

Abstract
Antibiotic resistance is a worldwide health issue spreading quickly among human and animal pathogens, as well as environmental bacteria. Misuse of antibiotics has an impact on the selection of resistant bacteria, thus contributing to an increase in the occurrence of resistant genotypes that emerge via spontaneous mutation or are acquired by horizontal gene transfer. There is a specific and urgent need not only to detect antimicrobial resistance but also to predict antibiotic resistance in silico. We now have the capability to sequence hundreds of bacterial genomes per week, including assembly and annotation. Novel and forthcoming bioinformatics tools can predict the resistome and the mobilome with a level of sophistication not previously possible. Coupled with bacterial strain collections and databases containing strain metadata, prediction of antibiotic resistance and the potential for virulence are moving rapidly toward a novel approach in molecular epidemiology. Here, we present a model system in antibiotic‐resistance prediction, along with its promises and limitations. As it is commonly multidrug resistant, Pseudomonas aeruginosa causes infections that are often difficult to eradicate. We review novel approaches for genotype prediction of antibiotic resistance. We discuss the generation of microbial sequence data for real‐time patient management and the prediction of antimicrobial resistance.
Keywords: antibiotic resistance, in silico prediction, emerging technologies, genomics
Introduction
The damaging effect of antimicrobial resistance (AMR) in infectious diseases is evident and a major preoccupation around the world. Bacterial infections are currently observed with ancient as well as emerging pathogens resistant to all classes of antibiotics. Bacterial infections claim at least 70,000 lives each year in Europe and 23,000 in the United States; at least two million infections occur each year worldwide.1 It is estimated that hundreds of thousands of people die each year in other areas of the world owing to AMR. Considerable variation is observed between countries, because data are simply not available or poorly described in the patterns of AMR observed. Different countries around the globe experience different problems in health care, public health and services, and overall hygiene. AMR may thrive in some environments, but solid data are virtually nonexistent. The rise of AMR has not gone unnoticed by governments around the world. For example, the UK government commissioned the O'Neill review in 2014. This report predicts that, without intervention, the number of deaths attributable to AMR will rise to 10 million in 2050, corresponding to one death every 3 seconds.2
In modern societies with excellent healthcare systems, the most recent technologies in diagnostics of infectious diseases, and an arsenal of therapeutics, the costs of treatment is continually increasing because of AMR. The mortality rate of patients caused by AMR is still very high. In aging populations, high‐level medical treatment with the most recent health care, including long periods in intensive care units and transplantation, and long‐term age‐related chronic diseases have dramatically augmented susceptibility to AMR bacterial infections. This has led to the emergence of pan‐resistant infections, meaning that no effective treatment is currently available.3
In many countries, antibiotics are often used excessively, in a very broad and unregulated fashion.2 They are often readily available over the counter, sometimes prescribed, and in other cases prescribed but with completely different prescribing habits, leading to excessive use and unnecessary overuse, which contributes to AMR. This is a worst‐case scenario in infectious diseases, as the global spread of AMR in bacterial pathogens is rapid, without borders, and amplified by the speed and volume of intercontinental travel.4 Moreover, the complexity of polymicrobial communities creates ideal conditions for what we propose to call the “AMR sprawl.”
Armed with an arsenal of new bacterial genomic and bioinformatic tools, coupled with a certain level of understanding of the biology of a bacterial pathogen, such as Pseudomonas aeruginosa, and its tendency to evolve AMR, there is an excellent opportunity to develop creative approaches in deciphering AMR.5, 6, 7, 8 The challenge is to have the capability to use predictive values that lead not only to rapid intervention and therapy but also to molecular epidemiological surveillance. Indeed, the O'Neill report also recommends increased surveillance and improved diagnostics in order to prevent unnecessary use of antibiotics.2
Here, we highlight recent creative concepts, hypothesis‐driven discoveries in predicting AMR, innovative strategies to limit its dissemination, and a way out of AMR. In specific cases, these novel approaches and disruptive technologies are promising and will certainly revolutionize the traditional view of using standards, such as the disk diffusion methods and the determination of minimal inhibitory concentrations (MICs), for predicting AMR. In other cases, development is still in its infancy, as was the case for polymerase chain reaction (PCR) in 1984. Overall, these new approaches are significant but limited advances in AMR prediction—a newly emerging field that will have a major impact in clinical microbiology and molecular epidemiology.9 AMR prediction presents an enormous challenge, and some of its current limitations are discussed.
Pseudomonas aeruginosa as a model system for the validation of AMR prediction
P. aeruginosa is an environmental Gram‐negative bacterium that exhibits extensive metabolic adaptability, enabling it to thrive in an extraordinary range of niches (for a review, see Ref. 10). It is also a highly successful opportunistic pathogen, causing a wide range of acute and chronic infections.11 Although transmission routes remain difficult to establish, it is generally accepted that P. aeruginosa is a ubiquitous opportunistic pathogen found in the environment. For example, most cystic fibrosis (CF) patients become infected with P. aeruginosa from the environment, and it is difficult to devise strategies to counter such infections.12 P. aeruginosa has inherent resistance to many antimicrobial classes. Its ability to acquire AMR via mutations to all relevant treatments and its frequent role in serious infections (i.e., bacteremia) with high mortality rates, have become true nightmares since the early 1990s.13
Molecular mechanisms of resistance in P. aeruginosa include prevention of access to target, increased efflux, changes in antibiotic targets by mutation, modification and protection of targets, and direct modification of antibiotics (for details, see Ref. 1). Even antibiotic‐susceptible strains of P. aeruginosa have considerable defenses due to intrinsic AMR. Ongoing international surveillance of P. aeruginosa AMR is fundamental to monitor trends in susceptibility patterns and to appropriately guide clinicians in choosing empirical or directed therapy.14 Whole‐genome shotgun (WGS) sequencing of P. aeruginosa provides a privileged perspective of the dramatic effect of mutator phenotypes on the accumulation of random mutations, most of which are transitions, as expected.15 Moreover, a frameshift mutagenic signature, consistent with error‐prone DNA polymerase activity as a consequence of SOS system induction, is also observed. This effect was observed for the evolution of resistance to all antibiotics tested, but it was higher for fluoroquinolones than for cephalosporins or carbapenems. Analysis of genotype versus phenotype confirmed expected resistance evolution trajectories, but also revealed new pathways. Qualitative RNA sequencing was used to identify the key genetic determinants of AMR in 135 clinical P. aeruginosa isolates from diverse geographic origins and infection sites.16 By applying transcriptome‐wide association studies, adaptive variations associated with AMR of fluoroquinolones, aminoglycosides, and β‐lactams were identified. Besides potential novel biomarkers with a direct correlation to AMR, global patterns of phenotype‐associated gene expression and sequence variations were identified by machine learning approaches.
