

Abstract
Scope
High‐level exposure to aflatoxin B1 (AFB1) is known to cause acute liver damage and fatality in animals and humans. The intakes actually causing this acute toxicity have so far been estimated based on AFB1 levels in contaminated foods or biomarkers in serum. The aim of the present study is to predict the doses causing acute liver toxicity of AFB1 in rats and humans by an in vitro–in silico testing strategy.
Methods and results
Physiologically based kinetic (PBK) models for AFB1 in rats and humans are developed. The models are used to translate in vitro concentration–response curves for cytotoxicity in primary rat and human hepatocytes to in vivo dose–response curves using reverse dosimetry. From these data, the dose levels at which toxicity would be expected are obtained and compared to toxic dose levels from available rat and human case studies on AFB1 toxicity. The results show that the in vitro–in silico testing strategy can predict dose levels causing acute toxicity of AFB1 in rats and human.
Conclusions
Quantitative in vitro in vivo extrapolation (QIVIVE) using PBK modeling‐based reverse dosimetry can predict AFB1 doses that cause acute liver toxicity in rats and human.
Keywords: acute liver toxicity, aflatoxin B1, physiologically based kinetic modeling, reverse dosimetry
A physiologically based kinetic (PBK) model simulating the concentration through time of aflatoxin B1 (AFB1) in blood is developed for humans and rats. By integrating a reverse dosimetry approach, oral doses corresponding to internal concentration of AFB1 are predicted. In vitro concentration–response curves for liver toxicity translated to dose–response curves are comparable with the available in vivo data.
1. Introduction
Aflatoxin B1 (AFB1) is a mycotoxin with well‐established chronic effects, including increased risk of liver cancer development in humans.[ 1 ] The effects induced by acute exposure to relatively higher dose levels of AFB1 have been explored less extensively.[ 2 ] In humans, acute exposure to AFB1 has been identified from described aflatoxicosis outbreaks, in which clinical symptoms could be associated with the consumption of AFB1 contaminated food.[ 3 , 4 ] More recently, biomarkers were used to confirm AFB1 exposure in individuals, for instance during the aflatoxicosis outbreak in Kenya in 2004 where 317 cases and 125 deaths were reported, or in the outbreak in Tanzania in 2016 with 68 cases and a 30% fatality rate.[ 5 , 6 , 7 ] Aflatoxicosis refers to the toxic liver injury caused by aflatoxin, featuring jaundice, hepatitis, vomiting, and abdominal pain.[ 3 , 5 , 6 ] Depending on the exposure and duration, two types of aflatoxicosis are observed, the first resulting in acute liver failure and subsequent illness or death, and the second being a sublethal intoxication with nutritional and immunological consequences.[ 8 , 9 ] From previously reported outbreaks, mortality rates in humans with acute aflatoxicosis are estimated to be between 27% and 60%, with children being more sensitive than adults.[ 4 , 7 , 8 , 9 ]
Based on the estimated intake of AFB1 and the onset of symptoms reported in three aflatoxicosis outbreaks, doses of AFB1 that could result in acute aflatoxicosis and present a risk of fatality in humans have been estimated to be around 20–100 µg kg−1 bw per day for a period of 1–3 weeks.[ 4 ] Joint FAO/WHO Expert Committee on Food Additives (JECFA)[ 9 ] reported a similar dose range, 20–120 µg kg−1 bw per day, derived from the AFB1 adduct concentrations per mg serum albumin detected in samples collected from victims of the 2004 outbreak in Kenya.[ 2 , 10 ] However, exposure assessments based on the AFB1 occurrence levels in food and/or the albumin‐adduct biomarker have uncertainty. The former may be affected by the variability in a food product with heterogeneous contamination, and the latter may rather be related to chronic instead of acute AFB1 exposure since for recent exposures the measurement of aflatoxin M1 or aflatoxin‐N7‐guanine adducts is considered a better indicator.[ 11 ]
In addition to human case studies, adverse effects upon acute AFB1 exposure have also been characterized in different animal models showing different sensitivities depending on the animal species, strain, sex, and age of the animals.[ 12 ] Given that the acute dose levels resulting in human liver toxicity have mainly been derived from human case studies estimating the corresponding dose levels based on levels in the contaminated food, it is of interest to investigate whether dose levels for acute toxicity can also be derived by using physiologically based kinetic (PBK) modeling‐based reverse dosimetry.[ 13 ] Within this context it is relevant to note that data on in vivo kinetics of AFB1 are available for rats and even for humans exposed to low dose levels,[ 14 , 15 ] although these data have, to the best of our knowledge, not yet been used to facilitate quantitative in vitro in vivo extrapolation (QIVIVE) predicting dose–response behavior for acute liver toxicity of AFB1. The use of PBK modeling‐based reverse dosimetry to perform QIVIVE has previously been shown to provide adequate predictions for in vivo acute liver toxicity of the pyrrolizidine alkaloids lasiocarpine and riddelliine, or for in vivo kidney toxicity of aristolochic acid.[ 16 , 17 , 18 ]
The aim of the present study was to use the PBK modeling‐based reverse dosimetry approach to perform QIVIVE to predict dose response data for acute liver toxicity of AFB1 in rats and humans. To this end, a PBK model for AFB1 was developed for both rats and humans. Using these PBK models concentration–response curves describing in vitro toxicity of AFB1 in rat and human primary hepatocytes, were translated into in vivo dose–response curves for acute liver toxicity by reverse dosimetry. Predictions made were evaluated against available in vivo toxicity data.
2. Experimental Section
2.1. Chemicals
AFB1 and MTT (3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide) were purchased from Sigma‐Aldrich (Zwijndrecht, The Netherlands). Cryopreserved pooled rat hepatocytes (Sprague Dawley), pooled human hepatocytes (pooled from 5‐donors, 2x lot HPP1834236 and 1x lot HPP1825348), cell maintenance supplement pack, fetal bovine serum (FBS), and Williams E Medium (WEM, A1217601) were purchased from ThermoFisher (Naarden, The Netherlands). Trypsin‐EDTA was purchased from Gibco (Paisley, Scotland, UK). DMSO was obtained from Acros Organics (Geel, Belgium).
