

Abstract
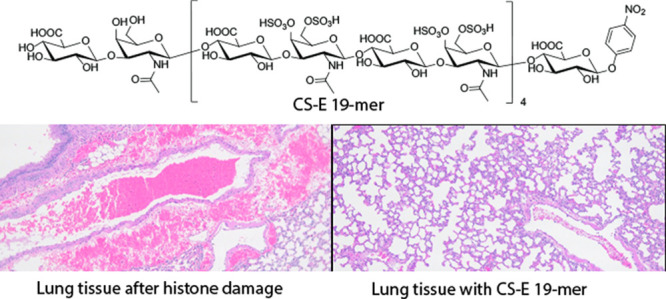
Chondroitin sulfate E (CS-E) is a sulfated polysaccharide that contains repeating disaccharides of 4,6-disulfated N-acetylgalactosamine and glucuronic acid residues. Here, we report the enzymatic synthesis of three homogeneous CS-E oligosaccharides, including CS-E heptasaccharide (CS-E 7-mer), CS-E tridecasaccharide (CS-E13-mer), and CS-E nonadecasaccharide (CS-E 19-mer). The anti-inflammatory effect of CS-E 19-mer was investigated in this study. CS-E 19-mer neutralizes the cytotoxic effect of histones in a cell-based assay and in mice. We also demonstrate that CS-E 19-mer treatment improves survival and protects against organ damage in a mouse model of endotoxemia induced by bacterial lipopolysaccharide (LPS). CS-E19-mer directly interacts with circulating histones in the plasma from LPS-challenged mice. CS-E 19-mer does not display anticoagulant activity nor react with heparin-induced thrombocytopenia antibodies isolated from patients. The successful synthesis of CS-E oligosaccharides provides structurally defined carbohydrates for advancing CS-E research and offers a potential therapeutic agent to treat life-threatening systemic inflammation.
Short abstract
Structurally homogeneous chondroitin sulfate E 19-mer protects against systematic inflammation by targeting to extracellular histone.
Introduction
Chondroitin sulfate (CS) is a glycosaminoglycan (GAG) found in all animals exhibiting essential physiological functions. The CS polysaccharide consists of a disaccharide repeating unit of →3) N-acetyl-β-d-galactosamine (GalNAc) (1 → 4) β-d-glucuronic acid (GlcA) (1 →, and both saccharide residues can carry sulfo groups. Four major subtypes of CS polysaccharides have been isolated: (1) chondroitin sulfate A (CS-A); (2) chondroitin sulfate C (CS-C); (3) chondroitin sulfate D (CS-D); and (4) chondroitin sulfate E (CS-E). Each subtype is characterized by a single sulfation pattern. CS-A contains 4-O(oxygen)-sulfated GalNAc residue, CS-C contains 6-O-sulfated residue, CS-D contains 2-O-sulfated GlcA, and CS-E contains 4,6-O-disulfated GlcNAc residue. CS plays important roles in neuroplasticity, cell communication, osteoblast differentiation, immunological response, and viral infection.1,2 Among CS subtypes, CS-E has a low abundance in mammals but plays a critical role in modulating angiogenesis through interacting with chemokines and growth factors3 and regulating tumor progression and metastasis.4,5 These activities of CS-E are governed by its unique sulfation pattern.6 CS-E isolated from biological tissues is a mixture of GAG chains with different lengths and sulfation patterns.7 The lack of homogeneous CS-E oligosaccharides of defined structure and size has limited in-depth investigation of the function and structure relationship of CS-E.
Systemic inflammation is a major contributor to sepsis8−10 impacting 31 million people globally and resulting in 5 million deaths annually.10 Currently, there are no specific drugs to treat sepsis. A murine model of endotoxemia, induced by administration of bacterial lipopolysaccharide (LPS), is widely used to study the systemic inflammatory responses that are a critical part of the complex pathology associated with sepsis.11 Upon administering bacterial LPS, neutrophils or injured cells release histones, nuclear DNA-binding proteins, into extracellular space and blood circulation.12−14 Extracellular histones activate immune cells and trigger the release of pro-inflammatory cytokines through toll-like receptors.15,16 Extracellular histones also mediate intravascular coagulation17 and cause endothelial dysfunction,18 leading to organ damage under hyper inflammatory conditions.19−21 Treatment with antibodies neutralizing histones resulted in an improved survival and reduced organ injury in septic mice.11 Therefore, targeting extracellular histones represents a promising strategy for the treatment of sepsis. Although heparin and heparan sulfate have been used to target to histone to attenuate systemic inflammation caused by LPS,22 the potential roles of CS-E in this disease model has not been investigated.
We sought to develop an efficient approach to synthesize homogeneous CS-E oligosaccharides and investigate their properties in biological systems. We were particularly interested in designing CS-E oligosaccharides to neutralize the pro-inflammatory effect of extracellular histones. The synthesis of CS-E oligosaccharides has been previously achieved through purely chemical synthesis.23,24 Such a synthetic approach is incredibly labor-intensive and costly, especially in the preparation of long CS-E chains resembling full-length GAGs. Recently, we developed an enzyme-based method for the highly efficient synthesis of CS-A and CS-C oligosaccharides.25 While enzymatic synthesis of structurally heterogeneous CS-E has been reported, the CS-E obtained consists of a mixture of sulfated polysaccharides26 offering few advantages to the CS-E natural product isolated from animal tissues. Here, we report an enzymatic synthesis of homogeneous CS-E oligosaccharides, up to a nonadecasaccharide (19-mer). This represents the longest CS oligosaccharide to have been synthesized. Nearly 400 mg of CS-E nonadecasaccharide (CS-E 19-mer) was synthesized for biological studies. In vivo evaluation in mice demonstrates that the CS-E 19-mer could reduce mortality and ameliorate organ damage in bacteria lipopolysaccharide (LPS)-induced endotoxemia, an animal model that mimics the symptoms of sepsis patients. Mechanistical studies show that CS-E 19-mer binds to extracellular histones and neutralizes histone toxicity in both in vitro and in vivo models. Our results reveal the possibility of using CS-E to treat systemic inflammation by neutralizing the cytotoxic activity of extracellular histones.
Results
Enzymatic Synthesis of CS-E Oligosaccharides
The enzymatic synthesis of CS-E oligosaccharides was initiated using a commercially available monosaccharide, p-nitrophenyl glucuronide (GlcA-pNP) (Figure 1A). The synthesis involved the serial elongation of this monosaccharide acceptor with uridine 5′-diphospho N-acetylgalactosamine (UDP-GalNAc) and uridine 5′-diphosphoglucuronic acid (UDP-GlcA) donors using a bacterial glycosyltransferase (KfoC). By the alternating addition of GalNAc and GlcA residues we accomplished the controlled extension of the nonsulfated chondroitin oligosaccharide backbone. The resulting unsulfated backbone underwent two rounds of sulfotransferase modifications in the presence of the sulfate donor 3′-phosphoadenosine 5′-phosphosulfate (PAPS) to install the sulfo groups. Sulfation catalyzed by chondroitin sulfate 4-O-sulfotransferase (CS 4OST) was first performed to install 4-O-sulfation on GalNAc residues, yielding CS-A. Subsequent modifications, catalyzed by N-acetylgalactosamine 4-sulfate 6-O-sulfotransferase (GalNAc4S-6OST), added 6-O-sulfo groups to the GalNAc4S residues to form CS-E oligosaccharide. GalNAc4S-6OST, the critical catalyst required for the synthesis of CS-E oligosaccharides, was expressed in insect cells (SF9 cells) using the baculovirus expression system and purified to homogeneity using heparin and nickel columns (Supporting Information Figure S1). Since the expression host cells produce a chondroitinase that degrades CS oligosaccharides,27 it was imperative to remove chondroitinase from the GalNAc4S-6OST preparation. We also tried to synthesize CS-E 19-mers without using GalNAc4S-6OST, but the synthesis failed (Supporting Information Figures S2–S4).
Figure 1.

Enzymatic synthesis of CS-E oligosaccharides. (A) The scheme to synthesize CS-E 7-mer, 13-mer, and 19-mer. The scheme is presented in shorthand symbols. (B) The chemical structures of four different CS oligosaccharides synthesized for the current study.
Three CS-E oligosaccharides, CS-E 7-mer, CS-E 13-mer, and CS-E 19-mer, were synthesized at a scale ranging from 86 to 393 mg (Supporting Information Figure S5). The nonsulfated chondroitin nonadecasaccharide backbone, CS-0S 19-mer, precursor was synthesized at the scale of 1.18 g and was used to serve as a negative control in our biological studies (Supporting Information Figure S2). The purity and structure of synthesized CS-E oligosaccharides were evaluated using high-resolution diethylaminoethyl (DEAE) high-performance liquid chromatography (HPLC), high-resolution mass spectrometry (MS), and nuclear magnetic resonance (NMR) spectroscopy. A representative example, CS-E 7-mer, was eluted a single symmetric peak from DEAE-HPLC, demonstrating a high degree of purity (Supporting Information Figure S6). High-resolution mass spectrometry analysis of CS-E 7-mer gave a mass of 1772.227 (calculated (calcd) value 1772.221) (Supporting Information Figure S7). 1H NMR and 13C NMR analyses further confirmed the purity of CS-E 7-mer (Supporting Information Figures S8 and S9). Two-dimensional NMR analyses were employed to confirm the nature of the glycosidic linkages between GalNAc and GlcA as well as the 4-O- and 6-O-sulfation on GalNAc residues (Figure 2A,B and Supporting Information Figure S10). The results from heteronuclear multiple bond coherence (HMBC) analysis confirmed that the linkages of G/F, E/D, and C/B are the 1 → 3 linkage, whereas the linkages between F/E, D/C, and B/A are 1 → 4 linkages. The anomeric proton–proton coupling constants (3JH–H) of residues B, C, D, E, F, and G were determined to be ∼8.0 Hz, indicative of the β-form of glycosidic linkages (Figure 2A). The results from heteronuclear single quantum coherence (HSQC) analysis permitted to locate the sulfo groups on GalNAc residues (Figure 2B). The chemical shifts of H-4 (4.74 and 4.76 ppm) and H-6 (4.17–4.24 ppm) from B and D residues were shifted downfield compared to the H-4 (4.11 ppm) and H-6 (3.69–3.74 ppm) signals of the F residue, demonstrating that both the 4- and 6-hydroxyl groups of residues B and D were sulfated (Figure 2B). The full assignment of 1H NMR for CS-E 7-mer is shown in Supporting Information Figure S11.
Figure 2.

NMR analysis of CS-E 7-mer. (A) The HMBC spectrum of CS-E 7-mer. Cross peaks between H-4 and C-1 or H-3 and C-1 are indicated in the chemical structure using curved arrows, where red color arrows indicate the cross peak between H-3 of GalNAc and C-1 of GlcA, and the blue arrows indicate the cross peak between H-4 of GlcA and C-1 of GalNAc. The results confirm the linkages between GalNAc and GlcA. An expanded region of HMBC between 110 and 100 ppm for 13C showing cross peaks between C-4 and H-1 or C-3 and H-1 are indicated in Supporting Information Figure S7. (B) The HSQC-NMR of CS-E 7-mer, in which the sulfation pattern at C-4 and C-6 of N-acetylgalactosamine can be characterized. The anomeric signals are indicated.
The purity of the CS-E 13-mer and CS-E 19-mer was similarly confirmed by DEAE-HPLC (Supporting Information Figures S12A and S17A), and the molecular masses of two oligosaccharides were determined to be 3389.304 (calcd value 3389.296) and 5006.367 (calcd value 5006.371) by high-resolution MS, respectively (Supporting Information Figures S12B and S17B). Full assignment of the NMR signals obtained from the CS-E 13-mer and CS-E 19-mer was more difficult than for CS-E 7-mer, since they contain a larger number of →3) GalNAc4S6S (1 → 4) GlcA (1→ repeating units resulting in severe signal overlap (Supporting Information Figures S13, S14, S18, and S19). Nevertheless, the C4 and C6 proton signals of GalNAc gave a range of chemical shifts from 4.76 to 4.81 (C4) and 4.18–4.30 ppm (C6), indicative of the GalNAc4S6S residue in the middle and at the reducing end of the oligosaccharide chain. In addition, the C-4 and C-6 proton of GalNAc signals of the nonreducing end GalNAc residue resonated from 4.10 to 4.17 ppm (C4) and 3.70–3.79 ppm (C6), consistent with the lack of sulfate groups on the nonreducing end GalNAc residue. The integration values for the CS-E 19-mer were consistent with their structure. In conclusion, the NMR results combined with the molecular mass measurement by high-resolution mass spectrometry (Supporting Information Figures S12–S16 and S17–S21) confirmed the structures of CS-E 13-mer and CS-E 19-mer. Notably, all the GalNAc residues, with the exception of the nonreducing end GalNAc residue, carried 4- and 6-O- sulfo groups. The failure of the enzymatic sulfation of the nonreducing terminal GalNAc residue results from the substrate specificity of two enzymes used in the synthesis. CS 4-OST is unable to sulfate the 4-hydroxyl group of the nonreducing end GalNAc residue.25 Since GalNAc4S-6OST can only transfer a sulfo group to the 6-hydroxyl group of a GalNAc4S residue the subsequent modification of the nonreducing terminal GalNAc by GalNAc4S-6OST cannot occur.28
CS-E 19-mer Neutralizes the Cytotoxic Effect of Histones
We next evaluated the binding of CS-E oligosaccharides to histones and their ability to protect against histone cytotoxicity. Extracellular histones alter cell membrane permeability by binding to phospholipid, therefore causing calcium influx and cell death.29 We examined the protective effect of CS-E oligosaccharides on histone-induced endothelial cell death using the EA.hy 926 cell line. Extensive cell death occurred when endothelial cells were challenged with histone H3 (30 μg /mL), consistent with previously published results.30 The addition of CS-E 19-mer (0.4–50 μg/mL) reduced the histone-induced cell death in a dose-dependent manner (Figure 3A). CS-E 13-mer tested at 10 and 50 μg/mL showed much smaller degrees of protection than the CS-E 19-mer, and incubation with CS-0S 19-mer showed no effect (Supporting Information Figure S27). These data demonstrate that the size and the sulfation of CS-E oligosaccharides determine their cytoprotective properties.
Figure 3.

CS-E 19-mer protects against cytotoxicity induced by histone in vitro and in vivo. (A) Cytotoxicity of histone H3 toward endothelial with or without CS-E 19-mer. Cell damage was measured by flow cytometry for propidium iodide (PI) staining, in EA.hy926 cell cultures with histone H3 (30 μg/mL) without or with different concentrations of CS-E 19-mer. (B) The binding affinity (KD) of different oligosaccharides to histone H3 as determined by SPR. No binding was detected for CS-E 7-mer and CS-0S 19-mer. (C) The survival plot of mice treated with histone (75 mg/kg) with CS-E 19-mer (75 mg/kg) or CS-0S 19-mer (75 mg/kg). The administration of histone and CS 19-mers was completed via retro-orbital injection. Seven animals were in histone/CS-E 19-mer, five were in the histone/CS-0S 19-mer cohorts, and nine animals were included in histone-treated cohort. Log-rank (Mantel-Cox) statistical analysis was performed to obtain P = 0.001. (D) The representative images and quantitation of hematoxylin and eosin (H&E) staining of formalin-fixed paraffin-embedded lung tissues. Extravascular hemorrhage and intravascular clots are indicated with arrows.
Next, we demonstrated that CS-E oligosaccharides directly bind to histones. Biotinylated CS-E oligosaccharides were used to perform the studies (the structures of biotinylated CS-E oligosaccharides are shown in Supporting Information Figures S22–S26). First, the biotinylated CS-E oligosaccharides were employed to pull down histone H3 from a buffer solution using an avidin-Sepharose affinity column (Supporting Information Figure S29). We discovered that both CS-E 13-mer and CS-E 19-mer bound to histone H3 as measured by western analysis. In contrast, CS-E 7-mer and CS-0S 19-mer did not bind histone H3. Second, we determined the binding affinity constants (KD) between biotinylated CS-E oligosaccharides and histone H3 using surface plasmon resonance (SPR) (Figure 3B). CS-E 19-mer was the tightest binder to histone H3 (44.7 nM) among the tested compounds (Figure 3B). The binding affinities of the four CS-E oligosaccharides toward a mixture of histone isoforms were also examined. A very similar binding affinity was observed to that of purified histone H3 (Supporting Information Figure S28). Taken together, our data suggest that the relative binding affinity between CS-E oligosaccharides and histone reflects their protective effects observed in histone-induced cell toxicity assay. Because CS-E 19-mer had the strongest binding affinity to histones in our in vitro experiments, the subsequent functional studies involving animals focused on CS-E 19-mer.
The cytoprotective effect CS-E 19-mer was next determined in mice that had been administered histones. We demonstrated that CS-E 19-mer treatment protects mice from histone-induced mortality (Figure 3C). In the untreated group, eight out of nine mice challenged with histone (75 mg/kg) died within 1 h. In contrast, pretreatment with CS-E 19-mer (75 mg/kg) before histone injection resulted in the survival of all seven mice (100%). Treatment with CS-0S 19-mer (75 mg/kg) before histone injection, however, did not improve survival. Unlike CS-E 19-mer, CS-0S 19-mer does not bind to histone or protect histone-induced endothelial cell death using EA.hy 926 cell line, suggesting that the protection effect requires the binding of the oligosaccharide to histone (Figure 3C). Histological analysis of lungs from histone-challenged mice showed intravascular thrombosis and intra-alveolar hemorrhage, resembling features of disseminated intravascular coagulation (DIC) associated with sepsis31 (Figure 3D). Importantly, treatment with CS-E 19-mer dramatically reduced these symptoms (Figure 3D). Similar effects of CS-E 19-mer on histone-induced coagulopathy were also observed in the mouse livers and kidneys (Supporting Information Figure S30). To further evaluate if the treatment with CS-E 19-mer can ameliorate DIC-like symptoms, we quantified the circulating platelets. The platelet counts in histone-challenged mice was dramatically decreased to 99 ± 52 × 103/μL (n = 3), compared to the healthy control group (1116 ± 59 × 103/μL, n = 3), indicating a consumptive coagulopathy and bleeding. Importantly, treatment of histone-challenged mice with CS-E 19-mer almost completely prevented the drop in platelet count (850 ± 39 × 103/μL, n = 3).
Treatment of CS-E 19-mer Attenuates Organ Damage in Endotoxemic Mice
The ability of CS-E 19-mer to neutralize the toxic effects of histones prompted us to examine whether this compound can attenuate organ damage in an animal model of systemic inflammation. In mice, LPS causes the release of histones, enhancing a hyper-inflammatory response and accelerating organ damage.11 Indeed, protease-cleaved histone H3 was first found in the mouse plasma 12 h after LPS administration, and full-length histone H3 was found 24 h after LPS administration, but they were absent in the control group (Figure 4A). Furthermore, we found that CS-E 19-mer forms a complex with circulated histone from the plasma of the endotoxemic mice (Figure 4B). Importantly, we found that the treatment with CS-E 19-mer reduced the LPS-induced mortality rate of endotoxemic mice from 92% to 30% (Figure 4C). These data suggest that CS-E 19-mer forms complexes with histone and neutralizes the toxicity, thereby displaying a protective effect against LPS-induced mortality.
Figure 4.

CS-E 19-mer protects against death and organ damages caused by bacterial lipopolysaccharides (LPS). (A) The image of western blot for the analysis of histone H3 in mice plasma after the administration of bacterial lipopolysaccharide (6 mg/kg). Two histone H3 bands were observed as reported previously.53 The top band is the intact protein, and the bottom band is a truncated form of histone H3 after protease cleavage. (B) The image of western analysis of mouse plasma samples with or without avidin-agarose affinity column purification. CS-E 19-mer forms a complex with histone to protect against histone-induced endothelial cell damage. The left two lanes are untreated mouse plasma incubated with biotinylated CS-E 19-mer after affinity purification. The right two lanes are LPS-treated mouse plasma with biotinylated CS-E 19-mer after affinity purification. (C) The survival plots of mice administered with LPS (6 mg/kg) with or without CS-E 19-mer (20 mg/kg). Ten animals were in LPS/CS-E 19-mer cohort, and 13 animals were included in LPS treated cohort. Log-rank (Mantel-Cox) statistical analysis was performed to obtain P = 0.003. (D–F) The plasma concentrations of different biomarkers, including creatinine, BUN, and AST in animals treated with phosphate-buffered saline, LPS, and LPS/CS-E 19-mer. (G) The concentrations of leaked Evans blue from the lung under the treatment of saline, LPS, or LPS/CS-E 19-mer. One-way ANOVA statistical analysis followed by Tukey’ multiple comparisons test was performed to obtain those P values. (*) P < 0.05, (**) P < 0.01, (****) P < 0.0001.
The extent of the LPS-induced organ damage was evaluated by measuring plasma levels of biomarkers as reported previously.32 Reductions in the plasma concentrations of creatinine and urine nitrogen (BUN) after CS-E 19-mer treatment indicates protection against loss of kidney function (Figure 4D,E). Concentrations of aspartate aminotransferase (AST), a marker for liver damage, were also reduced in the plasma of endotoxemic mice treated with CS-E 19-mer (Figure 4F). Disruption of endothelial cell barrier integrity and subsequent increase in vascular permeability is well-characterized in endotoxemic mice and contributes to end-organ damage.33 Treatment with CS-E 19-mer attenuated LPS-induced lung vascular permeability as measured by leakage of Evans Blue into lung tissue (Figure 4G), indicating protection against endothelial cell damage. The reduction of vascular permeability in kidneys and liver by CS-E 19-mer was not as obvious as that observed in the lung (Supporting Information Figure S31).
We examined two alternative possibilities allowing CS-E 19-mer to display its protection against LPS-induced organ damage. First, we tested whether CS-E 19-mer inhibits the expression of proinflammatory proteins, that is, tumor necrosis factor alpha (TNF-α), by acting on LPS/toll-like receptor 4 pathway using the THP-1 cell,34,35 a human monocytic cell line. We found that CS-E 19-mer does not affect the expression of TNF-α (Supporting Information Figure S32). As expected, a known toll-like receptor 4 inhibitor, TAK-242, inhibited the expression of TNF-α as reported previously.36 The data exclude the possibility that CS-E 19-mer disrupts the LPS/toll-like receptor 4 pathway. Second, we determined the direct binding of CS-E 19-mer and LPS using a centrifugal filtration method (Supporting Information Figure S33). We discovered that CS-E19-mer freely penetrated the membrane in the presence of LPS, but LPS did not. Our data suggest that there is no interaction between CS-E 19-mer and LPS. Taken together, our results are consistent with the proposed protection mechanism for CS-E 19-mer, namely, the compound neutralizes the cytotoxicity of histone.
CS-E 19-mer Does Not Display Anticoagulant Activity or Bind to Heparin-Induced Thrombocytopenia Antibodies
Bleeding risks are high in the setting of sepsis secondary to thrombocytopenia and other causes. Thus, we examined whether CS-E 19-mer had anticoagulant activity that could further worsen the bleeding risk by measuring the inhibition to both factor IIa (anti-FIIa) and factor Xa (anti-FXa). Compared with two anticoagulant drugs, namely, unfractionated heparin and fondaparinux, we did not find any detectable anti-FIIa and anti-FXa activities from CS-E 19-mer, suggesting that the protective effects of this compound do not involve a direct anticoagulant activity (Figure 5A,B).
Figure 5.
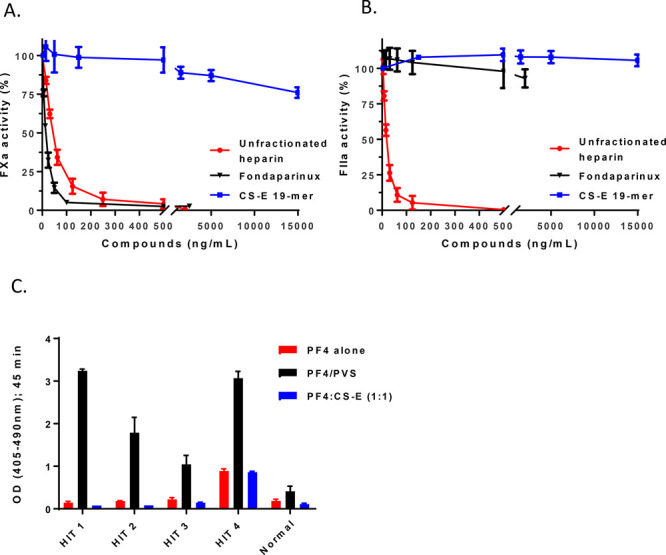
CS-E 19-mer does not exhibit anticoagulant or interact with HIT antibodies. (A) The inhibition curves of the activity of FXa (or anti-FXa activity) by unfractionated heparin, fondaparinux, and CS-E 19-mer. Both unfractionated heparin and fondaparinux are Food and Drug Administration-approved anticoagulant drug with anti-FXa activity. (B) The inhibition curves of the activity of FIIa (or anti-FIIa activity) by unfractionated heparin, fondaparinux, and CS-E 19-mer. Unlike unfractionated heparin, fondaparinux does not have anti-FIIa activity. (C) The binding of HIT antibodies from four patients and one normal patient to CS-E 19-mer/PF4 complex, PF4 alone, and poly(vinylsulfonate) (PVS)/PF4 complex. PF4 alone is the negative control, and PVS/PF4 complex is a positive control.
Next, we evaluated if CS-E 19-mer, by virtue of being a glycosaminoglycan (like heparin), is recognized by heparin-induced thrombocytopenia (HIT) antibodies when complexed to the target of these antibodies, platelet factor 4 (PF4). Enzyme-linked immunosorbent assay testing (Figure 5C) demonstrated that HIT antibodies bind strongly to PF4-polyanion complexes, as expected, but do not bind to CS-E-19-mer-PF4 complexes any better than with PF4 alone. This suggests that CS-E 19-mer will have a very low likelihood of stimulating an HIT immune response, as it does not expose HIT neoepitopes on PF4.
Discussion
In this manuscript, we report an efficient enzymatic method to synthesize structurally homogeneous CS-E oligosaccharides. The versatility of the enzymatic synthesis was demonstrated by completing the synthesis of the oligosaccharides in three different sizes, including short (CS-E 7-mer), intermediate (CS-E 13-mer), and long (CS-E 19-mer) chains. Although short CS-E oligosaccharides have been synthesized by chemical methods,6,23 the synthesis of longer than 12-mers had not yet been accomplished. It is noteworthy that the size of the synthesized long CS-E 19-mer represents 40–70% of full-length CS chains of the bikunan proteoglycan, a CS proteoglycan isolated from human urine.7 The ability to synthesize long, homogeneous CS oligosaccharides is critically important for their biological evaluation, as short CS oligosaccharides do not fully exhibit the functions of full-length CS polysaccharides. We also demonstrated the scalability of our process by synthesizing hundreds of milligrams of these target CS oligosaccharides to complete the animal studies. With a larger fermentation facility and liquid handling equipment, there are no clear obstacles to the synthesis of gram to hundreds of gram quantities of CS-E oligosaccharides required for extensive preclinical and clinical studies.
We also demonstrate that CS-E 19-mer protects against organ damage in endotoxemia mice by complexing with histones and neutralizing their cytotoxicity. Since histones are positively charged proteins, it is likely that negatively charged carbohydrates neutralize the cytotoxic effect of histones. Heparin is currently used as an anticoagulant in clinics and is a negatively charged polysaccharide that consists of distinct disaccharide repeating units from CS-E polysaccharide.37 Heparin also binds to histones and has been tested in sepsis patients,38 but the subsequent clinical trial failed to show clear benefits.39 Despite these disappointing outcomes, efforts to develop sulfated carbohydrate-based molecules to treat sepsis are still ongoing.22 The use of different sizes of heparin fragments to attenuate histone-induced lung injury was also published.40 It is also important to note that heparin and CS-E are distinctly different molecules. Whether heparin and CS-E follow the same mechanism to display the protection effect under systemic conditions is not fully understood.
The availability of structurally homogeneous CS oligosaccharides facilitates the investigation of the CS structure and biological function relationship. Whether a highly purified CS neutralizes histone’s cytotoxic effect is somewhat controversial,41,42 possibly attributed to different structures of CS polysaccharide from different sources. CS-containing extract isolated from skate cartilage reportedly reduces liver damage induced by lipopolysaccharide; however, the molecular target for the CS-containing extract was not known.43 Using the pure CS oligosaccharides, we demonstrate the protective effect against histone- or lipopolysaccharide-induced organ damage with certainty. In addition to histones, other proinflammatory proteins, like high mobility group box 1 (HMGB1), play important roles in the pathology of sepsis.44 It remains to be investigated whether the protective effects of CS-E 19-mer in the endotoxemia model can also be attributed to the interaction with HMGB1.
CS-E 19-mer has certain advantages over heparin and heparan sulfate, a less-sulfated form of heparin, for the treatment of systemic inflammation in sepsis patients. First, CS-E 19-mer does not contain anticoagulant activity as demonstrated by the absence of anti-FXa and anti-FIIa activities. The potential bleeding risk is a serious concern when heparin is given to septic patients.45 Nonanticoagulant CS-E 19-mer would eliminate this bleeding concern. Second, HIT is an immunological disorder resulting from the production of antibodies against a complex of heparin with platelet factor 4 (PF4)46 and is the major nonbleeding side effect associated with the use of heparin resulting in five to six deaths every day in the United States.47 The interaction of these antibodies with PF4-heparin/glycosaminoglycan complexes on the platelet surface48 activates platelet aggregation, triggering systemic thrombin generation that ultimately results in HIT-related thrombocytopenia and thrombosis. Recent studies have even suggested that septic patients may have higher levels of anti-PF4/heparin antibodies.49,50 While this association is preliminary, it poses the question of whether the use of any glycosaminoglycan, including CS-E 19-mer, is safe in this setting. Results presented here demonstrate clearly that HIT antibodies do not react with CS-E 19-mer-PF4 complexes reducing/eliminating the risk for HIT. Third, heparan sulfate and heparin are cleaved by heparanase and short oligosaccharides are released into circulation during sepsis. These short oligosaccharides may contribute to cognitive impairment in sepsis patients by deactivating hippocampal long-term potentiation.51,52CS-E 19-mer should not be susceptible to heparanase cleavage and is unlikely to enter the hippocampus causing cognitive impairment in sepsis patients. The access of structurally defined CS-E oligosaccharides synthesized by an enzymatic approach offers the essential reagents to further in-depth investigation of using CS oligosaccharides to treat diseases associated with systemic inflammation.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.0c00712.
Detailed experimental procedures, characterization data of all compounds, NMR and MS spectra, HPLC chromatograms, and additional experimental data (PDF)
Author Contributions
J.Li designed the studies, synthesized CS oligosaccharides, and tested their activities in both in vitro and in vivo models. E.M.S., J.L., and R.P. conducted the animal studies. G.S., P.H., S.B., and R.J.L. analyzed the structures of oligosaccharides by NMR. F.Z. performed the SPR analysis for the binding of oligosaccharides to histones. Y.X. prepared the biotinylated oligosaccharides. K.X. and R.J.L. performed the molecular weight analysis of CS oligosaccharides by high-resolution mass spectrometry. S.P. and A.P. determined the binding of CS oligosaccharides to HIT antibodies isolated from patients. J.L. and R.P. designed the studies. J. Liu, J. Li, R.P., R.J.L., and A.P. wrote the manuscript. All authors participated in the process of finalizing the manuscript.
The authors declare the following competing financial interest(s): JL and YX are founders of Glycan Therapeutics. Jine Li, GS, EMS, RP and JL are inventors for a US patent (PCT/US2018/040774). AP has equity ownership in Retham Technologies and is on the advisory board of Veralox Therapeutics. Other authors declare no competing financial interests.
Supplementary Material
References
- Miyata S.; Komatsu Y.; Yoshimura Y.; Taya C.; Kitagawa H. Persistent cortical plasticity by upregulation of chondroitin 6-sulfation. Nat. Neurosci. 2012, 15, 414–422. 10.1038/nn.3023. [DOI] [PubMed] [Google Scholar]
- Kwok J. C. F.; Warren P.; Fawcett J. W. Chondroitin sulfate: A key molecule in the brain matrix. Int. J. Biochem. Cell Biol. 2012, 44, 582–586. 10.1016/j.biocel.2012.01.004. [DOI] [PubMed] [Google Scholar]
- Kastana P.; Choleva E.; Poimenidi E.; Karamanos N.; Sugahara K.; Papadimitriou E. Insight into the role of chondroitin sulfate E in angiogenesis. FEBS J. 2019, 286, 2921–2936. 10.1111/febs.14830. [DOI] [PubMed] [Google Scholar]
- Vallen M. J. E.; Schmidt S.; Oosterhof A.; Bulten J.; Massuger L. F. A. G.; van Kuppevelt T. H. Primary Ovarian Carcinomas and Abdominal Metastasis Contain 4,6-Disulfated Chondroitin Sulfate Rich Regions, Which Provide Adhesive Properties to Tumour Cells. PLoS One 2014, 9, e111806. 10.1371/journal.pone.0111806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis C. M.; Klüppel M. Chondroitin Sulfate-E Is a Negative Regulator of a Pro-Tumorigenic Wnt/Beta-Catenin-Collagen 1 Axis in Breast Cancer Cells. PLoS One 2014, 9, e103966. 10.1371/journal.pone.0103966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gama C.; Tully S. E.; Sotogaku N.; Clark P. M.; Rawat M.; Vaidehi N.; Goddard W. A.; Nishi A.; Hsieh-Wilson L. C. Sulfation patterns of glycosaminoglycans encode molecular recognition and activity. Nat. Chem. Biol. 2006, 2, 467–473. 10.1038/nchembio810. [DOI] [PubMed] [Google Scholar]
- Ly M.; Leach F. E. III; Laremore T. N.; Toida T.; Amster I. J.; Linhardt R. J. The proteoglycan bikunin has sequence. Nat. Chem. Biol. 2011, 7, 827–833. 10.1038/nchembio.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaput C.; Zychlinsky A. Sepsis: the dark side of histones. Nat. Med. 2009, 15, 1245–1246. 10.1038/nm1109-1245. [DOI] [PubMed] [Google Scholar]
- Wheeler A. P.; Bernard G. R. Treating patients with server sepsis. N. Engl. J. Med. 1999, 340, 207–212. 10.1056/NEJM199901213400307. [DOI] [PubMed] [Google Scholar]
- Dickson K.; Lehmann C. Inflammatory response to different toxins in experiment sepsis models. Int. J. Mol. Sci. 2019, 20, 4341. 10.3390/ijms20184341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J.; Zhang X.; Pelayo R.; Monestier M.; Ammollo C. T.; Semeraro F.; Taylor F. B.; Esmon N. L.; Lupu F.; Esmon C. T. Extracellular histones are major mediators of death in sepsis. Nat. Med. 2009, 15, 1318–1321. 10.1038/nm.2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkmann V.; Reichard U.; Goosmann C.; Fauler B.; Uhlemann Y.; Weiss D. S.; Weinrauch Y.; Zychlinsky A. Neutrophil Extracellular Traps Kill Bacteria. Science 2004, 303, 1532–1535. 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- von Köckritz-Blickwede M.; Goldmann O.; Thulin P.; Heinemann K.; Norrby-Teglund A.; Rohde M.; Medina E. Phagocytosis-independentantimicrobialactivityofmastcellsbymeansof extracellulartrapformation. Blood 2008, 111, 3070–3080. 10.1182/blood-2007-07-104018. [DOI] [PubMed] [Google Scholar]
- Kang R.; Lotze M. T.; Zeh H. J.; Billiar T. R.; Tang D. Cell Death and DAMPs in Acute Pancreatitis. Mol. Med. 2014, 20, 466–477. 10.2119/molmed.2014.00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fattahi F.; Grailer J. J.; Lu H.; Dick R. S.; Parlett M.; Zetoune F. S.; Nuñez G.; Ward P. A. Selective Biological Responses of Phagocytes and Lungs to Purified Histones. J. Innate Immun. 2017, 9, 300–317. 10.1159/000452951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H.; Chen H.-W.; Evankovich J.; Yan W.; Rosborough B. R.; Nace G. W.; Ding Q.; Loughran P.; Beer-Stolz D.; Billiar T. R.; Esmon C. T.; Tsung A. Histones Activate the NLRP3 Inflammasome in Kupffer Cells during Sterile Inflammatory Liver Injury. J. Immunol. 2013, 191, 2665–2679. 10.4049/jimmunol.1202733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semeraro F.; Ammollo C. T.; Morrissey J. H.; Dale G. L.; Friese P.; Esmon N. L.; Esmon C. T. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood 2011, 118, 1952–1961. 10.1182/blood-2011-03-343061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meegan J. E.; Yang X.; Beard R. S.; Jannaway M.; Chatterjee V.; Taylor-Clark T. E.; Yuan S. Y. Citrullinated histone 3 causes endothelial barrier dysfunction. Biochem. Biophys. Res. Commun. 2018, 503, 1498–1502. 10.1016/j.bbrc.2018.07.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alhamdi Y.; Abrams S. T.; Cheng Z.; Jing S.; Su D.; Liu Z.; Lane S.; Welters I.; Wang G.; Toh C.-H. Circulating Histones Are Major Mediators of Cardiac Injury in Patients With Sepsis. Crit. Care Med. 2015, 43, 2094–2103. 10.1097/CCM.0000000000001162. [DOI] [PubMed] [Google Scholar]
- Tiwari R. L.; Singh V.; Singh A.; Barthwal M. K. IL-1R–Associated Kinase-1 Mediates Protein Kinase Cδ-Induced IL-1β Production in Monocytes. J. Immunol. 2011, 187, 2626–2631. 10.4049/jimmunol.1002526. [DOI] [PubMed] [Google Scholar]
- Kawai C.; Kotani H.; Miyao M.; Ishida T.; Jemail L.; Abiru H.; Tamaki K. Circulating Extracellular Histones Are Clinically Relevant Mediators of Multiple Organ Injury. Am. J. Pathol. 2016, 186, 829–843. 10.1016/j.ajpath.2015.11.025. [DOI] [PubMed] [Google Scholar]
- Wildhagen K. C.; García de Frutos P.; Reutelingsperger C. P.; Schrijver R.; Aresté C.; Ortega-Gómez A.; Deckers N. M.; Hemker H. C.; Soehnlein O.; Nicolaes G. A. Nonanticoagulant heparin prevents histone-mediated cytotoxicity in vitro and improves survival in sepsis. Blood 2014, 123, 1098–1101. 10.1182/blood-2013-07-514984. [DOI] [PubMed] [Google Scholar]
- Tamura J.-I.; Nakada Y.; Taniguchi K.; Yamane M. Synthesis of chondroitin sulfate E octasaccharide in a repeating region involving an acetamide auxiliary. Carbohydr. Res. 2008, 343, 39–47. 10.1016/j.carres.2007.09.009. [DOI] [PubMed] [Google Scholar]
- Tully S. E.; Mabon R.; Gama C. I.; Tsai S. M.; Liu X.; Hsieh-Wilson L. C. A chondroitin sulfate small molecule that stimulates neuronal growth. J. Am. Chem. Soc. 2004, 126, 7736–7737. 10.1021/ja0484045. [DOI] [PubMed] [Google Scholar]
- Li J.; Su W.; Liu J. Enzymatic synthesis of homogeneous chondroitin sulfate oligosaccharides. Angew. Chem., Int. Ed. 2017, 56, 11784–11787. 10.1002/anie.201705638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habuchi O.; Moroi R.; Ohtake S. Enzymatic synthesis of chondroitin sulfate E by N-acetylgalactosamine 4-sulfate 6-O-sulfotransferase purified from squid cartilage. Anal. Biochem. 2002, 310, 129–136. 10.1016/S0003-2697(02)00277-4. [DOI] [PubMed] [Google Scholar]
- Sugiura N.; Setoyama y.; Chiba M.; Kimata K.; Watanabe H. Baculovirus envelope protein ODV-E66 is a novel chondroitinase with distinct substrate specificity. J. Biol. Chem. 2011, 286, 29026–29034. 10.1074/jbc.M111.251157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito Y.; Habuchi O. Purification and characterization of N-acetylgalactosamine 4-sulfate 6-O-sulfotransferase from the squid cartilage. J. Biol. Chem. 2000, 275, 34728–34736. 10.1074/jbc.M909633199. [DOI] [PubMed] [Google Scholar]
- Abrams S. T.; Zhang N.; Dart C.; Wang S. S.; Thachil J.; Guan Y.; Wang G.; Toh C.-H. Human CRP Defends against the Toxicity of Circulating Histones. J. Immunol. 2013, 191, 2495–2502. 10.4049/jimmunol.1203181. [DOI] [PubMed] [Google Scholar]
- Xu J.; Zhang X.; Monestier M.; Esmon N. L.; Esmon C. T. Extracellular histones are mediators of death through TLR2 and TLR4 in mouse fatal liver injury. J. Immunol. 2011, 187, 2626–2631. 10.4049/jimmunol.1003930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semeraro N.; Ammollo C. T.; Semeraro F.; Colucci M. Coagulopathy of Acute Sepsis. Semin. Thromb. Hemostasis 2015, 41, 650–658. 10.1055/s-0035-1556730. [DOI] [PubMed] [Google Scholar]
- Hayashida K.; Parks W. C.; Park P. W. Syndecan-1 shedding facilitates the resolution of neutrophilic inflammation by removing sequestered CXC chemokines. Blood 2009, 114, 3033–3043. 10.1182/blood-2009-02-204966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakanpaa L.; Kiss E. A.; Jacquemet G.; Miinalainen I.; Lerche M.; Guzmán C.; Mervaala E.; Eklund L.; Ivaska J.; Saharinen P. Targeting β1-integrin inhibits vascular leakage in endotoxemia. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E6467–E6476. 10.1073/pnas.1722317115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand K.; Fowler B. J.; Edgington T. S.; Mackman N. Tissue factor mRNA in THP-1 monocytic cells is regulated at both transcriptional and posttranscriptional levels in response to lipopolysaccharide. Mol. Cell. Biol. 1991, 11, 4732–4738. 10.1128/MCB.11.9.4732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guha M.; O’Connell M. A.; Pawlinski R.; Hollis A.; McGovern P.; Yan S.-F.; Stern D. M.; Mackman N. Lipopolysaccharide activation of the MEK-ERK1/2 pathway in human monocytic cells mediates tissue factor and tumor necrosis factor a expreassion by inducing Elk-1 phosphorylation and Egr-1 expression. Blood 2001, 98, 1429–1439. 10.1182/blood.V98.5.1429. [DOI] [PubMed] [Google Scholar]
- Kawamoto T.; Ii M.; Kitazaki T.; Iizawa Y.; Kimura H. TAK-242 selectively suppresss toll-like receptor 4-signaling mediated by the intracellular domain. Eur. J. Pharmacol. 2008, 584, 40–48. 10.1016/j.ejphar.2008.01.026. [DOI] [PubMed] [Google Scholar]
- Liu J.; Linhardt R. J. Chemoenzymatic synthesis of heparan sulfate and heparin. Nat. Prod. Rep. 2014, 31, 1676–1685. 10.1039/C4NP00076E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson B. L.; Geerts W. H.; Lensing A. W. Low-dose heparin for severe sepsis. N. Engl. J. Med. 2002, 347, 1036–1037. 10.1056/NEJM200209263471316. [DOI] [PubMed] [Google Scholar]
- Jaimes F.; De La Rosa G.; Morales C.; Fortich F.; Arango C.; Aguirre D.; Muñoz A. Unfractioned heparin for treatment of sepsis: A randomized clinical trial (The HETRASE Study). Crit. Care Med. 2009, 37, 1185–1196. 10.1097/CCM.0b013e31819c06bc. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Haeger S. M.; Yang Y.; Dailey K. L.; Ford J. A.; Schmidt E. P. Circulating heparan sulfate fragments attenuate histone-induced lung injury independently of histone binding. Shock 2017, 48, 666–673. 10.1097/SHK.0000000000000907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaaban H.; Keshari R. S.; Silasi-Mansat R.; Popescu N. I.; Mehta-D’Souza P.; Lim Y.-P.; Lupu F. Inter-a inhibitor protein and its associated glycosaminoglycan protect against histone-induced injury. Blood 2015, 125, 2286–2296. 10.1182/blood-2014-06-582759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagano F.; Mizuno T.; Mizumoto S.; Yoshioka K.; Takahashi K.; Tsuboi N.; Maruyama S.; Yamada S.; Nagamatsu T. Chondroitin sulfate protects vascular endothelial cells from toxicities of extracellular histones. Eur. J. Pharmacol. 2018, 826, 48–55. 10.1016/j.ejphar.2018.02.043. [DOI] [PubMed] [Google Scholar]
- Song Y. O.; Kim M.; Woo M.; Baek J. M.; Kang K. H.; Kim S. H.; Roh S. S.; Park C. H.; Jeong K. S.; Noh J. S. Chondroitin sulfate-rich extract of skate cartilage attenuates lipopolysaccharide-induced liver damage in mice. Mar. Drugs 2017, 15, 178. 10.3390/md15060178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson U.; Yang H.; Harris H. E. Extrasceullar HMGB1 as a therapeutic target in flammatory diseases. Expert Opin. Ther. Targets 2018, 22, 263–277. 10.1080/14728222.2018.1439924. [DOI] [PubMed] [Google Scholar]
- van der Poll T.; Opal S. M. Should All Septic Patients Be Given Systemic Anticoagulation? No. Intensive Care Med. 2017, 43, 455–457. 10.1007/s00134-016-4607-x. [DOI] [PubMed] [Google Scholar]
- Greinacher A.; Fürll B.; Selleng S. Heparin-induced thrombocytopenia. Methods Mol. Biol. 2013, 992, 301–318. 10.1007/978-1-62703-339-8_23. [DOI] [PubMed] [Google Scholar]
- Dhakal B.; Kreuziger L. B.; Rein L.; Kleman A.; Fraser R.; Aster R. H.; Hari P.; Padmanabhan A. Disease burden, complication rates, and health-care costs of heparin-induced thrombocytopenia in the USA: a population-based study. Lancet Haematol. 2018, 5, e220–e231. 10.1016/S2352-3026(18)30046-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanabhan A. Why ″R″ HIT patients predisposed to thrombosis?. Blood 2015, 125, 2319–2320. 10.1182/blood-2015-02-629782. [DOI] [PubMed] [Google Scholar]
- Maharaj S.; Chang S. Anti-PF4/heparin antibodies are increased in hospitalized patients with bacterial sepsis. Thromb. Res. 2018, 171, 111–113. 10.1016/j.thromres.2018.09.060. [DOI] [PubMed] [Google Scholar]
- Mattioli A. V.; Manenti A.; Farinetti A. Heparin-induced thrombocytopenia and sepsis. Thromb. Res. 2018, 172, 119. 10.1016/j.thromres.2018.10.029. [DOI] [PubMed] [Google Scholar]
- Hippensteel J. A.; Anderson B. J.; Orfila J. E.; McMurtry S. A.; Dietz R. M.; Su G.; Ford J. A.; Oshima K.; Yang Y.; Zhang F.; Han X.; Yu Y.; Liu J.; Linhardt R. J.; Meyer N. L.; Herson P. S.; Schmidt E. P. Circulating heparan sulfate fragments mediate septic cognitive dysfunction. J. Clin. Invest. 2019, 129, 1779–1784. 10.1172/JCI124485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Han X.; Xia K.; Xu Y.; Yang Y.; Oshima K.; Haeger S. M.; Perez M. J.; McMurtry S. A.; Hippensteel J. A.; Ford J. A.; Herson P. S.; Liu J.; Schmidt E. P.; Linhardt R. J. Circulating heparin oligosaccharides rapidly target the hippocampus in sepsis, potentially impacting cognitive functions. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 9208–9213. 10.1073/pnas.1902227116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair R. R.; Mazza D.; Brambilla F.; Gorzanelli A.; Agresti A.; Bianchi M. E. LPS-Challenged Macrophages Release Microvesicles Coated With Histones. Front. Immunol. 2018, 9, 1463. 10.3389/fimmu.2018.01463. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.