

Summary
The blood vessel wall has a number of self‐healing properties, enabling it to minimize blood loss and prevent or overcome infections in the event of vascular trauma. Endothelial cells prepackage a cocktail of hemostatic, inflammatory and angiogenic mediators in their unique secretory organelles, the Weibel–Palade bodies (WPBs), which can be immediately released on demand. Secretion of their contents into the vascular lumen through a process called exocytosis enables the endothelium to actively participate in the arrest of bleeding and to slow down and direct leukocytes to areas of inflammation. Owing to their remarkable elongated morphology and their secretory contents, which span the entire size spectrum of small chemokines all the way up to ultralarge von Willebrand factor multimers, WPBs constitute an ideal model system for studying the molecular mechanisms of secretory organelle biogenesis, exocytosis, and content expulsion. Recent studies have now shown that, during exocytosis, WPBs can undergo several distinct modes of fusion, and can utilize fundamentally different mechanisms to expel their contents. In this article, we discuss recent advances in our understanding of the composition of the WPB exocytotic machinery and how, because of its configuration, it is able to support WPB release in its various forms.
Keywords: endothelial cells, exocytosis, von Willebrand disease, von Willebrand factor, Weibel–Palade bodies
Weibel–Palade bodies: secretory organelles of the endothelium
The main cargo of Weibel–Palade bodies (WPBs) is von Willebrand factor (VWF), which is a large multimeric adhesive protein that mediates platelet adhesion to the endothelium and to the subendothelial matrix 1, 2. VWF also acts as a chaperone for coagulation factor VIII in plasma, and prevents its premature clearance, which is critical for the maintenance of normal circulating FVIII levels. VWF undergoes a complex series of post‐translational modifications, including glycosylation, multimerization, and proteolytic processing, and is the driving force behind the formation of WPBs. Mutations in VWF that affect its synthesis or processing and subsequent storage in WPBs constitute the basis of von Willebrand disease (VWD), which is the most common inherited bleeding disorder, and is caused by quantitative or qualitative defects in VWF. The topics of VWF biosynthesis, WPB formation and VWD have been extensively covered in a number of excellent reviews 3, 4, 5, and are therefore not included in this review.
Alongside VWF, a considerable number of inflammatory and angiogenic mediators are copackaged into WPBs (Fig. 1C) 6, 7. Their simultaneous release from this vascular emergency package will also direct leukocytes to sites of inflammation and promote vessel repair. To tailor its secretory response to the prevailing vascular condition, the endothelium continuously transduces cues from the local microenvironment into dynamic control over the content of WPBs by selectively including or excluding certain cargoes. Conditions that mimic laminar flow lead to a reduced angiopoietin‐2 (Ang‐2) content in WPBs 8. Exposure to proinflammatory cytokines leads to upregulation of chemokines such as interleukin (IL)‐8, monocyte chemoattractant protein‐1, eotaxin‐3, and IL‐6, which are packaged into newly synthesized WPBs 9. Because WPBs are long‐lived storage organelles with a turnover of ~ 24 h 10, 11, 12, endothelial cells will accumulate distinct populations of granules that differ in their levels of cotargeted WPB cargo 9, 13. Whether the degree of inclusion of cotargeted WPB cargo reflects the endothelial activation state at the moment when the WPB was synthesized, or whether this is a result of stochastic variation following gradual redistribution of contents over the WPB population through content intermixing, as been reported for other long‐lived organelles such as lysosomes 14, is currently unclear. Interestingly, it has been suggested that subsets of granules can be subject to differential exocytosis 15, although the mechanism behind this differential release remains elusive.
Figure 1.
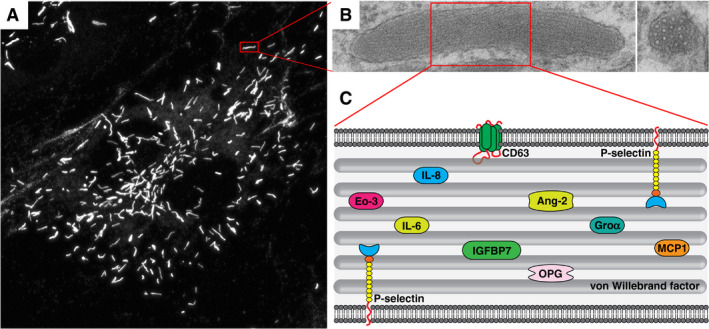
Weibel–Palade bodies (WPBs), secretory organelles of the endothelium. (A) Endothelial cells containing characteristic elongated WPBs visualized by von Willebrand factor (VWF) immunostaining. (B) WPB ultrastructure, with a longitudinal section (left) showing internal striations, and a cross‐section (right) showing bundles, which represent densely packed VWF tubules. (C) Cartoon representation of WPB cargo. Ang‐2, angiopoietin‐2; Eo‐3, eotaxin‐3; GROα, growth regulated oncogene α; IGFBP7, insulin‐like growth factor‐binding protein 7; IL‐6, interleukin‐6; IL‐8, interleukin‐8; MCP‐1, monocyte chemoattractant protein‐1; OPG, osteoprotegerin.
The endothelial secretory pathway
VWF secretion from endothelial cells occurs via three main routes: constitutive secretion and basal secretion, both of which occur in the absence of stimulation, and regulated secretion of WPBs in response to endothelial activation (Fig. 2). Following its journey through the early secretory pathway, VWF undergoes a sorting step at the level of the trans‐Golgi network (TGN): low molecular weight (LMW) VWF multimers enter the constitutive secretory pathway in small short‐lived anterograde carriers, which are immediately released at the plasma membrane 16. High molecular weight (HMW) VWF multimers enter the storage pathway by condensing into tubules, which are packaged in nascent WPBs that bud off the TGN 17. This compartment constitutes the releasable pool of HMW VWF that is secreted upon secretagogue stimulation. The proportion of VWF that is sorted into either direction has long been a matter of debate, partly because previous studies considered only constitutive and regulated secretion 16, 18. Careful kinetic monitoring of VWF trafficking has revealed that the storage pathway is also responsible for the majority of unstimulated VWF release through basal secretion 12. This most likely reflects gradual, stochastic turnover of WPBs, as VWF retention in this pathway correlates with the reported half‐life of these granules 10, 12. It suggests that the secretory machinery of WPBs at baseline is already in a certain degree of readiness that supports low‐level, spontaneous release. It also reconciles earlier and recent observations that the bulk of VWF released by resting endothelial cells is of a high multimeric nature 18, 19.
Figure 2.

The endothelial secretory pathway. von Willebrand factor (VWF) secretion occurs via three pathways: (i) constitutive secretion of low molecular weight VWF, which is primarily released at the basolateral side of the endothelium; and (ii) basal and (iii) regulated secretion of high molecular weight VWF from Weibel–Palade bodies (WPBs), which is primarily directed towards the apical surface. From the large number of WPBs that undergo exocytosis upon stimulated release, ultralarge VWF (UL‐VWF) multimers emerge that assemble into VWF strings on the apical side of the endothelium.
Apart from the degree of multimerization, another distinction between the constitutive and basal/stimulated secretion pathways can be made on the basis of the polarity of release. In vivo, the apical side of the endothelial cells faces the vascular lumen, which results in VWF secretion directly into the circulation. VWF unfurls and assembles into ultralarge VWF strings that can be up to several millimeters in length. Entanglement of several of these strings leads to the formation of spiderweb‐like networks that function as adhesive platforms for platelets 20. In this way, VWF strings act as polymeric force sensors that, upon flow, expose a shear‐dependent binding site for platelet glycoprotein Ib 4. Basolaterally released VWF is deposited in the subendothelial matrix, where it mediates platelet adhesion when the matrix is exposed following damage to the vessel wall, either directly or via self‐association with plasma VWF 21, 22. Over three decades ago, the idea had already arisen that secretion of such functionally distinct pools of VWF may differ in polarity, but previous studies addressing the polarity of endothelial VWF release remained inconclusive because they did not consider the contribution of spontaneous release of regulated secretory cargo via the basal route 23, 24. A recent study has now convincingly shown that constitutive release of LMW VWF mainly occurs basolaterally, whereas basal and stimulated release of HMW VWF from WPBs primarily occur at the apical face 19.
