

Summary
Hepatitis delta virus (HDV), as a defective sub‐virus that co‐infects with hepatitis B virus, imposes an emerging global health burden. However, genetic characteristics and molecular classification of HDV remain under investigated. In this study, we have systematically retrieved and analysed a large set of HDV full‐length genome sequences and identified novel recombinants. Based on phylogenetic and genetic analyses, we have established an updated classification system for HDV when recombinants were excluded. Furthermore, we have mapped the global distribution of different genotypes and subtypes. Finally, we have compiled a complete set of reference genomes for each subtype and proposed criteria for future identification of novel genotypes and subtypes. Of note, the global distribution map indicates that currently available HDV genetic data remain limited, and thus our proposed classification will likely evolve as future epidemiological data will accumulate. These results will facilitate the future research on the diagnosis, screening, epidemiology, evolution, prevention and clinical management of HDV infection.
Keywords: cRNA, genetic recombination, genotype, hepatitis delta virus, viral genome, viral RNA
Abbreviations
- bp
base pair
- cRNA
complementary RNA
- GEP
gap extension penalty
- GOP
gap‐opening penalty
- GTR
General Time Reversible
- HBsAg
hepatitis B surface antigen
- HBV
hepatitis B virus
- HCC
hepatocellular carcinoma
- HDAg
hepatitis delta antigen
- HDV
hepatitis delta virus/hepatitis D virus
- JTT
Jones‐Taylor‐Thornton
- ML
maximum‐likelihood
- mRNA
messenger RNA
- NJ
neighbour‐joining
- nts
nucleotides
- ORF
open reading frame
- ssRNA
single‐stranded RNA
- WHV
woodchuck hepatitis B virus
1. INTRODUCTION
Hepatitis delta virus (also known as hepatitis D virus, HDV) is a defective sub‐virus that requires hepatitis B virus (HBV) surface antigen (HBsAg) for virion assembly. The genome of HDV is a circular negative single‐stranded RNA (ssRNA) composed of approximately 1700 nucleotides (nt).1 It is considered the smallest RNA genome in all known animal viruses. There are three forms of HDV RNA without any DNA intermediate during viral replication, including circular genomic RNA (negative), circular complementary antigenomic RNA (cRNA, positive), and a short linear polyadenylated antigenomic RNA (positive).2 This linear form is the messenger RNA (mRNA) template encoding only one functional open reading frame (ORF) for the translation of the hepatitis delta antigen (HDAg).3 Though HDV RNA is single‐stranded, it is capable of undergoing self‐cleavage and ligation to generate circular RNA. Due to the high GC content of the nucleotide sequence, HDV RNA can also fold as an unbranched, double‐stranded, rod‐like structure with over 70% intra‐molecular base‐pairing.4
Since the identification of HDV in the 1970s, this peculiar pathogen has been neglected over the past decades,5, 6 and routine diagnosis is rare in clinical practice.7 However, co‐infection of HDV with HBV causes the most severe form of acute and chronic viral hepatitis in humans.1 It has been estimated that almost 5% of HBV infected patients have HDV co‐infection and up to 80% of these co‐infected patients can further progress to cirrhosis.8, 9 This long‐term co‐infection is associated with more rapid and severe progression to cirrhosis or hepatocellular carcinoma (HCC) than HBV infection alone.7 Worse yet, clustered outbreaks of HDV superinfection periodically occur across the world, imposing an emerging global health burden.1, 7
As a single‐stranded RNA, HDV is expected to evolve rapidly. Previous studies have indicated that HDV genotype plays an important role in pathogenesis and the efficiency of RNA editing can affect its natural history.10 However, the genetic features of HDV remain poorly characterized, and the current molecular classification systems are inconsistent.11, 12 Given that the epidemiology, virus evolution, infection course, clinical outcome and treatment response are likely associated with the different genotypes or subtypes, we aim to clarify the molecular classification of HDV and to propose standardized reference genomes.
2. MATERIALS AND METHODS
2.1. Sequence download
All HDV full‐length genome sequences available before 1 December 2017 were downloaded from NCBI Nucleotide Database. “Hepatitis delta virus” [Organism] NOT “patent” [title] was used as the search term, and the search results were filtered by sequence length from 1500 to 2000 nt. 357 full‐length sequences were retrieved from the search records but the final dataset comprised 345 sequences after removing duplication. Information on accession number‐strain/isolate‐collection date‐country/geographic origin, if available, was simultaneously retrieved from the database.