Studies of the genetic structure of microbial populations are central to understanding the evolution, ecology, and epidemiology associated with the origin of AMR. Numerous studies describing the genetic structure of P. aeruginosa populations are based on samples drawn mostly, even overwhelmingly, from clinical collections.17 This approach has resulted in a limited view of P. aeruginosa with respect to defining the potential evolutionary history of disease‐causing lineages, as well as the rapid development of AMR by the resistome and distribution by horizontal gene transfer via the mobilome.18, 19 A central question is: Can we define environmental and clinical P. aeruginosa isolates with distinct gene contents for virulence and AMR? To answer this question, we are developing a proof‐of‐concept approach based on bacterial genomics and a well‐defined collection of P. aeruginosa strains.5
Driven by the advent of next‐generation sequencing (NGS) coupled with bioinformatics, we are on the verge of significant technological advances at the interface between bacteriology and clinical practice.20, 21 Although we have made significant progress in understanding the variations within the species P. aeruginosa, including the identification of AMR transmissible strains associated with greater patient morbidity, there is still a lack of clarity and consensus. There are three key areas where improvements in our understanding of P. aeruginosa genomics would lead to benefits in infectious disease and human health: (1) given the clear link between infection with P. aeruginosa and patient morbidity/mortality, we need to better understand how patients become infected, including the role of cross infection; (2) given the limitations of current antibiotic therapy, especially in relation to chronic infections, we need to develop better and new therapeutic approaches; and (3) we need to identify better prognostic markers and detect AMR more rapidly to enable clinicians to make better evidence‐based decisions on patient care.
Identification of emerging P. aeruginosa transmissible strains and their AMR profiles
The experiences in the Liverpool, UK clinics and in Canada highlight the need to identify emerging transmissible strains early. Once the Liverpool Epidemic Strain (LES) isolates were identified, it was possible to design a simple PCR test, which is now used widely in UK CF clinics.22, 23 However, in reality, most CF patients at the Liverpool clinic were infected with the LES before it was discovered, and although segregation measures have led to a welcome decline in new cases, the adult unit remains dominated by this strain. Encouragingly, studies elsewhere in the world also show that patient segregation efficiently reduces the spread of epidemic clones.24, 25 Hence, we need strategies to make earlier interventions based on rapid identification of new transmissible strains. However, to achieve this, we require a greater understanding of the general population structure of P. aeruginosa and predictive capacity for AMR. Key to this is the identification of strain types that are not present in the CF population because they are of higher prevalence in the environment. There have also been examples of multiple CF patients in Germany carrying the same strain, known as P. aeruginosa Clone C.26 However, we know that Clone C is one of the most abundant clones of P. aeruginosa strains.27 Hence, it is likely that in any CF patient cohort, there will be multiple CF patients infected with this strain, because they acquired their infections from environmental sources. In contrast, the LES has not been identified from other environmental or clinical sources, clearly implicating its transmission route as patient‐to‐patient cross‐infection. These two examples are clear‐cut. However, to make informed decisions about the sources of other strains of P. aeruginosa identified in multiple CF patients (cross‐infection or environmental), we need to have a much better understanding of P. aeruginosa population structures and determine whether links exist with other types of P. aeruginosa opportunistic infections. To achieve this, we need to exploit bacterial WGS using NGS, predict AMR, and couple the data to transcriptomics via RNA sequencing (RNA‐Seq) in a well‐defined pipeline in as short a time frame as possible that would be amenable to the clinic. We must devise robust, unequivocal, and portable strategies for strain genotyping that will not only (1) enable the identification of P. aeruginosa strains present among isolates at higher than expected prevalence, but also (2) allow us to search for links between strain genotype, clinical outcome, and AMR and (3) design epidemiologically and clinically relevant strain panels for anti‐infective testing using clear, evidence‐based criteria.
P. aeruginosa strain diversity and AMR
Over the years, there have been many approaches to the typing of P. aeruginosa strains, driven by the availability of the technology, costs, and ease of use. Some of these approaches are poorly portable between laboratories (RAPDs and ERIC‐PCR) and therefore difficult to use for anything other than local outbreaks. Others (e.g., PFGE) have been considered as gold standards, but are again of limited use for interlaboratory comparisons and are susceptible to genetic instability, such as that exhibited during CF infections.22 There have also been a number of different methodological approaches to define the population structure of P. aeruginosa, each comparing collections of isolates from diverse geographical, environmental, and human sources, using methods that readily allow comparisons between laboratories.28, 29 The ArrayTube,30 MLST,31 and VNTR32 genotyping schemes have all indicated extensive diversity among P. aeruginosa isolates, but with dominant clones, clusters, or clonal complexes that are more widespread among the general P. aeruginosa population. However, each of these approaches has limitations. They can be technically demanding, time‐consuming, and expensive to carry out. There is clear evidence that NGS‐based approaches, such as single‐cell genome sequencing and portable real‐time sequencing (e.g., NanoPore MinIon), will eventually offer far better resolution and become cost‐effective solutions to clinical diagnostic laboratory practice.33 In addition to genotyping for strain surveillance, NGS approaches also generate a wealth of additional data that can be exploited for clinical impact, such as the identification of (1) markers for use in prognostic approaches, (2) novel therapeutic targets, and (3) genotypic markers for the prediction of AMR.
Exploiting P. aeruginosa genomics as proof of concept for AMR prediction
The basic principles on which clinical bacteriology practices are based have altered little over the past 50 years. In the context of chronic lung infections, such as those typical in CF and in opportunistic infections in intensive care units, there are severe limitations to these approaches, particularly with P. aeruginosa. Because P. aeruginosa populations are mostly found in biofilms34 and because strains diversify phenotypically and adapt rapidly to their hosts,35 antimicrobial susceptibility profiles based on MICs and disk diffusion methods applied to single isolates are poorly predictive of therapeutic efficacy.36 Additionally, highly resistant P. aeruginosa small‐colony variants (SCVs) are frequently isolated from patient samples, yet they are rarely reported or further analyzed by clinical microbiology services.37
Studies on the genetic structure of P. aeruginosa populations are central to understand the evolution, ecology, and epidemiology of opportunistic infections. NGS technology gives us the means to take such studies to new levels of sophistication, by incorporating data from worldwide, representative strain collections to generate new genomics‐based microbiology workflows for direct clinical benefit to patients and for epidemiological surveying. The challenge of this approach will be to take the genomic data through the translational pipeline using cutting‐edge bioinformatics and expertise from leading researchers worldwide and develop clinically oriented methodologies that are user‐friendly, evidence‐based, and lead to direct clinical impact. Namely, this should enhance prognostic marker development for AMR prediction. However, minimal metadata as well as international reporting standards will be required.
