

Abstract
Ependymoma with YAP1‐MAMLD1 fusion is a rare, recently described supratentorial neoplasm of childhood, with few cases published so far. We report on 15 pediatric patients with ependymomas carrying YAP1‐MAMLD1 fusions, with their characteristic histopathology, immunophenotype and molecular/cytogenetic, radiological and clinical features. The YAP1‐MAMLD1 fusion was documented by RT‐PCR/Sanger sequencing, and tumor genomes were studied by molecular inversion probe (MIP) analysis. Significant copy number alterations were identified by GISTIC (Genomic Identification of Significant Targets in Cancer) analysis. All cases showed similar histopathological features including areas of high cellularity, presence of perivascular pseudo‐rosettes, small to medium‐sized nuclei with characteristic granular chromatin and strikingly abundant cells with dot‐like cytoplasmic expression of epithelial membrane antigen. Eleven cases presented features of anaplasia, corresponding to WHO grade III. MRI showed large supratentorial multinodular tumors with cystic components, heterogeneous contrast enhancement, located in the ventricular or periventricular region. One of two variants of YAP1‐MAMLD1 fusions was detected in all cases. The MIP genome profiles showed balanced profiles, with focal alterations of the YAP1 locus at 11q22.1–11q21.2 (7/14), MAMLD1 locus (Xp28) (10/14) and losses of chromosome arm 22q (5/14). Most patients were female (13/15) and younger than 3 years at diagnosis (12/15; median age, 8.2 months). Apart from one patient who died during surgery, all patients are alive without evidence of disease progression after receiving different treatment protocols, three without postoperative further treatment (median follow‐up, 4.84 years). In this to date, largest series of ependymomas with YAP1‐MAMLD1 fusions we show that they harbor characteristic histopathological, cytogenetic and imaging features, occur mostly in young girls under 3 years and are associated with good outcome. Therefore, this genetically defined neoplasm should be considered a distinct disease entity. The diagnosis should be confirmed by demonstration of the specific fusion. Further studies on large collaborative series are warranted to confirm our findings.
Keywords: childhood, ependymoma, supratentorial, YAP1‐MAMLD1 fusion
Introduction
Supratentorial ependymomas are heterogeneous from clinical, molecular and morphological perspectives. In 2014, two studies independently reported on recurrent C11orf95‐RELA fusions as the most frequent recurrent genetic alteration in supratentorial ependymomas of childhood, occurring in more than 70% of cases 20, 21. RELA fusion‐positive ependymoma has already been included in the last amendment of the WHO classification of brain tumors as a novel, genetically defined disease entity 11. Most RELA fusion‐positive ependymomas have a characteristic morphological aspect, often displaying clear cells and branching capillaries, although some pleomorphic tumors can also be seen in this group 4. RELA fusions have been shown to be oncogenic by activation of the NFKB pathway. The tumors can be well detected by demonstration of the fusion or by immunohistochemistry for pathological accumulation of p65 RelA protein in the nucleus 4, 6, 21.
Recently, a tumor with fusion of YAP1 and MAMLD1 genes in a female infant with supratentorial ependymoma was described 20. Another publication identified a group of tumors with characteristic methylation profiles and frequent copy number alterations in chromosome 11 at the YAP1 locus (9/13) and almost exclusive location in the supratentorial compartment (one lesion was identified in the posterior fossa) 18. The authors sequenced seven of these tumors, confirmed the presence of a YAP1‐MAMLD1 fusion in six; one case carried a fusion of YAP1 and an uncharacterized gene, FAM118B. YAP1‐MAMLD1 fusion transcripts contained exons 1–5 or 1–6 (out of 9) of the YAP1 in frame fused to exons 2–7 or 3–7 of the MAMLD1 gene. In the same study no YAP1 fusion was identified by RNA sequencing in 48 additional ependymal tumors from other epigenetically defined ependymoma subgroups of the posterior fossa (PF) or the supratentorial (ST) region. Tumors in the YAP subgroup occurred mainly in young children, and predominantly female patients. There were both WHO grade II and III tumors in this group, according to the WHO tumor classification guidelines. From 10 patients with clinical follow‐up data available, only two patients had tumor progression and overall survival (OS) was 100% at 5 years 18. A recent study showed that three ependymoma with YAP1‐MAMLD1 fusions have characteristic superenhancer‐associated gene profiles, distinct from other types of ependymoma 12.
YES‐associated protein 1 (YAP1) is one of the main downstream effectors of the Hippo signaling pathway, a tumor suppressor pathway implicated in organ size control but which is also deregulated in different types of cancer such as ovarian carcinoma, non‐small cell lung carcinoma, esophageal squamous cell carcinoma and hepatocellular carcinoma, and also associated with metastatic potential in breast carcinoma and melanoma among others 2, 8. YAP1 has shown to promote resistance to RAF and MEK targeted therapies in melanoma and lung cancer cells 9. Furthermore, YAP1 can act as a coactivator, for instance, as an effector of the alternative Wnt signaling or also regulator of the canonical Wnt/β‐catenin pathways 19. YAP1 has shown to be a target of Wnt/β‐catenin in colon cancer cells, and promotes glioma cell proliferation through β‐catenin activation 7, 23. Characteristic fusions between YAP1 and Transcription Factor Binding to IGHM Enhancer 3 (TFE3) have been described in epithelioid hemangioendotheliomas 1. YAP1 amplifications have also been identified in several types of cancer including some medulloblastomas with Sonic hedgehog activation 10. Mamld1 (mastermind‐like domain containing 1) is a member of the mastermind‐like proteins, which are important regulators of transcriptional events in Notch signaling and other signal transduction pathways including MEF2C (muscle differentiation and myopathies), p53 tumor suppressor and ß‐catenin/WNT pathways (colon carcinoma) 5, 14. Few human diseases have been specifically associated with MAMLD1 mutations/polymorphisms, most notably disorders of sexual development 16.
At present, few clinical, histopathological and genetic data are available on ependymomas with YAP1‐MAMLD1 fusions, and although no population‐based study has addressed the issue so far, this disease seems to be rare. To our knowledge, only the single case originally reported by Parker and coworkers, the 13 cases from a cohort including 122 supratentorial ependymomas and one additional case in an adult patient have been published so far 13, 18, 20.
The aim of the present study was to define clinical, molecular and morphological characteristics in a cohort of patients with YAP1‐MAMLD1‐fused ependymomas.