Stimuli and signaling cascades
WPB exocytosis is triggered by a wide range of physiological (stress hormones, e.g. epinephrine; proteases, e.g. thrombin; biogenic amines, e.g. histamine and 5‐hydroxytryptamine; and shear stress) and pathological (e.g. bacterial toxins) signals (reviewed in 6) that use Ca2+ or cAMP as second messengers (Fig. 3). As part of an integrated response to vascular injury, Ca2+‐mediated secretagogues such as histamine and thrombin locally promote a prothrombotic, proinflammatory state by instantaneously causing release of large quantities of VWF and other WPB constituents, while simultaneously increasing endothelial permeability and vascular tone 25, 26. In contrast, cAMP‐mediated stimuli such as epinephrine and vasopressin act systemically, increase endothelial barrier function, and, when applied to cultured endothelial cells, induce a slow but sustained release of WPBs 27, 28, 29, 30. This pathway can be clinically exploited to correct prolonged bleeding times in patients with mild hemophilia A or VWD through administration of the vasopressin analog DDAVP, which mobilizes VWF from its endothelial stores following activation of vasopressin‐2 receptor (V2R) 28, 31. Despite their distinct kinetic and physiological profiles, Ca2+‐dependent and cAMP‐dependent pathways converge at the same effector pathways, which control actin remodeling and tethering and fusion of WPBs, albeit in some cases with different outcomes (Fig. 3).
Figure 3.
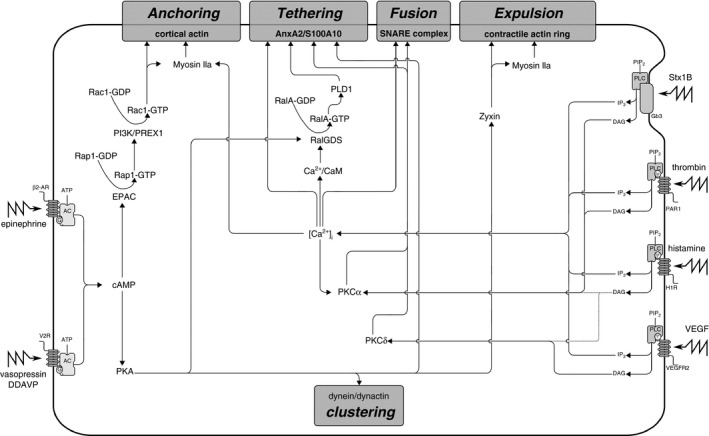
Signaling cascades in Weibel–Palade body (WPB) exocytosis. Ca2+‐mediated and cAMP‐mediated secretagogues that trigger WPB exocytosis use distinct and common signaling circuits that converge at effector pathways that control anchoring, tethering, vesicle fusion, and actin contractility. AC, adenylyl cyclase; ATP, adenosine triphosphate; CaM, calmodulin; [Ca2+]i, intracellular free Ca2+ concentration; DAG, diacylglycerol; EPAC, exchange protein that is directly activated by cAMP; Gb3, ceramide trihexoside; H1R, histamine H1 receptor; IP3, inositol 1,4,5‐triphosphate; PAR1, protease‐activated receptor 1; PI3K, phosphatidylinositol 3‐kinase; PIPa, phosphatidylinositol 4,5‐bisphosphate; PIP2, phosphatidylinositol 4,5‐bisphosphate; PKA, protein kinase A; PKC, protein kinase C; PLC, phospholipase C; PLD1, phospholipase D1; SNARE, soluble N‐ethylmaleimide‐sensitive factor attachment protein receptor; Stx1B, Shiga toxin 1B; V2F, vasopressin‐2 receptor; VEGF, vascular endothelial growth factor; VEGFR2, vascular endothelial growth factor receptor 2; β2‐AR, β2‐adrenergic receptor.
Increases in the intracellular free Ca2+ concentration ([Ca2+]i) occur after activation of phospholipase C (PLC) by ligand‐bound G‐protein‐coupled receptors (GPCRs) 32. Dose‐dependent, subsecond release of WPBs in response to ionophores or caged Ca2+ implies that sustained elevation of [Ca2+]i is a sufficient transduction signal to drive WPB fusion independently from additional receptor‐triggered signaling 26, 33. In nearly every regulated secretory system, a vesicle‐associated Ca2+ sensor is responsible for coupling transient elevations in [Ca2+]i to the soluble N‐ethylmaleimide‐sensitive factor attachment protein receptor (SNARE) fusion machinery, but, so far, such a WPB‐localized Ca2+ sensor has not been identified 34. However, a number of other Ca2+‐sensing mechanisms have been reported to couple cytosolic Ca2+ to the endothelial exocytotic response. When Ca2+ is associated with the Ca2+‐sensor calmodulin (CaM), the Ca2+–CaM complex binds to the N‐terminus of the guanine nucleotide exchange factor (GEF) RalGDS, thereby unleashing its GEF activity towards the small GTPase RalA 35. Activated RalA most likely coordinates a tethering step by promoting Arf6‐dependent phospholipase D1 (PLD1) activity 36. Local modification of phospholipids by PLD1 generates plasma membrane microdomains that recruit the Ca2+‐binding and phospholipid‐binding AnxA2–S100A10 complex, which, in its turn, links WPBs to membrane fusion sites through the WPB‐localized tethering factor Munc13‐4 37, 38, 39. Ca2+‐mediated VWF secretion is also dependent on rapid changes in the actin cytoskeleton that lead to the formation of parallel stress fibers, a process that is primarily regulated by GTPases of the Rho family 29, 40, 41.
Triggering of the GPCR‐coupled V2R and β2‐adrenergic receptors activates cAMP‐dependent protein kinase A (PKA) after conversion of ATP to cAMP 27, 28. PKA is critically involved in VWF secretion by activating a number of effector pathways 42, such as tethering of WPBs through the RalGDS–RalA pathway and the AnxA2–S100A10 tethering complex 30, 35, 38, and via phosphorylation of zyxin during contractile ring assembly 43. During cAMP‐mediated stimulation, a subset of WPBs avoid release by clustering together around the microtubule organizing center, as a result of dynein–dynactin‐dependent retrograde transport activated by PKA 42, 44. Whether this is a stochastic process that serves to limit the secretory response or whether these WPBs represent a specific subset of granules that are actively set aside, e.g. according to their recruited membrane components, state of maturation, or localization 15, is still unclear. The exchange protein that is directly activated by cAMP is also activated during cAMP‐mediated WPB release, and catalyzes the activation of the small GTPase Rap1 45, 46. Via a complex of its downstream effectors phosphatidylinositol 3‐kinase and the Rac exchange factor PREX1, activated Rap1, in its turn, promotes activation of the Rho GTPase Rac1 47. The Rap1–Rac1 pathway is thought to promote secretion by rearranging actin into thin cortical bundles in close apposition to the plasma membrane and/or by regulating the formation or contractility of actin on the fusing WPB membrane 43.
Protein kinase C (PKC) is a key signaling mediator for a number of endothelial agonists that trigger secretion of VWF. Phorbol esters, such as phorbol 12‐myristate 13‐acetate (PMA), are potent (non‐physiological) stimulators of VWF secretion that directly activate PKC by mimicking the action of diacylglycerol 25. Histamine, vascular endothelial growth factor (VEGF) and Shiga toxin 1B (Stx1B) elevate [Ca2+]i in a PLC‐dependent manner, and simultaneously either signal through the Ca2+‐independent PKCδ (VEGF and histamine) or utilize the Ca2+‐dependent PKCα (Stx1B and histamine) 32, 48, 49, 50, 51. Among its targets are components of the SNARE machinery, the assembly of which is partly regulated through PKC‐dependent phosphorylation events 52. Nevertheless, several ambiguities remain that suggest that PKC is not always essential for VWF secretion. Both VEGF and histamine activate PKCδ; however, broad‐range PKC inhibition or specific depletion of PKCδ significantly reduced secretory responses after VEGF but not after histamine administration 48. PKCα is activated by Stx1B as well as by histamine, but is only essential for Stx1B‐induced release; inhibition of PKCα did not block histamine‐induced VWF secretion and, in some studies, even enhanced it 48, 51, 53, 54, 55. Paradoxically, three independent studies showed that blocking Ca2+ signaling, which entirely abolishes VWF release induced by both histamine and PKCα‐dependent Stx1B, was not sufficient to fully inhibit VEGF‐induced VWF secretion 32, 48, 50, 53. The difference in requirement for PKC and Ca2+ raises intriguing questions about how common signaling pathways activated by different agonists are differentially utilized and integrated to control secretion.