2.2. Sequence alignment and recombinants identification
The original dataset was aligned by ClustalW (1.6) listed in the MEGA (version 7.0.26) using a gap‐opening penalty (GOP) of 15 and a gap extension penalty (GEP) of 6.66.13, 14, 15 RNA sequences were standardized to antigenomic cRNA form reading from normal initial site. The standardized dataset was realigned with woodchuck hepatitis B virus (WHV, accession number J04514, WHV8 strain)16 by ClustalW and went through minimum manual corrections. The well‐aligned dataset was used to construct the preliminary phylogenetic tree using different algorithm models listed in MEGA. Strains emerged as outlier branches or clustered as peculiar branches located at the crotch of different trees indicating the presence of recombinant sequences. These strains presenting the conflicting signals were regarded as potential recombinants and require further recombination identification. Recombination events were confirmed by Bootscanning analysis performed in Simplot v3.5.1 programs using Kimura 2‐parameter with a 160 base pair (bp) window, a 20 bp step increment, and 1000 bootstrap replicates. The recombination criterion is breakpoint high than >80% of the permuted tree.
2.3. Phylogenetic and genetic analyses
The new dataset without potential recombinants was realigned. Model Selection (ML) implemented in MEGA was used to find the best DNA/protein model. The best DNA and protein models were the General Time Reversible (GTR) model and the Jones‐Taylor‐Thornton (JTT) model, respectively. The maximum‐likelihood (ML) tree was reconstructed using the best model with 5 rate categories (G) and invariable sites (I). The neighbour‐joining (NJ) tree was reconstructed using p‐distance model with Bootstrap method test. Branch support was calculated using 1000 replicates, and only bootstrap values >70% were showed. Trees were rooted with WHV8 strain.
Hepatitis delta virus genotype distribution map was modified according to the free map templates (http://d-maps.com/carte.php?num_car=13180&lang=en) using Inkscape 0.92.2 software. Nucleotide similarities were calculated by the program Sequence Distances implemented in MegAlign software (Lasergene software; DNASTAR), and genetic distances were calculated by MEGA with the Kimura 2‐parameter/gamma model and 1000 bootstrap replicates.
3. RESULTS
3.1. Standardization of HDV full‐length genome sequences and identification of potential recombinants
Three hundred and fifty‐seven original full‐length sequences were retrieved, but 345 valid sequences were finally included after removing duplications. Our preliminary alignment of original dataset found that only a set of strains isolated from Brazil (accession number from KF786305 to KF786352) were cRNA form,17 and all the others were genomic RNA form. Furthermore, several strains from Turkey (accession number from HQ005364 to HQ005372) showed abnormal initial reading site, starting and ending at site 227.18 For further phylogenetic and genetic analysis, all original genomic RNA sequences were transformed to cRNA form and these Turkey strains were standardized to read from 1 to 1678.
Through preliminary phylogenetic analysis, 53 strains presenting conflicting signals were screened out for further Bootscanning analysis. 33 out of these 53 strains were finally confirmed that substantial recombination events occurred. Among these, two strains (AB118845 and KF660598) have been previously reported as recombinant,19, 20 and the other 31 strains were newly identified recombinants in this study (Figures S1 and S2). The recombination genotype component and corresponding breakpoint positions were summarized in Table S1.
3.2. Phylogenetic analysis and updated molecular classification
After removing the recombinant sequences, the phylogenetic trees were constructed using two different models, ML and NJ. The tree topologies obtained with the two models were similar. The ML tree was shown in Figure 1 and the NJ tree was shown in Figure S3. In our rooted trees, 312 full‐length strains were clustered as three big clades and further clearly grouped as eight small solid clades with 100% bootstrap value support. These eight clades were corresponding to eight genotypes, and in line with the classification previously descripted.12 Notably, genotype 2 and genotypes 4‐8 were consistently clustered as one big clade, and genotype 3 was located more close to the root of the trees.
Figure 1.