Bacterial strain and genome databases: first step in predicting P. aeruginosa AMR
The first Pseudomonas Genome Database (http://www.pseudomonas.com/) was built upon the Sanger sequenced genome of strain PAO138 with a community‐built annotation strategy and is now attempting to incorporate metadata.39 As of December 2016, the Pseudomonas Genome Database contained 2199 draft and 135 complete P. aeruginosa genomes, and the numbers are likely to continue to increase exponentially. This genome database is of immense value to the P. aeruginosa research community. In order to develop a strategy capable of predicting AMR in P. aeruginosa, a detailed collection of strains and metadata is also essential. In 2013, the International Pseudomonas Consortium funded by Cystic Fibrosis Canada was initiated to link genomics and metadata and create a repository of P. aeruginosa strains from around the world.5 The consortium has had remarkable success in acquiring strains and SCVs and is expanding its activities in AMR prediction.
The International Pseudomonas Consortium Database (IPCD) is a web application designed not only to store data for the P. aeruginosa collection, but also to provide access to each isolate's phenotypic and genomic data (http://ipcd.ibis.ulaval.ca/). Different levels of access may be granted, and data modification or updates are strictly reserved for the curators. The IPCD includes isolate identification; environmental origin; host; provider; date of isolation; geographical origin; phenotypic data with a strong emphasis on AMR when available; anonymized patient information; and technical NGS information, such as library preparation, type of NGS strategy, and details on the quality of genome assembly and software used. The IPCD currently contains NGS data and unpublished draft genomes from CF patients and from most of the other types of known human infections caused by P. aeruginosa. For comparative genomics purposes, the IPCD also contains animal infection isolates. Because of the limited information in the literature on P. aeruginosa environmental isolates encoding AMR, the IPCD has a large collection of strains from plants, soil, and water. Draft genomes of the IPCD are progressively made publicly available by the National Center for Biotechnology Information (NCBI), mainly in BioProject PRJNA325248 (see “Publicly available” in the IPCD's search engine).
The IPCD contains 1588 entries for P. aeruginosa isolates, with a time span of 135 years going back to 1880 and covering 85 locations in 35 countries on five continents. It includes previously described collections28, 31, 40 and was assembled with the aim of representing maximal genomic diversity. To this end, various criteria were taken into consideration, including geographic origin, previous genotyping, phenotype, and in vivo behavior. We envision that the collection could accommodate over 10,000 P. aeruginosa isolates.
To be a tool for predicting AMR, the quality of draft genomes must be exceptional. In the IPCD, for the 979 P. aeruginosa genomes sequenced and assembled to date, the median number of scaffolds is 43 (median number of contigs is 46), for a median coverage of 43× . The NCBI indicates, as of December 2016, a repertoire of 2104 P. aeruginosa sequenced genomes, with a median of 111 and up to 2797 scaffolds. Because this publicly available P. aeruginosa data set has been available since the early stages of NGS using different technologies, and because of the high variability in sequence quality and limited metadata information that is difficult to retrieve, it cannot be used for AMR prediction. In P. aeruginosa, predicting AMR can only be achieved using a strain and genome database such as the IPCD, where not only metadata but also an integrated approach for genome sequencing and annotation will first define the proof of concept in predicting AMR, its limitations, and how it may be used in other species.
Phylogeny of P. aeruginosa linked to the prediction of AMR
Although several P. aeruginosa genomes have been completely sequenced and annotated as a single circular chromosome,38, 41, 42, 43, 44 the species phylogeny has been clearly determined only recently.5, 45 Knowledge on this species’ diversity and evolution is essential in the development of tools for predicting AMR, because knowledge about diversity could be closely linked to the source of AMR, crucial information for surveillance and epidemiology.
P. aeruginosa is well known to have an adaptable genome between 5.5 and more than 7 Mbp. This large genome contains more than 550 transcriptional regulators, which presumably play exquisite roles in coordinating the colonization of a wide range of ecological niches.38 Comparative genomics approaches have identified changes in surface antigens, loss of virulence‐associated traits, increased AMR, overproduction of alginate, and the modulation of metabolic pathways. Its genome also has many regions that exhibit plasticity linked with AMR.46
The core genome phylogeny for the 390 genomes (Fig. 1) that constitute a key data set for predicting AMR in P. aeruginosa strains can be divided into three major groups,5 a result in agreement with what was previously observed45 but with enhanced resolution. Furthermore, the third major group, which includes strain PA7, has a genome sequence revealing considerable divergence from other P. aeruginosa strains.43 The PA7 genome was particularly useful for unraveling the genetic basis of AMR in this strain: point mutations in gyrA and parC conferred fluoroquinolone resistance, and efflux systems with conserved and unique oprA components were the primary modes of resistance. A more extensive survey of P. aeruginosa is required to further populate the tree. Still, these 390 strains are highly valuable for predicting AMR because they originate from a wide array of environmental, clinical, and animal sources.
Figure 1.
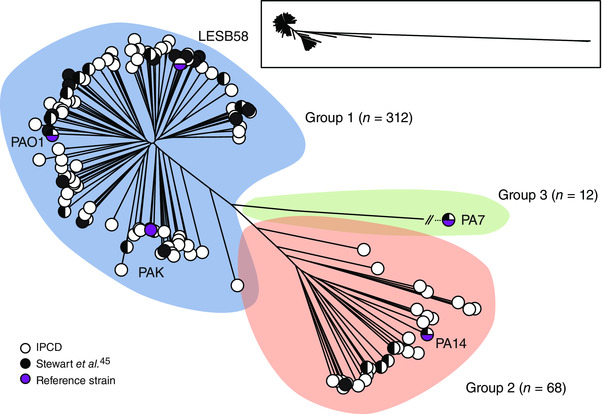
Unrooted tree of 390 P. aeruginosa genomes based on SNPs within the core genome as defined by Harvest (100 bootstraps). The total coverage of the core genome among all sequences was 17.5%. Strains are divided into three groups (blue, orange, and green). The number of strains for each group is shown. White circles represent one or more strains that were sequenced by the IPC, while black circles represent publicly available genomes. Adapted with permission from Ref. 5.