2.2. General Outline for PBK Modeling‐Based Reverse Dosimetry
The PBK model‐based reverse dosimetry steps consist of 1) establishing in vitro concentration–response curves for the toxicity, 2) defining a PBK model describing in vivo kinetics using kinetic parameters defined in vitro, 3) evaluation of the PBK model predictions against available in vivo data on kinetics, 4) translation of the in vitro concentration–response curves for toxicity into in vivo dose–response curves for toxicity using the PBK models, 5) deriving a point of departure (POD) from the in vivo dose–response curves obtained and 6) comparison of the POD to available in vivo data.[ 16 , 18 ]
2.3. In Vitro Cytotoxicity Concentration–Response Curves
Primary rat and human hepatocytes, and HepaRG cells (Biopredic International) were used to define the in vitro concentration–response curves for cytotoxicity. To this end, the hepatocytes were seeded at a density of 5 × 104 cells per well (5 × 105 cells mL−1) into 96‐well plates according to the supplier's protocol and cultured in plating medium for 6 h. Undifferentiated HepaRG cells were seeded at a density of 9 × 103 cells per well (9 × 104 cells mL−1) and were differentiated according to the supplier's protocol. Following the preincubation, medium was replaced by serum free medium containing different concentrations of AFB1 ranging from 0 to 50 µm for hepatocytes, and for HepaRG cells from 0 to 100 µm, added from 200 times concentrated stock solutions in DMSO (final DMSO concentration 0.5% v/v). Mitochondrial activity measured by the MTT assay was used as a viability endpoint. After 24 h treatment, 10 µL MTT in medium (5 mg mL−1) were added (final concentration 0.45 mg mL−1) and cells were incubated for an additional 3 h. Cells were lysed and the formazan crystals formed were dissolved by addition of 100 µL DMSO. The absorbance of the formazan solution was measured at 562 nm, and the absorbance values were corrected for the background absorbance measured at 620 nm using a microplate reader spectrophotometer (SpectraMax M2, USA). Relative cell viability (%) was calculated as (mean absorbance of sample/mean absorbance of vehicle control) × 100%. Each concentration was tested in at least three replicates, and three independent experiments were carried out.
2.4. Fraction Unbound (f ub) in Plasma and Plasma to Blood Conversion
For the reverse dosimetry, the unbound concentrations in vitro are set equal to the unbound concentrations in vivo, since the toxicity is ascribed to unbound AFB1. AFB1 can bind non‐covalently to serum albumin reducing the amount of free AFB1 in vitro and in vivo.[ 19 , 20 , 21 ] Thus, to perform reverse dosimetry, the in vitro concentrations were first corrected for the difference in protein binding in the in vitro and in vivo situation using Equation (1):
| (1) |
where C AFB1, blood is the total concentration of AFB1 in blood, C AFB1, in vitro the total AFB1 concentration in vitro, and f ub the fraction unbound in vitro or in blood. The exposure medium used in vitro was serum free, so the f ub, in vitro was considered to equal 1.0. The f ub, blood was assumed to be equal to the f ub for AFB1 in human plasma amounting to 0.16. This value was derived in silico using the fu,p Predictor software.[ 22 ] For rats, the f ub, blood was considered the same as for humans. Since the PBK models predict the AFB1 concentration in blood, for evaluation of the model performance, the plasma concentrations of AFB1 from in vivo kinetic studies were converted to blood concentrations to enable comparison to the model predictions, assuming that blood concentrations are 0.6 and 0.55 times the plasma concentrations in rats and humans, respectively.[ 23 , 24 , 25 ]
2.5. PBK Model for Rats and Humans
The conceptual PBK model used in the present study to describe AFB1 kinetics in rats and humans is shown in Figure 1 . The model follows previous models which include a separate compartment for liver as the metabolizing compartment, and compartments for stomach, intestine, fat, kidney, rapidly perfused tissue, slowly perfused tissue and blood.[ 26 , 27 ] In order to describe the transition from stomach to the small intestine and the transition within the small intestine the model contains a stomach compartment emptying into the small intestine compartment divided in 7 subcompartments.[ 26 , 27 ] AFB1 is absorbed by the small intestine following first order kinetics, after which it is transported to the liver via the hepatic portal vein.[ 28 , 29 , 30 , 31 , 32 ] The absorption rate constant in the small intestine (ka) for the rat PBK model was set at 5.84 h−1,[ 28 ] while for the human PBK model a ka of 5.05 h−1 was used.[ 15 ] The ka of 5.84 h−1 and of 5.05 h−1 was assumed to be the same for the 7 intestinal subcompartments in rats and humans, respectively. Physiological parameter values for the model, such as tissue volumes and blood flows, were obtained from literature.[ 33 ] Physicochemical parameters such as tissue/blood partition coefficients applied in the models were calculated based on the method reported by DeJongh et al.[ 34 ] using the logarithmic partition ratio of AFB1 for n‐octanol/water (log(K D)AFB1) of 1.15667 obtained using the software Chem3D (version 18, PerkinElmer, MA, USA). Both physiological and physiochemical parameters are presented in Table S1, Supporting Information. The PBK model equations were coded and numerically integrated into Berkeley Madonna (version 8.3.18, UC Berkeley, CA, USA) using Rosenbrock's algorithms for solving stiff differential equations. The model code describing the PBK models can be found in Supporting Information.
Figure 1.
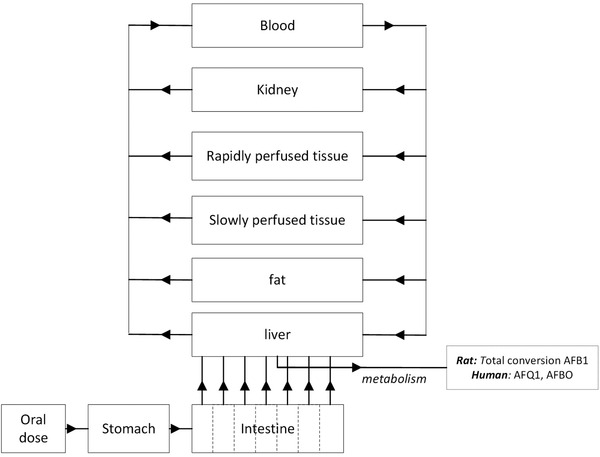
Schematic representation of the PBK model for AFB1 in rats and humans.
2.6. Kinetic Parameters
The kinetic parameters describing the clearance of AFB1 in rats and humans were obtained from literature (Table 1 ). In rats, the kinetic parameters, V max and K m, describing the hepatic clearance of AFB1 were obtained from incubations with rat liver S9 fraction from 28‐day old F344 rats.[ 35 ] In humans, the kinetic parameters for AFB1 were obtained from the metabolite formation in incubations with microsomes from 13 human livers, reporting the formation of aflatoxin Q1 (AFQ1) and AFB1 8,9‐epoxide (AFBO).[ 36 ] Given that AFQ1 is the major product of AFB1 metabolism in humans,[ 37 ] the geometric mean of the kinetic constants from the 13 human liver microsomes for the formation of AFQ1 and the geometric mean of the kinetic constants from the 13 human liver microsomes for the formation of AFBO were used to describe the clearance of AFB1 in humans. The kinetics in the rat model were set to follow Michalis–Menten kinetics (Equation (2)) while, and, in line with what was reported before, in humans the kinetics were set to follow sigmoidal kinetics (Hill Equation)[ 36 , 38 , 39 , 40 , 41 , 42 ] as seen in Equation (3):
| (2) |
| (3) |
where V max is the maximum velocity, K m is the Michaelis constant, S is the substrate concentration, S 50 is analogous to K m, the substrate concentration at half V max, and n is the Hill coefficient. The in vitro kinetic parameter V max was scaled from microsomal preparations to a whole liver considering an estimated S9 fraction protein yield of 125 mg S9 fraction protein g−1 liver for rat[ 43 ] and a microsomal protein yield of 39.46 mg microsomal protein g−1 liver for human.[ 44 ] The K m and S 50 parameters were assumed to be equal in vitro and in vivo.
