

Abstract
Cystinosis is an autosomal recessive storage disease due to impaired transport of cystine out of lysosomes. Since the accumulation of intracellular cystine affects all organs and tissues, the management of cystinosis requires a specialized multidisciplinary team consisting of pediatricians, nephrologists, nutritionists, ophthalmologists, endocrinologists, neurologists' geneticists, and orthopedic surgeons. Treatment with cysteamine can delay or prevent most clinical manifestations of cystinosis, except the renal Fanconi syndrome. Virtually all individuals with classical, nephropathic cystinosis suffer from cystinosis metabolic bone disease (CMBD), related to the renal Fanconi syndrome in infancy and progressive chronic kidney disease (CKD) later in life. Manifestations of CMBD include hypophosphatemic rickets in infancy, and renal osteodystrophy associated with CKD resulting in bone deformities, osteomalacia, osteoporosis, fractures, and short stature. Assessment of CMBD involves monitoring growth, leg deformities, blood levels of phosphate, electrolytes, bicarbonate, calcium, and alkaline phosphatase, periodically obtaining bone radiographs, determining levels of critical hormones and vitamins, such as thyroid hormone, parathyroid hormone, 25(OH) vitamin D, and testosterone in males, and surveillance for nonrenal complications of cystinosis such as myopathy. Treatment includes replacement of urinary losses, cystine depletion with oral cysteamine, vitamin D, hormone replacement, physical therapy, and corrective orthopedic surgery. The recommendations in this article came from an expert meeting on CMBD that took place in Salzburg, Austria, in December 2016.
Keywords: chronic kidney disease, CKD‐MBD, cystinosis, cystinosis metabolic bone disease, Fanconi syndrome, hypophosphatemic rickets, transplantation
1. INTRODUCTION
Cystinosis is an autosomal recessive lysosomal storage disorder caused by mutations in the CTNS gene.1, 2 CTNS encodes the lysosomal cystine transporter cystinosin, whose deficiency results in the accumulation of cystine in all organs and tissues. In the most common nephropathic form of cystinosis, infants present with Fanconi syndrome, a generalized dysfunction of the proximal tubule characterized by urinary wasting of water, electrolytes, minerals, bicarbonate, glucose, amino acids, and other molecules. The metabolic consequences include hypophosphatemic rickets and growth failure. Later in life, individuals with cystinosis also suffer from mineral and bone disorders related to chronic kidney disease (CKD‐MBD), including renal osteodystrophy, resulting in a complex bone phenotype termed cystinosis metabolic bone disease (CMBD).3, 4, 5 The treatment of cystinosis involves replacement of renal losses, symptomatic management of nonrenal complications and, most critically, cystine‐depleting therapy with oral cysteamine.6, 7 Both the early Fanconi syndrome and later CKD contribute to the bone changes of CMBD. In addition, a primary osteoblast and osteoclastic defect, abnormal thyroid metabolism, glucocorticoid treatment after renal transplantation and, rarely, cysteamine toxicity may further complicate CMBD.8 Nevertheless, there exists a paucity of specific recommendations for diagnosis and management of CMBD.
Here, we review the clinical features of cystinosis related to CMBD and present recommendations for management, developed during a meeting in Salzburg, Austria, in December 2016. They are aimed at nephrologists, metabolic physicians, and general practitioners who care for patients with cystinosis.
2. CYSTINOSIS BACKGROUND
Infants with nephropathic cystinosis appear normal at birth. However, failure to growth generally occurs at 6 to 9 months of age, when renal Fanconi syndrome develops.5 Early diagnosis is critical because cystinosis is treatable. Diagnosis is primary based on detection of elevated cystine concentration in polymorphonuclear leukocytes at any age, and identification of cystine crystals in the cornea on slit lamp examination in older children (age >2 years). If available, diagnosis should be confirmed by identification of biallelic pathogenic variants in CTNS on molecular genetic testing. In case of a positive family history increased cystine content in cultured fibroblasts or in the placenta at the time of birth are also proving.
The Fanconi syndrome in cystinosis causes severe polyuria (2‐10 L/d), polydipsia, dehydration (sometimes with fever), hypophosphatemia, hypokalemia, and hypochloremic metabolic acidosis.5 Treatment involves replacement of tubular losses of water, electrolytes, bicarbonate, phosphate, vitamin D, and other nutrients.5 Children should have free access to water, and intravenous delivery may be required in cases of dehydration. Potassium is typically supplemented 3 to 4 times per day as the citrate, bicarbonate, or chloride salts, and high doses (6‐10 mEq/kg/d) may only achieve a serum level of 3.0 mEq/mL. Most patients receive sufficient sodium from their diet and medications. Early gastric tube placement may be required to deliver nutrition or medications.
Indomethacin, which can decrease polyuria by 30% to 70% and improve weight gain in young patients,9 may decrease renal perfusion due to its suppressive effects on local production of prostaglandins, which are mandatory to maintain renal perfusion in states of dehydration. Therefore, indomethacin should be discontinued if a patient becomes dehydrated, hypotensive, or develops advanced CKD (>stage 3).
