

Summary
Objective
Add‐on cannabidiol (CBD) significantly reduced seizures associated with Dravet syndrome (DS) in a randomized, double‐blind, placebo‐controlled trial: GWPCARE1 Part B (NCT02091375). Patients who completed GWPCARE1 Part A (NCT02091206) or Part B, or a second placebo‐controlled trial, GWPCARE2 (NCT02224703), were invited to enroll in a long‐term open‐label extension trial, GWPCARE5 (NCT02224573). We present an interim analysis of the safety, efficacy, and patient‐reported outcomes from GWPCARE5.
Methods
Patients received a pharmaceutical formulation of highly purified CBD in oral solution (100 mg/mL), titrated from 2.5 to 20 mg/kg/d over a 2‐week period, with their existing medications. Based on response and tolerance, CBD could be reduced or increased up to 30 mg/kg/d.
Results
By November 2016, a total of 278 patients had completed the original randomized trials, and 264 (95%) enrolled in this open‐label extension. Median treatment duration was 274 days (range 1‐512) with a mean modal dose of 21 mg/kg/d, and patients received a median of 3 concomitant antiepileptic medications. Adverse events (AEs) occurred in 93.2% of patients and were mostly mild (36.7%) or moderate (39.0%). Commonly reported AEs were diarrhea (34.5%), pyrexia (27.3%), decreased appetite (25.4%), and somnolence (24.6%). Seventeen patients (6.4%) discontinued due to AEs. Twenty‐two of the 128 patients from GWPCARE1 (17.2%), all taking valproic acid, had liver transaminase elevations ≥3 times the upper limit of normal. In patients from GWPCARE1 Part B, the median reduction from baseline in monthly seizure frequency assessed in 12‐week periods up to week 48 ranged from 38% to 44% for convulsive seizures and 39% to 51% for total seizures. After 48 weeks of treatment, 85% of patients/caregivers reported improvement in the patient's overall condition on the Subject/Caregiver Global Impression of Change scale.
Significance
This trial shows that long‐term CBD treatment had an acceptable safety profile and led to sustained, clinically meaningful reductions in seizure frequency in patients with treatment‐resistant DS.
Keywords: cannabinoid, epileptic encephalopathy, seizures, treatment‐resistant epilepsy
1.
Key Points.
Two hundred sixty‐four patients with Dravet syndrome were treated with long‐term CBD (mean modal dose 21 mg/kg/d, median treatment 274 days)
The most common AEs were diarrhea, pyrexia, decreased appetite, and somnolence, and most were mild to moderate in severity
Sustained reductions in convulsive and total seizures were observed through 48 weeks
Eighty‐five percent of patients/caregivers reported improvement in overall condition after 48 weeks of treatment
2. INTRODUCTION
Dravet syndrome (DS) is a severe epileptic encephalopathy with onset during the first year of life.1 DS is characterized by the onset of recurrent febrile and/or afebrile generalized or hemiclonic seizures or status epilepticus on a background of normal development, followed by multiple seizure types, including myoclonic, absence, and focal impaired awareness seizures.1 Convulsive seizures (tonic–clonic and/or clonic seizures) can occur throughout patients' lives.1 Incidence estimates range from 1 in 15 700 to 40 000 infants,2, 3, 4, 5 and early mortality is high, with sudden unexplained death in epilepsy (SUDEP) and status epilepticus as the leading causes of death.6, 7 DS results from mutations in the gene encoding the α1 subunit of the voltage‐gated sodium channel (Nav1.1) encoded by SCN1A in 70%‐80% of cases, whereas mutations in other voltage‐gated sodium channel subunits as well as other ion channels can also cause DS.8, 9, 10, 11, 12
Seizures in DS are often drug resistant. First‐line antiseizure drugs, commonly referred to as antiepileptic drugs (AEDs), are clobazam and valproic acid. If seizure control is suboptimal, second‐line add‐on therapies include stiripentol, topiramate, or ketogenic diet, with clonazepam, levetiracetam, and zonisamide as third‐line therapies.13, 14 Stiripentol as adjunctive therapy with clobazam and valproic acid is the only therapy specifically approved for DS in Europe and the United States. GW Pharmaceuticals' formulation of highly purified cannabidiol (CBD; Epidiolex) was the first treatment that was approved by the US Food and Drug Administration (FDA) for patients with seizures associated with DS ≥2 years old in June 2018, followed by the FDA's approval of stiripentol as adjunctive therapy with clobazam in August 2018.
