

Abstract
The role of interferon (IFN)‐induced protein kinase R (PKR) in capripoxvirus (CaPV)‐infected cells remains unknown. In this study, we show that CaPV infection triggered PKR and eukaryotic translation initiation factor 2 alpha (eIF2α) protein phosphorylation in a dose‐dependent manner, and that this leads to decreased CaPV replication. Overexpression of PKR compromised viral gene expression and inhibited sheeppox virus (SPPV) replication. Downregulation of PKR with siRNAs significantly decreased eIF2α phosphorylation and reduced the mRNA level of IFN‐β, which increased virus replication. In luciferase assays, species‐different CaPVs K3L proteins inhibited sheep PKR (sPKR): goatpox virus K3L strongly inhibited sPKR and goat PKR (gPKR), but SPPV K3L only partially inhibited gPKR. These results are the first to show that SPPV infection induces phosphorylation of eIF2α through PKR activation, which then results in restriction of CaPV replication. Furthermore, our data show that CaPV K3L inhibits PKR in a species‐specific manner. The results presented are consistent with the hypothesis that different levels of PKR inhibition by K3L orthologs from various viruses could potentially contribute to the host range function of K3L.
Keywords: PKR, host range, species‐specific, K3L, poxvirus
We investigated whether infection of human and sheep cells with sheeppox virus could activate protein kinase R (PKR) and subsequently result in phosphorylation of eukaryotic translation initiation factor 2 alpha, production of interferon‐β, and reduced virus replication. Our data also show that capripoxvirus K3L inhibits PKR in a species‐specific manner. The results are consistent with the hypothesis that different levels of PKR inhibition by K3L orthologs from various viruses could potentially contribute to the host range function of K3L.
Introduction
Capripoxvirus (CaPV) is a genus of poxviruses that contain three closely related members: sheeppox virus (SPPV), goatpox virus (GTPV), and lumpy skin disease virus (LSDV), which naturally infect sheep, goats, and cattle, respectively. CaPVs are among the most serious of all animal poxviruses causing significant ecological and economic burdens worldwide.1 These viruses cause negative economic consequences by damaging hides and wool and forcing the establishment of trade restrictions in response to an outbreak.2 The geographic range of CaPV outbreaks is bounded within Asia and Africa. Both SPPV and GTPV are limited to North Africa, the Middle East, and continental Asia. In contrast, LSDV was only found in the Horn of Africa, but, recently, it has been introduced into Europe.3 The serological differentiation of CaPVs is technically impossible, due to heterologous cross‐protection and cross‐infection. Strains of GTPV, SPPV, and LSDV show genome identities at around 96%. Although GTPV, SPPV, and LSDV are generally considered to have very restricted host ranges, some recent reports suggest that some virus strains can cause cross‐species infections.4, 5, 6, 7, 8 Since the entry of poxviruses does not rely on any species‐specific receptors, the reproductive replication of poxviruses is often determined by their ability to counteract the innate immune responses in their host cells. However, the molecular mechanisms that determine host range differences among poxviruses are poorly understood.
A viral infection triggers innate immune responses in infected cells, which often recognize invasive pathogens by detecting pathogen‐associated molecular patterns (PAMPs). An example of a PAMP is double‐stranded RNA (dsRNA), which is abundantly produced during poxvirus infections as a result of bilateral transcription. The activation of innate immune responses by dsRNA leads to production of type I interferons (IFNs), which bind to IFN receptors to initiate a variety of antiviral pathways to antagonize virus infection. dsRNA‐dependent protein kinase R (PKR) is one of the key kinases induced by IFNs, which are activated by dsRNA, to restrict virus replication through a series of mechanisms including protein synthesis cessation, apoptosis induction, and enhancement of type I IFN production.9 Monomeric, inactivated PKR is activated by dsRNA binding, which triggers PKR dimerization and phosphorylation.10, 11, 12 Activated PKR binds to its substrate, eukaryotic translation initiation factor 2 alpha (eIF2α), and phosphorylates 51 serine of eIF2α, which leads to suppression of translation and activation of apoptosis, thereby inhibiting virus replication.13
Many viruses have evolved different factors to antagonize the antiviral effects of PKR. Examples of such factors include US11, which is a herpes‐simplex virus type 1 protein that can directly bind to PKR to block its activation.14 Most poxviruses possess two inhibitors of PKR that function through different mechanisms. Vaccinia virus (VACV) E3L protein mainly suppresses PKR activation by dsRNA sequestration through its dsRNA‐binding domain (dsRBD).15, 16, 17 VACV K3L exhibits structural similarity to eIF2α, which competes for the binding site with eIF2α on PKR, and therefore is designated a pseudosubstrate inhibitor18, 19, 20 of PKR. Interestingly, recent studies have shown that the inhibitory effect of K3L likely drives the rapid evolution of PKR, which suggests the importance of inhibiting PKR during poxvirus infection.21, 22 In addition, both VACV K3L and myxoma virus M156, a K3L ortholog, exhibit species‐specific inhibition of PKR.23, 24 However, currently no evidence is available on if and how CaPV K3L functions, despite the importance of CaPV.
In this study, we investigated whether infection of human and sheep cells with SPPV could activate PKR and subsequently result in phosphorylation of eIF2α. We also examined the role of PKR in CaPV replication by small interfering RNA (siRNA)–mediated inhibition and PKR overexpression. Using a very sensitive luciferase‐based assay, we identified that the inhibitory effect of CaPV K3L orthologs on PKR is highly species specific.
Materials and methods
Cells and viruses
The human cervical cancer cell line (HeLa) was purchased from the China Center for Type Culture Collection (Wuhan, China) and sheep testicular cell line (OA3) were grown and maintained as monolayers in Dulbecco's modified Eagle's medium (Gibco, C11995500BT) supplemented with 10% fetal bovine serum (FBS; Gibco, 10099–141), 10,000 U/mL penicillin, and 10,000 g/mL streptomycin (Quality Biological™). Cells were incubated at 37 °C with 5% CO2. SPPV (Gulang/2009 strain) was propagated in African green monkey kidney cells (Vero) and stored at −80 °C. Inactivation of SPPV was achieved by ultraviolet (UV) irradiation of the virus at room temperature for 2 hours. The infectivity of inactivated SPPV was determined by titration in Vero cells.
Reagents and antibodies
Crystal violet (C‐3886), mouse anti‐phospho‐PKR (pThr446) antibody, anti‐c‐Myc (C3956) antibody, sodium pyruvate (S8636), and 0.25 mg/mL trypsin were purchased from Sigma‐Aldrich and used at the indicated final concentrations. Methylcellulose (118‐084‐721) was purchased from MP Biomedical. Anti‐eIF2α (9722), anti‐phospho‐eIF2α (Ser 51) (9721), and β‐actin antibodies were purchased from Cell Signaling Technology®. Mouse anti‐PKR (K‐17) antibody, goat anti‐mouse IgG, F(ab′)2‐HRP (sc‐3696), and goat anti‐rabbit IgG, F(ab′)2‐HRP (sc‐3837) secondary antibodies were purchased from Santa Cruz Biotechnology. Rabbit anti‐HA antibody was purchased from ThermoFisher Scientific. SPPV L1 protein antiserum was prepared by our laboratory.
