

Abstract
Pancreatic β‐cells are the only source of insulin. Disturbances in β‐cell development or function may thus result in insulin deficiency or excess, presenting as hyper‐ or hypoglycemia. It is increasingly evident that common forms of diabetes (types 1 and 2) are pathogenically heterogeneous. Development of efficient therapies is dependent on reliable disease models. Although animal models are remarkably useful research tools, they present limitations because of species differences. As an alternative, human pluripotent stem cell technologies offer multiple possibilities for the study of human diseases in vitro. In the last decade, advances in the derivation of induced pluripotent stem cells from diabetic patients, combined with β‐cell differentiation protocols, have resulted in the generation of useful disease models for diabetes. First disease models have been focusing on monogenic diabetes. The development of genome editing technologies, more advanced differentiation protocols and humanized mouse models based on transplanted cells have opened new horizons for the modeling of more complex forms of β‐cell dysfunction. We present here the incremental progress made in the modeling of diabetes using pluripotent stem cells. We discuss the current challenges and opportunities of these approaches to dissect β‐cell pathology and devise new pharmacological and cell replacement therapies. stem cells 2019;37:33–41
Keywords: Pluripotent stem cells, Diabetes, β‐Cells, Genome editing, CRISPR, Pancreatic differentiation, Disease modeling
Significance Statement.
This concise review summarizes the evolving progress in the modeling of diabetes using human pluripotent stem cells. Recent studies are discussed, focusing on those interrogating β‐cell dysfunction using stem cell‐based models and highlight the implementation of novel technologies like genome editing to further improve the disease models.
Introduction
Diabetes is one of the main global health problems, affecting 415 million people by 2015 and predicted to increase to 642 million by 2040 1. Diabetes is diagnosed when blood glucose is sustained at abnormally high levels, reflecting the incapacity of pancreatic β‐cells to secrete sufficient amounts of insulin to cope with its demand. Type 2 diabetes (T2D) is by far the most common form of diabetes, most often associated with obesity‐induced insulin resistance. However, it is evident that T2D is a very heterogeneous condition and failing β‐cell function is the primary determinant of diabetes development 2. Type 1 diabetes (T1D), which is characterized by autoimmunity against pancreatic β‐cells, constitutes approximately 10% of the cases in western countries, but its incidence is highly variable between different countries and populations 3. In addition, several monogenic forms of diabetes have been identified, presenting in young adults as maturity onset diabetes of the young (MODY) 4. These diabetes types are caused by dominantly inherited mutations in genes important for normal β‐cell function 5. Permanent neonatal diabetes mellitus (PNDM) is a rare form of monogenic diabetes that is diagnosed before 6 months of age. It is caused by mutations in genes important for β‐cell development or function 6, 7, 8. Mutations in genes involved in β‐cell insulin secretion can also lead to congenital hyperinsulinism (CHI), characterized by inappropriately high insulin secretion which often presents as severe neonatal hypoglycemia 9. In many cases, the hyperinsulinism is not responsive to currently available drugs and new therapeutic options are needed.
In order to understand the pathogenic role of diabetes‐associated genetic variants, experimental β‐cell models are needed. Transgenic mice have provided a lot of important information 10. However, mouse models have their limitations because of species differences between mice and men 11, 12. The only possibility to experimentally study primary human β‐cells is based on islets isolated from cadaveric organ donors. This material is associated with several problems, such as scarce availability, high variability, poor maintenance of cell functionality in vitro and limited possibilities to genetically manipulate the cells. As an alternative, human β‐cell lines have been developed. The EndoC‐βH1 line is a valuable glucose‐responsive human β‐cell model 13. However, these are aneuploid immortalized highly proliferative cells and thus not directly equivalent with primary cells. In addition, it is not possible to study developmental aspects with these cells and their low clonal efficiency has limited possibilities of their genetic modification.
The remaining option for the modeling of human β‐cell biology is based on the use of human pluripotent stem cells (hPSC). Human embryonic stem cells (hESC) are derived from the inner cell mass of human blastocyst stage embryos 14. Many hESC lines are widely used for research around the world and also for clinical application of stem‐cell derived pancreatic cells for diabetes therapy 15. Human induced pluripotent stem cells (hiPSC) offer an alternative that is not facing many of the ethical problems associated with hESC. These cells are derived from somatic cells by transcription factor‐based reprogramming 16, 17, 18. The first iPSC were generated with genome integrating retroviruses, but later several nonintegrative methods have been developed 19. An obvious benefit of hiPSC is that they can be derived from patient cells, enabling the establishment of cell models with the desired disease‐associated genetic background.
Furthermore, iPSC are an ideal material for the biobanking of living cells because of their unlimited expansion and differentiation capacity. Biobanks of iPSC derived from genetically characterized cohorts could be used for the functional validation of diabetes‐associated genotypes 20. Several iPSC biobanks have already been established in Europe (https://www.ebisc.org, www.hipsci.org).
In this concise review, we summarize the progress in utilizing hPSC as a model to study diseases affecting the pancreatic β‐cell.
Pancreatic Differentiation of hPSC
Pancreatic differentiation protocols are based on the knowledge from decades of careful study of pancreatic development in animal models 21, 22. They have been devised by attempting to replicate in vitro the developmental stages occurring in vivo 23, 24. This is achieved by using empirically determined cocktails of growth factors and small molecules that create an optimal signaling environment, enabling the progression from the pluripotent stage toward the pancreatic lineage 11.