CBD is a phytocannabinoid derived from Cannabis sativa 15 and has antiseizure activity in vitro, in animal seizure models, and in a mouse model of DS.16, 17, 18 CBD is unique and structurally distinct in comparison to currently available AEDs, with potentially novel multimodal mechanisms of action including reducing neuronal hyperexcitability through the transient receptor potential vanilloid 1 (TRPV1), antagonism of the G‐protein coupled receptor 55 (GPR55), and modulation of adenosine reuptake.18, 19, 20 Inhibition of the orphan receptor GPR55 by CBD is a lead candidate mechanism of antiseizure activity.18 At physiologically achievable concentrations, CBD does not bind directly to or activate cannabinoid receptors CB1 or CB2.20, 21, 22
In 214 pediatric patients with childhood‐onset treatment‐resistant epilepsy (20% of whom had DS) treated with GW Pharmaceuticals' formulation of CBD in physician‐initiated expanded‐access programs, add‐on CBD reduced seizure frequency with an acceptable safety profile,23 with a second, recently completed analysis extending the findings through 96 weeks in 607 patients.24 A 14‐week, double‐blind, placebo‐controlled trial (GWPCARE1 Part B) in patients with DS demonstrated a significant reduction in convulsive seizure frequency with CBD vs placebo, with higher rates of adverse events (AEs; including somnolence and diarrhea) and reversible liver enzyme elevations.25
GWPCARE5 is an ongoing open‐label extension trial of add‐on CBD in patients with DS who completed GWPCARE1 or GWPCARE2 (a placebo‐controlled trial in patients with DS ongoing at the time of this analysis) and patients with Lennox–Gastaut syndrome (LGS) who completed treatment in one of 2 phase 3 trials (GWPCARE3 and GWPCARE4). Herein we report the safety, efficacy, and patient‐reported outcomes for patients with DS enrolled in GWPCARE5.
3. METHODS
Patients who completed the treatment period in trials GWPCARE1 (Part A, NCT02091206; Part B, NCT02091375) or GWPCARE2 (NCT02224703) were eligible for enrollment in GWPCARE5 (NCT02224573), an open‐label extension trial. All patients had a clinical diagnosis of DS, confirmed by the Epilepsy Study Consortium, that was inadequately controlled by ≥1 current AED. Patients from GWPCARE1 Part B and GWPCARE2 were 2‐18 years of age with ≥4 convulsive seizures in the 4‐week baseline period, whereas patients from the dose‐ranging study GWPCARE1 Part A were 4‐10 years of age with <4 convulsive seizures during the 4‐week baseline period. Convulsive seizures in the trials were defined as tonic–clonic, tonic, clonic, and atonic seizures.
No trial procedures were carried out until written consent was obtained from patients or their parent, caregiver, or legal representative, and, when possible, written assent had been obtained from the patient. The informed consent form, protocol, and amendments for this trial were submitted to and approved by the institutional review board or independent ethics committee at each trial site.
Patients received a pharmaceutical formulation of highly purified, plant‐derived CBD (100 mg/mL) as an oral solution (GW Research Ltd), titrated from 2.5 mg/kg/d to 20 mg/kg/d and administered in 2 divided doses over a 2‐week period, and they continued to receive this dose during the maintenance period. Patients received CBD in addition to their current AEDs. Investigators could decrease the dose of CBD and/or concomitant AEDs if a patient experienced intolerance or could increase the dose to a maximum of 30 mg/kg/d, if thought to be of benefit by the physician. Patients could receive treatment for up to 1 year (United Kingdom, Spain, The Netherlands, Israel) or up to 3 years (United States, France, Poland). Upon completion of the open‐label extension, the dose of CBD was tapered to 10% per day for 10 days for patients not continuing treatment. A follow‐up visit was performed 4 weeks (±3 days) after the last dose of CBD in patients completing or withdrawing from the trials (including the last tapered dose, where applicable). The data cut for this interim analysis was November 3, 2016.
The primary objective of this open‐label extension was to evaluate the long‐term safety and tolerability of adjunctive CBD treatment, based on treatment‐emergent AEs (occurring at any time during the open‐label extension from enrollment through the follow‐up visit), vital signs, 12‐lead electrocardiograms, and clinical laboratory parameters. Secondary objectives were to evaluate the efficacy of CBD as determined by changes in convulsive seizure and total seizure frequency, seizure reduction responder rates (proportion of patients with ≥25%, ≥50%, ≥75%, and 100% reductions in convulsive and total seizure frequency), episodes of status epilepticus, and patient‐reported outcomes based on changes in the Subject/Caregiver Global Impression of Change (S/CGIC) scale.
Caregivers completed a paper diary daily to record AEs and daily use of CBD, concomitant AEDs, and rescue medications. Information on seizure number and type was collected through an interactive voice recording system telephone diary, completed weekly. Blood and urine sampling for clinical laboratory assessments was carried out at all clinic visits. The 7‐point S/CGIC scale (Appendix S1) was assessed at weeks 24, 36, and 48; if both caregiver and patient completed the S/CGIC, the caregiver score was used (Appendix S1).
No formal sample size calculations were performed; only patients who completed treatment in placebo‐controlled trials were eligible for inclusion. Safety analyses (except for liver transaminase analyses) included all enrolled patients (n = 264). Patients who originally enrolled in GWPCARE2 were excluded from efficacy analyses and liver transaminase analyses, as that trial was ongoing and not yet unblinded at the time of this interim analysis; these patients were included in patient‐reported outcome analyses, which were not dependent on baseline values. Patients originally enrolled in GWPCARE1 Part A were excluded from both efficacy and patient‐reported outcomes analyses, as that trial had randomization criteria that was different from GWPCARE1 Part B and GWPCARE2 (<4 convulsive seizures vs ≥4 convulsive seizures in the baseline period, respectively). Thus, the safety population included 264 patients, and the efficacy population included 104 patients (Figure 1).
Figure 1.