Exocytotic machinery of WPBs
After emerging from the TGN, immature WPBs initially are secretion‐incompetent because they have yet to acquire (parts of) the exocytotic machinery 56. In a maturation‐dependent manner, WPBs recruit several GTPases of the Rab family, including Rab27A, several Rab3 isoforms, and Rab15 37, 56, 57, 58. Rab GTPases are molecular switches that cycle between a GDP‐bound ‘off’ state and a membrane‐associated, GTP‐bound ‘on’ state, and generally contribute to defining organelle identity 59. In their active GTP‐bound state, the Rabs are responsible for the subsequent recruitment of a set of effector proteins (MyRIP, Munc13‐4, and Slp4‐a) to the WPB. This process correlates with the acquisition of secretion competence 56, and enables WPBs to interact with the cytoskeleton and/or plasma membrane (Fig. 4) 56, 58, 60. MyRIP is a Rab27A‐specific effector that binds actin, directly and via the actin motor protein myosin Va, and tethers WPBs to the actin cytoskeleton in the cell periphery 41, 60, 61. The actin cytoskeleton plays opposing roles in the release of WPBs. On the one hand, it is necessary for peripheral distribution of WPBs, which is important for releasability and depends on myosin IIa 62. On the other hand, actin acts as a barrier 29, 40, and, by anchoring WPBs to the actin cytoskeleton, MyRIP acts as a brake during exocytosis 41, 56, 60. Munc13‐4 binds both Rab27A and Rab15, and promotes exocytosis by tethering WPBs to release sites on the plasma membrane, which contain the annexin A2–S100A10 complex 39, 58. Slp4‐a interacts with Rab3 isoforms and Rab27A, in an activity‐insensitive manner with the latter, and promotes WPB exocytosis 56 by providing the link between the WPB and members of the SNARE complex 63. From this all, it becomes clear that, during acquisition of secretion competence, WPBs recruit a cocktail of Rabs and Rab effectors that individually perform opposing functions during secretion. Upon exocytosis, they engage in a tug of war in which the balance of effectors, their levels of activation by upstream signaling events and the efficacy of their downstream mechanisms decide the probability of release 56.
Figure 4.

The Weibel–Palade body (WPB) exocytotic machinery. Rab effector complexes mediate anchoring of WPBs to the cytoskeleton, tethering to the plasma membrane, and interactions with the soluble N‐ethylmaleimide‐sensitive factor attachment protein receptor (SNARE) fusion machinery. The Rab27A–MyRIP–myosin Va complex anchors WPBs to the actin cytoskeleton. Munc13‐4 can be recruited by Rab GTPase‐dependent (Rab15/Rab27A) or by Rab‐independent mechanisms, and tethers WPBs to membrane fusion sites via the annexin A2–S100A10 complex. The Rab27A–Slp4‐a complex docks WPBs and forms the link between the WPB and the SNARE complex via members of the syntaxin‐binding protein (STXBP) family. VAMP, vesicle‐associated membrane protein.
In the final phase of exocytosis, the WPB fuses with the plasma membrane; this is catalyzed by a ternary complex of SNARE proteins that are positioned on the two opposing membranes. SNARE proteins can be functionally classified as t‐SNAREs, which are found on the target membrane, and v‐SNAREs, which are found on the donor/vesicle membrane. The exocytotic SNARE complex consists of a v‐SNARE of the vesicle‐associated membrane protein (VAMP) family on the donor (i.e. WPB) membrane, and two t‐SNAREs (one SNARE helix from a syntaxin, and two SNARE helices from a SNAP25 homolog) on the acceptor (i.e. plasma) membrane (Fig. 5). Together, they assemble into a four‐helix bundle that, when it rolls up, brings donor and acceptor membranes together in a zipper‐like motion, thereby largely overcoming the energy barrier that normally prevents fusion of membranes. WPBs contain two v‐SNAREs, VAMP3 and VAMP8, of which only VAMP3 has so far been shown to support stimulus‐induced WPB release 64, 65. t‐SNAREs that take part in WPB release include the SNAP25 homolog SNAP23, syntaxin‐3, syntaxin‐4, and most likely also syntaxin‐2 52, 63, 66, 67. Owing to their promiscuous nature, various combinations of these SNAREs exist: in endothelial cells, syntaxin‐4 engages with SNAP23 and VAMP3 or VAMP8 65, 67, and syntaxin‐3 primarily interacts with VAMP8 and SNAP23 67. The SNARE partners of syntaxin‐2 are still unknown. This suggests that endothelial cells can employ at least three (syntaxin‐4–SNAP23–VAMP3, syntaxin‐4–SNAP23–VAMP8, and syntaxin‐3–SNAP23–VAMP8) and possibly more distinct SNARE complexes for release of WPBs. Interestingly, whereas syntaxin‐4 is localized on the plasma membrane, syntaxin‐3 is found on WPBs 67, suggesting that, in certain cases, WPBs can also function as the acceptor compartment during fusion.
Figure 5.

Secretory modes of Weibel–Palade body (WPB) exocytosis. (A) Cartoon of a WPB with WPB‐localized v‐SNAREs (blue) engaging with target membrane‐localized t‐SNAREs (pink and green), thereby bringing together donor and acceptor membranes. Top: a narrow fusion pore is formed that permits release of small cargo. Lingering‐kiss fusion resulting from premature closure of the fusion pore results in large cargo (von Willebrand factor [VWF]) being retained in a collapsed granule, whereas expansion of the fusion pore results in full fusion followed by explosive release of ultralarge VWF strings. Top right: compound fusion of WPBs, possibly including lingering‐kiss end‐products, leads to the formation of an enlarged secretory pod that eventually undergoes fusion at the plasma membrane. Bottom left: cumulative or sequential fusion, in which a primary granule undergoes fusion at the plasma membrane, which is followed by secondary fusion events in the postfusion WPB, ultimately leading to extensive, ginger root‐like fusion structures. (B) Two models of SNARE‐mediated cumulative fusion. (Bi) Sequential fusion events are supported by cognate SNARE assemblies on individual, prefusion WPBs. (Bii) Cumulative release is supported by rapid membrane mixing, recruiting plasma membrane SNARE components into the postfusion WPB. SNARE, soluble N‐ethylmaleimide‐sensitive factor attachment protein receptor; VAMP, vesicle‐associated membrane protein.
The formation of SNARE complexes is controlled by syntaxin‐binding proteins (STXBPs), several of which take part in WPB release. The Slp4‐a interactor STXBP1 promotes VWF secretion and interacts with syntaxin‐2 or syntaxin‐3 63, potentially controlling two separate downstream mechanisms for exocytosis. Munc18c (also known as STXBP3) controls the assembly of syntaxin‐4‐containing SNARE complexes. Depending on a (de)phosphorylation switch controlled by PKCα and protein phosphatase 2B, phosphorylated Munc18c releases its grip on syntaxin‐4 and allows the latter to engage with VAMP3 52, 68. STXBP5 was initially implicated in the WPB exocytotic machinery as a genetic modifier of circulating VWF: a polymorphism encoding the non‐synonymous N436S substitution in STXBP5 is linked to lower VWF plasma levels 69. In endothelial cells, STXBP5 is a negative regulator of WPB release, and interacts with syntaxin‐4 but not with SNAP23 70. Possibly, the C‐terminal VAMP‐like domain acts as a decoy for syntaxin‐4, producing a non‐fusogenic dead end, thereby lowering the number of syntaxin‐4‐containing SNARE complexes that can support exocytosis. Interestingly, the N436S variant of STXBP5 attenuates VWF secretion in endothelial cells 70, although the mechanism through which this substitution inhibits release is still unclear. The SNARE machinery is therefore highly dynamic and, according to its configuration, may promote or attenuate secretion, but exactly how all of these individual components together orchestrate WPB release remains unclear. Moreover, it raises the question of why endothelial cells utilize so many different SNARE complexes for exocytosis of the same secretory organelle.
Secretory modes of WPB exocytosis
Several different modes of stimulus‐induced WPB exocytosis have been reported (Fig. 5A). Initially, a lipidic fusion pore is formed at the interface of vesicle and plasma membrane that establishes an aqueous channel between the WPB lumen and the extracellular space 71. Recordings of fusion pore dynamics of serotonin‐loaded WPBs by he use of amperometry revealed that this transient structure exists for only a limited time (usually < 200 ms) before it rapidly expands within tens of milliseconds 72. With its pore expanded fully, the WPB undergoes full fusion, resulting in the delivery of soluble granule cargo into the lumen and collapse of the vesicle membrane together with its associated membrane components into the plasma membrane 26, 73. In a subset of cases, the WPB fusion pore does not expand fully, but the pore lingers in a restricted state for some seconds before closing prematurely. The restricted diameter of the pore during this ‘lingering’ mode of fusion, so‐called lingering‐kiss exocytosis, acts as a molecular size filter: small cargoes, such as chemokines or the integral membrane protein CD63, are able to traverse the narrow pore that prevents the passage of larger cargo proteins such as VWF, VWF propeptide, and P‐selectin. After resealing of the unexpanded fusion pore, these proteins are selectively retained in spherical WPBs, which probably owe their rounded morphology and the disruption of the orderly bundling of VWF tubules to the hydration of their paracrystalline interior 74.