Materials and Methods
Fifteen cases of YAP1‐MAMLD1 ependymomas were retrieved from the archives of the DGNN Reference Center for Brain Tumors at the Institute of Neuropathology at the University of Bonn, Ste Anne/Necker Hospitals in Paris, University of Vienna and Seoul National University Children's hospital. The cases were identified by retrospective evaluation of supratentorial ependymomas of childhood and adolescents (0–21 years) and subsequent exclusion of RELA‐related ependymomas; residual cases were checked for the presence of YAP1‐MAMLD1 fusions. Ependymomas with YAP1‐MAMLD1 fusions correspond to approximately 4% of all supratentorial ependymomas in the age range of 0–14 years in the series of the DGNN brain tumor reference center, which covers all newly diagnosed pediatric brain tumors in Germany in a population‐based fashion. Retrospective histological re‐review and neuropathological classification were performed according to the recently revised 2016 WHO classification of tumors of the CNS 11.
Preoperative MRI scans and clinical data were collected including information on extent of surgery and postoperative treatment. Kaplan–Meier survival plots were derived from event‐free survival (EFS) and overall survival (OS) data using Sigma‐Plot 12.5 (Systat, Erkrath, Germany) software package.
Immunohistochemistry
Immunohistochemical (IHC) analysis was performed using standard protocols on an automated Ventana Benchmark XT immunostaining system (Roche‐Ventana, Darmstadt, Germany). We used primary antibodies against glial fibrillary acidic protein (GFAP; rabbit polyclonal, Agilent/Dako, Glostrup, Denmark), microtubule‐associated protein 2 (Map2; mouse monoclonal (HM‐2), Sigma‐Aldrich, St. Louis, MO, USA), p53 protein (mouse monoclonal (DO‐7), Agilent/Dako, Glostrup, Denmark), Olig‐2 (goat polyclonal, R&D Systems, Abingdon, UK), epithelial membrane antigen (mouse monoclonal (E‐29), Agilent/Dako, Glostrup, Denmark), p65 RelA (rabbit monoclonal (D14E12), Cell signaling, Danvers, U.S.A.), L1CAM (mouse monoclonal, (UJ127.11), Sigma–Aldrich, St Louis, MO, USA), Claudin‐1 (mouse monoclonal (ab56417), Abcam, Cambridge, UK), Ki67 (mouse monoclonal (Ki‐67P), Dianova, Hamburg, Germany), phospho‐histone‐3 (rabbit polyclonal, Bioclare Medical, Hague, Netherlands) and NF (mouse monoclonal (2F11), Agilent/Dako).
Electron microscopy
One case could be studied by electron microscopy. One small fragment of formalin‐fixed, paraffin‐embedded material was postfixed and processed for electron microscopy as described. The sections were examined with an electron microscope (Zeiss, EM10) 24.
Reverse‐transcription PCR for YAP1‐MAMLD1 fusion mRNA
PCR was performed on total RNA extracted from formalin‐fixed, paraffin‐embedded (FFPE) material as previously described 21. Total RNA from 15 tumor samples was extracted from FFPE tissue using the AllPrep DNA/RNA FFPE Kit from Qiagen (Hilden, Germany), according to the manufacturer's instructions. 100–500 nanograms of RNA (as measured by 260‐nm extinction) were then reverse‐transcribed using the Superscript III First‐Strand Synthesis System for RT‐PCR (Invitrogen/Thermofisher, Waltham, MA, USA) and random primers. PCR of cDNA was performed with the following primers: The forward primer was located in exon 5 of YAP1 5′‐AACTGCAGATGGAGAAGGAG‐3′, the reverse primers in exon 2 of MAMLD1 R1, 5′‐TGTCTGGAAACTGGAAGTGG‐3′ and R2, 5′‐GTGACATCTTCAAGGCAAGG‐3′ resulting in products sized 227 base pairs (R1) and 282 base pairs (R2) in the case of a YAP1‐exon 6/MAMLD1‐exon 2 fusion and 105 bp (R1) and 160 bp (R2) in the case of a YAP1‐exon 5/MAMLD1‐exon 3. PCR was performed according to the following parameters: denaturation 94°C for 30 s, annealing temperature 60°C, for 30‐s elongation, 72°C for 30 s, 50 cycles. The generated PCR fragments were analyzed on a 2% agarose gel. PCR products were visualized and documented on a Gel Doc 1000 system (Bio‐Rad, Düsseldorf, Germany). The PCR purification kit from Qiagen (Hilden, Germany) was used for purifying PCR products. Direct Sanger sequencing reactions were performed in duplicate (forward and reverse; Eurofins MWG Operon, Ebersberg, Germany).
Molecular inversion probe analysis
To identify genomic copy number gains and losses a molecular inversion probe (MIP) assay (OncoScan Version 3; Affymetrix, Santa Clara, CA, USA) was used. MIP assay of 15 supratentorial ependymoma samples was performed as previously described, using 80‐ng DNA from each case 22. The raw MIP data file was analyzed with Nexus Copy Number 7.0 Discovery Edition software (BioDiscovery, El Segundo, CA, USA). BioDiscovery's SNP‐FASST2 segmentation algorithm was used to make copy number and loss of heterozygosity calls. Genomic Identification of Significant Targets in Cancer (GISTIC) analysis was used to distinguish significant chromosomal aberrations from random background 3.
FISH
FISH study was performed on interphase nuclei according to the standard procedures and the manufacturer's instructions. Break‐apart fluorescent FISH of the YAP1 locus was performed using the Dual Color YAP1 Break Apart probe (Empire Genomics, New York City, NY, USA). Signals were scored in at least 100 nonoverlapping intact interphase nuclei per case. FISH for gene rearrangement was considered positive if at least 20% of analyzed cells showed a split of at least one set of red and green signals or an isolated (red or green) signal. Results were recorded using a DM600 imaging fluorescence microscope (Leica Biosystems, Richmond, IL, USA) fitted with appropriate filters, a CCD camera and digital imaging software from Leica (Cytovision, v7.4).
Results
Imaging features
We were able to retrieve preoperative MRI data for 10 patients. Main findings in imaging are summarized in supplementary Table S1 and depicted in Figure 1. All tumors analyzed were large at presentation with prominent cystic components. Location was intraventricular/paraventricular in nine patients and paraventricular in one patient. The solid component showed a multinodular pattern and radiological sings of hemorrhage were present. All tumors showed heterogeneous contrast enhancement. Most tumors were isointense on T1 and T2 images, compared to cerebral cortex. Peritumoral edema was variable.