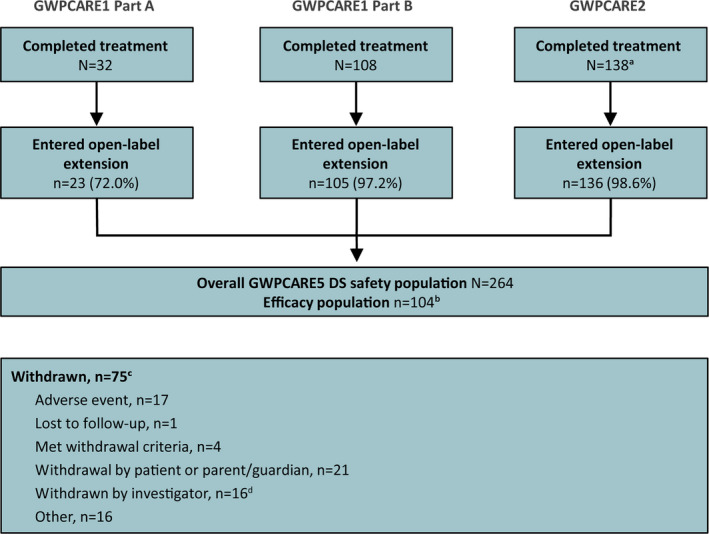
Patient disposition. aOngoing trial, number of patients completed as of data cutoff of November 3, 2016. bPatients originally enrolled in GWPCARE1 Part A (different enrollment criteria) and GWPCARE2 (ongoing and blinded) were excluded from efficacy analyses. Of the 105 patients from GWPCARE1 Part B, one patient discontinued GWPCARE5 prior to reporting seizure frequency data. cWithdrawals are shown by the primary reason reported for each patient and encompass full follow‐up period. dBased on additional free‐text information entered by the investigator, most were judged to be efficacy‐related. DS, Dravet syndrome
Seizure frequencies (per 28 days) were determined for each 12‐week period of treatment. Percentage change in seizure frequency was calculated relative to the prerandomization baseline period from the parent placebo‐controlled trial. Analyses of seizure frequency and seizure reduction responder rates were repeated using inclusion of a last observation carried forward (LOCF) step, described in detail in Appendix S1. Analyses were descriptive, and there was no formal hypothesis testing.
4. RESULTS
4.1. Patients
A total of 140 patients with DS completed treatment in GWPCARE1, of whom 128 enrolled in GWPCARE5. At the time of the data cut, 138 patients had completed treatment in the ongoing GWPCARE2 trial, of whom 136 enrolled in GWPCARE5, for a total of 264 enrolled patients with DS in GWPCARE5 (Figure 1) across 49 centers in the United States, Europe, and Israel. Seventy‐five patients (28.4%) had withdrawn from treatment, most commonly because of patient or parent/guardian decision (n = 21 [8.0%]), AEs (n = 17 [6.4%]), or investigator decision (n = 16 [6.1%]). Withdrawals during each 12‐week treatment period are shown in Table S1. Thirty‐four patients (12.9%) had completed treatment (per country‐specific protocols that limited treatment to 1 year), and 155 patients (58.7%) had ongoing treatment; median CBD treatment duration was 274 days (range 1‐512 days). There were 7 patients with treatment <14 days, of whom only 2 reported seizure data and were included in the efficacy analysis for the first 1‐ to 12‐week visit window. The retention rate at 1 year, calculated as the number of patients who reached the 49‐60 week visit window divided by the total number of patients who could have reached it at the time of this analysis, was 73% (120/165).
The mean modal dose was 21.2 mg/kg/d over the treatment period for all patients. Over each 12‐week reporting interval, and for the duration of the trial period to the data cut, the daily dose remained stable: the mean modal dose per 12‐week reporting interval ranged from 19.9 to 22.7 mg/kg/d over the first 48 weeks of treatment, and during the last 12 weeks of treatment, the mean modal dose was 21.5 mg/kg/d (n = 257).
The patient population was well balanced between genders, and approximately 70% of patients were receiving ≥3 concurrent AEDs during the trial, with more than 60% of patients taking clobazam and/or valproic acid (Table 1). Patients who were originally enrolled in GWPCARE1 discontinued a median of 4 AEDs before receiving study drug in that trial.
Table 1.
Patient demographics and baseline characteristics (safety population)
| Parameter | CBD (N = 264) |
|---|---|
| Age at entry to OLE, y | |
| Mean (SD) | 9.8 (4.4) |
| Median (range) | 9.3 (2.5‐19.3) |
| Age group (years), n (%) | |
| 2‐5 | 64 (24) |
| 6‐11 | 117 (44) |
| 12‐17 | 75 (28) |
| 18‐55 | 8 (3) |
| Gender | |
| Male, n (%) | 133 (50) |
| Geographic region, n (%) | |
| United States | 147 (56) |
| Rest of world | 117 (44) |
| Body mass index at entry to OLE, mean (SD) | 18.3 (4.2) |
| Number of concomitant AEDs, median (range) | 3.0 (1‐6) |
| Concomitant AEDs (>20%), n (%) | |
| Clobazam | 180 (68) |
| Valproic acid | 168 (64) |
| Stiripentol | 101 (38) |
| Levetiracetam | 72 (27) |
| Topiramate | 66 (25) |
| Time on CBD treatment, median (range), d | 274 (1‐512) |
| Modal CBD dose, mean (SD) mg/kg/d | 21.2 (5.2) |
AED, antiepileptic drug; CBD, cannabidiol; OLE, open‐label extension; SD, standard deviation.