Apart from these heterotypic fusion modes (fusion between different compartments, i.e. WPB and plasma membrane), homotypic fusion modes (i.e. WPB–WPB) have also been described. During compound fusion, several individual WPBs coalesce intracellularly prior to release, and then collectively undergo exocytosis as a single entity. The first evidence for this mode of fusion in endothelial cells was provided by measurements of membrane capacitance, which is proportional to cell surface area, stepwise increases of which are attributable to the addition of membrane during the fusion of secretory organelles. A significant proportion of discrete membrane capacitance increases observed after evoking of WPB exocytosis with high levels of [Ca2+]i were too large to be accounted for by the fusion of a single WPB, and could only be explained by fusion of an accumulated structure made up of several WPBs 33. Morphological studies of such intermediate structures have shown that they consist of enlarged, rounded structures, termed secretory pods, which contain disordered VWF tubules 75. Secretory pods are prominent features following extended stimulation with PMA, but the occurrence of similar structures has also been demonstrated in vivo in response to activated FX or histamine in toads and rats, respectively 76, 77. Despite the loss of tubular organization of VWF multimers in the spherical secretory pods, VWF strings can eventually emerge from the extruded material 78. Finally, by monitoring of the redistribution of EGFP–CD63 through the membranes of fusing granules, a related but mechanistically distinct mode of homotypic WPB fusion, termed sequential or cumulative exocytosis, was observed 79. In this mode, a postfusion WPB is used as a membrane fusion site for subsequent cumulative fusion of additional WPBs, culminating in the ginger root‐like fusion structures that were observed in ultrastructural studies 76, 80.
A number of questions remain. First, what is the molecular basis for all of these different release mechanisms? The specificity of membrane fusion during exocytosis is mediated by the interaction of SNAREs on secretory organelles with their cognate SNAREs on the target membrane. In cases of homotypic fusion, such as during secretory pod or cumulative exocytosis, this would require the formation of a trans‐complex consisting of v‐SNAREs and t‐SNAREs that are positioned on opposing WPBs (Fig. 5Bi). Within the extensive set of SNAREs that have, in one way or another, been implicated in the WPB exocytotic machinery, the pair composed of VAMP8 and syntaxin‐3 has been shown to sustain homotypic fusion modes in cells in which both were found on secretory organelles 65, 67, 81, 82. An alternative explanation is offered by the rapid transfer of plasma membrane proteins to fusing WPBs through membrane mixing, such as was observed for Rab35 79. Possibly, this confers plasma membrane identity to the postfusion WPB, including the acquisition of components of the fusion machinery from the plasma membrane, such as the cognate SNAREs for WPB‐localized VAMPs (Fig. 5Bii). This also raises the possibility that secretory pods represent an intermediate state during cumulative fusion into WPBs that have previously undergone lingering‐kiss exocytosis.
Second, what is the use of all these distinct modes of exocytosis? Indirect or incomplete fusion modes are, in certain cases, energetically favorable. Homotypic fusion modes can serve to concentrate cargo release at hotspots when access to fusion sites on the membrane becomes limiting. It may also augment the secretory response by providing indirect access to the plasma membrane to a pool of WPBs situated deeper into the cytoplasm 80. Lingering‐kiss fusion not only saves vesicle reformation and retrieval of vesicle membrane proteins, but may also help in preserving a delicate balance between endocytosis and exocytosis by limiting membrane expenditure. Exocytosis–endocytosis coupling is pertinent to the choice of secretory mode, as pharmacological blocking of compensatory endocytosis resulted in a shift from full fusion towards cumulative exocytosis, possibly because of the inability to retrieve membrane components from the postfusion WPBs 80. Distinct fusion modes also allow the endothelium to fine‐tune its secretory response to specific vascular conditions: lingering‐kiss fusion releases only a subset of molecules, such as chemokines, whereas prothrombotic cargo is not released 74. Postfusion entanglement of VWF tubules in both compound and cumulative fusion structures does not preclude the eventual formation of VWF strings, but results in shorter strings when cumulative fusion is promoted 78, 80.
Content expulsion
Secretory cargo expulsion is dependent on lateral tension in the plasma membrane; relaxation of this tension through the addition of new membrane from fusing granules opens the fusion pore and provides a force to drive secretory cargo out; this is followed by full collapse of the vesicle into the plasma membrane 83. However, the near‐solid VWF paracrystal presents a significant barrier to release, as its enormous size and state will naturally resist discharge and membrane collapse. Recent studies have shown that endothelial cells, depending on their type of activation, can utilize fundamentally different modes of secretory content expulsion.
The rapid process of content expulsion during Ca2+‐mediated full fusion of WPBs bears the hallmarks of a jack‐in‐the‐box mechanism that is often observed when condensed, polyionic molecules such as VWF need to undergo a phase transition before release 84. Exchange of ions and entry of water molecules into the vesicle interior through the stalk‐shaped fusion pore leads to hydration of the vesicle core and rapid neutralization of the acidic milieu of the WPB 26. Dissolution of the proton gradient and other cationic species such as Ca2+ that are crucial for the condensed tubular packing of VWF 85, 86 will result in loss of shielding of negative charges in VWF. The consequential charge repulsion within and between VWF multimers leads to decondensation of cargo, as shown by the swelling of the granule and increased separation between VWF tubules 71. This is followed by an explosive discharge of ultralarge VWF multimers and other cargo on a subsecond timescale, consistent with a mechanism in which VWF tubules act as electrostatic springs 26, 73, 87, 88.
An alternative mechanism of VWF expulsion has been observed in response to Ca2+‐independent secretagogues, such as cAMP‐mediated agonists or phorbol esters. Prior to fusion, local rearrangements of cortical actin lead to the formation of actin frameworks that appear to encage WPBs. The formation of such frameworks is dependent on the tension‐sensitive focal adhesion protein zyxin following its phosphorylation by PKA, and positions WPBs close to the membrane 43. Then, shortly after opening of the fusion pore, an actomyosin ring is assembled coating the distal end of the fusing granule, a process that involves de novo actin polymerization 43, 89. Inhibition of the myosin IIa‐dependent contractility of the actomyosin ring prevented VWF release from fusing WPBs, while not affecting the secretory fate of P‐selectin 89 (but not in 62), leading to the hypothesis that contraction of such rings provides the necessary physical force to squeeze VWF out of the fusing granule. It is currently unclear what initiates actomyosin ring formation on the postfusion WPB, but the involvement of the tension sensor zyxin 43 suggests that changes in local (membrane) tension, such as on a swelling granule, may be an important factor in this process. Actomyosin ring contractility is regulated by phosphorylation of myosin IIa, which has been shown to involve zyxin‐dependent recruitment of casein kinase II 62. Interestingly, zyxin depletion did not impair VWF release by thrombin or histamine, which is consistent with the observation that actomyosin rings do not play an important role in expulsion of VWF during Ca2+‐mediated WPB exocytosis 43, 87.