Without cysteamine treatment, renal glomerular damage progresses inexorably, culminating in end‐stage kidney disease (ESKD) by approximately 10 years of age and requiring dialysis or kidney transplantation; patients with cystinosis do well following renal transplantation. Oral cysteamine therapy drastically lowers intracellular cystine and, while it does not ameliorate the Fanconi syndrome, slows the progression of CKD,6, 7 delays the need for renal replacement therapy,10, 11, 12 enhances growth, and prevents late complications of the disease.13 The recommended dosage is 60 to 90 mg/kg/d or 1.3‐1.95 g/m2/d, intended to achieve a leukocyte cystine level <1.0 nmol half‐cystine/mg of protein. Oral cysteamine has an unpleasant taste and smell, and induces nausea and other digestive complaints, so only one third of patients strictly comply with the dosage regimen, particularly in adolescence.11, 14 Patients with gastric side effects of cysteamine therapy can benefit from medications such as omeprazole or ranitidine.15
Other tissue and organ damage occur in cystinosis due to the accumulation of intracellular cystine. Corneal crystals are visible by 16 months of age and can be dissolved with topical cysteamine eyedrops, that is, Cystaran16 or Cystadrops.17 Hypothyroidism results from direct damage to the thyroid gland, the first sign usually being elevated thyroid‐stimulating hormone (TSH). Growth retardation represents a major problem in cystinosis. Typically, infants are at the third percentile for height at 1 year of age, and without adequate treatment, have low growth velocity.4 While nutrition, phosphate supplementation, and oral cysteamine therapy can achieve a normal growth rate, the addition of growth hormone (GH) therapy can provide catch‐up growth, particularly when used in children prior to dialysis.18
Gonadal dysfunction, that is, hypergonadotropic hypogonadism, largely affects males, often results in delayed puberty, and is characterized by low levels of testosterone and very high levels of luteinizing hormone (LH) and follicle‐stimulating hormone (FSH). Testosterone supplementation can be used to manage gonadal dysfunction. Since adult males with cystinosis are infertile but show viable epidydimal sperm,19 sperm preservation can be considered. Puberty is generally delayed by 1 to 2 years in males and some females with cystinosis, but ovarian function is preserved and several women with cystinosis have delivered healthy babies.20
Other complications of cystinosis include a distal vacuolar myopathy (Figure 1), exocrine and endocrine pancreatic dysfunction, benign intracranial hypertension, and retinal blindness,21 often causing restrictive lung disease and/or swallowing difficulty.22 Most complications can be prevented with early and lifelong cystine‐depleting therapy.
Figure 1.
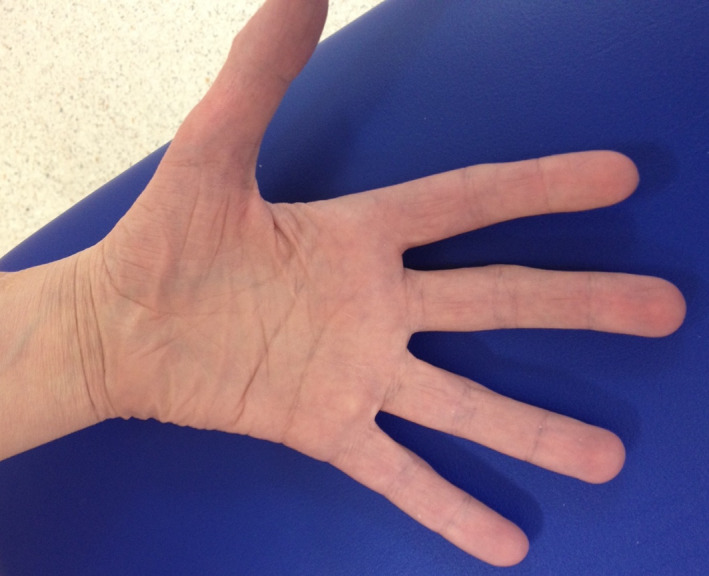
Distal myopathy of cystinosis, with atrophy of the thenar and hypothenar eminences
3. CYSTINOSIS METABOLIC BONE DISEASE
3.1. Clinical characteristics
CMBD can manifest as rickets (Figure 2) and renal osteodystrophy, including growth failure, bone pain, deformations, osteomalacia, and osteoporosis. Patients may have short stature spontaneous fractures (Table 1). In infants and young children, rickets causes bone deformities, including genu valgum (Figure 3) and genu varus, making walking painful enough to delay ambulation.5, 23 Clinical signs of rickets typically include widening of the forearm at the wrist and thickening of the costochondral junctions. In addition, a rachitic rosary and Harrison's groove may also develop. Another radiographic sign of rickets is increased thickness of the growth plates of the long bones, with irregular, hazy appearance at the diaphyseal line.
Figure 2.
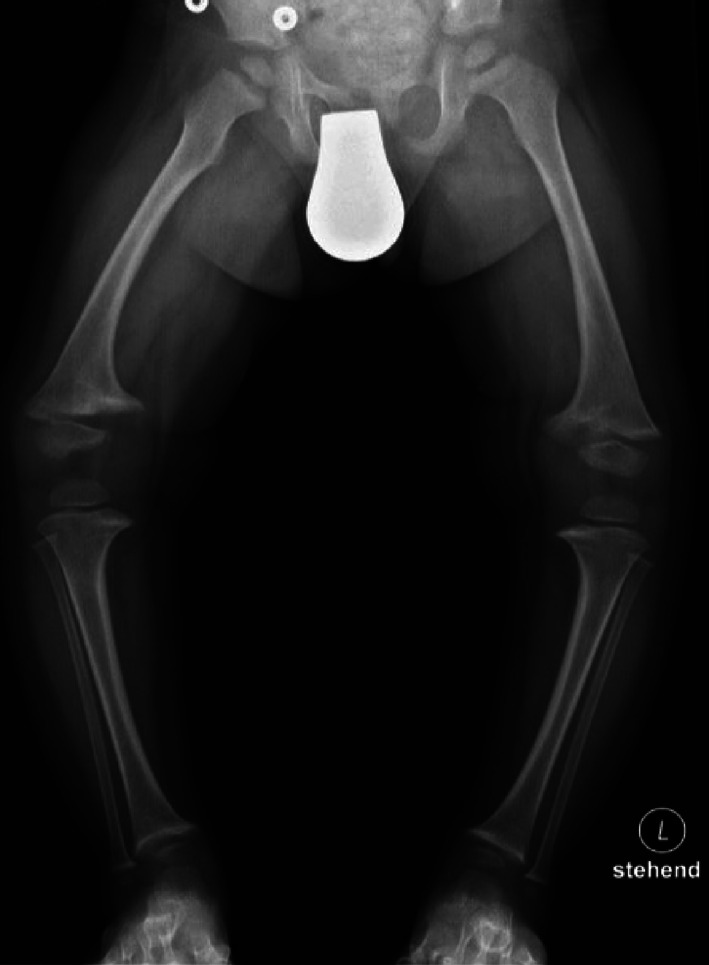
Active rachitic bone disease on X‐ray of both legs. Note reduced bone density, widening of the metaphyses, and fraying of the epiphyses
Table 1.