4.2. Safety
Treatment‐emergent AEs were reported by 246 of 264 patients (93.2%), with a slightly higher incidence in patients with a modal dose >20 vs ≤20 mg/kg/d (Table 2).
Table 2.
Adverse events (safety population)
| CBD modal dose | CBD (N = 264) | ||
|---|---|---|---|
| ≤20 mg/kg/d (n = 190) | >20 mg/kg/d (n = 74) | ||
| All‐causality treatment‐emergent AEs, n (%) | 173 (91.1) | 73 (98.6) | 246 (93.2) |
| AEs leading to withdrawal,a n (%) | 17 (8.9) | 2 (2.7) | 19 (7.2) |
| Serious AEs, n (%) | 57 (30.0) | 20 (27.0) | 77 (29.2) |
| AEs reported in >10% of patients, n (%) | |||
| Diarrhea | 60 (31.6) | 31 (41.9) | 91 (34.5) |
| Pyrexia | 49 (25.8) | 23 (31.1) | 72 (27.3) |
| Decreased appetite | 46 (24.2) | 21 (28.4) | 67 (25.4) |
| Somnolence | 48 (25.3) | 17 (23.0) | 65 (24.6) |
| Nasopharyngitis | 25 (13.2) | 16 (21.6) | 41 (15.5) |
| Convulsion | 25 (13.2) | 15 (20.3) | 40 (15.2) |
| Vomiting | 24 (12.6) | 13 (17.6) | 37 (14.0) |
| Upper respiratory tract infection | 25 (13.2) | 11 (14.9) | 36 (13.6) |
| Status epilepticus | 17 (8.9) | 12 (16.2) | 29 (11.0) |
| Fatigue | 20 (10.5) | 7 (9.5) | 27 (10.2) |
| Serious AEs reported in >1% of patients, n (%) | |||
| Status epilepticus | 17 (8.9) | 12 (16.2) | 29 (11.0) |
| Convulsion | 8 (4.2) | 5 (6.8) | 13 (4.9) |
| Pyrexia | 8 (4.2) | 2 (2.7) | 10 (3.8) |
| Pneumonia | 4 (2.1) | 3 (4.1) | 7 (2.7) |
| AST increased | 4 (2.1) | 1 (1.4) | 5 (1.9) |
| Dehydration | 3 (1.6) | 1 (1.4) | 4 (1.5) |
| Influenza | 3 (1.6) | 1 (1.4) | 4 (1.5) |
| Generalized tonic–clonic seizure | 3 (1.6) | 1 (1.4) | 4 (1.5) |
| Diarrhea | 2 (1.1) | 1 (1.4) | 3 (1.1) |
AE, adverse event; AST, aspartate aminotransferase; CBD, cannabidiol.
Includes all patients with an AE listed as one of the reasons for withdrawal.
Maximum AE severity was mild (36.7%) or moderate (39.0%) in most patients. Diarrhea, pyrexia, decreased appetite, and somnolence were the most commonly reported AEs. Decreased body weight was reported as an AE in 14 patients (5.3%), and the majority of AEs (13 of 14) were of mild or moderate severity. Serious AEs were reported in 77 patients (29.2%), with status epilepticus, convulsion, and pyrexia the most commonly reported (Table 2). There were 19 discontinuations due to AEs (7.2%), most commonly (>1%) due to increased alanine aminotransferase (ALT) and/or aspartate aminotransferase (AST) levels (n = 10 [3.8%]), convulsion (n = 4 [1.5%]), and liver function test abnormality (n = 3 [1.1%]); patients could discontinue due to multiple AEs. There were 2 deaths during the trial, both due to SUDEP and considered unrelated to treatment by the investigator.
Of the 128 patients who enrolled from GWPCARE1, 71 (55%) were receiving concomitant valproic acid, and of these, 22 (31%) had increases in ALT or AST levels >3 times the upper limit of normal (ULN). In 10 of these 22 patients (45%), the elevations occurred within 1 month of initiating the open‐label extension. No patient met the Hy's law criteria for serious drug‐induced liver injury (AST or ALT >3 times ULN and total bilirubin >2 times ULN). At the time of this analysis, increased ALT/AST level had resolved in 18 of 22 patients, either spontaneously (n = 6), following treatment discontinuation (n = 6), or after dose reduction of CBD or concomitant AEDs (n = 6; n = 4 patients after dose reduction of valproic acid). In the patients not on valproic acid (57/128 [45%]), no ALT/AST elevations were observed.
4.3. Efficacy
The decrease in the number of patients at the later 12‐week treatment periods was due to both withdrawals (Table S1) and patients with ongoing treatment not having reached the later treatment periods at the time of this interim analysis. Efficacy outcomes are shown through week 48, as a sufficient number of patients had completed 48 weeks of treatment to enable meaningful data interpretation.