The human pancreas develops from the endoderm‐derived dorsal and ventral pancreatic bud cells of the posterior foregut, which at gestational week 5, start to express PDX1, the master transcription factor for pancreatic development 25. Pancreatic buds are formed by stratified epithelium that stochastically gets polarized, forming microlumens that then coalesce into a plexus of tubular structures. This plexus eventually gives rise to the pancreatic ductal tree. Within the developing ductal tree, the peripheral tip domains are specified into the acinar cell lineage, marked by the expression of PTF1A together with SOX9 12, 26. The centrally located trunk domain cells will give rise to ductal and endocrine cells. Some of the trunk cells upregulate NEUROG3, triggering endocrinogenesis. This process is controlled by different signaling mechanism in which NOTCH pathway plays a critical role. The NEUROG3 expressing cells commit to the endocrine lineage, delaminating from the pancreatic epithelium to form aggregates representing primitive pancreatic islets. NEUROG3 induces the expression of other transcription factors that establish the endocrine cell gene regulatory network (e.g., NKX2.2, NEUROD1, PAX4, PAX6, ARX, etc.) and it is thus required for the development of all pancreatic endocrine subtypes 27. The pancreatic islets of Langerhans are complex miniorgans where the interactions between different cell types are functionally important. They consist of five major endocrine cell types: glucagon expressing α‐cells, insulin expressing β‐cells, somatostatin expressing δ‐cells, pancreatic polypeptide expressing PP‐cells and ghrelin expressing ε‐cells. In mature human islets, approximately 60% of the cells represent β‐cells and 30% α‐cells, leaving the remaining 10% to the other endocrine cell types. Ghrelin cells are mainly found in embryonic and neonatal islets and are rare in the adult pancreas 28, 29, 30. The different endocrine cell types establish paracrine and autocrine regulatory interactions that control hormone secretion 31, 32. Therefore, the attempts to generate functional human islets from pluripotent stem cells should reproduce these islet cell proportions, intraislet interactions and cytoarchitecture, which are likely critical to achieve proper physiological function.
First protocols to differentiate ES cells to islet cells were based on embryoid body differentiation 33, 34. Although some insulin positive cells were derived, these protocols did not produce true pancreatic endocrine cells. In some cases it was not clear whether the cells actually were capable of synthesizing insulin or it was an artifact resulting from insulin media uptake 35.
It is now evident that the key to successful differentiation protocols is to mimic normal pancreatic development, based on the inductive signals which guide development in vivo 23, 36, 37. The essential basis was established by the group of investigators led by Emmanuel Baetge in the laboratory of CyThera (later Novocell) Inc., San Diego, CA, USA. They were the first to describe a robust protocol for the derivation of definitive endoderm cells from hESC 21. Induction of definitive endoderm has become a routine in many laboratories and depending on the fine‐tuning of the subsequent differentiation stages, this can give rise to progenitors of several endodermal organs, such as thyroid, lung 38, liver 39, pancreas, and intestine 40.
The Novocell group published the first differentiation protocol mimicking pancreatic development signals 22. It was based on a monolayer cell culture following a sequential five‐stage path through definitive endoderm, gut‐tube endoderm, pancreatic endoderm, endocrine precursor to finally yield endocrine hormone expressing cells. The group further showed that these stem cell derived β‐like cells can rescue streptozotocin‐induced diabetes in mice. After transplantation to immunocompromised mice, the cells were able to develop into single‐hormone expressing functional endocrine cells 41. Using an INS‐GFP hES reporter cell line and a monolayer differentiation protocol, Basford et al. were able to purify INS expressing cells from the differentiation cultures and characterize their transcriptome and functionality and compare with human islets. They concluded that these cells were often polyhormonal, with defective glucose‐stimulated insulin secretion (GSIS), and with a transcriptomic profile resembling immature endocrine cells. Also, they observed that the INS+ cells were a heterogeneous population, which could result in important functional differences 42.
Characterization at the transcriptomic and epigenetic levels of the different stages of hPSC‐differentiation to β‐cells in vitro and after in vivo maturation showed how chromatin architecture is remodeled upon differentiation 43. These results indicated that polycomb group‐mediated repression is an important mechanism to control transcription of developmental regulators. Interestingly, aberrant histone modifications were identified in genes that were not properly induced during in vitro differentiation, suggesting that the protocol used to differentiate the cells was not able to faithfully replicate all the signals necessary for proper islet cell differentiation.
The poor functionality of hPSC‐derived β‐cells, together with polyhormonality, has been a common problem of the first monolayer differentiation protocols. Bruin et al. characterized the endocrine cells after 1 month of monolayer differentiation, concluding that they do not represent functional mature β‐cells because of the lack of GSIS in perifusion assay. In comparison with human islets, these cells lacked protein expression of glucose transporter GLUT1, together with imbalance expression of the potassium channel subunits SUR1 and KIR6.2, which could explain their poor functionality 44.
RNA‐sequencing of sorted hPSC‐derived β‐cells, human fetal and adult β‐cells enabled their transcriptomic profile comparison 45. These analyses showed that hPSC‐derived β‐cells resemble more closely human fetal β‐cells at the transcriptomic level, with the consequent functional differences in terms of poorer GSIS for both cell types.
The derivation of pancreatic progenitor cells that express not only PDX1, but also the β‐cell programming factor NKX6.1, is crucial for the successful differentiation into monohormonal β‐cells 46, 47. This can be significantly enhanced by the introduction of EGF and nicotinamide into the pancreatic progenitor specification stage 48.
In 2014 to 2015, three groups published “next generation” differentiation protocols where several stages had been significantly modified achieving more efficient monohormonal β‐cell differentiation with functionality in vitro 49, 50, 51. Consisting of 6 to 7 differentiation stages, the protocols were optimized to generate homogeneous populations of pancreatic progenitors (PDX1+NKX6.1+) with low NEUROG3 and controlled induction into the endocrine lineage. β‐Cell lineage differentiation was induced by adding the EGFR ligand betacellulin and simultaneous ALK5 and NOTCH signaling inhibition 52. In the later stages, cells were cultured in air–liquid interphase or as aggregates in suspension in order to increase oxygen transfer and reproduce better the cytoarchitecture of pancreatic islets. The transcription factor MAFA, critical for fetal β‐cell maturation, was induced by stimulating thyroid receptor signaling using triiodothyronine (T3) 53.
The transcriptome of the β‐like cells derived through these differentiation protocols closely resembled human β‐cells 50. In static incubation, the stem cell derived islet‐like cells also demonstrated moderate GSIS that was not significantly different from the responses of isolated human islets. However, dynamic GSIS in perifusion assays revealed minimal responses, whereas suboptimal Ca2+ fluxes further highlighted the immaturity of the β‐like cells 49.
Despite major progress, it is evident that functionally mature β‐cells have not been robustly derived in vitro until now. Importantly, transplantation of the immature human islet‐like cells to immunodeficient mice enables significant further maturation of the islet cells. This is evidenced by human C‐peptide secretion in glucose tolerance tests and by morphological and ultrastructural studies 49, 50, 54. It is important to understand what the critical factors in the in vivo milieu that favor functional maturation are. These could be related with local signals provided by the in vivo niche at the transplant site. Particularly interaction with endothelial cells may play an important role 55. Interestingly, it was recently shown that maturation occurs faster and more efficiently in female recipient mice, pointing to the potential role of estrogen receptor signaling in maturation 56.
