

Abstract
Aim
Neurodegeneration is associated with dysfunction of calcium buffering capacity and thereby sustained cellular and mitochondrial calcium overload. Paraneoplastic cerebellar degeneration (PCD), characterized by progressive Purkinje neurone degeneration following paraneoplastic Yo antibody internalization and binding to cerebellar degeneration‐related protein CDR2 and CDR2L, has been linked to intracellular calcium homeostasis imbalance due to calbindin D28k malfunction. Therefore, we hypothesized that Yo antibody internalization affects not only calbindin calcium binding capacity, but also calcium‐sensitive mitochondrial‐associated signalling, causing mitochondrial calcium overload and thereby Purkinje neurone death.
Methods
Immunohistochemically, we evaluated cerebellar organotypic slice cultures of rat brains after inducing PCD through the application of Yo antibody‐positive PCD patient sera or purified antibodies against CDR2 and CDR2L how pharmacologically biased mitochondrial signalling affected PCD pathology.
Results
We found that Yo antibody internalization into Purkinje neurons caused depletion of Purkinje neurone calbindin‐immunoreactivity, cannabinoid 1 receptor over‐activation and alterations in the actions of the mitochondria permeability transition pore (MPTP), voltage‐dependent anion channels, reactive oxygen species (ROS) and Na+/Ca2+ exchangers (NCX). The pathological mechanisms caused by Yo antibody binding to CDR2 or CDR2L differed between the two targets. Yo‐CDR2 binding did not alter the mitochondrial calcium retention capacity, cyclophilin D‐independent opening of MPTP or activity of NCX.
Conclusion
These findings suggest that minimizing intracellular calcium overload toxicity either directly with cyclosporin‐A or indirectly with cannabidiol or the ROS scavenger butylated hydroxytoluene promotes mitochondrial calcium homeostasis and may therefore be used as future neuroprotective therapy for PCD patients.
Keywords: calcium homeostasis, calcium‐sensitive mitochondrial‐associated signalling, cerebellar degeneration‐related proteins CDR2 and CDR2L, paraneoplastic cerebellar degeneration, paraneoplastic Yo antibody
Introduction
Paraneoplastic neurological syndromes are autoimmune‐mediated neurodegenerative diseases caused by autoantibodies and autoreactive T cells against specific tumour types 1. Paraneoplastic cerebellar degeneration (PCD) is associated with severe and progressive ataxia, dysarthria and nystagmus due to the loss of cerebellar Purkinje neurons 1, 2, 3. PCD associated with the paraneoplastic autoantibody Yo occurs mainly in breast or ovarian cancer patients 2. The pathogenesis of Yo‐PCD is not completely understood, but Purkinje neurone loss may occur due to autoreactive T cells 3, 4, 5, 6, 7 and Yo autoantibodies 8, 9, 10, 11. Autoantibody Yo cross‐react with the cerebellar degeneration‐related proteins, CDR2 and CDR2Like (CDR2L) 1, 12, 13, which have approximately 50% sequence identity 12, 13. CDR2 and CDR2L are widely expressed in gynaecological tumours 14, 15, normal tissue 15 and brain 16, 17, 18, where Yo recognizes the leucine zipper motif of CDR2 19. In the cerebellum, CDR2 and CDR2L are present in neuronal cytoplasm and proximal dendrites of Purkinje neurons but little is known about their neuronal functions 16, 17, 18, 20. Functionally, CDR2 is connected to c‐myc and the expression of calcium modulator and buffer calbindin D28k 17, 20, 21, whereas CDR2L is linked to plasma membrane signalling involving voltage‐gated calcium channel‐ (VGCC) or AMPA receptor‐mediated calcium flux regulation 20.
Maintenance of intracellular calcium homeostasis, signal transduction regulation and ATP production are critical for normal cell metabolism and mitochondrial homeostasis, and if these processes are deregulated or dysfunctional, neurodegeneration eventually occurs 22, 23, 24, 25. Dysfunction of voltage‐dependent anion channels (VDAC) or Na+/Ca2+ exchangers (NCX) 26, deregulation of the mitochondrial calcium buffering capacity through mitochondria permeability transition pore (MPTP) opening 22, 25, or the increase in cytochrome‐C and reactive oxygen species (ROS) signalling and production 23, 27 can all lead to excessive mitochondrial calcium overload, which in turn increases the intracellular calcium levels and can thereby induce cell death 24. Calcium homeostasis is not only regulated by influx and efflux of calcium but also by the modulation of the availability of free intracellular calcium through calcium buffer molecules like calbindin D28k 28. Calbindin D28k depletion has been correlated to neurodegeneration in PCD 29, Parkinson's disease 21 and Alzheimer's disease 30. Furthermore, in Purkinje neurons, calbindin D28k serves as a functional biomarker, as it modulates intracellular calcium and regulates Purkinje neurone motor coordination precision 28, 31, 32, which is affected in Yo‐PCD patients. Recent experimental evidence suggests that Yo antibodies affect intracellular calcium homeostasis 20; however, the detailed mechanisms are still largely unknown. As current antineoplastic and immunosuppressive PCD treatment is inadequate 33, greater understanding of the mechanisms behind the Yo driven calcium homeostasis imbalance could potentially provide crucial knowledge for development of effective neuroprotective therapies. We, therefore, investigated whether binding of autoantibody Yo to CDR2 and CDR2L affected mitochondrial homeostasis by investigating the activity of the MPTP, VDAC, NCX and ROS, and the deregulated activity of cannabinoid 1 receptor (CB1R) in Purkinje neurons by using a rat‐based ex vivo PCD model of cerebellar organotypic slice culture (cOTSC). Clinically relevant compounds were used to modulate the assumed dysregulation of the calcium‐sensitive mitochondrial‐associated signalling.
Materials and methods
Patient sera
Sera were obtained from four patients with gynaecological cancer and PCD who had Yo autoantibodies against CDR2 and CDR2L (anti‐Yo1–4) 12 but lacked P/Q‐type VGCC antibodies 20. Four sex‐ and age‐matched nonparaneoplastic neurological syndrome sera (non‐hCDR1–4) and a pool of sera from 100 healthy donors (non‐hCDR100p) were used as controls. Sera were not heat‐inactivated before use. Control and anti‐Yo patient sera were collected before patients were treated for cancer or PCD and were stored at the PND Biobank #133/2015 or the Biobank for diagnostic cancer marker #188.05 with approval of the regional ethics committee, Western‐Norway.
Cerebellar organotypic slice culture
All procedures were performed according to the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals Norway (FOTS 20135149/20157494). Wistar Hannover GLAST rat pups (n = 224), age P10 to P15, were used for cOTSC preparation as previously described 20 (Figure 1 A). Briefly, following anaesthesia and decapitation, the cerebellum was rapidly transferred into ice‐cold EBSS solution (#24010043; Gibco‐ThermoFisher Scientific, Waltham, MA, USA) containing 0.5% glucose (#G8769; Sigma‐Aldrich, St. Louis, Mo, USA) and 10 mM HEPES (#15630056; Gibco). Four cerebellar parasagittal slices (400‐μm thick) from the vermis were cut on NVSLM1 motorized advance vibroslice (WPI) and transferred onto membranes of 12‐mm2 Millipore cell culture inserts with 0.4‐μm pore size (# PICMO1250, Millicell; Merck ‐ Millipore, Burlington, MA, USA) for each cerebellum (Figure 1 A). Slices were maintained in 24‐well plates at the air/culture media interface consisting of 30% advanced DMEM/F12 solution (#126340010; Gibco), 20% MEM solution (#41090028; Gibco), 25% EBSS solution, 25% heat‐inactivated horse serum (#H1138; Sigma), 1 mM l‐glutamine (#35050038; Gibco), 5 mg/ml d‐glucose and 2% B‐27 serum‐free supplement (#17504044, Gibco) and were incubated with 5% CO2 at 35°C. All culture medium was removed and replaced 24 h postslicing, and 75% of the medium was replaced every 48 h thereafter. The slices were allowed to recover for 7 days before commencement of treatment.
Figure 1.