AMR databases: the second step in predicting AMR in P. aeruginosa
Promising application of NGS technology includes hospital infection‐control surveillance programs and community outbreak investigations.9 In addition to bacterial WGS sequencing and identification at the subspecies and strain levels based on single‐nucleotide polymorphisms (SNPs) and average nucleotide identity for species confirmation,47 we entertain the possibility that a critical approach should include a clear focus on predicting AMR. Conducting AMR testing to guide therapy is one of the major critical tests in a clinical laboratory. Bacterial WGS has an excellent potential to predict known gene content encoding AMR (i.e., the resistome). WGS of Escherichia coli and Klebsiella pneumoniae clinical isolates and BLASTn used to identify the presence of relevant AMR genes demonstrated 96% and 97% sensitivity and specificity for predicting AMR, respectively.48
Predicting AMR will require two complimentary approaches to have some degree of success: comprehensive databases and software capable of giving statistical prediction with a high level of confidence. In addition, there is no a priori evidence that all AMR can be successfully predicted in all bacterial species. It has not been established whether WGS coupled with AMR prediction can be broadly applied to the full spectrum of pathogenic bacteria, particularly those with a diverse armamentarium of resistance mechanisms. P. aeruginosa is certainly a case in point described here.
The three major extensive AMR databases are the Comprehensive Antibiotic Resistance Database (CARD),7 Antibiotic Resistance Gene‐ANNOTation (ARG‐ANNOT),49 and ResFinder.50 The CARD is one of the more sophisticated, highly detailed, and comprehensive databases (http://arpcard.mcmaster.ca), because it is a manually curated resource containing high‐quality reference data on the molecular basis of AMR, with an emphasis on the genes, proteins, and mutations involved. The CARD is ontologically structured, model centric, and spans the breadth of AMR drug classes and resistance mechanisms, including intrinsic, mutation‐driven, and acquired resistance. Its design allows the development of novel genome analysis tools, such as the Resistance Gene Identifier (RGI) for resistome prediction from raw genome sequences. Recent improvements include extensive curation of additional reference sequences and mutations, development of a unique ontology and accompanying AMR detection models to power sequence analysis, new visualization tools, and expansion of the RGI for detection of emergent threats.6
As a proof of concept in predicting AMR in P. aeruginosa, the RGI was used to detect AMR genes and specific gene variants from WGS of 390 Pseudomonas strains (Fig. 2).5 Approximately one‐third of the 46 detected resistance genes were found in most strains. On the other hand, the other two thirds were found only in a restricted group of strains, highlighting the great variability of P. aeruginosa strains with respect to AMR genes. This variation can only be unraveled by extensive sampling. This information can be exploited to study the pool of resistance genes present in clinical strains and understand the links between clinical and environmental strains in the context of AMR. Remarkably, as depicted in Figure 2, AMR genes or mutants were found for the major families of antibiotics used, including aminoglycosides, β‐lactams, fluoroquinolones, fosfomycin, macrolides, polymyxin, rifampin, sulfonamide, and tetracycline. On the basis of our own work, as well as machine learning attempts from another team,51, 52 the only single gene that can be directly associated with a resistance phenotype is gyrA, for which specific variants are associated with quinolone resistance.53 This highlights the complex nature of the molecular basis of AMR in P. aeruginosa and the challenge that is AMR prediction for this organism.
Figure 2.

Heat map showing unique resistomes for 390 P. aeruginosa strains. The heat map was obtained by performing tblastn searches using the sequences present on the Comprehensive Antibiotic Resistance Database (CARD)7 as queries and the genomes as subjects. Green, perfect match to a sequence in the CARD; red, similar to a sequence in the CARD, within a curated e‐value cutoff (gene specific); and black, absent. The bar plot represents absolute frequency of each resistome. On the left of the heat map, genes, proteins, and specific variants (*) are grouped according to their biological function or the resistance they confer. Adapted with permission from Ref. 5.
As depicted in Figure 2, an impressive repertoire of efflux genes encoding multidrug resistance (MDR) efflux pumps were also identified in these 390 P. aeruginosa genomes. This opens major questions: What is the significance of efflux in AMR and what is a resistance gene? MDR efflux pumps will be a major challenge in predicting AMR, particularly in P. aeruginosa. MDR efflux pumps are chromosomally encoded and capable of mediating resistance to toxic compounds in several life forms. Hence, they exhibit multiple functions relevant to bacterial physiology in addition to mediating AMR.54 MDR efflux pumps contribute to both intrinsic and acquired resistance to toxic compounds, including in humans, where they have a role in resistance to anticancer drugs,55 and in bacteria, where they are involved in resistance to antibiotics.56, 57 MDR efflux pumps are highly conserved in a given bacterial species and are presumably implicated in bacterial virulence,58 trafficking of quorum‐sensing molecules,59, 60 and detoxification processes from metabolic intermediates and toxic compounds, such as heavy metals, solvents, or antimicrobials produced by other microorganisms.54 It is relatively well documented, however, that efflux gene expression is a better predictor of efflux‐mediated AMR than the presence of efflux pump–encoding genes alone.61, 62 Regulatory genes that are known to have an impact on AMR are part of the CARD; eight of them are represented at the bottom of Figure 2. In P. aeruginosa, an experimental study of 108 clinical isolates revealed complex associations, where only certain combinations of multiple regulatory mutations and overexpressed efflux genes were associated with MDR phenotypes.63 Although multiple efflux systems have been characterized, namely MexAB‐OprM and MexXY complexes, their expression is controlled by an intricate network of regulators and modulators, not to mention the impact of environmental conditions. 64 Hence, knowledge of each regulatory gene and its role(s) does not necessarily ease the task of predicting phenotype from genotype. Additionally, it has come to our attention that the CARD nomenclature sometimes differs from what is expected in terms of gene names. For instance, with the current version, strain PAO1's mexB is called mexF. Moreover, some efflux genes tend to be identified in multiple copies per genome by the RGI. Therefore, it is difficult to ascertain whether we are really comparing the same locus when comparing genetically diverse isolates of Pseudomonas.
The third step: software for predicting AMR––a route out of resistance?
The Mykrobe Predictor software was able to detect resistance to the five first‐line antibiotics in over 99% of Staphylococcus aureus cases, matching the performance of traditional drug‐sensitivity testing.65 For Mycobacterium tuberculosis, where the genetic basis for drug resistance is less well understood, the Mykrobe Predictor still detected 82.6% of resistant infections around 5–16 weeks faster than traditional drug susceptibility testing. Mykrobe Predictor can be rapidly updated with a simple software upgrade that allows researchers to detect new resistance mutations as they evolve. A further advantage of Mykrobe is that it can identify infections where a patient's body contains a mixture of both drug‐resistant and drug‐susceptible bacteria.65 The ability to distinguish between these bacterial subpopulations is an advantage over conventional testing in detecting resistance to second‐line tuberculosis (TB) drugs. This is important for diagnosing infections like extensively drug‐resistant TB, which is resistant to at least four of the core TB drugs and is considered a global threat to public health by the World Health Organization.
ARG‐ANNOT is a bioinformatics tool that was created to detect existing and putative AMR genes in bacterial genomes.49 ARG‐ANNOT uses a local BLAST program in the Bio‐Edit software that allows the user to analyze sequences without a Web interface. All AMR genetic determinants were collected from published works and online resources; nucleotide and protein sequences were retrieved from the NCBI GenBank database. After building a database that includes 1689 AMR genes, the software was tested in a blind manner using 100 random sequences selected from the database to verify that the sensitivity and specificity were at 100%, even when partial sequences were queried. It will be interesting to observe the potential of ARG‐ANNOT and its limitations when using a well‐defined set of P. aeruginosa genomes encoding multiple AMR genes.
The Integrated Rapid Infectious Disease Analysis (IRIDA) platform combines PHYLoVIZ (enhanced phylogenomics visualization), IslandViewer and IslandCompare (genomic island, virulence factor, and antibiotic‐resistance gene identification and comparison), GenGIS (phylogeography analysis and visualization), CARD, and SNVPhyl phylogenomics analysis (http://www.irida.ca/tools/). The IRIDA platform is a promising integrated approach but needs to enhance some of its capabilities in Island and in virulence factor prediction with NGS data.
OSPREY was able to predict the most likely mutations to come out of certain bacteria, and clinicians were then able to test treatment with antimicrobials that are still in the experimental phase.66 Identifying the most likely mutations while antimicrobials are still under development indicates a better position for success when these compounds become available for general use. OSPREY was used to specifically pinpoint methicillin‐resistant S. aureus, a common cause of hospital infection. Developing preemptive strategies while antimicrobials are still in the design phase will provide a head start on the next line of compounds that will be effective despite the AMR mutations. If we can somehow predict how bacteria might respond to a particular antibiotic ahead of time, we can change the regimen for treatment, plan for the next one, or rule out therapies that are unlikely to remain effective for long.
Machine learning to predict AMR
A major drawback of database approaches is that, by definition, they will never detect what is unknown. This is why machine learning is such a promising approach for predicting AMR. To date, however, machine learning applied to P. aeruginosa AMR has had mitigated success51, 52 and will need to be better adapted to the genomic complexity of the species. Currently, there are two major approaches that could be amenable to AMR prediction: rules‐based methods and machine learning methods. The rules‐based algorithm makes predictions based on current, curated knowledge of AMR genes. The machine learning algorithm predicts AMR and susceptibility based on a model built from a training set of resistant and susceptible isolates. In general, machine learning algorithms work by finding the relevant features in a complex data set that enables the ability to make a strong prediction.
In one of the largest phenotypic studies ever performed on biocides and on AMR, a total of 1632 worldwide clinical strains of S. aureus were analyzed.67 S. aureus is a major human pathogen, a major cause of nosocomial infections, and a significant cause of foodborne infection. By combining different machine learning methodologies, namely decision trees and clustering, to explore the data in order to find biologically and statistically significant results, it was demonstrated that reduced susceptibility to two common biocides, chlorhexidine and benzalkonium chloride, which belong to different structural families, is associated with multi‐AMR. An MIC greater than 2 mg/L for both biocides is related to AMR in S. aureus.
A machine learning approach was used to predict bacterial susceptibility using the presence or absence of over 500 SNPs found in a data set of 652 M. tuberculosis isolates and used as features for a number of classification algorithms.68 Susceptibility and AMR were defined on the basis of phenotypic growth patterns, and the results from the machine learning method were compared to predictions based on the presence of a set of known AMR mutations. Misclassified isolates were also examined for commonalities, revealing 11 potentially new AMR mutations. The prediction of antibiotic susceptibility gave a classification accuracy of 93% for predicting AMR to isoniazid. Machine learning was capable of particularly high sensitivity, ranging between 95% and 100% across the four antibiotics examined.
WGS data were used from 78 clinical Enterobacteriaceae isolates identified to represent a variety of phenotypes, from fully susceptible to pan‐resistant strains for the antibiotics tested. The predictions of the rules‐based and machine learning algorithms for these isolates were compared to results of phenotype‐based diagnostics.8 The rules‐based and machine learning predictions achieved agreement with standard‐of‐care phenotypic diagnostics of 89.0% and 90.3%, respectively, across 12 antibiotic agents from six major antibiotic classes. Several sources of disagreement between the algorithms were identified. Novel variants of known resistance factors and incomplete genome assembly confounded the rules‐based algorithm, resulting in predictions based on gene family rather than on knowledge of the specific variant found. Low‐frequency resistance caused errors in the machine learning algorithm because those genes were not seen or seen infrequently in the test set. They also identified an example of variability in the phenotype‐based results that led to disagreement with both genotype‐based methods.