WPBs in health and disease
Circulating levels of VWF vary widely across the human population 90. The clinical manifestations at the far ends of this distribution highlight the pivotal role that VWF plays in vascular health. Low levels of plasma VWF lead to bleeding, such as in the inherited bleeding disorder VWD 5. In some cases, this is the result of mutations in VWF that lead to altered WPB morphology and decreased releasability, such as is evident in VWD blood outgrowth endothelial cells 91. Increased levels of circulating VWF are associated with cardiovascular morbidities, such as coronary heart disease, ischemic stroke, and venous and arterial thrombosis 92, 93, which are pathologies for which the risks are reduced in VWD patients 94. The variation in VWF plasma levels across the population is largely genetically determined 95. Population‐based linkage analyses in healthy individuals have identified a number of genetic loci that are associated with VWF levels, including the ABO locus and the clearance receptors CLEC4M and STAB2, but also genes encoding several components of the SNARE machinery (STX2 and STXBP5) 69, 96. One of the SNPs in STXBP5 that was linked with higher VWF levels is also associated with venous thrombosis 97. Variants in both STX2 and STXBP5 are also associated with VWF levels and disease severity in VWD patients 98. Together, these findings show that SNARE‐mediated secretion from WPBs is an important determinant of VWF plasma levels. This fits with the current consensus that most circulating VWF, which primarily consists of high‐multimer VWF, originates from the storage pathway rather than from constitutive secretion 12, 18, 19. Whether this arises from the basal compartment or stimulated compartment may be less evident. Several of the Rab‐associated and SNARE‐associated regulators of stimulated WPB exocytosis also affect the basal secretion of VWF 56, 63, 67, suggesting that the same SNARE fusion machinery is also responsible for basal release. Basal secretion accounts for the bulk of VWF that endothelial cells produce and release over time. How reflective this is of the situation in vivo, in which some areas of the vasculature may be under the continuous influence of low‐level stimulation by hormones such as epinephrine or vasopressin, is unclear. However, on the basis of extrapolation of the ratio of basal to stimulated release from cultured endothelial cells, the area of vascular bed that needs to be to be maximally activated at any given time to match what is secreted basally makes it unlikely that stimulated release contributes significantly to steady‐state VWF levels 19.
Another pathology that may arise from disturbed release from WPBs is angiodysplasia, which is a common complication in VWD patients that causes recurrent gastrointestinal bleeding through small vascular malformations in the gut. Recent studies have shown that VWF regulates angiogenesis, extracellularly by controlling the cell surface expression of αVβ3 integrin and/or indirectly by influencing the release of one of its WPB coresidents, the angiogenic mediator Ang‐2 99. In the absence of its storage compartment, such as occurs in severe type 3 VWD, Ang‐2 is secreted constitutively, promoting vessel destabilization. Apart from Ang‐2, a number of other angiogenic mediators, such as insulin‐like growth factor‐binding protein 7, galectin‐1, and galectin‐3, are found in WPBs 7, 100, and may be subject to similar dysregulated release in the absence of VWF, potentially further contributing to pathological angiogenesis.
Conclusions and future directions
Endothelial cells control secretion from their WPBs by using a remarkably complex exocytotic machinery, the outlines of which are now starting to become clear. However, our knowledge of the exact composition of this machinery is incomplete and, importantly, we are still a long way from understanding how all of these components are able to direct the different modes of WPB fusion. Also, we currently do not understand how expansion of the fusion pore is regulated or what drives content expulsion in response to different endothelial activation states. To obtain a deeper understanding of these processes, we will need to determine the changes to the characteristic ultrastructure of WPBs during fusion and content expulsion, and to link these changes to individual components of the exocytotic machinery. Understanding the mechanism and purpose of the endothelium's ability to fine‐tune its secretory response to vascular events would potentially allow us to exploit this mechanism for therapeutic benefit in the treatment of hematological and cardiovascular diseases.
Disclosure of Conflict of Interests
The authors state that they have no conflict of interest.
Acknowledgements
We apologize to all authors whose work has been omitted owing to space restrictions, and for not always citing primary literature. Work in our laboratory is supported by grants from the Landsteiner Stichting voor Bloedtransfusie Research (LSBR‐1244 and LSBR‐1707), the Netherlands Ministry of Health (PPOC‐2015‐24P and PPOC‐2018‐21), the Dutch Thrombosis Foundation (TSN‐56‐2015 and TSN‐2017‐01), and a Research Fellowship from the European Hematology Association (EHA).
Schillemans M, Karampini E, Kat M, Bierings R. Exocytosis of Weibel–Palade bodies: how to unpack a vascular emergency kit. J Thromb Haemost 2019; 17: 6–18.
Manuscript handled by: P. H. Reitsma
Final decision: P. H. Reitsma, 15 October 2018
References
- 1. Weibel ER, Palade GE. New cytoplasmic components in arterial endothelia. J Cell Biol 1964; 23: 101–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ruggeri ZM. Von Willebrand factor, platelets and endothelial cell interactions. J Thromb Haemost 2003; 1: 1335–42. [DOI] [PubMed] [Google Scholar]
- 3. Valentijn KM, Sadler JE, Valentijn JA, Voorberg J, Eikenboom J. Functional architecture of Weibel–Palade bodies. Blood 2011; 117: 5033–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Springer TA. von Willebrand factor, Jedi knight of the bloodstream. Blood 2014; 124: 1412–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Leebeek FWG, Eikenboom JCJ. Von Willebrand's disease. N Engl J Med 2016; 375: 2067–80. [DOI] [PubMed] [Google Scholar]
- 6. Rondaij MG, Bierings R, Kragt A, van Mourik JA, Voorberg J. Dynamics and plasticity of Weibel–Palade bodies in endothelial cells. Arterioscler Thromb Vasc Biol 2006; 26: 1002–7. [DOI] [PubMed] [Google Scholar]
- 7. van Breevoort D, van Agtmaal EL, Dragt BS, Gebbinck JK, Dienava‐Verdoold I, Kragt A, Bierings R, Horrevoets AJG, Valentijn KM, Eikenboom JC, Fernandez‐Borja M, Meijer AB, Voorberg J. Proteomic screen identifies IGFBP7 as a novel component of endothelial cell‐specific Weibel–Palade bodies. J Proteome Res 2012; 11: 2925–36. [DOI] [PubMed] [Google Scholar]
- 8. van Agtmaal EL, Bierings R, Dragt BS, Leyen TA, Fernandez‐Borja M, Horrevoets AJG, Voorberg J. The shear stress‐induced transcription factor KLF2 affects dynamics and angiopoietin‐2 content of Weibel–Palade bodies. PLoS ONE 2012; 7: e38399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Knipe L, Meli A, Hewlett L, Bierings R, Dempster J, Skehel P, Hannah MJ, Carter T. A revised model for the secretion of tPA and cytokines from cultured endothelial cells. Blood 2010; 116: 2183–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kobayashi T, Vischer UM, Rosnoblet C, Lebrand C, Lindsay M, Parton RG, Kruithof EK, Gruenberg J. The tetraspanin CD63/lamp3 cycles between endocytic and secretory compartments in human endothelial cells. Mol Biol Cell 2000; 11: 1829–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fiedler U, Scharpfenecker M, Koidl S, Hegen A, Grunow V, Schmidt JM, Kriz W, Thurston G, Augustin HG. The Tie‐2 ligand angiopoietin‐2 is stored in and rapidly released upon stimulation from endothelial cell Weibel–Palade bodies. Blood 2004; 103: 4150–6. [DOI] [PubMed] [Google Scholar]