Table 1.
Kinetic parameters used for the PBK model for rats and humans
| Liver tissue | AFB1 total Conversion | AFQ1 formation | AFBO formation | Reference | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| V max a) | K m b) | V max a) | S 50 c) | n d) | V max a) | S 50 c) | n d) | |||
| Rat | S9 fraction | 0.0027 | 13 | — | — | — | — | — | — | [ 35 ] |
| Human | microsomes | — | — | 0.003469 | 427 | 1.2 | 0.000542 | 197 | 1.1 | [ 36 ] |
V max in µmol mg−1 S9 fraction or microsomal protein min−1;
Michaelis–Menten constant in µm;
Analogous to K m but used in the Hill equation for sigmoidal kinetics in µm (Equation (3));
n, Hill coefficient.
2.7. Sensitivity Analysis and PBK Model Evaluation
To identify the parameters that affect the predicted maximal liver blood concentrations (CVLmax) to the largest extent, sensitivity coefficients (SC)[ 45 ] were determined as follows:
| (4) |
where P is the initial PBK model parameter, C the initial output of the model calculation, and is the modified parameter considering a 5% increase, and the modified output obtained by the model upon the 5% increment in parameter P. The sensitivity analysis was conducted at oral dose levels of 0.4 µg kg−1 bw and 5 mg kg−1 bw for the rat model, and of 3.74 × 104 µg kg−1 bw per day and 120 µg kg−1 bw per day for humans, representing in rats an intake level used in studies with repeated relatively low dose levels and an acute high dose intake, and in humans a low and a high dietary intake of AFB1.[ 9 , 11 , 15 , 46 ] Each parameter was analyzed individually, keeping the other parameters to their initial values. Only parameters with an SC above an absolute value of 0.1 are presented.
To evaluate the performance of the PBK model, AFB1 concentrations in human or rat plasma reported in in vivo studies available in the literature[ 15 , 47 ] were transformed to concentrations in whole blood assuming that blood concentrations are 0.6 and 0.55 times the plasma concentrations in rats and humans. This enabled comparison to the PBK model predictions for blood concentrations (CB). In rats, in vivo kinetic data for AFB1 levels in plasma after oral exposure were reported in two studies (Table S2, Supporting Information). Although in one of the studies, AFB1 was co‐administered with 0.5 mg kg−1 bw T‐2,[ 47 ] the data on AFB1 quantified in plasma by LC‐MS were used for comparison assuming T‐2 would not affect AFB1 bioavailability. The study by Coulombe Jr and Sharma,[ 14 ] used radiolabelled AFB1 and quantified the radioactivity, reporting the level of both AFB1 and its metabolites in plasma samples. Both studies used Sprague Dawley rats, and exposed them orally to 0.5 mg kg−1 bw and 0.6 mg kg−1 bw, respectively. In vivo kinetic data for AFB1 available for the evaluation of the human PBK model, were taken from the study of Jubert et al.[ 15 ] reporting levels of AFB1 equivalents in plasma of 4 human volunteers who were exposed to an oral low‐dose (30 ng) of 14C labelled AFB1 (3.74 × 10−7 mg kg−1 bw). For this comparison of the PBK model‐based predictions the body weight in the human PBK model was set at 82.4 kg, the average body weight of the 4 volunteers. (Table S3, Supporting Information).
2.8. Translation of the In Vitro Concentration–Response Curves to In Vivo Dose–Response Curves by Reverse Dosimetry
The PBK modeling‐based reverse dosimetry approach was applied for translating the in vitro concentration–response curves to in vivo dose–response curves. In vitro concentrations were corrected for differences in protein binding in the in vitro and in vivo situation (Equation (1)). The C AFB1, blood thus obtained was assumed to equal the maximum concentration of AFB1 in liver blood (CLVmax) in the PBK models. To obtain the corresponding oral dose levels of AFB1 in the process denoted as reverse dosimetry, a parameter plot of the maximum concentration of AFB1 in blood liver (CVLmax) against the oral dose (mg kg−1 bw) was used.
2.9. Analysis of In Vitro Concentration–Response Data and of Predicted In Vivo Dose–Response Data
From the in vitro concentration–response curves the EC50 values for in vitro toxicity were obtained using the dose–response stimulation non‐linear regression model by GraphPad Prism, 5.0 software (San Diego, CA). For the predicted in vivo dose–response curves, the lower and upper bounds of the benchmark dose resulting in a 10% extra response above background (BMDL10‐BMDU10) were determined using the model averaging of the fitted dose–response models using the PROAST software (version 38.9, The Dutch National Institute for Public Health and the Environment, The Netherlands).
2.10. Evaluation of the Combined In Vitro PBK Approach
To evaluate the outcome from the PBK model‐based reverse dosimetry approach, the predicted BMDL10‐BMDU10 ranges for in vivo liver toxicity in rats were compared with PODs derived from in vivo acute liver toxicity data from literature[ 48 , 49 ] and with reported estimated median lethal doses (LD50) of AFB1 for rats[ 50 , 51 , 52 , 53 ] (both detailed in Tables S4 and S5, Supporting Information). Comparison to the LD50 assumes that the acute liver toxicity would be the cause of the fatal outcome and the results of the comparison should be considered keeping this in mind. For humans, the PBK model‐based reverse dosimetry predictions were evaluated with the suspected doses that cause acute aflatoxicosis and possibly death of 20–100 µg kg−1 bw per day estimated by Wild and Gong (2009) and of 20–120 µg kg−1 bw per day reported by JECFA (2018).
3. Results
3.1. In Vitro Cytotoxicity
The concentration–response curves for the in vitro cytotoxicity of AFB1 assessed by the MTT assay performed with serum‐free medium using cryopreserved rat hepatocytes, cryopreserved human hepatocytes and HepaRG cells are shown in Figure 2 . Figure 2 also includes concentration–response curves from literature obtained using serum‐free medium.