PATRIC (Pathosystems Resource Integration Center (patricbrc.org)) is a National Institutes of Health–supported bioinformatics resource center that was built to enable comparative genomic analysis of bacterial pathogens.69 The PATRIC file‐transfer protocol server enables access to genomes that are binned by their AMR phenotypes, as well as metadata including MICs. The custom‐built AdaBoost (adaptive boosting) machine learning classifiers can identify carbapenem resistance in Acinetobacter baumannii, methicillin resistance in S. aureus, and β‐lactam and cotrimoxazole resistance in Streptococcus pneumoniae with accuracies ranging from 88% to 99%. AdaBoost can predict isoniazid, kanamycin, ofloxacin, rifampicin, and streptomycin resistance in M. tuberculosis, achieving accuracies ranging from 71% to 88%. This set of classifiers has been used to provide an initial framework for species‐specific AMR and genomic feature prediction in the RAST and PATRIC annotation services.70
The functional study of AMR
Understanding the mechanisms responsible for AMR will continue to remain a central aspect of AMR research. Like AMR prediction, this field also benefits greatly from the most recent NGS technologies. For instance, a transposon‐sequencing (Tn‐seq) methodology was used to screen large numbers of transposon mutants in P. aeruginosa.71 This method was benchmarked with the identification of mutations reducing intrinsic resistance to tobramycin, a phenotype that had been previously analyzed at the genome scale using mutant‐by‐mutant screening. The results show the effectiveness of the Tn‐seq method in defining the genetic basis of a complex resistance trait in P. aeruginosa. Harnessing evolutionary biology is also a promising route out of AMR. Experimental evolution of P. aeruginosa in the presence of an antibiotic monitored using WGS sequencing allows not only the direct identification of the mutations that occur, but also an investigation of the fitness cost associated with these mutations, which is likely to affect their success in a host environment.72, 73
Conclusions
The clinical and basic science literature suggest that bacterial WGS can be successfully applied to rapid diagnostics in infectious diseases and the prediction of AMR. Predicting AMR may be useful for clinical cases that fail or challenge the limits of traditional laboratory testing. WGS analysis coupled with AMR prediction could be particularly beneficial for slow‐growing SCVs of P. aeruginosa or for other difficult‐to‐culture organisms and organisms that elude phenotypic testing altogether. As for species or strains that grow normally and for which current laboratory methods are relatively efficient, we argue that sequencing‐based approaches offer the potential to provide much more clinically relevant information in a similar time‐frame.
However, there are clear limitations in predicting AMR without phenotypic confirmation; caution needs to be taken in attributing relevance to any genes hitherto not shown to confer drug resistance. Four AMR postulates have been proposed, and an AMR trait should be only described as such if the criteria of these four postulates are met.74 These include the following: (1) show that the genetic trait is present in drug‐resistant microbes; (2) demonstrate that the genetic trait is not present or not expressed in drug‐susceptible strains; (3) replace the genetic trait with wild‐type DNA sequence and show that it confers drug susceptibility; and (4) introduce the genetic trait into a drug‐susceptible strain and show that it confers AMR. These postulates are supported by the biology of AMR, where we know the microbiological effects of sublethal doses of antibiotics, including in P. aeruginosa; 75 the relative contribution of recombination and point mutation to the diversification of P. aeruginosa clones;76 the impact of AMR caused by gene amplification and its implication on the evolution of AMR;77 the ramifications of SNPs associated with AMR;78 and the challenge of pinpointing which MDR efflux pumps are involved in AMR. However, one additional genomics approach to support AMR prediction and its implementation could include data on the transcriptome of AMR for the bacterial species to be tested. In P. aeruginosa, RNA‐Seq has defined a transcriptional landscape known to be shaped by environmental heterogeneity and genetic variation.79 In specific cases, such as in P. aeruginosa CF lung infections, single–bacterial cell genome sequencing could assist in predicting AMR.80 Single‐cell genomics and transcriptomics are advancing rapidly and could interrogate AMR through a novel, complimentary approach.
Although many potential impediments to the utilization of NGS‐based diagnostics and prediction of AMR exist, it would be a loss to the medical community if this technology could not be applied to predict AMR and assist in patient care in some capacity. The rapid evolution of NGS challenges both the regulatory framework and the development of laboratory standards and will certainly contribute to further research using P. aeruginosa and the IPCD toward tangible improvement and progress.
Competing interests
The authors declare no competing interests.
Acknowledgments
We express our gratitude to members of the Plateforme d'analyses génomiques and the Plateforme de bioinformatique at IBIS. A special thanks to Andrew McArthur for his support and encouragement in using the Comprehensive Antibiotic Resistance Database. J. Jeukens was supported by a Cystic Fibrosis Canada postdoctoral fellowship. R. C. Levesque is funded by Cystic Fibrosis Canada (Grant ID: 2610), a CIHR‐Joint Programming Initiative on Antimicrobial Resistance (JPIAMR) team grant, and by Genome Québec and Genome Canada.
References
- 1. Blair, J.M. et al 2015. Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 13: 42–51. [DOI] [PubMed] [Google Scholar]
- 2. O'Neill, J. 2016. Tackling drug‐resistant infections globally: final report and recommendations. Accessed March 22, 2017. https://amr-review.org/sites/default/files/160518_Final%20paper_with%20cover.pdf.
- 3. O'Neil, J. 2014. Antimicrobial resistance: tackling a crisis for the health and wealth of nations In Review on Antimicrobial Resistance: Tackling Drug‐Resistant Infections Globally. HM Government, Welcome Trust; Accessed April 28, 2017. https://amr-review.org/sites/default/files/160518_Final%20paper_with%20cover.pdf. [Google Scholar]
- 4. Little, T.J. et al 2012. Harnessing evolutionary biology to combat infectious disease. Nat. Med. 18: 217–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Freschi, L. et al 2015. Clinical utilization of genomics data produced by the international Pseudomonas aeruginosa consortium. Front. Microbiol. 6: 1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jia, B. et al 2016. CARD 2017: expansion and model‐centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 45: D566–D573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McArthur, A.G. et al 2013. The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother. 57: 3348–3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pesesky, M.W. et al 2016. Evaluation of machine learning and rules‐based approaches for predicting antimicrobial resistance profiles in Gram‐negative bacilli from whole genome sequence data. Front. Microbiol. 7: 1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Goldberg, B. et al 2015. Making the leap from research laboratory to clinic: challenges and opportunities for next‐generation sequencing in infectious disease diagnostics. mBio 6: e01888‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Silby, M.W. et al 2011. Pseudomonas genomes: diverse and adaptable. FEMS Microb. Rev. 35: 652–680. [DOI] [PubMed] [Google Scholar]