Figure 1.
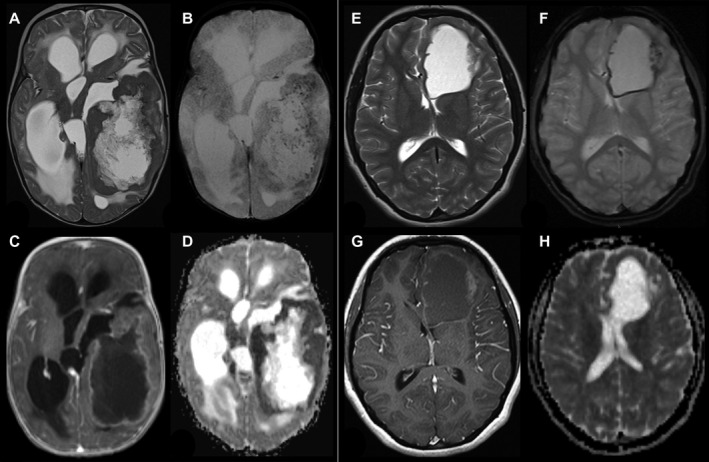
Radiological findings in two patients with YAP1‐MAMLD1‐fused ependymoma. (A–D; Patient #9) 4 months old infant with a huge left temporal tumor, a large cystic part surrounded by solid tumor, which is isointense to gray matter on T2‐weigthed image (A) and punctuated hemorrhages in the solid part a small rim along the cyst wall (T2*, B). The solid part of the tumor shows a garland‐shaped enhancement (C) and ADC values are decreased compared to gray matter. T2 and ADC changes are compatible with a higher cell density of the tumor. (E–H, patient # 6) 15‐year‐old girl with a left frontal tumor without edema consisting of a large cyst and a small solid part at the lateral border. The solid part is nearly isointense to gray matter in T2‐weighted image (E). T2* (F) reveals subtle punctuated hemorrhages in the solid part a small rim along the cyst wall. The solid part of the tumor shows a faint enhancement on T1‐weighted image (G) and the apparent diffusion coefficient (ADC) is comparable to the gray matter.
Histopathological analysis
Main histopathological features are depicted in Figures 2 and 3 and are summarized in Supplementary Table S2. All tumors showed remarkable morphological similarity. Cellularity was moderate to high, and perivascular pseudorosettes were observed in all cases (Figure 2a,b). Tumor cell nuclei were of small to medium size. Nuclear contours were often not exclusively round. Twelve cases displayed nuclei with polygonal angulated contours, in five of which as a predominant feature and in seven cases focally (Figures 2c,d and 3e). In two cases, a focal marked nuclear pleomorphism was identified, without increased mitotic activity in these areas. The nuclear chromatin texture appeared relatively dense and homogeneous. Tumor cell cytoplasms were eosinophilic with mostly indistinct borders. Nevertheless, in eight cases tumor cells with well‐demarcated, granular eosinophilic cytoplasms, some reminiscent of mini‐gemistocytes, often grouped in small clusters were seen (Figure 3c). Interestingly, eight tumors displayed eosinophilic granular bodies, either very focally 2, focally 4 or conspicuously 2 (Figure 3d). While the granular bodies showed variable PAS positivity, only focal cytoplasmic granular positivity was observed in most cases. There was no evidence of neuronal, in particular neurocytic or gangliocytic differentiation.
Figure 2.
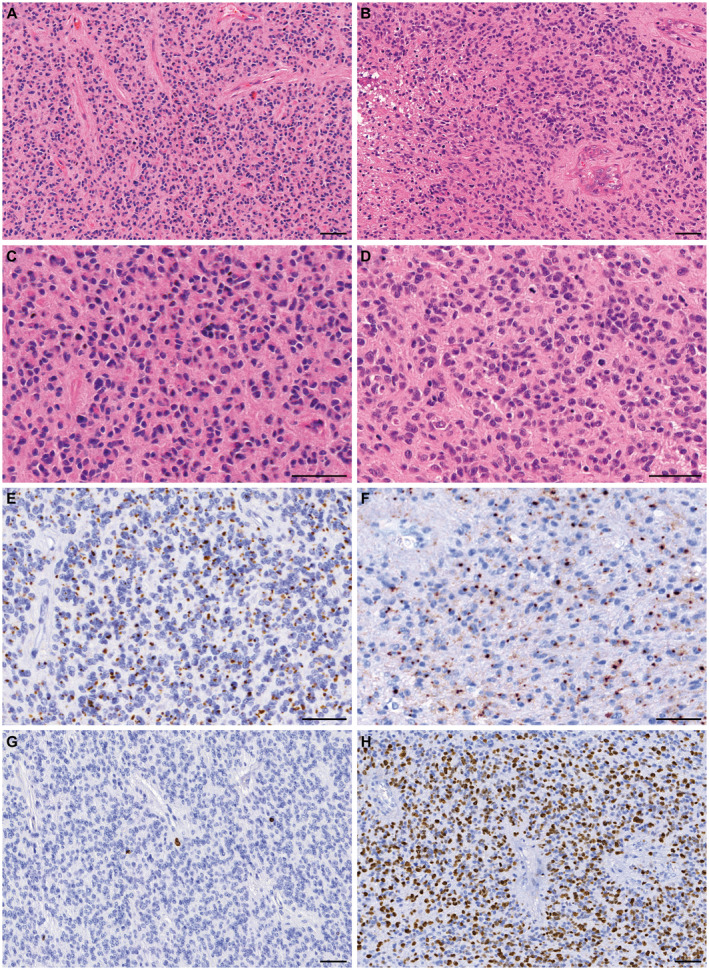
Representative morphology and immunophenotype of ependymomas with YAP1‐MAMLD1 fusion. Panels on the left (A, C, E, G) are from patient #1, and panels on the right (B, D, F, H) from patient #10. All tumors in this series showed densely populated areas and perivascular pseudorosettes, as can be observed in H&E stained slides from the two tumors (A) and (B). In (B) vascular proliferation and necrosis can be observed. At higher magnification (C–D) the characteristic tumor cells with small nuclei, and eosinophilic cytoplasms are seen, as well as a brisk mitotic activity in (D). Panels (E) and (F) show the strong and diffuse dot‐like epithelial membrane antigen (EMA) expression. Panels (G) and (H) highlight the strikingly variable proliferation index (Ki67 expression), very low in the areas shown from tumor on the left (G) and high in the tumor on the right (H). bar, 50 µm.
Figure 3.
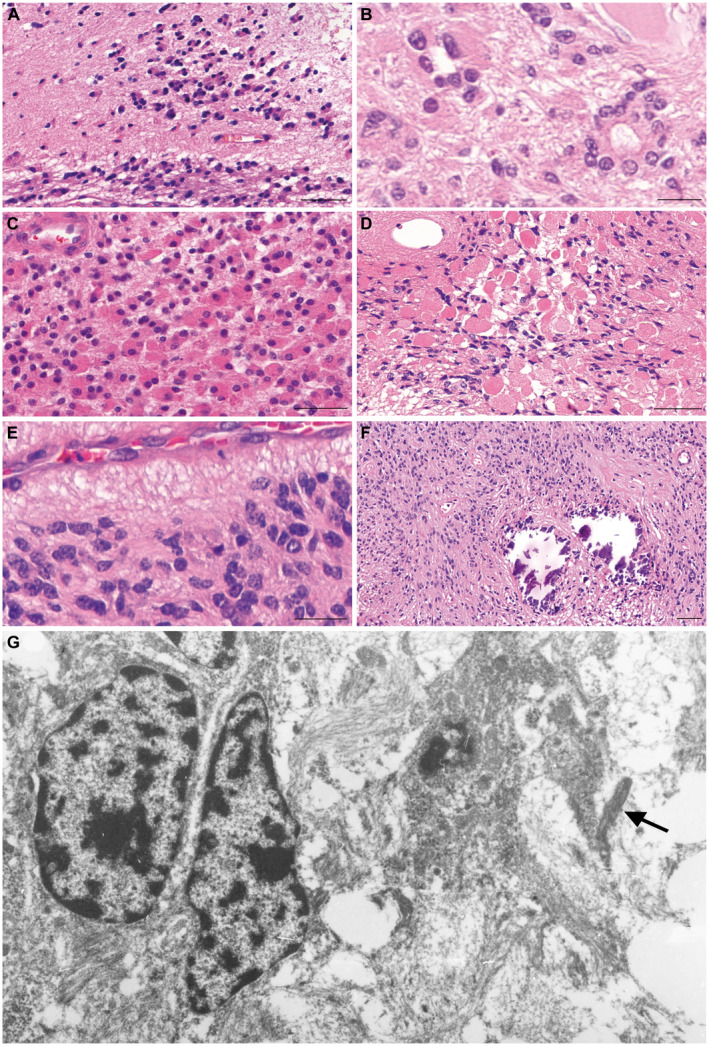
Morphological and ultrastructural features of YAP1‐MAMLD1‐fused ependymomas. Although generally well demarcated from surrounding brain parenchyma, 7/15 tumors showed focally invasive tumor cells or tumor cell clusters as depicted in panel (A). True ependymal rosettes were also often present (B). Another common feature were tumor cells with eosinophilic, granular cytoplasms, sometimes with well‐defined boundaries, peripherally placed nuclei and tendency to form small groupings. (C). Eosinophilic granular bodies were also found, in the case shown in (D) these structures were very abundant. In (E), one observes the small nuclei with homogenous chromatin, yet pleomorphic often angulated contours. Calcifications were also frequent, as shown in (F). Similar nuclear features to those depicted in (E) are observed by electron microscopy performed in a sample from patient #4 (G). In addition, abortive cilia (arrow) can be observed as an indicator of ependymal differentiation. Bars in (A), (C), (D), (F) 50 µm, in (B), (E), 20 µm.
According to the revised 2016 WHO classification of tumors of the CNS, a WHO grade II was assigned to 4 tumors and a WHO grade III to 11. Mitotic activity ranged from 0 to 1 mitotic figure/10 high power fields (HPF) in WHO grade II tumors and from 10 to 61 mitoses/10 HPF in WHO grade III tumors. The proliferation index assessed by Ki67 staining of nuclei was less than 5% in all WHO grade II tumors and was higher than 10% in the most proliferative areas of WHO grade III tumors. The Ki67 labeling index typically varied in different areas within individual tumors. Four tumors exhibited discrete nodules with both high cell density and high proliferative index as compared to other tumor regions. In accordance with the variable mitotic index (see also Supplementary Table S2) the proliferation index was quite variable (Figure 2g,h).
True ependymal rosettes were detectable in seven cases, always focally (Figure 3b). Papillary areas, clear cell morphology or tanycytic differentiation was absent. Necrosis was present in 13 cases, including two WHO grade II tumors, and was often extensive. Calcifications were present in 9/15 tumors (Figure 3f). Microvascular endothelial proliferation was seen in 13 cases. Tumors were generally well demarcated from surrounding brain parenchyma which often showed prominent reactive gliosis. However, microscopic foci of peritumoral invasion of surrounding brain parenchyma were frequently observed (7/15 cases) (Figure 3a).
Most tumors displayed perivascular expression of GFAP and variable positivity for neural MAP2. Positivity for the transcription factor Olig2 was seen in two cases only and was restricted to rare isolated tumor cells in these. Interestingly, all tumors depicted striking widespread and strong positivity for EMA, all with a dot‐like sometimes also ring‐like intracytoplasmatic pattern (Figure 2e,f). EMA expression was restricted to such structures in most cases; only in two cases, a diffuse granular cytoplasmic or cell membrane‐oriented pattern was observed in addition. Cell nuclei were negative for p65 RelA protein. No positivity for L1CAM was observed in any of the 11 cases which could be tested for this antigen. Cytoplasmic neurofilaments were only found in single tumor cells in two cases. The essentially solid nature of the tumors in this series was also documented by the absence of preexisting neurofilament‐positive axons in the tumor tissue. Only two tumors showed focally entrapped axons, exclusively located in their periphery.
As increased expression of Claudin‐1 has been reported in YAP fusion‐positive ependymomas 18 we analyzed ímmunohistochemical expression of Claudin‐1. In 9 of 13 cases Claudin‐1 was found widely expressed. Two further cases showed only focal expression; two were negative. We used five posterior fossa group A ependymomas and five RELA fusion‐positive ependymomas as control; most cases were positive for Claudin‐1 (data not shown).