Figure 2.
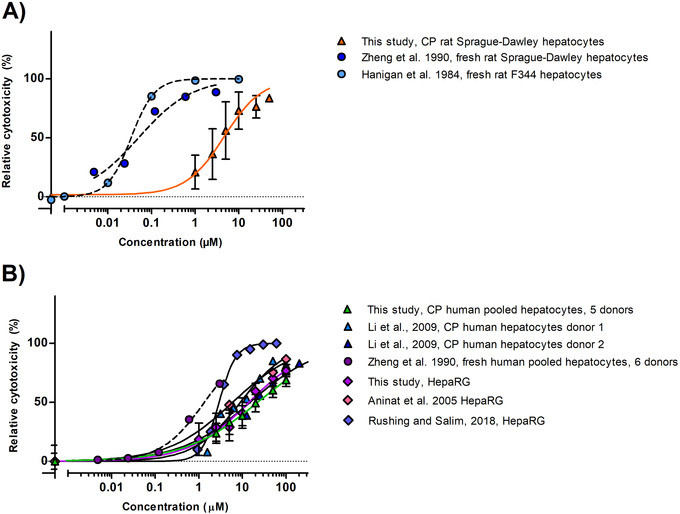
Concentration response curves for the cytotoxicity of AFB1 (µm) in A) rat and B) human cell models upon 24 h exposure in medium without serum (mean values + SD) as measured in the present study and reported in literature. Circle symbols with dotted curves correspond to fresh hepatocytes. Triangle symbols to cryopreserved hepatocytes, and diamonds to HepaRG cells. CP: cryopreserved.
Figure 2A shows that the in vitro concentration–response curves obtained in the present study for rat cryopreserved hepatocytes, result in EC50 values that are about 100‐fold higher than those reported for fresh hepatocytes.[ 54 , 55 ]
Figure 2B shows that concentration–response curves obtained in the present study for cryopreserved human hepatocytes and HepaRG cells are comparable. Figure 2B, also presents data reported in the literature for cytotoxicity of AFB1 in human liver models as reported in other studies. The EC50 values derived from the curves reveal that for the human hepatocytes the cryopreserved hepatocytes result in cytotoxicity curves with EC50 values that are about 5–13‐fold higher than those obtained with fresh hepatocytes, indicating the fresh hepatocytes to be more sensitive, with the difference being less than what was observed for the rat hepatocytes. Comparison of data from studies using HepaRG cells exposed to AFB1 reveals some variability between studies as the EC50 obtained in the different studies varies up to 4.7‐fold with especially the value reported by Rushing and Selim[ 56 ] measured by flow cytometry instead of the MTT assay, being lower. The EC50 of all the curves are presented in Table S6, Supporting Information.
3.2. PBK Model Description, Prediction, and Evaluation
Figure 3 shows the PBK model‐based predictions for the time dependent AFB1 concentrations in blood of rat (Figure 3A) and human (Figure 3B). In rats, the predicted C max (10.08 ng mL−1) was only 1.01‐fold higher than the C max in blood upon exposure of rats to 0.5 mg kg−1 bw.[ 47 ] Also, the predicted T max, the time to reach the C max, of 20 min, is close to the value reported by Han et al.[ 47 ] of 10.2 min (0.17 h). For the human model, the C max for the 4 human volunteers reported by Jubert et al.[ 15 ] was about 1.2–2.1‐fold higher than the C max predicted (0.39 pg mL−1). The predicted T max (≈35 min) is 1.7‐fold lower than what was observed for the 4 human individuals, but since for the reverse dosimetry only C max values are used, this deviation will not affect the subsequent use of the model for reverse dosimetry.
Figure 3.
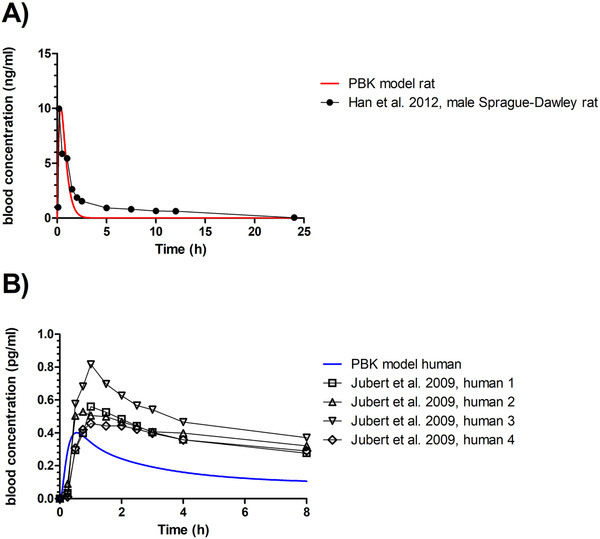
PBK model‐predicted time‐dependent blood concentration of AFB1 in A) rats and B) humans upon an oral dose of 0.5 mg kg−1 bw oral (red line) and 0.374 ng kg−1 bw (blue line) respectively. Reported time‐dependent blood concentrations depicted as symbols, were calculated from reported plasma concentrations assuming that blood concentrations are 0.6 and 0.55 times the plasma concentrations in rats and humans, respectively.[ 23 , 24 , 25 ]
3.3. Sensitivity Analysis
Figure 4 shows the results of the sensitivity analysis presenting the parameters that affect the PBK model prediction for C max to the highest extent. In both the rat and human models, the volume of the liver (VLc), the stomach emptying rate (ksto), the absorption constant rate (ka), the scaling factors to whole liver (in rats the mg S9 fraction protein per g liver tissue [S9PL] and in humans the mg microsomal protein per g liver tissue [MPL]), as well as the kinetic parameters for AFB1 conversion or metabolite formation, influence the model. Additionally, in humans, the body weight (BW), the blood flow to the liver (QLc), the blood flow to the rapidly perfused tissues (QRc), the blood flow to the slowly perfused tissues (QSc), as well as the Hill coefficient used to describe the formation of AFQ1 (nAFQ1) and the formation of AFBO (nAFBO) also appeared to be influential parameters.
Figure 4.
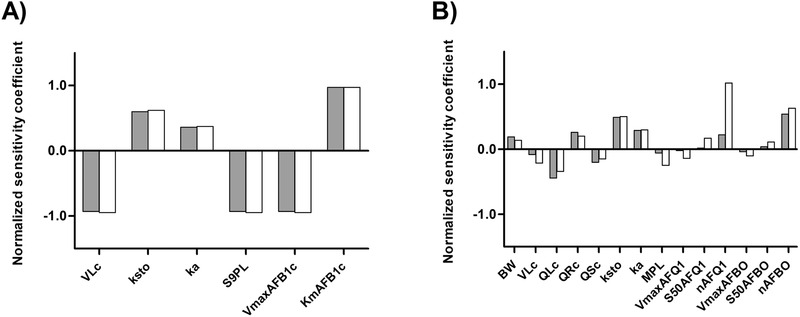
Normalized sensitivity coefficients for the parameters of A) rat and B) human PBK model for AFB1. The sensitivity analysis for the rat model (A) was performed at single oral doses of 0.4 µg kg−1 bw (gray bars) and 5 mg kg−1 bw (white bars). The sensitivity analysis for the human model (B) was performed at single oral doses of 3.74 × 10−4 µg kg−1 bw (gray bars) and 120 µg kg−1 bw (white bars). VLc = volume of the liver, ksto = stomach emptying rate, ka = absorption rate constant, S9PL = scaling factor from liver S9 fraction to whole liver, VmaxAFB1c = V max of AFB1 conversion, KmAFB1c = K m of AFB1 conversion, BW = body weight, QLc = blood flow to liver, QRc = blood flow to rapidly perfused tissues, QSc = blood flow to slowly perfused tissues, MPL = scaling factor from liver microsomes to whole liver, V max AFQ1 = V max of AFQ1 formation, S50AFQ1 = S 50 of AFQ1 formation, V max AFBO = V max of AFBO formation, S50AFBO = S 50 of AFBO formation, nAFQ1 = Hill coefficient for AFQ1 formation, nAFBO = Hill coefficient for AFBO formation.
3.4. Translation of the In Vitro Concentration Response Curves to In Vivo Dose–Response Curves
Figure 5 shows the predicted in vivo dose–response curves for acute liver toxicity of AFB1 in rats and humans. The dose–response curves were obtained using PBK model‐based facilitated reverse dosimetry converting all in vitro concentration response curves depicted in Figure 2 (Table S7, Supporting Information).
Figure 5.
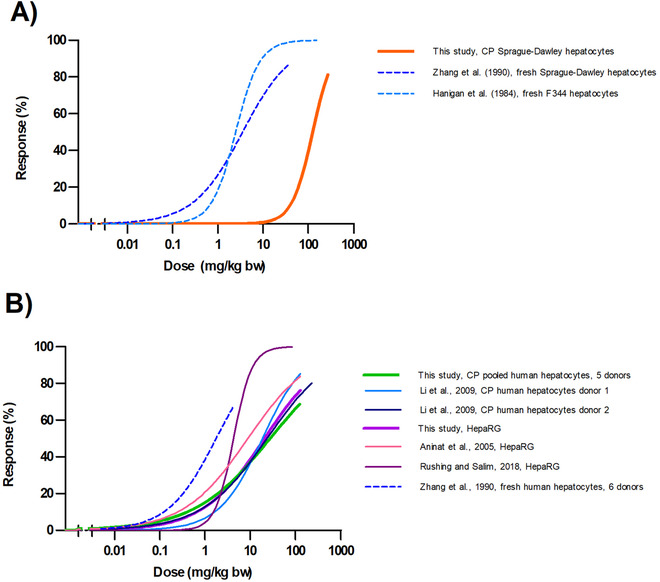
Predicted in vivo dose–response curves for acute liver toxicity of aflatoxin B1 in A) male rat and B) human obtained by PBK modeling facilitated reverse dosimetry of the concentration response curves presented in Figure 2. Dotted lines correspond to fresh hepatocytes. CP = cryopreserved.
3.5. Evaluation of the Predicted In Vivo Liver Toxicity
Due to the differences between cryopreserved and fresh hepatocytes in both rats and humans, reflected by the different EC50 values (Figure 2; Table S6, Supporting Information), the PODs used for the evaluation were derived from the dose–response curves predicted based on the data obtained with fresh hepatocytes and not from the data obtained with cryopreserved hepatocytes or HepaRG cells since these can be expected to result in an underprediction of the in vivo AFB1 toxicity (Table S8, Supporting Information). Figure 6 , presents a visual comparison of the predicted BMDL10‐BMDU10 values for acute AFB1 toxicity in rats compared with the POD values obtained from data on liver toxicity markers after a single acute exposure to AFB1[ 48 , 57 ] and after a repeated exposure (daily over 7 days).[ 49 ] Taken together, the predicted BMDL10‐BMDU10 values from the fresh F344 and Sprague Dawley rat hepatocytes match the POD values derived from the single and repeated exposure data on AFB1 induced acute liver toxicity quite well. Figure 6 also presents the LD50 values for lethality upon a single oral dose of AFB1. These LD50 values are generally somewhat higher than the observed and predicted POD values for acute liver toxicity, which may be in line with the fact that the lethality endpoint likely requires higher dose levels than liver damage as such.
Figure 6.
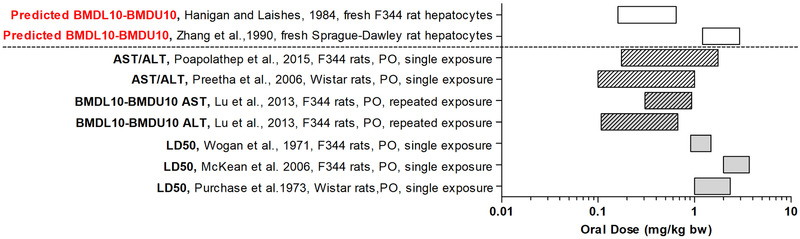
Predicted BMDL10‐BMDU10 values for acute liver toxicity of AFB1 in male rat obtained by the PBK modeling‐facilitated reverse dosimetry approach using data from fresh hepatocytes by Zhang et al.[ 54 ] and Hanigan and Laishes[ 55 ] (white rectangle bars). The predicted BMDL10‐BMDU10 are compared to POD for ALT or AST response after a single exposure (striped rectangle bars) or repeated exposure (striped gray rectangle bars) and in vivo male median lethal doses (LD50) (gray rectangle bars). PO = oral, ALT = aminotransferase, and AST = aspartate aminotransferase.
Figure 7 , shows the comparison of the predicted BMDL10‐BMDU10 values for acute liver toxicity in humans derived from data obtained in fresh human hepatocytes.[ 54 ] The predicted BMDL10‐BMDU10 values amount to 77–519 µg kg−1 bw with the BMDL10 of 76 µg kg−1 bw being in line with the values of 20–120 µg kg−1 bw per day and of 20–100 µg kg−1 bw per day reported in the literature for the onset of acute toxicity of AFB1 in humans.[ 4 , 9 ]
Figure 7.