- 11. Winstanley, C. , O'Brien S. & Brockhurst M.A.. 2016. Pseudomonas aeruginosa evolutionary adaptation and diversification in cystic fibrosis chronic lung infections. Trends Microb. 24: 327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kidd, T.J. et al 2013. Shared Pseudomonas aeruginosa genotypes are common in Australian cystic fibrosis centres. Eur. Respir. J. 41: 1091–1100. [DOI] [PubMed] [Google Scholar]
- 13. Livermore, D.M. 2002. Multiple mechanisms of antimicrobial resistance in Pseudomonas aeruginosa: our worst nightmare? Clin. Infect. Dis. 34: 634–640. [DOI] [PubMed] [Google Scholar]
- 14. Sharma, S. & Srivastava P.. 2016. Resistance of antimicrobial in Pseudomonas aeruginosa . Int. J. Curr. Microbiol. Appl. Sci. 5: 121–128. [Google Scholar]
- 15. Cabot, G. et al 2016. Evolution of Pseudomonas aeruginosa antimicrobial resistance and fitness under low and high mutation rates. Antimicrob. Agents Chemother. 60: 1767–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Khaledi, A. et al 2016. Transcriptome profiling of antimicrobial resistance in Pseudomonas aeruginosa . Antimicrob. Agents Chemother. 60: 4722–4733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Selezska, K. et al 2012. Pseudomonas aeruginosa population structure revisited under environmental focus: impact of water quality and phage pressure. Environ. Microbiol. 14: 1952–1967. [DOI] [PubMed] [Google Scholar]
- 18. D'Costa, V.M. et al 2006. Sampling the antibiotic resistome. Science 311: 374–377. [DOI] [PubMed] [Google Scholar]
- 19. Perry, J.A. & Wright G.D.. 2013. The antibiotic resistance “mobilome”: searching for the link between environment and clinic. Front. Microbiol. 4: 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Goodwin, S. , McPherson J.D. & McCombie W.R.. 2016. Coming of age: ten years of next‐generation sequencing technologies. Nat. Rev. Genet. 17: 333–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Loman, N.J. & Pallen M.J.. 2015. Twenty years of bacterial genome sequencing. Nat. Rev. Microbiol. 13: 787–794. [DOI] [PubMed] [Google Scholar]
- 22. Fothergill, J.L. et al 2010. Impact of Pseudomonas aeruginosa genomic instability on the application of typing methods for chronic cystic fibrosis infections. J. Clin. Microbiol. 48: 2053–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Parsons, Y.N. et al 2002. Use of subtractive hybridization to identify a diagnostic probe for a cystic fibrosis epidemic strain of Pseudomonas aeruginosa . J. Clin. Microbiol. 40: 4607–4611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Aaron, S.D. et al 2010. Infection with transmissible strains of Pseudomonas aeruginosa and clinical outcomes in adults with cystic fibrosis. JAMA 304: 2145–2153. [DOI] [PubMed] [Google Scholar]
- 25. van Mansfeld, R. et al 2016. The effect of strict segregation on Pseudomonas aeruginosa in cystic fibrosis patients. PLoS One 11: e0157189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Romling, U. & Tummler B.. 1994. Bacterial genome mapping. J. Biotechnol. 35: 155–164. [DOI] [PubMed] [Google Scholar]
- 27. Romling, U. et al 2005. Worldwide distribution of Pseudomonas aeruginosa clone C strains in the aquatic environment and cystic fibrosis patients. Environ. Microbiol. 7: 1029–1038. [DOI] [PubMed] [Google Scholar]
- 28. Pirnay, J.P. et al 2009. Pseudomonas aeruginosa population structure revisited. PLoS One 4: e7740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pirnay, J.P. et al 2002. Pseudomonas aeruginosa displays an epidemic population structure. Environ. Microbiol. 4: 898–911. [DOI] [PubMed] [Google Scholar]
- 30. Wiehlmann, L. et al 2007. Population structure of Pseudomonas aeruginosa . Proc. Natl. Acad. Sci. U.S.A. 104: 8101–8106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kidd, T.J. et al 2012. Pseudomonas aeruginosa exhibits frequent recombination, but only a limited association between genotype and ecological setting. PLoS One 7: e44199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Turton, J.F. et al 2010. Evaluation of a nine‐locus variable‐number tandem‐repeat scheme for typing of Pseudomonas aeruginosa . Clin. Microbiol. Infect. 16: 1111–1116. [DOI] [PubMed] [Google Scholar]
- 33. Didelot, X. et al 2012. Transforming clinical microbiology with bacterial genome sequencing. Nat. Rev. Genet. 13: 601–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Singh, P.K. et al 2000. Quorum‐sensing signals indicate that cystic fibrosis lungs are infected with bacterial biofilms. Nature 407: 762–764. [DOI] [PubMed] [Google Scholar]
- 35. Mowat, E. et al 2011. Pseudomonas aeruginosa population diversity and turnover in cystic fibrosis chronic infections. Am. J. Respir. Crit. Care Med. 183: 1674–1679. [DOI] [PubMed] [Google Scholar]
- 36. Keays, T. et al 2009. A retrospective analysis of biofilm antibiotic susceptibility testing: a better predictor of clinical response in cystic fibrosis exacerbations. J. Cyst. Fibros. 8: 122–127. [DOI] [PubMed] [Google Scholar]
- 37. Johns, B.E. et al 2015. Phenotypic and genotypic characteristics of small colony variants and their role in chronic infection. Microbiol. Insights 8: 15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Stover, C.K. et al 2000. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406: 959–964. [DOI] [PubMed] [Google Scholar]
- 39. Winsor, G.L. et al 2016. Enhanced annotations and features for comparing thousands of Pseudomonas genomes in the Pseudomonas genome database. Nucleic Acids Res. 44: D646–D653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Stewart, R.M. et al 2011. Genetic characterization indicates that a specific subpopulation of Pseudomonas aeruginosa is associated with keratitis infections. J. Clin. Microbiol. 49: 993–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. He, J. et al 2004. The broad host range pathogen Pseudomonas aeruginosa strain PA14 carries two pathogenicity islands harboring plant and animal virulence genes. Proc. Natl. Acad. Sci. U.S.A. 101: 2530–2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mathee, K. et al 2008. Dynamics of Pseudomonas aeruginosa genome evolution. Proc. Natl. Acad. Sci. U.S.A. 105: 3100–3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Roy, P.H. et al 2010. Complete genome sequence of the multiresistant taxonomic outlier Pseudomonas aeruginosa PA7. PLoS One 5: e8842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Winstanley, C. et al 2009. Newly introduced genomic prophage islands are critical determinants of in vivo competitiveness in the Liverpool Epidemic Strain of Pseudomonas aeruginosa . Genome Res. 19: 12–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Stewart, L. et al 2014. Draft genomes of 12 host‐adapted and environmental isolates of Pseudomonas aeruginosa and their positions in the core genome phylogeny. Pathog. Dis. 71: 20–25. [DOI] [PubMed] [Google Scholar]