Virus infection
HeLa or OA3 cells were seeded in 6‐well plates or T75 flask, and grown to ∼90% confluence prior to infection. Cells were then gently washed twice with prewarmed phosphate‐buffered saline (PBS) and were either mock infected with PBS or infected with SPPV at a different multiplicity of infection (MOI). After 1 h of infection, inoculum was removed and replaced with fresh DMEM containing 2.5% FBS. In order to test the importance of PKR in SPPV‐infected cells, HeLa cells were transfected with pCMV‐HA‐SPPV‐K3L or/and pcDNA‐sheep PKR (sPKR) or siRNAs by Lipofectamine® 3000 transfection reagent (Invitrogen). Cells were then either mock infected or infected with SPPV at an MOI of 3 at 24 or 48 h posttransfection (hpt). Total proteins were harvested for analyses at 6, 12, and 24 h postinfection (hpi). Viral titers were determined by plaque assay in Vero cells.
Western blot analyses
Cells were washed three times with ice‐cold PBS and then lysed with RIPA cell lysis buffer (Beyotime, China, P0013B) in the presence of protease inhibitor PMSF (Beyotime, Beijing, China, ST506‐1) and phosphatase inhibitors (ThermoFisher Scientific, 78420). Cells lysates were then incubated on ice for 30 min with gentle shaking. Collected cell lysates were centrifuged at 12,000 rpm, 4 °C for 15 min prior to mixing with 5 × SDS‐PAGE loading buffer (Beyotime, Beijing), followed by boiling at 95 °C for 5 minutes. Protein samples were subjected to 12% polyacrylamide gel electrophoresis and subsequently transferred to nitrocellulose membrane, using a semidry method. Membranes were blocked with 5% skim milk in Tris‐buffered saline containing 0.1% Tween 20 (TBST) at room temperature for 2 h, and then were incubated with specific primary antibody in TBST at 4 °C overnight. Membranes were washed three times with TBST at room temperature for 10 min and incubated with horseradish peroxidase–labeled secondary antibody in TBST for 1 h at room temperature, followed by washing three times. Membranes were exposed in a darkroom after treatment with Pierce® ECL western blotting substrate (ThermoFisher Scientific, 32109).
Plasmids construction and transfection
Human PKR (hPKR) was cloned from HeLa cDNA, and sPKR was cloned from OA3 cDNA. cPKR was cloned from MDBK cDNA (ATCC‐4243) and goat PKR (gPKR) was cloned from epithelial cells of Shaneng milk goat, which were provided by Baohua Ma, Northwest A&F University. PKR from various species were cloned into the eukaryotic expression vector pcDNA3.1(–)/Myc‐His(B). VACV K3L, LSDV K3L, SPPV K3L, and GTPV K3L were cloned into pCMV‐HA. All plasmids for mammalian cell transfection were purified with an Endo‐Free® plasmid preparation kit (QIAGEN, 12362). Transfections were performed with Lipofectamine 3000 (Invitrogen, L3000) following the manufacturer's instructions. Briefly, 2.5 μg plasmids and 5 μL Lipofectamine 3000 were diluted in Opti‐MEM™ medium (Gibco), followed by mixing and incubating at room temperature for 5 minutes. The DNA‐liposome complexes were then added into HeLa cell monolayers at 80% confluency. Cells were incubated at 37 °C with 5% CO2 for 24 h prior to the subsequent procedures, as described.
siRNA transfection
In order to inhibit expression of endogenous PKR, RNA interference sequences (siPKR: sense 5′‐GGUGAAGGUAGAUCAAAGATT‐3′, antisense 5′‐UCUUUGAUCUACCUUCACCTT‐3′; scramble sense 5′‐UUCUCCGAACGUGUCACGUTT‐3′, scramble antisense 5′‐ACGUGACACGUUCGGAGAATT‐3′)25 targeting hPKR, and a nonsense control (5′‐UUCUCCGAACGUGUCACGU‐3′) were ordered from GenePharma Co., Ltd. (Shanghai, China). HeLa cells were seeded in 6‐well plates and transfected at around 80% confluency. SiPKR and control siRNA were transfected with Lipofectamine 3000 following the manufacturer's instructions (75 pmol/well). The medium from transfected wells was replaced with fresh DMEM (2% FBS) 5 hpt. Cells were infected as previously described 48 hpt.
Colocalization of PKR and K3L
HeLa cells were seeded on slides and cotransfected with plasmids pCMV‐SPPV K3L and pcDNA‐sPKR with Lipofectamine 3000 at around 80% confluency. Single transfections with pCMV‐SPPV K3L or pcDNA‐sPKR were performed simultaneously as controls. Cells were gently washed with prewarmed PBS three times 24 h later and were then fixed with 4% paraformaldehyde for 30 min and then permeabilizated with 1% Triton X‐100 for 20 min at room temperature. Fixed cells were then incubated overnight at 4 °C with anti‐Myc monoclonal antibody (1:200) and murine HA‐tag antibody (1:1000); the cells transfected with a single plasmid were incubated with corresponding antibody. Alexa Fluor® 488 goat anti‐rabbit (ThermoFisher Scientific, A11070) and Alexa Fluor 546 goat anti‐mouse secondary antibodies (ThermoFisher Scientific, A11018) (1:500) were added in a darkroom. Finally, the nuclei of cells were stained with DAPI (Beyotime, C1005) for 10 min and then the cells were mounted with antifade mounting medium (Beyotime, P0126). Fluorescence was directly observed under a fluorescence microscope.
Immunoprecipitation
HeLa cells were seeded in 10 cm cell culture dishes and cotransfected with plasmids containing SPPV K3L and sPKR using Lipofectamine 3000 when the cells were approximately 80% confluent. Cell culture medium was removed at 48 hpt and the cells were washed twice with ice‐cold PBS prior to addition of ice‐cold immunoprecipitation lysis buffer (Thermo Scientific Pierce Co‐Immunoprecipitation Kit, 26149). Cell lysates were transferred into 1.5 mL centrifuge tubes after 10 min incubation on ice and then centrifuged at 13,000 × g for 10 min at 4 °C. Supernatants were collected and cell lysates were pretreated with a control agarose resin to remove nonspecific binding proteins during immunoprecipitation. The treated protein samples were added to the appropriate resins that had been incubated with antibody and then were incubated on the rotary device at 4 °C overnight. Subsequently, the samples were washed several times following the instructions and the protein samples were obtained after the resin had been eluted with elution buffer. The protein samples were then prepared for western blot after adding 5 × SDS loading buffer and incubating with a dry incubator for 10 min at 100 °C.