During weeks 1‐12, the median reduction in monthly convulsive seizure frequency from the baseline period was 37.5% (a reduction from 12.4 to 7.5 seizures per month), and this reduction remained consistent at weeks 13‐24, 25‐36, and 37‐48 (42.9%‐44.3%; Figure 2A). Percentage reductions were similar in the LOCF analysis (Figure S1A). Five of 104 patients (4.8%) were convulsive seizure‐free in their last 12 weeks of treatment. More than 40% of patients had convulsive seizure‐frequency reductions of ≥50% at each visit window; ≥25%, ≥50%, ≥75%, and 100% responder rates are shown in Figure 3A. Responder rates were generally similar in the LOCF analysis (Figure S2A).
Figure 2.
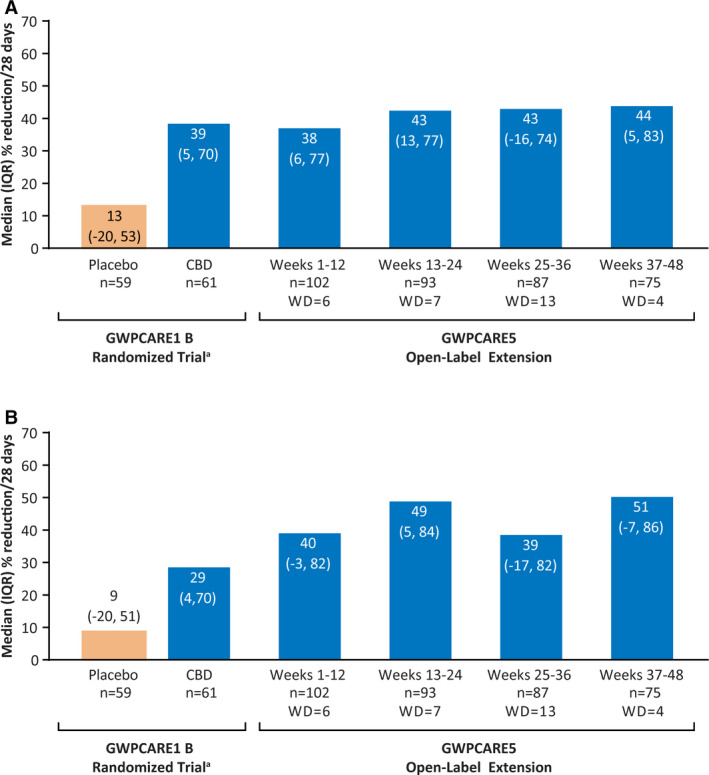
Reduction in (A) convulsive seizure frequency and (B) total seizure frequency (efficacy population). aData previously published25 is provided here for context. CBD, cannabidiol; IQR, interquartile range; n, number of patients with available data during visit window; WD, number of withdrawals during visit window
Figure 3.
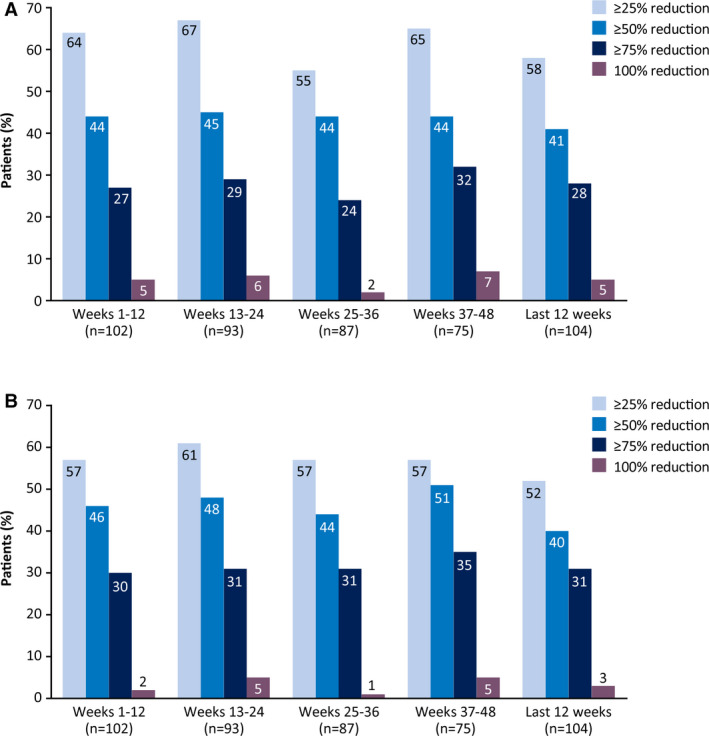
Responder rates for (A) convulsive and (B) total seizures (efficacy population)
Median reduction in total seizure frequency from baseline at weeks 1‐12 was 39.5% (a reduction from 32.4 to 14.5 seizures per month), and reductions during the subsequent 3 treatment windows ranged from 39.0% to 50.7% (Figure 2B). LOCF analysis findings were similar (Figure S1B). Three patients (2.9%) were totally seizure‐free in their last 12 weeks of treatment. No patients were seizure‐free over the full duration of the treatment period to the time of the data cut. Responder rates of ≥25%, ≥50%, ≥75%, and 100% remained consistent over time (Figure 3B), and rates were generally similar in the LOCF analysis (Figure S2B).
At each 12‐week visit window, 2%‐4% of patients reported convulsive status epilepticus and 3%‐5% reported nonconvulsive status epilepticus (Table S2); baseline incidence was 1% and 5%, respectively.
Of the 89 patients/caregivers from GWPCARE1 Part B and GWPCARE2 who had completed the S/CGIC after 48 weeks of treatment, 85% reported an overall improvement in the patient's condition (Figure 4A). Similar proportions of patients/caregivers reported improvement at weeks 24 and 38.
Figure 4.
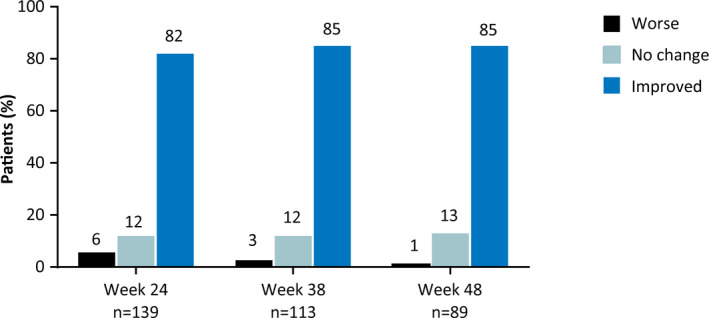
Patient/caregiver ratings of change in overall condition on the S/CGIC scale. S/CGIC, Subject/Caregiver Global Impression of Change
5. DISCUSSION
Our long‐term trial data confirm and extend previous findings, demonstrating that add‐on CBD treatment in patients with DS had an acceptable safety profile and reduced the frequency of total and convulsive seizures up to 48 weeks of treatment. Overall, the safety profile of CBD was similar to that observed in the previous 14‐week, randomized controlled trial, GWPCARE1 Part B, with no new safety signals emerging.25 Median CBD exposure in this extension study was approximately 39 weeks, over twice the duration of the parent randomized‐controlled studies, with some patients treated for 73 weeks. When taking into consideration the duration of follow‐up, the withdrawal rate of 28% is not surprising, and within the range of other long‐term AED studies of 20%‐40% at approximately 1 year of follow‐up.26, 27 Study burden and the addition of new drug trials for DS (fenfluramine [NCT02826863], ataluren [NCT02758626]) likely contributed to the withdrawals in the current study.
Although a greater incidence of AEs would be expected given the extended follow‐up period, the most commonly reported AEs in this trial (diarrhea, pyrexia, decreased appetite, and somnolence) had incidence similar to that seen in the completed parent trial. Despite AEs of diarrhea and decreased appetite being reported in many patients, an AE of decreased weight was reported by fewer patients (5.3%) and was usually mild or moderate in severity. The incidence of status epilepticus as an AE was twice that observed in GWPCARE1 Part B (11% vs 6%, respectively), and there was a greater proportion of patients with serious AEs (29% vs 16%) and withdrawals (28% vs 13%). These findings may be explained by the longer treatment exposure in the present study.
It is known that coadministration of CBD and clobazam produces an approximately threefold increase in plasma concentrations of N‐desmethylclobazam, the active metabolite of clobazam (a substrate of cytochrome P450 2C19). This may increase the risk of clobazam‐related adverse reactions. In patients taking clobazam and CBD who experience bothersome sedation, a reduction of the clobazam (or CBD) dose may be considered.28 All patients with liver enzyme elevations were receiving concomitant valproic acid, as was observed in GWPCARE1, although most patients on valproic acid (69%) did not have elevations. A possible interaction between CBD and valproic acid leading to liver enzyme elevations in patients with treatment‐resistant epilepsies was also reported in a CBD expanded‐access program.29 Based on the findings of this and previous studies, patients taking valproate and CBD (especially 20 mg/kg/d) are at greatest risk of liver enzyme elevations; discontinuation or reduction of CBD and/or concomitant valproate should be considered. Higher doses of CBD, elevated transaminase levels at baseline, and—to a lesser degree—concomitant clobazam are also risk factors for liver enzyme elevations.28 Two deaths occurred during follow‐up, both due to SUDEP; this translates to a rate of ≈11 deaths/1000 patient‐years, consistent with the reported ≈16 deaths/1000 patient‐years among patients with DS.6
Recurrent convulsive seizures are characteristic of DS and persist throughout life despite pharmacotherapy.1, 14, 30 Elimination or significant reduction in the frequency of convulsive seizures is the highest priority in DS treatment.14 In GWPCARE1 Part B, CBD reduced convulsive seizure frequency by 41% after achieving a stable CBD dose in the 12‐week maintenance period25; this reduction was maintained in the present trial. This finding is notable when considered in the context of the number of concomitant therapies received by enrolled patients (median 3.0 AEDs) and the number of therapies discontinued by patients before CBD treatment (median 4.0 AEDs in all patients enrolled in GWPCARE1 Part B [data on file]).
Other common seizure types in DS include myoclonic and absence seizures and focal seizures with loss of awareness, and these are included in the total seizure endpoint.1, 14 The reductions in total seizure frequency observed in GWPCARE1 Part B25 were maintained in this open‐label extension and were similar to reductions in convulsive seizure frequency, suggesting that CBD may have sustained, broad‐spectrum antiseizure properties.
The S/CGIC scale, a patient‐ and caregiver‐reported outcome measure, assesses improvement in the patient's overall condition. The proportion of patients/caregivers reporting improvement was >80% at all time points assessed, strongly suggesting that the reduced seizure frequencies were clinically meaningful for most patients/caregivers.
The mean modal CBD dose was generally consistent across each 12‐week period as well as in the last 12 weeks of data for each patient, indicating that there was no development of tolerance to treatment; seizure frequency reductions were sustained without increased CBD dose.
The major limitation of our data, as for all open‐label extension trials, was the lack of a placebo arm. Efficacy and patient/caregiver reported outcome data were determined as percentage changes from the pretreatment baseline from the original randomized trials. This is a potential confounding factor due to the different duration of exposure to CBD between those patients originally randomized to placebo and those randomized to CBD; however, this approach has advantages over using baselines from the beginning of the open‐label extension, which would be confounded by the improvements already observed in those patients initially randomized to CBD. For this interim analysis, patients had different durations of exposure to drug, such that not all patients had completed the later treatment windows. An additional limitation of this trial is that patients were instructed to take the drug consistently with respect to meal times, but the meal composition and time of meal in relation to dosing were not recorded. However, patients were not instructed to fast; therefore patients were likely taking their medication close to meal times, consistent with a bi‐daily administration (morning and evening meal). The prandial state of patients overall in the study was likely to be closer to a fed state than fasting conditions, with most patients consuming standard meals.
In this open‐label extension trial, long term add‐on CBD treatment had an acceptable safety profile in patients with treatment‐resistant DS. Reductions in convulsive and total seizure frequency observed in the original placebo‐controlled trial were maintained with continued CBD treatment up to 48 weeks, with more than 80% of patients/caregivers reporting an improvement in overall condition. Our interim analysis supports the use of add‐on CBD as a long‐term treatment in patients with DS.
CONFLICTS OF INTEREST
Orrin Devinsky has served as a consultant/advisor to GW Pharmaceuticals and Pairnomix, and as a study investigator for GW Pharmaceuticals, and has equity interest in Tevard, Empatica, Privateer Holdings, and Receptor Life Sciences. Rima Nabbout has served as a consultant/advisor/lecturer for Novartis, Zogenix, Nutricia, Advicennes, Eisai, GW Pharmaceuticals and as a study investigator for GW Pharmaceuticals, Advicennes, UCB, Eisai, and Zogenix. Ian Miller has served as a consultant/advisor to GW Pharmaceuticals, Insys Therapeutics, Visualase, and NeuroPace, and as a study investigator for GW Pharmaceuticals. Linda Laux is a study investigator for GW Pharmaceuticals and Zogenix. Marta Zolnowska is a study investigator for GW Pharmaceuticals. Stephen Wright is employed by GW Research Ltd. Claire Roberts was employed by GW Research Ltd at the time of this study, and is now affiliated with Eisai Ltd. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Supporting information
ACKNOWLEDGMENTS
The authors would like to thank the patients, their families, and the staff at sites that participated in this study, as well as Blake Agnew, ATC‐L, of Greenwich Biosciences, Inc., for his contributions as clinical study manager. Medical writing support was provided to authors by Jeremy Kennard, PhD, and Dena McWain of Ashfield Healthcare Communications, Middletown, CT, and funded by Greenwich Biosciences, Inc.
Devinsky O, Nabbout R, Miller I, et al. Long‐term cannabidiol treatment in patients with Dravet syndrome: An open‐label extension trial. Epilepsia. 2019;60:294–302. 10.1111/epi.14628
Funding information
This study was sponsored by GW Research Ltd, Cambridge, UK.
REFERENCES
- 1. Dravet C, Bureau M, Oguni H, et al. Dravet syndrome (severe myoclonic epilepsy in infancy) In: Bureau M, Genton P, Dravet C, et al. editors. Epileptic syndromes in infancy, childhood and adolescence. Montrouge, France: John Libbey Eurotext, 2012; p. 125–56. [Google Scholar]
- 2. Bayat A, Hjalgrim H, Moller RS. The incidence of SCN1A‐related Dravet syndrome in Denmark is 1:22,000: a population‐based study from 2004 to 2009. Epilepsia. 2015;56:e36–9. [DOI] [PubMed] [Google Scholar]
- 3. Brunklaus A, Ellis R, Reavey E, et al. Prognostic, clinical and demographic features in SCN1A mutation‐positive Dravet syndrome. Brain. 2012;135:2329–36. [DOI] [PubMed] [Google Scholar]
- 4. Rosander C, Hallbook T. Dravet syndrome in Sweden: a population‐based study. Dev Med Child Neurol. 2015;57:628–34. [DOI] [PubMed] [Google Scholar]
- 5. Wu YW, Sullivan J, McDaniel SS, et al. Incidence of Dravet syndrome in a US Population. Pediatrics. 2015;136:e1310–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cooper MS, McIntosh A, Crompton DE, et al. Mortality in Dravet syndrome. Epilepsy Res. 2016;128:43–7. [DOI] [PubMed] [Google Scholar]
- 7. Shmuely S, Sisodiya SM, Gunning WB, et al. Mortality in Dravet syndrome: a review. Epilepsy Behav. 2016;64:69–74. [DOI] [PubMed] [Google Scholar]
- 8. Claes L, Del‐Favero J, Ceulemans B, et al. De novo mutations in the sodium‐channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet. 2001;68:1327–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Depienne C, Trouillard O, Saint‐Martin C, et al. Spectrum of SCN1A gene mutations associated with Dravet syndrome: analysis of 333 patients. J Med Genet. 2009;46:183–91. [DOI] [PubMed] [Google Scholar]
- 10. Harkin LA, McMahon JM, Iona X, et al. The spectrum of SCN1A‐related infantile epileptic encephalopathies. Brain. 2007;130:843–52. [DOI] [PubMed] [Google Scholar]
- 11. Catarino CB, Liu JY, Liagkouras I, et al. Dravet syndrome as epileptic encephalopathy: evidence from long‐term course and neuropathology. Brain. 2011;134:2982–3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Steel D, Symonds JD, Zuberi SM, et al. Dravet syndrome and its mimics: beyond SCN1A. Epilepsia. 2017;58:1807–16. [DOI] [PubMed] [Google Scholar]
- 13. Wirrell EC. Treatment of Dravet syndrome. Can J Neurol Sci. 2016;43(Suppl 3):S13–8. [DOI] [PubMed] [Google Scholar]
- 14. Wirrell EC, Laux L, Donner E, et al. Optimizing the diagnosis and management of Dravet syndrome: recommendations from a North American Consensus Panel. Pediatr Neurol. 2017;68:18–34e3. [DOI] [PubMed] [Google Scholar]
- 15. Mechoulam R, Shvo Y. Hashish. I. The structure of cannabidiol. Tetrahedron. 1963;19:2073–8. [DOI] [PubMed] [Google Scholar]
- 16. Jones NA, Glyn SE, Akiyama S, et al. Cannabidiol exerts anti‐convulsant effects in animal models of temporal lobe and partial seizures. Seizure. 2012;21:344–52. [DOI] [PubMed] [Google Scholar]
- 17. Jones NA, Hill AJ, Smith I, et al. Cannabidiol displays antiepileptiform and antiseizure properties in vitro and in vivo. J Pharmacol Exp Ther. 2010;332:569–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kaplan JS, Stella N, Catterall WA, et al. Cannabidiol attenuates seizures and social deficits in a mouse model of Dravet syndrome. Proc Natl Acad Sci U S A. 2017;114:11229–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Carrier EJ, Auchampach JA, Hillard CJ. Inhibition of an equilibrative nucleoside transporter by cannabidiol: a mechanism of cannabinoid immunosuppression. Proc Natl Acad Sci U S A. 2006;103:7895–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Devinsky O, Cilio MR, Cross H, et al. Cannabidiol: pharmacology and potential therapeutic role in epilepsy and other neuropsychiatric disorders. Epilepsia. 2014;55:791–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ibeas Bih C, Chen T, Nunn AV, et al. Molecular targets of cannabidiol in neurological disorders. Neurotherapeutics. 2015;12:699–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McPartland JM, Duncan M, Di Marzo V, et al. Are cannabidiol and Delta(9) ‐tetrahydrocannabivarin negative modulators of the endocannabinoid system? A systematic review Br J Pharmacol. 2015;172:737–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Devinsky O, Marsh E, Friedman D, et al. Cannabidiol in patients with treatment‐resistant epilepsy: an open‐label interventional trial. Lancet Neurol. 2016;15:270–8. [DOI] [PubMed] [Google Scholar]
- 24. Szaflarski JP, Bebin EM, Comi AM, et al. Long‐term safety and treatment effects of cannabidiol in children and adults with treatment‐resistant epilepsies: Expanded access program results. Epilepsia. 2018;59:1540‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Devinsky O, Cross JH, Laux L, et al. Trial of cannabidiol for drug‐resistant seizures in the Dravet syndrome. N Engl J Med. 2017;376:2011–20. [DOI] [PubMed] [Google Scholar]
- 26. Kwok CS, Johnson EL, Krauss GL. Comparing safety and efficacy of “third‐generation” antiepileptic drugs: long‐term extension and post‐marketing treatment. CNS Drugs. 2017;31:959–74. [DOI] [PubMed] [Google Scholar]
- 27. Toledo M, Beale R, Evans JS, et al. Long‐term retention rates for antiepileptic drugs: a review of long‐term extension studies and comparison with brivaracetam. Epilepsy Res. 2017;138:53–61. [DOI] [PubMed] [Google Scholar]
- 28. Epidiolex® (cannabidiol) oral solution [Prescribing information]. Carlsbad, CA: Greenwich Biosciences; 2018. [Google Scholar]
- 29. Gaston TE, Bebin EM, Cutter GR, et al. Interactions between cannabidiol and commonly used antiepileptic drugs. Epilepsia. 2017;58:1586–92. [DOI] [PubMed] [Google Scholar]
- 30. Dravet C, Oguni H. Dravet syndrome (severe myoclonic epilepsy in infancy). Handb Clin Neurol. 2013;111:627–33. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials