

Abstract
p62 is a stress‐inducible protein able to change among binding partners, cellular localizations and form liquid droplet structures in a context‐dependent manner. This protein is mainly defined as a cargo receptor for selective autophagy, a process that allows the degradation of detrimental and unnecessary components through the lysosome. Besides this role, its ability to interact with multiple binding partners allows p62 to act as a main regulator of the activation of the Nrf2, mTORC1, and NF‐κB signaling pathways, linking p62 to the oxidative defense system, nutrient sensing, and inflammation, respectively. In the present review, we will present the molecular mechanisms behind the control p62 exerts over these pathways, their interconnection and how their deregulation contributes to cancer progression.
Keywords: autophagy, cancer, Keap1, mTORC1, NF‐κB, Nrf2, p62/SQSTM1
p62/SQSTM1 interacts with key regulator proteins for diverse signal transduction pathways including mTORC1 activation, NF‐κB signaling, the Keap1‐Nrf2 system, and selective autophagy and serves as a signaling hub for anabolism, inflammatory response, antioxidant response, and catabolism. In this State‐of‐the‐Art Review, we discuss about diverse roles of p62/SQSTM1 and about their deregulation in cancer.
Abbreviations
- ARE
antioxidant response element
- bZIP
leucine zipper
- ccRCC
clear cell renal cell carcinoma
- CK2
casein kinase 2
- CLEAR
coordinated lysosomal expression regulation
- CNC
cap‘n'collar
- EpRE
electrophile response element
- HCC
hepatocellular carcinoma
- KIR
Keap1‐interacting region
- LIR
LC3‐interacting region
- mTORC1
mechanistic target of rapamycin complex 1
- PB1
Phox1 and Bem1p
- PDAC
pancreatic ductal adenocarcinoma
- ROS
reactive oxygen species
- TAK1
TGF‐β‐activated kinase
- TBK1
TANK‐binding kinase 1
- TB
TRAF6 binding
- TRAF6
tumor necrosis factor receptor‐associated factor 6
- UBA
ubiquitin‐associated
- ZZ
zinc finger
Introduction
SQSTM1, the human gene encoding for p62/SQSTM1 (hereafter as p62), localizes in chromosome 5 and comprises 8 exons distributed through 16 kb. The gene is conserved throughout the metazoans and its expression is ubiquitously observed 1. In response to various stresses, master gene regulators Nrf2, NF‐κB, and MiT/TFE upregulate the expression of SQSTM1 2, 3, 4 (Fig. 1A). Missense mutations in SQSTM1 have been primarily associated with Paget's disease of bone, amyotrophic lateral sclerosis, and frontotemporal lobar degeneration 5. In addition, copy gains in chromosome 5q lead to clear cell renal cell carcinoma (ccRCC) at least partially due to increased expression levels of p62 6 and p62 accumulation has been observed in multiple forms of cancer (Table 1). Different p62‐positive structures such as Mallory–Denk bodies and intracellular hyaline bodies have been identified in patients suffering from steatohepatitis, alcoholic hepatitis, and hepatocellular carcinoma 7, 8.
Figure 1.
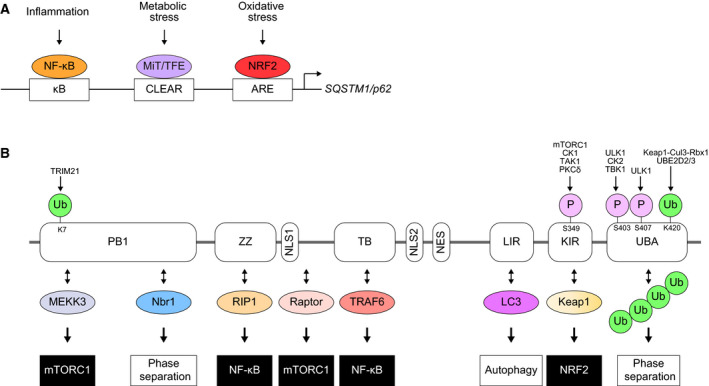
(A) Gene expression of SQSTM1/p62 through stress‐responsible transcription factors. CLEAR: coordinated lysosomal expression regulation. ARE: antioxidant response element. (B) Domain structure of the p62 protein. Through the interaction with multiple proteins, p62 serves as a signaling hub that modulates a variety of cellular functions (anabolism and catabolism) and/or protein property (phase separation), which are regulated by post‐translational modifications of p62. PB1: Phox and Bem1p. ZZ: Zinc finger. NLS: nuclear localization signal. TB: TRAF6‐binding domain. NES: nuclear export signal. LIR: LC3‐interacting region. KIR: Keap1‐interacting region. UBA: ubiquitin‐associated.
Table 1.
Cancer forms with described p62 accumulation
| Type of cancer | References |
|---|---|
| Human hepatocellular carcinoma | 42, 46, 53, 54, 90, 101, 102 |
| Intrahepatic cholangiocarcinoma | 103 |
| Melanoma | 104 |
| Pancreatic | 105 |
| Lung | 106, 107 |
| Esophageal | 108 |
| Gastric | 105, 109 |
| Colon | 105, 110 |
| Breast | 111, 112, 113, 114 |
| Prostate | 115, 116 |
| Endometrial | 117 |
| Ovarian | 118 |
| Kidney | 6 |
| Squamous cell carcinoma of head and neck | 119 |
| Oral | 120 |
p62, initially identified as a 62‐kDa protein 9, has multiple functional domains which include an N‐terminal Phox1 and Bem1p (PB1) domain, a zinc finger (ZZ), a tumor necrosis factor receptor‐associated factor 6 (TRAF6)‐binding (TB) motif, an LC3‐interacting region (LIR), a Keap1‐interacting region (KIR), and a ubiquitin‐associated (UBA) domain 10, 11, 12 (Fig. 1B). The protein localization is not limited to the cytoplasm, but can also be observed in the nucleus, in autophagosomes, and on lysosomes 13, 14, 15, 16. Due to its role as a receptor for selective autophagy, p62 also localizes to cargos that will be degraded during the process, such as ubiquitin‐positive protein aggregates 14, 17, 18, 19, damaged mitochondria, and invading bacteria 20. Under certain circumstances p62 can also be degraded by the proteasome 21 or endosomal‐related autophagy 22, but it is primarily degraded during selective autophagy.
p62 is known as a multifunctional signaling hub, as it participates in the activation of mTORC1 (mechanistic target of rapamycin complex 1) in nutrient sensing 15, NF‐κB activation during inflammation and apoptosis 23 and the activation of the Keap1‐Nrf2 pathway for antioxidant response 24, as well as its mentioned receptor role in selective autophagy 25. Alterations in all these pathways have been associated with cancer development 26, 27, 28, 29. Here, we describe how the interplay between p62 and the Nrf2, mTORC1, NF‐κB, and autophagy pathways plays a key cellular role both in homeostatic conditions and during cancer development.
p62‐mediated Nrf2 activation
Nrf2 is a cap‘n'collar (CNC), leucine zipper (bZIP) transcription factor which controls the basal and inducible expression of over 200 genes involved in the antioxidant stress response. While Nrf2 is expressed in all cell types, its basal protein levels are usually kept low during unstressed conditions because its binding partner, Keap1, is an adaptor of Cullin3‐based ubiquitin ligases. A Keap1 homodimer recognizes a Nrf2 monomer through a two‐site ‘hinge and latch’ binding: a tight interaction with the Nrf2‐ETGE motif and a weaker one with the Nrf2‐DLGex motif, which are indispensable for sufficient ubiquitination of Nrf2. Consequently, Nrf2 undergoes proteasomal degradation, and this mechanism of regulation of Nrf2 is known as the Keap1‐Nrf2 pathway 30 (Fig. 2).
Figure 2.
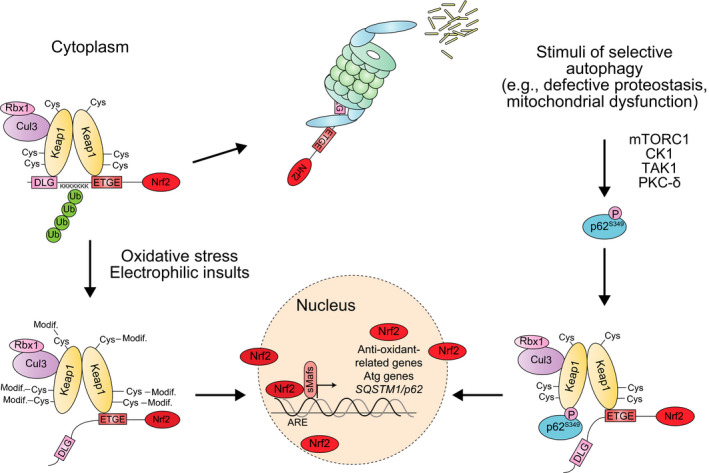
Mechanism of Nrf2 activation dependent on S349 phosphorylation of p62. Left: under basal conditions, Keap1, in collaboration with the Cul3/Rbx1 E3 ubiquitin ligase complex, promotes the proteasomal degradation of Nrf2. Upon oxidative stress, Keap1 oxidation results in the liberation of Nrf2 and its translocation to the nucleus, where it promotes the expression of a battery of target genes encoding antioxidant proteins and anti‐inflammatory enzymes. Right: phosphorylated p62 interacts with the Nrf2‐binding site of Keap1 and competitively inhibits the Keap1–Nrf2 interaction, resulting in the expression of Nrf2 target genes, which include p62. Therefore, Ser349 phosphorylated p62 causes the constitutive activation of Nrf2.
Keap1 is a cysteine‐rich protein that, upon exposure to oxidative stress or electrophilic insults, is oxidized, resulting in a conformational change in the Keap1 dimer. As a result, Nrf2 escapes from Keap1 interaction and translocates into the nucleus to induce the expression of a battery of target genes. These Nrf2 targets contain antioxidant response elements (AREs) or electrophile response elements (EpREs) in their regulatory region and encode for antioxidant and anti‐inflammatory enzymes 29 (Fig. 2).
An aberrant activation of this pathway has been observed in multiple forms of cancer 31. There are at least six alterations that can result in this activation. First, somatic mutations in the Cullin3 gene, in the coding region of Keap1 or in the DLGex and ETGE motifs of Nrf2 hamper Nrf2 recognition and degradation by the Keap1/Cul3 complex 32, 33, 34. Second, aberrant transcription of the Nrf2 gene lacking the second exon results in protein forms that cannot interact with Keap1 and constitutively localize to the nucleus 35. Third, promoter methylation of the Keap1 gene 36. Fourth, accumulation of Keap1‐binding proteins that interfere in the Keap1–Nrf2 interaction 24, 37. This mechanism will be discussed in more detail later in this section. Fifth, Keap1 oxidation by oncometabolites such as fumarate 38, 39. Sixth, transcriptional activation of the Nrf2 gene by an oncogenic KRAS mutant 40.
Besides providing increased resistance to oxidative stress, cancer cells also benefit from a metabolic reprogramming resulting from Nrf2‐persistent activation. Nrf2 regulates the expression of genes involved in glutaminolysis, purine nucleotide and glutathione synthesis, and the pentose phosphate pathway 41, 42. This establishes a feedback loop between the Keap1‐Nrf2 and the phosphatidylinositol 3‐kinase‐Akt pathways 41, 43. The latter pathway is also commonly affected in cancer 44.
As mentioned before, Nrf2 activation can also be achieved through Keap1‐binding proteins that interfere with Nrf2 degradation. p62 presents a Keap1‐interacting region (KIR) that binds to the Keap1 domain responsible for Nrf2 interaction: the Keap1 DC domain 3, 24, 45. Under basal conditions, the interaction between p62‐KIR and Keap1‐DC is too weak to compete with the interaction between Keap1‐DC and the ETGE and DLGex domains of Nrf2 46. However, p62‐KIR can undergo phosphorylation in Ser349 by multiple kinases, including mTORC1 and TGF‐β‐activated kinase (TAK1) 46, 47, 48. This modification prevents the two‐site binding between Keap1 and Nrf2, resulting in the release of Nrf2 and its translocation to the nucleus 46 (Fig. 2). Since p62 is phosphorylated at Ser349 on autophagic cargos such as ubiquitinated proteins, the Keap1‐Nrf2 pathway and selective autophagy are coupled with each other (Fig. 2 and Fig. 5C).
Figure 5.
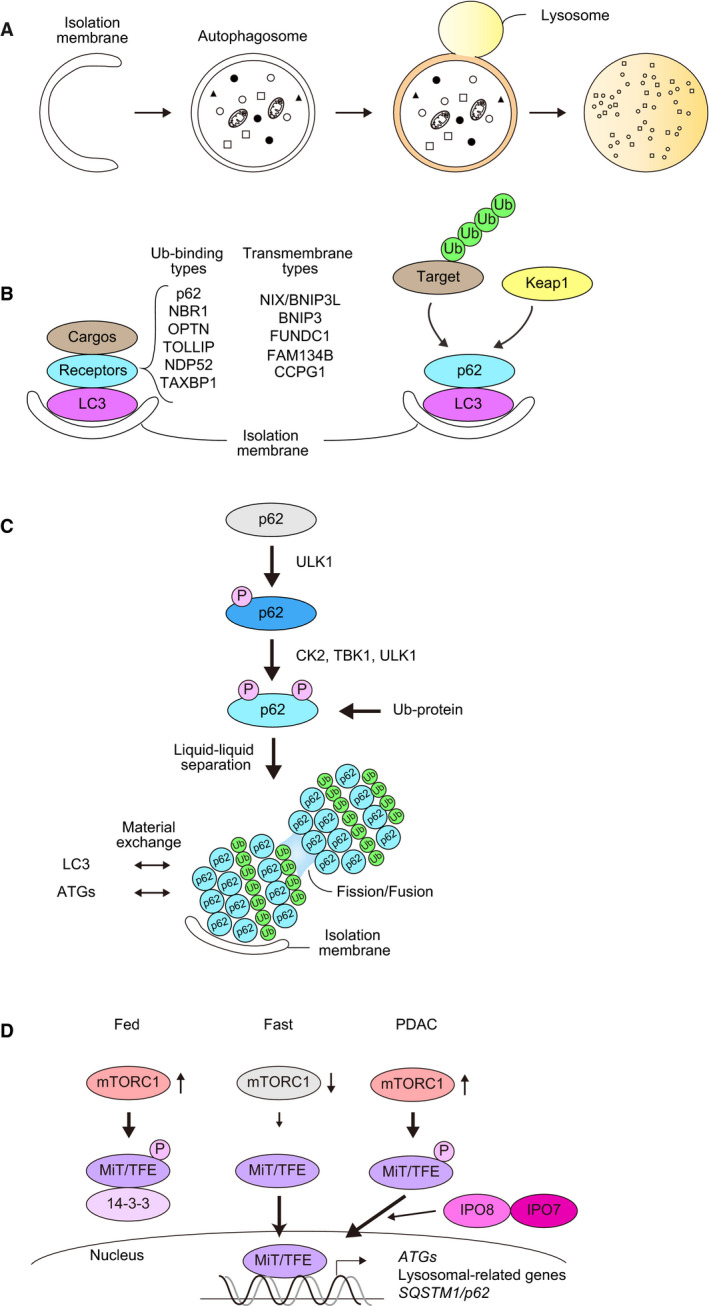
(A) Macroautophagy is accompanied by dynamic membrane biogenesis and autophagosome formation. The autophagosome sequesters a portion of the cytoplasm and fuses with the lysosome, where its contents are degraded. (B) Receptor‐mediated selective autophagy. Receptor proteins are divided into two groups: ubiquitin‐binding type and transmembrane type. Both have the ability to bind to Atg8 family proteins (e.g., LC3s and GABARAPs). p62 is a representative Ub‐binding type receptor, and it also mediates autophagic degradation of certain proteins (e.g., Keap1) through their direct interaction without prior ubiquitination. (C) p62‐mediated selective autophagy. When selective autophagic cargos such as misfolded proteins appear in the cytoplasm, they are ubiquitinated. Concomitantly, Ser407, located at the UBA domain of p62, is initially phosphorylated by ULK1 kinase. This phosphorylation destabilizes the UBA dimer interface and subsequently casein kinase 2 (CK2), TANK‐binding kinase 1 (TBK1), or ULK1 phosphorylate Ser403 of the UBA domain, which increases the binding affinity of p62 for the ubiquitin chain. The p62 forming a complex with ubiquitin chains has liquid‐like properties and forms droplets in which LC3 and other Atg proteins assemble to form the isolation membrane. Keap1, a binding protein for p62 may also shuttle from cytoplasm to the droplets. (D) Regulation of the nuclear translocation of the MiT‐TFE family by mTORC1‐mediated phosphorylation. Under nutrient‐rich conditions, the MiT‐TFE family is phosphorylated by mTORC1 and subsequently trapped by 14‐3‐3 protein. As a result, the MiT‐TFE family is kept in the cytoplasm. Once mTORC1 is inactivated by metabolic stresses such as nutrient deprivation, the MiT‐TFE family translocate into the nucleus to induce lysosomal biogenesis, Atg genes and p62/SQSTM1. In PDAC, increased level of Importin8 and 7 bypasses the regulation of mTORC1.
After its release from Keap1‐mediated degradation, Nrf2 promotes the expression of two isoforms of p62: the full‐length protein and a splicing variant lacking the KIR domain 49. We recently showed that this splicing form is still functional for autophagy but is unable to interfere in the interaction between Keap1 and Nrf2. Then, the variant form stabilizes Keap1 and suppresses the expression of Nrf2 targets (including p62) 49. Keap1 can also downregulate the p62‐Keap1‐Nrf2 pathway by promoting the ubiquitination and subsequent autophagic degradation of p62 50. Although Keap1 degradation is mainly achieved through autophagy 51, possibly in a p62‐dependent manner 3, 52, it is not clear whether the ubiquitination of p62 by Keap1 affects the autophagic degradation of Keap1 or not.
Despite these two possible mechanisms of downregulation of the p62‐Keap1‐Nrf2 pathway, the fact that p62 is a target gene of Nrf2 3 (Fig. 1A) opens the possibility for a positive feedback loop that is exploited in many forms of cancer. Table 1 shows that accumulation of p62 is a common observation in cancer cells. Phosphorylated p62 is also present in these accumulations 53, thus resulting in Nrf2 upregulation and the aforementioned metabolic reprogramming. Recent studies showed that p62 accumulation is enough to induce HCC development and confer cancer cells with enhanced proliferation capability and anticancer drug resistance due to persistent Nrf2 activation 42, 54. p62 is also a key driver of neoplastic progression in pancreas 55. Nrf2 activation derived from p62 accumulation induces MDM2, which then acts through p53‐dependent and ‐independent mechanisms to promote pancreatic ductal adenocarcinoma 55.
mTORC1 activation by p62
The mechanistic target of rapamycin is a serine/threonine protein kinase which belongs to the family of PI3K‐related kinases family. It constitutes the catalytic subunit of two distinct protein complexes, named mTOR Complex 1 (mTORC1) and 2 (mTORC2). In mTORC1, mTOR associates with Raptor, which is responsible for mTORC1 subcellular localization and substrate recruitment, and mLST8, which stabilizes the kinase activation loop of mTOR. The complex also includes two inhibitory subunits: PRAS40 and DEPTOR (Fig. 3A). Depending on the environmental conditions, cells have to fine‐tune the balance between catabolism and anabolism. In favorable conditions, mTORC1 stimulates protein, lipid, and nucleotide production to promote cell growth, concurrently blocking catabolic pathways such as autophagy. mTORC1‐dependent anabolic induction is achieved through the phosphorylation of the downstream kinase S6K1, as well as 4EBP (protein synthesis induction), Lipin1 (lipid synthesis), ATF4 (nucleotide synthesis), and HIF1α (glycolysis) (Fig. 3A). Conversely, phosphorylation of ULK1, an autophagy‐initiating kinase, by mTORC1 suppresses autophagy 26 (Fig. 3A).
Figure 3.
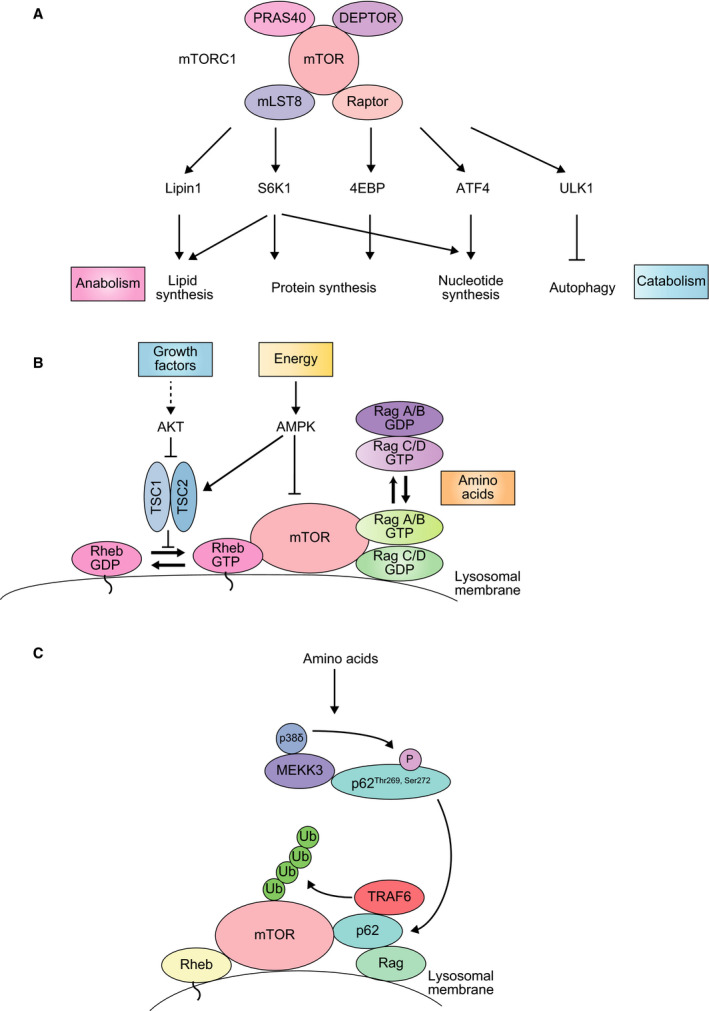
(A) The mTORC1 pathway. The mTORC1 complex is formed by the mTOR kinase, the regulator subunits Raptor and PRAS40 and the inhibitors mLST8 and DEPTOR. Upon mTORC1 stimulation, downstream effectors promote anabolism and suppress catabolism. (B) The activity of mTORC1 is inhibited by the TSC complex, which is itself inactivated in the presence of growth factors and upregulated upon energy deprivation. In the later situation, AMPK inhibits mTORC1 both directly and through TSC activation. Increased amino acid availability converts RagA/B GDP and RagC/D GTP into RagA/B GTP and RagC/D GDP respectively, assembling mTORC1 on the lysosome, where Rheb activates mTORC1. (C) mTORC1 activation by p62. Amino acid stimulation promotes the phosphorylation of p62 at Thr269 and Ser272 by MEKK3/p38delta. This results in the formation of a signaling hub over the lysosomal membrane through the interaction of p62 with Raptor, TRAF6 and Rag proteins. As a consequence, mTOR is ubiquitinated by TRAF6, and then activated by Rheb.
Multiple signals, including growth factors, hormones, energy, and amino acid levels can regulate mTORC1 activity. In the case of growth factors, this regulation is achieved through the inhibition of mTORC1‐negative regulator TSC, resulting in mTORC1 activation by the small GTPase Rheb (Fig. 3B). Energy reduction, such as glucose deprivation, activates the metabolic regulator AMPK, which inhibits mTORC1 both directly and through TSC. On the other hand, high amino acid levels promote mTORC1 activation through Rag GTPases. These proteins, together with Rheb and the Ragulator complex, are located on the lysosomal membrane. Upon amino acid stimulation, a Rag heterodimer binds to Raptor and recruits mTORC1 to the lysosome, where it can be activated by Rheb 56, 57.
p62 was identified as an interacting partner for Raptor 15 (Fig. 1B). Further research on the topic showed that, in response to amino acid abundance, p62 is phosphorylated at Thr269 and Ser272 by its PB1‐mediated interaction with MEKK3 58. Next, p62 forms a signaling hub on the lysosomal membrane through its interaction with Raptor and the Rags proteins which recruits the ubiquitin ligase TRAF6 and mTORC1 to the surface of the organelle and, ultimately, results in K63‐linked ubiquitination of mTOR (Fig. 3C). This ubiquitination driven by p62‐TRAF6 under amino acid‐rich conditions results in mTORC1 activation, followed by coupling nutrient availability to anabolism and cell growth 15, 58, 59.
The enhanced proliferative rate of cancer cells is highly benefited by mTORC1 upregulation 26. Thus, upstream regulators of mTOR, such as PTEN and Akt, are frequently mutated in human tumors, and MTOR mutations are also found in some cancer subtypes 26, 60. p62 is accumulated in multiple forms of cancer (Table 1), and it is likely exerting its carcinogenic role through enhanced mTORC1 activation. In agreement with this, HCC developed by TSC depletion (which results in constitutive mTORC1 activation) is reverted by concurrent deletion of p62 54. But, while tumor cells benefit from increased levels of p62, they also take profit from p62 downregulation in the surrounding stromal cells, a phenomenon commonly observed in prostate and liver 11. In prostate cancer, loss of p62 in the stromal fibroblasts impairs mTORC1 activation, which results in a reduced metabolic detoxification capacity and the secretion of the prosurvival inflammatory cytokine IL‐6, which sustains the growth and invasiveness of the neighboring prostate cancer cells 61. In addition, this loss of p62 in stromal cells also promotes the accumulation of ATF4 (a downstream effector of mTORC1 that undergoes proteasomal degradation in a p62‐dependent manner), starting a transcriptional program that delivers asparagine to the cancer cells, allowing them to survive in conditions of glutamine deprivation 62. This asparagine also ensures mTORC1 activation in the tumor, sustaining the proliferative anabolic signaling in cancer cells 63, 64.
Regulation of NF‐κB signaling through p62
The NF‐κB is a key family of regulators of inflammation and the immune response that is composed by five members combined into homo‐ or heterodimers. There are two main kinds of NF‐κB signaling pathways: the classical, in which proinflammatory cytokines promote a rapid and transient induction, and the alternative, which depends on de novo synthesis of the NF‐κB‐inducing kinase NIK, and then requires a longer activation time. In both cases, the activation is achieved through the degradation of specific inhibitors, the IκB proteins, once they are phosphorylated by an IKK complex 65 (Fig. 4A).
Figure 4.
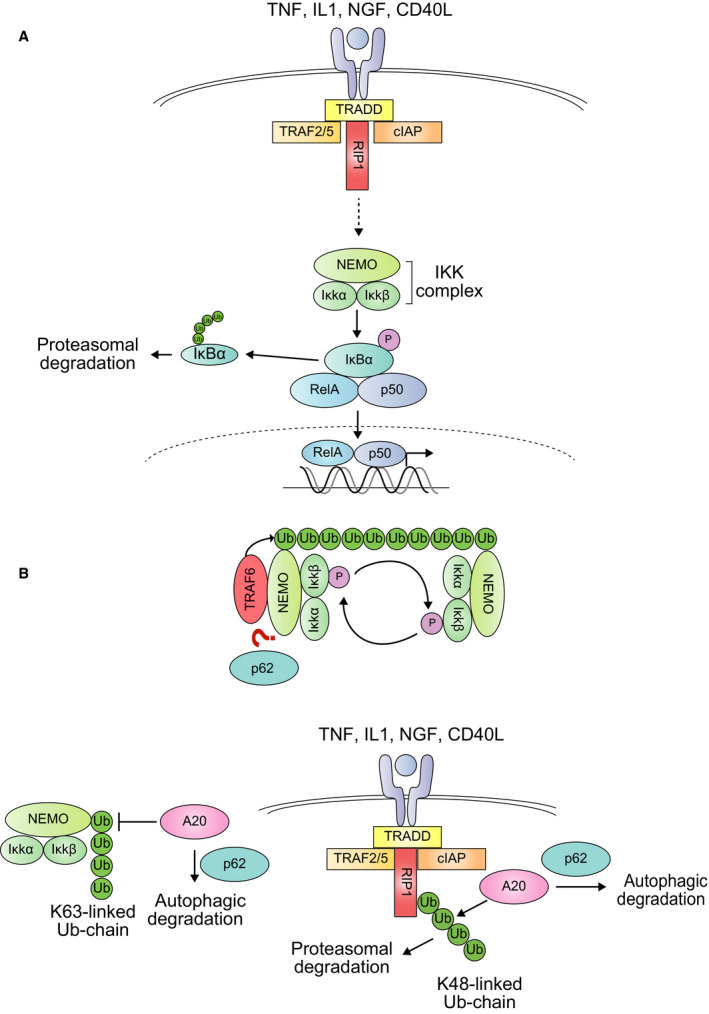
(A) The NF‐κB pathway. Multiple signals, such as proinflammatory cytokines or growth factors, can induce NF‐κB activation. Following the ligand‐receptor binding, the activation of the IKK complex releases NF‐κB (RelA–p50 complex) from its inhibitor IκB. This is achieved through the phosphorylation and subsequent proteasomal degradation of the inhibitor. Then, NF‐κB translocates to the nucleus and induces the expression of target genes, including p62. (B) Two mechanisms of p62‐mediated NF‐κB activation. First, p62 seems to be required for NEMO ubiquitination by TRAF6. This ubiquitination is recognized by the NEMO subunits of other IKK complexes, bringing them into the close proximity required for their cross‐phosphorylation and activation. Second, p62 promotes the autophagic degradation of the ubiquitin‐editing enzyme A20, which otherwise inhibits NF‐κB activation by promoting RIP1 degradation and reversing NEMO ubiquitination.
Classically, the IKK complex is composed by the catalytic subunits IKKα and IKKβ together with the regulatory subunit NEMO (or IKKγ). The activation of the complex requires the trans‐autophosphorylation between two adjacent IKK catalytic subunit dimers 27 (Fig. 4B). These dimers are brought together by the nondegradative ubiquitination of their NEMO subunits, which can bind to the ubiquitin chain of the other regulatory subunit 66. This nondegradative ubiquitination, mainly composed of K63‐ and M1‐linked chains, is catalyzed by E3 ubiquitin ligases such as TRAF6 and LUBAC, respectively. In the case of TRAF6, p62 seems to participate in the K63‐linked ubiquitination of NEMO 67 (Fig. 4B). An additional role of p62 in NF‐κB activation is to promote the degradation of A20, a ubiquitin‐editing enzyme that prevents excessive NF‐κB activation by counteracting K63‐linked ubiquitination of NEMO and promoting K48‐linked ubiquitination of RIP1, followed by its degradation by the 26S proteasome 68, 69. At the same time, p62 is upregulated by NF‐κB, creating a positive feedback loop 2 (Fig. 1A).
The NF‐κB pathway participates in every step of tumor formation. First, it promotes the production of reactive oxygen species (ROS) that induce DNA damage and oncogenic mutations. Second, it enhances the proliferation of these cells, while preventing p53‐induced apoptosis. Finally, inflammation and NF‐κB support malignant progression by stimulating the expression of genes that enhance neoangiogenesis and the invasive behavior of the cancer cells 70, 71. Pancreatic ductal adenocarcinoma (PDAC) and lung cancer are two examples in which KRAS mutation results in a feedforward loop between NF‐κB and p62 that promotes tumorigenesis through the aforementioned mechanisms 2, 72.
During cancer development, macrophages are recruited to the tumor microenvironment to protect the organism by engulfing the damaged cells. Induction of p62 through NF‐κB in macrophages seems to be required to limit inflammasome‐mediated IL‐1β release. Then, p62 protects macrophages from excessive NF‐κB activation, and genetic ablation of p62 in these cells results in increased tissue damage and IL‐1β‐dependent inflammation, which is likely to promote tumorigenesis 73, 74. Likewise, as mentioned in a previous section, cancer‐associated fibroblasts develop an aberrant phenotype due to their reduced levels of p62. These fibroblasts maintain a prosurvival transcriptional program that increases the secretion of proinflammatory cytokines. These paracrine signals stimulate tumorigenesis and cancer progression at least partly in an NF‐κB‐dependent manner 61, 75, 76.
Autophagy and p62
Autophagy is defined as an intracellular lysosomal degradation pathway. Among its different subtypes, macroautophagy (thereafter referred to as autophagy) is the most characterized form of autophagy and is defined by the formation of a double membrane structure called the autophagosome that will later fuse with the lysosome 77 (Fig. 5A). There are two modes of autophagy: nonselective and selective autophagy, in which the autophagosome either randomly sequesters cytoplasmic components or targets specific cargos, respectively. When specific autophagic cargos such as damaged or excess organelles appear in the cytoplasm, they are tagged with ubiquitin, leading to the assembly of receptor proteins that bind to the ubiquitin chain (Ub‐binding type receptors, Fig. 5B). Alternatively, transmembrane type of receptor proteins directly localize on cargos (transmembrane‐type receptors, Fig. 5B). In the latter case, molecular markers like ubiquitin are not needed. Most receptor proteins have LC3‐interacting regions, called LIRs, to interact with ATG8 family proteins 25, 78. Thus, cargo labeling and the transfer of receptor proteins to cargos mainly regulate selective autophagy (Fig. 5B). Several receptor proteins are delivered to the lumen of autophagosomes together with ATG8 family proteins and degraded in lysosomes, being themselves targets of selective autophagy.
p62 is the best characterized receptor protein for selective autophagy of ubiquitinated cargos 10 (Fig. 5B). When its activity as an autophagy receptor is not required, p62 is held inactive through homodimerization of its UBA domain, which prevents it from interacting with ubiquitin 79, 80. Phosphorylation events drive the liberation of the UBA domain from dimeric inactivation; in particular, phosphorylation of serine 407 of p62 by ULK1 has been shown to facilitate the transition from dimer to monomer 81. This modification is followed by phosphorylation of p62 on serine 403, either by ULK1, casein kinase 2 (CK2) or TANK‐binding kinase 1 (TBK1), which enhances its binding to ubiquitin chains 82, 83, 84 (Fig. 5C). Once p62 binds to ubiquitin chains, it acquires liquid‐like properties 85, 86. Such phase‐separated droplets allow the exchange of their components, including ubiquitin and LC3, with the surrounding environment. Consequently, the droplets can also function as nodes from which signaling cascades can be activated in the context of selective autophagy (Fig. 5C). The droplet formation is rendered more efficient by the presence of NBR1, another autophagic receptor that cooperates with p62 in cells 86. Considering the analogy of APE1 transport into vacuole through selective autophagy in yeast 87 and the cases of parkin‐mediated mitophagy 88 and CCPG1‐mediated ER‐phagy 89, upstream autophagy factors such as the ULK1‐kinase complex as well as p62 binders such as Keap1 might also be translocated onto the droplets. In agreement with this hypothesis, Keap1, a p62‐interacting protein involved in Nrf2 signaling 50, has been found to localize to these droplets 3, 24, 52. Once the pathway is activated, p62‐positive structures together with the recruited cargo are degraded by autophagy.