Electron microscopy
Ependymal differentiation could be confirmed by the presence of abortive cilia. The angulated nuclei contours observed on H&E slides were also well depicted by electron microscopy (Figure 3g).
Detection of YAP1‐MAMLD1 fusion mRNA
In all cases, YAP1‐MAMLD1 fusions were confirmed by RT‐PCR and sequencing. In 14/15 tumors a fusion between YAP1‐exon 5 and MAMLD1‐exon 3 as detected. In a single case (case #6), a fusion between YAP1‐exon 5 and exon 2 of MAMLD1 (transcript variant 2) was detected (Figure 4a).
Figure 4.
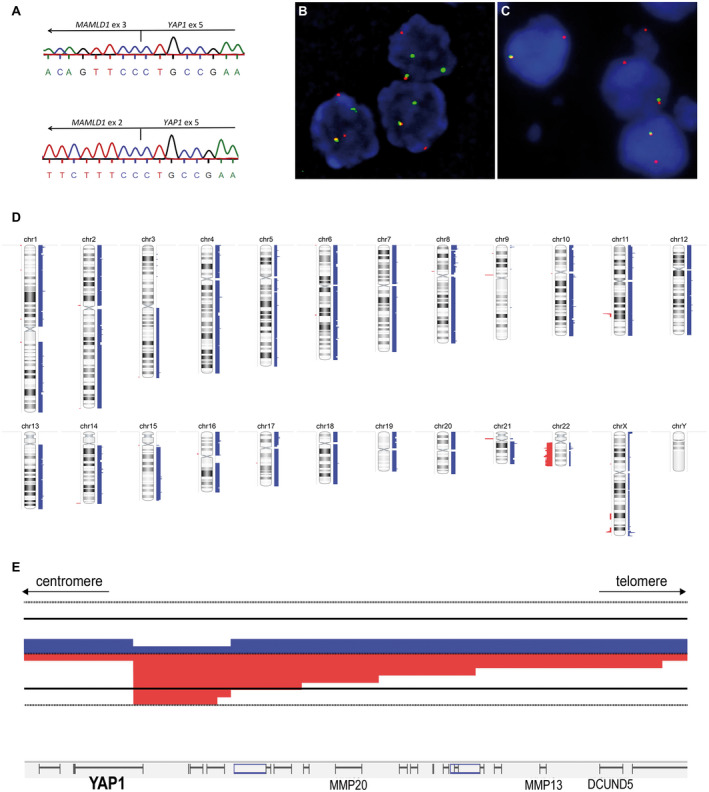
Molecular and cytogenetic features of ependymomas with YAP1‐MAMLD1 fusions. (A) shows cDNA sequencing of two distinct fusions. On the top the most common type is shown, between YAP1‐exon 5 and MAMLD1‐exon3 seen in 14 cases and on the bottom the fusion YAP1‐exon 5 and MAMLD1‐exon 2 detected in a single case (#6). In (B) and (C), two different patterns observed by YAP1 FISH, confirming the YAP1‐rearrangement. In (B) a classic rearrangement with one fused (normal) and one break‐apart signal in each nucleus (3′YAP1: green signal; 5′YAP1: red signal) is shown (patient #4). In (C) images of a rearrangement with one fused signal and a single 5′YAP1 (unbalanced rearrangement with loss of the 3′locus during the translocation; 3′YAP1: green signal; 5′YAP1: red signal) (patient #14). In (D) a virtual karyotype (summary plot) of 14 ependymoma with YAP1‐MAMLD1 fusion analyzed by MIP assay is shown. Gains are indicated by blue bars on the right side of each chromosome, losses are shown in red on the left side. Thickness of the bars indicates the frequency of alterations. chr, chromosome. In (E), a detailed view on the YAP1 locus is shown at higher magnification revealing focal copy number alterations. Six cases had a breakpoint within the YAP1 gene with copy number loss of 3′ regions of YAP1.
FISH analysis
By FISH analysis, 4 of 9 cases tested showed a rearrangement of YAP1. In one case, a classical rearrangement with one fused and one break‐apart signal was found in each nucleus. In three cases, unbalanced rearrangements with one fused and one 5′YAP1 signal was identified, while the signal for the 3′ probe of YAP1 was lost indicating a deletion of the 3′ end of YAP1. Examples of both patterns of alterations are shown in Figure 4b,c. Five cases were not informative because of failed FISH analysis, most likely due to insufficient quality of the archival FFPE material. FISH results are summarized in supplementary Table S3.
Genome‐wide copy number analysis by molecular inversion probe assay
MIP yielded interpretable results for 14/15 patients tested. In one case, the assay failed due to extensive fragmentation of the DNA extracted. A summary plot is depicted in Figure 4d and most frequent recurrent alterations are summarized in Supplementary Table S3. Most tumors showed stable genomic profiles. Two tumors (from patients #5 and #6) appeared polyploid. Copy number alterations of the YAP locus (11q22) were present in 8/14 patients, of which 7 displaying losses, with indication of a break in YAP1 in 6 patients. One patient had loss of the YAP1 locus without evidence for a break within the YAP1 gene and one patient had a gain of the whole chromosome 11, including the YAP1 locus. A summary plot showing the YAP1 locus alterations is shown in Figure 4e. Focal copy number alterations in the MAMLD1 locus (Xp28) were seen in 10 cases, of which 5 showed gains and 5 showed losses in this region. Eight of 14 cases showed an indication for a break in MAMLD1. In addition, 4/14 cases exhibited monosomy 22. One further case had regional losses of chromosome 22. The common region of loss encompasses 22q12.3–22q13.3.
Clinical characteristics
Clinical data are summarized in Table 1. From 15 studied patients, 13 were female and 2 were male. Twelve patients were younger than 3 years at diagnosis. Median age was 8.2 months. Information regarding treatment and clinical follow‐up was available for all patients (mean duration of follow‐up was 6.4 years, median 4.8 years, range 0.6–16 years). One patient died very early due to surgical complications and was therefore not included in the outcome analysis. All patients were initially treated by surgical excision of the tumor. Complete resection was achieved for 11/14 patients after one (n = 6), two (n = 4) or three (n = 1) surgical procedures. Three patients had residual disease after surgery: patient #1 who has stable residue with a follow‐up of 15 years, patient #4 without detectable residue after chemo‐/radiotherapy and patient #15 with a follow‐up of 15 months. Two patients had progressive disease. Patient #10 underwent a total resection followed by adjuvant chemotherapy and showed a relapse 6 months later, which was treated by surgery and proton therapy. Two years later there was no evidence of disease. Patient #14 had residual disease after first surgery, which progressed 4 months later during chemotherapy. The tumor was completely resected in a second surgical procedure and postsurgical follow‐up in two months was uneventful. Four patients were treated by a combination of chemotherapy and radiotherapy, five by postoperative chemotherapy only and two by radiotherapy only, according to the age‐ and risk‐adapted protocols. Notably, three patients did not receive any postoperative treatment, of which two had WHO grade II tumors and one an anaplastic WHO grade III tumor. Apart from the patient who died during surgery, all patients were alive, without evidence of progressive disease at latest follow‐up (Figure 5).
Table 1.
Clinical data of the patient cohort.
| Patient | Sex | Age at dx (y) | WHO grade | Number of surgeries performed | Residual disease | Chemotherapy | Relapse | Radiotherapy | Death | OS (Y) | EFS (Y) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | f | 0.26 | III | 1 | Yes | E‐HIT2000 | No (constant residual) | Yes (total 56 Gy) | No | 15.00 | 15.00 |
| 2 | f | 0.56 | III | 2 | No | E‐HIT2000 | No | No | No | 11.28 | 11.28 |
| 3 | f | 1.80 | III | 1 | No | E‐HIT 2000 | No | Yes (total 54Gy) | No | 9.03 | 9.03 |
| 4 | m | 0.97 | III | 1 | Yes | E‐HIT2000 | No | Yes (proton, total 54 Gy) | No | 5.57 | 5.57 |
| 5 | f | 6.62 | II | 1 | No | No | No | Yes (total 68Gy) | No | 1.36 | 1.36 |
| 6 | f | 14.59 | II | 1 | No | No | No | Yes (total 68 Gy) | No | 3.66 | 3.66 |
| 7 | m | 0.11 | III | 1 | died during surgery | No | ‐ | No | Yes | 0 | 0 |
| 8 | f | 0.68 | III | 2 | No | E‐HIT2000 | No | No | No | 4.11 | 4.11 |
| 9 | f | 0.34 | III | 1 | No | E‐HIT2000 (3 cycles), 6 cycles temozolamide | No | No | No | 3.40 | 3.40 |
| 10 | f | 0.33 | III | 1 | No | I‐HIT‐MED | Yes | Yes (proton, total 54 Gy) | No | 2.18 | 0.49 |
| 11 | f | 1.53 | II | 2 | No | No | No | No | No | 16.01 | 16.01 |
| 12 | f | 0.04 | III | 3 | No | No | No | No | No | 7.12 | 7.12 |
| 13 | f | 6.01 | II | 1 | No | No | No | No | No | 9.53 | 9.53 |
| 14 | f | 1.18 | III | 2 | No | I‐HIT‐MED | Yes (progression after first surgery) | No | No | 0.61 | 0.35 |
| 15 | f | 0.28 | III | 1 | Yes | E‐II/5 cycles PEI | No | No | No | 1.28 | 1.28 |
Figure 5.
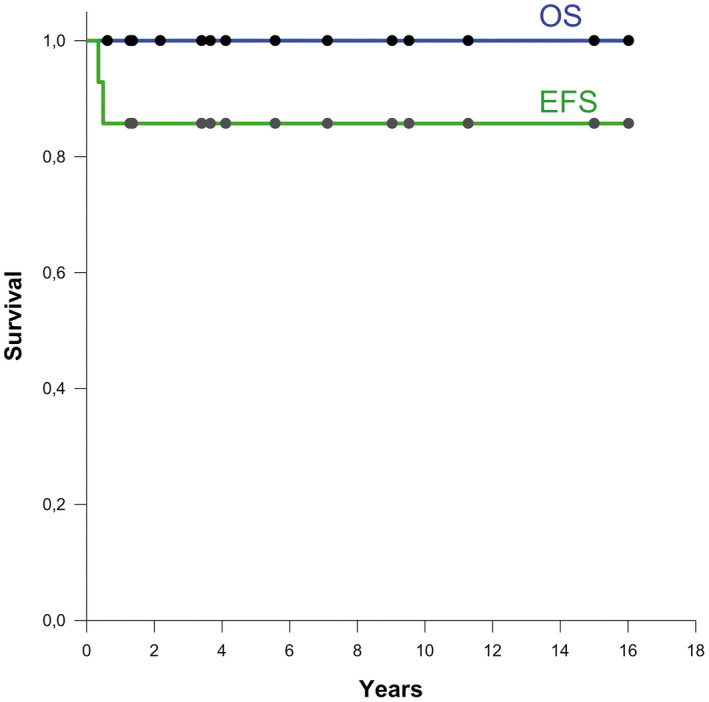
Overall survival (OS) and event‐free survival (EFS) (n = 14).
Discussion
It has been postulated for a long time that supratentorial ependymomas of childhood may have a different biology from ependymomas in other locations, but the underlying biology was unclear until the recurrent C11orf95‐RELA fusion was identified in the majority of supratentorial ependymomas. The entity “RELA fusion‐positive ependymoma” was included in the revised WHO classification in 2016. A second recurrent fusion was found involving the genes encoding the transcriptional cofactors Yap1 and Mamld1. Its biological significance was not clear since YAP1 is known to be involved in various genetic abnormalities such as amplifications in some tumors and fusions with different partner genes. In addition, several other fusions have been identified in ependymomas 17, some of which were not recurrent and some occurred on the RNA level without underlying DNA mutations. The recurrent YAP1‐MAMLD1 fusion has been demonstrated to occur on the genomic level 20, also confirmed in this study by FISH.
We studied the histopathology of ependymomas with YAP1‐MAMLD1 fusion, and observed characteristic features in every single case: areas of high cellularity, perivascular pseudorosettes, relatively monomorphic small tumor cells, often angulated or at least focally irregularly shaped nuclei with mottled and dense chromatin. Despite the presence of focal clusters of infiltrating cells in some cases, a diffuse infiltrative growth pattern was absent at the tumor margins. This is in line with the sharp demarcation seen in other ependymoma variants. Interestingly, several cases displayed eosinophilic granular bodies, which are not typical for ependymal tumors. A characteristic feature of YAP1‐MAMLD1 fusion‐positive tumors was a strikingly strong and widespread dot‐like and sometimes ring‐like intracytoplasmic EMA expression. At lower density, such structures frequently occur in other ependymoma variants and are believed to represent microlumina. As expected, none of the YAP1‐MAMLD1 fusion‐positive ependymomas showed a nuclear p65 RelA accumulation typical for NFkB‐activated, RELA fusion‐positive supratentorial ependymoma 6, 21.
As Claudin‐1 overexpression has been shown in ependymomas with YAP1‐MAMLD1 fusion 18, we investigated whether immunohistochemical detection of Claudin‐1 could be of diagnostic value in its differential diagnoses. Although Claudin‐1 was positive in most tumors with YAP1‐MAMLD1 fusions, positivity was also seen in other ependymoma variants, and thus proved to be nonspecific.
Cytogenetically, our data are also in accordance with the previous reports on few YAP1‐MAMLD1‐fused ependymomas showing rare chromosomal imbalances and frequent local aberrations occurring around the YAP1 locus in 9/13 patients in the series by Pajtler et al 18. In the present series, focal YAP1 aberrations were found in 7/14 patients. Focal loss in the telomeric regions on chromosome 11 (YAP1 locus) with a break within YAP1 were detected in six cases. The FISH results were in total concordance with the MIP results. The cases showing a typical FISH break‐apart signal and in addition, a loss of one telomeric (green) signal showed also loss of this region in MIP. Furthermore, we identified frequent alterations of the MAMLD1 locus on Xq28 in the majority of patients as a prominent genetic feature. While most cases showed a stable genome with few chromosomal alterations (with chromosome 22 loss as the most frequent whole chromosomal copy number aberration), two cases showed polyploidy; both were WHO grade II tumors that occurred in older children (6 and 14.6 years at diagnosis). Extensive microchromothripsis proposed as a hallmark of RELA fusion‐positive ependymomas was not identified in this cohort. Therefore, chromosomal alterations are clearly different between RELA fusion‐positive and YAP1‐MAMLD1 fusion‐positive ependymomas.
Our results confirm some of the epidemiological findings described in retrospective cohorts, showing a predominance of ependymomas with YAP1‐MAMLD1 fusion among young female children. In the present series, 13/15 patients were female and only 3/15 were older than 3 years at diagnosis.
In contrast to supratentorial ependymomas with RELA fusion which were described to be preferentially located in the cerebral cortex carrying predominant cystic components 4, 15, YAP1‐MAMLD1‐fused ependymomas showed intra‐/periventricular location and prominent multinodular components, irrespective of cystic changes. The T2‐signal isointense to the cortex matched well with the high cellularity of YAP1‐MAMLD1‐fused ependymomas.
Although the few YAP1‐MAMLD1 cases described so far seem to have a more favorable prognosis compared to RELA fusion‐positive ependymomas, a detailed outcome analysis of these patients was not available. As patients in our series were analyzed retrospectively, and treated with different protocols it is hard to draw definitive conclusions on the behavior of the tumors. Yet, a very striking finding in our cohort was an excellent outcome with 100% of children alive at a median follow‐up of 4.8 years, although one patient (#14) had to be reoperated and one patient (#10) was irradiated after tumor progression. One patient (#1) with a WHO grade III tumor has residual stable disease after irradiation and chemotherapy under German E‐HIT2000 protocol. Most interestingly, three patients did not receive any postoperative oncological treatment, two patients (#11, #13) with WHO grade II tumors and one (#12) with a WHO grade III tumor. The favorable clinical outcome in this case is surprising, as this tumor was histopathologically highly malignant showing abundant necrosis, vascular proliferation and mitotic activity (more than 100 mitoses in 10 high power fields, labeled with phospho‐histone 3 as a mitotic marker). In fact, two of three patients with tumors corresponding to WHO grade II were older than 6 years, but the third patient (#11) was 18 months old at diagnosis. Although the number of patients analyzed is too small to allow conclusions, it is tempting to speculate if there could be a possible relation between patient age and proliferation activity in these tumors.
Further studies are urgently needed to determine if a specific treatment protocol should be offered to children with YAP1‐MAMLD1‐fused ependymomas. Current standard treatment for ependymomas is surgery followed by high‐dose radiotherapy for children over 12 months without evidence of disseminated disease. Patients with YAP‐MAMLD1 fusion‐positive ependymomas may qualify for a reduction of treatment intensity in clinical trials with the aim to avoid the deleterious late effects of radiotherapy. As these tumors are rare, prospective clinical studies with a significant number of patients will be difficult to achieve and collaborative work will be important for further assessment of clinical behavior and optimal treatment.
Regarding the diagnosis we recommend to test supratentorial RELA fusion‐negative ependymomas of childhood for this recurrent fusion. As the fusion transcript is highly expressed, RT‐PCR and subsequent sequencing represents a simple and robust technology applicable on standard diagnostic FFPE material. Direct identification of the fusion transcripts by specific probes by Nanostring technology may represent an alternative assay. In addition, FISH analysis with break‐apart probes for YAP1 and MAMLD1 may be employed or methylation profiling because such tumors show a characteristic epigenetic signature. However, the last two assays provide only indirect evidence for this entity. In diagnostic neuropathology, direct tests for specific genetic events should be preferred. Since specific and sensitive immunohistological surrogate markers (comparable with p65‐RelA nuclear staining for RELA fusion‐positive ependymomas) are still lacking, a prescreening assay for these cases is currently not available; we therefore recommend to test all RELA fusion‐negative supratentorial ependymomas for YAP1‐MAMLD1 fusions.
Our data show that ependymomas carrying YAP1‐MAMLD1 fusions represent a distinct disease entity showing characteristic epidemiological, neuroradiological, histopathological, genetic and clinical features.
Supporting information
Table S1. Preoperative neuroradiological features.
Table S2. Morphology and immunophenotype.
Table S3. FISH and MIP results.
Acknowledgments
This study was supported by funding on the German Childrens Cancer Foundation (grant DKS 2014_17).
Contributor Information
Felipe Andreiuolo, Email: felipe.andreiuolo@ukbonn.de.
Torsten Pietsch, Email: t.pietsch@uni-bonn.de.
References
- 1. Antonescu CR, Le Loarer F, Mosquera JM, Sboner A, Zhang L, Chen CL et al (2013) Novel YAP1‐TFE3 fusion defines a distinct subset of epithelioid hemangioendothelioma. Genes Chrom Cancer 52:775–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bao Y, Hata Y, Ikeda M, Withanage K (2011) Mammalian Hippo pathway: from development to cancer and beyond. J Biochem (Tokyo) 149:361–379. [DOI] [PubMed] [Google Scholar]
- 3. Beroukhim R, Getz G, Nghiemphu L, Barretina J, Hsueh T, Linhart D et al (2007) Assessing the significance of chromosomal aberrations in cancer: methodology and application to glioma. Proc Natl Acad Sci U S A 104:20007–20012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Figarella‐Branger D, Lechapt‐Zalcman E, Tabouret E, Jünger S, de Paula AM, Bouvier C et al (2016) Supratentorial clear cell ependymomas with branching capillaries demonstrate characteristic clinicopathological features and pathological activation of nuclear factor‐kappaB signaling. Neuro‐Oncol 18:919–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fukami M, Wada Y, Okada M, Kato F, Katsumata N, Baba T et al (2008) Mastermind‐like domain‐containing 1 (MAMLD1 or CXorf6) transactivates the Hes3 promoter, augments testosterone production, and contains the SF1 target sequence. J Biol Chem 283:5525–5532. [DOI] [PubMed] [Google Scholar]
- 6. Gessi M, Giagnacovo M, Modena P, Elefante G, Gianno F, Buttarelli FR et al (2017) Role of immunohistochemistry in the identification of supratentorial C11ORF95‐RELA fused ependymoma in routine neuropathology. Am J Surg Pathol 2017 Dec 20. [Epub ahead of print]. doi: 10.1097/PAS.0000000000000979. [DOI] [PubMed] [Google Scholar]
- 7. Konsavage WM, Kyler SL, Rennoll SA, Jin G, Yochum GS (2012) Wnt/β‐catenin signaling regulates Yes‐associated protein (YAP) gene expression in colorectal carcinoma cells. J Biol Chem 287:11730–11739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lamar JM, Stern P, Liu H, Schindler JW, Jiang Z‐G, Hynes RO (2012) The Hippo pathway target, YAP, promotes metastasis through its TEAD‐interaction domain. Proc Natl Acad Sci U S A 109:E2441–E2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lin L, Sabnis AJ, Chan E, Olivas V, Cade L, Pazarentzos E et al (2015) The Hippo effector YAP promotes resistance to RAF‐ and MEK‐targeted cancer therapies. Nat Genet 47:250–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lorenzetto E, Brenca M, Boeri M, Verri C, Piccinin E, Gasparini P et al (2014) YAP1 acts as oncogenic target of 11q22 amplification in multiple cancer subtypes. Oncotarget 5:2608–2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Louis D, Ohgaki H, Wiestler OD, Cavenee WK (eds) (2016) WHO Classification of Tumours of the Central Nervous System, 4th edn. IARC Press: Lyon. [Google Scholar]
- 12. Mack SC, Pajtler KW, Chavez L, Okonechnikov K, Bertrand KC, Wang X, et al (2018) Therapeutic targeting of ependymoma as informed by oncogenic enhancer profiling. Nature 553:101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Malgulwar PB, Nambirajan A, Pathak P, Faruq M, Rajeshwari M, Singh M, et al (2018) C11orf95‐RELA fusions and upregulated NF‐KB signalling characterise a subset of aggressive supratentorial ependymomas that express L1CAM and nestin. J Neurooncol 138:29–39. [DOI] [PubMed] [Google Scholar]
- 14. McElhinny AS, Li J‐L, Wu L (2008) Mastermind‐like transcriptional co‐activators: emerging roles in regulating cross talk among multiple signaling pathways. Oncogene 27:5138–5147. [DOI] [PubMed] [Google Scholar]
- 15. Nowak J, Jünger ST, Huflage H, Seidel C, Hohm A, Vandergrift LA et al (2018) MR imaging phenotype of RELA‐fused pediatric supratentorial ependymomas. Clin Neuroradiol [Epub ahead of print]. doi: 10.1007/s00062-018-0704-2. [DOI] [PubMed] [Google Scholar]
- 16. Ogata T, Sano S, Nagata E, Kato F, Fukami M (2012) MAMLD1 and 46, XY disorders of sex development. Semin Reprod Med 30:410–416. [DOI] [PubMed] [Google Scholar]
- 17. Olsen TK, Panagopoulos I, Gorunova L, Micci F, Andersen K, Kilen Andersen H et al (2016) Novel fusion genes and chimeric transcripts in ependymal tumors. Genes Chromosomes Cancer 55:944–953. [DOI] [PubMed] [Google Scholar]
- 18. Pajtler KW, Witt H, Sill M, Jones DTW, Hovestadt V, Kratochwil F, et al (2015) Molecular classification of ependymal tumors across all CNS compartments, histopathological grades, and age groups. Cancer Cell 27:728–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Park HW, Kim YC, Yu B, Moroishi T, Mo J‐S, Plouffe SW et al (2015) Alternative Wnt signaling activates YAP/TAZ. Cell 162:780–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Parker M, Mohankumar KM, Punchihewa C, Weinlich R, Dalton JD, Li Y et al (2014) C11orf95‐RELA fusions drive oncogenic NF‐κB signalling in ependymoma. Nature 506:451–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pietsch T, Wohlers I, Goschzik T, Dreschmann V, Denkhaus D, Dörner E et al (2014) Supratentorial ependymomas of childhood carry C11orf95‐RELA fusions leading to pathological activation of the NF‐κB signaling pathway. Acta Neuropathol (Berl) 127:609–611. [DOI] [PubMed] [Google Scholar]
- 22. Wang Y, Cottman M, Schiffman JD (2012) Molecular inversion probes: a novel microarray technology and its application in cancer research. Cancer Genet 205:341–355. [DOI] [PubMed] [Google Scholar]
- 23. Wang Y, Pan P, Wang Z, Zhang Y, Xie P, Geng D et al (2017) β‐catenin‐mediated YAP signaling promotes human glioma growth. J Exp Clin Cancer Res CR 36:136–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yun Y, Bergmann M, Klein H, Sternowsky HJ (1995) Congenital myopathy with focal loss of cross striations: a case report with morphologic and immunohistochemical study. Gen Diagn Pathol 141:155–160. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Preoperative neuroradiological features.
Table S2. Morphology and immunophenotype.
Table S3. FISH and MIP results.