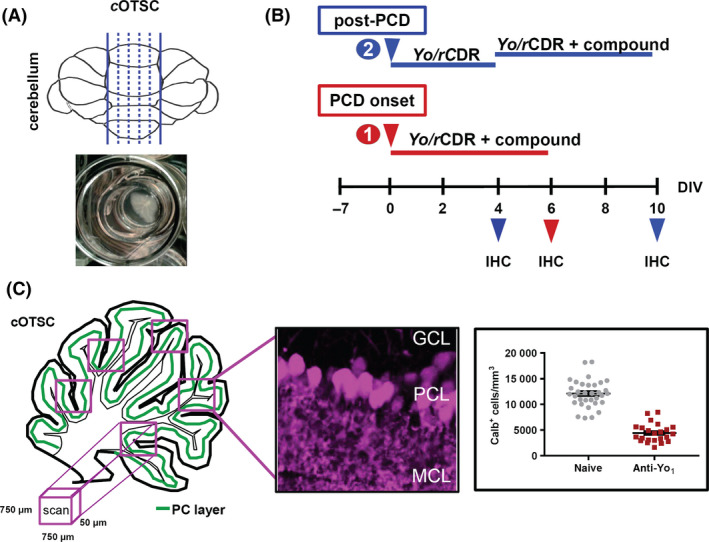
Ex vivo paraneoplastic cerebellar degeneration (PCD) model. (A) The illustration shows the cutting planes of the cerebellar vermis including the incubation chamber to maintain the rat cerebellar organotypic slice culture (cOTSC), the basis of the ex vivo PCD model system. (B) Experimental design: 7 days’ postpreparation, anti‐Yo/rCDR and compounds were co‐applied either immediately at PCD onset [(1), red arrow] or 4 days after PCD induction [(2), post‐PCD, blue arrow] for 6 days. The pathological progression of anti‐Yo/rCDR internalization was determined by immunohistochemical (IHC) staining of Purkinje neurons with biomarker calbindin at 4, 6 and 10 days of treatment. (C) Three to eight confocal images of 750 × 750 × 50 μm in each cOTSC slice for each treatment were scanned and the Yo antibody incorporation‐dependent slice pathology (4, 6 and 10 days) was analysed by counting Purkinje neurone somata in the Purkinje cell layer (PCL) immunohistochemically visualized by biomarker calbindin D28K (magenta). Calbindin‐positive cells (Calb+) were calculated as number of Calb+ cells/mm3 and plotted for each group.
Ex vivo PCD model
Seven days postslicing, the cOTSC medium was replaced with medium containing; human serum positive for autoantibody Yo (anti‐Yo; hCDR2/2L; 4 μl/ml) or heat‐inactivated (56°C, 45 min) polyclonal affinity‐purified rabbit CDR antibodies (anti‐rCDR; 100 ng/ml) (Figure 1 B). The hCDR model involved anti‐Yo and two controls, non‐hCDR1–4 and non‐hCDR100p. The rCDR model involved affinity‐purified rabbit polyclonal antibody against CDR2 (#HPA023870; Sigma), CDR2L (#HPA022015; Sigma), or CDR2/2L (1:1 mixture), and the antibody control rabbit immunoglobulin G (rIgG; #12370, Millipore). Slices were collected 4, 6 or 10 days after commencement of treatment to evaluate the antibody effects (Figure 1 B). Each independent experiment included positive (non‐hCDR1–4; non‐hCDR100p; rIgG) and negative (naive) controls to account for variations in cell survival between slice preparations and the experimental approaches (with and without sera, antibodies and compounds). All treatments were performed in triplicate or quadruplicate.
Purkinje neurone‐specific antibodies
As a control to evaluate the specificity of the cytotoxic nature of anti‐Yo internalization in PCD, we studied two Purkinje neurone‐specific antigens, GAD65 and PCP2 (also known as L7). cOTSC was treated with 1000 ng/ml mouse anti‐GAD65 (#559931; BD Bioscience, Franklin Lakes, NJ, USA) or 1000 ng/ml rabbit anti‐PCP2 (#M194; Takara Bio Inc., Kusatsu, Shinga, Japan) for 6 days. These concentrations were 10‐fold higher than the anti‐rCDR concentration used in the rCDR PCD model.
Neuropharmacology
Approaches 1 and 2 (Figure 1 B): To evaluate the neuroprotective potential of compounds that modulate calcium‐sensitive mitochondrial‐associated signalling, we co‐applied the compounds with anti‐Yo/rCDR immediately at PCD onset (approach 1) or 4 days after PCD induction (approach 2); cultures were treated for 6 days. Compounds tested were AM281 [IC50: 12 nM (CB1R), 4.2 μM (CB2R); #1115, Tocris Bioscience, Bristol, UK], benzamil [IC50: approximately 100 nM (NCX); #3380; Tocris], butylated hydroxytoluene [BHT; IC50: 3.5 μM (ROS); #W218405; Sigma], cannabidiol [CBD; IC50: 3.35 μM (CB1R), 27.5 μM (CB2R); #1570; Tocris], CGP37157 [IC50: 400 nM (NCX); #1114; Tocris], cyclosporin‐A [CsA; IC50: 5 nM (MPTP); #1101; Tocris] and KB‐R7943 [IC50: 700 nM (NCXrev), 5.5 μM (MCU); #1244; Tocris]. As this PCD model system is based on an interface hydrophilic PTFE membrane‐diffusion approach where the cOTSC is not submerged, the system behaves like an in vivo brain. Therefore, several test dosages were evaluated starting with 2.5‐fold of the IC50 value to determine effective, nontoxic doses if there was no literature guided initial dose selection available for this kind of culture system. Depending on the hydrophilic or hydrophobic nature of the used compounds, the effective dosage was determined between 2.5 to 200 times of the IC50 value.
MitoTracker
cOTSC was incubated at 37°C in culture media with MitoTracker Deep Red FM (MTDR, 500 nM; #M22426; Invitrogen, Carlsbad, CA, USA) or MitoTracker Red CM‐H2xROS (MTROS, 1 μM; #M7513; Invitrogen) for 30 min before fixation to evaluate the mitochondrial membrane potential (Δψ) and ROS output under anti‐Yo as well as neuropharmacological treatment. As a positive control for increased O2 consumption, the mitochondrial protonophore uncouplers FCCP (2 μM; IC50: 11.5 μM; #0453; Tocris) 34 and BAM15 (500 nM; EC50: 270 nM; #5737; Tocris) 35 were added 15 min after MitoTracker application, respectively. Unbound/unreactive MitoTracker was removed by wash with prewarm 0.1 M PBS (37°C, 4 × 1 min) before fixation. The relative fluorescence intensity was quantified using Fiji Measurement tool by creating a Plot Profile of a selected region of interest and corrected the integrated density to mean fluorescence background.
Immunohistochemistry
To evaluate Purkinje neurone health at the end of the treatment, cOTSC was washed with prewarmed 0.1 M PBS (1 × PBS; #70013016; Gibco) and fixed with 4% paraformaldehyde (PFA‐PBS, pH 7.2, #28908, ThermoFisher Scientific) containing 0.5% sucrose for 4 h at 4°C. Slices were quenched with 1 × PBS containing 50 mM NH4Cl (PBSN, #A9434, Sigma), permeabilized with 1% Triton X‐100 (#T9284; Sigma) in PBSN (60 min, 22°C), rinsed with PBSN (3 × 15 min), and incubated with 2 μg/ml primary antibody to calbindin D28K (#C9848, mouse, Sigma; #214005, guinea pig, SySy ‐ Synaptic Systems, Göttingen, Germany) for 2 days at 4°C in PBSN containing 5% bovine serum albumin (BSA; #A2153; Sigma), 0.2% Triton X‐100 and 100 μM glycine (#G7126; Sigma). To visualize not only Purkinje neurons by biomarker calbindin, but also the 4–10 days lasting live uptake of anti‐Yo and anti‐rCDR, the slices were rinsed with PBSN (3 × 15 min) and incubated with highly cross‐absorbed secondary antibodies: donkey anti‐mouse or anti‐guinea pig (Calb), donkey anti‐rabbit (rCDR) and donkey anti‐human (Yo) conjugated to CF™488/594/647‐Dye (1:400; #20014, #20115, #20046, #20015, #20152, #20047, #20074, #20075, #20169, #20170; Biotium, Fremont, CA, USA) for 2 days at 4°C in PBSN containing 2.5% BSA. To remove unbound secondary antibody, slices were rinsed with PBSN (3 × 15 min), and briefly tipped into MilliQ water before mounting in hardening PromoFluor Antifade Reagent (#PK‐PF‐AFR1; Promikine ‐ PromoCell, Heidelberg, Germany). After 2 days of hardening at 18–21°C in the dark, slices were stored at 4°C until imaging as previously described 20. All slices for each experimental setup were stained simultaneously to minimize variations in immunoreactivity levels within the investigated groups.
Imaging
Z‐stack images were collected at 0.5–1 μm intervals with a DMI6000‐CS‐TCS‐SP5 microscope using a HC‐PL‐APO 20 × 0.75 IMM‐CORR‐CS2 objective to detect CF™488/594/647 dye emission and superimposed with LASAF software V2.5.1 (Leica Microsystems GmbH, Wetzlar, Germany). The fluorescence intensity was adjusted to 75% of the maximum in untreated naive control tissue for each experiment.
Quantification of Purkinje neurone pathology
The anti‐Yo‐induced pathological state was evaluated by assessment of functional biomarker calbindin, neurodegeneration marker Fluoro‐Jade‐C (0.001%, 30 min prior mounting; #AG325; Millipore), and cell death marker propidium iodide (2 μM, 48 h prior to fixation; #P3566; Invitrogen) as described by Brana and Noraberg 36, 37. The pathological progress was quantified by counting calbindin‐immunoreactive Purkinje neurone somata (Calb+ cells) manually (but blind) and automatically in 3–8 images of 750 × 750 × 50 μm in each slice for each treatment, experiment, and group with counts reported per mm3 (Figure 1 C). The automatic count showed a discrepancy of approximately 5% compared to manual count; therefore, each experimental set was counted solely manually or automatically. The automatic count was performed with Fiji as previously described 20.
Data analysis and statistics
All treatments were performed in triplicate or quadruplicate. Data were evaluated using the nonparametric Mann–Whitney U‐test (Graph‐Pad‐Prism 4.0 software, San Diego, CA, USA) with P < 0.05 considered statistically significant.
Results
Rat cOTSC‐based ex vivo PCD model
Yo antibody‐positive sera from four PCD patients, and purified rabbit polyclonal antibodies against the Yo antigens CDR2 and CDR2L were used to induce Purkinje neurone degeneration. The antibody‐induced Purkinje neurone pathology was evaluated by visualizing the targeted neurons with the functional biomarker calbindin D28k (Calb). Confocal imaging 6 days after application of Yo antibody showed that the tested Yo+ sera reduced the Calb+ cell count by 70.7 ± 7.8% (Yo1) and 67.1 ± 7.0% (Yo2) relative to naive controls (Figure 2 A). This effect was specific to anti‐Yo as none of the control sera (non‐hCDR1–4 and non‐hCDR100p) caused depletion of calbindin‐immunoreactive cells (Figure 2 A). Similar results were obtained when purified rabbit polyclonal antibodies against CDR2 and CDR2L were used. Antibody against CDR2 reduced the Calb+ cell count by 62.4 ± 5.5% and antibody against CDR2L by 67.7 ± 5.7%, whereas antibodies against other Purkinje neurone‐specific proteins such as anti‐PCP2 or ataxia‐related anti‐GAD65 showed no loss of calbindin immunostaining (Figure 2 B). To verify that the depletion of calbindin‐immunoreactive cells reflects neurodegeneration, we employed the marker Fluoro‐Jade‐C. After Yo antibody treatment, the few remaining Calb+ Purkinje neurons were neither positive for Yo incorporation nor the neurodegeneration marker Fluoro‐Jade‐C (Figure 2 C). In contrast, the calbindin depleted Purkinje neurons showed strong Yo internalization, Jade‐C staining and shrunken cell bodies (Figure 2 C1), but were negative for cell death marker propidium iodide (Figure 2 C2), indicating that neurodegeneration had begun but that cell death had not yet occurred.
Figure 2.
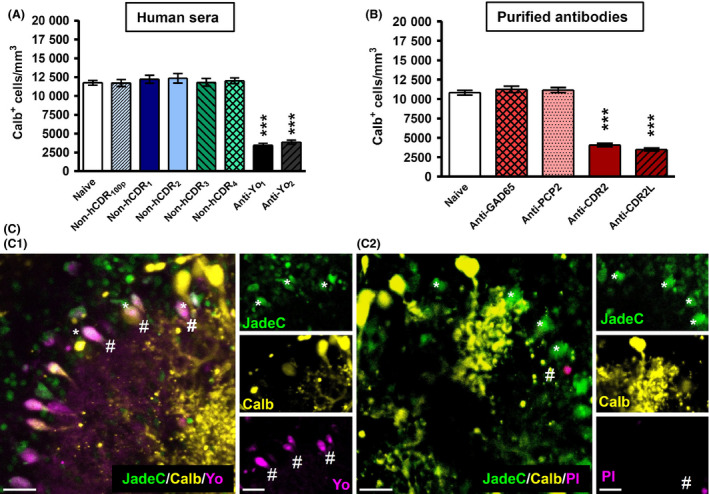
Yo and rCDR antibody internalization depletes calbindin D28k in rat Purkinje neurons. Significant depletion of calbindin‐immunoreactive cells was observed after (A) Yo antibody and (B) rCDR antibody incorporation, whereas control sera (non‐hCDR1–4,100p) but IgG controls showed no impact on the Calb+ cell count. (B) The incubation with antibodies against Purkinje neurone specific protein 2 (PCP2) or ataxia‐related GAD65 (1000 ng, 6 days) revealed no reduction in the Calb+ cell count. (C) Calbindin depleted Purkinje neurons were positive for the neurodegeneration marker Fluoro‐Jade‐C (*, green) and showed high incorporation of Yo antibodies (#, magenta) (C1). The cell death marker propidium iodine (#) (C2, magenta) was not found either in Calb+ (yellow) or Jade‐C+ (green) Purkinje neurons after 6 days of Yo antibody incorporation. Scale bar 25 μm.
Yo antibody binding to CDR2/2L leads to deregulation of cyclophilin D‐dependent MPTP opening and increased ROS production
Immunohistochemical staining revealed that CsA (1 μM), which inhibits the cyclophilin D‐dependent opening of the MPTP 22, abolished the CDR antibody associated loss of calbindin immunoreactivity when cultures were co‐treated at PCD onset (Figure 3 A,B) and after PCD was established (Figure 3 C; post‐PCD). CsA showed similar neuroprotective effects whether PCD was induced by human Yo+ PCD sera or by purified CDR antibodies and did not affect the counts of Calb+ cells in the control slices (Figure 3 A–C) nor the Yo antibody internalization (Figure 3 D). CsA co‐treatment reduced the numbers of shrunken Purkinje neurone cell bodies that showed high Yo antibody internalization. When cultures were treated with anti‐Yo and CsA simultaneously, most Yo+ cells had normal cell body size and were positive for calbindin (Figure 3 D). BHT (10 μM), a ROS scavenger 38, was tested to confirm that Yo antibody internalization leads to deregulation of the mitochondrial calcium buffering capacity, indicated by the CsA data. At PCD onset, BHT completely prevented the depletion of calbindin‐immunoreactive cells caused by anti‐Yo2–4 and anti‐rCDR2/2L internalization but was less effective against anti‐Yo1, anti‐rCDR2 and anti‐rCDR2L‐induced pathology (Figure 3 A,B). After PCD was established (post‐PCD), BHT treatment was able to reduce the loss of calbindin immunoreactivity caused by anti‐Yo1–2 and anti‐rCDR2/2L internalization, almost to baseline (Figure 3 C). BHT had no impact on control tissue (Figure 3 A–C) and did not prevent the internalization of Yo antibodies (Figure 3 D). Under BHT treatment, the cell count of Purkinje neurons positive for both calbindin and anti‐Yo increased and Yo‐positive Purkinje neurons did not display shrunken cell bodies. In support of the results obtained with CsA, these data indicate that Yo antibody binding to CDR2 and CDR2L induces ROS over‐production potentially through calcium overload and effect on respiratory chain complex I 27.
Figure 3.
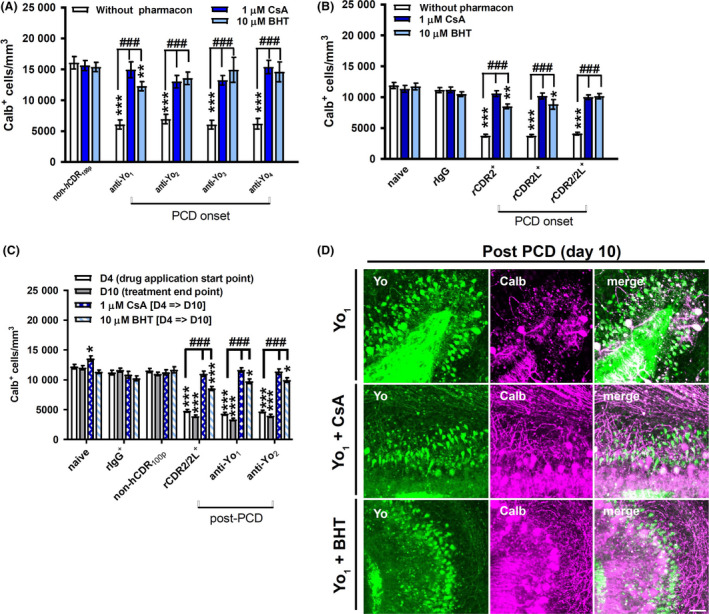
Yo antibody binding to CDR2 and CDR2L leads to deregulation of mitochondria permeability transition pore opening and reactive oxygen species (ROS) production. (A–C) Calb+ cells per mm3 in cerebellar organotypic slice culture in response to treatment with anti‐Yo/rCDR and co‐treatment with cyclosporin‐A (CsA, n = 5) and ROS scavenger butylated hydroxytoluene (BHT, n = 4). The antagonistic effects on anti‐Yo‐induced pathology are represented in panels A, C, and for anti‐rCDRs‐induced pathology in panels B, C. Panels A, B, are showing the data collected when the antagonists were directly applied at paraneoplastic cerebellar degeneration (PCD) onset and panels C when applied at day 4 after PCD pathology was established. The neuroprotective capacity of the tested compounds was evaluated after 6 days for both treatment strategies. CsA and BHT significantly reduced or abolished neurodegeneration‐related depletion of calbindin‐immunoreactive cells in both treatment strategies. Data are collected at treatment day 6 (onset‐PCD) or 4 and 10 (post‐PCD) and expressed as mean ± SEM [*P < 0.05; **P < 0.01; ***P < 0.001; nonparametric two‐tailed paired Mann–Whitney's U‐test; asterisk (*): significance to naive control; hash (#): significance to anti‐Yo/rCDR]. (D) z‐stack confocal micrographs collected at day 10 of treatment showed that CsA (middle row) and BHT (bottom row) co‐applied for 6 days from day 4 of Yo antibody incorporation (post‐PCD) were not affecting the uptake of anti‐Yo (green, left column) but reduced depletion of calbindin‐immunoreactive cells (magenta, middle column) in comparison to anti‐Yo incorporation alone (top row). Under CsA and BHT treatment, two types of Yo+ cells were observed: Yo+ cells (green) negative for calbindin (magenta) with shrunken Purkinje neurone somata and Yo+ cells (green) positive for calbindin (magenta, merge white) without Purkinje neurone somata shrinkage (right column). Scale bar 25 μm.
Yo antibody binding to CDR2 impacts CB1R activity
CB1Rs are highly expressed in the cerebellar molecular layer 39, 40. The CB1R antagonist AM281 (10 μM) completely rescued the depletion of calbindin‐immunoreactive cells induced by anti‐Yo1/2 and partially by anti‐Yo3/4 (anti‐Yo3: 24.0 ± 7.9%, P = 0.0010; anti‐Yo4: 28.0 ± 6.1%, P < 0.0001; Figure 4 A). However, AM281 caused a profound loss of calbindin‐positive cells in control cOTSC (50.6 ± 8.1%, P < 0.001; Figure 4 A). Besides AM281, we also tested the clinically used low‐potency CB1R inverse agonist CBD (10 μM) 41. CBD prevented the depletion of calbindin‐immunoreactive cells when PCD was induced by human Yo+ PCD sera (Figure 4 A). The depletion of calbindin‐immunoreactive cells induced by anti‐rCDR was only partially rescued, and there were differences in the CBD protection level for the anti‐rCDR2, anti‐rCDR2L and anti‐rCDR2/2L treatments (Figure 4 B). At PCD onset, CBD was less effective against anti‐rCDR2L than anti‐rCDR2 or anti‐rCDR2/2L (Figure 4 B). cOTSC treated with a 1:1 mixture of anti‐rCDR2 and anti‐rCDR2L responded more favourably to the CBD treatment than cultures treated with anti‐rCDRs individually (CDR2 + CBD vs. CDR2/2L + CBD P = 0.0014; CDR2L + CBD vs. CDR2/2L + CBD P < 0.0001; CDR2 + CBD vs. CDR2L + CBD P = 0.0874). In PCD free control slices, CBD caused a slight reduction in calbindin‐positive Purkinje neurons [22.8 ± 4.1% (naive), 16.9 ± 3.1% (rIgG); P < 0.001; Figure 4 B]. When CBD was applied after PCD was established, at day 4, depletion of calbindin‐immunoreactive cells was reduced in anti‐Yo and anti‐rCDR2/2L‐treated cultures but levels did not return to baseline control levels (P < 0.001; Figure 4 C). Furthermore, as seen for CsA and BHT also CBD reduced the anti‐Yo‐induced depletion of calbindin‐immunoreactive cells, including the anti‐Yo‐associated cell shrinkage but without preventing the antibody uptake in itself (Figure 4 D).
Figure 4.
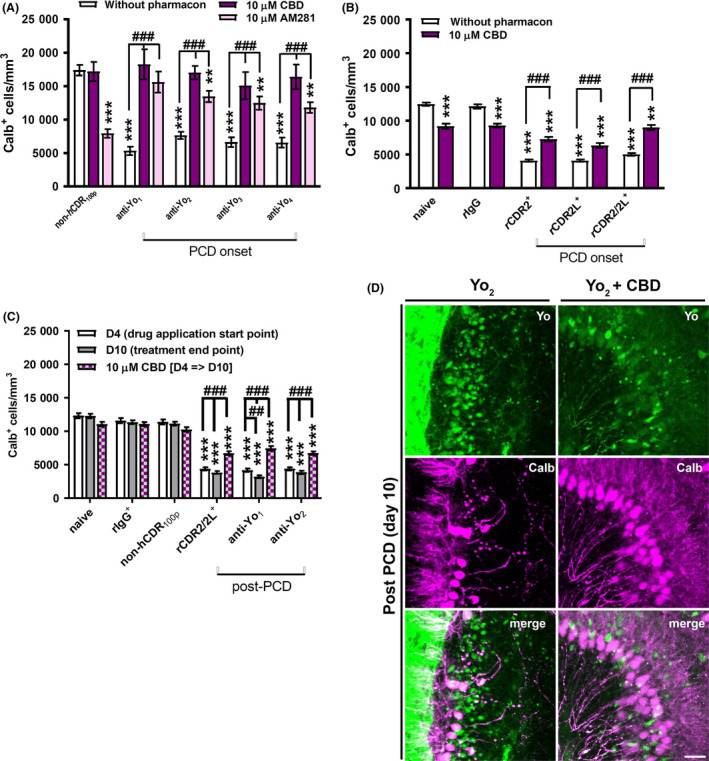
Yo antibody binding to CDR2 impacts cannabinoid 1 receptor activity. (A–C) Calb+ cells per mm3 in cerebellar organotypic slice culture in response to treatment with anti‐Yo/rCDR and co‐treatment with AM281 (n = 3) and cannabidiol (CBD, n = 6). The antagonistic effects on anti‐Yo‐induced pathology are represented in panels A, C, and for anti‐rCDRs‐induced pathology in panels B, C. Panels A and B are showing the data collected when the antagonists were directly applied at paraneoplastic cerebellar degeneration (PCD) onset and panels C when applied at day 4 after PCD pathology was established. The neuroprotective capacity of the tested compounds was evaluated after 6 days for both treatment strategies. AM281 and CBD minimized the Yo‐induced depletion of calbindin‐immunoreactive cells but caused depletion of calbindin‐immunoreactive cells in naive, IgG and serum controls, respectively. Data are collected at treatment day 6 (onset‐PCD) or 4 and 10 (post‐PCD) and expressed as mean ± SEM [*P < 0.05; **P < 0.01; ***P < 0.001; nonparametric two‐tailed paired Mann–Whitney's U‐test; asterisk (*): significance to naive control; hash (#): significance to anti‐Yo/rCDR]. (D) z‐stack confocal micrographs collected at day 10 of treatment showed that CBD co‐applied for 6 days from day 4 of Yo antibody incorporation (post‐PCD) was not affecting the uptake of anti‐Yo (green) itself (top row) but reduced depletion of calbindin‐immunoreactive cells (magenta, middle row) in comparison to Yo incorporation alone. Yo+ cells (green) negative for calbindin with shrunken Purkinje neurone somata as well as Yo+ cells (green) positive for calbindin (white) without Purkinje neurone somata shrinkage were observed under CBD treatment (bottom row). Scale bar 25 μm.
Yo antibody binding reduces mitochondrial membrane potential and increases ROS activity
To support the pharmacological data, we performed a semiquantitative fluorescence intensity analysis of the mitochondrial membrane potential by using MitoTracker deep red (MTDR) and also ROS activity by using MitoTracker Red CM‐H2xROS (MTROS) (Figure 5 A,C). MTDR signal was reduced by 71.76 ± 5.5% after 10 days of anti‐Yo internalization (P < 0.001; Figure 5 B). This anti‐Yo‐induced MTDR level reduction was similar to MTDR levels observed under protonophore uncoupler FCCP or BAM15 [78.59 ± 10.9% (FCCP), 77.78 ± 14.5% (BAM15); P < 0.001; Figure 5 B]. Following PCD initiation (post‐PCD condition), loss of MTDR fluorescence was decreased but not entirely prevented by CsA, BHT and CBD, whereas BHT showed the highest effectiveness, followed by CBD and then CsA [MTDR fluorescence reduction: 48.87 ± 4.7% (Yo/CsA), 10.97 ± 4.9% (Yo/BHT), 26.21 ± 4.1% (Yo/CBD); P < 0.001; Figure 5 B]. MTROS staining showed a 2‐fold increase after 10 days of anti‐Yo internalization [1.904 ± 0.084 (Yo); P < 0.001; Figure 5 D]. Under post‐PCD experimental settings, the anti‐Yo increased MTROS level was not altered by CsA (1.771 ± 0.098; P = 0.2977); it was reduced to control level by BHT (0.956 ± 0.063; P < 0.0001) and potentiated by CBD (2.701 ± 0.1404; P < 0.001) (Figure 5 D). Interestingly, the MTROS signal was not affected by protonphore uncoupler BAM15 but significantly increased by FCCP (1.816 ± 0.075; P < 0.001).
Figure 5.
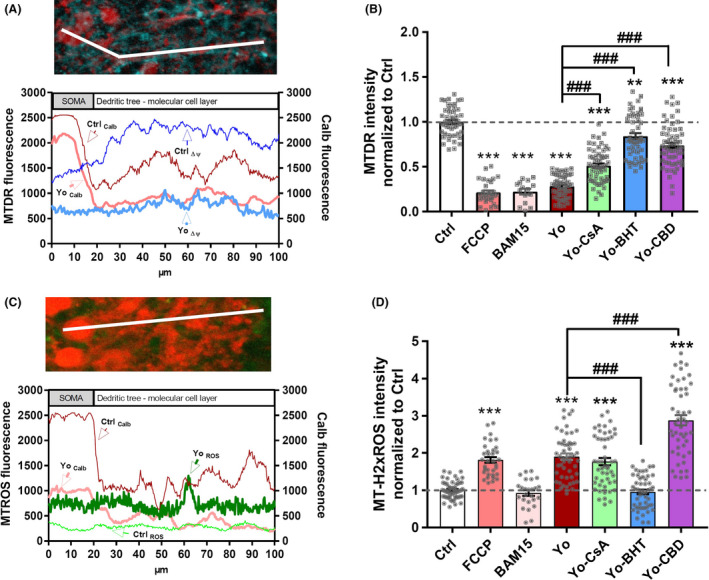
Yo antibody binding to CDR2 and CDR2L reduces mitochondrial membrane potential and boosts reactive oxygen species (ROS) production. At day 10 of postparaneoplastic cerebellar degeneration treatment, Purkinje neurons were loaded with (A,B) MitoTracker Deep Red (MTDR) or (C,D) MitoTracker Red CM‐H2xROS (MTROS) and stained for calbindin D28k (Calb). (A,C) Representative line‐scan plot across a single Purkinje neurone from soma to dendritic tree in the molecular cell layer showing the fluorescence dissipation of (A) MTDR (blue) and (C) MTROS (green) neuroanatomy‐dependent indicated by the calbindin (red) fluorescence course in an untreated control (ctrl) slice vs. a slice that had internalized anti‐Yo (Yo). (B,D) Relative fluorescence intensity of (B) MTDR and (D) MTROS normalized to control in response to treatment with anti‐Yo and co‐treatment with cyclosporin‐A (CsA, n = 3), ROS scavenger butylated hydroxytoluene (BHT, n = 3) and cannabidiol (CBD, n = 3). (B) Anti‐Yo internalization reduced the mitochondrial membrane potential (Δψ) similar to protonophore uncoupler FCCP or BAM15. CSA, BHT and CBD prevented partially the anti‐Yo induced Δψ reduction (D) Anti‐Yo internalization enhanced ROS production which was suppressed by BHT treatment, unaffected by CSA and potentiated by CBD. Data are collected at treatment day 10, expressed as mean ± SEM and normalized to control [*P < 0.05; **P < 0.01; ***P < 0.001; nonparametric two‐tailed paired Mann–Whitney's U‐test; asterisk (*): significance to naive control; hash (#): significance to anti‐Yo].
Yo antibody binding to CDR2L deregulates NCX signalling
As there is a tight coupling of intracellular calcium homeostasis and mitochondrial‐associated signalling, we evaluated whether Yo antibody internalization alters the function of NCX. NCX exchange of Ca2+ for Na+ is essential for neurone survival and associated with plasma and mitochondrial membranes 25, 26, 42. Under certain pathological conditions, NCX goes into reverse mode (NCXrev) and increases calcium entry into the cell by 30% 26 with deleterious consequences for cellular homeostasis 25. We found that at PCD onset NCXrev antagonist KB‐R7943 (KB‐R, 10 μM) 43, 44, or NCX antagonists CGP37157 (CGP, 10 μM) or benzamil (Benz, 250 nM) 42, reduced depletion of calbindin‐immunoreactive cells by up to 60% when co‐applied with Yo+ PCD sera or antibody against CDR2L (Figure 6 A,B). Interestingly, the depletion of calbindin‐immunoreactive cells induced by CDR2 antibody binding was not affected by KB‐R7943 and CGP37157. In control slices, however, blocking NCXrev with KB‐R7943 did cause strong depletion of calbindin‐immunoreactive cells (53.2 ± 6.2%; P < 0.001), whereas the blockage of NCX with CGP37157 or benzamil did not affect the Calb+ cell count (Figure 6 B).
Figure 6.
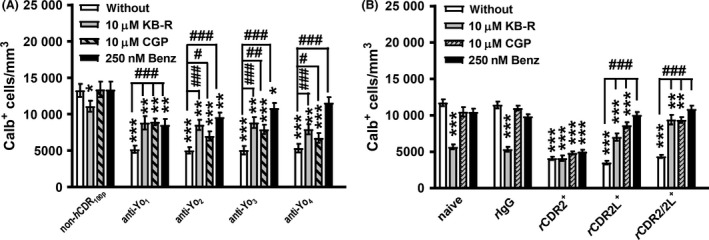
Yo antibody binding to CDR2L deregulates Na+/Ca2+ exchanger (NCX) signalling. (A,B) Calb+ cells per mm3 in cerebellar organotypic slice culture in response to treatment with anti‐Yo/rCDR and co‐treatment with KB‐R7943 (KB‐R, n = 4), CGP37157 (CGP, n = 4) and benzamil (Ben, n = 3). The anti‐Yo/rCDR pathology and its correlation to the antagonistic effects on NCX (CGP, Benz) itself or its reverse mode (KB‐R) showed a significant reduction of the depletion of calbindin‐immunoreactive cells for (A) Yo‐positive sera as well as (B) anti‐rCDR2L and anti‐rCDR2/2L but not for anti‐rCDR2 treatment. NCX inhibitor benzamil abolished Calb+ cell loss completely under anti‐Yo4 as well as anti‐rCDR2L and anti‐rCDR2/2L. Data are collected at treatment day 6 and expressed as mean ± SEM [*P < 0.05; **P < 0.01; ***P < 0.001; nonparametric two‐tailed paired Mann–Whitney's U‐test; asterisk (*): significance to naive control; hash (#): significance to anti‐Yo/rCDR].
Discussion
Ex vivo PCD model
The ex vivo PCD model used, makes it possible to study in vivo Yo antibody‐induced pathology 45, 46 without the complexities of the blood–brain barrier and an active immune system 45, 47. The Yo antibody‐positive sera contained active complement factors, whereas the affinity‐purified rabbit CDR antibodies (rCDR) were used to perform neutral IgG antibody assay without exposing the cOTSC to any related peptides which could activate brain naive‐resident T cells 47. Both of these approaches led to identical depletion of the functional biomarker calbindin 28, 29, 31, 32, 48, 49 in Purkinje neurons indicating that PCD is predominantly an antibody‐driven neurodegenerative disease. The Yo/CDR antibodies were clearly internalized by Purkinje neurons and caused a loss of calbindin immunoreactivity, whereas age‐/sex‐matched sera without Yo antibody or antibodies against Purkinje cell‐specific protein 2 (PCP2) or ataxia‐related GAD65 were not internalized and did not affect the immunoreactivity of biomarker calbindin.
Yo antibody‐induced pathology can be linked to the disturbance of calcium‐sensitive mitochondrial‐associated signalling
Calbindin is the major modulator and buffer of Purkinje neurone intracellular calcium 28, 32, and calbindin depletion has been linked to neurodegeneration in PCD 29, Parkinson's disease 21 and Alzheimer's disease 30. We have shown that calbindin co‐immunoprecipitated with CDR2, but not with CDR2L 20, which suggests that calbindin could stabilize both CDR2 expression and function. This has been recently confirmed by others 21. We have also demonstrated that Yo antibody binding to CDR2 and CDR2L leads to dysregulation of the calcium‐dependent VGCC‐PKC‐calbindin signalling pathway and calpain‐2 over‐activation, suggesting that cellular calcium overload plays an important role in Purkinje neurone degeneration and cerebellar atrophy in PCD 20. In Parkinson's disease, CDR2 expression is modulated by calpain‐ and ROS‐activity 21. PKC‐related kinase 1, an important factor in Alzheimer's disease and amyotrophic lateral sclerosis 50, interacts with CDR2 by phosphorylation 51.
As intracellular calcium homeostasis and mitochondrial‐associated signalling are strongly interdependent, sustained perturbations in this network can have devastating consequences to the cell. As neurons have high energy requirements, they are especially vulnerable to mitochondrial dysfunction, and we have found that Purkinje neurons display the highest mitochondria density of cells in the cerebellum (unpublished results). In the present study, we found that anti‐Yo internalization enhanced ROS production and lowered the mitochondrial membrane potential (ΔΨm) to similar levels as the protonophore uncoupler FCCP and BAM15. The rate of ROS production, ΔΨm and the activity of the electron transport chain complex are highly interdependent 27. We believe that anti‐Yo internalization not only downregulates ΔΨm and increases ROS generation through inhibition of respiration, it is also affecting the cation inward and anion outward transport of mitochondrial and plasma membrane to counteract calcium overload 52. Protonophore uncoupler FCCP does depolarize both mitochondrial and plasma membranes and thus causes and rapid collapse of calcium homeostasis within neurons 34, 35, as was indicated by the increased MTROS fluorescence levels under FCCP in our study. The specific depolarization of only the mitochondrial membrane by BAM15 did not show such an effect on the ROS output.
In the present study, we used the ex vivo PCD model to evaluate clinically used compounds that could possibly modulate the Purkinje neurone calcium homeostasis, as mitochondrial calcium overload, ROS signalling/production and MPTP opening are several key steps in neurodegeneration 22, 23, 25, 26. Mitochondrial calcium overload can be caused by increased VDAC and NCXrev mitochondrial calcium influx, reduced NCX mitochondrial calcium efflux, and changes in mitochondrial calcium buffering capacity through MPTP opening 23. Prolonged opening of MPTP, a voltage and calcium‐dependent, CsA‐sensitive, high conductance channel is associated with increased inner mitochondrial membrane permeability. Depolarization of the inner mitochondrial membrane leads to respiratory chain dysfunction and is associated with increased ROS production 27 as we have recorded with MTDR and MTROS after anti‐Yo was internalized into Purkinje neurons.
We found that targeting the MPTP with CsA and KB‐R7943 22, 44, the respiratory chain with CBD and KB‐R7943 41, 43, VDAC and cytochrome‐C with CBD 41 and ROS over‐production with BHT 38 provided graded neuroprotective capacity during exposure and accumulation of Yo antibodies in Purkinje neurons (Figure 7) albeit these compounds did not prevent the internalization of Yo antibody into Purkinje neurons.
Figure 7.
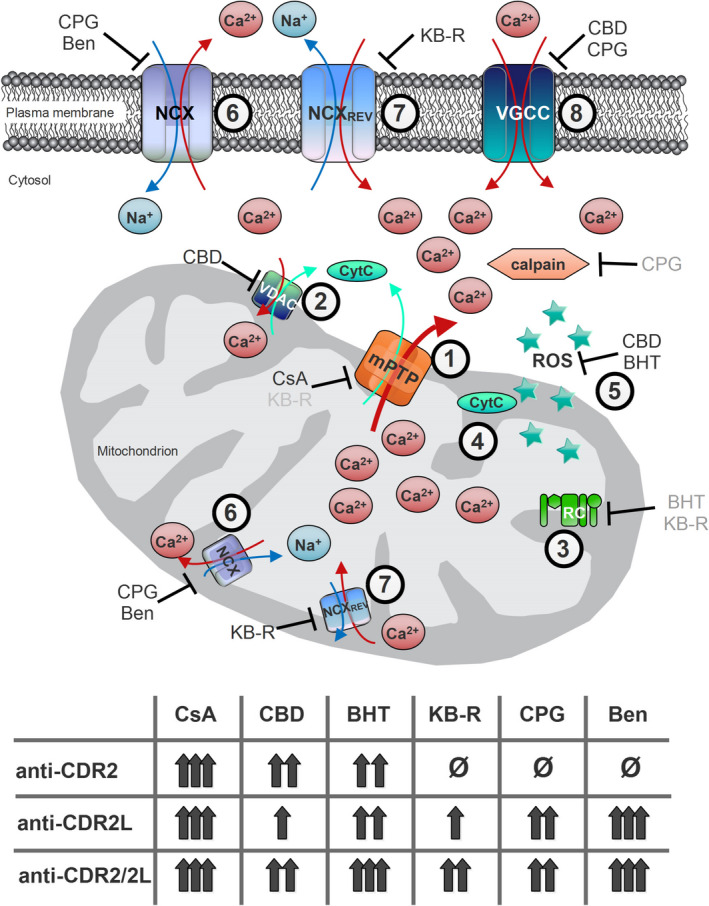
Calcium flux regulation in Purkinje neurons. Schematic diagram illustrates important calcium flux regulators in Purkinje neurone calcium homeostasis and how the six used antagonists modulate calcium flux and reveal neuroprotective capacity in antibody‐dependent manner. Some of the used antagonists have multiple targets, and a total of eight calcium flux regulating processes were subdue: (1) mitochondria permeability transition pore opening by cyclosporin‐A (cyclophilin D‐dependent) and KB‐R7943 (cyclophilin D‐independent, grey), (2) voltage‐dependent anion channels (VDAC) activity by cannabidiol (CBD), (3) respiratory chain (RC) regulation indirectly by butylated hydroxytoluene (BHT) and KB‐R7943 (grey), (4) cytochrome‐c (Cyt C) production by CBD, (5) reactive oxygen species production by BHT and CBD, (6) Na+/Ca2+ exchanger (NCX) activity by CGP37157 and benzamil, (7) NCX rev activity by KB‐R7943 and (8) voltage‐gated calcium channels (VGCC) activity by CGP37157 and CBD. Balancing these eight calcium‐sensitive targets resulted in moderate (single arrow) to high (triple arrows) neuroprotective effects during anti‐Yo/rCDR internalization, with one exception. In Purkinje neurone degeneration caused by anti‐rCDR2, inhibition of NCX (process 6) or its reverse mode (process 7) showed no beneficial effect (ø). Indirect inhibitory effects are marked in grey.
Although CsA is used clinically, there are caveats to its use especially as it does not efficiently cross the blood–brain barrier 53. It is unlikely therefore to be an effective neuroprotective agent in the cerebellum in vivo. However, we show that CsA has a beneficial effect on the mitochondrial membrane potential without affecting the ROS release when anti‐Yo was internalized in Purkinje neurons. Furthermore, because of its immunosuppressive properties and ability to induce necrosis in breast cancer cells by downregulating PKM2 expression and ATP synthesis 54, CsA may still have a potential for use in PCD therapy.
The seconded tested clinical compound, CBD, is not only a low potency CB1R inverse agonist, but potentially targets multiple calcium signalling modulators such as the respiratory chain, VDACs and cytochrome‐C 41, 55, 56. Furthermore, it has anti‐inflammatory and antioxidant properties, and interferes with tumour neovascularization, cancer cell migration, adhesion and invasion 57. We found that CBD can minimize the downgrading of the mitochondrial membrane potential if Yo pathology occurs; however, CBD also potentiated the anti‐Yo‐induced ROS production boost probably due to its modulatory property towards the respiratory chain. Furthermore, CBD can easily cross the blood–brain barrier because of its lipophilic nature. CB1Rs are highly expressed in the cerebellum 40 and their activation reduces neurotransmitter release at all major classes of Purkinje neurone synapses 58. The results obtained with CBD and AM281 treatment suggest that Yo antibody binding mainly to CDR2 causes a reduced neurotransmitter release 58 and sparse neural activity due to CB1R over‐activation 59. Under normal physiological conditions, blocking CB1Rs can, however, disrupt neuronal activity and cause neurodegeneration 59 as we observed in the experimental controls. Currently, an oromucosal spray containing CBD is in clinical use to treat moderate to severe spasticity in multiples sclerosis 60 which might be used as a first step towards a CBD‐based PCD therapy (Figure 7).
The effect of CsA and CBD both support mitochondrial dysfunction as a major factor in PCD, which is also supported by the effect of ROS scavenger BHT data. Similar to observations in Parkinson's disease, where CDR2 expression is modulated by calpain and ROS activity 21 we showed earlier that Yo antibody internalization alters the activity of calpain 20 and now the ROS output. These findings indicate that binding of Yo antibodies to CDR2 and CDR2L potentially affects the complex I of the respiratory chain, a known site for increased ROS production 27.
Targeting the calcium efflux regulator NCX that regulates not only cytosolic calcium efflux, but also mitochondrial calcium homeostasis with CGP37157 26, 42 or KB‐R7943 43, 44 revealed deregulation after anti‐Yo internalization and a significant difference in the neuropathological processes induced by Yo antibody binding to CDR2 and CDR2L, respectively. NCX blockage failed to block the loss of calbindin immunoreactivity induced by Yo antibody binding to CDR2 but did reduce the depletion of calbindin‐immunoreactive cells induced by Yo binding to CDR2L. This supports our previous findings that functionally linked CDR2L to membrane‐associated signalling 20. Although KB‐R7943 is mainly known as an NCXrev antagonist, it is also reported to modulate calcium flux by modulating the opening of MPTPs independently of cyclophilin D, L‐type VGCC, ryanoidine receptors, store‐operated calcium entry 44 and mitochondrial complex I 43. KB‐R7943 influences mitochondrial calcium handling by increasing the calcium retention capacity, which can protect mitochondrial function from pathological calcium overload 44. Therefore, the nonresponse of anti‐CDR2 pathology to KB‐R7943 indicates that CDR2 binding does not alter the mitochondrial calcium retention capacity or the cyclophilin D‐independent opening of MPTPs.
We show that the binding of the Yo antibody (ies) to the assigned antigens CDR2 or CDR2L initiates different pathological cascades in the cerebellum, especially in Purkinje neurons. Our data indicate that not so much the antibody concentration 20 more like the ratio of Yo binding to CDR2 or CDR2L influences the effectiveness of each antagonistic neurodegenerative treatment strategy.
Conclusion
Taken together, our data indicate that Yo antibody incorporation into Purkinje neurons causes multiple alterations regarding mitochondrial and cytosolic calcium influx/efflux regulation: CB1R activity, MPTP opening, mitochondrial membrane potential, ROS output or NCX calcium influx/efflux regulation. Thus, anti‐Yo‐induced pathology can be beneficially modulated by clinical used compounds such as CsA, CBD and ROS scavenger BHT.
However, our study is solely based on neuropharmacological manipulation where the used compounds can interfere with more than one signalling pathway, for that reason we have provided only a first indication that mitochondrial dysfunction occurs if autoantibody Yo is internalized into Purkinje neurons. Further molecular biological studies have to be done to characterize the precise mechanistic alterations, for example, alteration of the complex I of the respiratory chain, to deepen our knowledge on mitochondrial‐associated autoimmune‐mediated neurodegeneration.
As current immunomodulatory therapies for Yo antibody‐mediated PCD are not very effective 33, 61, additional therapeutic strategies addressing in particular the Purkinje neurone degeneration are important for future PCD therapy and our findings demonstrate that treatment addressing calcium homeostasis imbalance and ROS over‐production seems to provide neuroprotective potential.
Author contributions
Study conception and design were done by D.P., C.A.V. and M.S.; Data acquisition and analysis were done by D.P. and M.S.; PCD patients were examined by C.A.V; Manuscript drafting was done by D.P., C.A.V. and M.S.
Conflict of interest
No conflict of interest.
Acknowledgement
Financial support was received from HelseVest grant #911.976. The confocal imaging was performed at the Molecular Imaging Centre (MIC) and was thus supported by the Department of Biomedicine and the Faculty of Medicine and Dentistry, at the University of Bergen, and its partners.
Panja D., Vedeler C. A. and Schubert M. (2019) Neuropathology and Applied Neurobiology 45, 141–156 Paraneoplastic cerebellar degeneration: Yo antibody alters mitochondrial calcium buffering capacity
References
- 1. Storstein A, Vedeler CA. Paraneoplastic neurological syndromes and onconeural antibodies: clinical and immunological aspects. Adv Clin Chem 2007; 44: 143–85 [DOI] [PubMed] [Google Scholar]
- 2. Jarius S, Wildemann B. ‘Medusa head ataxia’: the expanding spectrum of Purkinje cell antibodies in autoimmune cerebellar ataxia. Part 3: anti‐Yo/CDR2, anti‐Nb/AP3B2, PCA‐2, anti‐Tr/DNER, other antibodies, diagnostic pitfalls, summary and outlook. J Neuroinflammation 2015; 12: 166 10.1186/s12974-015-0358-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Storstein A, Krossnes BK, Vedeler CA. Morphological and immunohistochemical characterization of paraneoplastic cerebellar degeneration associated with Yo antibodies. Acta Neurol Scand 2009; 120: 64–7 [DOI] [PubMed] [Google Scholar]
- 4. Albert ML, Darnell JC, Bender A, Francisco LM, Bhardwaj N, Darnell RB. Tumor‐specific killer cells in paraneoplastic cerebellar degeneration. Nat Med 1998; 4: 1321–4 [DOI] [PubMed] [Google Scholar]
- 5. Carpenter EL, Vance BA, Klein RS, Voloschin A, Dalmau J, Vonderheide RH. Functional analysis of CD8+ T cell responses to the onconeural self protein cdr2 in patients with paraneoplastic cerebellar degeneration. J Neuroimmunol 2008; 193: 173–82 [DOI] [PubMed] [Google Scholar]
- 6. Santomasso BD, Roberts WK, Thomas A, Williams T, Blachère NE, Dudley ME, Houghton AN, Posner JB, Darnell RB. A T‐cell receptor associated with naturally occurring human tumor immunity. Proc Natl Acad Sci U S A 2007; 104: 19073–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sutton IJ, Steele J, Savage CO, Winer JB, Young LS. An interferon‐gamma ELISPOT and immunohistochemical investigation of cytotoxic T lymphocyte‐mediated tumour immunity in patients with paraneoplastic cerebellar degeneration and anti‐Yo antibodies. J Neuroimmunol 2004; 150: 98–106 [DOI] [PubMed] [Google Scholar]
- 8. Dahm L, Ott C, Steiner J, Stepniak B, Teegen B, Saschenbrecker S, Hammer C, Borowski K, Begemann M, Lemke S, Rentzsch K. Seroprevalence of autoantibodies against brain antigens in health and disease: brain‐Targeting Autoantibodies. Ann Neurol 2014; 76: 82–94 [DOI] [PubMed] [Google Scholar]
- 9. Diamond B, Huerta PT, Mina‐Osorio P, Kowal C, Volpe BT. Losing your nerves? Maybe it's the antibodies. Nat Rev Immunol 2009; 9: 449–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Roberts WK, Darnell RB. Neuroimmunology of the paraneoplastic neurological degenerations. Curr Opin Immunol 2004; 16: 616–22 [DOI] [PubMed] [Google Scholar]
- 11. Storstein A, Monstad SE, Honnorat J, Vedeler CA. Paraneoplastic antibodies detected by isoelectric focusing of cerebrospinal fluid and serum. J Neuroimmunol 2004; 155: 150–4 [DOI] [PubMed] [Google Scholar]
- 12. Eichler TW, Totland C, Haugen M, Qvale TH, Mazengia K, Storstein A, Haukanes BI, Vedeler CA. CDR2L antibodies: a new player in paraneoplastic cerebellar degeneration. PLoS ONE 2013; 8: e66002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Totland C, Aarskog NK, Eichler TW, Haugen M, Nøstbakken JK, Monstad SE, Salvesen HB, Mørk S, Haukanes BI, Vedeler CA. CDR2 antigen and Yo antibodies. Cancer Immunol Immunother 2011; 60: 283–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Darnell JC, Albert ML, Darnell RB. Cdr2, a target antigen of naturally occuring human tumor immunity, is widely expressed in gynecological tumors. Cancer Res 2000; 60: 2136–9 [PubMed] [Google Scholar]
- 15. Raspotnig M, Haugen M, Thorsteinsdottir M, Stefansson I, Salvesen HB, Storstein A, Vedeler CA. Cerebellar degeneration‐related proteins 2 and 2‐like are present in ovarian cancer in patients with and without Yo antibodies. Cancer Immunol Immunother 2017; 66: 1463–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rodriguez M, Truh LI, O'Neill BP, Lennon VA. Autoimmune paraneoplastic cerebellar degeneration: ultrastructural localization of antibody‐binding sites in Purkinje cells. Neurology 1988; 38: 1380–6 [DOI] [PubMed] [Google Scholar]
- 17. Okano HJ, Park WY, Corradi JP, Darnell RB. The cytoplasmic Purkinje onconeural antigen cdr2 down‐regulates c‐Myc function: implications for neuronal and tumor cell survival. Genes Dev 1999; 13: 2087–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Corradi JP, Yang C, Darnell JC, Dalmau J, Darnell RB. A post‐transcriptional regulatory mechanism restricts expression of the paraneoplastic cerebellar degeneration antigen cdr2 to immune privileged tissues. J Neurosci 1997; 17: 1406–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sakai K, Mitchell DJ, Tsukamoto T, Steinman L. Isolation of a complementary DNA clone encoding an autoantigen recognized by an anti‐neuronal cell antibody from a patient with paraneoplastic cerebellar degeneration. Ann Neurol 1991; 30: 738 [DOI] [PubMed] [Google Scholar]
- 20. Schubert M, Panja D, Haugen M, Bramham CR, Vedeler CA. Paraneoplastic CDR2 and CDR2L antibodies affect Purkinje cell calcium homeostasis. Acta Neuropathol 2014; 128: 835–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hwang JY, Lee J, Oh CK, Kang HW, Hwang IY, Um JW, Park HC, Kim S, Shin JH, Park WY, Darnell RB. Proteolytic degradation and potential role of onconeural protein cdr2 in neurodegeneration. Cell Death Dis 2016; 7: e2240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bhosale G, Sharpe JA, Sundier SY, Duchen MR. Calcium signaling as a mediator of cell energy demand and a trigger to cell death: mitochondrial calcium signaling. Ann N Y Acad Sci 2015; 1350: 107–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Brini M, Calì T, Ottolini D, Carafoli E. Neuronal calcium signaling: function and dysfunction. Cell Mol Life Sci 2014; 71: 2787–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lee J. Mitochondrial drug targets in neurodegenerative diseases. Bioorg Med Chem Lett 2016; 26: 714–20 [DOI] [PubMed] [Google Scholar]
- 25. Graier WF, Frieden M, Malli R. Mitochondria and Ca2+ signaling: old guests, new functions. Pflüg Arch – Eur J Physiol 2007; 455: 375–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Araújo IM, Carreira BP, Pereira T, Santos PF, Soulet D, Inácio â, Bahr BA, Carvalho AP, Ambrósio AF, Carvalho CM. Changes in calcium dynamics following the reversal of the sodium‐calcium exchanger have a key role in AMPA receptor‐mediated neurodegeneration via calpain activation in hippocampal neurons. Cell Death Differ 2007; 14: 1635–46 [DOI] [PubMed] [Google Scholar]
- 27. Görlach A, Bertram K, Hudecova S, Krizanova O. Calcium and ROS: a mutual interplay. Redox Biol 2015; 6: 260–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schwaller B, Meyer M, Schiffmann S. ‘New’ functions for ‘old’ proteins: the role of the calcium‐binding proteins calbindin D‐28k, calretinin and parvalbumin, in cerebellar physiology. Studies with knockout mice. Cerebellum Lond Engl 2002; 1: 241–58 [DOI] [PubMed] [Google Scholar]
- 29. Laure‐Kamionowska M, Maślińska D. Calbindin positive Purkinje cells in the pathology of human cerebellum occurring at the time of its development. Folia Neuropathol 2009; 47: 300–5 [PubMed] [Google Scholar]
- 30. Palop JJ, Jones B, Kekonius L, Chin J, Yu G‐Q, Raber J, Masliah E, Mucke L. Neuronal depletion of calcium‐dependent proteins in the dentate gyrus is tightly linked to Alzheimer's disease‐related cognitive deficits. Proc Natl Acad Sci U S A 2003; 100: 9572–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Barski JJ, Hartmann J, Rose CR, Hoebeek F, Mörl K, Noll‐Hussong M, De Zeeuw CI, Konnerth A, Meyer M. Calbindin in cerebellar Purkinje cells is a critical determinant of the precision of motor coordination. J Neurosci 2003; 23: 3469–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bastianelli E. Distribution of calcium‐binding proteins in the cerebellum. Cerebellum Lond Engl 2003; 2: 242–62 [DOI] [PubMed] [Google Scholar]
- 33. Greenlee JE. Treatment of paraneoplastic cerebellar degeneration. Curr Treat Options Neurol 2013; 15: 185–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ward MW, Huber HJ, Weisova P, Dussmann H, Nicholls DG, Prehn JHM. Mitochondrial and plasma membrane potential of cultured cerebellar neurons during glutamate‐induced necrosis, apoptosis, and tolerance. J Neurosci 2007; 27: 8238–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kenwood BM, Weaver JL, Bajwa A, Poon IK, Byrne FL, Murrow BA, Calderone JA, Huang L, Divakaruni AS, Tomsig JL, Okabe K. Identification of a novel mitochondrial uncoupler that does not depolarize the plasma membrane. Mol Metab 2014; 3: 114–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Brana C, Benham C, Sundstrom L. A method for characterising cell death in vitro by combining propidium iodide staining with immunohistochemistry. Brain Res Brain Res Protoc 2002; 10: 109–14 [DOI] [PubMed] [Google Scholar]
- 37. Noraberg J, Kristensen BW, Zimmer J. Markers for neuronal degeneration in organotypic slice cultures. Brain Res Brain Res Protoc 1999; 3: 278–90 [DOI] [PubMed] [Google Scholar]
- 38. Zou JY, Crews FT. TNF alpha potentiates glutamate neurotoxicity by inhibiting glutamate uptake in organotypic brain slice cultures: neuroprotection by NF kappa B inhibition. Brain Res 2005; 1034: 11–24 [DOI] [PubMed] [Google Scholar]
- 39. Petitet F, Jeantaud B, Reibaud M, Imperato A, Dubroeucq MC. Complex pharmacology of natural cannabinoids: evidence for partial agonist activity of delta9‐tetrahydrocannabinol and antagonist activity of cannabidiol on rat brain cannabinoid receptors. Life Sci 1998; 63: PL1–6 [DOI] [PubMed] [Google Scholar]
- 40. Ashton JC, Appleton I, Darlington CL, Smith PF. Immunohistochemical localization of cannabinoid CB1 receptor in inhibitory interneurons in the cerebellum. Cerebellum Lond Engl 2004; 3: 222–6 [DOI] [PubMed] [Google Scholar]
- 41. Ibeas Bih C, Chen T, Nunn AVW, Bazelot M, Dallas M, Whalley BJ. Molecular targets of cannabidiol in neurological disorders. Neurother J Am Soc Exp Neurother 2015; 12: 699–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ruiz A, Alberdi E, Matute C. CGP37157, an inhibitor of the mitochondrial Na+/Ca2+ exchanger, protects neurons from excitotoxicity by blocking voltage‐gated Ca2+ channels. Cell Death Dis 2014; 5: e1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Brustovetsky T, Brittain MK, Sheets PL, Cummins TR, Pinelis V, Brustovetsky N. KB‐R7943, an inhibitor of the reverse Na+/Ca2+ exchanger, blocks N‐methyl‐d‐aspartate receptor and inhibits mitochondrial complex I. Br J Pharmacol 2011; 162: 255–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wiczer BM, Marcu R, Hawkins BJ. KB‐R7943, a plasma membrane Na+/Ca2+ exchanger inhibitor, blocks opening of the mitochondrial permeability transition pore. Biochem Biophys Res Commun 2014; 444: 44–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cho S, Wood A, Bowlby MR. Brain slices as models for neurodegenerative disease and screening platforms to identify novel therapeutics. Curr Neuropharmacol 2007; 5: 19–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lossi L, Alasia S, Salio C, Merighi A. Cell death and proliferation in acute slices and organotypic cultures of mammalian CNS. Prog Neurobiol 2009; 88: 221–45 [DOI] [PubMed] [Google Scholar]
- 47. Ling C, Verbny YI, Banks MI, Sandor M, Fabry Z. In situ activation of antigen‐specific CD8+ T cells in the presence of antigen in organotypic brain slices. J Immunol 2008; 180: 8393–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kim JH. Overexpression of calbindin‐D28K in hippocampal progenitor cells increases neuronal differentiation and neurite outgrowth. FASEB J 2006; 20: 109–11. 10.1096/fj.05-4826fje [DOI] [PubMed] [Google Scholar]
- 49. Ghoumari AM, Dusart I, El‐Etr M, Tronche F, Sotelo C, Schumacher M, Baulieu E‐E. Mifepristone (RU486) protects Purkinje cells from cell death in organotypic slice cultures of postnatal rat and mouse cerebellum. Proc Natl Acad Sci U S A 2003; 100: 7953–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kawamata T, Taniguchi T, Mukai H, Kitagawa M, Hashimoto T, Maeda K, Ono Y, Tanaka C. A protein kinase, PKN, accumulates in Alzheimer neurofibrillary tangles and associated endoplasmic reticulum‐derived vesicles and phosphorylates tau protein. J Neurosci 1998; 18: 7402–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Takanaga H, Mukai H, Shibata H, Toshimori M, Ono Y. PKN interacts with a paraneoplastic cerebellar degeneration‐associated antigen, which is a potential transcription factor. Exp Cell Res 1998; 241: 363–72 [DOI] [PubMed] [Google Scholar]
- 52. Angelova PR, Abramov AY. Role of mitochondrial ROS in the brain: from physiology to neurodegeneration. FEBS Lett 2018; 592: 692–702. 10.1002/1873-3468.12964 [DOI] [PubMed] [Google Scholar]
- 53. Gajavelli S, Sinha VK, Mazzeo AT, Spurlock MS, Lee SW, Ahmed AI, Yokobori S, Bullock RM. Evidence to support mitochondrial neuroprotection, in severe traumatic brain injury. J Bioenerg Biomembr 2015; 47: 133–48 [DOI] [PubMed] [Google Scholar]
- 54. Wang Q. Cyclosporine A inhibits breast cancer cell growth by downregulating the expression of pyruvate kinase subtype M2. Int J Mol Med 2012; 30: 302–8. 10.3892/ijmm.2012.989 [DOI] [PubMed] [Google Scholar]
- 55. Rimmerman N, Ben‐Hail D, Porat Z, Juknat A, Kozela E, Daniels MP, Connelly PS, Leishman E, Bradshaw HB, Shoshan‐Barmatz V, Vogel Z. Direct modulation of the outer mitochondrial membrane channel, voltage‐dependent anion channel 1 (VDAC1) by cannabidiol: a novel mechanism for cannabinoid‐induced cell death. Cell Death Dis 2013; 4: e949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ryan D, Drysdale AJ, Lafourcade C, Pertwee RG, Platt B. Cannabidiol targets mitochondria to regulate intracellular Ca2+ levels. J Neurosci 2009; 29: 2053–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Massi P, Solinas M, Cinquina V, Parolaro D. Cannabidiol as potential anticancer drug: cannabidiol and cancer. Br J Clin Pharmacol 2013; 75: 303–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Takahashi KA, Linden DJ. Cannabinoid receptor modulation of synapses received by cerebellar Purkinje cells. J Neurophysiol 2000; 83: 1167–80 [DOI] [PubMed] [Google Scholar]
- 59. Bernard C, Milh M, Morozov YM, Ben‐Ari Y, Freund TF, Gozlan H. Altering cannabinoid signaling during development disrupts neuronal activity. Proc Natl Acad Sci U S A 2005; 102: 9388–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Keating GM. Delta‐9‐tetrahydrocannabinol/cannabidiol oromucosal spray (Sativex®): a review in multiple sclerosis‐related spasticity. Drugs 2017; 77: 563–74 [DOI] [PubMed] [Google Scholar]
- 61. Orange D, Frank M, Tian S, Dousmanis A, Marmur R, Buckley N, Parveen S, Graber JJ, Blachère N, Darnell RB. Cellular immune suppression in paraneoplastic neurologic syndromes targeting intracellular antigens. Arch Neurol 2012; 69: 1132–40 [DOI] [PMC free article] [PubMed] [Google Scholar]