Predicted BMDL10‐BMDU10 values for acute liver toxicity of AFB1 in human obtained by the PBK modeling‐facilitated reverse dosimetry approach using data from fresh human hepatocytes.[ 54 ] The predicted BMDL10‐BMDU10 (white rectangle bars) are compared to the estimated doses that can cause a risk of acute aflatoxicosis and possibly fatality in humans[ 4 , 9 ] (gray rectangle bars).
4. Discussion
In humans, acute liver toxicity caused by AFB1 has been reported but specific aflatoxin exposure doses casing toxicity are unknown due to the difficulty to relate disease outcomes with a specific oral AFB1 consumption.[ 11 , 58 ] In this study we aimed to use an in vitro PBK model‐based reverse dosimetry approach to predict oral dose levels of AFB1 causing in vivo acute liver toxicity in rats and humans. Previous work has shown agreement between predicted QIVIVE data and data from in vivo studies in experimental animal studies for different toxicity endpoints.[ 18 , 26 , 59 , 60 ] With respect to the liver as target organ, the acute liver toxicity of the pyrrolizidine alkaloids lasiocarpine and riddelliine could be predicted based on in vitro toxicity studies in cryopreserved rat and human hepatocytes and PBK models for rats and humans to facilitate reverse dosimetry based QIVIVE.[ 16 , 17 ]
In the present study, cryopreserved pooled Sprague Dawley rat hepatocytes, cryopreserved pooled human hepatocytes, and HepaRG cells were used to assess the cytotoxicity of AFB1 in vitro, while data obtained in similar models and fresh hepatocytes, reported in literature, were included for comparison. The EC50 values obtained for AFB1 cytotoxicity revealed that fresh hepatocytes of both rats and humans are more sensitive than cryopreserved hepatocytes from rats and humans. In rats, the EC50 values obtained with the cryopreserved Sprague Dawley rat hepatocytes used in the present study displayed a 60–146‐fold higher EC50 compared with the reported EC50 values obtained with fresh rat hepatocytes.[ 54 , 55 ] The differences between the different models might best be ascribed to differences in the activity of the cytochromes P450 (CYP) responsible for bioactivation of AFB1 in fresh and cryopreserved hepatocytes.[ 61 ] In rats, various CYP enzymes are reported to be involved in AFB1 metabolism, including CYP1A1/2, CYP3A1/2, CYP2C11, and CYP2B1/2,[ 62 ] and although the activity of some CYP enzymes, like CYP2A in cryopreserved rat hepatocytes can be comparable to that in fresh rat hepatocytes,[ 63 , 64 ] the activity of other CYP enzymes, like CYP3A, can be lower.[ 65 ]
In humans, CYP1A2, 2B6, 3A4, 3A5, 3A7, and 2A13 are involved in AFB1 phase I metabolism.[ 42 ] Although their expression can largely vary between individuals, CYP3A4 and CYP1A2 are regarded as the main enzymes involved in both detoxification and biotransformation of AFB1.[ 36 , 42 ] In human, basal expression of some CYP enzymes is lower in cryopreserved hepatocytes than in freshly isolated hepatocytes.[ 66 ] CYP1A2 is 2–13‐fold lower in cryopreserved hepatocytes, while CYP3A4 is comparable in both, fresh and cryopreserved, hepatocytes, yet for both CYP enzymes a large batch‐to‐batch variation in their expression and inducibility may occur depending on the donors.[ 36 , 66 , 67 , 68 ] Hence, the observed differences between fresh and cryopreserved human hepatocytes observed in the present study may be due to the difference between fresh and cryopreserved hepatocytes but may also (in part) be due to the interindividual variability in the expression of CYP enzymes between the respective donors.
In this study, the developed PBK models describing the kinetics of AFB1 for rats and humans, were shown to result in predictions that were comparable to available in vivo kinetic data for rats and humans.[ 15 , 47 ] Based on the prediction of C max within 1.01–2.1‐fold accuracy, the model was considered suitable for QIVIVE. In the next step, the in vitro concentration–response curves for AFB1 cytotoxicity were translated using PBK modeling‐based reverse dosimetry to dose–response curves, enabling prediction of PODs for comparison with in vivo data. The predicted BMDL10‐BMDU10 from the rat model were in line with the PODs for liver toxicity derived from in vivo studies (Figure 6). The LD50 values derived from data on AFB1 in rats were somewhat higher than the predicted and observed PODs for liver toxicity like AST and ALT[ 48 , 49 , 57 ] which is in line with the expected role of the enzyme biomarkers as earlier markers of AFB1 induced adverse effects. Differences observed between the different values predicted and observed for liver toxicity may be due to differences in the sensitivity of the different rat models with F344 rats being more sensitive than Sprague Dawley rats[ 46 , 69 ] and older rats being more sensitive than younger ones.[ 50 , 53 , 70 ]
In humans, the BMDL10 of 77 µg kg−1 bw predicted based on data from fresh human hepatocytes was in line with the AFB1 dose levels estimated to result in the onset of acute symptoms and possible fatality by Wild and Gong (20–100 µg kg−1 bw per day) and by JECFA (20–120 µg kg−1 bw per day).[ 4 , 9 ] It is of interest to note that the current study used pooled liver samples of 13 individuals.[ 36 ] This implies that interindividual differences are not reflected. To account for interindividual variability in AFB1 kinetics, combining the PBK modeling with Monte Carlo analysis as recently done by Ning et al. to predict interindividual variation in bioactivation and liver toxicity of lasiocarpine[ 71 ] could be considered in a future study.
In conclusion, the PBK modeling‐based reverse dosimetry for QIVIVE used in this study could adequately predict in vivo kinetics. The study also showed a proof‐of‐principle for QIVIVE by integrating in vitro data with in silico PBK modeling‐based reverse dosimetry to predict doses that may cause acute liver toxicity of AFB1 in rats and humans.
Conflict of Interest
The authors declare no conflict of interest.
Author Contributions
I.G.S. and S.W. performed the research. I.G.S. and I.M.C.M.R. designed the research study. I.G.S. and I.M.C.M.R. analyzed the data. I.G.S. and I.M.C.M.R. wrote and edited the manuscript.
Supporting information
Supporting Information
Supporting Information
Acknowledgements
This work was financed by the Mexican National Council for Science and Technology (CONACYT) via the scholarship granted to I.G.S. (No. 570901).