- 12. Giblin JP, Hewlett LJ, Hannah MJ. Basal secretion of von Willebrand factor from human endothelial cells. Blood 2008; 112: 957–64. [DOI] [PubMed] [Google Scholar]
- 13. Bierings R, van den Biggelaar M, Kragt A, Mertens K, Voorberg J, van Mourik JA. Efficiency of von Willebrand factor‐mediated targeting of interleukin‐8 into Weibel–Palade bodies. J Thromb Haemost 2007; 5: 2512–19. [DOI] [PubMed] [Google Scholar]
- 14. Deng YP, Storrie B. Animal cell lysosomes rapidly exchange membrane proteins. Proc Natl Acad Sci USA 1988; 85: 3860–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cleator JH, Zhu WQ, Vaughan DE, Hamm HE. Differential regulation of endothelial exocytosis of P‐selectin and von Willebrand factor by protease‐activated receptors and cAMP. Blood 2006; 107: 2736–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sporn LA, Marder VJ, Wagner DD. Inducible secretion of large, biologically potent von Willebrand factor multimers. Cell 1986; 46: 185–90. [DOI] [PubMed] [Google Scholar]
- 17. Zenner HL, Collinson LM, Michaux G, Cutler DF. High‐pressure freezing provides insights into Weibel–Palade body biogenesis. J Cell Sci 2007; 120: 2117–25. [DOI] [PubMed] [Google Scholar]
- 18. Tsai HM, Nagel RL, Hatcher VB, Seaton AC, Sussman II. The high molecular weight form of endothelial cell von Willebrand factor is released by the regulated pathway. Br J Haematol 1991; 79: 239–45. [DOI] [PubMed] [Google Scholar]
- 19. Lopes da Silva M, Cutler DF. von Willebrand factor multimerization and the polarity of secretory pathways in endothelial cells. Blood 2016; 128: 277–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. De Ceunynck K, De Meyer SF, Vanhoorelbeke K. Unwinding the von Willebrand factor strings puzzle. Blood 2013; 121: 270–7. [DOI] [PubMed] [Google Scholar]
- 21. Stel HV, Sakariassen KS, de Groot PG, van Mourik JA, Sixma JJ. Von Willebrand factor in the vessel wall mediates platelet adherence. Blood 1985; 65: 85–90. [PubMed] [Google Scholar]
- 22. Savage B, Sixma JJ, Ruggeri ZM. Functional self‐association of von Willebrand factor during platelet adhesion under flow. Proc Natl Acad Sci USA 2002; 99: 425–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. van Buul‐Wortelboer MF, Brinkman HJ, Dingemans KP, de Groot PG, van Aken WG, van Mourik JA. Reconstitution of the vascular wall in vitro. A novel model to study interactions between endothelial and smooth muscle cells. Exp Cell Res 1986; 162: 151–8. [DOI] [PubMed] [Google Scholar]
- 24. Sporn LA, Marder VJ, Wagner DD. Differing polarity of the constitutive and regulated secretory pathways for von Willebrand factor in endothelial cells. J Cell Biol 1989; 108: 1283–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Loesberg C, Gonsalves MD, Zandbergen J, Willems C, van Aken WG, Stel HV, Van Mourik JA, De Groot PG. The effect of calcium on the secretion of factor VIII‐related antigen by cultured human endothelial cells. Biochim Biophys Acta 1983; 763: 160–8. [DOI] [PubMed] [Google Scholar]
- 26. Erent M, Meli A, Moisoi N, Babich V, Hannah MJ, Skehel P, Knipe L, Zupancic G, Ogden D, Carter TD. Rate, extent and concentration‐dependence of histamine‐evoked Weibel–Palade body exocytosis determined from individual fusion events in human endothelial cells. J Physiol 2007; 583: 195–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vischer UM, Wollheim CB. Epinephrine induces von Willebrand factor release from cultured endothelial cells: involvement of cyclic AMP‐dependent signalling in exocytosis. Thromb Haemost 1997; 77: 1182–8. [PubMed] [Google Scholar]
- 28. Kaufmann JE, Oksche A, Wollheim CB, Günther G, Rosenthal W, Vischer UM, Gunther G. Vasopressin‐induced von Willebrand factor secretion from endothelial cells involves V2 receptors and cAMP. J Clin Invest 2000; 106: 107–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vischer UM, Barth H, Wollheim CB. Regulated von Willebrand factor secretion is associated with agonist‐specific patterns of cytoskeletal remodeling in cultured endothelial cells. Arterioscler Thromb Vasc Biol 2000; 20: 883–91. [DOI] [PubMed] [Google Scholar]
- 30. Rondaij MG, Sellink E, Gijzen KA, ten Klooster JP, Hordijk PL, van Mourik JA, Voorberg J. Small GTP‐binding protein Ral is involved in cAMP‐mediated release of von Willebrand factor from endothelial cells. Arterioscler Thromb Vasc Biol 2004; 24: 1315–20. [DOI] [PubMed] [Google Scholar]
- 31. Mannucci PM, Ruggeri ZM, Pareti FI, Capitanio A. 1‐Deamino‐8‐d‐arginine vasopressin: a new pharmacological approach to the management of haemophilia and von Willebrands’ diseases. Lancet 1977; 1: 869–72. [DOI] [PubMed] [Google Scholar]
- 32. Xiong Y, Huo Y, Chen C, Zeng H, Lu X, Wei C, Ruan C, Zhang X, Hu Z, Shibuya M, Luo J. Vascular endothelial growth factor (VEGF) receptor‐2 tyrosine 1175 signaling controls VEGF‐induced von Willebrand factor release from endothelial cells via phospholipase C‐gamma 1‐ and protein kinase A‐dependent pathways. J Biol Chem 2009; 284: 23217–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zupancic G, Ogden D, Magnus CJ, Wheeler‐Jones C, Carter TD. Differential exocytosis from human endothelial cells evoked by high intracellular Ca(2+) concentration. J Physiol 2002; 544: 741–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pang ZP, Südhof TC. Cell biology of Ca2+‐triggered exocytosis. Curr Opin Cell Biol 2010; 22: 496–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rondaij MG, Bierings R, van Agtmaal EL, Gijzen KA, Sellink E, Kragt A, Ferguson SSG, Mertens K, Hannah MJ, van Mourik JA, Fernandez‐Borja M, Voorberg J. Guanine exchange factor RalGDS mediates exocytosis of Weibel–Palade bodies from endothelial cells. Blood 2008; 112: 56–63. [DOI] [PubMed] [Google Scholar]
- 36. Disse J, Vitale N, Bader M‐FF, Gerke V. Phospholipase D1 is specifically required for regulated secretion of von Willebrand factor from endothelial cells. Blood 2009; 113: 973–80. [DOI] [PubMed] [Google Scholar]
- 37. Knop M, Aareskjold E, Bode G, Gerke V. Rab3D and annexin A2 play a role in regulated secretion of vWF, but not tPA, from endothelial cells. EMBO J 2004; 23: 2982–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brandherm I, Disse J, Zeuschner D, Gerke V. cAMP‐induced secretion of endothelial von Willebrand factor is regulated by a phosphorylation/dephosphorylation switch in annexin A2. Blood 2013; 122: 1042–51. [DOI] [PubMed] [Google Scholar]
- 39. Chehab T, Santos NC, Holthenrich A, Koerdt SN, Disse J, Schuberth C, Nazmi AR, Neeft M, Koch H, Man KNM, Wojcik SM, Martin TFJ, van der Sluijs P, Brose N, Gerke V. A novel Munc13‐4/S100A10/annexin A2 complex promotes Weibel–Palade body exocytosis in endothelial cells. Mol Biol Cell 2017; 28: 1688–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Klarenbach SW, Chipiuk A, Nelson RC, Hollenberg MD, Murray AG. Differential actions of PAR2 and PAR1, in stimulating human endothelial cell exocytosis and permeability: the role of Rho‐GTPases. Circ Res 2003; 92: 272–8. [DOI] [PubMed] [Google Scholar]
- 41. Conte IL, Hellen N, Bierings R, Mashanov GI, Manneville J‐B, Kiskin NI, Hannah MJ, Molloy JE, Carter T. Interaction between MyRIP and the actin cytoskeleton regulates Weibel–Palade body trafficking and exocytosis. J Cell Sci 2016; 129: 592–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vischer UM, Wollheim CB. Purine nucleotides induce regulated secretion of von Willebrand factor: involvement of cytosolic Ca2+ and cyclic adenosine monophosphate‐dependent signaling in endothelial exocytosis. Blood 1998; 91: 118–27. [PubMed] [Google Scholar]
- 43. Han X, Li P, Yang Z, Huang X, Wei G, Sun Y, Kang X, Hu X, Deng Q, Chen L, He A, Huo Y, Li D, Betzig E, Luo J. Zyxin regulates endothelial von Willebrand factor secretion by reorganizing actin filaments around exocytic granules. Nat Commun 2017; 8: 14639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rondaij MG, Bierings R, Kragt A, Gijzen KA, Sellink E, van Mourik JA, Fernandez‐Borja M, Voorberg J. Dynein–dynactin complex mediates protein kinase A‐dependent clustering of Weibel–Palade bodies in endothelial cells. Arterioscler Thromb Vasc Biol 2006; 26: 49–55. [DOI] [PubMed] [Google Scholar]
- 45. Øynebråten I, Barois N, Hagelsteen K, Johansen F, Bakke O, Haraldsen G. Characterization of a novel chemokine‐containing storage granule in endothelial cells: evidence for preferential exocytosis mediated by protein kinase A and diacylglycerol. J Immunol 2005; 175: 5358–69. [DOI] [PubMed] [Google Scholar]
- 46. van Hooren KWEM, van Agtmaal EL, Fernandez‐Borja M, van Mourik JA, Voorberg J, Bierings R. The Epac–Rap1 signaling pathway controls cAMP‐mediated exocytosis of Weibel–Palade bodies in endothelial cells. J Biol Chem 2012; 287: 24713–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. van Hooren KWEM, van Breevoort D, Fernandez‐Borja M, Meijer AB, Eikenboom J, Bierings R, Voorberg J. Phosphatidylinositol‐3,4,5‐triphosphate‐dependent Rac exchange factor 1 regulates epinephrine‐induced exocytosis of Weibel–Palade bodies. J Thromb Haemost 2014; 12: 273–81. [DOI] [PubMed] [Google Scholar]
- 48. Lorenzi O, Frieden M, Villemin P, Fournier M, Foti M, Vischer UM. Protein kinase C‐delta mediates von Willebrand factor secretion from endothelial cells in response to vascular endothelial growth factor (VEGF) but not histamine. J Thromb Haemost 2008; 6: 1962–9. [DOI] [PubMed] [Google Scholar]
- 49. Wheeler‐Jones C, Abu‐Ghazaleh R, Cospedal R, Houliston RA, Martin J, Zachary I. Vascular endothelial growth factor stimulates prostacyclin production and activation of cytosolic phospholipase A2 in endothelial cells via p42/p44 mitogen‐activated protein kinase. FEBS Lett 1997; 420: 28–32. [DOI] [PubMed] [Google Scholar]
- 50. Matsushita K, Yamakuchi M, Morrell CN, Ozaki M, O'Rourke B, Irani K, Lowenstein CJ. Vascular endothelial growth factor regulation of Weibel–Palade‐body exocytosis. Blood 2005; 105: 207–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Huang J, Haberichter SL, Sadler JE. The B subunits of Shiga‐like toxins induce regulated VWF secretion in a phospholipase D1‐dependent manner. Blood 2012; 120: 1143–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Fu J, Naren AP, Gao X, Ahmmed GU, Malik AB. Protease‐activated receptor‐1 activation of endothelial cells induces protein kinase Calpha‐dependent phosphorylation of syntaxin 4 and Munc18c: role in signaling p‐selectin expression. J Biol Chem 2005; 280: 3178–84. [DOI] [PubMed] [Google Scholar]
- 53. Liu F, Huang J, Sadler JE. Shiga toxin (Stx)1B and Stx2B induce von Willebrand factor secretion from human umbilical vein endothelial cells through different signaling pathways. Blood 2011; 118: 3392–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Carew MA, Paleolog EM, Pearson JD. The roles of protein kinase C and intracellular Ca2+ in the secretion of von Willebrand factor from human vascular endothelial cells. Biochem J 1992; 286(Pt 2): 631–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Huwiler A, Döll F, Ren S, Klawitter S, Greening A, Römer I, Bubnova S, Reinsberg L, Pfeilschifter J. Histamine increases sphingosine kinase‐1 expression and activity in the human arterial endothelial cell line EA.hy 926 by a PKC‐α‐dependent mechanism. Biochim Biophys Acta Mol Cell Biol Lipids 2006; 1761: 367–76. [DOI] [PubMed] [Google Scholar]
- 56. Bierings R, Hellen N, Kiskin N, Knipe L, Fonseca A‐V, Patel B, Meli A, Rose M, Hannah MJ, Carter T. The interplay between the Rab27A effectors Slp4‐a and MyRIP controls hormone‐evoked Weibel–Palade body exocytosis. Blood 2012; 120: 2757–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hannah MJ, Hume AN, Arribas M, Williams R, Hewlett LJ, Seabra MC, Cutler DF. Weibel–Palade bodies recruit Rab27 by a content‐driven, maturation‐dependent mechanism that is independent of cell type. J Cell Sci 2003; 116: 3939–48. [DOI] [PubMed] [Google Scholar]
- 58. Zografou S, Basagiannis D, Papafotika A, Shirakawa R, Horiuchi H, Auerbach D, Fukuda M, Christoforidis S. A complete Rab screening reveals novel insights in Weibel–Palade body exocytosis. J Cell Sci 2012; 125: 4780–90. [DOI] [PubMed] [Google Scholar]
- 59. Barr FA. Rab GTPases and membrane identity: causal or inconsequential? J Cell Biol 2013; 202: 191–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Nightingale TD, Pattni K, Hume AN, Seabra MC, Cutler DF. Rab27a and MyRIP regulate the amount and multimeric state of VWF released from endothelial cells. Blood 2009; 113: 5010–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rojo Pulido I, Nightingale TD, Darchen F, Seabra MC, Cutler DF, Gerke V. Myosin Va acts in concert with Rab27a and MyRIP to regulate acute von‐Willebrand factor release from endothelial cells. Traffic 2011; 12: 1371–82. [DOI] [PubMed] [Google Scholar]
- 62. Li P, Wei G, Cao Y, Deng Q, Han X, Huang X, Huo Y, He Y, Chen L, Luo J. Myosin IIa is critical for cAMP‐mediated endothelial secretion of von Willebrand factor. Blood 2017; 131: 686–98. [DOI] [PubMed] [Google Scholar]
- 63. van Breevoort D, Snijders AP, Hellen N, Weckhuysen S, van Hooren KWEM, Eikenboom J, Valentijn K, Fernandez‐Borja M, Ceulemans B, De Jonghe P, Voorberg J, Hannah M, Carter T, Bierings R. STXBP1 promotes Weibel–Palade body exocytosis through its interaction with the Rab27A effector Slp4‐a. Blood 2014; 123: 3185–94. [DOI] [PubMed] [Google Scholar]
- 64. Matsushita K, Morrell CN, Cambien B, Yang SX, Yamakuchi M, Bao C, Hara MR, Quick RA, Cao W, O'Rourke B, Lowenstein JM, Pevsner J, Wagner DD, Lowenstein CJ. Nitric oxide regulates exocytosis by S‐nitrosylation of N‐ethylmaleimide‐sensitive factor. Cell 2003; 115: 139–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Pulido IR, Jahn R, Gerke V. VAMP3 is associated with endothelial Weibel–Palade bodies and participates in their Ca(2+)‐dependent exocytosis. Biochim Biophys Acta 2011; 1813: 1038–44. [DOI] [PubMed] [Google Scholar]
- 66. Zhu QM, Zhu Q, Yamakuchi M, Lowenstein CJ. SNAP23 regulates endothelial exocytosis of von Willebrand factor. PLoS ONE 2015; 10: e0118737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Schillemans M, Karampini E, van den Eshof BL, Gangaev A, Hofman M, van Breevoort D, Meems H, Janssen H, Mulder AA, Jost CR, Escher JC, Adam R, Carter T, Koster AJ, van den Biggelaar M, Voorberg J, Bierings R. Weibel–Palade body localized syntaxin‐3 modulates Von Willebrand factor secretion from endothelial cells. Arterioscler Thromb Vasc Biol 2018; 38: 1549–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Nolasco LH, Gushiken FC, Turner NA, Khatlani TS, Pradhan S, Dong J‐F, Moake JL, Vijayan KV. Protein phosphatase 2B inhibition promotes the secretion of von Willebrand factor from endothelial cells. J Thromb Haemost 2009; 7: 1009–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Smith NL, Chen MH, Dehghan A, Strachan DP, Basu S, Soranzo N, Hayward C, Rudan I, Sabater‐Lleal M, Bis JC, De Maat MPM, Rumley A, Kong X, Yang Q, Williams FMK, Vitart V, Campbell H, Mälarstig A, Wiggins KL, Van Duijn CM, et al Novel associations of multiple genetic loci with plasma levels of factor VII, factor VIII, and von Willebrand factor: the CHARGE (Cohorts for Heart and Aging Research in Genome Epidemiology) Consortium. Circulation 2010; 121: 1382–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Zhu Q, Yamakuchi M, Ture S, de la Luz Garcia‐Hernandez M, Ko KA, Modjeski KL, LoMonaco MB, Johnson AD, O'Donnell CJ, Takai Y, Morrell CN, Lowenstein CJ. Syntaxin‐binding protein STXBP5 inhibits endothelial exocytosis and promotes platelet secretion. J Clin Invest 2014; 124: 4503–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Berriman JA, Li S, Hewlett LJ, Wasilewski S, Kiskin FN, Carter T, Hannah MJ, Rosenthal PB. Structural organization of Weibel–Palade bodies revealed by cryo‐EM of vitrified endothelial cells. Proc Natl Acad Sci USA 2009; 106: 17407–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Cookson EA, Conte IL, Dempster J, Hannah MJ, Carter T. Characterisation of Weibel–Palade body fusion by amperometry in endothelial cells reveals fusion pore dynamics and the effect of cholesterol on exocytosis. J Cell Sci 2013; 126: 5490–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hannah MJ, Skehel P, Erent M, Knipe L, Ogden D, Carter T. Differential kinetics of cell surface loss of von Willebrand factor and its propolypeptide after secretion from Weibel–Palade bodies in living human endothelial cells. J Biol Chem 2005; 280: 22827–30. [DOI] [PubMed] [Google Scholar]