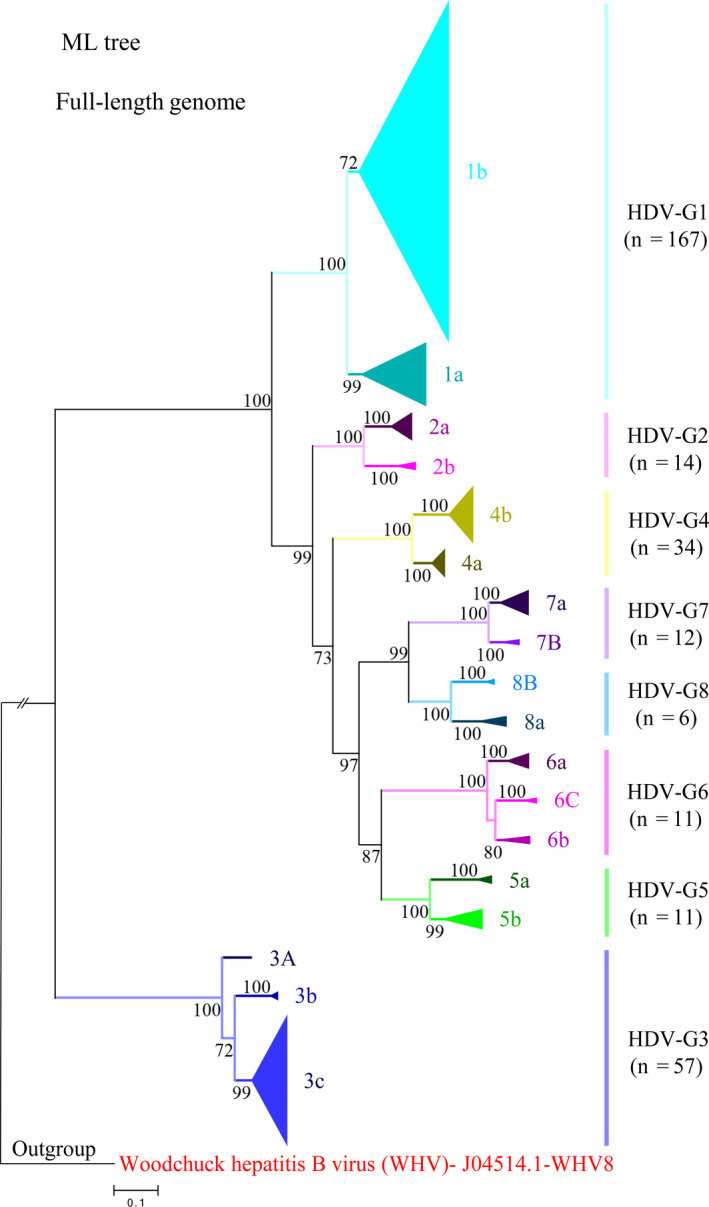
Phylogenetic analysis of 312 hepatitis delta virus (HDV) full‐length antigenomic RNA (cRNA) sequences. The maximum‐likelihood (ML) tree of eight HDV genotypes with subtypes showing the overall classification framework. All original HDV full‐length genomic RNA sequences were transformed to cRNA sequences and standardized to read from 1 to 1678. The tree was reconstructed using the best DNA model, General Time Reversible (GTR) model of evolution with 5 rate categories (G) and invariable sites (I). Potential recombinants were excluded from the tree. Branch support was calculated using 1000 replications, and only bootstrap values >70% are shown. The tree was rooted with woodchuck hepatitis B virus (WHV)
As shown in the trees, eight genotypes were respectively further grouped into two (a, b) or three (a‐c) subtypes (Figures 1 and S3). Genotype 3 and 6 were segregated into three subtypes, and genotype 1, 2, 4, 5, 7 and 8 were only grouped into two subtypes. Referring to the nomenclature used for HCV,21 capital in the nomenclature of HDV subtypes was used to indicate the unconfirmed status due to the available sequences less than three. Furthermore, we performed the same phylogenetic analysis using subgenomic HDAg coding gene fragment (600 nt) and the corresponding amino acid sequences. However, though some genotypes could be classified faithfully by using subgenomic fragment or HDAg amino acid, the classification of many other genotypes and subtypes were variable (Figure S4A,B).
To further support our classification system, we compared the nucleotide similarities and genetic distances between different groups (Figures 2 and S5). Comparative genetic analyses showed that HDV genotype 3 was distantly separated from other genotypes. The intergenotypic nucleotide similarities and genetic distances between genotype 3 and other genotypes were 61.79%‐63.97% and 0.334‐0.355, respectively, outside the ranges of intergenotypic nucleotide similarities and genetic distances between other genotypes (69.64%‐79.96%, 0.207‐0.285). In addition, HDV genotype 1 showed higher divergence than other genotypes. The nucleotide similarity of intersubtype between subtype 1a and 1b was 82.58%, which was lower than the intersubtype nucleotide similarity range within other genotypes (85.82%‐90.49%). The genetic distance of intersubtype between subtype 1a and 1b was 0.162, higher than the range within other genotypes (0.094‐0.139).
Figure 2.