Gilbert‐Sandoval I., Wesseling S., Rietjens I. M. C. M., Predicting the Acute Liver Toxicity of Aflatoxin B1 in Rats and Humans by an In Vitro–In Silico Testing Strategy. Mol. Nutr. Food Res. 2020, 64, 2000063 10.1002/mnfr.202000063
References
- 1. IARC , A Review of Human Carcinogens. Part F: Chemical Agents and Related Occupations/IARC Working Group on the Evaluation of Carcinogenic Risks to Humans, IARC, Lyon, France: 2012, p. 225. [PMC free article] [PubMed] [Google Scholar]
- 2. Groopman J. D., Egner P. A., Schulze K. J., Wu L. S.‐F., Merrill R., Mehra S., Shamim A. A., Ali H., Shaikh S., Gernand A., Food Chem. Toxicol. 2014, 74, 184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Peraica M., Radić B., Lucić A., Pavlović M., Bull W. H O. 1999, 77, 754. [PMC free article] [PubMed] [Google Scholar]
- 4. Wild C. P., Gong Y. Y., Carcinogenesis 2010, 31, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Azziz‐Baumgartner E., Lindblade K., Gieseker K., Rogers H. S., Kieszak S., Njapau H., Schleicher R., McCoy L. F., Misore A., DeCock K., Environ. Health Perspect. 2005, 113, 1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kensler T. W., Roebuck B. D., Wogan G. N., Groopman J. D., Toxicol. Sci. 2011, 120, S28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kamala A., Shirima C., Jani B., Bakari M., Sillo H., Rusibamayila N., De Saeger S., Kimanya M., Gong Y., Simba A., World Mycotoxin J. 2018, 11, 311. [Google Scholar]
- 8. Williams J. H., Phillips T. D., Jolly P. E., Stiles J. K., Jolly C. M., Aggarwal D., Am. J. Clin. Nutr. 2004, 80, 1106. [DOI] [PubMed] [Google Scholar]
- 9. Safety Evaluation of Certain Contaminants in Food , Prepared by the eighty‐third meeting of the Joint FAO/WHO Expert Committee on Food Additives (JECFA), WHO Food Additives Series: 74, FAO JECFA Monographs 19 bis, Food and Agriculture Organization of the United Nations/World Health Organization, Geneva 2018.
- 10. Cupid B., Lightfoot T., Russell D., Gant S., Turner P., Dingley K., Curtis K., Leveson S., Turteltaub K. W., Garner R., Food Chem. Toxicol. 2004, 42, 559. [DOI] [PubMed] [Google Scholar]
- 11. EFSA Panel on Contaminants in the Food Chain (CONTAM) , EFSA Journal 2020, 18, 6040. [Google Scholar]
- 12. Cullen J. M., Newberne P. M., The Toxicology of Aflatoxins, Elsevier, New York: 1994, p. 3. [Google Scholar]
- 13. Louisse J., Beekmann K., Rietjens I. M. C. M., Chem. Res. Toxicol. 2017, 30, 114. [DOI] [PubMed] [Google Scholar]
- 14. R. A. Coulombe, Jr. , Sharma R., Food Chem. Toxicol. 1985, 23, 827. [DOI] [PubMed] [Google Scholar]
- 15. Jubert C., Mata J., Bench G., Dashwood R., Pereira C., Tracewell W., Turteltaub K., Williams D., Bailey G., Cancer Prev. Res. 2009, 2, 1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen L., Ning J., Louisse J., Wesseling S., Rietjens I. M. C. M., Food Chem. Toxicol. 2018, 116, 216. [DOI] [PubMed] [Google Scholar]
- 17. Ning J., Chen L., Strikwold M., Louisse J., Wesseling S., Rietjens I. M. C. M., Arch. Toxicol. 2019, 93, 801. [DOI] [PubMed] [Google Scholar]
- 18. Abdullah R., Alhusainy W., Woutersen J., Rietjens I. M. C. M., Punt A., Food Chem. Toxicol. 2016, 92, 104. [DOI] [PubMed] [Google Scholar]
- 19. Dirr H. W., Schabort J. C., Biochim. Biophys. Acta, Gen. Subj. 1986, 881, 383. [DOI] [PubMed] [Google Scholar]
- 20. Poór M., Bálint M., Hetényi C., Gődér B., Kunsági‐Máté S., Kőszegi T., Lemli B., Toxins 2017, 9, 339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bagheri M., Fatemi M. H., J. Lumin. 2018, 202, 345. [Google Scholar]
- 22. Watanabe R., Esaki T., Kawashima H., Natsume‐Kitatani Y., Nagao C., Ohashi R., Mizuguchi K., Mol. Pharmaceutics 2018, 15, 5302. [DOI] [PubMed] [Google Scholar]
- 23. Probst R. J., Lim J. M., Bird D. N., Pole G. L., Sato A. K., Claybaugh J. R., J. Am. Assoc. Lab. Anim. Sci. 2006, 45, 49. [PMC free article] [PubMed] [Google Scholar]
- 24. Billett H. H., Clinical Methods: The History, Physical, and Laboratory Examinations, 3rd ed., Butterworths, Waltham, MA: 1990. [PubMed] [Google Scholar]
- 25. Yang J., Jamei M., Yeo K. R., Rostami‐Hodjegan A., Tucker G. T., Drug Metab. Dispos. 2007, 35, 501. [DOI] [PubMed] [Google Scholar]
- 26. Li H., Zhang M., Vervoort J., Rietjens I. M. C. M., van Ravenzwaay B., Louisse J., Toxicol. Lett. 2017, 266, 85. [DOI] [PubMed] [Google Scholar]
- 27. Louisse J., Bosgra S., Blaauboer B. J., Rietjens I. M. C. M., Verwei M., Arch. Toxicol. 2015, 89, 1135. [DOI] [PubMed] [Google Scholar]
- 28. Ramos A., Hernandez E., Mycopathologia 1996, 134, 27. [DOI] [PubMed] [Google Scholar]
- 29. Busbee D., Norman J., Ziprin R., Arch. Toxicol. 1990, 64, 285. [DOI] [PubMed] [Google Scholar]
- 30. Kumagai S., Toxicol. Appl. Pharmacol. 1989, 97, 88. [DOI] [PubMed] [Google Scholar]
- 31. Gratz S., Wu Q., El‐Nezami H., Juvonen R., Mykkänen H., Turner P., Appl. Environ. Microbiol. 2007, 73, 3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Versantvoort C. H., Oomen A. G., Van de Kamp E., Rompelberg C. J., Sips A. J., Food Chem. Toxicol. 2005, 43, 31. [DOI] [PubMed] [Google Scholar]
- 33. Brown R. P., Delp M. D., Lindstedt S. L., Rhomberg L. R., Beliles R. P., Toxicol. Ind. Health 1997, 13, 407. [DOI] [PubMed] [Google Scholar]
- 34. DeJongh J., Verhaar H. J., Hermens J. L., Arch. Toxicol. 1997, 72, 17. [DOI] [PubMed] [Google Scholar]
- 35. Roebuck B. D., Wogan G. N., Cancer Res. 1977, 37, 1649. [PubMed] [Google Scholar]
- 36. Kamdem L. K., Meineke I., Gödtel‐Armbrust U., Brockmöller J., Wojnowski L., Chem. Res. Toxicol. 2006, 19, 577. [DOI] [PubMed] [Google Scholar]
- 37. Mykkänen H., Zhu H., Salminen E., Juvonen R. O., Ling W., Ma J., Polychronaki N., Kemiläinen H., Mykkänen O., Salminen S., Int. J. Cancer 2005, 115, 879. [DOI] [PubMed] [Google Scholar]
- 38. Houston J. B., Kenworthy K. E., Drug Metab. Dispos. 2000, 28, 246. [PubMed] [Google Scholar]
- 39. Ueng Y.‐F., Kuwabara T., Chun Y.‐J., Guengerich F. P., Biochemistry 1997, 36, 370. [DOI] [PubMed] [Google Scholar]
- 40. Gallagher E. P., Wienkers L., Stapleton P., Kunze K., Eaton D. L., Cancer Res. 1994, 54, 101. [PubMed] [Google Scholar]
- 41. Gallagher E. P., Kunze K. L., Stapleton P. L., Eaton D. L., Toxicol. Appl. Pharmacol. 1996, 141, 595. [DOI] [PubMed] [Google Scholar]
- 42. Eaton D., Beima K., Bammler T., Riley R., Voss K., Compr. Toxicol. 2017, 2, 483. [Google Scholar]
- 43. Chiu W. A., Ginsberg G. L., Toxicol. Appl. Pharmacol. 2011, 253, 203. [DOI] [PubMed] [Google Scholar]
- 44. Zhang H., Gao N., Tian X., Liu T., Fang Y., Zhou J., Wen Q., Xu B., Qi B., Gao J., Sci. Rep. 2015, 5, 17671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Evans M. V., Andersen M. E., Toxicol. Sci. 2000, 54, 71. [DOI] [PubMed] [Google Scholar]
- 46. Wogan G., Paglialunga S., Newberne P., Food Cosmet. Toxicol. 1974, 12, 681. [DOI] [PubMed] [Google Scholar]
- 47. Han Z., Zhao Z., Song S., Liu G., Shi J., Zhang J., Liao Y., Zhang D., Wu Y., De Saeger S., Anal. Methods 2012, 4, 3708. [Google Scholar]
- 48. Poapolathep S., Imsilp K., Machii K., Kumagai S., Poapolathep A., Biocontrol Sci. 2015, 20, 171. [DOI] [PubMed] [Google Scholar]
- 49. Lu X., Hu B., Shao L., Tian Y., Jin T., Jin Y., Ji S., Fan X., Food Chem. Toxicol. 2013, 55, 444. [DOI] [PubMed] [Google Scholar]
- 50. Roebuck B. D., Maxuitenko Y. Y., in The Toxicology of Aflatoxins (Eds: Eaton D., Groopman J. D.), Academic Press, San Diego, CA: 1994, p. 27. [Google Scholar]
- 51. Wogan G., Edwards G., Newberne P., Cancer Res. 1971, 31, 1936. [PubMed] [Google Scholar]
- 52. Purchase I., Steyn M., Gilfillan T., Chem.‐Biol. Interact. 1973, 7, 283. [DOI] [PubMed] [Google Scholar]
- 53. McKean C., Tang L., Tang M., Billam M., Wang Z., Theodorakis C., Kendall R., Wang J.‐S., Food Chem. Toxicol. 2006, 44, 868. [DOI] [PubMed] [Google Scholar]
- 54. Zhang S.‐Z., Lipsky M. M., Trump B. F., Hsu I.‐C., Cell Biol. Toxicol. 1990, 6, 219. [DOI] [PubMed] [Google Scholar]
- 55. Hanigan H. M., Laishes B. A., Toxicology 1984, 30, 185. [DOI] [PubMed] [Google Scholar]
- 56. Rushing B. R., Selim M. I., Oncotarget 2018, 9, 4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Preetha S., Kanniappan M., Selvakumar E., Nagaraj M., Varalakshmi P., Comp. Biochem. Physiol. C: Toxicol. Pharmacol. 2006, 143, 333. [DOI] [PubMed] [Google Scholar]
- 58. Groopman J. D., Kensler T. W., Carcinogenesis 1999, 20, 1. [DOI] [PubMed] [Google Scholar]
- 59. Louisse J., de Jong E., van de Sandt J. J., Blaauboer B. J., Woutersen R. A., Piersma A. H., Rietjens I. M. C. M., Verwei M., Toxicol. Sci. 2010, 118, 470. [DOI] [PubMed] [Google Scholar]
- 60. Strikwold M., Spenkelink B., de Haan L. H., Woutersen R. A., Punt A., Rietjens I. M. C. M., Arch. Toxicol. 2017, 91, 2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Griffin S. J., Houston J. B., Drug Metab. Dispos. 2004, 32, 552. [DOI] [PubMed] [Google Scholar]
- 62. Stresser D., Bailey G., Williams D., Drug Metab. Dispos. 1994, 22, 383. [PubMed] [Google Scholar]
- 63. Zaleski J., Richburg J., Kauffman F. C., Biochem. Pharmacol. 1993, 46, 111. [DOI] [PubMed] [Google Scholar]
- 64. Pearce R., Greenway D., Parkinson A., Arch. Biochem. Biophys. 1992, 298, 211. [DOI] [PubMed] [Google Scholar]
- 65. Silva J. M., Day S. H., Nicoll‐Griffith D. A., Chem.‐Biol. Interact. 1999, 121, 49. [DOI] [PubMed] [Google Scholar]
- 66. Roymans D., Van Looveren C., Leone A., Parker J. B., McMillian M., Johnson M. D., Koganti A., Gilissen R., Silber P., Mannens G., Biochem. Pharmacol. 2004, 67, 427. [DOI] [PubMed] [Google Scholar]
- 67. Peter J., Chan K., Silber P. M., Chem.‐Biol. Interact. 2004, 150, 97. [DOI] [PubMed] [Google Scholar]
- 68. Yokoyama Y., Sasaki Y., Terasaki N., Kawataki T., Takekawa K., Iwase Y., Shimizu T., Sanoh S., Ohta S., Biol. Pharm. Bull. 2018, 41, 722. [DOI] [PubMed] [Google Scholar]
- 69. Rogers A. E., Kula N. S., Newberne P. M., Cancer Res. 1971, 31, 491. [PubMed] [Google Scholar]
- 70. Dohnal V., Wu Q., Kuča K., Arch. Toxicol. 2014, 88, 1635. [DOI] [PubMed] [Google Scholar]
- 71. Ning J., Rietjens I. M. C. M., Strikwold M., Arch. Toxicol. 2019, 93, 2943. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information