- 74. Babich V, Meli A, Knipe L, Dempster JE, Skehel P, Hannah MJ, Carter T. Selective release of molecules from Weibel–Palade bodies during a lingering kiss. Blood 2008; 111: 5282–90. [DOI] [PubMed] [Google Scholar]
- 75. Valentijn KM, van Driel LF, Mourik MJ, Hendriks G‐J, Arends TJ, Koster AJ, Valentijn JA. Multigranular exocytosis of Weibel Palade bodies in vascular endothelial cells. Blood 2010; 116: 1807–16. [DOI] [PubMed] [Google Scholar]
- 76. Fujimoto S. Degranulation of endothelial specific granules of the toad aorta after treatment with compound 48/80. Anat Rec 1982; 203: 197–204. [DOI] [PubMed] [Google Scholar]
- 77. Richardson M, Tinlin S, De Reske M, Webster S, Senis Y, Giles AR. Morphological alterations in endothelial cells associated with the release of von Willebrand factor after thrombin generation in vivo. Arterioscler Thromb 1994; 14: 990–9. [DOI] [PubMed] [Google Scholar]
- 78. Mourik MJ, Valentijn JA, Voorberg J, Koster AJ, Valentijn KM, Eikenboom J. von Willebrand factor remodeling during exocytosis from vascular endothelial cells. J Thromb Haemost 2013; 11: 2009–19. [DOI] [PubMed] [Google Scholar]
- 79. Kiskin NI, Babich V, Knipe L, Hannah MJ, Carter T. Differential cargo mobilisation within Weibel–Palade bodies after transient fusion with the plasma membrane. PLoS ONE 2014; 9: e108093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Stevenson NL, White IJ, McCormack JJ, Robinson C, Cutler DF, Nightingale TD. Clathrin‐mediated post‐fusion membrane retrieval influences the exocytic mode of endothelial Weibel–Palade bodies. J Cell Sci 2017; 130: 2591–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Behrendorff N, Dolai S, Hong W, Gaisano HY, Thorn P. Vesicle‐associated membrane protein 8 (VAMP8) is a SNARE (soluble N‐ethylmaleimide‐sensitive factor attachment protein receptor) selectively required for sequential granule‐to‐granule fusion. J Biol Chem 2011; 286: 29627–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Zhu D, Koo E, Kwan E, Kang Y, Park S, Xie H, Sugita S, Gaisano HY. Syntaxin‐3 regulates newcomer insulin granule exocytosis and compound fusion in pancreatic beta cells. Diabetologia 2013; 56: 359–69. [DOI] [PubMed] [Google Scholar]
- 83. Kozlov MM, Chernomordik LV. Membrane tension and membrane fusion. Curr Opin Struct Biol 2015; 33: 61–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Verdugo P. Goblet cells secretion and mucogenesis. Annu Rev Physiol 1990; 52: 157–76. [DOI] [PubMed] [Google Scholar]
- 85. Wagner DD, Mayadas T, Marder VJ. Initial glycosylation and acidic pH in the Golgi apparatus are required for multimerization of von Willebrand factor. J Cell Biol 1986; 102: 1320–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Huang R‐H, Wang Y, Roth R, Yu X, Purvis AR, Heuser JE, Egelman EH, Sadler JE. Assembly of Weibel–Palade body‐like tubules from N‐terminal domains of von Willebrand factor. Proc Natl Acad Sci USA 2008; 105: 482–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Conte IL, Cookson E, Hellen N, Bierings R, Mashanov G, Carter T. Is there more than one way to unpack a Weibel–Palade body? Blood 2015; 126: 2165–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Michaux G, Abbitt KB, Collinson LM, Haberichter SL, Norman KE, Cutler DF. The physiological function of von Willebrand's factor depends on its tubular storage in endothelial Weibel–Palade bodies. Dev Cell 2006; 10: 223–32. [DOI] [PubMed] [Google Scholar]
- 89. Nightingale TD, White IJ, Doyle EL, Turmaine M, Harrison‐Lavoie KJ, Webb KF, Cramer LP, Cutler DF. Actomyosin II contractility expels von Willebrand factor from Weibel–Palade bodies during exocytosis. J Cell Biol 2011; 194: 613–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Sadler JE. von Willebrand factor: two sides of a coin. J Thromb Haemost 2005; 3: 1702–9. [DOI] [PubMed] [Google Scholar]
- 91. Wang J‐W, Bouwens EAM, Pintao MC, Voorberg J, Safdar H, Valentijn KM, de Boer HC, Mertens K, Reitsma PH, Eikenboom J. Analysis of the storage and secretion of von Willebrand factor in blood outgrowth endothelial cells derived from patients with von Willebrand disease. Blood 2013; 121: 2762–72. [DOI] [PubMed] [Google Scholar]
- 92. Koster T, Blann AD, Briet E, Vandenbroucke JP, Rosendaal FR. Role of clotting factor VIII in effect of von Willebrand factor on occurrence of deep‐vein thrombosis. Lancet 1995; 345: 152–5. [DOI] [PubMed] [Google Scholar]
- 93. Sonneveld MAH, De Maat MPM, Leebeek FWG. Von Willebrand factor and ADAMTS13 in arterial thrombosis: a systematic review and meta‐analysis. Blood Rev 2014; 28: 167–78. [DOI] [PubMed] [Google Scholar]
- 94. Sanders YV, Eikenboom J, de Wee EM, van der Bom JG, Cnossen MH, Degenaar‐Dujardin MEL, Fijnvandraat K, Kamphuisen PW, Laros‐van Gorkom BAP, Meijer K, Mauser‐Bunschoten EP, Leebeek FWG, Kors A, Zweegman S, Goverde GJ, Jonkers MH, Dors N, Nijziel MR, Meijer K, Tamminga RYJ, et al Reduced prevalence of arterial thrombosis in von Willebrand disease. J Thromb Haemost 2013; 11: 845–54. [DOI] [PubMed] [Google Scholar]
- 95. Desch KC, Ozel AB, Siemieniak D, Kalish Y, Shavit J, Thornburg CD, Sharathkumar AA, McHugh CP, Laurie CC, Crenshaw A, Mirel DB, Kim Y, Cropp CD, Molloy AM, Kirke PN, Bailey‐Wilson JE, Wilson AF, Mills JL, Scott JM, Brody LC, et al Linkage analysis identifies a locus for plasma von Willebrand factor undetected by genome‐wide association. Proc Natl Acad Sci USA 2013; 110: 588–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. van Loon JE, Leebeek FWG, Deckers JW, Dippel DWJ, Poldermans D, Strachan DP, Tang W, O'Donnell CJ, Smith NL, de Maat MPM. Effect of genetic variations in syntaxin‐binding protein‐5 and syntaxin‐2 on von Willebrand factor concentration and cardiovascular risk. Circ Cardiovasc Genet 2010; 3: 507–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Smith NL, Rice KM, Bovill EG, Cushman M, Bis JC, McKnight B, Lumley T, Glazer NL, van Hylckama Vlieg A, Tang W, Dehghan A, Strachan DP, O'Donnell CJ, Rotter JI, Heckbert SR, Psaty BM, Rosendaal FR. Genetic variation associated with plasma von Willebrand factor levels and the risk of incident venous thrombosis. Blood 2011; 117: 6007–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Sanders YV, van der Bom JG, Isaacs A, Cnossen MH, de Maat MPM, Laros‐van Gorkom BAP, Fijnvandraat K, Meijer K, van Duijn CM, Mauser‐Bunschoten EP, Eikenboom J, Leebeek FWG, Fijnvandraat K, Coppens M, Kors A, de Meris J, Nijziel MR, Meijer K, Tamminga RYJ, Ypma PF, et al CLEC4M and STXBP5 gene variations contribute to von Willebrand factor level variation in von Willebrand disease. J Thromb Haemost 2015; 13: 956–66. [DOI] [PubMed] [Google Scholar]
- 99. Starke RD, Ferraro F, Paschalaki KE, Dryden NH, McKinnon TAJ, Sutton RE, Payne EM, Haskard DO, Hughes AD, Cutler DF, Laffan MA, Randi AM. Endothelial von Willebrand factor regulates angiogenesis. Blood 2011; 117: 1071–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Saint‐Lu N, Oortwijn BD, Pegon JN, Odouard S, Christophe OD, De Groot PG, Denis CV, Lenting PJ. Identification of galectin‐1 and galectin‐3 as novel partners for von Willebrand factor. Arterioscler Thromb Vasc Biol 2012; 32: 894–901. [DOI] [PubMed] [Google Scholar]