Pathological conditions of CMBD
| Pathophysiology | Histology | Radiography | Clinical picture | |
|---|---|---|---|---|
| Osteomalacia | Deficit of mineralization of bone matrix:
|
|
|
|
| Osteoporosis |
|
|
|
|
Figure 3.
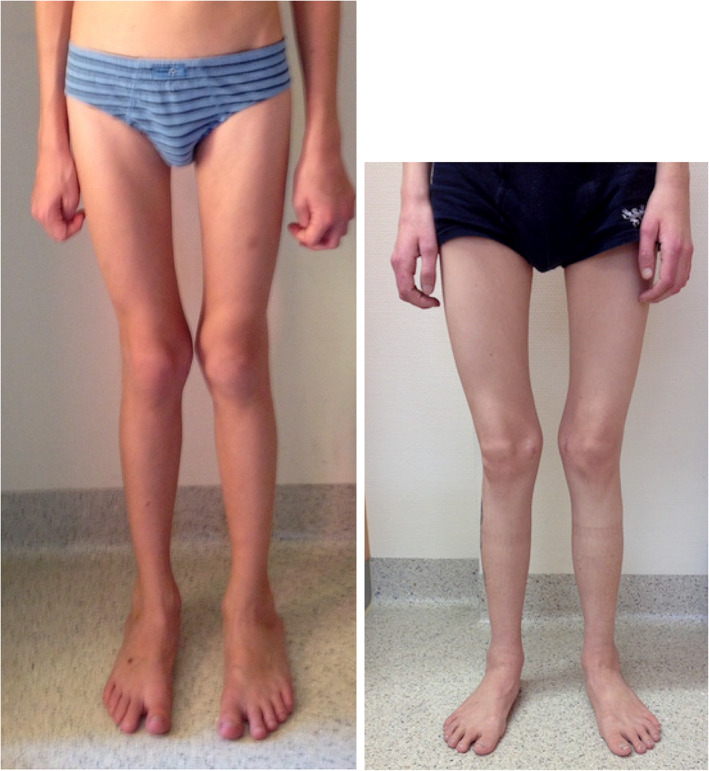
Muscular atrophy and bone deformation, that is, genu valgum
As children with cystinosis lose glomerular function, CKD‐MBD becomes prominent.24, 25 CKD‐MBD is a systemic disorder of mineral and bone metabolism manifested by one or a combination of the following: (a) abnormalities of calcium, phosphorus, intact serum parathyroid hormone (PTH), or vitamin D metabolism; (b) abnormalities in bone turnover, mineralization, volume, linear growth, or strength (renal osteodystrophy); and (c) vascular or other soft‐tissue calcification.24, 25
3.2. Pathogenesis of CMBD
Many factors contribute to the bone disease of cystinosis (Figure 4). Perhaps the most devastating is early‐onset Fanconi syndrome, with its renal losses of phosphate, calcium, and bicarbonate and diminished synthesis of active vitamin D causing hypophosphatemia, acidosis, and sometimes hypocalcemia, leading to rickets and osteomalacia.26, 27 Acidosis due to tubular losses of bicarbonate impairs bone mineralization, and caloric and protein malnutrition due to ingestion of excess fluids lacking calories, along with CKD, may further contribute to poor bone health. Later in childhood, progressive loss of glomerular function results in CKD‐MBD, as observed in patients with other causes of CKD.
Figure 4.
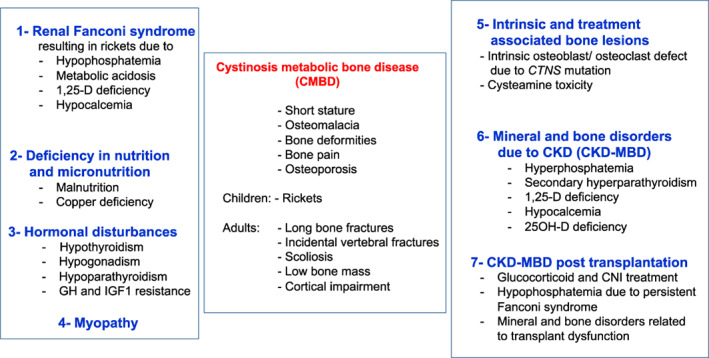
Current understanding of the abnormalities leading to cystinosis metabolic bone disease (CMBD). Virtually all individuals with classical, nephropathic cystinosis suffer from CMBD, related to the renal Fanconi syndrome in infancy and progressive chronic kidney disease (CKD) later in life inducing CKD‐associated mineral and bone disorders (CKD‐MBD). Malnutrition and copper deficiency, but also hormonal disturbances, myopathy, and transplantation may worsen the clinical picture. The cystinosin defect also induces intrinsic bone defects such as osteoblastic and osteoclastic dysfunction. The impact of cysteamine on bone deserves further studies, but high doses of cysteamine may contribute to CMBD. Taken together, all these mechanisms can lead to bone deformities and pains, osteoporosis, fractures, cortical impairment, and short stature in teenagers and young adults
In CMBD, interactions among bone, joints, and muscle are critical. Muscle and bone form a functional unit, with mechanical stimulation through muscle activity driving bone development.28 Consequently, impaired muscle function, which is often observed in cystinosis, leads to disturbances of bone development. Furthermore, reduced plasma and muscle levels of carnitine in pretransplant patients with cystinosis29 may impair the functionality of the muscle/bone unit.21
GH, LH, FSH, androgens/estrogens, insulin‐like growth factor (IGF)‐1, insulin, amylin, and TSH/thyroxine all contribute to maintaining normal bone metabolism. This balance is disrupted in cystinosis. Cystine accumulation in thyroid follicular cells causes fibrosis and atrophy.9 Moreover, decreased thyroglobulin synthesis due to endoplasmic reticulum stress/unfolded protein response and impaired lysosomal processing has been demonstrated in a mouse model of cystinosis.30 Consequently, hypothyroidism often appears in the first decade of life and can contribute to growth retardation.15 In addition, poor nutrition may result in reduced IGF‐1 serum levels and, in advanced CKD, lack of sensitivity to endogenous GH and IGF‐1 can further impede growth.31, 32 Cystine crystals in bone may also impair growth. Finally, treatment with glucocorticoids limits catch‐up growth after renal transplantation and may cause osteoporosis with increased fracture risk, as seen in other patients with renal allografts.8
Interestingly, knockout of Ctns in mice did not result in full‐blown Fanconi syndrome, yet these animals showed osteopenia with decreased mineralization and cortical thickness (long bones and vertebrae), raising the hypothesis of a specific underlying bone defect in cystinosis.33 Indeed, preclinical studies suggest that mutations in CTNS may reduce the ability of osteoblast precursor cells to transform into mature osteoblasts capable of synthesizing osteoid, further contributing to defective mineralization and rickets.34 Moreover, low doses of cysteamine in vitro stimulate osteoblastic differentiation and mineralization, with an inhibitory effect at higher doses, possibly explaining the bone toxicity observed in patients receiving high doses of cysteamine. Cystinosin may also be required for proper osteoclastic and osteoblastic activity.35, 36
3.3. Assessing CMBD
Serum mineral and enzyme values provide measures of bone health in both children and adults with cystinosis (Table 2). In children with hypophosphatemic rickets, serum phosphate, bicarbonate, and potassium reflect renal losses and the efficacy of replacement therapy, and alkaline phosphatase (ALP) is a biomarker of rickets and osteomalacia.37 Elevated ALP also indicates increased bone turnover as a feature of CKD‐MBD. Measurement of serum PTH, calcium, 25(OH) vitamin D, and phosphate also serve as the mainstays of monitoring for CKD‐MBD.38 Nevertheless, infants and adolescents with cystinosis may develop bone disease despite largely “normal” blood levels of phosphate, calcium and ALP; normal blood levels have a wide standard range and daily fluctuations, are not always biochemically evident, and may have a large cumulative effect upon bone metabolism. Therefore, urine losses for calcium and phosphate should be monitored as well.
Table 2.
Recommended tests for CMBD
| Assessment | Methods and frequency |
|---|---|
| Growth |
|
| Bone metabolism |
|
| Bone deformities |
|
| Growth hormone |
|
| Thyroid function |
|
| Gonadal function |
|
| Muscle function |
|
| Other |
|
Abbreviations: ALP, alkaline phosphatase; CKD, chronic kidney disease; FSH, follicle‐stimulating hormone; GH, growth hormone; IGF‐1, Insulin‐like growth factor 1; iPTH, intact serum parathyroid hormone; LH, luteinizing hormone; TSH, thyroid‐stimulating hormone.
New biomarkers may better reflect bone cell activity in CMBD,39 and pediatric reference values are available.40 Bone ALP, an osteoblast enzyme, is a sensitive and specific marker of bone formation and remodeling during periods of rapid longitudinal growth and in cases of rickets.24, 38 TRAP5b is considered a specific marker of late osteoclast differentiation,38 and sclerostin is an osteocyte‐derived inhibitor of bone formation.38, 41, 42 Fibroblast growth factor 23 (FGF23), released by osteocytes and osteoblasts, helps maintain mineral and vitamin D homeostasis, and is the earliest detectable abnormality in bone mineral metabolism in CKD patients.38, 43 However, FGF23 levels are typically normal in patients with cystinosis prior to dialysis, possibly due to hypophosphatemia.44
Radiographic imaging plays an important role in assessing CMBD. Although dual‐energy X‐ray absorptiometry (DXA) allows quantification of bone mineral density,45 it cannot distinguish the different stages of CKD.46 DXA studies are of not recommended in the clinical management of cystinosis, since the results are influenced by bone size and body height, do not distinguish between a mineralization defect (osteomalacia) and loss of bone tissue (osteoporosis),47 and have poor predictive value for fractures (eg, in stage 3‐5 CKD). In fact, bone mineral density is often normal in pediatric CKD patients, when data are corrected for reduced height.48, 49
Quantitative computed tomography (QCT) and peripheral QCT (pQCT) provide separate measures of cortical and trabecular bone in the central and peripheral skeleton and true volumetric density, without being confounded by body size.50 High‐resolution pQCT can assess trabecular microarchitecture and bone biomechanical properties (finite element analysis). However, there are limited reference data in young children,45 the measurement requires children to sit still for 2 to 3 minutes, and the results only provide a “window” into the bone at a single time point.51
Iliac crest bone biopsies can help establish a diagnosis in an individual with an unclear bone disorder.52 Biopsies provide dynamic histomorphometric measures, such as mineralizing surface, bone formation rate, and mineral apposition rate. Standardized nomenclature,53 along with normative data for iliac bone histomorphometry,54 allow comparison with patient data. However, bone biopsy is invasive, cannot be repeated frequently, and requires prior tetracycline labeling for optimal evaluation.55
3.4. Management of CMBD
The treatment of Fanconi syndrome largely involves oral replacement of urinary losses plus nutrition that provides the recommended daily requirements of protein and calories (Table 3). Phosphate repletion aims to resolve rickets and normalize ALP activity and serum phosphate levels, which are measured immediately prior to dosing. A typical starting dose is 30 to 40 mg/kg/d based on elemental phosphorus in 3 to 5 doses equally spaced throughout the day and administered at the same times daily. However, treatment should be increased (up to 80 mg/kg/d) to treat acute rickets or lowered to avoid abdominal discomfort and diarrhea. For small children, phosphate tablets can be crushed and dissolved in tea or water, and intravenous phosphate solutions can be given orally. Phosphate should not be administered at the same time as calcium, since this can lead to precipitation, although providing phosphate with milk products is acceptable. Phosphate administration can contribute to nephrocalcinosis, but is still required. Oral phosphate supplements may be reduced in patients with advanced CKD.
Table 3.
Treatment of CMBD
| Treatment | Dosing |
|---|---|
| Phosphate |
|
| Citrate/bicarbonate |
|
| Calcium/active and native vitamin D |
|
| GH |
|
| Parathyroid levels |
|
| Sex hormone replacement therapy |
|
| L‐Thyroxine |
|
| Cysteamine |
|
Abbreviations: CKD, chronic kidney disease; GH, growth hormone; HbA1c, glycated hemoglobin; PTH, parathyroid hormone; TSH, thyroid‐stimulating hormone.
Calcitriol or alfacalcidol, can treat and prevent deficiency of active vitamin D and hypocalcemia. In addition, both agents improve phosphate reabsorption from the gut and prevent phosphate‐driven secondary hyperparathyroidism. Initially, a calcitriol or alfacalcidol dose of 0.1 to 1 μg is used to cure rickets, but this can be reduced at a later stage, when laboratory, radiological and clinical findings are normalized. High doses of active vitamin D may increase hypercalciuria and nephrocalcinosis, and can promote extraskeletal (vascular) calcifications as reported in other patients with advanced CKD.56 Supplementation of native vitamin D (eg, cholecalciferol) should be titrated to achieve normal values.57 For most patients, calcium requirements can be met by adequate nutrition and vitamin D administration, but calcium supplementation can serve as “insurance” against imperceptible daily losses of calcium that eventually lower bone density. However, calcium supplements may contribute to the development of ectopic (vascular) calcifications in patients with advanced CKD and cumulative calcium intake should be within the recommended daily allowance as recommended for other patients with CKD.58
Left wrist and/or knee once a year radiographs will gauge the presence/recurrence/resolution of rickets (based on increased thickness of the growth plates of the long bones, with irregular, hazy appearance at the diaphyseal line). Bone biopsies appear justified only when obtained during corrective orthopedic surgery or to determine the etiology and treatment of longstanding refractory bone deformities.
Orthopedic surgery (temporary hemiepiphysiodesis or osteotomy) can correct persistent leg bowing. Surgery during puberty is preferred, with metabolic control optimized prior to surgery to prevent recurrence of leg bowing. Active vitamin D may be paused during prolonged immobilization to prevent hypercalcemia.
Initiation of GH treatment may be considered at any stage of CKD in the presence of persistent short stature (<3rd percentile) and low height velocity (<25th percentile), despite adequate nutritional intake, metabolic control, and cysteamine treatment. Before starting treatment, bone age (X‐ray of the left hand) should be assessed in children aged >5 years to confirm open epiphyses and to determine bone age. IGF‐1 and thyroid hormone levels should be obtained to rule out GH deficiency and hypothyroidism.59, 60 During treatment, calcium, phosphate, HbA1c, fasting glucose, PTH, and thyroid hormone levels should be monitored, and the presence of rickets and scoliosis should be evaluated. Persistent headaches or visual acuity loss should prompt an ophthalmological examination to rule out intracranial hypertension. GH treatment should be discontinued if progressive scoliosis or intracranial hypertension occurs.
For patients with cystinosis and CKD stage 1 to 2, we suggest keeping PTH levels within the normal range. However, such patients may have suppressed PTH levels caused by treatment with active vitamin D. Therefore, PTH levels should be checked at least every 6 to 12 months. In patients with CKD stages 3 to 5, PTH levels should be maintained in the target range recommended for other renal diseases, using dietary measures, active/native vitamin D, calcimimetics, and/or oral phosphate binders.24, 58, 61, 62 As CKD progresses, phosphate supplements should be reduced or even stopped. Some patients with cystinosis and ESKD may only require a low dose of oral phosphate binders, if any, due to the ongoing renal phosphate wasting.
In patients on renal replacement therapy, persistent Fanconi syndrome may impair bone health,63 and native kidney nephrectomy might be considered. Minimal glucocorticoid exposure should be considered to permit attainment of a normal adult height.64, 65 Preservation of transplant function and optimizing metabolic control can further improve growth.64 GH may be started in case of persistent short stature (>12 months after transplantation). In addition, early and diligent physical and rehabilitation therapy, including muscle strengthening and targeted exercise, can prevent and/or improve skeletal deformities, and possibly eliminate the need for extensive orthopedic surgeries.
4. SUMMARY
For any patient with an established diagnosis of nephropathic cystinosis, regular assessments of growth, skeletal status, bone deformities, and walking difficulties should be performed. Rickets should be treated with phosphate, bicarbonate/citrate, and vitamin D replacement. Treatment with cysteamine helps to minimize possible metabolic bone disease related to CKD by slowing deterioration of renal function. Management of bone disease should be undertaken by a multidisciplinary team including a nephrologist, dietician, physiotherapist, and experienced orthopedic surgeon. Future studies should focus on the mechanisms of cystinosis bone disease, specifically the functional interactions among bone, muscle, and joints.
CONTRIBUTING PATIENT SUPPORT GROUPS
Asociación Cistinosis, Barcelona, Espaῆa, Cystinosis Group Netherlands, Amstelveen, Netherlands, Cystinosis Foundation UK, Leyland Lancashire, England, Cystinosis Research Network US, Lake Forest, USA, Cystinosis Foundation, Moraga, CA, USA,Cystinosis Foundation Ireland, Cavendish Row, Ireland, Cystinose‐Selbsthilfe e.V., Enningerlohe, Germany, Cystinosis Network Europe, Ratingen, Germany.
CONFLICT OF INTEREST
Atif Awan, Justine Bacchetta, Frank Rauch, Erik Harms, Bernd Hoppe, Nadine Herzig, Bernd Hoppe, Ewa Elenberg, William A. Gahl, Christian Koeppl, Elena Levtchenko, Malcolm Lewis, Galina Nesterova, Fernando Santos, Karl P. Schlingmann, Aude Servais, Neveen A. Soliman, Guentehr Steidle, Rezan Topaloglu, Ulrike Treikauskas, Alexey Tsygin, Koenraad Veys, Josef Zustin, Rodo v. Vigier have no conflict of interest. Gema Ariceta received speaker honorarium from Chiesi, Orphan and Horizon and consulting fee from Chiesi. Susanne Bechtold received speaker honorarium from Sandoz and consulting fee from Alexion. Carsten Bergmann received speaker honorarium from Alexion. George Deschennes received consulting honorarium from Chiesi. Dieter Haffner received speaker honorarium from Horizon, Chiesi and Orphan. Katharina Hohenfellner received speaker fee from Orphan and consulting honorarium from Leadiant. This article does not contain any studies with human or animal subjects performed by the any of the authors.
ACKNOWLEDGMENTS
We highly appreciate the help of Vanessa Schneider in organizing the meeting and coordinating the paper. The expert meeting on CMBD took place in Salzburg, Austria, in December 2016, and was organized by Cystinosis Foundation Germany. The content of the article has not been influenced by any sponsors.
Hohenfellner K, Rauch F, Ariceta G, et al. Management of bone disease in cystinosis: Statement from an international conference. J Inherit Metab Dis. 2019;42:1019–1029. 10.1002/jimd.12134
Communicating Editor: Gregory M. Pastores
Funding information Cystinosis Foundation Germany ; Cystinosis Foundation
REFERENCES
- 1. Gahl WA, Bashan N, Tietze F, Bernardini I, Schulman JD. Lysosomal cystine transport is defective in cystinosis. Science. 1982;217:1263‐1265. [DOI] [PubMed] [Google Scholar]
- 2. Town M, Jean G, Cherqui S, et al. A novel gene encoding an integral membrane protein is mutated in nephropathic cystinosis. Nat Genet. 1998;18:319‐324. [DOI] [PubMed] [Google Scholar]
- 3. Cherqui S, Courtoy PJ. The renal Fanconi syndrome in cystinosis: pathogenic insights and therapeutic perspectives. Nat Rev Nephrol. 2017;13:115‐131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Elmonem MA, Veys KR, Soliman NA, van Dyck M, van den Heuvel LP, Levtchenko E. Cystinosis: a review. Orphanet J Rare Dis. 2016;11:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nesterova G, Gahl WA. In: Pagon RA, Adam MP, Ardinger HH, et al., eds. GeneReviews(®) [Internet]. Seattle (WA): University of Washington, Seattle; 2001:1993‐2017. [Google Scholar]
- 6. Gahl WA, Reed GF, Thoene JG, et al. Cysteamine therapy for children with nephropathic cystinosis. N Engl J Med. 1987;316:971‐977. [DOI] [PubMed] [Google Scholar]
- 7. Markello TC, Bernardini IM, Gahl WA. Improved renal function in children with cystinosis treated with cysteamine. N Engl J Med. 1993;328:1157‐1162. [DOI] [PubMed] [Google Scholar]
- 8. Zimakas PJ, Sharma AK, Rodd CJ. Osteopenia and fractures in cystinotic children post renal transplantation. Pediatr Nephrol. 2003;18:384‐390. [DOI] [PubMed] [Google Scholar]
- 9. Emma F, Nesterova G, Langman C, et al. Nephropathic cystinosis: an international consensus document. Nephrol Dial Transplant. 2014;29(suppl 4):iv87‐iv94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brodin‐Sartorius A, Tête M‐J, Niaudet P, et al. Cysteamine therapy delays the progression of nephropathic cystinosis in late adolescents and adults. Kidney Int. 2012;81:179‐189. [DOI] [PubMed] [Google Scholar]
- 11. Kleta R, Bernardini I, Ueda M, et al. Long‐term follow‐up of well treated nephropathic cystinosis patients. J Pediatr. 2004;145:555‐560. [DOI] [PubMed] [Google Scholar]
- 12. Nesterova G, Williams C, Bernardini I, Gahl WA. Cystinosis: renal glomerular and renal tubular function in relation to compliance with cystine‐depleting therapy. Pediatr Nephrol. 2015;30(6):945‐951. [DOI] [PubMed] [Google Scholar]
- 13. Gahl WA, Balog JZ, Kleta R. Nephropathic cystinosis in adults: natural history and effects of oral cysteamine therapy. Ann Intern Med. 2007;147:242‐250. [DOI] [PubMed] [Google Scholar]
- 14. Ariceta G, Lara E, Camacho JA, et al. Cysteamine (Cystagon®) adherence in patients with cystinosis in Spain: successful in children and a challenge in adolescents and adults. Nephrol Dial Transplant. 2015;30(3):475‐480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nesterova G, Gahl WA. Cystinosis: the evolution of a treatable disease. Pediatr Nephrol. 2013;28:51‐59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kaiser‐Kupfer MI, Fujikawa L, Kuwabara T, Gahl WA. Removal of corneal crystals by topical cysteamine in nephropathic cystinosis. N Engl J Med. 1987;316:775‐779. [DOI] [PubMed] [Google Scholar]
- 17. Labbe A, Baudoin C, Deschenenes G, et al. A new gel formulation of topical cysteamine for the treatment of corneal cystine crystals in cystinosis: the Cystadrops OCT‐1 study. Mol Genet Metab. 2014;111(3):314‐320. [DOI] [PubMed] [Google Scholar]
- 18. Wühl E, Haffner D, Offner G, et al. Long‐term treatment with growth hormone in short children with nephropathic cystinosis. J Pediatr. 2001;138(6):880‐887. [DOI] [PubMed] [Google Scholar]
- 19. Veys KR, D'Hauwers KW, van Dongen AJCM, et al. First successful conception induced by a male cystinosis patient. JIMD Rep. 2018;38:1‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Reiss RE, Kuwabara T, Smith ML, Gahl WA. Successful pregnancy despite placental cystine crystals in a woman with nephropathic cystinosis. N Engl J Med. 1988;319:223‐226. [DOI] [PubMed] [Google Scholar]
- 21. Gahl WA, Dalakas M, Charnas L, et al. Myopathy and cystine storage in muscles in a patient with nephropathic cystinosis. N Engl J Med. 1988;319:1461‐1464. [DOI] [PubMed] [Google Scholar]
- 22. Sonies B, Ekman EF, Andersson H, et al. Swallowing dysfunction in nephropathic cystinosis. N Engl J Med. 1990;323:565‐570. [DOI] [PubMed] [Google Scholar]
- 23. Langmann CB. Bone complications of cystinosis. J Pediatr. 2017;183S(4):S2‐S4. [DOI] [PubMed] [Google Scholar]
- 24. Kidney Disease: Improving Global Outcomes (KDIGO) CKD‐MBD Work Group . KDIGO clinical practice guideline for the diagnosis, evaluation, prevention, and treatment of chronic kidney disease‐mineral and bone disorder (CKD‐MBD). Kidney Int. 2016;89(6):1192‐1203.27181776 [Google Scholar]
- 25. National Kidney Foundation . K/DOQI clinical practice guidelines for bone metabolism and disease in children with chronic kidney disease. Am J Kidney Dis. 2005;46:1‐121. [PubMed] [Google Scholar]
- 26. Asplin JR, Donahue S, Kinder J, Coe FL. Urine calcium excretion predicts bone loss in idiopathic hypercalciuria. Kidney Int. 2006;70(8):1463‐1467. [DOI] [PubMed] [Google Scholar]
- 27. García‐Nieto V, Ferrández C, Monge M, de Sequera M, Rodrigo MD. Bone mineral density in pediatric patients with idiopathic hypercalciuria. Pediatr Nephrol. 1997;11(5):578‐583. [DOI] [PubMed] [Google Scholar]
- 28. Fricke O, Beccard R, Semler O, Schoenau E. Analyses of muscular mass and function: the impact on bone mineral density and peak muscle mass. Pediatr Nephrol. 2010;25(12):2393‐2400. [DOI] [PubMed] [Google Scholar]
- 29.Besouw M, Cornelissen E, Cassiman D, Kluijtmans L, van den Heuvel L, Levtchenko E. Carnitine Profile and Effect of Suppletion in Children with Renal Fanconi Syndrome due to Cystinosis. JIMD Rep. 2014;16:25‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gaide Chevronnay HP, Janssens V, Van Der Smissen P, et al. A mouse model suggests two mechanisms for thyroid alterations in infantile cystinosis: decreased thyroglobulin synthesis due to endoplasmic reticulum stress/unfolded protein response and impaired lysosomal processing. Endocrinology. 2015;156(6):2349‐2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mehls O, Wühl E, Tönshoff B, Schaefer F, Nissel R, Haffner D. Growth hormone treatment in short children with chronic kidney disease. Acta Paediatr. 2008;97(9):1159‐1164. [DOI] [PubMed] [Google Scholar]
- 32. Rabkin R, Sun DF, Chen Y, Tan J, Schaefer F. Growth hormone resistance in uremia, a role for impaired JAK/STAT signaling. Pediatr Nephrol. 2005;20(3):313‐318. [DOI] [PubMed] [Google Scholar]
- 33. Cherqui S, Sevin C, Hamard G, et al. Intralysosomal cystine accumulation in mice lacking cystinosin, the protein defective in cystinosis. Mol Cell Biol. 2002;22(21):7622‐7632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Conforti A, Tranta A, Biagini S, et al. Cysteamine treatment restores the in vitro ability to differentiate along the osteoblastic lineage of mesenchymal stromal cells isolated from bone marrow of a cystinotic patient. J Transl Med. 2015;13:143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Battafarano G, Rossi M, Rega LR, et al. Intrinsic bone defects in cystinotic mice. Am J Pathol. 2019;189(5):1053‐1064. [DOI] [PubMed] [Google Scholar]
- 36. Claramunt‐Taberner D, Flammier S, Gaillard S, et al. Bone disease in nephropathic cystinosis is related to cystinosin‐induced osteoclastic dysfunction. Nephrol Dial Transplant. 2018;33(9):1525‐1532. [DOI] [PubMed] [Google Scholar]
- 37. Ros I, Alvarez L, Guañabens N, et al. Hypophosphatemic osteomalacia: a report of five cases and evaluation of bone markers. J Bone Miner Metab. 2005;23(3):266‐269. [DOI] [PubMed] [Google Scholar]
- 38. Doyon A, Fischer DC, Bayazit AK, et al. Markers of bone metabolism are affected by renal function and growth hormone therapy in children with chronic kidney disease. PLoS One. 2015;10(2):e0113482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Moe S, Drueke T, Cunningham J, et al. Definition, evaluation, and classification of renal osteodystrophy: a position statement from kidney disease: improving global outcomes (KDIGO). Kidney Int. 2006;69:1945‐1953. [DOI] [PubMed] [Google Scholar]
- 40. Fischer DC, Mischek A, Wolf S, et al. Paediatric reference values for the C‐terminal fragment of fibroblast‐growth factor‐23, sclerostin, bone‐specific alkaline phosphatase and isoform 5b of tartrate‐resistant acid phosphatase. Ann Clin Biochem. 2012;49(Pt 6):546‐553. [DOI] [PubMed] [Google Scholar]
- 41. Kanbay M, Siriopol D, Saglam M, et al. Serum sclerostin and adverse outcomes in nondialyzed chronic kidney disease patients. J Clin Endocrinol Metab. 2014;99(10):E1854‐E1861. [DOI] [PubMed] [Google Scholar]
- 42. Qureshi AR, Olauson H, Witasp A, et al. Increased circulating sclerostin levels in end‐stage renal disease predict biopsy‐verified vascular medial calcification and coronary artery calcification. Kidney Int. 2015;88(6):1356‐1364. [DOI] [PubMed] [Google Scholar]
- 43. Portale AA, Wolf M, Jüppner H, et al. Disordered FGF23 and mineral metabolism in children with CKD. Clin J Am Soc Nephrol. 2014;9(2):344‐353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Florenzano P, Ferreira C, Nesterova G, et al. Skeletal consequences of nephropathic cystinosis. J Bone Miner Res. 2018;33(10):1870‐1880. [DOI] [PubMed] [Google Scholar]
- 45. Leonard MB, Bachrach LK. Non‐invasive techniques for bone mass measurement Pediatric Bone. 2nd ed San Diego, CA: Elsevier (Academic Press); 2012. [Google Scholar]
- 46. Fidan N, Inci A, Coban M, Ulman C, Kursat S. Bone mineral density and biochemical markers of bone metabolism in predialysis patients with chronic kidney disease. J Investig Med. 2016;64(4):861‐866. [DOI] [PubMed] [Google Scholar]
- 47. Leonard MB. A structural approach to skeletal fragility in chronic kidney disease. Semin Nephrol. 2009;29:133‐143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Griffin LM, Kalkwarf HJ, Zemel BS, et al. Assessment of dual‐energy X‐ray absorptiometry measures of bone health in pediatric chronic kidney disease. Pediatr Nephrol. 2012;27(7):1139‐1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Weber LT, Mehls O. Limitations of dual x‐ray absorptiometry in children with chronic kidney disease. Pediatr Nephrol. 2010;25(1):3‐5. [DOI] [PubMed] [Google Scholar]
- 50. Adams JE et al. Radiology Pediatric Bone. 2nd ed San Diego, CA: Elsevier (Academic Press); 2012. [Google Scholar]
- 51. Moorthi RN, Moe SM. Recent advances in the noninvasive diagnosis of renal osteodystrophy. Kidney Int. 2013;84(5):886‐894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rauch F. Pediatric Bone. 2nd ed San Diego, CA: Elsevier (Academic Press); 2012. [Google Scholar]
- 53. Dempster DW, Compston JE, Drezner MK, et al. Standardized nomenclature, symbols, and units for bone histomorphometry: a 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res. 2013;28(1):2‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Glorieux FH, Travers R, Taylor A, et al. Normative data for iliac bone histomorphometry in growing children. Bone. 2000;26(2):103‐109. [DOI] [PubMed] [Google Scholar]
- 55. Rauch F. Bone biopsy: indications and methods. Endocr Dev. 2009;16:49‐57. [DOI] [PubMed] [Google Scholar]
- 56. Pilz S, Iodice S, Zittermann A, Grant WB, Gandini S. Vitamin D status and mortality risk in CKD: a meta‐analysis of prospective studies. Am J Kidney Dis. 2011;58(3):374‐382. [DOI] [PubMed] [Google Scholar]
- 57. Shroff R, Wan M, Nagler EV, et al. Clinical practice recommendations for native vitamin D therapy in children with chronic kidney disease stages 2–5 and on dialysis. Nephrol Dial Transplant. 2017;32(7):1098‐1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ketteler M, Elder GJ, Evenepoel P, et al. Revisiting KDIGO clinical practice guideline on chronic kidney disease‐mineral and bone disorder: a commentary from a kidney disease: improving global outcomes controversies conference. Kidney Int. 2015;87(3):502‐528. [DOI] [PubMed] [Google Scholar]
- 59. Besouw MT, Van Dyck M, Francois I, Van Hoyweghen E, Levtchenko EN. Detailed studies of growth hormone secretion in cystinosis patients. Pediatr Nephrol. 2012;27(11):2123‐2127. [DOI] [PubMed] [Google Scholar]
- 60. Jens D, Mandy W, Marjolein B, et al. Clinical practice recommendations for growth hormone treatment in children with chronic kidney disease. Nat Rev Nephrol. 2019. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ketteler M, Block GA, Evenepoel P, et al. Executive summary of the 2017 KDIGO chronic kidney disease‐mineral and bone disorder (CKD‐MBD) guideline update: what's changed and why it matters. Kidney Int. 2017;92(1):26‐36. [DOI] [PubMed] [Google Scholar]
- 62. Klaus G, Watson A, Edefonti A, et al. Prevention and treatment of renal osteodystrophy in children on chronic renal failure: European guidelines. Pediatr Nephrol. 2006;21(2):151‐159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Haffner D, Schüler U. Metabolic bone disease after renal transplantation. Curr Opin Pediatr. 2014;26(2):198‐206. [DOI] [PubMed] [Google Scholar]
- 64. Franke D, Thomas L, Steffens R, et al. Patterns of growth after kidney transplantation among children with ESRD. Clin J Am Soc Nephrol. 2015;10(1):127‐134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Klare B, Montoya CR, Fischer DC, Stangl MJ, Haffner D. Normal adult height after steroid‐withdrawal within 6 months of pediatric kidney transplantation: a 20 years single center experience. Transpl Int. 2012;25(3):276‐282. [DOI] [PubMed] [Google Scholar]