Overall, the attempts to produce bona fide functional β‐cells in vitro and in vivo need to be characterized with detailed studies of the functional maturity of the cells, and truly evaluate them in comparison with primary high‐quality functional β‐cells (reviewed in 57). Detailed comparison of the differentiation protocols have been reviewed elsewhere 37, 58.
Human Pancreatic Development Studied through Pluripotent Stem Cell Differentiation
A challenge to develop new modifications to improve the differentiation protocols is the lack of understanding of human fetal β‐cell development. The study of human pancreatic development has been limited by the scarcity of human fetal tissues donated to research. The studies conducted on this material have shed light on the similarities and differences between human and mouse development in terms of gene expression 25, 59, 60 and functionality 61, 62. Additional new knowledge about human pancreatic development, together with the extensive available data on human adult islets, can now be compared with the in vitro hPSC‐derived β‐cells to determine which stage of human pancreatic development they represent. A good example is the characterization by flow cytometry of human fetal pancreatic cells at defined developmental stages that was later correlated with in vitro derived β‐cells 63.
With the new wave of single‐cell “omics,” it is becoming clear that β‐cells do not constitute a homogeneous cell population, a fact to observe when comparing with the hPSC‐derived β‐cells. β‐Cell heterogeneity arises already during pancreatic development and has been documented in rodent and human islets 64. For example, single cell transcriptomic analysis of human islets has revealed different subpopulations within α‐ and β‐cells 65, 66, 67, 68, 69. Heterogeneity was also observed at the protein expression level when using single cell mass cytometry, with at least three different subpopulations of β‐cells observed, including a proliferative one 29.
A closer study of the differentiation cultures with novel techniques might result in better understanding of the human pancreatic development and the elucidation of useful markers that could be utilized to generate better β‐cells. Using transcriptomic analysis of hPSC‐derived pancreatic progenitors, Ameri et al. identified GP2 as a surface marker of pancreatic progenitors and described that GP2+ isolated progenitors gave rise to functional β‐cells in vitro 70. This was also reported by Cogger et al., who used a different approach based in mass spectrometry analysis of the N‐glycome of hPSC‐derived pancreatic progenitors and further confirmed that GP2+ cells give rise to monohormonal β‐cells 71.
By studying single‐cell transcripts using qRT‐PCR of different stages of hPSC‐differentiation toward β‐cell, Petersen et al. reported the identification of two different pancreatic progenitor populations giving rise to monohormonal β‐cells 72. They described that NKX6.1 expression in the progenitor cells that will give rise to β‐cells can occur before or after NEUROG3 expression commitment to the endocrine lineage. We have similar observations when performing single‐cell RNA sequencing using inDROP platform on hPSC‐derived islet‐like clusters at the end of the differentiation. Using pseudotemporal ordering of the single‐cell transcriptomic profiles, we can identify two different pancreatic progenitor populations giving rise to the β‐cell lineage (D. Balboa et al., manuscript in publication). Whether these findings represent the heterogeneity of pancreatic development dynamics or an in vitro artifact will require further examination of human fetal pancreas development 60.
At the moment, the characteristics of the differentiation protocols allow the interrogation of mechanistic questions that previously could only be studied in mouse models. Löf‐Öhlin et al. used this approach to study the control of pancreatic progenitor polarity regulated by EGFR signaling at different stages of mouse development, validating it in hPSC‐derived pancreatic progenitors as model of human pancreas development 73. Furthermore, the current protocols enable to some extent the modeling of monogenic diabetes forms affecting pancreatic development or β‐cell function.
iPSC Differentiation and Genome Editing to Understand Human β‐Cell Dysfunction
Induced pluripotent stem cell technology makes possible the generation of stem cells from diabetic patients. Combined with the differentiation protocols to β‐cells, this constitutes an approach to generate patient‐specific models of β‐cell dysfunction associated with diabetes. Table 1 presents the studies so far reported on the modeling of β‐cell dysfunction using hPSC. Shang and colleagues provided the first example of this approach. They derived iPSC from patients with diabetes caused by a mutation in the WFS1 gene (Wolfram syndrome) and differentiated them in parallel with healthy‐donor control iPSC 74. They observed that patient iPSC presented elevated endoplasmic reticulum (ER) stress and unfolded protein response, and this disease phenotype could be rescued in vitro by overexpressing WFS1 gene or treatment with chemical chaperones.
Table 1.
Summary of human pluripotent stem cell‐based diabetes disease models
| Type of diabetes | Genes mutated | Key findings | Cell lines used | Differentiation | Transplantation | References |
|---|---|---|---|---|---|---|
| PNDM | WFS1 | Increased ER‐stress, reduced insulin content, processing and GSIS | Patient and control hiPSC | Monolayer | Kidney capsule. NSG mice. 12 weeks | Shang et al. 74 |
| NEUROG3 | Abrogated endocrine cell differentiation | Genome edited hESC (H1) | Monolayer | Kidney capsule, splenic lobe. NSG mice. 6 weeks | McGrath et al. 75 | |
| PDX1, RFX6, PTF1A, GLIS3, MNX1, NEUROG3 | Impairment of pancreatic progenitor and endocrine cell differentiation | Genome edited hESC (HUES8) | Monolayer + air–liquid interphase | NA | Zhu et al. 76 | |
| KCNJ11 | Impaired insulin secretion in vitro and in vivo | Genome edited hESC (HES3‐INS‐GFP) | Monolayer + air–liquid interphase | Kidney capsule. SCID‐beige mice + STZ. 2 to 10 weeks | Zeng et al. 77 | |
| STAT3 | Premature endocrine cell differentiation via NEUROG3 induction | Mutation corrected patient‐hiPSC, control iPSC, hESC | Monolayer + suspension | Kidney capsule. NSG mice. 12 weeks | Saarimäki‐Vire et al. 78 | |
| GATA4, GATA6 | GATA6 and GATA4 dosages are critical for pancreatic differentiation | Genome edited hESC (HUES8, H1) | Monolayer + air–liquid interphase | Kidney capsule. SCID‐beige mice. 4 to 16 weeks | Shi et al. 79 | |
| GATA6 | Impaired definitive endoderm differentiation and β‐cell function in vitro | Mutation corrected patient‐hiPSC, genome edited hESC (MEL1‐INS‐GFP) | Monolayer | NA | Tiyaboonchai et al. 80 | |
| INS | Increased ER‐stress, reduced insulin content, β‐cell proliferation, size and mTORC1 signaling | Mutation corrected patient‐hiPSC | Monolayer + suspension | Kidney capsule. NSG mice. 4 to 24 weeks | Balboa et al. 81 | |
| MODY | HNF1B (MODY5) | Compensatory increase in endoderm and pancreatic progenitor markers and decrease in PAX6 | Patient and control hiPSC | Monolayer | NA | Teo et al. 82 |
| HNF4A (MODY1) | No differences on HNF4A mutant differentiated β‐cells | Patient and control hiPSC | Monolayer + air–liquid interphase | NA | Vethe et al. 83 | |
| T1D | NA | No differences on T1D differentiated β‐cells | Patient and control hiPSC | Suspension | Kidney capsule. SCID‐beige mice + Alloxan. 2‐20 weeks | Millman et al. 84 |
| NA | T1D differentiated showed increased Caspase 3 after cytokines treatment. Differential expression of immune‐related genes | Patient and control hiPSC | Monolayer | NA | Hosokawa et al. 85 | |
| NA | No differences on T1D differentiated β‐cells | Nuclear transfer hESC, hESC, and hiPSC | Monolayer + suspension | Subcutaneous and kidney capsule. NSG mice + STZ. 2 to 12 weeks | Sui et al. 86 | |
| T2D | CDKAL1, KCNQ1, KCNJ11 | Impaired insulin secretion in vitro and in vivo. CDKAL KO cells sensitive to glucolipotoxicity | Genome edited hESC (HES3‐INS‐GFP) | Monolayer + air–liquid interphase | Kidney capsule. SCID‐beige mice + STZ. 2 to 10 weeks | Zeng et al. 77 |
| CDKAL1 | CDKAL knockout results in downregulation of metallothionein genes, causing increased ER‐stress | Genome edited hESC (HES3‐INS‐GFP) | Monolayer + air–liquid interphase | Kidney capsule. SCID‐beige mice + STZ. 6 weeks | Guo et al. 87 | |
| CHI | ABCC8 | Increased insulin secretion in vitro | Genome edited hESC (H1) | Monolayer | NA | Guo et al. 87 |
| ABCC8 | Increased insulin secretion in vitro and in vivo | Mutation corrected patient‐hiPSC and control iPSC | Monolayer + suspension | Kidney capsule. NSG mice. 4 to 24 weeks | Lithovius et al. (manuscript in preparation) | |
| Other | ARX | Reduced β‐, α‐ and γ‐cell differentiation and increased delta cell differentiation | Genome edited hESC (CA1) | Monolayer | NA | Gage et al. 88 |
| HES1, ARX | HES1 knockout increased endocrine cell differentiation. ARX knockout reduced β‐cell and abrogated α‐cell differentiation | Genome edited hESC (HUES8) | Monolayer + air–liquid interphase | NA | Zhu et al. 76 |
Table reproduced from Reference 81. Abbreviations: PNDM, Permanent Neonatal Diabetes Mellitus; MODY, Maturity Onset Diabetes of the Young; T1D, Type 1 Diabetes; T2D, Type 2 Diabetes; CHI, Congential Hyperinsulinism; GSIS, Glucose‐Stimulated Insulin Secretion; KO, Knock‐out; hiPSC, Human induced pluripotent stem cells; hESC, human embryonic stem cells; NSG, NOD‐SCID gamma mouse; SCID, Severe Combined Immunodeficiency mouse; STZ, Streptozotocin.
Other types of monogenic diabetes have been modeled using direct comparison of patient‐derived and healthy‐donor control iPSC differentiated to β‐cells. A particular mutation of the HNF1B gene causing MODY was interrogated by Teo and colleagues, showing that it resulted in a compensatory increase in endoderm and pancreatic markers and reduced PAX6 expression 82. A mutation in HNF4A MODY gene was modeled with a similar approach using iPSC derived from several members of an affected family. The differentiated β‐cells were examined by quantitative proteomics, but no differences were found between the control and patient iPSC‐derived cells 83. This may indicate that the mutation‐associated pathogenic mechanism is beyond the stage of differentiation that can be reproduced in vitro.
T1D is associated with β‐cell targeted autoimmunity. Genome‐wide association studies have identified a number of genes associated with T1D risk. However, with the exception of human leukocyte antigen and INS, the relative risk associated with these genes is rather low 89. Different studies have generated iPSC and nuclear transfer hES cells from diabetic patients and differentiated them into β‐cells. Although two of them found no differences between the T1D and control hPSC‐derived β‐cells 84, 86, a third one showed increased sensitivity of the β‐like cells to cytokine treatment using iPSC derived from a patient with a fulminant form of T1D which is more common in Japan 85. Overall, it is evident that T1D is not an optimal disease for iPSC‐based modeling because of its complex autoimmune pathogenesis, which involves a strong environmental component. In order to recapitulate the interactions between the immune system and β‐cells, new disease models are required in which this is facilitated. This could be achieved by coculturing immune cells and iPSC‐derived β‐cells from the same patient. Another alternative would be the generation of humanized mice with immune system reconstituted from T1D‐patient hematopoietic stem cells, in which iPSC‐derived β‐cells are transplanted 90.
The greatly variable differentiation propensity of iPSC lines is a major problem for their use in disease modeling. In the case of pancreatic differentiation this is clearly illustrated by Sui and colleagues, who compared 12 different iPSC, NT‐hESC and hESC lines and showed a range of 8%‐75% efficiency in c‐peptide+ cell generation using the same protocol 85. This variation is likely explained by the inherent different capability of the hPSC lines to respond to the differentiation protocols, which only reproduce partially the in vivo milieu of the developing pancreas. A wider understanding of the processes involved in controlling pancreatic development and its implementation into the differentiation protocols will likely reduce the variability. Meanwhile, this obstacle can be solved by the generation of isogenic cell lines, in which the parental cell lines is genome edited to generate or correct a particular mutation of interest. Genome editing technologies (Zinc Finger Nuclease [ZFN], Transcription Activator‐Like Effector Nucleases [TALEN], and CRISPR‐Cas9) enable the efficient modification of the genome, and they have been used to generate isogenic stem cell lines for disease modeling (reviewed in 91).
Genome editing of a particular cell line that differentiates efficiently to β‐cells can be exploited to generate knock‐out models of genes involved in pancreatic development and elucidate their role. Following this approach, ARX KO hESC generated with ZFN were shown to have reduced differentiation potential to α‐, β‐ and γ‐cells, and increased to δ‐cells 88. Similarly, NEUROG3 KO hESC generated with CRISPR‐Cas9 were used to show that this gene is required for human endocrine differentiation 75, whereas dosage of GATA6 and GATA4 was shown to be crucial for pancreatic development and β‐cell function 79, 80. In a remarkable effort, Zhu and colleagues used an inducible Cas9 hESC line to generate KOs for PDX1, RFX6, PTF1A, GLIS3, MNX1, NEUROG3, HES1, and ARX using CRISPR‐Cas9, and studied their role in pancreatic differentiation 76, showing how these genes are essential at different stages of development.
In a pioneer attempt of modeling the role of T2D risk genes KCNJ11, KCNQ1 and CDKAL1, Zeng and colleagues generated knockout hESC‐lines, differentiated them to β‐cells and generated humanized mouse models. Although inactivation of KCNJ11 and KCNQ1 resulted in impaired in vitro and in vivo GSIS, CDKAL1 knockout β‐cells were more sensitive to glucolipotoxicity 77. A follow‐up study by the same group showed that CDKAL1 knockout results in metallothionein gene downregulation and increased sensitivity to ER‐stress 87. However, it is obvious that efficient in vitro modeling of β‐cell failure in T2D will require achieving full functional maturation, as well as ability to reproduce β‐cell responses to long‐term exposure to metabolic stress agents. Currently, these remain major challenges.
Another approach is to correct the diabetes causative point mutation in patient‐derived iPSC. The generation of these mutation‐correction isogenic cell lines preserves the genetic background of the diabetic patient and enables faithfully interrogation of the mutation role in the disease. This approach was utilized to investigate the mechanism of STAT3 mutation in a rare case of neonatal diabetes, revealing an unexpected mechanism based on premature differentiation of multipotent pancreatic progenitors 78. Genome editing was also used to correct the GATA6 mutation in iPSC from a patient with pancreatic agenesis to demonstrate the essential function of GATA6 in pancreatic development and β‐cell function 80. Mutation corrected patient iPSC have also been used to dissect the impact of INS mutations for β‐cell development (D. Balboa et al., manuscript in publication).
Finally, the stem‐cell based approaches can also be used to study β‐cell defects behind CHI. There is an obvious need for a relevant model to study the defects in insulin secretion in this disease, because there are no effective drugs for many of the patients who suffer from severe neonatal hypoglycemia caused by mutations in genes encoding ATP‐sensitive potassium channels. The disease phenotype can be recapitulated in β‐like cells differentiated from patient iPSC, providing a platform for drug screening 92.
Conclusion
The establishment of an in vitro method for the differentiation of pluripotent stem cells into a highly specialized endocrine tissue such as pancreatic islets is a formidable challenge. Looking back, it is evident that progress in this area has been remarkable (Fig. 1). The essential steps of the process; commitment into definitive endoderm, then into pancreatic progenitors and further into immature islet‐like cells, have been firmly established in multiple laboratories. This has already allowed the use of this method to reveal mechanisms of monogenic diabetes and even of T2D susceptibility genes that act at the level of β‐cell differentiation. The remaining challenge is to achieve metabolic maturation of the endocrine cells, required for the β‐cells to fine‐tune their insulin secretion to nutrient stimuli. This is essential for the detection of phenotypes associated with genetic defects impairing metabolic regulation of insulin secretion. During the next few years we can expect to see the realization of many of the promises introduced by the combination of the pluripotent stem cells and genome editing also in the diabetes research field. This will include the functional analysis of many risk alleles associated with common forms of diabetes using physiological studies in isogenic pairs of stem‐cell derived β‐like cells discordant just for the variant of interest. Importantly, this will not be limited to coding sequences because functional validation of the noncoding genome is equally feasible. At the same time, large‐scale drug screens will become possible using β‐like cells with diabetes patients genotypes. It is also likely that the rapidly developing organoid technologies will allow more physiological and stable models of pancreatic islet function based on patient‐derived cells (Fig. 1). β‐Cell replacement therapeutic applications will also finally become clinically efficient, potentially revolutionizing the treatment of T1D. However, even before that, it is evident that major clinical benefits will be gained by using in vitro disease models to increase the understanding of pathogenic mechanisms behind β‐cell dysfunction, with the consequent development of more efficient and specific drugs for various forms of diabetes.
Figure 1.
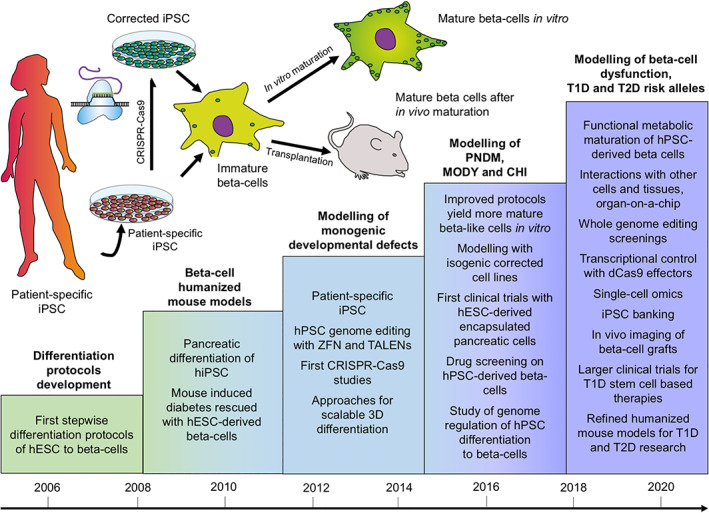
Human pluripotent stem cells for the modeling of pancreatic β‐cell pathology. Schematic representation of the achieved and future milestones on the road to establish pluripotent stem cell‐based models for β‐cell dysfunction.
Author Contributions
D.B.: Literature review, manuscript writing, manuscript revision, table and figures design. J.S‐V.: Literature review, manuscript writing and figures design. T.O.: Manuscript writing, manuscript revision and final approval.
Disclosure of Potential Conflicts of Interest
The authors indicated no potential conflicts of interest.
Acknowledgments
This research was supported by the Academy of Finland, The Innovative Medicines Initiative 2 Joint Undertaking under grant agreement No 115797 (INNODIA), which receives support from the European Union's Horizon 2020 research and innovation programme and “EFPIA”, “JDRF” and “The Leona M. and Harry B. Helmsley Charitable Trust”, the Sigrid Juselius Foundation, the Novo Nordisk Foundation, and the Juvenile Diabetes Research Foundation.
References
- 1. Ogurtsova K, da Rocha Fernandes JD, Huang Y et al. IDF diabetes atlas: Global estimates for the prevalence of diabetes for 2015 and 2040. Diabetes Res Clin Pract 2017;128:40–50. [DOI] [PubMed] [Google Scholar]
- 2. Ahlqvist E, Storm P, Käräjämäki A et al. Novel subgroups of adult‐onset diabetes and their association with outcomes: A data‐driven cluster analysis of six variables. Lancet Diabetes Endocrinol 2018;6:361–369. [DOI] [PubMed] [Google Scholar]
- 3. Atkinson MA, Eisenbarth GS, Michels AW. Type 1 diabetes. Lancet 2014;383:69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Teo AK, Wagers AJ, Kulkarni RN. New opportunities: Harnessing induced pluripotency for discovery in diabetes and metabolism. Cell Metab 2013;18:775–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Teo AK, Windmueller R, Johansson BB et al. Derivation of human induced pluripotent stem cells from patients with maturity onset diabetes of the young. J Biol Chem 2013;288:5353–5356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yang Y, Chan L. Monogenic diabetes: What it teaches us on the common forms of type 1 and type 2 diabetes. Endocr Rev 2016;37:190–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Aguilar‐Bryan L, Bryan J. Neonatal diabetes mellitus. Endocr Rev 2008;29:265–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Murphy R, Ellard S, Hattersley AT. Clinical implications of a molecular genetic classification of monogenic beta‐cell diabetes. Nat Clin Pract Endocrinol Metab 2008;4:200–213. [DOI] [PubMed] [Google Scholar]
- 9. De León DD, Stanley CA. Mechanisms of disease: Advances in diagnosis and treatment of hyperinsulinism in neonates. Nat Clin Pract Endocrinol Metab 2007;3:57–68. [DOI] [PubMed] [Google Scholar]
- 10. Chatzigeorgiou A, Halapas A, Kalafatakis K et al. The use of animal models in the study of diabetes mellitus. In Vivo 2009;23:245–258. [PubMed] [Google Scholar]
- 11. Balboa D, Otonkoski T. Human pluripotent stem cell based islet models for diabetes research. Best Pract Res Clin Endocrinol Metab 2015;29:899–909. [DOI] [PubMed] [Google Scholar]
- 12. Nair G, Hebrok M. Islet formation in mice and men: Lessons for the generation of functional insulin‐producing β‐cells from human pluripotent stem cells. Curr Opin Genet Dev 2015;32:171–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tsonkova VG, Sand FW, Wolf XA et al. The Endo C‐βH1 cell line is a valid model of human beta cells and applicable for screenings to identify novel drug target candidates. Mol Metab 2018;8:144–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Thomson JA, Itskovitz‐Eldor J, Shapiro SS et al. Embryonic stem cell lines derived from human blastocysts. Science 1998;282:1145–1147. [DOI] [PubMed] [Google Scholar]
- 15. Espes D, Lau J, Carlsson P‐O. Towards the clinical translation of stem cell therapy for type 1 diabetes. Eur J Endocrinol 2017;177:R159–R168. [DOI] [PubMed] [Google Scholar]
- 16. Takahashi K, Tanabe K, Ohnuki M et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007;131:861–872. [DOI] [PubMed] [Google Scholar]
- 17. Yu J, Vodyanik MA, Smuga‐Otto K et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007;318:1917–1920. [DOI] [PubMed] [Google Scholar]
- 18. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006;126:663–676. [DOI] [PubMed] [Google Scholar]
- 19. De Vos J, Bouckenheimer J, Sansac C et al. Human induced pluripotent stem cells: A disruptive innovation. Curr Res Transl Med 2016;64:91–96. [DOI] [PubMed] [Google Scholar]
- 20. Beer NL, Gloyn AL. Genome‐edited human stem cell‐derived beta cells: A powerful tool for drilling down on type 2 diabetes GWAS biology. F1000Research 2016;5:1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. D'Amour KA, Agulnick AD, Eliazer S et al. Efficient differentiation of human embryonic stem cells to definitive endoderm. Nat Biotechnol 2005;23:1534–1541. [DOI] [PubMed] [Google Scholar]
- 22. D'Amour KA, Bang AG, Eliazer S et al. Production of pancreatic hormone‐expressing endocrine cells from human embryonic stem cells. Nat Biotechnol 2006;24:1392–1401. [DOI] [PubMed] [Google Scholar]
- 23. Pan FC, Wright C. Pancreas organogenesis: From bud to plexus to gland. Dev Dyn 2011;240:530–565. [DOI] [PubMed] [Google Scholar]
- 24. Larsen HL, Grapin‐Botton A. The molecular and morphogenetic basis of pancreas organogenesis. Semin Cell Dev Biol 2017;66:51–68. [DOI] [PubMed] [Google Scholar]
- 25. Jennings RE, Berry AA, Kirkwood‐Wilson R et al. Development of the human pancreas from foregut to endocrine commitment. Diabetes 2013;62:3514–3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schaffer AE, Freude KK, Nelson SB et al. Nkx6 transcription factors and Ptf1a function as antagonistic lineage determinants in multipotent pancreatic progenitors. Dev Cell 2010;18:1022–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vieira A, Druelle N, Avolio F et al. β‐Cell replacement strategies: The increasing need for a “β‐cell dogma”. Front Genet 2017;8:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pisania A, Weir GC, O'Neil JJ et al. Quantitative analysis of cell composition and purity of human pancreatic islet preparations. Lab Invest 2010;90:1661–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang YJ, Golson ML, Schug J et al. Single‐cell mass cytometry analysis of the human endocrine pancreas. Cell Metab 2016;24:616–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. In't Veld P, Smeets S. Microscopic Anatomy of the Human Islet of Langerhans. Islets of Langerhans, Dordrecht: Springer Netherlands, 2015:19–38. [Google Scholar]
- 31. Gylfe E, Tengholm A. Neurotransmitter control of islet hormone pulsatility. Diabetes Obes Metab 2014;16:102–110. [DOI] [PubMed] [Google Scholar]
- 32. Rorsman P, Ashcroft FM. Pancreatic β‐cell electrical activity and insulin secretion: Of mice and men. Physiol Rev 2018;98:117–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Roche E, Reig JA, Campos A et al. Insulin‐secreting cells derived from stem cells: Clinical perspectives, hypes and hopes. Transpl Immunol 2005;15:113–129. [DOI] [PubMed] [Google Scholar]
- 34. Assady S, Maor G, Amit M et al. Insulin production by human embryonic stem cells. Diabetes 2001;50:1691–1697. [DOI] [PubMed] [Google Scholar]
- 35. Quiskamp N, Bruin JE, Kieffer TJ. Differentiation of human pluripotent stem cells into β‐cells: Potential and challenges. Best Pract Res Clin Endocrinol Metab 2015;29:833–847. [DOI] [PubMed] [Google Scholar]
- 36. Collombat P, Hecksher‐Sørensen J, Serup P et al. Specifying pancreatic endocrine cell fates. Mech Dev 2006;123:501–512. [DOI] [PubMed] [Google Scholar]
- 37. Loo LSW, Lau HH, Jasmen JB et al. An arduous journey from human pluripotent stem cells to functional pancreatic β‐cells. Diabetes Obes Metab 2017;20:3–13. [DOI] [PubMed] [Google Scholar]
- 38. Longmire TA, Ikonomou L, Hawkins F et al. Efficient derivation of purified lung and thyroid progenitors from embryonic stem cells. Cell Stem Cell 2012;10:398–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hay DC, Fletcher J, Payne C et al. Highly efficient differentiation of hESCs to functional hepatic endoderm requires activin A and Wnt 3a signaling. Proc Natl Acad Sci USA 2008;105:12301–12306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Spence JR, Mayhew CN, Rankin SA et al. Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature 2011;470:105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kroon E, Martinson LA, Kadoya K et al. Pancreatic endoderm derived from human embryonic stem cells generates glucose‐responsive insulin‐secreting cells in vivo. Nat Biotechnol 2008;26:443–452. [DOI] [PubMed] [Google Scholar]
- 42. Basford CL, Prentice KJ, Hardy AB et al. The functional and molecular characterisation of human embryonic stem cell‐derived insulin‐positive cells compared with adult pancreatic beta cells. Diabetologia 2012;55:358–371. [DOI] [PubMed] [Google Scholar]
- 43. Xie R, Everett LJ, Lim H‐W et al. Dynamic chromatin remodeling mediated by polycomb proteins orchestrates pancreatic differentiation of human embryonic stem cells. Cell Stem Cell 2013;12:224–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bruin JE, Erener S, Vela J et al. Characterization of polyhormonal insulin‐producing cells derived in vitro from human embryonic stem cells. Stem Cell Res 2014;12:194–208. [DOI] [PubMed] [Google Scholar]
- 45. Hrvatin S, O'Donnell CW, Deng F et al. Differentiated human stem cells resemble fetal, not adult, β cells. Proc Natl Acad Sci USA 2014;111:3038–3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rezania A, Bruin JE, Xu J et al. Enrichment of human embryonic stem cell‐derived NKX6.1‐expressing pancreatic progenitor cells accelerates the maturation of insulin‐secreting cells in vivo. Stem Cells 2013;31:2432–2442. [DOI] [PubMed] [Google Scholar]
- 47. Schaffer AE, Taylor BL, Benthuysen JR et al. Nkx6.1 controls a gene regulatory network required for establishing and maintaining pancreatic beta cell identity. PLoS Genet 2013;9:e1003274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nostro MC, Sarangi F, Yang C et al. Efficient generation of NKX6‐1+ pancreatic progenitors from multiple human pluripotent stem cell lines. Stem Cell Rep 2015;4:591–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rezania A, Bruin JE, Arora P et al. Reversal of diabetes with insulin‐producing cells derived in vitro from human pluripotent stem cells. Nat Biotechnol 2014;32:1121–1133. [DOI] [PubMed] [Google Scholar]
- 50. Pagliuca FW, Millman JR, Gürtler M et al. Generation of functional human pancreatic β cells in vitro. Cell 2014;159:428–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Russ HA, Parent AV, Ringler JJ et al. Controlled induction of human pancreatic progenitors produces functional beta‐like cells in vitro. EMBO J 2015;34:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Huotari MA, Miettinen PJ, Palgi J et al. Erb B signaling regulates lineage determination of developing pancreatic islet cells in embryonic organ culture. Endocrinology 2002;143:4437–4446. [DOI] [PubMed] [Google Scholar]
- 53. Aguayo‐Mazzucato C, DiIenno A, Hollister‐Lock J et al. MAFA and T3 drive maturation of both fetal human islets and insulin‐producing cells differentiated from hESC. J Clin Endocrinol Metab 2015;100:3651–3659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yoshihara E, Wei Z, Lin CS et al. ERRγ is required for the metabolic maturation of therapeutically functional glucose‐responsive β cells. Cell Metab 2016;23:622–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kao D‐I, Lacko LA, Ding B‐S et al. Endothelial cells control pancreatic cell fate at defined stages through EGFL7 signaling. Stem Cell Rep 2015;4:181–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Saber N, Bruin JE, O'Dwyer S et al. Sex differences in maturation of human embryonic stem cell‐derived beta cells in mice. Endocrinology 2018;159:1827–1841. [DOI] [PubMed] [Google Scholar]
- 57. Chen C, Cohrs CM, Stertmann J et al. Human beta cell mass and function in diabetes: Recent advances in knowledge and technologies to understand disease pathogenesis. Mol Metab 2017;6:943–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Santosa MM, Low BSJ, Pek NMQ et al. Knowledge gaps in rodent pancreas biology: Taking human pluripotent stem cell‐derived pancreatic beta cells into our own hands. Front Endocrinol 2016;6:194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Salisbury RJ, Blaylock J, Berry AA et al. The window period of NEUROGENIN3 during human gestation. Islets 2014;6:e954436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Jennings RE, Berry AA, Gerrard DT et al. Laser capture and deep sequencing reveals the transcriptomic programmes regulating the onset of pancreas and liver differentiation in human embryos. Stem Cell Rep 2017;9:1387–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Otonkoski T, Andersson S, Knip M et al. Maturation of insulin response to glucose during human fetal and neonatal development. Studies with perifusion of pancreatic isletlike cell clusters. Diabetes 1988;37:286–291. [DOI] [PubMed] [Google Scholar]
- 62. Otonkoski T. Insulin and glucagon secretory responses to arginine, glue agon, and theophylline during perifusion of human fetal islet‐like cell clusters. J Clin Endocrinol Metab 1988;67:734–740. [DOI] [PubMed] [Google Scholar]
- 63. Ramond C, Glaser N, Berthault C et al. Reconstructing human pancreatic differentiation by mapping specific cell populations during development. Elife 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Roscioni SS, Migliorini A, Gegg M et al. Impact of islet architecture on β‐cell heterogeneity, plasticity and function. Nat Rev Endocrinol 2016;12:695–709. [DOI] [PubMed] [Google Scholar]
- 65. Carrano AC, Mulas F, Zeng C et al. Interrogating islets in health and disease with single‐cell technologies. Mol Metab 2017;6:991–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Segerstolpe Å, Palasantza A, Eliasson P et al. Single‐cell transcriptome profiling of human pancreatic islets in health and type 2 diabetes. Cell Metab 2016;24:593–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Baron M, Veres A, Wolock SLL et al. A single‐cell transcriptomic map of the human and mouse pancreas reveals inter‐ and intra‐cell population structure. Cell Syst 2016;3:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Muraro MJ, Dharmadhikari G, Grün D et al. A single‐cell transcriptome atlas of the human pancreas. Cell Syst 2016;3:385–394.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wang YJ, Schug J, Won K‐J et al. Single cell transcriptomics of the human endocrine pancreas. Diabetes 2016;65:3028–3038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ameri J, Borup R, Prawiro C et al. Efficient generation of glucose‐responsive beta cells from isolated GP2 + human pancreatic progenitors. Cell Rep 2017;19:36–49. [DOI] [PubMed] [Google Scholar]
- 71. Cogger KF, Sinha A, Sarangi F et al. Glycoprotein 2 is a specific cell surface marker of human pancreatic progenitors. Nat Commun 2017;8:331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Petersen MBK, Azad A, Ingvorsen C et al. Single‐cell gene expression analysis of a human ESC model of pancreatic endocrine development reveals different paths to β‐cell differentiation. Stem Cell Rep 2017;9:1246–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Löf‐Öhlin ZM, Nyeng P, Bechard ME et al. EGFR signalling controls cellular fate and pancreatic organogenesis by regulating apicobasal polarity. Nat Cell Biol 2017;19:1313–1325. [DOI] [PubMed] [Google Scholar]
- 74. Shang L, Hua H, Foo K et al. β‐Cell dysfunction due to increased ER stress in a stem cell model of Wolfram syndrome. Diabetes 2014;63:923–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. McGrath PS, Watson CL, Ingram C et al. The basic helix‐loop‐helix transcription factor NEUROG3 is required for development of the human endocrine pancreas. Diabetes 2015;64:2497–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Zhu Z, Li QV, Lee K et al. Genome editing of lineage determinants in human pluripotent stem cells reveals mechanisms of pancreatic development and diabetes article genome editing of lineage determinants in human pluripotent stem cells reveals mechanisms of pancreatic development and diabetes. Cell Stem Cell 2016;18:755–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Zeng H, Guo M, Zhou T et al. An isogenic human esc platform for functional evaluation of genome‐wide‐association‐study‐identified diabetes genes and drug discovery. Cell Stem Cell 2016;19:326–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Saarimäki‐Vire J, Balboa D, Russell MA et al. An activating STAT3 mutation causes neonatal diabetes through premature induction of pancreatic differentiation. Cell Rep 2017;19:281–294. [DOI] [PubMed] [Google Scholar]
- 79. Shi Z, Lee K, Yang D et al. Genome editing in hPSCs reveals GATA6 haploinsufficiency and a genetic interaction with GATA4 in human pancreatic development article genome editing in hPSCs reveals GATA6 haploinsufficiency and a genetic interaction with GATA4 in human pancreatic develop. Stem Cell 2017;20:675–688.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Tiyaboonchai A, Cardenas‐Diaz FL, Ying L et al. GATA6 plays an important role in the induction of human definitive endoderm, development of the pancreas, and functionality of pancreatic β cells. Stem Cell Rep 2017;8:589–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Balboa D. Human Pluripotent Stem Cells and CRISPR‐Cas9 Genome Editing to Model Diabetes. Thesis. University of Helsinki, 2018. ISBN: 978‐951‐51‐4437‐9. [Google Scholar]
- 82. Teo AKK, Lau HH, Valdez IA et al. Early developmental perturbations in a human stem cell model of MODY5/HNF1B pancreatic hypoplasia. Stem Cell Rep 2016;6:357–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Vethe H, Bjørlykke Y, Ghila LM et al. Probing the missing mature β‐cell proteomic landscape in differentiating patient iPSC‐derived cells. Sci Rep 2017;7:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Millman JR, Xie C, Van Dervort A et al. Generation of stem cell‐derived β‐cells from patients with type 1 diabetes. Nat Commun 2016;7:11463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Hosokawa Y, Toyoda T, Fukui K et al. Insulin‐producing cells derived from “induced pluripotent stem cells” of patients with fulminant type 1 diabetes: Vulnerability to cytokine insults and increased expression of apoptosis‐related genes. J Diabetes Investig 2017;9:481–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Sui L, Danzl N, Campbell SR et al. β‐Cell replacement in mice using human type 1 diabetes nuclear transfer embryonic stem cells. Diabetes 2018;67:26–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Guo M, Zhang T, Dong X et al. Using hESCs to probe the interaction of the diabetes‐associated genes CDKAL1 and MT1E. Cell Rep 2017;19:1512–1521. [DOI] [PubMed] [Google Scholar]
- 88. Gage BK, Asadi A, Baker RK et al. The role of ARX in human pancreatic endocrine specification. PLoS ONE 2015;10:e0144100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Anjos S, Polychronakos C. Mechanisms of genetic susceptibility to type I diabetes: Beyond HLA. Mol Genet Metab 2004;81:187–195. [DOI] [PubMed] [Google Scholar]
- 90. Walsh NC, Kenney LL, Jangalwe S et al. Humanized mouse models of clinical disease. Annu Rev Pathol 2017;12:187–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Millette K, Georgia S. Gene editing and human pluripotent stem cells: Tools for advancing diabetes disease modeling and beta‐cell development. Curr Diab Rep 2017;17:116. [DOI] [PubMed] [Google Scholar]
- 92. Guo D, Liu H, Ruzi A et al. Modeling congenital hyperinsulinism with ABCC8‐deficient human embryonic stem cells generated by CRISPR/Cas9. Sci Rep 2017;7:3156. [DOI] [PMC free article] [PubMed] [Google Scholar]