- 46. Klockgether, J. et al 2011. Pseudomonas aeruginosa genomic structure and diversity. Front. Microbiol. 2: 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Konstantinidis, K.T. & Tiedje J.M.. 2005. Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. U.S.A. 102: 2567–2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Stoesser, N. et al 2013. Predicting antimicrobial susceptibilities for Escherichia coli and Klebsiella pneumoniae isolates using whole genomic sequence data. J. Antimicrob. Chemother. 68: 2234–2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gupta, S.K. et al 2014. ARG‐ANNOT, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrob. Agents Chemother. 58: 212–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zankari, E. et al 2012. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 67: 2640–2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Drouin, A. et al 2016. Predictive computational phenotyping and biomarker discovery using reference‐free genome comparisons. BMC Genomics 17: 754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Drouin, A. et al 2016. Large scale modeling of antimicrobial resistance with interpretable classifiers. arXiv: 1612.01030. [Google Scholar]
- 53. Yonezawa, M. et al 1995. DNA gyrase gyrA mutations in quinolone‐resistant clinical isolates of Pseudomonas aeruginosa . Antimicrob. Agents Chemother. 39: 1970–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Alvarez‐Ortega, C. , Olivares J. & Martinez J.L.. 2013. RND multidrug efflux pumps: what are they good for? Front. Microbiol. 4: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wu, C.P. , Hsieh C.H. & Wu Y.S.. 2011. The emergence of drug transporter‐mediated multidrug resistance to cancer chemotherapy. Mol. Pharm. 8: 1996–2011. [DOI] [PubMed] [Google Scholar]
- 56. Poole, K. 2007. Efflux pumps as antimicrobial resistance mechanisms. Ann. Med. 39: 162–176. [DOI] [PubMed] [Google Scholar]
- 57. Webber, M.A. & Piddock L.J.. 2003. The importance of efflux pumps in bacterial antibiotic resistance. J. Antimicrob. Chemother. 51: 9–11. [DOI] [PubMed] [Google Scholar]
- 58. Alcalde‐Rico, M. et al 2016. Multidrug efflux pumps at the crossroad between antibiotic resistance and bacterial virulence. Front. Microbiol. 7: 1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Evans, K. et al 1998. Influence of the MexAB‐OprM multidrug efflux system on quorum sensing in Pseudomonas aeruginosa . J. Bacteriol. 180: 5443–5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kohler, T. et al 2001. Overexpression of the MexEF‐OprN multidrug efflux system affects cell‐to‐cell signaling in Pseudomonas aeruginosa . J. Bacteriol. 183: 5213–5222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Li, X.‐Z. & Nikaido H.. 2009. Efflux‐mediated drug resistance in bacteria. Drugs 69: 1555–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Piddock, L.J.V. 2006. Clinically relevant chromosomally encoded multidrug resistance efflux pumps in bacteria. Clin. Microbiol. Rev. 19: 382–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kiser, T.H. et al 2010. Efflux pump contribution to multidrug resistance in clinical isolates of Pseudomonas aeruginosa . Pharmacotherapy 30: 632–638. [DOI] [PubMed] [Google Scholar]
- 64. Li, X.‐Z. & Plésiat P.. 2016. Antimicrobial drug efflux pumps in Pseudomonas aeruginosa In Efflux‐Mediated Antimicrobial Resistance in Bacteria: Mechanisms, Regulation and Clinical Implications. Li X.‐Z., Elkins C.A. & Zgurskaya H.I., Eds.: 359–400. Cham: Springer International Publishing. [Google Scholar]
- 65. Bradley, P. et al 2015. Rapid antibiotic‐resistance predictions from genome sequence data for Staphylococcus aureus and Mycobacterium tuberculosis . Nat. Commun. 6: 10063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Reeve, S.M. et al 2015. Protein design algorithms predict viable resistance to an experimental antifolate. Proc. Natl. Acad. Sci. U.S.A. 112: 749–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Coelho, J.R. et al 2013. The use of machine learning methodologies to analyse antibiotic and biocide susceptibility in Staphylococcus aureus . PLoS One 8: e55582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Niehaus, K.E. et al 2014. Machine learning for the prediction of antibacterial susceptibility in Mycobacterium tuberculosis In IEEE‐EMBS International Conference on Biomedical and Health Informatics (BHI). 618–621. 10.1109/BHI.2014.6864440 [DOI] [Google Scholar]
- 69. Wattam, A.R. et al 2014. PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res. 42: D581–D591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Davis, J.J. et al 2016. Antimicrobial resistance prediction in PATRIC and RAST. Sci. Rep. 6: 27930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Gallagher, L.A. , Shendure J. & Manoil C.. 2011. Genome‐scale identification of resistance functions in Pseudomonas aeruginosa using Tn‐seq. mBio 2: e00315‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Melnyk, A.H. , Wong A. & Kassen R.. 2015. The fitness costs of antibiotic resistance mutations. Evol. Appl. 8: 273–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wong, A. , Rodrigue N. & Kassen R.. 2012. Genomics of adaptation during experimental evolution of the opportunistic pathogen Pseudomonas aeruginosa . PLoS Genet. 8: e1002928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Piddock, L.J. 2016. Assess drug‐resistance phenotypes, not just genotypes. Nat. Microbiol. 1: 16120. [DOI] [PubMed] [Google Scholar]
- 75. Andersson, D.I. & Hughes D.. 2014. Microbiological effects of sublethal levels of antibiotics. Nat. Rev. Microbiol. 12: 465–478. [DOI] [PubMed] [Google Scholar]
- 76. Spratt, B.G. , Hanage W.P. & Feil E.J.. 2001. The relative contributions of recombination and point mutation to the diversification of bacterial clones. Curr. Opin. Microbiol. 4: 602–606. [DOI] [PubMed] [Google Scholar]
- 77. Sandegren, L. & Andersson D.I.. 2009. Bacterial gene amplification: implications for the evolution of antibiotic resistance. Nat. Rev. Microbiol. 7: 578–588. [DOI] [PubMed] [Google Scholar]
- 78. Chewapreecha, C. et al 2014. Comprehensive identification of single nucleotide polymorphisms associated with beta‐lactam resistance within pneumococcal mosaic genes. PLoS Genet. 10: e1004547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Dotsch, A. et al 2015. The Pseudomonas aeruginosa transcriptional landscape is shaped by environmental heterogeneity and genetic variation. mBio 6: e00749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Gawad, C. , Koh W. & Quake S.R.. 2016. Single‐cell genome sequencing: current state of the science. Nat. Rev. Genet. 17: 175–188. [DOI] [PubMed] [Google Scholar]