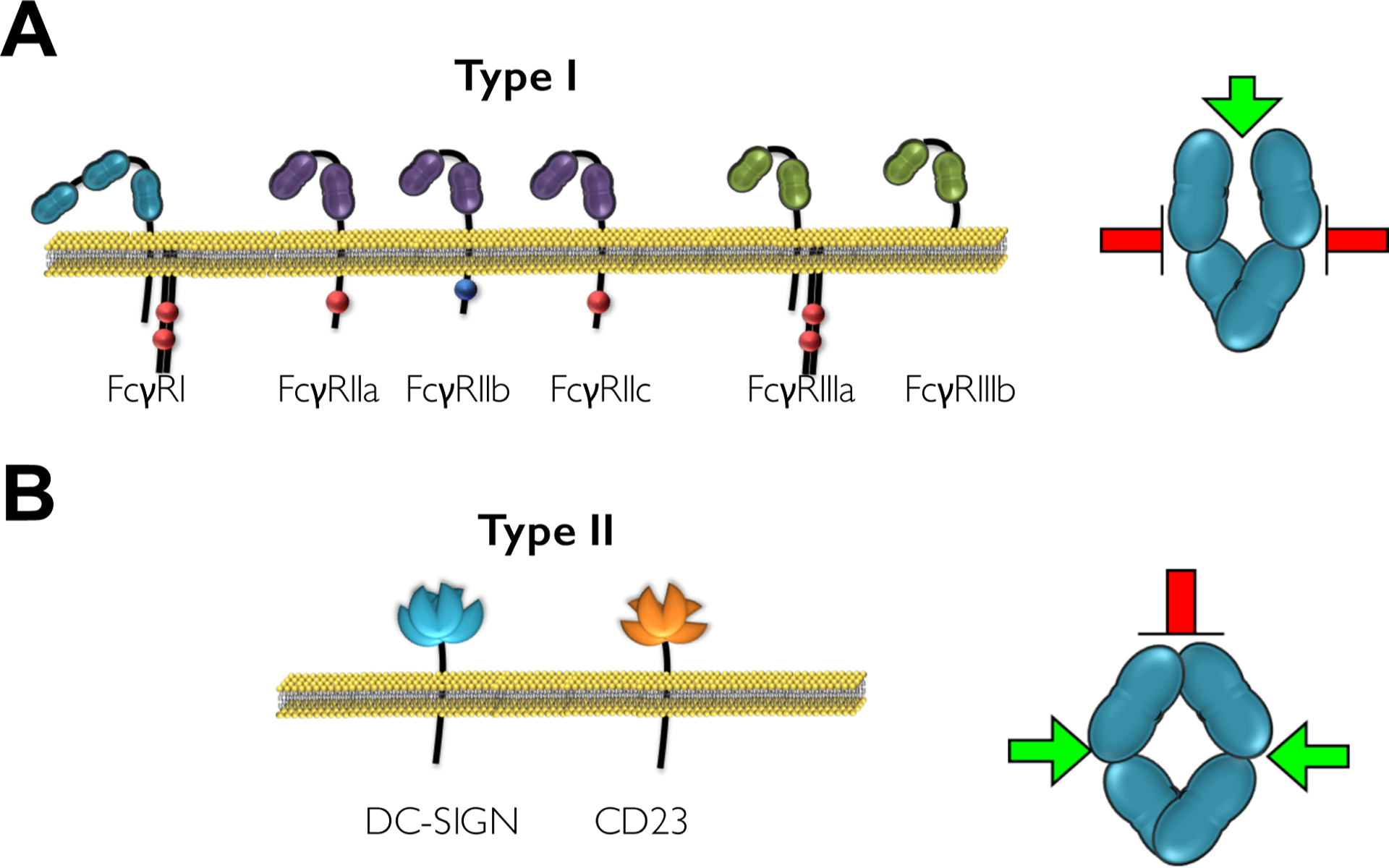
(A) Human type I FcγRs belong to the immunoglobulin (Ig) receptor superfamily and comprise of two or three extracellular Ig domains that mediate IgG binding. They are functionally divided into activating or inhibitory based on the presence of an intracellular ITAM or ITIM motif that transduce immunostimulatory or inhibitory signals following receptor crosslinking. Binding of type I FcγRs is mediated at the hinge proximal CH2 region (green arrow); a site that is accessible when the Fc domain exhibits the “open” conformation. (B) Type II FcγRs include DC-SIGN and CD23 that are both C-type lectin receptors. Binding of the Fc domain to these receptors is mediated at the CH2-CH3 interface following a conformational change that is induced upon sialylation of the Fc-associated glycan structure. At this conformation (“closed”), the type II FcγR binding sites become accessible (green arrows), whereas binding of type I FcγRs is inhibited (red arrow).
Type I FcγR Family
Type I FcγRs belong to the immunoglobulin (Ig) superfamily of immunoreceptors, as their extracellular domain is comprised of two (or three for FcγRI) Ig domains (20, 21)(Figure 1A). These domains mediate IgG binding through interactions with the hinge-CH2 interface in a 1:1 stoichiometric complex (22, 23). In humans, type I FcγRs are encoded by eight different genes, each with multiple transcriptional isoforms. With the exception of the high affinity FcγRI, all FcγR genes are mapped at a common locus located at 1q23 (24, 25). This locus is highly conserved among mammalian species and emerged very early in mammalian evolutionary history. Despite the high degree of homology between mammalian species, sequential non-homologous recombination events followed by gain-of-function mutations late in human and chimpanzee divergence from the common non-human primate ancestor, generated additional type I FcγR-encoding genes that are uniquely found in humans and chimpanzees, but not in other non-human primates, like rhesus macaques. These genes include FCGR2C and FCGR3B, which encode FcγRIIc and FcγRIIIb, respectively. FcγRIIc sequence analysis indicates that FCGR2C gene is the result of a non-homologous recombination event that created a chimeric receptor, which comprises the extracellular domain of FcγRIIb, whereas the transmembrane and intracellular domains have originated from the FCGR2A gene. Likewise, FCGR3B gene resulted from gene duplication of FCGR3A followed by a point mutation (Phe203Ser) at the membrane proximal region of the extracellular domain of FcγRIIIb that created a post-translational modification signal sequence that processes FcγRIIIb as GPI-anchored protein (26).
Type I FcγRs are classified into activating or inhibitory based on their capacity to transduce immunostimulatory or immunosuppressive signals following receptor engagement by IgG immune complexes and crosslinking (27, 28). Activating type I FcγRs follow a distinct pattern of signaling cascades that are initiated following IgG-FcγR interactions through the presence of intracellular immunoreceptor tyrosine-based activation motifs (ITAMs). These motifs are located in the receptor α chain or in the associated FcR γ-chain (or ζ chain) and become phosphorylated upon binding of multimeric IgG immune complexes to activating FcγRs (29–31). Indeed, Fc-FcγR interactions trigger the phosphorylation of the ITAM tyrosine residues, which in turn induces the subsequent activation of a number of cytoplasmic tyrosine kinases of the Src and Syk family of kinases (26, 29, 32–36). Src and Syk kinase activation sequentially activates the PI3K-PKC pathway, leading to Ca2+ mobilization and cellular activation (37–40). Additionally, MEK and MAP family kinases become activated, leading to the transcription of pro-inflammatory cytokines and chemokines with effects on cellular survival and differentiation (38, 41, 42). In humans, activating type I FcγRs comprise FcγRI, FcγRIIa/c and FcγRIIIa. Whereas FcγRI and FcγRIIIa require the FcR γ-chain for expression, assembly to the cell membrane, and signaling (43–45), FcγRIIa and FcγRIIc exhibit an ITAM domain at the intracellular region, and are therefore capable of signal transduction upon receptor engagement, without relying on FcR γ-chain for expression and function.
FcγRI is the only high affinity FcγR and is capable of binding monomeric IgG with high affinity (KD 10−9-10−10 for human IgG1); a property attributed to FcγRI unique structure. In contrast to other type I FcγRs, FcγRI consists of three Ig domains at the extracellular, IgG binding region. The presence of this third domain acts as a spacer for the Fab domain during Fc-FcγR interactions, greatly enhancing the binding strength of these interactions (46). Under steady-state conditions, FcγRI is ubiquitously expressed by monocytes and macrophages; however, several pro-inflammatory cytokines, predominantly interferon-γ and to a lesser extent granulocyte-colony stimulating factor (G-CSF), interferon-α and interleukin-12 have the capacity to stimulate FcγRI expression in several myeloid leukocytes, including neutrophils, eosinophils, and dendritic cells (47, 48). In contrast to FcγRI, FcγRII and FcγRIII represent low affinity IgG receptors that are incapable of binding monomeric IgG, exhibiting low affinity for IgG (KD for human IgG1: 10−5-10−7). Despite their low affinity for monomeric IgG, these FcγRs can be engaged by IgG immune complexes through multimeric, high avidity interactions (49). This property represents a key homeostatic mechanism to ensures that FcγR crosslinking and downstream signaling can only be accomplished by IgG immune complexes that are generated during an immune response, but not by monomeric IgG under physiological conditions, preventing thereby inappropriate or excessive activation of FcγR pathways. Both FcγRIIa and FcγRIIc encompass ITAM domains at their cytoplasmic tail and are therefore capable of transducing immunostimulatory signals upon receptor crosslinking by IgG complexes. Despite this similarity, these FcγRs follow a unique pattern of expression among the various leukocyte populations. Whereas FcγRIIc expression is restricted to NK cells, FcγRIIa is widely expressed by the majority of myeloid cell populations, including neutrophils, monocytes, eosinophils, dendritic cells, macrophages, and platelets (20, 24, 25). Lastly, FcγRIIIa, an activating FcγR that is expressed by macrophages, NK cells and a subset of monocytes, associates with the FcR γ (and/or ζ in NK cells)-chain for expression and signaling (26, 44, 50).
The signaling function of activating FcγRs is directly antagonized by FcγRIIb, the only inhibitory FcγR. In contrast to activating FcγRs, FcγRIIb is characterized by the presence of an immunoreceptor tyrosine-based inhibitory motif (ITIM) at the intracellular domain. Following receptor aggregation, crosslinking of ITIMs and phosphorylation by Src family kinases trigger the recruitment of SHIP and SHP2 phosphates (51–53). Such phosphatases induce the hydrolysis of phosphatidylinositol 3,4,5-triphosphate to phosphatidylinositol 4,5-biphosphate, inhibiting thereby the recruitment and activation of PLCγ and BTK (51, 54, 55). These intracellular signals counterbalance any immunostimulatory signals mediated through activating FcγR engagement, effectively limiting cellular activation and effector responses. FcγRIIb is widely expressed by several leukocyte types of the lymphoid and myeloid lineage, and it is processed as two splicing variants (FcγRIIb1 and FcγRIIb2) that differ in their capacity for internalization following receptor crosslinking (56, 57). Lastly, contrary to activating or inhibitory type I FcγRs, FcγRIIIb represents the sole FcγR that is incapable of transducing activating or inhibitory signals upon engagement, as it lacks any intracellular signaling motifs. However, FcγRIIIb has the capacity -though limited- to mediate signal transduction by associating and acting synergistically with other receptors, such as FcγRIIa and complement receptors or signaling subunits, like FcR γ-chain or ζ-chain (58). FcγRIIIb expression is restricted to neutrophil granulocytes, and can be induced under certain conditions (IFNγ) in eosinophils (30, 59).
Type II FcγR Family
A characteristic common to all type II FcγRs is their structure and their capacity to engage the IgG Fc domain in a conformation (‘closed’) that is not permissive for type I FcγR binding (Figure 1B). In contrast to type I FcγRs, type II FcγRs mediate binding at the CH2-CH3 interface in a 2:1 complex (receptor:IgG)(30, 60, 61) and belong to the C-type lectin family of receptors. Until now, two type II FcγRs, DC-SIGN and CD23, have been characterized with the capacity to mediate in vivo effector activity upon receptor binding. However, several other C-type lectin receptors have been described to exhibit binding capacity for the IgG Fc domain, such as CD22 and DCIR, without a yet-defined, clear in vivo role and effector functions induced upon Fc interactions (62–64).
Consistent with their common functional properties, the genes encoding DC-SIGN and CD23, CD209 and FCER2, respectively, are mapped at a common locus on chromosome 19 (19p13). DC-SIGN is expressed predominantly by dendritic cells and by subsets of monocytes and macrophages (65, 66). Its binding specificity is not limited to IgG Fc, but also extends to other ligands, primarily mannose-rich glycan structures. In contrast to IgG Fc binding to DC-SIGN that is accomplished through protein-protein interactions, engagement of DC-SIGN by these carbohydrate ligands is exclusively mediated through interactions between the carbohydrate residues present on the heavily glycosylated DC-SIGN. Given the diversity of the ligands and the differential mode of interactions with DC-SIGN, the precise signaling events that follow DC-SIGN engagement appear to be dependent upon the type and the nature of the DC-SIGN – ligand interaction, as well as the effector leukocyte types involved. In the case of IgG Fc-DC-SIGN interactions, receptor engagement on regulatory macrophages induces the expression of interleukin 33 (IL-33) that limits T cell- and IgG-mediated inflammation (67, 68).
The other type II FcγR, CD23, is also a heavily glycosylated C-type lectin, which apart from the Fc domain of IgG, it has also the capacity to interact with IgE. Indeed, CD23 has been initially identified as the low affinity IgE receptor (61, 69, 70) with CD23-IgE interactions playing a key role in regulating IgE production by B cells and controlling IgE-meditated responses (71, 72). Molecular modeling of the CD23 interaction with IgE and IgG revealed a common binding pattern to CD23 between these two ligands that can be attributed to the intrinsic flexibility of the Fc domain structure (61). CD23 exists in two splicing isoforms (CD23a and CD23b), that differ in their expression pattern and responsiveness to IL-4. In particular, CD23a is constitutively expressed by mature B cells, whereas CD23b expression is only induced by IL-4 in several leukocyte types, including T cells, monocytes, macrophages and granulocytes (73). Although the precise signaling events following CD23 engagement by IgG complexes have not been fully elucidated yet, CD23 crosslinking on B cells induces the upregulation of the inhibitory type I FcγR, FcγRIIb that in turn regulates IgG affinity responses (74).
Regulation of FcγR effector functions
As discussed in the previous section, engagement of type I and type II FcγRs initiates a cascade of pro-inflammatory, anti-inflammatory and immunomodulatory signaling pathways with the capacity to impact several downstream effector responses. Similar to other immune processes, the activity of FcγR-mediated pathways is tightly regulated and several homeostatic mechanisms exist to control FcγR-mediated effector responses, avoiding thereby inappropriate or excessive cellular activation. Indeed, small perturbations in FcγR activity are often associated with chronic inflammatory and autoimmune pathologies, or influence the therapeutic responsiveness of passively-infused IgG-based therapeutics.
Although a number of determinants have been described to influence IgG-mediated effector functions, the structure of the Fc domain represents the major determinant that regulates Fc-FcγR interactions, and subsequent downstream responses. Despite considered the invariant domain of the IgG molecule, substantial heterogeneity has been described for the Fc domain, stemming predominantly from amino acid differences among the various subclasses and Gm allotypes as well as from the structure and composition of the Fc-associated N-glycan. This glycan structure plays a key role in maintaining the Fc domain at a conformation permissive for FcγR interactions and fine-tuning specific FcγR binding interactions. Indeed, the presence of the central N-linked glycan is required to maintain the Fc domain at a characteristic horseshoe-like conformation, in which the two CH3 domains of the heavy chains are tightly associated, whereas CH2 domains remain further apart (75). This conformation is essential for FcγR interactions, as enzymatic cleavage or deletion of the site at which the N-linked glycan is attached (Asn297 for human IgG1) readily results in loss of Fc-FcγR interactions (28, 76–78). In addition to regulating Fc domain structure and FcγR binding, the Fc-associated glycan also determines binding to the different types of FcγRs through modulation of the glycan structure and composition. Indeed, the presence of terminal sialic acid residues at the core glycan structure influences the flexibility of the Fc domain and allows it to adopt a “closed” conformation that is associated with exposure of the type II binding interface at the CH2-CH3 region, whereas the hinge-proximal region of CH2, where the type I FcγR binding site is mapped, becomes occluded. Indeed, crystallographic studies revealed that sialylation of the Fc-associated glycan induces a “closed” conformation, whereas in the absence of sialic acid residues, the Fc domain remains at an “open” conformation, permissive for type I FcγRs binding (30, 60). This mechanism represents a key homeostatic strategy that effectively switches the binding specificity of the Fc domain to different types of FcγRs through a simple modulation of the composition of the Fc-associated glycan. Differences in the primary amino acid backbone of the IgG Fc domain also influence the binding affinity and specificity for the different FcγRs, influencing thereby the in vivo effector activity of antibodies. For example, in seminal studies using a mouse model of metastatic melanoma, IgG subclass variants of a cytotoxic antibody targeting a surface glycoprotein (gp75) expressed by melanocytes exhibited differential therapeutic activity in vivo, correlating precisely with the capacity of these subclasses to preferentially engage activating classes of FcγRs (28).
Apart from the affinity of the IgG Fc domain for particular types of FcγRs, which is regulated by the primary amino acid sequence of the different IgG Fc subclasses and the Fc-associated N-linked glycan structure, several additional determinants regulate the in vivo effector function of antibodies and influence the responsiveness of effector leukocytes upon FcγR crosslinking. The majority of leukocyte populations co-express more than one FcγR type, with often opposing signaling activities, as in the case of activating and inhibitory type I FcγRs. Additionally, the levels as well as the pattern of FcγR expression are dynamically regulated during the different stages of leukocyte development and differentiation and can be influenced by particular chemokines and cytokines that are present at sites of infection, inflammation or tissue injury (48, 67, 79–81).
Given the complexity of the FcγR expression pattern and downstream signaling events that are induced upon Fc-FcγR interactions, the outcome of IgG-mediated inflammation and the induction of distinct immunomodulatory pathways reflect a finely-tuned balance between the different types of FcγRs expressed by the various leukocyte populations. This balanced activity is dynamically regulated during the different stages of the immune response to mediate a range of effector functions with potent immunomodulatory activity (Figure 2). Such functions include cellular activation, antibody-dependent cellular cytotoxicity and phagocytosis, as well as the release of cytokines and chemokines. In addition, FcγR-mediated pathways have the capacity to modulate several aspects of leukocyte functional activity, influencing thereby immune responses. For example, FcγR-crosslinking on neutrophils and other granulocytes is associated with cellular activation, degranulation and generation of reactive oxygen and nitrogen intermediates with potent antimicrobial activity (82–87). Similarly, FcγRIIa engagement on platelets is associated with platelet degranulation and aggregation (88, 89), whereas FcγRIIIa-mediated activation of NK cells induces the release of perforin and granzymes in close proximity to IgG-coated targets to stimulate the formation of pores at the cell membrane and the induction of pro-apoptotic pathways that lead to cell death (26, 90–92).
Figure 2: Overview of the diversity of Fc effector functions.
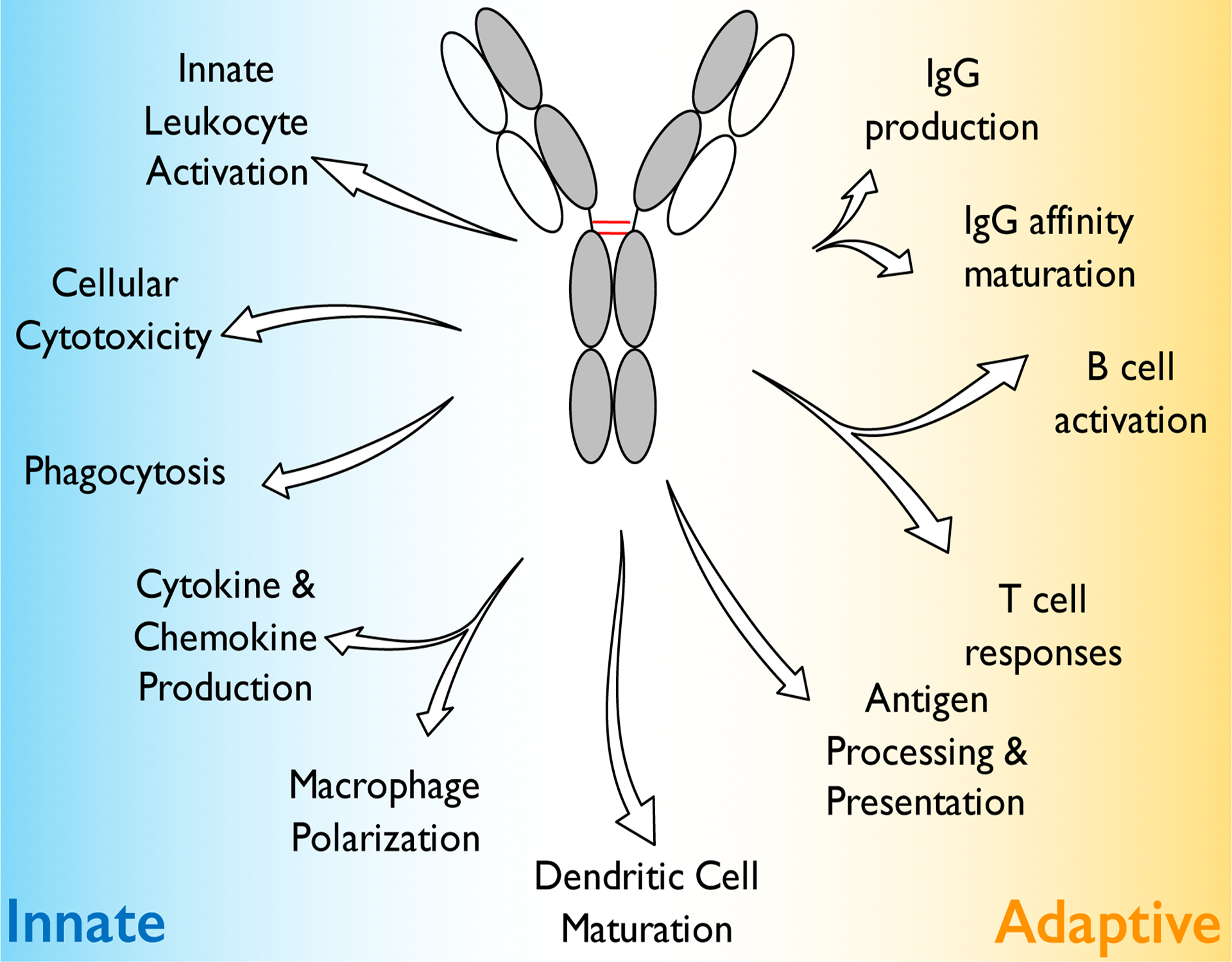
Although IgG antibodies represent molecules of the adaptive immune system, their capacity to interact with type I and type II FcγRs expressed on the surface of effector leukocytes enables them to mediate pleiotropic effector functions through the activation of distinct downstream signaling pathways. These FcγR-mediated pathways have diverse consequences that regulate several aspects of innate and adaptive immunity, regulating thereby inflammation and immune responses.
FcγR-mediated signaling on antigen-presenting cells influence several aspects of cellular and humoral immunity by directly modulating antigen presenting cell maturation and differentiation, antigen processing and presentation to MHC class II molecules, as well as subsequent T cell responses. For example, signaling through FcγRs on macrophages affects cellular polarization and downstream effector responses. Indeed, activating FcγR signaling coupled with stimulation through TLRs, like TLR4 triggers the induction of a specific polarization state that is characterized by augmented IL-10, IL-1 and IL-6 expression as well as by increased migratory and phagocytic capacity (80, 93, 94). Likewise, engagement of DC-SIGN by sialylated IgG Fc on regulatory macrophages and dendritic cells triggers the expression of IL-33, which in turn, induces the expansion of IL-4-producing basophils that upregulate the expression of the inhibitory FcγR, FcγRIIb, on effector leukocytes, raising the threshold for IgG-mediated inflammation (67). In addition, phagocytic uptake of IgG-coated targets and signaling through activating FcγRs is associated with enhanced endosomal maturation and lysosomal fusion, leading consequently to more efficient antigen processing and presentation on MHC class II (31, 95–97) that in turn results in the induction of more robust T cell responses (98–101). Similarly, dendritic cell maturation and antigen presentation are regulated by the contrasting activity of the activating FcγRIIa and the inhibitory FcγRIIb, that are co-expressed on the surface of immature dendritic cells. Skewing the balance between these two FcγRs to specifically engage the activating FcγRIIa over FcγRIIb stimulates dendritic cell maturation and the expression of co-stimulatory molecules that in turn induce robust T cell responses (79, 100–104).
It is therefore well-appreciated that for many FcγR-mediated effector activities, the balance of activating and inhibitory FcγRs is a key determinant for the outcome of IgG-mediated inflammation and immunity. Minor deviations in this balance readily influence the in vivo effector activity of antibodies, as demonstrated in several studies using mice deficient for either activating or inhibitory FcγRs. For example, FcγRIIb knock-out mice present a more severe phenotype in models of immune complex-induced shock, arthritis, and alveolitis, due to lower threshold of activation upon challenge (90, 93, 105). Likewise, Fcgr2b−/− mice exhibit improved bacterial clearance in models of pneumococcal peritonitis (106, 107), as pro-inflammatory signals of activating FcγRs are not subject to FcγRIIb-mediated inhibition. In contrast, overexpression of FcγRIIb is associated with increased mortality upon challenge with Streptococcus pneumoniae (106, 107); an effect attributed to impaired macrophage phagocytic capacity.
For the majority of leukocyte populations, predominantly those of the myeloid lineage, the expression of the inhibitory FcγRIIb is coupled with activating FcγRs, providing thereby a negative inhibition mechanism to control pro-inflammatory signals induced upon activating FcγR crosslinking. However, for cell types, like B cells, which express only the inhibitory FcγRIIb, but not any activating FcγR, FcγRIIb acts as a direct inhibitor of the signals transduced upon B-cell receptor (BCR) crosslinking. Indeed, in an analogy to activating FcγR signaling inhibition, FcγRIIb plays a central role in B cell responses by regulating B cell function and survival. Indeed, FcγRIIb engagement by IgG immune complexes induce pro-apoptotic signals, which are attenuated in the presence of BCR-FcγRIIb co-engagement (51, 55, 108, 109). This mechanism ensures that only B cells with high affinity BCRs survive, whereas those with low or no antigenic affinity are effectively eliminated through apoptosis. Likewise, in plasma cells, which are negative for surface BCR expression, FcγRIIb cross-linking by IgG immune complexes induces cellular apoptosis to restrict IgG production by plasma cells, terminating effectively humoral immune responses (51, 54, 110). The key role of FcγRIIb in B cell function is best exemplified by the clinical association of autoimmune diseases, like SLE with FCGR2B alleles that impact receptor expression (111–113) or activity (106, 114). Similarly, Fcgr2b deficient mice are characterized by spontaneous induction of autoimmunity, and failure to induce high affinity IgG responses (108, 109).
In addition to FcγRIIb, mature B cells also express CD23, a type II FcγR, which also regulates B cell function and antibody responses. Since CD23 is also a low affinity receptor for IgE, early studies have shown that CD23 functions similarly to FcγRIIb to control IgE responses on B cells and regulate antigen capture and presentation to dendritic cells (72). In addition, given its capacity to also engage sialylated Fc domain of IgG antibodies, CD23 also participates in the regulation of IgG responses (61, 74). More specifically, sialylated Fc-CD23 interactions on B cells trigger the upregulation of FcγRIIb expression on B cells, which in turn raises the threshold for B cell selection, leading consequently in the generation of high affinity IgG responses (74). These findings highlight the synergistic activity of the different types of FcγRs to regulate B cell function and consequently antibody affinity and specificity.
Anti-HIV Fc effector activities
In the previous section, we presented an overview of the role of FcγR-mediated pathways in the regulation of inflammation and immunity, as well as the diversity of effector functions initiated upon Fc-FcγR interactions (Figure 2). Among the various effector leukocytes, engagement of the different types of FcγRs has diverse immunomodulatory consequences, including antibody-dependent cellular cytotoxicity or, phagocytosis, release of cytokines and chemokines, leukocyte differentiation and survival, as well as modulation of T and B cell responses (27, 30). For several antibodies against viral, bacterial, and fungal pathogens, substantial experimental evidence suggests that their in vivo activity depends on Fc-FcγR interactions, since disruption of these interactions readily results in reduced in vivo activity (115). Likewise, the in vivo protective activity of these antibodies is highly dependent on their capacity to preferentially engage activating FcγRs, highlighting the importance of the balanced activity of activating and inhibitory FcγRs in the regulation of IgG-mediated immunity (115).
Similar to antibodies against other infectious microbes, anti-HIV-1 antibodies are also expected to utilize FcγR-mediated pathways to achieve full protective activity in vivo. Indeed, early studies using b12, one of the first broadly neutralizing anti-Env antibodies characterized, revealed that its activity to protect rhesus monkeys from SHIV infection is dependent upon Fc-FcγR interactions (13). More specifically, in a pre-exposure prophylaxis model, the activity of Fc domain variants of b12 with differential activity to activate complement and FcγR-mediated pathways was assessed, revealing that Fc domain variants with minimal capacity for FcγR engagement provided no protection against SHIV challenge (13). In contrast, b12 Fc variants defective for C1q binding and complement activation exhibited activity comparable to that of wild-type b12, indicating that FcγRs, but not complement, are required for optimal in vivo antibody activity to protect against SHIV infection.
The recent description of several highly potent, broadly neutralizing anti-Env antibodies with the potential for clinical use for the prevention and treatment of HIV-1 infection in humans, prompted the characterization of the role of FcγR effector activity in their in vivo protective activity. Indeed, understanding of the precise FcγR-mediated pathways that are required for optimal in vivo activity could lead to the generation of antibody-based therapeutics with improved in vivo efficacy through modulating their capacity to interact with FcγRs. However, a major drawback in the study of FcγR effector activity of anti-Env antibodies is the lack of a robust in vivo HIV-1 infection model that would faithfully recapitulate the complete virus infection cycle, human effector cells, and FcγR diversity to provide useful insights into the precise contribution of FcγR-mediated pathways to the in vivo antibody activity. Although in vitro systems are commonly used to determine the activity of anti-HIV-1 antibodies, they fail to reproduce the diversity of receptors, effector cells, and microenvironments that contribute to protection during in vivo conditions.
To overcome these limitations, several complementary in vivo models are commonly employed to provide the full picture of the precise role of FcγR-mediated interactions in the in vivo activity of antibodies (18, 59, 116, 117). For example, using a mouse model for HIV-1 entry (116), assessment of the in vivo protective activity of a panel of broadly neutralizing antibodies targeting different HIV-1 env epitopes revealed that genetic deletion of FcγR encoding genes or mutation of the Fc domain to abrogate Fc-FcγR interactions resulted in reduced in vivo neutralization activity (116, 118). Likewise, IgG subclass variants of several broadly neutralizing anti-Env antibodies exhibited differential activity in vivo, which correlated with their capacity to engage activating FcγRs. More specifically, increased activity for the mouse IgG2a subclass compared to mouse IgG1 was evident, consistent with the capacity of IgG2a to interact preferentially with activating FcγRs, inducing thereby clearance of antibody-opsonized viral particles through activating FcγR engagement (28). Similar findings were also observed in a model of intrasplenic passive transfer of ex vivo HIV-1-infected cells in mice that have been treated with anti-Env antibodies (117). Assessment of the capacity of anti-Env antibodies to induce clearance of infected cells in vivo revealed that their activity is totally dependent on Fc-FcγR interactions. In particular, Fc domain variants of anti-Env antibodies with diminished capacity to engage FcγRs or administration of function-blocking anti-FcγR antibodies completely abrogated the activity of anti-Env antibodies to eliminate infected cells in vivo (117).
Consistent with these observations, a role for FcγR-mediated pathways in the in vivo protective activity of anti-Env antibodies has also been observed in studies using HIV-1-infected humanized mice (118, 119). In models of antibody-mediated post-exposure prophylaxis and therapy of established HIV infection, comparison of the in vivo protective activity of anti-Env antibodies with differential FcγR binding capacity revealed the contribution of FcγR-mediated interactions to the in vivo protective activity of anti-Env antibodies. More specifically, contrary to wild-type hIgG1 anti-Env antibodies, Fc domain variants with diminished FcγR binding capacity failed to suppress viremia in a model of post-exposure prophylaxis using HIV-1-infected humanized mice (119). Likewise, in a model of antibody-mediated therapeutic suppression of virus replication in humanized mice with established HIV infection, Fc variants of anti-Env antibodies optimized for enhanced affinity to activating FcγRs exhibited improved in vivo protective activity, as evidenced by prolonged and durable suppression of viremia (118). These findings highlight the contribution of diverse FcγR-mediated mechanisms to the in vivo antiviral activity of anti-HIV-1 antibodies. Indeed, apart from directly inhibiting viral entry and infection, broadly neutralizing anti-HIV-1 antibodies achieve potent antiviral activity in vivo though pleiotropic Fc effector functions. These include the opsonization of viral particles, leading to their clearance by FcγR-expressing leukocytes, as well as elimination of HIV-infected cells through the recruitment of FcγR+ effector leukocytes, like macrophages and NK cells to mediate cytotoxicity against these cells (Figure 3). These functions contribute substantially to the protective activity of anti-HIV-1 antibodies, suggesting that enhancement of these effector functions through augmented activation of FcγR-mediated pathways could lead to anti-Env antibodies with improved in vivo efficacy.
Figure 3: FcγR-mediated effector activities mediated by anti-HIV-1 antibodies.
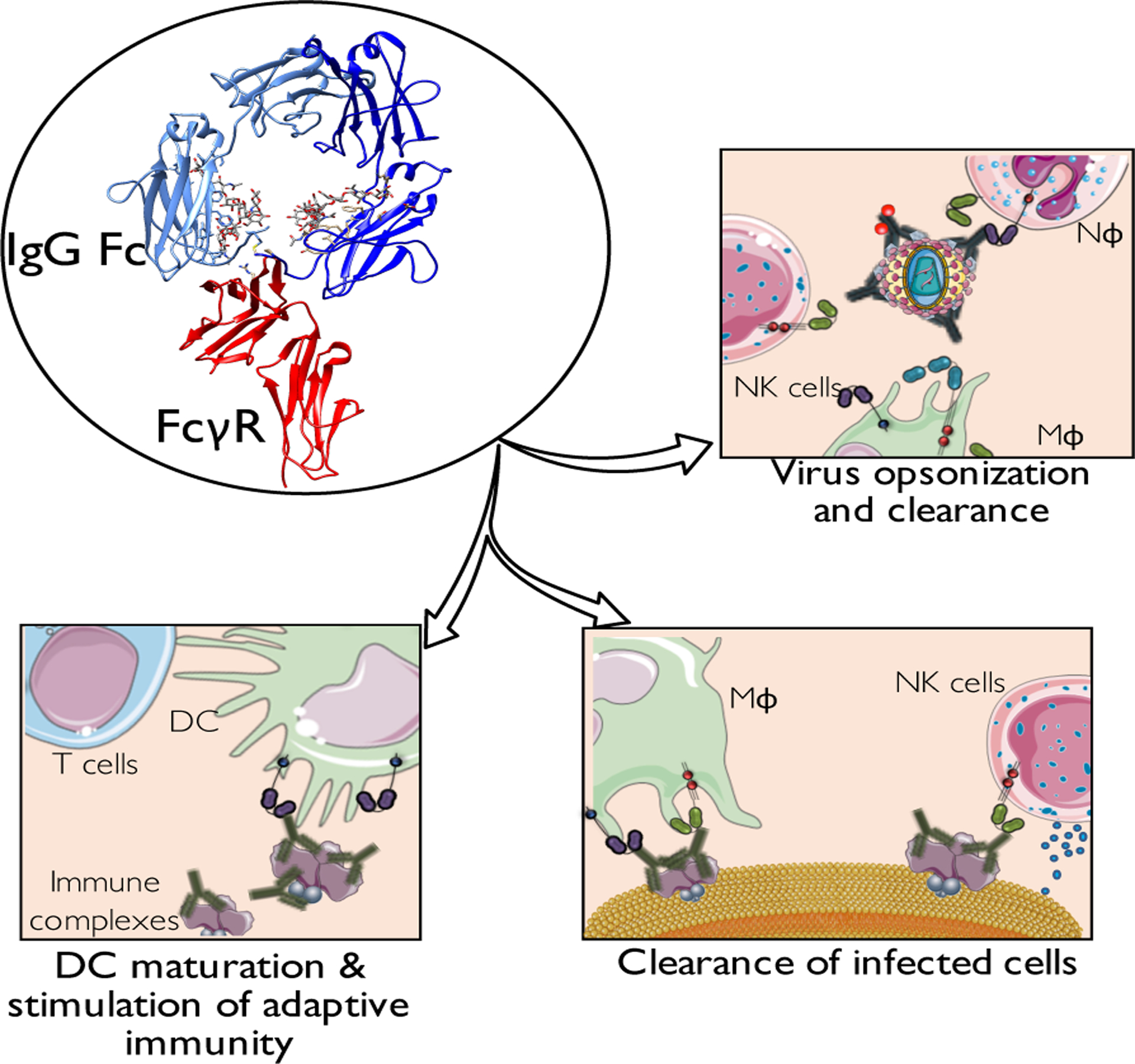
In addition to the Fab-mediated function to block viral entry and fusion to target cells, the in vivo antiviral activity of anti-HIV-1 antibodies also depends on effector functions mediated through the interaction of their Fc domains with FcγRs (depicted here is the interaction of FcγRIIIa with human IgG1 Fc (PDB: 1E4K)). These effector functions include: opsonization and clearance of viral particles, as well as elimination of HIV-infected cells, limiting thereby the viral reservoir and preventing further infection spreading. Additionally, given the capacity of FcγR-mediated interactions to modulate the functional activity of antigen-presenting cells, like dendritic cells, anti-HIV-1 immune complexes can also stimulate adaptive immune responses, through the induction of dendritic cell maturation and enhanced antigen processing and presentation.
Given the complexity of FcγR-mediated pathways to induce pleiotropic effects on several aspects of adaptive immunity, including antigen processing and presentation, modulation of antigen presenting cell function and regulation of T and B-cell responses, FcγR engagement by IgG immune complexes generated following passive administration of anti-Env antibodies have also the capacity to stimulate host immunity. Indeed, this opportunity for the induction of long-term antibody and T cell responses represents a major advantage of antibody-based therapeutics over conventional antiretroviral pharmacologic mediators. Indirect evidence for the role of Fc- FcγR interactions in modulating host immune responses following anti-retroviral antibody administration is provided by two studies using non-human primates. More specifically, passive administration of the anti-CD4bs antibody b12 in SHIV-infected rhesus monkeys affected their B cell responses, leading to the induction of neutralizing antibody responses. Indeed, serum obtained from these animals demonstrated potent neutralizing activity against SHIV and SIV and conferred protection against SHIV infection when passively transferred to naïve animals (120, 121). Likewise, enhanced antibody responses were evident upon administration of anti-HIV-1 broadly neutralizing antibodies in chronically SHIV-infected rhesus monkeys (16). More importantly, a similar observation has also been noted in chronically HIV-1-infected patients treated with the anti-CD4bs antibody 3BNC117, where a single antibody infusion significantly improved neutralizing antibody responses against heterologous tier 2 viruses in almost all study participants (122). These findings highlight the diversity of FcγR-mediated pathways to confer antiviral activity in vivo by modulating the function of FcγR-expressing effector leukocytes and stimulating host immune responses to provide long-term antiviral immunity (Figure 3).
Experience from other systems and future perspectives
Substantial evidence from different in vivo models of HIV-1 infection clearly supports a key role for Fc-FcγR interactions in their in vivo protective activity. Understanding the FcγR-mediated mechanisms that are required for optimal antiviral activity can lead to the development of anti-HIV-1 antibodies optimized for enhanced Fc effector activity by specifically augmenting their capacity to engage particular FcγR types. Similar approaches have already been used to increase the efficacy of several antibody-based therapeutics. In particular, experience from the clinical use of therapeutic antibodies, especially against neoplastic diseases, clearly suggests that Fc-FcγR interactions contribute substantially to the in vivo antibody activity. Indeed, shortly after the introduction of antibody-based therapeutics into the clinic, it was noted that carriers of particular allelic variants of FCGR2A and FCGR3A genes with increased affinity for IgG demonstrated improved therapeutic responsiveness to anti-tumor antibody therapy in cases of chronic lymphocytic leukemia, breast and colorectal cancers (123–125). These observations confirmed previous findings from pre-clinical models that defined a major role for FcγR-mediated interactions in the in vivo activity of therapeutic anti-tumor antibodies, like rituximab and trastuzumab, using genetically-modified mouse strains, deficient for particular types of FcγR. Indeed, genetic deletion of the inhibitory FcγRIIb resulted in improved therapeutic responsiveness to antibody therapy (99), whereas Fc domain engineering for enhanced affinity to activating FcγRs was associated with significantly improved in vivo effector function (59, 104). Based on these observations, the second generation of anti-tumor antibodies have been developed with optimized FcγR binding profile to increase their capacity to induce Fc effector activities. A prime example of this approach is the development of the anti-CD20 antibody, obinutuzumab, which has been engineered for enhanced binding to FcγRIIIa through modification of its glycan structure (afucosylated). Comparison of the activity of obinutuzumab, with the non-modified, first-generation anti-CD20 antibody, rituximab revealed superior therapeutic activity for obinutuzumab, extending progression-free survival for over 10 months (126). These findings clearly highlight the importance of Fc effector activity in antibody-based therapies and provide a paradigm for the development of anti-HIV-1 antibodies with improved Fc effector functions. Indeed, given the contribution of FcγR-mediated interactions to the in vivo antiviral activity of anti-HIV-1 antibodies, enhancing their capacity to interact and activate distinct FcγR pathways could lead to the development of antibodies with improved in vivo efficacy for the control of HIV-1 infection in humans.
Acknowledgements
We wish to thanks and acknowledge support from the Rockefeller University, the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (P01AI100148) and the Bill & Melinda Gates Foundation (OPP1124068). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. SB is an amfAR Mathilde Krim Fellow in Basic Biomedical Research (108977-57-RKVA). The authors have no conflicting financial interests.
References
- 1.Casadevall A, Dadachova E, Pirofski LA. Passive antibody therapy for infectious diseases. Nat Rev Microbiol.2004;2:695–703. [DOI] [PubMed] [Google Scholar]
- 2.Yoshihara S, et al. Effect of palivizumab prophylaxis on subsequent recurrent wheezing in preterm infants. Pediatrics.2013;132:811–818. [DOI] [PubMed] [Google Scholar]
- 3.Migone TS, et al. Raxibacumab for the treatment of inhalational anthrax. N Engl J Med.2009;361:135–144. [DOI] [PubMed] [Google Scholar]
- 4.Qiu X, et al. Sustained protection against Ebola virus infection following treatment of infected nonhuman primates with ZMAb. Sci Rep.2013;3:3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gogtay N, et al. Safety and pharmacokinetics of a human monoclonal antibody to rabies virus: a randomized, dose-escalation phase 1 study in adults. Vaccine.2012;30:7315–7320. [DOI] [PubMed] [Google Scholar]
- 6.Lowy I, et al. Treatment with monoclonal antibodies against Clostridium difficile toxins. N Engl J Med.2010;362:197–205. [DOI] [PubMed] [Google Scholar]
- 7.Lynch RM, et al. Virologic effects of broadly neutralizing antibody VRC01 administration during chronic HIV-1 infection. Sci Transl Med.2015;7:319ra206. [DOI] [PubMed] [Google Scholar]
- 8.Caskey M, et al. Viraemia suppressed in HIV-1-infected humans by broadly neutralizing antibody 3BNC117. Nature.2015;522:487–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scheid JF, et al. HIV-1 antibody 3BNC117 suppresses viral rebound in humans during treatment interruption. Nature.2016;535:556–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klein F, Mouquet H, Dosenovic P, Scheid JF, Scharf L, Nussenzweig MC. Antibodies in HIV-1 vaccine development and therapy. Science.2013;341:1199–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Simek MD, et al. Human immunodeficiency virus type 1 elite neutralizers: individuals with broad and potent neutralizing activity identified by using a high-throughput neutralization assay together with an analytical selection algorithm. J Virol.2009;83:7337–7348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Balazs AB, Chen J, Hong CM, Rao DS, Yang L, Baltimore D. Antibody-based protection against HIV infection by vectored immunoprophylaxis. Nature.2012;481:81–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hessell AJ, et al. Fc receptor but not complement binding is important in antibody protection against HIV. Nature.2007;449:101–104. [DOI] [PubMed] [Google Scholar]
- 14.Mascola JR, et al. Protection of macaques against vaginal transmission of a pathogenic HIV-1/SIV chimeric virus by passive infusion of neutralizing antibodies. Nat Med.2000;6:207–210. [DOI] [PubMed] [Google Scholar]
- 15.Gautam R, et al. A single injection of anti-HIV-1 antibodies protects against repeated SHIV challenges. Nature.2016;533:105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barouch DH, et al. Therapeutic efficacy of potent neutralizing HIV-1-specific monoclonal antibodies in SHIV-infected rhesus monkeys. Nature.2013;503:224–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Horwitz JA, et al. HIV-1 suppression and durable control by combining single broadly neutralizing antibodies and antiretroviral drugs in humanized mice. Proc Natl Acad Sci U S A.2013;110:16538–16543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klein F, et al. HIV therapy by a combination of broadly neutralizing antibodies in humanized mice. Nature.2012;492:118–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shingai M, et al. Antibody-mediated immunotherapy of macaques chronically infected with SHIV suppresses viraemia. Nature.2013;503:277–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bournazos S, Woof JM, Hart SP, Dransfield I. Functional and clinical consequences of Fc receptor polymorphic and copy number variants. Clin Exp Immunol.2009;157:244–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nimmerjahn F, Ravetch JV. Fcgamma receptors: old friends and new family members. Immunity.2006;24:19–28. [DOI] [PubMed] [Google Scholar]
- 22.Shields RL, et al. High resolution mapping of the binding site on human IgG1 for Fc gamma RI, Fc gamma RII, Fc gamma RIII, and FcRn and design of IgG1 variants with improved binding to the Fc gamma R. J Biol Chem.2001;276:6591–6604. [DOI] [PubMed] [Google Scholar]
- 23.Sondermann P, Huber R, Oosthuizen V, Jacob U. The 3.2-A crystal structure of the human IgG1 Fc fragment-Fc gammaRIII complex. Nature.2000;406:267–273. [DOI] [PubMed] [Google Scholar]
- 24.Qiu WQ, de Bruin D, Brownstein BH, Pearse R, Ravetch JV. Organization of the human and mouse low-affinity Fc gamma R genes: duplication and recombination. Science.1990;248:732–735. [DOI] [PubMed] [Google Scholar]
- 25.Su K, Wu J, Edberg JC, McKenzie SE, Kimberly RP. Genomic organization of classical human low-affinity Fcgamma receptor genes. Genes Immun.2002;3 Suppl 1:S51–56. [DOI] [PubMed] [Google Scholar]
- 26.Selvaraj P, Carpén O, Hibbs ML, Springer TA. Natural killer cell and granulocyte Fc gamma receptor III (CD16) differ in membrane anchor and signal transduction. J Immunol.1989;143:3283–3288. [PubMed] [Google Scholar]
- 27.Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol.2008;8:34–47. [DOI] [PubMed] [Google Scholar]
- 28.Nimmerjahn F, Ravetch JV. Divergent immunoglobulin g subclass activity through selective Fc receptor binding. Science.2005;310:1510–1512. [DOI] [PubMed] [Google Scholar]
- 29.Swanson JA, Hoppe AD. The coordination of signaling during Fc receptor-mediated phagocytosis. J Leukoc Biol.2004;76:1093–1103. [DOI] [PubMed] [Google Scholar]
- 30.Pincetic A, et al. Type I and type II Fc receptors regulate innate and adaptive immunity. Nat Immunol.2014;15:707–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Amigorena S, Salamero J, Davoust J, Fridman WH, Bonnerot C. Tyrosine-containing motif that transduces cell activation signals also determines internalization and antigen presentation via type III receptors for IgG. Nature.1992;358:337–341. [DOI] [PubMed] [Google Scholar]
- 32.Durden DL, Liu YB. Protein-tyrosine kinase p72syk in Fc gamma RI receptor signaling. Blood.1994;84:2102–2108. [PubMed] [Google Scholar]
- 33.Durden DL, Kim HM, Calore B, Liu Y. The Fc gamma RI receptor signals through the activation of hck and MAP kinase. J Immunol.1995;154:4039–4047. [PubMed] [Google Scholar]
- 34.Eiseman E, Bolen JB. Engagement of the high-affinity IgE receptor activates src protein-related tyrosine kinases. Nature.1992;355:78–80. [DOI] [PubMed] [Google Scholar]
- 35.Jouvin MH, Adamczewski M, Numerof R, Letourneur O, Vallé A, Kinet JP. Differential control of the tyrosine kinases Lyn and Syk by the two signaling chains of the high affinity immunoglobulin E receptor. J Biol Chem.1994;269:5918–5925. [PubMed] [Google Scholar]
- 36.Pignata C, Prasad KV, Robertson MJ, Levine H, Rudd CE, Ritz J. Fc gamma RIIIA-mediated signaling involves src-family lck in human natural killer cells. J Immunol.1993;151:6794–6800. [PubMed] [Google Scholar]
- 37.García-García E, Sánchez-Mejorada G, Rosales C. Phosphatidylinositol 3-kinase and ERK are required for NF-kappaB activation but not for phagocytosis. J Leukoc Biol.2001;70:649–658. [PubMed] [Google Scholar]
- 38.Sánchez-Mejorada G, Rosales C. Fcgamma receptor-mediated mitogen-activated protein kinase activation in monocytes is independent of Ras. J Biol Chem.1998;273:27610–27619. [DOI] [PubMed] [Google Scholar]
- 39.Kanakaraj P, Duckworth B, Azzoni L, Kamoun M, Cantley LC, Perussia B. Phosphatidylinositol-3 kinase activation induced upon Fc gamma RIIIA-ligand interaction. J Exp Med.1994;179:551–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ninomiya N, et al. Involvement of phosphatidylinositol 3-kinase in Fc gamma receptor signaling. J Biol Chem.1994;269:22732–22737. [PubMed] [Google Scholar]
- 41.Bracke M, Coffer PJ, Lammers JW, Koenderman L. Analysis of signal transduction pathways regulating cytokine-mediated Fc receptor activation on human eosinophils. J Immunol.1998;161:6768–6774. [PubMed] [Google Scholar]
- 42.Aramburu J, Azzoni L, Rao A, Perussia B. Activation and expression of the nuclear factors of activated T cells, NFATp and NFATc, in human natural killer cells: regulation upon CD16 ligand binding. J Exp Med.1995;182:801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Duchemin AM, Ernst LK, Anderson CL. Clustering of the high affinity Fc receptor for immunoglobulin G (Fc gamma RI) results in phosphorylation of its associated gamma-chain. J Biol Chem.1994;269:12111–12117. [PubMed] [Google Scholar]
- 44.Indik ZK, et al. The high affinity Fc gamma receptor (CD64) induces phagocytosis in the absence of its cytoplasmic domain: the gamma subunit of Fc gamma RIIIA imparts phagocytic function to Fc gamma RI. Exp Hematol.1994;22:599–606. [PubMed] [Google Scholar]
- 45.Ernst LK, Duchemin AM, Anderson CL. Association of the high-affinity receptor for IgG (Fc gamma RI) with the gamma subunit of the IgE receptor. Proc Natl Acad Sci U S A.1993;90:6023–6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kiyoshi M, et al. Structural basis for binding of human IgG1 to its high-affinity human receptor FcγRI. Nat Commun.2015;6:6866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li Y, et al. Increased expression of FcgammaRI/CD64 on circulating monocytes parallels ongoing inflammation and nephritis in lupus. Arthritis Res Ther.2009;11:R6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Uciechowski P, Schwarz M, Gessner JE, Schmidt RE, Resch K, Radeke HH. IFN-gamma induces the high-affinity Fc receptor I for IgG (CD64) on human glomerular mesangial cells. Eur J Immunol.1998;28:2928–2935. [DOI] [PubMed] [Google Scholar]
- 49.Sondermann P, Kaiser J, Jacob U. Molecular basis for immune complex recognition: a comparison of Fc-receptor structures. J Mol Biol.2001;309:737–749. [DOI] [PubMed] [Google Scholar]
- 50.Hibbs ML, et al. Mechanisms for regulating expression of membrane isoforms of Fc gamma RIII (CD16). Science.1989;246:1608–1611. [DOI] [PubMed] [Google Scholar]
- 51.Pearse RN, Kawabe T, Bolland S, Guinamard R, Kurosaki T, Ravetch JV. SHIP recruitment attenuates Fc gamma RIIB-induced B cell apoptosis. Immunity.1999;10:753–760. [DOI] [PubMed] [Google Scholar]
- 52.Amigorena S, et al. Cytoplasmic domain heterogeneity and functions of IgG Fc receptors in B lymphocytes. Science.1992;256:1808–1812. [DOI] [PubMed] [Google Scholar]
- 53.Muta T, Kurosaki T, Misulovin Z, Sanchez M, Nussenzweig MC, Ravetch JV. A 13-amino-acid motif in the cytoplasmic domain of Fc gamma RIIB modulates B-cell receptor signalling. Nature.1994;368:70–73. [DOI] [PubMed] [Google Scholar]
- 54.Ono M, Bolland S, Tempst P, Ravetch JV. Role of the inositol phosphatase SHIP in negative regulation of the immune system by the receptor Fc(gamma)RIIB. Nature.1996;383:263–266. [DOI] [PubMed] [Google Scholar]
- 55.Ono M, Okada H, Bolland S, Yanagi S, Kurosaki T, Ravetch JV. Deletion of SHIP or SHP-1 reveals two distinct pathways for inhibitory signaling. Cell.1997;90:293–301. [DOI] [PubMed] [Google Scholar]
- 56.Lewis VA, Koch T, Plutner H, Mellman I. A complementary DNA clone for a macrophage-lymphocyte Fc receptor. Nature.1986;324:372–375. [DOI] [PubMed] [Google Scholar]
- 57.Brooks DG, Qiu WQ, Luster AD, Ravetch JV. Structure and expression of human IgG FcRII(CD32). Functional heterogeneity is encoded by the alternatively spliced products of multiple genes. J Exp Med.1989;170:1369–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhou MJ, Brown EJ. CR3 (Mac-1, alpha M beta 2, CD11b/CD18) and Fc gamma RIII cooperate in generation of a neutrophil respiratory burst: requirement for Fc gamma RIII and tyrosine phosphorylation. J Cell Biol.1994;125:1407–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Smith P, DiLillo DJ, Bournazos S, Li F, Ravetch JV. Mouse model recapitulating human Fcgamma receptor structural and functional diversity. Proc Natl Acad Sci U S A.2012;109:6181–6186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ahmed AA, et al. Structural characterization of anti-inflammatory immunoglobulin G Fc proteins. J Mol Biol.2014;426:3166–3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sondermann P, Pincetic A, Maamary J, Lammens K, Ravetch JV. General mechanism for modulating immunoglobulin effector function. Proc Natl Acad Sci U S A.2013;110:9868–9872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jellusova J, Nitschke L. Regulation of B cell functions by the sialic acid-binding receptors siglec-G and CD22. Front Immunol.2011;2:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schwab I, Seeling M, Biburger M, Aschermann S, Nitschke L, Nimmerjahn F. B cells and CD22 are dispensable for the immediate antiinflammatory activity of intravenous immunoglobulins in vivo. Eur J Immunol.2012;42:3302–3309. [DOI] [PubMed] [Google Scholar]
- 64.Böhm S, Kao D, Nimmerjahn F. Sweet and sour: the role of glycosylation for the anti-inflammatory activity of immunoglobulin G. Curr Top Microbiol Immunol.2014;382:393–417. [DOI] [PubMed] [Google Scholar]
- 65.Schwab I, Nimmerjahn F. Intravenous immunoglobulin therapy: how does IgG modulate the immune system? Nat Rev Immunol.2013;13:176–189. [DOI] [PubMed] [Google Scholar]
- 66.Anthony RM, Wermeling F, Karlsson MC, Ravetch JV. Identification of a receptor required for the anti-inflammatory activity of IVIG. Proc Natl Acad Sci U S A.2008;105:19571–19578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Anthony RM, Kobayashi T, Wermeling F, Ravetch JV. Intravenous gammaglobulin suppresses inflammation through a novel T(H)2 pathway. Nature.2011;475:110–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fiebiger BM, Maamary J, Pincetic A, Ravetch JV. Protection in antibody- and T cell-mediated autoimmune diseases by antiinflammatory IgG Fcs requires type II FcRs. Proc Natl Acad Sci U S A.2015;112:E2385–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Borthakur S, Andrejeva G, McDonnell JM. Basis of the intrinsic flexibility of the Cε3 domain of IgE. Biochemistry.2011;50:4608–4614. [DOI] [PubMed] [Google Scholar]
- 70.Dhaliwal B, et al. Crystal structure of IgE bound to its B-cell receptor CD23 reveals a mechanism of reciprocal allosteric inhibition with high affinity receptor FcεRI. Proc Natl Acad Sci U S A.2012;109:12686–12691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fujiwara H, et al. The absence of IgE antibody-mediated augmentation of immune responses in CD23-deficient mice. Proc Natl Acad Sci U S A.1994;91:6835–6839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Henningsson F, et al. IgE-mediated enhancement of CD4+ T cell responses in mice requires antigen presentation by CD11c+ cells and not by B cells. PLoS One.2011;6:e21760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yokota A, et al. Two species of human Fc epsilon receptor II (Fc epsilon RII/CD23): tissue-specific and IL-4-specific regulation of gene expression. Cell.1988;55:611–618. [DOI] [PubMed] [Google Scholar]
- 74.Wang TT, et al. Anti-HA Glycoforms Drive B Cell Affinity Selection and Determine Influenza Vaccine Efficacy. Cell.2015;162:160–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Narciso JE, et al. Analysis of the antibody structure based on high-resolution crystallographic studies. N Biotechnol.2011;28:435–447. [DOI] [PubMed] [Google Scholar]
- 76.Albert H, Collin M, Dudziak D, Ravetch JV, Nimmerjahn F. In vivo enzymatic modulation of IgG glycosylation inhibits autoimmune disease in an IgG subclass-dependent manner. Proc Natl Acad Sci U S A.2008;105:15005–15009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lux A, Nimmerjahn F. Impact of differential glycosylation on IgG activity. Adv Exp Med Biol.2011;780:113–124. [DOI] [PubMed] [Google Scholar]
- 78.Nimmerjahn F, Anthony RM, Ravetch JV. Agalactosylated IgG antibodies depend on cellular Fc receptors for in vivo activity. Proc Natl Acad Sci U S A.2007;104:8433–8437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Boruchov AM, Heller G, Veri MC, Bonvini E, Ravetch JV, Young JW. Activating and inhibitory IgG Fc receptors on human DCs mediate opposing functions. J Clin Invest.2005;115:2914–2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dhodapkar KM, et al. Selective blockade of the inhibitory Fcgamma receptor (FcgammaRIIB) in human dendritic cells and monocytes induces a type I interferon response program. J Exp Med.2007;204:1359–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.te Velde AA, de Waal Malefijt R, Huijbens RJ, de Vries JE, Figdor CG. IL-10 stimulates monocyte Fc gamma R surface expression and cytotoxic activity. Distinct regulation of antibody-dependent cellular cytotoxicity by IFN-gamma, IL-4, and IL-10. J Immunol.1992;149:4048–4052. [PubMed] [Google Scholar]
- 82.Cowland JB, Johnsen AH, Borregaard N. hCAP-18, a cathelin/pro-bactenecin-like protein of human neutrophil specific granules. FEBS Lett.1995;368:173–176. [DOI] [PubMed] [Google Scholar]
- 83.Sørensen O, Arnljots K, Cowland JB, Bainton DF, Borregaard N. The human antibacterial cathelicidin, hCAP-18, is synthesized in myelocytes and metamyelocytes and localized to specific granules in neutrophils. Blood.1997;90:2796–2803. [PubMed] [Google Scholar]
- 84.Egesten A, Breton-Gorius J, Guichard J, Gullberg U, Olsson I. The heterogeneity of azurophil granules in neutrophil promyelocytes: immunogold localization of myeloperoxidase, cathepsin G, elastase, proteinase 3, and bactericidal/permeability increasing protein. Blood.1994;83:2985–2994. [PubMed] [Google Scholar]
- 85.Gabay JE, Almeida RP. Antibiotic peptides and serine protease homologs in human polymorphonuclear leukocytes: defensins and azurocidin. Curr Opin Immunol.1993;5:97–102. [DOI] [PubMed] [Google Scholar]
- 86.Owen CA, Campbell MA, Boukedes SS, Campbell EJ. Inducible binding of bioactive cathepsin G to the cell surface of neutrophils. A novel mechanism for mediating extracellular catalytic activity of cathepsin G. J Immunol.1995;155:5803–5810. [PubMed] [Google Scholar]
- 87.Fouret P, du Bois RM, Bernaudin JF, Takahashi H, Ferrans VJ, Crystal RG. Expression of the neutrophil elastase gene during human bone marrow cell differentiation. J Exp Med.1989;169:833–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cerletti C, Tamburrelli C, Izzi B, Gianfagna F, de Gaetano G. Platelet-leukocyte interactions in thrombosis. Thromb Res.2012;129:263–266. [DOI] [PubMed] [Google Scholar]
- 89.Martini F, Riondino S, Pignatelli P, Gazzaniga PP, Ferroni P, Lenti L. Involvement of GD3 in platelet activation. A novel association with Fcgamma receptor. Biochim Biophys Acta.2002;1583:297–304. [DOI] [PubMed] [Google Scholar]
- 90.Takai T, Li M, Sylvestre D, Clynes R, Ravetch JV. FcR gamma chain deletion results in pleiotrophic effector cell defects. Cell.1994;76:519–529. [DOI] [PubMed] [Google Scholar]
- 91.Guillerey C, Huntington ND, Smyth MJ. Targeting natural killer cells in cancer immunotherapy. Nat Immunol.2016;17:1025–1036. [DOI] [PubMed] [Google Scholar]
- 92.Vivier E, et al. Innate or adaptive immunity? The example of natural killer cells. Science.2011;331:44–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Clynes R, Maizes JS, Guinamard R, Ono M, Takai T, Ravetch JV. Modulation of immune complex-induced inflammation in vivo by the coordinate expression of activation and inhibitory Fc receptors. J Exp Med.1999;189:179–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sutterwala FS, Noel GJ, Clynes R, Mosser DM. Selective suppression of interleukin-12 induction after macrophage receptor ligation. J Exp Med.1997;185:1977–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bergtold A, Desai DD, Gavhane A, Clynes R. Cell surface recycling of internalized antigen permits dendritic cell priming of B cells. Immunity.2005;23:503–514. [DOI] [PubMed] [Google Scholar]
- 96.Hoffmann E, Kotsias F, Visentin G, Bruhns P, Savina A, Amigorena S. Autonomous phagosomal degradation and antigen presentation in dendritic cells. Proc Natl Acad Sci U S A.2012;109:14556–14561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bonnerot C, et al. syk protein tyrosine kinase regulates Fc receptor gamma-chain-mediated transport to lysosomes. EMBO J.1998;17:4606–4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Desai DD, et al. Fc gamma receptor IIB on dendritic cells enforces peripheral tolerance by inhibiting effector T cell responses. J Immunol.2007;178:6217–6226. [DOI] [PubMed] [Google Scholar]
- 99.Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat Med.2000;6:443–446. [DOI] [PubMed] [Google Scholar]
- 100.Dhodapkar KM, et al. Selective blockade of inhibitory Fcgamma receptor enables human dendritic cell maturation with IL-12p70 production and immunity to antibody-coated tumor cells. Proc Natl Acad Sci U S A.2005;102:2910–2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kalergis AM, Ravetch JV. Inducing tumor immunity through the selective engagement of activating Fcgamma receptors on dendritic cells. J Exp Med.2002;195:1653–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Diaz de Ståhl T, Heyman B. IgG2a-mediated enhancement of antibody responses is dependent on FcRgamma+ bone marrow-derived cells. Scand J Immunol.2001;54:495–500. [DOI] [PubMed] [Google Scholar]
- 103.Regnault A, et al. Fcgamma receptor-mediated induction of dendritic cell maturation and major histocompatibility complex class I-restricted antigen presentation after immune complex internalization. J Exp Med.1999;189:371–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.DiLillo DJ, Ravetch JV. Differential Fc-Receptor Engagement Drives an Anti-tumor Vaccinal Effect. Cell.2015;161:1035–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yuasa T, et al. Deletion of fcgamma receptor IIB renders H-2(b) mice susceptible to collagen-induced arthritis. J Exp Med.1999;189:187–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Brownlie RJ, et al. Distinct cell-specific control of autoimmunity and infection by FcgammaRIIb. J Exp Med.2008;205:883–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Clatworthy MR, Smith KG. FcgammaRIIb balances efficient pathogen clearance and the cytokine-mediated consequences of sepsis. J Exp Med.2004;199:717–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bolland S, Ravetch JV. Spontaneous autoimmune disease in Fc(gamma)RIIB-deficient mice results from strain-specific epistasis. Immunity.2000;13:277–285. [DOI] [PubMed] [Google Scholar]
- 109.Bolland S, Yim YS, Tus K, Wakeland EK, Ravetch JV. Genetic modifiers of systemic lupus erythematosus in FcgammaRIIB(−/−) mice. J Exp Med.2002;195:1167–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Xiang Z, et al. FcgammaRIIb controls bone marrow plasma cell persistence and apoptosis. Nat Immunol.2007;8:419–429. [DOI] [PubMed] [Google Scholar]
- 111.Blank MC, et al. Decreased transcription of the human FCGR2B gene mediated by the −343 G/C promoter polymorphism and association with systemic lupus erythematosus. Hum Genet.2005;117:220–227. [DOI] [PubMed] [Google Scholar]
- 112.Su K, et al. A promoter haplotype of the immunoreceptor tyrosine-based inhibitory motif-bearing FcgammaRIIb alters receptor expression and associates with autoimmunity. I. Regulatory FCGR2B polymorphisms and their association with systemic lupus erythematosus. J Immunol.2004;172:7186–7191. [DOI] [PubMed] [Google Scholar]
- 113.Tackenberg B, et al. Impaired inhibitory Fcgamma receptor IIB expression on B cells in chronic inflammatory demyelinating polyneuropathy. Proc Natl Acad Sci U S A.2009;106:4788–4792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Floto RA, et al. Loss of function of a lupus-associated FcgammaRIIb polymorphism through exclusion from lipid rafts. Nat Med.2005;11:1056–1058. [DOI] [PubMed] [Google Scholar]
- 115.Bournazos S, DiLillo DJ, Ravetch JV. The role of Fc-FcγR interactions in IgG-mediated microbial neutralization. J Exp Med.2015;212:1361–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Pietzsch J, et al. A mouse model for HIV-1 entry. Proc Natl Acad Sci U S A.2012;109:15859–15864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lu CL, et al. Enhanced clearance of HIV-1-infected cells by broadly neutralizing antibodies against HIV-1 in vivo. Science.2016;352:1001–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bournazos S, Klein F, Pietzsch J, Seaman MS, Nussenzweig MC, Ravetch JV. Broadly Neutralizing Anti-HIV-1 Antibodies Require Fc Effector Functions for In Vivo Activity. Cell.2014;158:1243–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Halper-Stromberg A, et al. Broadly Neutralizing Antibodies and Viral Inducers Decrease Rebound from HIV-1 Latent Reservoirs in Humanized Mice. Cell.2014;158:989–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Haigwood NL, et al. Passive immunotherapy in simian immunodeficiency virus-infected macaques accelerates the development of neutralizing antibodies. J Virol.2004;78:5983–5995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ng CT, et al. Passive neutralizing antibody controls SHIV viremia and enhances B cell responses in infant macaques. Nat Med.2010;16:1117–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Schoofs T, et al. HIV-1 therapy with monoclonal antibody 3BNC117 elicits host immune responses against HIV-1. Science.2016;352:997–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bibeau F, et al. Impact of Fc{gamma}RIIa-Fc{gamma}RIIIa polymorphisms and KRAS mutations on the clinical outcome of patients with metastatic colorectal cancer treated with cetuximab plus irinotecan. J Clin Oncol.2009;27:1122–1129. [DOI] [PubMed] [Google Scholar]
- 124.Zhang W, et al. FCGR2A and FCGR3A polymorphisms associated with clinical outcome of epidermal growth factor receptor expressing metastatic colorectal cancer patients treated with single-agent cetuximab. J Clin Oncol.2007;25:3712–3718. [DOI] [PubMed] [Google Scholar]
- 125.Tamura K, et al. FcγR2A and 3A polymorphisms predict clinical outcome of trastuzumab in both neoadjuvant and metastatic settings in patients with HER2-positive breast cancer. Ann Oncol.2011;22:1302–1307. [DOI] [PubMed] [Google Scholar]
- 126.Goede V, et al. Obinutuzumab plus chlorambucil in patients with CLL and coexisting conditions. N Engl J Med.2014;370:1101–1110. [DOI] [PubMed] [Google Scholar]