Real‐time PCR
The real‐time fluorescent quantitative PCR (qPCR) method was used to detect the levels of SPPV DNA, IFN‐β, and K3L messenger RNAs (mRNAs). The total DNA of HeLa cells infected with SPPV Gulang/2009 strain was extracted by the MiniBEST Viral RNA/DNA Extraction Kit (TaKaRa, Dalian, 9766). The real‐time fluorescent qPCR probe and primer sequence for SPPV were the following: 5′‐CAATGGGTAAAAGATTTCTA‐3′; SPPV Q‐F: 5′‐GGCGATGTCCATTCCCTG‐3′; and SPPV Q‐R: 5′‐AGCATTTCATTTCCGTGAGGA‐3′. The β‐actin gene was used as an internal reference. Total RNAs from OA3 cells treated with SPPV or poly (I:C) were extracted by the TRIzol® reagent (TaKaRa, Dalian) and then reverse‐transcribed into cDNA by the PrimeScript™ RT reagent Kit with gDNA Eraser (Perfect Real Time) (RR047A; Takara Bio Inc). Primer sequences for PCR amplification were K3L 1: 5′‐ATGTCATCGAATAGCGATTTGG‐3′; K3L 2: 5′‐GTTCATCCTTACAATTTGCACA‐3′; β‐actin 1: 5′‐GATCTGGCACCAC ACCTTCT‐3′; and β‐actin 2: 5′‐GGGGTGTTGAAGGTCTC AAA‐3′). Quantitative RT‐PCR reactions were performed with SYBR Premix Ex Taq II (DRR081; Takara Bio Inc.). The forward primer sequence of IFN‐β is 5′‐ACGACAGCTCTTTCCATGA‐3′ and the IFN‐β reverse primer sequence is 5′‐AGCCAGTGCTCGATGAATCT‐3′. The forward primer of K3L is 5′‐ATGTCATCGAATAGCGATTTGG‐3′ and the reverse primer of K3L is 5′‐GTTCATCCTTACAATTTGCACA‐3′. The forward primer of β‐actin is 5′‐ACGACAGCTCTTTCCATGA‐3′ and the reverse primer is 5′‐AGCCAGTGCTCGATGAATCT‐3′. The expression level of target mRNA was analyzed by the Schmitten method,26 and the relative content of a target gene was calculated according to the average relative content (F) = 2−ΔΔCt: ΔΔCt = ((control group CT value of gene of interest − control group reference gene CT value) − (detected group gene CT value − detected group reference gene CT)).
RT‐PCR
To confirm the efficacy of RNAi targeting of PKR in OA3 cells, RT‐PCR was used to amplify the target gene in the transfected cells. Total RNA was extracted from OA3 cells with TRIzol and incubated for 1 h at 37 °C with DNase RQ1 (TaKaRa, Dalian). To detect PKR mRNA expression in OA3 cells, RT‐PCR was conducted using 2.0 μg of RNA with the SuperScript™ One‐Step RT‐PCR system (Gibco, BRL). Retrotranscription of β‐actin was the control. PCR was run for 30 cycles with 95 °C for 30, 56 °C for 45, and 72 °C for 45 seconds. In order to verify primer specificity, a melting curve was analyzed and RT‐PCR products were further cloned into pMD18‐T for sequencing.
Luciferase assay
The procedure for luciferase assay was previously described.24 Briefly, 5 × 104 HeLa cells were seeded in 24‐well plates 24 h before transfection. For each transfection, pCMV‐Gluc minus SS (0.05 μg, Nanolight) and pcDNA‐3.1 plasmids encoding PKR (0.2 μg) or K3L (0.4 μg) were transfected using the FuGENE® HD transfection reagent (Promega). For titration experiments, amounts of transfected plasmids are indicated in the figures. For controls, an empty pcDNA3.1 vector of the same amount was transfected. Each transfection was conducted in triplicate. After 48 h, the cells were harvested and luciferase activities were determined using luciferase detection reagents (Promega) in a luminometer.
Statistical analysis
The data were expressed as mean ± SD. Significance was determined with the two‐tailed independent Student's t‐test (P < 0.05) between two groups. One‐way analysis of variance (ANOVA) followed by the Tukey's test were used to compare multiple groups (>2). The data were represented as mean ± SEM and were analyzed by two‐way ANOVA using the SPSS software version 17.0 (SPSS Inc., Chicago, IL). P < 0.05 was considered statistically significant.
Results
SPPV infection triggers phosphorylation of hPKR and eIF2α protein
To test if PKR can be activated by SPPV infection, HeLa cells were infected with SPPV at an MOI of 3 and endogenous PKR and eIF2α phosphorylation were evaluated by western blot analyses. Both PKR and eIF2α were phosphorylated at 12 and 24 hpi, while the total amount of PKR and eIF2α did not change (Fig. 1A). To test if activation of PKR was due to virus infection, we analyzed PKR and eIF2α phosphorylation following SPPV infection at various MOIs (1, 2, and 3); the levels of both PKR and eIF2α phosphorylation increased in response to increased viral input (Fig. 1B). To test if activation of PKR observed following virus infection requires productive viral replication, we performed a similar experiment but with UV‐inactivated SPPV. UV exposure effectively reduced SPPV infectivity (Fig. 1C), and prevented PKR phosphorylation and the subsequent phosphorylation of eIF2α.
Figure 1.
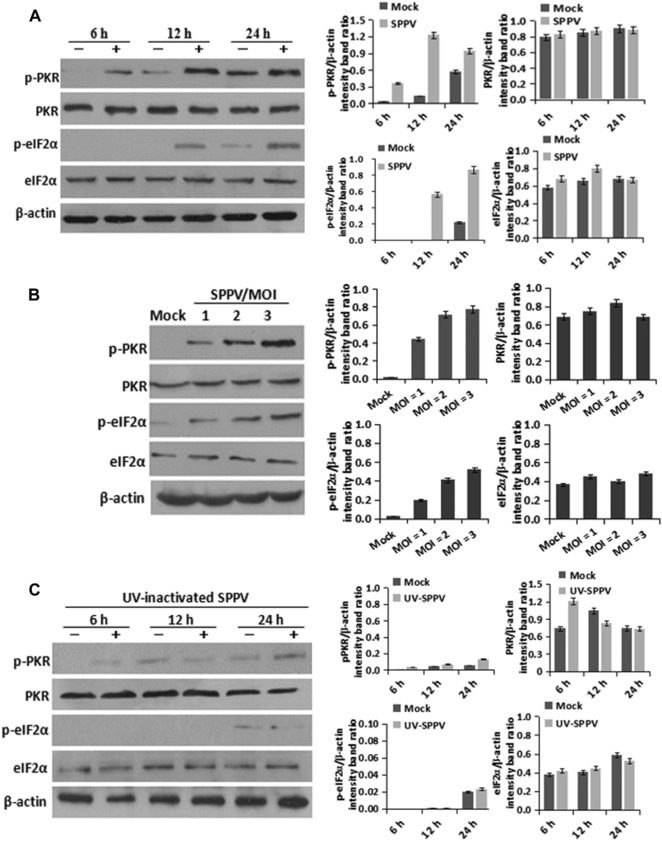
SPPV infection triggers phosphorylation of PKR and eIF2α. (A). HeLa cells were infected with SPPV Gulang/2009 at an MOI of 3. PKR, phosphorylated PKR, eIF2α, and phosphorylated eIF2α expression were detected by western blotting at 6, 12, and 24 hpi, respectively. (B) HeLa cells were infected with different doses (MOI of 1, 2, and 3) of SPPV Gulang/2009. PKR, phosphorylated PKR, eIF2α, and phosphorylated eIF2α expression were detected by western blotting at 12 hpi. (C) HeLa cells were infected with UV‐inactivated SPPV Gulang/2009. PKR, phosphorylated PKR, eIF2α, and phosphorylated eIF2α expression were detected by western blotting at 6, 12, and 24 hpi, respectively. The β‐actin gene was used as a protein loading control. Data are represented as mean ± SEM; n = 3. Representative of three independent experiments. **P < 0.01; ***P < 0.001. ns, not significant.
Exogenous PKR expression inhibits SPPV replication
To determine the effect of overexperssion of hPKR on SPPV replication, a hPKR‐expressing plasmid, or an empty vector (pcDNA3.1 alone) as a negative control, was transfected into HeLa cells. The expression level of hPKR in the transfected cells was fourfold higher than that in cells transfected with the empty vector; and phosphorylation of eIF2α was significantly increased in cells expressing exogenous hPKR (Fig. 2A). HeLa cells expressing hPKR were then infected with SPPV Gulang/2009; SPPV L1 protein, an early protein of virus replication, was decreased (Fig. 2A) in the infected cells. Intracellular SPPV DNA (Fig. 2B) and virus titer (Fig. 2C) were also reduced in the hPKR‐expressing cells, compared with vector‐transfected and mock‐treated cells. Consistent with these results, intracellular SPPV DNA (Fig. 2D) and virus titer in OA3 cells transfected with hPKR‐expressing pcDNA3.1 were also reduced compared with the empty vector−transfected and mock‐infected cells at 6, 12, 24, and 48 hpi (Fig. 2E).
Figure 2.
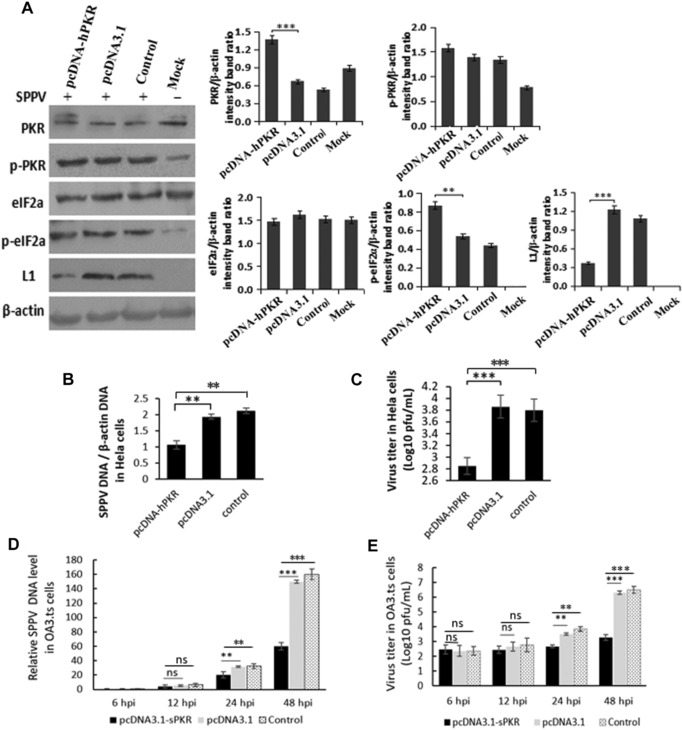
PKR inhibits SPPV replication in HeLa cells. (A) HeLa cells were transfected with pcDNA3.1 or pcDNA‐PKR. HeLa cells were transfected with Lipo3000 as control. After 24 h of incubation, cells were infected with SPPV Gulang/2009 at an MOI of 3. Uninfected cells as MOCK. PKR, phosphorylated PKR, eIF2α, phosphorylated eIF2α, and L1 expression were detected by western blotting. β‐Actin was used as a protein loading control. (B) SPPV genome DNA levels in HeLa cells were quantified by real‐time PCR at 12 hpi, which were transfected with pcDNA3.1 or pcDNA‐PKR and then infected with SPPV at 24 hpt. HeLa cells were transfected with Lipo3000 as control. (C) HeLa cells transfected with pcDNA3.1 or pcDNA‐PKR were infected with SPPV Gulang/2009 at 48 hpt, and cells and virus were harvested at 48 hpi. (D) SPPV genome DNA levels in OA3 were quantified by real‐time PCR at 6, 12, 24, and 48 hpi, which were transfected with pcDNA3.1 or pcDNA‐PKR and then infected with SPPV at 24 hpt. OA3 were transfected with Lipo3000 as control. (E) OA3 transfected with pcDNA3.1 or pcDNA‐PKR were infected with SPPV Gulang/2009 at 48 hpt, and cells and virus were harvested at 6, 12, 24, and 48 hpi. SPPV titer in cells was assayed by plaque assay. HeLa or OA3.ts cells were transfected with Lipo3000 as control. Data are represented as mean ± SEM; n = 3. Representative of three independent experiments. Significance was analyzed using two‐tailed Student's t‐test. **P < 0.01; ***P < 0.001. ns, not significant.
Effects of suppression of PKR expression on SPPV replication in HeLa and OA3 cells
To further examine the effects of PKR on SPPV replication, PKR‐specific siRNA was transfected into HeLa cells to suppress endogenous PKR expression transiently. Western blot showed that the expression of PKR in cells that were transfected with PKR‐specific siRNA was downregulated compared with control cells (Fig. 3A, upper panel), and that phosphorylation of eIF2α was significantly decreased concomitantly.
Figure 3.
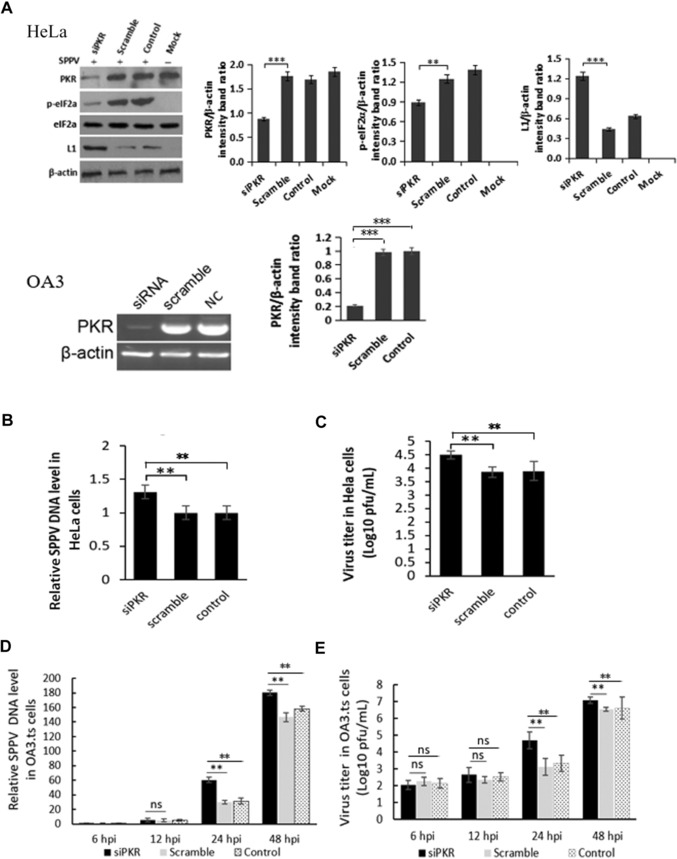
PKR knockdown increases SPPV replication. (A) HeLa cells were transfected with the siRNA specific for PKR (75 pmol/mL) or control siRNA. Take HeLa cells transfected with Lipo3000 as control. After 48 h of incubation, cells were infected with SPPV Gulang/2009. Take uninfected cells as MOCK. PKR, phosphorylated eIF2α, and SPPV L1 expression were detected by western blotting. β‐Actin was used as a protein loading control. (B) SPPV genome DNA levels in the cells were quantified by real‐time PCR at 12 hpi, which were transfected with siPKR or control siRNA and then infected with SPPV at 48 hpt. HeLa cells were transfected with Lipo3000 as control. (C) HeLa cells were transfected with siPKR or control siRNA and infected with SPPV Gulang/2009 at 48 hpt. (D) SPPV genome DNA levels in the sheep OA3 were quantified by real‐time PCR at 6, 12, 24, and 48 hpi, which were transfected with siPKR or control siRNA and then infected with SPPV at 48 hpt. OA3 were transfected with Lipo3000 as control. (E) OA3 cells were transfected with siPKR or control siRNA, infected with SPPV Gulang/2009 at an MOI of 3 at 48 hpt. Cells and virus were collected at 6, 12, 24, and 48 hpi. SPPV titer in cells was assayed by plaque assay. Data are represented as mean ± SEM; n = 3. Representative of three independent experiments. Significance was analyzed using two‐tailed Student's t‐test. **P < 0.01; ***P < 0.001. ns, not significant.
HeLa cells transfected with PKR‐specific siRNA were infected at 48 hpt with SPPV Gulang/2009 strain and then total DNA was harvested at 12 hpi. SPPV genome DNA in cells was detected by qPCR. Viral DNA in cells transfected with PKR‐specific siRNA was significantly increased (∼2.2‐fold) compared with control cells (Fig. 3B). SPPV titers were also examined by plaque assay; SPPV titers at 24 hpi in cells with PKR‐specific siRNA were significantly increased (Fig. 3C).
To examine the role of sPKR in SPPV replication, OA3 cells were transfected with sPKR‐specific siRNA and then the amount of sPKR mRNA was evaluated by semiquantitative RT‐PCR (as no antibody for sPKR is available). sPKR mRNA was reduced by about 80% (Fig. 3A, lower panel). OA3 transfected with sPKR‐specific siRNA were infected with SPPV and then total DNA was harvested at 6, 12, 24, and 48 hpi. SPPV genome DNA in cells was detected by qPCR. Consistent with the results observed in HeLa cells, viral DNA in cells transfected with sPKR‐specific siRNA was significantly increased (Fig. 3D) at 24 and 48 hpi compared with controls cells.
Figure 1 shows that virus infection stimulates PKR in a dose‐dependent manner. To determine the effect of siRNA knockdown of PKR on SPPV viral replication, OA3 cells were infected with SPPV Gulang/2009 at an MOI of 3. Cells and virus were collected at 6, 12, 24, and 48 hpi and then SPPV titer was assayed by plaque assay. SPPV titers in OA3 with siRNA knockdown of PKR were also significantly increased at 24 and 48 hpi (Fig. 3E). SPPV titers in OA3 with siRNA knockdown of PKR was ∼12 times higher compared with the control group at 24 hpi, and five times higher compared with the control group at 48 hpi (Fig. 3E).
PKR promotes IFN‐ production in response to SPPV infection
Activation of PKR leads to the induction of type I IFN in many types of cells, which can further increase PKR transcriptionally in a positive feedback loop.27 To test if SPPV‐induced PKR activation can promote the production of type I IFN, we evaluated activation of PKR upon SPPV infection and mRNA levels of type I IFNs in HeLa cells; as a control, cells were treated in parallel with the polyinosinic‐polycytidylic acid [poly (I:C)], an analogue of viral ds‐RNA widely used to mimic viral infection and known to induce type I IFNs. Phosphorylation of both PKR and eIF2α significantly increased in cells either infected with SPPV or treated with poly (I:C) (Fig. 4A), compared with mock‐infected cells. In cells infected with SPPV Gulang/2009, IFN‐β mRNA levels were increased by more than 30‐fold; in cells treated with poly (I:C), 70‐fold (Fig. 4B). To confirm that induction of IFN‐β upon SPPV infection was due to activated PKR, we measured mRNA of IFN‐β in cells with endogenous PKR transiently knocked down by siRNA. Figure 4C and D show that IFN‐β mRNA induction was reduced in cells with the PKR knocked down by PKR‐specific siRNA, but not in cells with control siRNA.
Figure 4.
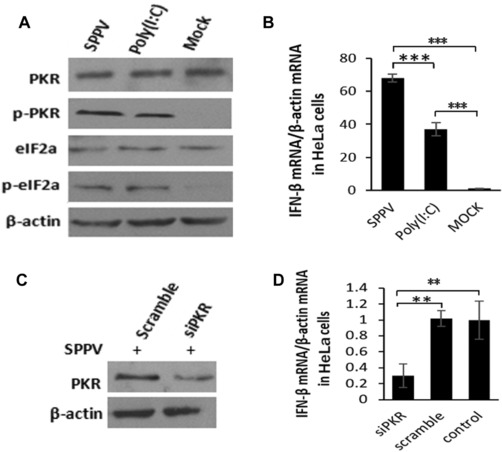
PKR promotes IFN‐β production in response to SPPV infection in HeLa cells. (A) HeLa cells were treated with poly (I:C) (10 μg/mL) or infected with SPPV Gulang/2009 at an MOI of 3 for 12 hours. Take uninfected cells as MOCK. PKR, phosphorylated PKR, eIF2α, and phosphorylated eIF2α expression were detected by western blotting. β‐Actin was used as a protein loading control. (B) HeLa cells were treated with poly (I:C) (10 μg/mL) or infected with SPPV Gulang/2009 at an MOI of 3 for 12 hours. IFN‐β mRNA levels in the cell lysates were quantified by real‐time PCR. (C) HeLa cells were transfected with the siRNA specific for PKR (75 pmol/mL) or control siRNA for 48 h and then infected with SPPV Gulang/2009 at an MOI of 3. RNA interference effects for PKR in cells were detected by western blot. (D) HeLa cells transfected with siRNAs were infected with SPPV Gulang/2009 at an MOI of 3. IFN‐β mRNA levels in the cell lysates were quantified by real‐time PCR at 12 hpi. Data are represented as mean ± SEM; n = 3. Representative of three independent experiments. Significance was analyzed using two‐tailed Student's t‐test. **P < 0.01; ***P < 0.001. ns, not significant.
Antiviral effects mediated by PKR can be inhibited by SPPV K3L
VACV K3L is a potent inhibitor of PKR in many cell types. To test if SPPV K3L can inhibit PKR, we first examined SPPV K3L expression in HeLa cells during SPPV infection. HeLa cells were inoculated with SPPV and total RNA from the cells was extracted at 2, 4, 6, 8, 10, and 12 hpi. The total RNA was reverse transcribed to cDNA and then the quantity of K3L mRNA was determined by the relative fluorescence quantitative RT‐PCR. K3L mRNA increased rapidly in the early stage of SPPV infection 4−6 hpi (Fig. 5).
Figure 5.
K3L mRNA in SPPV infected cells. HeLa cells were inoculated with SPPV Gulang/2009 strain and cellular total RNA from the cells was extracted at 2, 4, 6, 8, 10, and 12 hpi. Total RNA was then reverse transcribed to cDNA and the quantity of K3L was determined by relative fluorescence quantitative RT‐PCR. Data are represented as mean ± SEM; n = 3. Representative of three independent experiments.
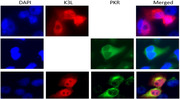
To test if SPPV K3L interacts with host cell PKR, pCMV‐SPPV K3L plasmid was cotransfected with a sPKR‐expressing plasmid into HeLa cells; interactions between K3L and PKR were then tested by indirect immunofluorescence or western blot with HA‐tag and Myc polyclonal antibodies. SPPV K3L (red, upper panel of Fig. 6A) was expressed in both cytoplasm and nucleus, and sPKR (green, middle panel of Fig. 6A) was expressed only in the cytoplasm; colocalized SPPV K3L (red) and sPKR (green) were expressed mainly in the cytoplasm (lower panel of Fig. 6A). We harvested proteins from the cells, performed immunoprecipitation with anti‐Myc antibody, and then blotted with anti‐HA antibody; K3L co‐immunoprecipitated with PKR, consistent with the conclusion that the two proteins interact with each other in HeLa cells (Fig. 6B and C). We also performed immunoprecipitation with anti‐HA antibody, and then blotted with anti‐Myc antibody; PKR was co‐immunoprecipitated with K3L, again consistent with the conclusion that the two proteins interact with each other in HeLa cells (Fig. 6D and E).
Figure 6.
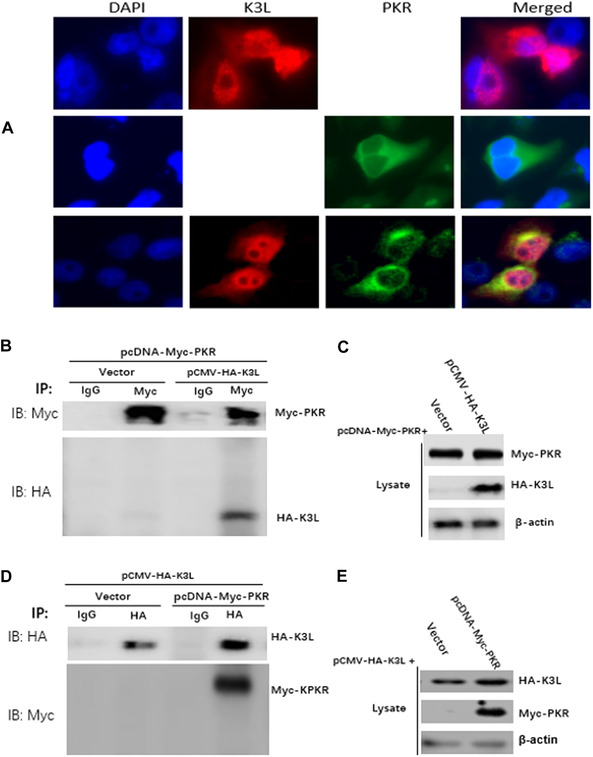
K3L interaction with PKR. (A) The colocalization of PKR and K3L in HeLa cells. HeLa cells was transfected with pCMV‐SPPV K3L plasmids (2 μg, upper panel) and pcDNA‐sPKR plasmid (1 μg, middle panel) for 36 h, respectively. HeLa cells were cotransfected with pCMV‐SPPV K3L plasmids (2 μg) and pcDNA‐sPKR plasmid (1 μg) for 36 h (lower panel). Expression of pCMV‐SPPV K3L and pcDNA‐sPKR was detected by immunofluorescence assay. Cells were double‐immunostained for pCMV‐HA‐SPPV K3L (red) and pcDNA‐sPKR (green); the cellular nuclei were counterstained with DAPI (blue). (B) HeLa cells were cotransfected with pcDNA‐Myc‐sPKR and pCMV‐HA‐K3L plasmids for 36 hours. Whole cell lysates were immune‐precipitated with mouse normal IgG antibody or mouse anti‐Myc antibody and subjected to western blotting. (C) Whole cell lysates were also detected with western blotting to validate the expression of the proteins of interesting. (D) HeLa cells were cotransfected with pcDNA‐Myc‐sPKR and pCMV‐HA‐K3L plasmids for 36 hours. Whole cell lysates were immune‐precipitated with mouse normal IgG antibody or mouse anti‐HA antibody and subjected to western blotting. (E) Whole cell lysates were also detected with western blotting to validate the expression of the proteins of interesting.
CaPV K3L orthologs inhibit PKR in a species‐specific manner
The structure‐based sequence alignment of K3L showed that each K3L of poxvirus forms its own conserved residues, which are consistent with the host, and most of them are identical. However, different residues are endemic to all viruses (Fig. 7A). The alignment of PKR orthologs showed that PKR is a 551 amino acid protein that contains an N‐terminal dsRBD (aa 6−169). This binding domain comprises two dsRNA‐binding motifs (dsRBMs, aa 6−79), which are spaced by a flexible 20 amino acid (79−96) linker, which is depicted below the dsRBMs (aa 96−169), and a C‐terminal kinase domain (aa 274−551), with small and large N‐ and C‐terminal domains. The sequence alignment showed that cPKR, gPKR, and sPKR have a similar sequence structure (Fig. 7B). The percentage of identity between hPKR and cPKR, sPKR, or gPKR is 61%, 60%, and 61%, respectively; between cPKR and sPKR or gPKR is 86% and 86%, respectively; and between sPKR and gPKR is 97%, using the Geneious alignment method in Geneious R10 software.
Figure 7.
CaPV K3L orthologs inhibit PKR from different species. (A) Structure‐based sequence alignment of K3L orthologs. In PKR recognition motif, VACV Pro9, Asp13, Tyr27, Leu30, Tyr33, Glu37, Gly60, Val67, Arg69, Asp71, and Lys74 are conservative residues (green)18. Compared with the rate/human eIF2a sequence, the researchers found that the SPPV Pro14 (VACV Pro9), Tyr32(VACV Tyr27), Leu35(VACV Leu30), Tyr38 (VACV Tyr33), Glu41 (VACV Glu37), Arg69, Lys74, Tyr76, and Asp78 are conservative, indicating K3L sequences of GTPV, SPPV, LSDV, and VACV strains are conserved in human eIF2a (green); Ly60, al67, Asp71, and Arg83 are different. While the K3L sequences of GTPV, SPPV, and LSDV are almost identical, C24 and I84 of SPPV K3L are F24, K26, and V84 in K3L of GTPV and LSDV (gray), and N50 of SPPV/LSDV K3L is D50 in GTPV K3L. VACV and CaPV share many sequences (green and blue); VACV Asp13 and V84 are conserved in VACV K3L and eIF2a, but not GTPV (orange). (B) Sequence alignment of PKR orthologs. (C) Expression of PKR orthologs and K3L orthologs in HeLa cells. PKRs (upper) and K3Ls (middle) expression were detected with western blotting. (D) CaPV K3L orthologs inhibit PKR from different species. PKR from different species were cotransfected with VACV K3L or LSDV K3L or SPPV K3L or GTPV K3L ortholog at a ratio of 1:4 into HeLa cells, which were transfected with siRNA targeting the endogenous hPKR. All light units were normalized to PKR only, and the relative Gaussia luciferase value are shown (graph). Representative of three independent experiments. Significance was analyzed using two‐tailed Student's t‐test. **P < 0.01; ***P < 0.001. ns, not significant.
Studies have shown that K3L of VACV and rabbit poxvirus can bind to PKR and inhibit eIF2α, thereby blocking transcription of downstream genes.23 To test whether K3L inhibits PKR in a species‐specific manner, PKR from human, sheep, cattle, and goat were cloned into pcDNA3.1 and VACV K3L, LSDV K3L, SPPV K3L, and GTPV K3L were cloned into the pCMV‐HA, and then combinations of plasmids were transfected in HeLa cells (Fig. 7C). Cotransfections of plasmids encoding Gaussia luciferase, PKR, and K3L were carried out in cells whose endogenous PKR was knocked down by siRNA (in this assay, PKR inhibits translation of luciferase, which is represented by a reduced level of light that cell lysates emit; cotransfection of PKR inhibitors can suppress PKR activity and, as a result, enhance light units; published work has shown that the combination of PKR and K3L is based on the ratio of 1:2, see Ref. 18). For our study, the ratio of PKR to K3L was selected as 1:4. Figure 7D shows that all CaPVs K3L orthologs inhibited sPKR; GTPV K3L significantly inhibited PKR from sheep and goat; while SPPV and LSDV K3L orthologs weakly inhibited gPKR.
Discussion
dsRNA is a common PAMP that is known to activate PKR and other nucleic sensors, such as RIG‐I and MDA5, in many types of cells. Most dsRNA produced during a CaPV infection is generated in the period of intermediate and late gene convergent transcription.28 Many viruses have evolved mechanisms to inhibit the antiviral effect of PKR. Viral inhibitors of PKR include dsRNA‐binding proteins (VACV E3L and influenza virus NS1), RNA inhibitors (hepatitis C virus IRES), competitive inhibitors (VACV K3L and HIV Tat), PKR degradation enzymes (poliovirus protease), and those that interact with PKR directly (influenza virus P58, VACV K3L, and hepatitis C virus NS5A and E2).29, 30 Previous studies have found that cells can inhibit the replication of VACV through the activated PKR signaling pathway. E3L and K3L, which are VACV host range genes, inhibit the antiviral effect of PKR through different mechanisms.31, 32 Even though the interactions between PKR and VACV K3L have been extensively researched, CaPV orthologs of K3L are still poorly studied.
Our study focused on the relationship PKR and CaPV during a virus infection in cells. We found that SPPV infection led to phosphorylation of both PKR and eIF2α in HeLa cells in a dose‐dependent manner, which indicates that hPKR in HeLa cells can be activated by SPPV infection and is dependent on productive virus replication. We found that the amount of viral DNA, the expression of viral L1 protein, and SPPV titer were reduced in HeLa and OA3 cells in which PKR was overexpressed; in contrast, viral DNA, L1 protein, and virus titer increased in OA3 and HeLa cells in which PKR was transiently suppressed with siRNA. These data suggest that overexpression of hPKR phosphorylates human eIF2α, leading to reduced viral DNA replication, viral protein expression, and virus production in HeLa cells. Overexpression of sPKR in OA3 cells also reduced DNA replication and virus production, indicating that exogenous PKR can restrict SPPV replication in OA3 cell lines, probably also by phosphorylating sheep eIF2a. As far as we know, ours is the first report indicating that CaPV can activate PKR and its downstream antiviral activities, such as eIF2α phosphorylation and IFN‐β induction.
K3L is one of the most important host range factors for VACV.33 Studies have shown that VACV K3L mimics eIF2α and interacts with PKR to inhibit the phosphorylation of eIF2α.20, 34 Sequence homology analysis and a VACV K3L and eIF2α‐N‐terminus competitive binding assay showed that PKR recognizes eIF2α and K3L by a similar mechanism.35 Deletion of K3L increases virus sensitivity to type I IFNs.20 K3L is also able to inhibit the phosphorylation of eIF2α induced by other eIF2α protein kinases, including yeast GCN2, HRI, and PEK.36 Genome analysis has shown that the CaPV K3L homologous gene is a virus host range factor.37, 38, 39 However, how K3L of SPPV and GTPV regulate host range and cellular antiviral signaling is largely unknown. In cells cotransfected with both SPPV K3L and PKR, we assayed for interaction and localization; PKR colocalized with K3L in the cytoplasm when expressed exogenously in HeLa cells, and immunoprecipitation experiments showed that PKR and SPPV K3L interact physically with each other. These results suggest that SPPV K3L may have a similar function as VACV K3L.
Studies have shown that VACV K3L can bind to PKR and induce eIF2α phosphorylation, which blocks the translation of downstream genes.23 Cotransfections of plasmids encoding luciferase, PKR, and SPPV K3L were carried out in cells whose PKR was knocked down by siRNA. In our experiments, all CaPVs K3L orthologs inhibited sPKR: GTPV K3L strongly inhibited sheep and gPKR, while SPPV K3L and LSDV K3L inhibited gPKR weakly; these results are no doubt affected by differences in the key amino acids of the PKRs and K3Ls in sheep and goat cells and viruses. More generally, these results suggest that GTPV may be infectious in sheep, whereas SPPV may not establish productive infections in goats. In other words, GTPV may have a wider host range than SPPV, partially owing to PKR sensitivity to viral inhibitors; this may explain why some GTPV strains can be isolated from sheep in the wild.6, 8, 40 The data further suggest that SPPV and GTPV may be exceptional poxvirus models for studying host range variation and evolution.
In conclusion, our study demonstrated that SPPV activates PKR, which then inhibits the replication of SPPV by suppressing general translation and stimulating the production of IFN‐β. Our data also indicate that CaPV K3L directly interacts with PKR and antagonize PKR. More importantly, our results also show for the first time that all CaPVs K3L orthologs inhibit sPKR, but not cPKR and gPKR. GTPV K3L strongly inhibited sheep and gPKR, and SPPV K3L only partially inhibited gPKR. These results indicate the possibility that the various degrees of PKR inhibition by CaPV K3L orthologs might correlate with the different host ranges that CaPVs display in nature.
Competing interests
The authors declare no competing interests.
Author contributions
Zhixun Zhao assisted in experimental design of the study, wrote this manuscript, and prepared figures for publication. Zhixun Zhao, Xueliang Zhu, Na Wu, and Caiyun Huang conducted most of the experiments. Guohua Wu helped to construct partial plasmids and analyzed data. Qiang Zhang and Zhidong Zhang analyzed data and modified the manuscript. All authors read and approved the final manuscript.
Acknowledgments
This project was supported by Grants from the National Natural Science Foundation of China (31201892, 31872449, and 31502096). The work was supported by the National Key Research and Development Program of China (2016YFD0500907). We thank Dr. Chen Peng, Rockefeller University, for his support in technical details and suggestions of experimental design, and Dr. Zhilong Yang, Kansas State University, for his advice during the revision of the manuscript.
Contributor Information
Qiang Zhang, Email: zhangqiang@caas.cn.
Zhidong Zhang, Email: zhangzhidong@caas.cn.
References
- 1. Babiuk, S. , Bowden T.R., Parkyn G., et al 2009. Yemen and Vietnam capripoxviruses demonstrate a distinct host preference for goats compared with sheep. J. Gen. Virol. 90: 105–114. [DOI] [PubMed] [Google Scholar]
- 2. Yeruham, I. , Yadin H., Van Ham M., et al 2007. Economic and epidemiological aspects of an outbreak of sheeppox in a dairy sheep flock. Vet. Rec. 160: 236–237. [DOI] [PubMed] [Google Scholar]
- 3. Tuppurainen, E.S.M. , Venter E.H., Shisler J.L., et al 2017. Capripoxvirus diseases: current status and opportunities for control. Transbound Emerg. Dis. 64: 729–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kitching, R.P. & Taylor W.P.. 1985. Transmission of capripoxvirus. Res. Vet. Sci. 39: 196–199. [PubMed] [Google Scholar]
- 5. Lefkowitz, E.J. , Upton C., Changayil S.S., et al 2005. Poxvirus Bioinformatics Resource Center: a comprehensive Poxviridae informational and analytical resource. Nucleic Acids Res. 33: D311–D316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ramakrishnan, M.A. , Santhamani R. & Pandey A.B.. 2017. Capripox outbreak in a mixed flock of sheep and goats in India. Transbound Emerg. Dis. 64: 27–30. [DOI] [PubMed] [Google Scholar]
- 7. Santhamani, R. , Venkatesan G., Minhas S.K., et al 2015. Detection and characterization of atypical capripoxviruses among small ruminants in India. Virus Genes 51: 33–38. [DOI] [PubMed] [Google Scholar]
- 8. Yan, X.M. , Chu Y.F., Wu G.H., et al 2012. An outbreak of sheep pox associated with goat poxvirus in Gansu province of China. Vet. Microbiol. 156: 425–428. [DOI] [PubMed] [Google Scholar]
- 9. Garcia, M.A. , Meurs E.F. & Esteban M.. 2007. The dsRNA protein kinase PKR: virus and cell control. Biochimie 89: 799–811. [DOI] [PubMed] [Google Scholar]
- 10. Kostura, M. & Mathews M.B.. 1989. Purification and activation of the double‐stranded RNA‐dependent eIF‐2 kinase DAI. Mol. Cell. Biol. 9: 1576–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Langland, J.O. , Pettiford S., Jiang B., et al 1994. Products of the porcine group C rotavirus NSP3 gene bind specifically to double‐stranded RNA and inhibit activation of the interferon‐induced protein kinase PKR. J. Virol. 68: 3821–3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Patel, R.C. , Stanton P., Mcmillan N.M., et al 1995. The interferon‐inducible double‐stranded RNA‐activated protein kinase self‐associates in vitro and in vivo . Proc. Natl. Acad. Sci. USA 92: 8283–8287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rothenburg, S. , Chinchar V.G. & Dever T.E.. 2011. Characterization of a ranavirus inhibitor of the antiviral protein kinase PKR. BMC Microbiol. 11: 56 https://doi.org.10.1186/1471-2180-11-56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cassady, K.A. & Gross M.. 2002. The herpes simplex virus type 1 U(S)11 protein interacts with protein kinase R in infected cells and requires a 30‐amino‐acid sequence adjacent to a kinase substrate domain. J. Virol. 76: 2029–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chang, H.W. , Watson J.C. & Jacobs B.L.. 1992. The E3L gene of vaccinia virus encodes an inhibitor of the interferon‐induced, double‐stranded RNA‐dependent protein kinase. Proc. Natl. Acad. Sci. USA 89: 4825–4829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rice, A.P. & Kerr I.M.. 1984. Interferon‐mediated, double‐stranded RNA‐dependent protein kinase is inhibited in extracts from vaccinia virus‐infected cells. J. Virol. 50: 229–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Whitaker‐Dowling, P. & Youngner J.S.. 1984. Characterization of a specific kinase inhibitory factor produced by vaccinia virus which inhibits the interferon‐induced protein kinase. Virology 137: 171–181. [DOI] [PubMed] [Google Scholar]
- 18. Dar, A.C. & Sicheri F.. 2002. X‐ray crystal structure and functional analysis of vaccinia virus K3L reveals molecular determinants for PKR subversion and substrate recognition. Mol. Cell. 10: 295–305. [DOI] [PubMed] [Google Scholar]
- 19. Davies, M.V. , Chang H.W., Jacobs B.L., et al 1993. The E3L and K3L vaccinia virus gene products stimulate translation through inhibition of the double‐stranded RNA‐dependent protein kinase by different mechanisms. J. Virol. 67: 1688–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kawagishi‐Kobayashi, M. , Silverman J.B., Ung T.L., et al 1997. Regulation of the protein kinase PKR by the vaccinia virus pseudosubstrate inhibitor K3L is dependent on residues conserved between the K3L protein and the PKR substrate eIF2alpha. Mol. Cell. Biol. 17: 4146–4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Elde, N.C. , Child S.J., Geballe A.P., et al 2009. Protein kinase R reveals an evolutionary model for defeating viral mimicry. Nature 457: 485–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rothenburg, S. , Seo E.J., Gibbs J.S., et al 2009. Rapid evolution of protein kinase PKR alters sensitivity to viral inhibitors. Nat. Struct. Mol. Biol. 16: 63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Langland, J.O. & Jacobs B.L.. 2002. The role of the PKR‐inhibitory genes, E3L and K3L, in determining vaccinia virus host range. Virology 299: 133–141. [DOI] [PubMed] [Google Scholar]
- 24. Peng, C. , Haller S.L., Rahman M.M., et al 2016. Myxoma virus M156 is a specific inhibitor of rabbit PKR but contains a loss‐of‐function mutation in Australian virus isolates. Proc. Natl. Acad. Sci. USA 113: 3855–3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. He, B. , Gross M. & Roizman B.. 1997. The gamma (1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double‐stranded RNA‐activated protein kinase. Proc. Natl. Acad. Sci. USA 94: 843–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Livak, K.J. & Schmittgen T.D.. 2001. Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) Method. Methods 25: 402–408. 26. [DOI] [PubMed] [Google Scholar]
- 27. Samuel, C.E. 2001. Antiviral actions of interferons. Clin. Microbiol. Rev. 14: 778–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ludwig, S. , Pleschka S., Planz O., et al 2006. Ringing the alarm bells: signalling and apoptosis in influenza virus infected cells. Cell. Microbiol. 8: 375–386. [DOI] [PubMed] [Google Scholar]
- 29. Cheng, G. , Feng Z. & He B.. 2005. Herpes simplex virus 1 infection activates the endoplasmic reticulum resident kinase PERK and mediates eIF‐2alpha dephosphorylation by the gamma (1)34.5 protein. J. Virol. 79: 1379–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vyas, J. , Elia A. & Clemens M.J.. 2003. Inhibition of the protein kinase PKR by the internal ribosome entry site of hepatitis C virus genomic RNA. RNA 9: 858–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rice, A.D. , Turner P.C., Embury J.E., et al 2011. Roles of vaccinia virus genes E3L and K3L and host genes PKR and RNase L during intratracheal infection of C57BL/6 mice. J. Virol. 85: 550–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Thakur, M. , Seo E.J. & Dever T.E.. 2014. Variola virus E3L Zalpha domain, but not its Z‐DNA binding activity, is required for PKR inhibition. RNA 20: 214–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Beattie, E. , Tartaglia J. & Paoletti E.. 1991. Vaccinia virus‐encoded eIF‐2 alpha homolog abrogates the antiviral effect of interferon. Virology 183: 419–422. [DOI] [PubMed] [Google Scholar]
- 34. Taylor, S.S. , Haste N.M. & Ghosh G.. 2005. PKR and eIF2alpha: integration of kinase dimerization, activation, and substrate docking. Cell 122: 823–825. [DOI] [PubMed] [Google Scholar]
- 35. Dar, A.C. , Dever T.E. & Sicheri F.. 2005. PKR and eIF2α: integration of kinase dimerization, activation, and substrate docking. Cell 122: 823–834. [DOI] [PubMed] [Google Scholar]
- 36. Davies, M.V. , Elroy‐Stein O., Jagus R., et al 1992. The vaccinia virus K3L gene product potentiates translation by inhibiting double‐stranded‐RNA‐activated protein kinase and phosphorylation of the alpha subunit of eukaryotic initiation factor 2. J. Virol. 66: 1943–1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Elde, N.C. , Child S.J., Eickbush M.T., et al 2012. Poxviruses deploy genomic accordions to adapt rapidly against host antiviral defenses. Cell 150: 831–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Haller, S.L. , Peng C., Mcfadden G., et al 2014. Poxviruses and the evolution of host range and virulence. Infect. Genet. Evol. 21: 15–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tulman, E.R. , Afonso C.L., Lu Z., et al 2002. The genomes of sheeppox and goatpox viruses. J. Virol. 76: 6054–6061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhou, T. , Jia H., Chen G., et al 2012. Phylogenetic analysis of Chinese sheeppox and goatpox virus isolates. J. Virol. 9: 25 10.1186/1743-422X-9-25. [DOI] [PMC free article] [PubMed] [Google Scholar]