Autophagy impairment caused by the loss of Atg5 or Atg7 in mouse liver is usually accompanied by accumulation of degenerated protein aggregates, damaged mitochondria, peroxisomes, and lipid droplets, as well as persistent activation of Nrf2 due to the sequestration of Keap1 into p62‐positive structures (refer to the Nrf2 section), leading to benign adenoma 90, 91. Mice haploinsufficient for the autophagy gene Beclin 1 are also cancer prone; they develop spontaneous tumors, including HCC, and are highly susceptible to HCC upon infection with HBV 92, 93. The primary cause of such tumorigenesis is probably ROS derived from degenerated mitochondria and/or peroxisomes, leading to genomic instability and ultimately to spontaneous oncogenesis. Simultaneous loss of p62 or Nrf2 in mice with livers deficient in Atg7 or Atg5 suppresses tumor development 91, 94, implying that the p62‐mediated activation of Nrf2 contributes to tumor growth in autophagy‐deficient mouse livers.
During the first steps of cancer initiation, autophagy is thought to be tumor suppressive through its cytoprotective function 95. However, once the tumor has originated, autophagy is induced and protects cancer cells from metabolic stress, inflammation, and genotoxic and therapy‐induced stresses. Indeed, loss of autophagy suppresses tumor development in various mouse models including nonsmall‐cell lung cancer, pancreatic ductal adenocarcinoma, melanoma, and prostatic carcinoma 95. While dephosphorylation of ULK1 through inactivation of mTORC1 is needed for the induction of autophagy in response to nutrient deprivation, transcriptional activation of genes involved in lysosomal biogenesis and Atg genes is also indispensable for prolonged stimulation of autophagy, which is usually observed in tumor cells. The expression of such genes is mainly regulated by the basic helix‐loop‐helix transcription factors of the MiT‐TFE family (MITF, TFE, and TFEB) 4. In nutrient‐rich conditions, the MiT‐TFE family is phosphorylated by mTORC1 and retained in the cytoplasm 96 (Fig. 5D). When mTORC1 is inactivated under nutrient‐poor conditions, the MiT‐TFE family is dephosphorylated and then translocated into the nucleus in order to trans‐activate the genes harboring a coordinated lysosomal expression regulation (CLEAR) element in their promoter regions (Fig. 5D). In PDAC, the MiT‐TFE family is constantly localized in the nucleus even in mTORC1‐activating conditions. This has been attributed to enhanced transport of the MiT‐TFE family to the nucleus due to increased level of the nuclear transporters Importin 7 and 8 97 (Fig. 5D). In addition, phosphorylation of ULK1 by mTORC1, which leads to suppression of autophagy, is inhibited due to hyperactivation of the responsible phosphatase, B55α‐PP2A, in PDAC 98. Since p62/SQSTM1 is also a target gene for the TiF‐TFE family as well as Nrf2 and NF‐κB, both of which are usually activated in tumor cells, the p62 protein might be increased in certain types of cancers even under autophagy‐inducing conditions.
Concluding remarks
Evaluation of biological complexity has been an elusive question for decades. It is now increasingly clear that the connectivity of regulatory networks rather than the sheer number of genes or the genomic size is what determines how complex an organism is 99. In the case of p62 it is not the many pathways in which it is involved, but how they are entangled what truly represents the beauty of molecular complexity. Focusing on the processes described here, p62 upregulation of mTORC1 upon amino acid abundance provokes an inhibition of autophagy, which results in increased levels of p62 100. At the same time, mTORC1 is responsible for p62 activation by phosphorylation during stress conditions, which directly results in Nrf2 activation 46. p62 and TRAF6 seem to collaborate in the activation of mTORC1 59. p62 and TRAF6 also cooperate in the activation of the NF‐κB pathway. In addition, both NF‐κB and Nrf2 upregulate the expression of p62, and can enter as a consequence in a feedforward loop 2, 3. Whether the four major pathways described here are the only ones subjected to regulation by p62 or not still requires further research. What is increasingly clear is that, just by functioning as an adaptor of protein signaling and controlling the autophagic degradation of some of its binding partners, p62 masters a wide biological network affecting multiple and sometimes unrelated processes.
Cancer cells benefit from increased levels of p62 (that may be caused by autophagy impairment), as it provides them with an improved antioxidant and detoxifying response (Nrf2 activation) and proangiogenic and prosurvival signals (NF‐κB activation), both of which promote tumor progression. Accumulation of p62 may also cause a switch toward anabolism (mTORC1 activation), although this connection needs to be further explored. At the same time, loss of p62 in the surrounding stromal cells, and thereby downregulation of these pathways, results in the secretion of metabolites and paracrine signals that further help cancer thriving. Curiously, although the mechanisms discussed in this review support that both up‐ and downregulation of p62 can promote cancer, p62 mutations are primarily linked to skeletal and neurodegenerative diseases, with only one report linking copy gains of p62 with kidney cancer 6. Still, being at the crossroads of so many cellular pathways, any temptation of employing p62 as a therapeutic target has to be counterbalanced with the myriad of possible side effects in nonconsidered scenarios.
Author contributions
MK and TS designed the review contents. TS and PS‐M created the Figures. All authors wrote the manuscript.
Acknowledgements
PS‐M is supported by a Grant‐in‐Aid for JSPS Research Fellows (P18099). TS is supported by a Grant‐in‐Aid for JSPS Research Fellows (17J05623). MK is supported by a Grant‐in‐Aid for Scientific Research on Innovative Areas (JP25111006), a Grant‐in‐Aid for Scientific Research (B) (18H02611), the Japan Society for the Promotion of Science (an A3 foresight program), and the Takeda Science Foundation (to MK).
References
- 1. Svenning S, Lamark T, Krause K & Johansen T (2011) Plant NBR1 is a selective autophagy substrate and a functional hybrid of the mammalian autophagic adapters NBR1 and p62/SQSTM1. Autophagy 7, 993–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ling J, Kang Y, Zhao R, Xia Q, Lee DF, Chang Z, Li J, Peng B, Fleming JB, Wang H et al (2012) KrasG12D‐induced IKK2/beta/NF‐kappaB activation by IL‐1alpha and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell 21, 105–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jain A, Lamark T, Sjottem E, Larsen KB, Awuh JA, Overvatn A, McMahon M, Hayes JD & Johansen T (2010) p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element‐driven gene transcription. J Biol Chem 285, 22576–22591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P et al (2011) TFEB links autophagy to lysosomal biogenesis. Science 332, 1429–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rea SL, Majcher V, Searle MS & Layfield R (2014) SQSTM1 mutations–bridging Paget disease of bone and ALS/FTLD. Exp Cell Res 325, 27–37. [DOI] [PubMed] [Google Scholar]
- 6. Li L, Shen C, Nakamura E, Ando K, Signoretti S, Beroukhim R, Cowley GS, Lizotte P, Liberzon E, Bair S et al (2013) SQSTM1 is a pathogenic target of 5q copy number gains in kidney cancer. Cancer Cell 24, 738–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zatloukal K, French SW, Stumptner C, Strnad P, Harada M, Toivola DM, Cadrin M & Omary MB (2007) From Mallory to Mallory‐Denk bodies: what, how and why? Exp Cell Res 313, 2033–2049. [DOI] [PubMed] [Google Scholar]
- 8. Zatloukal K, Stumptner C, Fuchsbichler A, Heid H, Schnoelzer M, Kenner L, Kleinert R, Prinz M, Aguzzi A & Denk H (2002) p62 Is a common component of cytoplasmic inclusions in protein aggregation diseases. Am J Pathol 160, 255–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Joung I, Strominger JL & Shin J (1996) Molecular cloning of a phosphotyrosine‐independent ligand of the p56lck SH2 domain. Proc Natl Acad Sci USA 93, 5991–5995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lamark T, Svenning S & Johansen T (2017) Regulation of selective autophagy: the p62/SQSTM1 paradigm. Essays Biochem 61, 609–624. [DOI] [PubMed] [Google Scholar]
- 11. Reina‐Campos M, Shelton PM, Diaz‐Meco MT & Moscat J (2018) Metabolic reprogramming of the tumor microenvironment by p62 and its partners. Biochim Biophys Acta 1870, 88–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sánchez‐Martín P & Komatsu M (2018) p62/SQSTM1: steering the cell through health and disease. J Cell Sci 131, jcs222836. [DOI] [PubMed] [Google Scholar]
- 13. Clausen TH, Lamark T, Isakson P, Finley K, Larsen KB, Brech A, Overvatn A, Stenmark H, Bjorkoy G, Simonsen A et al (2010) p62/SQSTM1 and ALFY interact to facilitate the formation of p62 bodies/ALIS and their degradation by autophagy. Autophagy 6, 330–344. [DOI] [PubMed] [Google Scholar]
- 14. Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H & Johansen T (2005) p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin‐induced cell death. J Cell Biol 171, 603–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Duran A, Amanchy R, Linares JF, Joshi J, Abu‐Baker S, Porollo A, Hansen M, Moscat J & Diaz‐Meco MT (2011) p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol Cell 44, 134–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pankiv S, Lamark T, Bruun JA, Overvatn A, Bjorkoy G & Johansen T (2010) Nucleocytoplasmic shuttling of p62/SQSTM1 and its role in recruitment of nuclear polyubiquitinated proteins to promyelocytic leukemia bodies. J Biol Chem 285, 5941–5953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G & Johansen T (2007) p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 282, 24131–24145. [DOI] [PubMed] [Google Scholar]
- 18. Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S et al (2007) Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy‐deficient mice. Cell 131, 1149–1163. [DOI] [PubMed] [Google Scholar]
- 19. Ichimura Y, Kumanomidou T, Sou YS, Mizushima T, Ezaki J, Ueno T, Kominami E, Yamane T, Tanaka K & Komatsu M (2008) Structural basis for sorting mechanism of p62 in selective autophagy. J Biol Chem 283, 22847–22857. [DOI] [PubMed] [Google Scholar]
- 20. Zheng YT, Shahnazari S, Brech A, Lamark T, Johansen T & Brumell JH (2009) The adaptor protein p62/SQSTM1 targets invading bacteria to the autophagy pathway. J Immunol 183, 5909–5916. [DOI] [PubMed] [Google Scholar]
- 21. Song P, Li S, Wu H, Gao R, Rao G, Wang D, Chen Z, Ma B, Wang H, Sui N et al (2016) Parkin promotes proteasomal degradation of p62: implication of selective vulnerability of neuronal cells in the pathogenesis of Parkinson's disease. Protein Cell 7, 114–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mejlvang J, Olsvik H, Svenning S, Bruun JA, Abudu YP, Larsen KB, Brech A, Hansen TE, Brenne H, Hansen T et al (2018) Starvation induces rapid degradation of selective autophagy receptors by endosomal microautophagy. J Cell Biol 217, 3640–3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sanz L, Diaz‐Meco MT, Nakano H & Moscat J (2000) The atypical PKC‐interacting protein p62 channels NF‐kappaB activation by the IL‐1‐TRAF6 pathway. EMBO J 19, 1576–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI et al (2010) The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol 12, 213–223. [DOI] [PubMed] [Google Scholar]
- 25. Rogov V, Dotsch V, Johansen T & Kirkin V (2014) Interactions between autophagy receptors and ubiquitin‐like proteins form the molecular basis for selective autophagy. Mol Cell 53, 167–178. [DOI] [PubMed] [Google Scholar]
- 26. Saxton RA & Sabatini DM (2017) mTOR signaling in growth, metabolism, and disease. Cell 168, 960–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Taniguchi K & Karin M (2018) NF‐kappaB, inflammation, immunity and cancer: coming of age. Nat Rev Immunol 18, 309–324. [DOI] [PubMed] [Google Scholar]
- 28. Kimmelman AC & White E (2017) Autophagy and tumor metabolism. Cell Metab 25, 1037–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rojo de la Vega M, Chapman E & Zhang DD (2018) NRF2 and the hallmarks of cancer. Cancer Cell 34, 21–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Suzuki T & Yamamoto M (2017) Stress‐sensing mechanisms and the physiological roles of the Keap1‐Nrf2 system during cellular stress. J Biol Chem 292, 16817–16824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kitamura H & Motohashi H (2018) NRF2 addiction in cancer cells. Cancer Sci 109, 900–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Martinez VD, Vucic EA, Thu KL, Pikor LA, Lam S & Lam WL (2015) Disruption of KEAP1/CUL3/RBX1 E3‐ubiquitin ligase complex components by multiple genetic mechanisms: association with poor prognosis in head and neck cancer. Head Neck 37, 727–734. [DOI] [PubMed] [Google Scholar]
- 33. Romero R, Sayin VI, Davidson SM, Bauer MR, Singh SX, LeBoeuf SE, Karakousi TR, Ellis DC, Bhutkar A, Sanchez‐Rivera FJ et al (2017) Keap1 loss promotes Kras‐driven lung cancer and results in dependence on glutaminolysis. Nat Med 23, 1362–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shibata T, Ohta T, Tong KI, Kokubu A, Odogawa R, Tsuta K, Asamura H, Yamamoto M & Hirohashi S (2008) Cancer related mutations in NRF2 impair its recognition by Keap1‐Cul3 E3 ligase and promote malignancy. Proc Natl Acad Sci USA 105, 13568–13573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Goldstein LD, Lee J, Gnad F, Klijn C, Schaub A, Reeder J, Daemen A, Bakalarski CE, Holcomb T, Shames DS et al (2016) Recurrent loss of NFE2L2 exon 2 is a mechanism for Nrf2 pathway activation in human cancers. Cell Rep 16, 2605–2617. [DOI] [PubMed] [Google Scholar]
- 36. Fabrizio FP, Costantini M, Copetti M, la Torre A, Sparaneo A, Fontana A, Poeta L, Gallucci M, Sentinelli S, Graziano P et al (2017) Keap1/Nrf2 pathway in kidney cancer: frequent methylation of KEAP1 gene promoter in clear renal cell carcinoma. Oncotarget 8, 11187–11198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chen W, Sun Z, Wang XJ, Jiang T, Huang Z, Fang D & Zhang DD (2009) Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2‐mediated antioxidant response. Mol Cell 34, 663–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Adam J, Hatipoglu E, O'Flaherty L, Ternette N, Sahgal N, Lockstone H, Baban D, Nye E, Stamp GW, Wolhuter K et al (2011) Renal cyst formation in Fh1‐deficient mice is independent of the Hif/Phd pathway: roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell 20, 524–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ooi A, Wong JC, Petillo D, Roossien D, Perrier‐Trudova V, Whitten D, Min BW, Tan MH, Zhang Z, Yang XJ et al (2011) An antioxidant response phenotype shared between hereditary and sporadic type 2 papillary renal cell carcinoma. Cancer Cell 20, 511–523. [DOI] [PubMed] [Google Scholar]
- 40. DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, Mangal D, Yu KH, Yeo CJ, Calhoun ES et al (2011) Oncogene‐induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 475, 106–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H, Yamamoto M & Motohashi H (2012) Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 22, 66–79. [DOI] [PubMed] [Google Scholar]
- 42. Saito T, Ichimura Y, Taguchi K, Suzuki T, Mizushima T, Takagi K, Hirose Y, Nagahashi M, Iso T, Fukutomi T et al (2016) p62/Sqstm1 promotes malignancy of HCV‐positive hepatocellular carcinoma through Nrf2‐dependent metabolic reprogramming. Nat Commun 7, 12030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hayes JD, Chowdhry S, Dinkova‐Kostova AT & Sutherland C (2015) Dual regulation of transcription factor Nrf2 by Keap1 and by the combined actions of beta‐TrCP and GSK‐3. Biochem Soc Trans 43, 611–620. [DOI] [PubMed] [Google Scholar]
- 44. Yuan TL & Cantley LC (2008) PI3K pathway alterations in cancer: variations on a theme. Oncogene 27, 5497–5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lau A, Wang XJ, Zhao F, Villeneuve NF, Wu T, Jiang T, Sun Z, White E & Zhang DD (2010) A noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62. Mol Cell Biol 30, 3275–3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ichimura Y, Waguri S, Sou YS, Kageyama S, Hasegawa J, Ishimura R, Saito T, Yang Y, Kouno T, Fukutomi T et al (2013) Phosphorylation of p62 activates the Keap1‐Nrf2 pathway during selective autophagy. Mol Cell 51, 618–631. [DOI] [PubMed] [Google Scholar]
- 47. Watanabe Y, Tsujimura A, Taguchi K & Tanaka M (2017) HSF1 stress response pathway regulates autophagy receptor SQSTM1/p62‐associated proteostasis. Autophagy 13, 133–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hashimoto K, Simmons AN, Kajino‐Sakamoto R, Tsuji Y & Ninomiya‐Tsuji J (2016) TAK1 regulates the Nrf2 antioxidant system through modulating p62/SQSTM1. Antioxid Redox Signal 25, 953–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kageyama S, Saito T, Obata M, Koide RH, Ichimura Y & Komatsu M (2018) Negative regulation of the Keap1‐Nrf2 pathway by a p62/Sqstm1 splicing variant. Mol Cell Biol 38, e00642–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lee Y, Chou TF, Pittman SK, Keith AL, Razani B & Weihl CC (2017) Keap1/Cullin3 modulates p62/SQSTM1 activity via UBA domain ubiquitination. Cell Rep 19, 188–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Taguchi K, Fujikawa N, Komatsu M, Ishii T, Unno M, Akaike T, Motohashi H & Yamamoto M (2012) Keap1 degradation by autophagy for the maintenance of redox homeostasis. Proc Natl Acad Sci USA 109, 13561–13566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cloer EW, Siesser PF, Cousins EM, Goldfarb D, Mowrey DD, Harrison JS, Weir SJ, Dokholyan NV & Major MB (2018) p62‐dependent phase separation of patient‐derived KEAP1 mutations and NRF2. Mol Cell Biol 38, e00644–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Shimizu T, Inoue K, Hachiya H, Shibuya N, Aoki T & Kubota K (2016) Accumulation of phosphorylated p62 is associated with NF‐E2‐related factor 2 activation in hepatocellular carcinoma. J Hepatobiliary Pancreat Sci 23, 467–471. [DOI] [PubMed] [Google Scholar]
- 54. Umemura A, He F, Taniguchi K, Nakagawa H, Yamachika S, Font‐Burgada J, Zhong Z, Subramaniam S, Raghunandan S, Duran A et al (2016) p62, upregulated during Preneoplasia, induces hepatocellular carcinogenesis by maintaining survival of stressed HCC‐initiating cells. Cancer Cell 29, 935–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Todoric J, Antonucci L, Di Caro G, Li N, Wu X, Lytle NK, Dhar D, Banerjee S, Fagman JB, Browne CD et al (2017) Stress‐activated NRF2‐MDM2 cascade controls neoplastic progression in pancreas. Cancer Cell 32 (824–839), e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kim YC & Guan KL (2015) mTOR: a pharmacologic target for autophagy regulation. J Clin Invest 125, 25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wolfson RL & Sabatini DM (2017) The Dawn of the age of amino acid sensors for the mTORC1 pathway. Cell Metab 26, 301–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Linares JF, Duran A, Reina‐Campos M, Aza‐Blanc P, Campos A, Moscat J & Diaz‐Meco MT (2015) Amino acid activation of mTORC1 by a PB1‐domain‐driven kinase complex cascade. Cell Rep 12, 1339–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Linares JF, Duran A, Yajima T, Pasparakis M, Moscat J & Diaz‐Meco MT (2013) K63 polyubiquitination and activation of mTOR by the p62‐TRAF6 complex in nutrient‐activated cells. Mol Cell 51, 283–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Grabiner BC, Nardi V, Birsoy K, Possemato R, Shen K, Sinha S, Jordan A, Beck AH & Sabatini DM (2014) A diverse array of cancer‐associated MTOR mutations are hyperactivating and can predict rapamycin sensitivity. Cancer Discov 4, 554–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Valencia T, Kim JY, Abu‐Baker S, Moscat‐Pardos J, Ahn CS, Reina‐Campos M, Duran A, Castilla EA, Metallo CM, Diaz‐Meco MT et al (2014) Metabolic reprogramming of stromal fibroblasts through p62‐mTORC1 signaling promotes inflammation and tumorigenesis. Cancer Cell 26, 121–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Linares JF, Cordes T, Duran A, Reina‐Campos M, Valencia T, Ahn CS, Castilla EA, Moscat J, Metallo CM & Diaz‐Meco MT (2017) ATF4‐induced metabolic reprograming is a synthetic vulnerability of the p62‐deficient tumor stroma. Cell Metab 26 (817–829), e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gwinn DM, Lee AG, Briones‐Martin‐Del‐Campo M, Conn CS, Simpson DR, Scott AI, Le A, Cowan TM, Ruggero D & Sweet‐Cordero EA (2018) Oncogenic KRAS regulates amino acid homeostasis and asparagine biosynthesis via ATF4 and alters sensitivity to L‐asparaginase. Cancer Cell 33 (91–107), e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Krall AS, Xu S, Graeber TG, Braas D & Christofk HR (2016) Asparagine promotes cancer cell proliferation through use as an amino acid exchange factor. Nat Commun 7, 11457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Vallabhapurapu S & Karin M (2009) Regulation and function of NF‐kappaB transcription factors in the immune system. Annu Rev Immunol 27, 693–733. [DOI] [PubMed] [Google Scholar]
- 66. Hu H & Sun SC (2016) Ubiquitin signaling in immune responses. Cell Res 26, 457–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zotti T, Scudiero I, Settembre P, Ferravante A, Mazzone P, D'Andrea L, Reale C, Vito P & Stilo R (2014) TRAF6‐mediated ubiquitination of NEMO requires p62/sequestosome‐1. Mol Immunol 58, 27–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kanayama M, Inoue M, Danzaki K, Hammer G, He YW & Shinohara ML (2015) Autophagy enhances NFkappaB activity in specific tissue macrophages by sequestering A20 to boost antifungal immunity. Nat Commun 6, 5779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Shembade N & Harhaj EW (2012) Regulation of NF‐kappaB signaling by the A20 deubiquitinase. Cell Mol Immunol 9, 123–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Grivennikov SI, Greten FR & Karin M (2010) Immunity, inflammation, and cancer. Cell 140, 883–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wu Y & Zhou BP (2009) Inflammation: a driving force speeds cancer metastasis. Cell Cycle 8, 3267–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Duran A, Linares JF, Galvez AS, Wikenheiser K, Flores JM, Diaz‐Meco MT & Moscat J (2008) The signaling adaptor p62 is an important NF‐kappaB mediator in tumorigenesis. Cancer Cell 13, 343–354. [DOI] [PubMed] [Google Scholar]
- 73. Zhong Z, Umemura A, Sanchez‐Lopez E, Liang S, Shalapour S, Wong J, He F, Boassa D, Perkins G, Ali SR et al (2016) NF‐kappaB restricts inflammasome activation via elimination of damaged mitochondria. Cell 164, 896–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Apte RN, Dotan S, Elkabets M, White MR, Reich E, Carmi Y, Song X, Dvozkin T, Krelin Y & Voronov E (2006) The involvement of IL‐1 in tumorigenesis, tumor invasiveness, metastasis and tumor‐host interactions. Cancer Metastasis Rev 25, 387–408. [DOI] [PubMed] [Google Scholar]
- 75. Erez N, Truitt M, Olson P, Arron ST & Hanahan D (2010) Cancer‐associated fibroblasts are activated in incipient neoplasia to orchestrate tumor‐promoting inflammation in an NF‐kappaB‐dependent manner. Cancer Cell 17, 135–147. [DOI] [PubMed] [Google Scholar]
- 76. Kalluri R (2016) The biology and function of fibroblasts in cancer. Nat Rev Cancer 16, 582–598. [DOI] [PubMed] [Google Scholar]
- 77. Dikic I & Elazar Z (2018) Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol 19, 349–364. [DOI] [PubMed] [Google Scholar]
- 78. Khaminets A, Behl C & Dikic I (2016) Ubiquitin‐dependent and independent signals in selective autophagy. Trends Cell Biol 26, 6–16. [DOI] [PubMed] [Google Scholar]
- 79. Isogai S, Morimoto D, Arita K, Unzai S, Tenno T, Hasegawa J, Sou YS, Komatsu M, Tanaka K, Shirakawa M et al (2011) Crystal structure of the ubiquitin‐associated (UBA) domain of p62 and its interaction with ubiquitin. J Biol Chem 286, 31864–31874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Long J, Garner TP, Pandya MJ, Craven CJ, Chen P, Shaw B, Williamson MP, Layfield R & Searle MS (2010) Dimerisation of the UBA domain of p62 inhibits ubiquitin binding and regulates NF‐kappaB signalling. J Mol Biol 396, 178–194. [DOI] [PubMed] [Google Scholar]
- 81. Lim J, Lachenmayer ML, Wu S, Liu W, Kundu M, Wang R, Komatsu M, Oh YJ, Zhao Y & Yue Z (2015) Proteotoxic stress induces phosphorylation of p62/SQSTM1 by ULK1 to regulate selective autophagic clearance of protein aggregates. PLoS Genet 11, e1004987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Matsumoto G, Shimogori T, Hattori N & Nukina N (2015) TBK1 controls autophagosomal engulfment of polyubiquitinated mitochondria through p62/SQSTM1 phosphorylation. Hum Mol Genet 24, 4429–4442. [DOI] [PubMed] [Google Scholar]
- 83. Matsumoto G, Wada K, Okuno M, Kurosawa M & Nukina N (2011) Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol Cell 44, 279–289. [DOI] [PubMed] [Google Scholar]
- 84. Pilli M, Arko‐Mensah J, Ponpuak M, Roberts E, Master S, Mandell MA, Dupont N, Ornatowski W, Jiang S, Bradfute SB et al (2012) TBK‐1 promotes autophagy‐mediated antimicrobial defense by controlling autophagosome maturation. Immunity 37, 223–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Sun D, Wu R, Zheng J, Li P & Yu L (2018) Polyubiquitin chain‐induced p62 phase separation drives autophagic cargo segregation. Cell Res 28, 405–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Zaffagnini G, Savova A, Danieli A, Romanov J, Tremel S, Ebner M, Peterbauer T, Sztacho M, Trapannone R, Tarafder AK et al (2018) p62 filaments capture and present ubiquitinated cargos for autophagy. EMBO J 37, e98308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Farre JC & Subramani S (2016) Mechanistic insights into selective autophagy pathways: lessons from yeast. Nat Rev Mol Cell Biol 17, 537–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI & Youle RJ (2015) The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524, 309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Smith MD, Harley ME, Kemp AJ, Wills J, Lee M, Arends M, von Kriegsheim A, Behrends C & Wilkinson S (2018) CCPG1 is a non‐canonical autophagy cargo receptor essential for ER‐phagy and pancreatic ER proteostasis. Dev Cell 44 (217–232), e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Inami Y, Waguri S, Sakamoto A, Kouno T, Nakada K, Hino O, Watanabe S, Ando J, Iwadate M, Yamamoto M et al (2011) Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J Cell Biol 193, 275–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, Eishi Y, Hino O, Tanaka K & Mizushima N (2011) Autophagy‐deficient mice develop multiple liver tumors. Genes Dev 25, 795–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y et al (2003) Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest 112, 1809–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Yue Z, Jin S, Yang C, Levine AJ & Heintz N (2003) Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci USA 100, 15077–15082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Ni HM, Woolbright BL, Williams J, Copple B, Cui W, Luyendyk JP, Jaeschke H & Ding WX (2014) Nrf2 promotes the development of fibrosis and tumorigenesis in mice with defective hepatic autophagy. J Hepatol 61, 617–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Amaravadi R, Kimmelman AC & White E (2016) Recent insights into the function of autophagy in cancer. Genes Dev 30, 1913–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Puertollano R, Ferguson SM, Brugarolas J & Ballabio A (2018) The complex relationship between TFEB transcription factor phosphorylation and subcellular localization. EMBO J 37, e98804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Perera RM, Stoykova S, Nicolay BN, Ross KN, Fitamant J, Boukhali M, Lengrand J, Deshpande V, Selig MK, Ferrone CR et al (2015) Transcriptional control of autophagy‐lysosome function drives pancreatic cancer metabolism. Nature 524, 361–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Wong PM, Feng Y, Wang J, Shi R & Jiang X (2015) Regulation of autophagy by coordinated action of mTORC1 and protein phosphatase 2A. Nat Commun 6, 8048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Szathmary E, Jordan F & Pal C (2001) Molecular biology and evolution – Can genes explain biological complexity? Science 292, 1315–1316. [DOI] [PubMed] [Google Scholar]
- 100. Kim J, Kundu M, Viollet B & Guan KL (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13, 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Bao L, Chandra PK, Moroz K, Zhang X, Thung SN, Wu T & Dash S (2014) Impaired autophagy response in human hepatocellular carcinoma. Exp Mol Pathol 96, 149–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Jin GZ, Dong H, Yu WL, Li Y, Lu XY, Yu H, Xian ZH, Dong W, Liu YK, Cong WM et al (2013) A novel panel of biomarkers in distinction of small well‐differentiated HCC from dysplastic nodules and outcome values. BMC Cancer 13, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Aishima S, Fujita N, Mano Y, Iguchi T, Taketomi A, Maehara Y, Oda Y & Tsuneyoshi M (2010) p62+ Hyaline inclusions in intrahepatic cholangiocarcinoma associated with viral hepatitis or alcoholic liver disease. Am J Clin Pathol 134, 457–465. [DOI] [PubMed] [Google Scholar]
- 104. Ellis RA, Horswell S, Ness T, Lumsdon J, Tooze SA, Kirkham N, Armstrong JL & Lovat PE (2014) Prognostic impact of p62 expression in cutaneous malignant melanoma. J Invest Dermatol 134, 1476–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Mohamed A, Ayman A, Deniece J, Wang T, Kovach C, Siddiqui MT & Cohen C (2015) P62/Ubiquitin IHC expression correlated with clinicopathologic parameters and outcome in gastrointestinal carcinomas. Front Oncol 5, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Inoue D, Suzuki T, Mitsuishi Y, Miki Y, Suzuki S, Sugawara S, Watanabe M, Sakurada A, Endo C, Uruno A et al (2012) Accumulation of p62/SQSTM1 is associated with poor prognosis in patients with lung adenocarcinoma. Cancer Sci 103, 760–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Schlafli AM, Adams O, Galvan JA, Gugger M, Savic S, Bubendorf L, Schmid RA, Becker KF, Tschan MP, Langer R et al (2016) Prognostic value of the autophagy markers LC3 and p62/SQSTM1 in early‐stage non‐small cell lung cancer. Oncotarget 7, 39544–39555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Adams O, Dislich B, Berezowska S, Schlafli AM, Seiler CA, Kroll D, Tschan MP & Langer R (2016) Prognostic relevance of autophagy markers LC3B and p62 in esophageal adenocarcinomas. Oncotarget 7, 39241–39255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Masuda GO, Yashiro M, Kitayama K, Miki Y, Kasashima H, Kinoshita H, Morisaki T, Fukuoka T, Hasegawa T, Sakurai K et al (2016) Clinicopathological correlations of autophagy‐related proteins LC3, Beclin 1 and p62 in gastric cancer. Anticancer Res 36, 129–136. [PubMed] [Google Scholar]
- 110. Park JM, Huang S, Wu TT, Foster NR & Sinicrope FA (2013) Prognostic impact of Beclin 1, p62/sequestosome 1 and LC3 protein expression in colon carcinomas from patients receiving 5‐fluorouracil as adjuvant chemotherapy. Cancer Biol Ther 14, 100–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Thompson HG, Harris JW, Wold BJ, Lin F & Brody JP (2003) p62 overexpression in breast tumors and regulation by prostate‐derived Ets factor in breast cancer cells. Oncogene 22, 2322–2333. [DOI] [PubMed] [Google Scholar]
- 112. Luo RZ, Yuan ZY, Li M, Xi SY, Fu J & He J (2013) Accumulation of p62 is associated with poor prognosis in patients with triple‐negative breast cancer. Onco Targets Ther 6, 883–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Rolland P, Madjd Z, Durrant L, Ellis IO, Layfield R & Spendlove I (2007) The ubiquitin‐binding protein p62 is expressed in breast cancers showing features of aggressive disease. Endocr Relat Cancer 14, 73–80. [DOI] [PubMed] [Google Scholar]
- 114. Choi J, Jung W & Koo JS (2013) Expression of autophagy‐related markers beclin‐1, light chain 3A, light chain 3B and p62 according to the molecular subtype of breast cancer. Histopathology 62, 275–286. [DOI] [PubMed] [Google Scholar]
- 115. Kitamura H, Torigoe T, Asanuma H, Hisasue SI, Suzuki K, Tsukamoto T, Satoh M & Sato N (2006) Cytosolic overexpression of p62 sequestosome 1 in neoplastic prostate tissue. Histopathology 48, 157–161. [DOI] [PubMed] [Google Scholar]
- 116. Giatromanolaki A, Sivridis E, Mendrinos S, Koutsopoulos AV & Koukourakis MI (2014) Autophagy proteins in prostate cancer: relation with anaerobic metabolism and Gleason score. Urol Oncol 32 (39), e11–e18. [DOI] [PubMed] [Google Scholar]
- 117. Iwadate R, Inoue J, Tsuda H, Takano M, Furuya K, Hirasawa A, Aoki D & Inazawa J (2015) High expression of p62 protein is associated with poor prognosis and aggressive phenotypes in endometrial cancer. Am J Pathol 185, 2523–2533. [DOI] [PubMed] [Google Scholar]
- 118. Ju LL, Zhao CY, Ye KF, Yang H & Zhang J (2016) Expression and clinical implication of Beclin1, HMGB1, p62, survivin, BRCA1 and ERCC1 in epithelial ovarian tumor tissues. Eur Rev Med Pharmacol Sci 20, 1993–2003. [PubMed] [Google Scholar]
- 119. Kuo WL, Sharifi MN, Lingen MW, Ahmed O, Liu J, Nagilla M, Macleod KF & Cohen EE (2014) p62/SQSTM1 accumulation in squamous cell carcinoma of head and neck predicts sensitivity to phosphatidylinositol 3‐kinase pathway inhibitors. PLoS One 9, e90171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Inui T, Chano T, Takikita‐Suzuki M, Nishikawa M, Yamamoto G & Okabe H (2013) Association of p62/SQSTM1 excess and oral carcinogenesis. PLoS One 8, e74398. [DOI] [PMC free article] [PubMed] [Google Scholar]