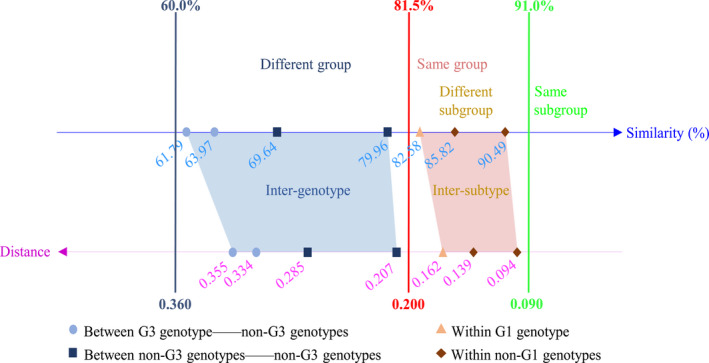
Genetic analysis of 312 hepatitis delta virus (HDV) full‐length antigenomic RNA (cRNA) sequences. Cartoon representation of the identification criteria of HDV novel genotype or subtype. The comparisons of mean intergenotypic and intersubtypic nucleotide similarity and genetic distance were based on 312 HDV full‐length cRNA sequences. Axes show the percentage similarity and genetic distance, respectively. The blue dash line indicates the lowest range of intergenotypic nucleotide similarity and genetic distance, the red dash line indicates the cut‐off range between genotype and subtype, and the green dash line indicates the highest range of intersubtypic nucleotide similarity and genetic distance. The detailed results of calculation were shown in Figure S5A,B
3.3. Diversity of HDV genotype distribution
The global distribution of HDV genotype varies geographically (Figure 3). Genotype 1 is common globally; genotype 2 and 4 are mainly in Asia; genotype 3 is in South America; whereas genotype 5‐8 are in Africa. It is noteworthy that several genotypes or subtypes concurrently prevail in most endemic regions, such as Asia and Africa. Particularly, HDV strains circulating in Africa exhibit extremely high genotypic diversity.
Figure 3.
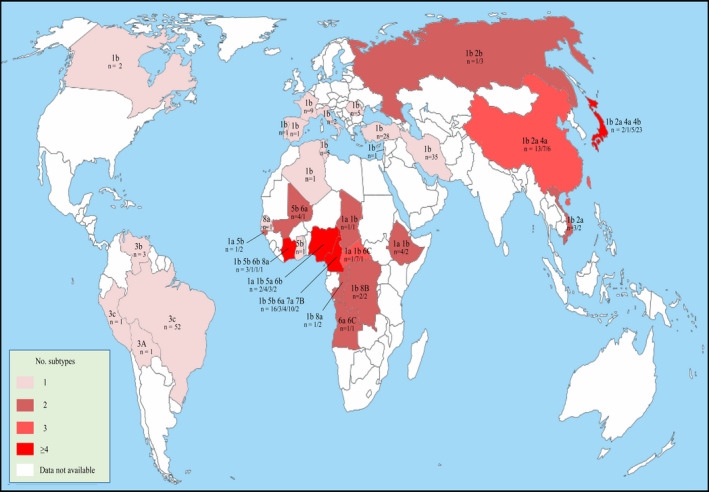
Worldwide distribution of hepatitis delta virus (HDV) genotypes and subtypes. Data were based on HDV full‐length genomic sequences. This map was modified according to the free map templates (http://d-maps.com/carte.php?num_car=13180&lang=en) using Inkscape 0.92.2 software
3.4. Proposed criteria for identifying novel HDV genotype or subtype
Based on our phylogenetic and genetic analysis, we hereby proposed the identification criteria for novel HDV genotype and subtype. Firstly, standardized HDV full‐length antigenomic sequence is recommended to be used. Secondly, potential recombination should be excluded and phylogenetically cluster together as a solid group or subgroup is essentially required. Finally, phylogenetic grouping should be supported by nucleotide similarity and genetic distance of intergenotype or intersubtype. The demarcation of a novel genotype is the intergenotypic nucleotide similarity at the range of 60.0%‐81.5% and the corresponding intergenotypic genetic distance at the range of 0.200‐0.360. For novel subtype, the intersubtypic nucleotide similarity and the corresponding intersubtypic genetic distance are at the range of 81.5%‐91.0% and 0.090‐0.200, respectively (Figure 2).
3.5. Proposed reference genomes for HDV subtypes
In order to facilitate the communication between researchers and help clarify the epidemiology of HDV, we proposed a standard reference set of full‐length genome sequences. These reference sequences were selected according to the following detailed criteria. Firstly, only full‐length genome sequences were considered, and subgenomic fragments or potential full‐length recombinants were eliminated. Secondly, to ensure the clarity of the strain origin and to minimize disruption of previous prototype notification, priority was given to the sequences with full information, but previous noted prototype strains12 were also taken into consideration at the same time. Thirdly, if there was no sequence with complete information, priority was given to the prototype strain(s), if prototype strain(s) was/were noted. Fourthly, if no sequence was available with complete information or noted as prototype strain for a subtype, or more than one sequences were available for a subtype, priority was firstly given to the sequence with earlier collection date, then to the sequence earlier submitted to GenBank. If the collection or submission dates were identical, the sequence with lowest alphabetic/numeric accession number was proposed.
Finally, 21 full‐length genome sequences were proposed as the references for eighteen HDV subtypes (Table 1). We proposed one reference sequence for each subtype of genotypes 2‐8. But 5 reference sequences were proposed for two subtypes of genotype 1. This is because HDV genotype 1 showed more introgenotypic divergence than other genotypes. As for 1a, a strain of ETH2170 (accession no. KY463677) collected in 2013 was noted as the prototype strain of genotype 1.12 However, another 1a strain named 36011‐NIE1150 (accession no. JX888100) was collected in 2006, which was earlier than the prototype strain. Thus, both strains were proposed as the reference sequences of 1a. For 1b, 141 full‐length genome sequences were available to date. Because it was previously further classified as three subtypes,12 thus three sequences were proposed to these three previously assigned subtypes. Notably, all the proposed reference sequences were RNA form and only a few of them have collection date available.
Table 1.
Reference sequences for hepatitis delta virus subtypes
| Genotype | Subtypea | Accession no. | Strain/Isolateb | Datec | Countryd |
|---|---|---|---|---|---|
| 1 | 1a | JX888100 | 36011‐NIE1150 | 2006 | Nigeria |
| 1a | KY463677 | ETH2170 | 2013 | Ethiopia | |
| 1b | JX888098 | 24187‐NIE | 2004 | Nigeria | |
| 1b | KJ744242 | D34 | 2008 | Iran | |
| 1b | KJ744255 | D66 | 2003 | Iran | |
| 2 | 2a | X60193 | 7/18/83 | 2005 | Japan |
| 2b | AJ309879 | Yakut26 | 2016 | Russia | |
| 3 | 3A | LT604954 | dFr6727 | 2017 | Bolivia |
| 3b | AB037947 | VnzD8375 | 2001 | Venezuela | |
| 3c | KC590319 | Brazil‐1 | 2011 | Brazil | |
| 4 | 4a | AF018077 | TW‐2b (Taiwan isolate) | 1998 | China |
| 4b | AB118818 | Miyako (JA‐M2) | 2006 | Japan | |
| 5 | 5a | JX888103 | 36102‐NIE875 | 2006 | Nigeria |
| 5b | AM183331 | dFr2005 | 2007 | GW | |
| 6 | 6a | AJ584847 | dFr48 | 2005 | Cameroon |
| 6b | JX888102 | 36036‐NIE464 | 2006 | Nigeria | |
| 6C | AM183332 | dFr2139 | 2007 | CAR | |
| 7 | 7a | AJ584844 | dFr‐45 | 2005 | Cameroon |
| 7B | AM183333 | dFr2158 | 2017 | Cameroon | |
| 8 | 8a | AJ584849 | dFr644 | 2005 | CGO |
| 8B | LT594488 | dFr7707 | 2017 | COD |
Capital standing for the unconfirmed status due to the available sequences less than 3.
Previous designated prototype strains are indicated in bold.
Sequences with clear collection date are highlighted in bold, sequences with collection date missed and replaced by GenBank release date are shown in italic.
GW, Guinea‐Bissau; CAR, Central African Republic; CGO, Republic of the Congo; COD, Democratic Republic of the Congo.
4. DISCUSSION
With the introduction of HBV vaccine, although the prevalence of HDV has declined in some sporadic areas, the global prevalence of HDV is still high and even increasing in most of the endemic areas, such as Central and Northern Africa, the Amazon Basin, Eastern and Mediterranean Europe, the Middle East and parts of Asia.5, 7, 22, 23 In sub‐Saharan Africa, the estimated prevalence of anti‐HDV has exceeded the global prevalence and around 7 million people are infected by HDV.7 HBsAg‐positive patients with HDV co‐infection showed higher risk to progress to liver fibrosis or HCC compared to asymptomatic controls.7, 22, 24 Thus, for better management or prevention of HDV infection, diagnosis and screening for high‐risk populations are recommended.5, 7 This in turn requires a unified HDV genotype classification system. Due to the confusion as a negative and circular genome, both genomic and antigenomic RNA forms of HDV genome sequences have been submitted to the GenBank database by different research groups and a substantial set of sequences were even read from abnormal initial site.17, 18 Furthermore, two different genotype classification systems have been previously proposed.11, 12 Thus, it is urgent to clarify these inconsistencies, in order to facilitate the future research in this field.
Previous studies have demonstrated that HDV homologous recombination may occur both in nature (patient with mixed infection) and the laboratory (cotransfection in cell culture system).25, 26, 27 Through phylogenetic and Bootscaning analysis, we confirmed two previous reported recombinants and identified 31 new potential recombinants. The two recombinants, one intra‐genotypic recombination (AB118845, 4a/4b)20 and the other inter‐genotypic recombination (KF660598, 2a/1b),19 have the same recombination pattern with only one crossover. However, the other 31 newly identified recombinants have another predominant pattern with two crossovers. The formation of different recombination patterns may be associated with the distinct replication mechanisms of HDV genome.27, 28, 29 Recombination events were detected among several genotypes, but more frequent in genotype 1 and 5. For genotype 1, it may be explained by its global distribution (Figure 3).12 Although genotype 5 is mainly present in Western Africa, the high recombination frequency may be associated with the intergenotypic evolutional relationship and the African origin.12, 30 Analysis of the recombination junctions has indicated that recombination events occurred at four regions throughout the whole genome. Among these regions, nt 694‐872 at genomic RNA corresponding to nt 807‐985 at antigenomic RNA is the hotspot of HDV RNA recombination. This genome region serves as the pseudoknot ribozyme domain of HDV genome. It is identical with the hotspot fragment “D” previously shown in the HDV‐1/HDV‐4 recombination map.28 A model has been proposed to illustrate the mechanism of HDV recombination, which is via a viral‐RNA‐structure‐promoted template‐switching mechanism driven by the host RNA polymerase,28 although further validation is required.
For molecular classification of HDV, eight clades have been proposed a decade ago, but recently designated as eight genotypes.12 In contrast, a latest study has proposed to group the eight HDV genotypes into three large genogroups by grouping clade 2 and clade 4‐6 as one.11 In our study, we have applied the ML and NJ models to reconstruct phylogenetic trees based on standardized full‐length antigenomic sequences excluding the potential recombinants. Indeed, all the HDV strains were clustered as three big clades, but further grouped into eight groups with high bootstrap value support. Even though the three big clades shared some characteristics as described, three genotype classification has neglected HDV genotypic divergence. Besides, this classification system only divided HDV strains as genotype, but not further into subtypes. Thus, we agree with the classification system of eight genotypes.12 However, the subtype classification of genotype 1, 3 and 6 in our system showed clear differences, compared to the previous study.12 We grouped genotype 1 into two subtypes, 1a and 1b. Because the previously classified strains of the HDV‐1b, HDV‐1c and HDV‐1d subtypes12 do not always cluster as independent groups but rather as one big branch supported by over 70% Bootstrap value in our trees (Figures 1 and S3). Genotype 3 was segregated into 3a, 3b and 3c three subtypes by including more sequences; whereas this was not clear in the previous study due to the lacking of sufficient sequences.12 We classified genotype 6 into three subtypes; whereas only two were previously defined.12 Consequently, 312 strains were further classified as eight genotypes with eighteen subtypes in our updated classification system supported by high Bootstrap values, nucleotide identity and genetic distance. Of note, neither HDAg gene fragments nor amino acid sequences can classify HDV strains into subtypes faithfully (Figure S4), because many genotypic characteristics are located outside of the HDAg gene.11, 12
Among these eight genotypes, genotype 1 is the most predominant with highest divergence. This may be resulted from prolonged wide‐spread transmission (Figure 3).12 Genotype 3 is located close to the root of phylogenetic tree, showing distantly nucleotide similarity and genetic distance with other genotypes. Given that genotype 3 was only found in South America (Figure 3), it is plausible whether this genotype is an independent lineage or the early HDV progenitor. The exact evolutional relationship between genotype 3 and other genotypes requires further investigate. However, the current genetic data are still insufficient to address this question (Figure 3).5, 7 Importantly, based on our phylogenetic and genetic results, we have proposed detailed criteria for identifying novel genotype or subtype.
Finally, we have compiled a complete set of reference genome sequences for HDV subtypes. The main criteria were based on previous study of hepatitis E virus reference genomes31 and the unique features of HDV. We have proposed one reference sequence for each subtype of genotypes 2‐8. But five reference sequences were proposed for the two subtypes of genotype 1, because of the huge divergent. When generating these 21 reference sequences, we found that the majority HDV full‐length sequences have missing information, in particular the collection date. Thus, we strongly recommend researchers to provide the essential information, when submitting their sequence data to the online database.
In summary, we have systematically retrieved and analysed a large set of HDV full‐length genome sequences and identified novel recombinants. Based on phylogenetic and genetic analyses, we have established an updated classification system for HDV when recombinants were excluded. Furthermore, we have mapped the global distribution of different genotypes and subtypes. Finally, we have compiled a complete set of reference genomes for each subtype and proposed criteria for future identification of novel genotypes and subtypes. Of note, our global distribution map indicates that currently available HDV genetic data remain limited, and thus, the proposed classification will likely evolve as future epidemiological data will accumulate. Overall, these results shall facilitate the future research on the diagnosis, screening, epidemiology, evolution, prevention and clinical management of HDV infection.
Supporting information
ACKNOWLEDGEMENTS
This work was financially supported by the Gansu Provincial Science and Technology Grant (grant number 17YF1WA166, 1504WKCA094); Ministry of Science and Technology Assistance Project Grant (grant number KY201501005); Characteristic discipline of bioengineering construction for the special guide project of the “world‐class universities and world‐class disciplines” of Northwest Minzu University (grant number 10018703) and Changjiang Scholars and Innovative Research Team in University (grant number IRT_17R88). Z. Miao was supported by China Scholarship Council (no. 201708530234).
Miao Z, Zhang S, Ma Z, et al. Recombinant identification, molecular classification and proposed reference genomes for hepatitis delta virus. J Viral Hepat. 2019;26:183–190. 10.1111/jvh.13010
REFERENCES
- 1. Hughes SA, Wedemeyer H, Harrison PM. Hepatitis delta virus. Lancet. 2011;378(9785):73‐85. [DOI] [PubMed] [Google Scholar]
- 2. Luan BS, Santos ADOD, Salcedo JMV, Vieira DS. Hepatitis delta: virological and clinical aspects. Virol J. 2017;14(1):177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pascarella S, Negro F. Hepatitis D virus: an update. Liver Int. 2011;31(1):7‐21. [DOI] [PubMed] [Google Scholar]
- 4. Kuo MY, Goldberg J, Coates L, Mason W, Gerin J, Taylor J. Molecular cloning of hepatitis delta virus RNA from an infected woodchuck liver: sequence, structure, and applications. J Virol. 1988;62(6):1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ahn J, Gish RG. Hepatitis D virus: a call to screening. Gastroenterol Hepatol. 2014;10(10):647‐686. [PMC free article] [PubMed] [Google Scholar]
- 6. Rizzetto M, Canese MG, Aricò S, et al. Immunofluorescence detection of new antigen‐antibody system (delta/anti‐delta) associated to hepatitis B virus in liver and in serum of HBsAg carriers. Gut. 1977;18(12):997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Stockdale AJ, Chaponda M, Beloukas A, et al. Prevalence of hepatitis D virus infection in sub‐Saharan Africa: a systematic review and meta‐analysis. Lancet Glob Health. 2017;5(10):e992‐e1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Taghavi SA, Sedighi S, Mehrabani D, Khademolhosseini F. Hepatitis D in chronic active hepatitis B: prevalence, liver enzyme levels and histopathology – an Epidemiological Study in Shiraz, Southern Iran, 2003–2004. Hepat Monthly. 2008;8(4):248‐251. [Google Scholar]
- 9. Yamaguchi Y, Deléhouzée S, Handa H. HIV and hepatitis delta virus: evolution takes different paths to relieve blocks in transcriptional elongation. Microbes Infect. 2002;4(11):1169‐1175. [DOI] [PubMed] [Google Scholar]
- 10. Hsu SC, Syu WJ, Sheen IJ, Liu HT, Jeng KS, Wu JC. Varied assembly and RNA editing efficiencies between genotypes I and II hepatitis D virus and their implications. Hepatology. 2002;35(3):665‐672. [DOI] [PubMed] [Google Scholar]
- 11. Delfino CM, Cerrudo CS, Biglione M, Oubiña JR, Ghiringhelli PD, Mathet VL. A comprehensive bioinformatic analysis of hepatitis D virus (HDV) full‐length genomes. J Viral Hepat. 2018;25:860‐869. [DOI] [PubMed] [Google Scholar]
- 12. Gal FL, Brichler S, Drugan T, et al. Genetic diversity and worldwide distribution of the deltavirus genus: a study of 2,152 clinical strains. Hepatology. 2017;66:1826‐1841. [DOI] [PubMed] [Google Scholar]
- 13. Radjef N, Gordien E, Ivaniushina V, et al. Molecular phylogenetic analyses indicate a wide and ancient radiation of African hepatitis delta virus, suggesting a deltavirus genus of at least seven major clades. J Hepatol. 2004;78(5):2537‐2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kumar S, Stecher G, Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol Biol Evol. 2016;33(7):1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nei M, Kumar S. Molecular evolution and phylogenetics. Heredity. 2013;86(86):385. [Google Scholar]
- 16. Hung CC, Wu SM, Lin PH, et al. Increasing incidence of recent hepatitis D virus infection in HIV‐infected patients in an area hyperendemic for hepatitis B virus infection. Clin Infect Dis. 2014;58(11):1625‐1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cicero MF, Pena NM, Santana LC, et al. Is hepatitis delta infections important in Brazil? BMC Infect Dis. 2016;16(1):525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Celik I, Karatayli E, Cevik E, et al. Complete genome sequences and phylogenetic analysis of hepatitis delta viruses isolated from nine Turkish patients. Adv Virol. 2011;156(12):2215‐2220. [DOI] [PubMed] [Google Scholar]
- 19. Sy BT, Nguyen HM, Toan NL, et al. Identification of a natural intergenotypic recombinant hepatitis delta virus genotype 1 and 2 in Vietnamese HBsAg‐positive patients. J Viral Hepat. 2015;22(1):55‐63. [DOI] [PubMed] [Google Scholar]
- 20. Lin CC, Lee CC, Lin SH, et al. RNA recombination in Hepatitis delta virus: Identification of a novel naturally occurring recombinant. J Microbiol Immunol Infect. 2015;50(6):771‐780. [DOI] [PubMed] [Google Scholar]
- 21. Simmonds P, Bukh J, Combet C, et al. Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hepatology. 2005;42(4):962‐973. [DOI] [PubMed] [Google Scholar]
- 22. Wranke A, Pinheiro Borzacov LM, Parana R, et al. Clinical and virological heterogeneity of hepatitis delta in different regions world‐wide: the Hepatitis Delta International Network (HDIN). Liver Int. 2017;38(5):842‐850. [DOI] [PubMed] [Google Scholar]
- 23. Han M, Littlejohn M, Yuen L, et al. Molecular epidemiology of hepatitis delta virus in the Western Pacific region. J Clin Virol. 2014;61(1):34‐39. [DOI] [PubMed] [Google Scholar]
- 24. Béguelin C, Moradpour D, Sahli R, et al. Hepatitis delta‐associated mortality in HIV/HBV‐coinfected patients. J Hepatol. 2017;66:297‐303. [DOI] [PubMed] [Google Scholar]
- 25. Chao M, Wang TC, Lee SE. Detection of hepatitis delta virus recombinants in cultured cells co‐transfected with cloned genotypes I and IIb DNA sequences. J Virol Methods. 2006;137(2):252‐258. [DOI] [PubMed] [Google Scholar]
- 26. Wang TC, Chao M. RNA recombination of hepatitis delta virus in natural mixed‐genotype infection and transfected cultured cells. J Virol. 2005;79(4):2221‐2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wu JC, Chiang TY, Shiue WK, et al. Recombination of hepatitis D virus RNA sequences and its implications. Mol Biol Evol. 1999;16(11):1622‐1632. [DOI] [PubMed] [Google Scholar]
- 28. Chao M, Wang TC, Lin CC, et al. Analyses of a whole‐genome inter‐clade recombination map of hepatitis delta virus suggest a host polymerase‐driven and viral RNA structure‐promoted template‐switching mechanism for viral RNA recombination. Oncotarget. 2017;8(37):60841‐60859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chao M. RNA recombination in hepatitis delta virus: implications regarding the abilities of mammalian RNA polymerases. Virus Res. 2007;127(2):208‐215. [DOI] [PubMed] [Google Scholar]
- 30. Le Gal F, Badur S, Hawajri NA, et al. Current hepatitis delta virus type 1 (HDV1) infections in central and eastern Turkey indicate a wide genetic diversity that is probably linked to different HDV1 origins. Arch Virol. 2012;157(4):647‐659. [DOI] [PubMed] [Google Scholar]
- 31. Smith DB, Simmonds P, Izopet J, et al. Proposed reference sequences for Hepatitis E virus subtypes. J Gen Virol. 2016;97(3):537. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials