

Abstract
Objective
Acute myeloid leukemia (AML) can be subtyped based on recurrent cytogenetic and molecular genetic abnormalities with diagnostic and prognostic significance. Although cytogenetic characterization classically involves conventional chromosome and/or fluorescence in situ hybridization (FISH) assays, limitations of these techniques include poor resolution and the inability to precisely identify breakpoints.
Method
We evaluated whether an NGS‐based methodology that detects structural abnormalities and copy number changes using mate pair sequencing (MPseq) can enhance the diagnostic yield for patients with AML.
Results
Using 68 known abnormal and 20 karyotypically normal AML samples, each recurrent primary AML‐specific abnormality previously identified in the abnormal samples was confirmed using MPseq. Importantly, in eight cases with abnormalities that could not be resolved by conventional cytogenetic studies, MPseq was utilized to molecularly define eight recurrent AML‐fusion events. In addition, MPseq uncovered two cryptic abnormalities that were missed by conventional cytogenetic studies. Thus, MPseq improved the diagnostic yield in the detection of AML‐specific structural rearrangements in 10/88 (11%) of cases analyzed.
Conclusion
Utilization of MPseq represents a precise, molecular‐based technique that can be used as an alternative to conventional cytogenetic studies for newly diagnosed AML patients with the potential to revolutionize the diagnosis of hematologic malignancies.
Keywords: acute myeloid leukemia, molecular cytogenetics, MPseq
1. INTRODUCTION
Acute myeloid leukemia (AML) is the most common acute leukemia in adults with an incidence of approximately 3‐5 cases per 100 000 individuals.1, 2, 3 Approximately 20 000 new cases are diagnosed annually, half of which will die from this disease.2 The World Health Organization groups AML with recurrent genetic abnormalities into 11 subtypes based on specific chromosomal rearrangements and genetic mutations.4 An additional category characterized by recurrent cytogenetic abnormalities, including unbalanced copy number variation (CNVs), is defined as “AML with myelodysplasia‐related changes”.3 The identification of these well‐characterized recurrent genomic abnormalities provides important diagnostic, prognostic, and treatment‐related information.
Currently, most genomic testing of bone marrow or blood specimens from AML patients occurs by karyotype analysis, fluorescence in situ hybridization (FISH), or reverse transcription‐polymerase chain reaction (RT‐PCR) targeting chimeric fusion genes as well as Next‐Generation Sequencing (NGS) for the detection of point mutations. While these techniques collectively provide the current gold standard for AML genetic characterization, there are significant limitations including poor and variable resolution of conventional chromosome studies. In addition, cryptic balanced rearrangements may be missed. Karyotyping requires dividing cells arrested in metaphase, is labor‐intensive, subjective, requires highly skilled technologists and can result in long turnaround times. While FISH studies address some of these limitations, FISH is limited to the interrogation of only the regions for which FISH probes are available, which requires a priori knowledge of a specific rearrangement or CNV. FISH panels for AML also need to be quite large to be comprehensive. For example, the current Mayo Clinic AML FISH panel contains 29 probe sets (Table S1), and each probe set requires an independent, costly, and time‐consuming validation.5, 6 Despite their size, these panels still have the potential to miss cryptic gene fusions resulting from insertional events. While FISH has a higher resolution (100‐200 kb compared to >5 Mb) than karyotyping, its resolution is inferior compared to newer methodologies such as chromosomal microarray and NGS technologies, which have the potential to provide more precise characterization of chromosomal abnormalities.
We explored the ability of an NGS methodology, mate pair sequencing (MPseq), to overcome the limitations of conventional karyotyping and FISH studies. MPseq is a whole‐genome sequencing assay that utilizes long input DNA (2‐5 Kb) that is circularized and fragmented to the size of paired‐end fragments (200‐500 bp). This modification to traditional NGS sequencing enables the detection of structural rearrangements and copy number changes throughout the genome with significantly reduced sequencing depth, resulting in a more cost‐effective strategy. MPseq has higher resolution than karyotyping and FISH, does not require dividing cells or a priori knowledge of specific abnormalities. In addition, MPseq provides an alternative technology to comprehensively evaluate a sample for chromosomal rearrangements and copy number changes in a single assay rather than large panels of independent FISH probes. Here, we compare the performance of MPseq in 68 abnormal and 20 normal samples previously characterized by standard clinical cytogenetic studies to detect chromosome rearrangements and copy number changes in patients with AML. The results described here demonstrate the utility of MPseq as a single assay replacement for conventional karyotyping and FISH studies for diagnostic AML samples and highlight the potential to increase diagnostic yield and clarity.
2. METHODS
2.1. Patient samples
All samples were obtained and evaluated as part of an Institutional Review Board approved study. Fresh diagnostic bone marrow (BM), peripheral blood (PB), or fixed cell pellet (FCP) samples from patients with a reason for referral (RFR) of AML referred to the Mayo Clinic Genomics Laboratory were selected based on reported abnormalities previously tested as part of routine clinical care by conventional karyotyping and/or FISH. Due to the scarcity of BCR/ABL1‐positive AML patients, we included seven patients with CML in order to evaluate BCR/ABL1 fusions.
2.2. Conventional chromosome analysis
A conventional G‐banded chromosome evaluation was performed as part of routine clinical testing. First, a cell count is performed on the specimen to establish a plating volume and based on the cell count, a corresponding volume of bone marrow is added to two culture flasks containing culture medium and incubated for 24‐48 hours at 37°C. In the harvest process, the cells are exposed to colcemid and hypotonic solution, and are fixed with glacial acid and methanol. Metaphase cells are dropped onto microscope slides and are stained by G‐banding. All cells analyzed are captured using a computerized imaging system, and one or more karyograms from each clone are prepared to document the type of abnormality and to permit systematic interpretation of the anomalies. Minimal evidence for the presence of an abnormal clone is defined as two or more metaphases with the same structural abnormality or chromosome gain (trisomy), or three or more metaphases lacking the same chromosome. Twenty metaphases are analyzed by qualified clinical cytogenetic technologists and interpreted by a board‐certified clinical cytogeneticist.
2.3. Fluorescence in situ hybridization (FISH)
Commercial and “laboratory‐developed” (LD) dual‐color dual‐fusion (D‐FISH), break‐apart and enumeration FISH probes were utilized to detect AML‐specific abnormalities (Table S1). All specimens were subjected to standard FISH pretreatment, hybridization, and fluorescence microscopy according to specimen‐specific protocols. Methods were described in the manuscripts by Aypar et al5 and Keefe et al.6 FISH analysis was performed by qualified clinical cytogenetic technologists and interpreted by a board‐certified clinical cytogeneticist.
2.4. DNA extraction and library preparation
The DNA extraction and mate pair library preparation methods were described in the manuscripts by Johnson et al7 and Smadbeck et al.8 Briefly, DNA was isolated from BM and PB samples using the Qiagen Puregene extraction kit when the sample volume was <2 mL, Autopure LS Automated high quality DNA extraction for those samples more than 2 mL, and the QIAmp Tissue kit for fixed cell pellet samples. DNA was processed using the Illumina Nextera Mate Pair library preparation kit and sequenced on the Illumina HiSeq 2500 in rapid run mode. Pooled libraries were hybridized onto a flow cell (two samples per lane) and sequenced using 101‐basepair reads and paired‐end sequencing.
2.5. Structural variant bioinformatics pipeline
These and the remaining methods were described in the manuscripts by Johnson et al7 and Smadbeck et al.8 MPseq data were processed using BIMA to map to the reference genome and SVAtools for breakpoint detection of both junctions in chromosomal rearrangements and copy number changes (process outlined in Figure 1). BIMA is a binary indexing mapping algorithm for simultaneous mapping of both reads in a mate pair fragment9 and was used to map all MPseq fragments to reference genome GRCh38 using default settings. BIMA is tuned to detect reads that map to two discontinuous genomic areas, such as when a read crosses a breakpoint or a biotin‐junction (common in NGS mate pair library preparation).
Figure 1.
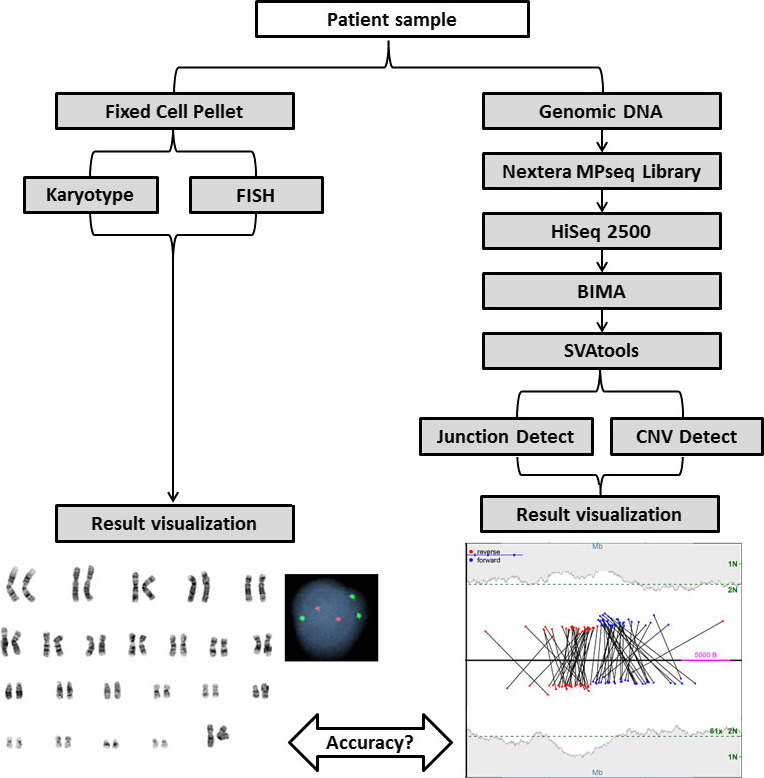
Schematic of MPseq AML workflow. Blood or bone marrow samples are processed into fixed cells or extracted for genomic DNA. Fixed cell pellets are processed for karyotype and/or FISH analysis. Genomic DNA is extracted, prepared using the Illumina Nextera Mate Pair library preparation kit and sequenced on a HiSeq 2500. Reads are aligned to the reference genome with BIMA and variants are detected using SVAtools. Two algorithms are utilized for variant detection; junction detection and CNVDetect which also incorporates aneuploidy detection and resulting data are visualized
Structural variation (SV) was detected by SVAtools, which utilizes the BIMA output to detect and report the breakpoints of structural variants. SVAtools combines three algorithmic approaches: read‐pair, split‐read, and read depth/count. SVAtools detects junctions of chromosomal rearrangements by clustering the discordant and split‐read fragments. Masking and filtering reduce the false‐positive clusters to generate a reliable and meaningful list of junctions. Each cluster with at least three fragments, and passing the mask and filter criteria, is called by SVAtools and is considered a putative junction. Copy number variations were detected by SVAtools using the read count of concordant fragments within non‐overlapping bins using CNVDetect and Aneuploidy Detection algorithms. CNVDetect uses the list of detected junctions discovered in SVAtools to supplement the edge detection in order to improve resolution and sensitivity. The results from CNVDetect provide calculated normalized read depth (NRD) scores for each region of the genome segmented during the edge detection step. The NRD score estimates the copy number level as compared to the expected normal 2 N copy number level. The Aneuploidy Detection algorithm performs the same calculations used in CNVDetect, but on a chromosome arm level. Instead of segmenting the genome into regions of similar copy number prior to evaluation, each chromosome arm is evaluated as a whole in order to calculate a normalized read depth to compare to the expected 2 N copy number level.
Coverage estimations provide thresholds to confidently call variants. Base coverage (often referred to as depth of coverage, read depth, or coverage) is dependent on the count and length of reads sequenced, and therefore, on total sequenced nucleotides. SVAtools uses a count of the number of fragments spanning a given position, “bridged coverage” to establish confidence for each SV detected. Bridged coverage will depend on the number of fragments and the length the fragments (read lengths plus insert length). Given the bridged coverage of these samples, the reported breakpoints are estimated to be within 200 bps when a split read is not present.7
2.6. Structural variation visualization
Junctions and CNVs were graphically illustrated using genome, junction and region plots as described in Johnson, et al.7 We restricted our analysis to classic‐AML rearrangements involving the genes ABL1, BCR, CBFB, CREBBP, DEK, KAT6A, MECOM, MLF1, KMT2A (MLL), MYH11, NPM1, NUP214, NUP98, PML, RARA, RPN1, RUNX1, RUNX1T1, and copy number changes on chromosomes 5, 7, 8, 13, 17, and 20 (Figure 2). This restriction was necessary for the validation of the MPseq assay by comparing it to the gold standard FISH assay and FISH probes were available for the above‐listed targets. In addition, interstitial CNV regions with no supporting junction, and losses <200 kb and gains <500 kb were not reported. Location of breakpoints identified in each gene from MPseq is indicated along with the specific transcript determined by the consensus transcript from the St. Jude's PeCan Data Portal (Table S3).
Figure 2.
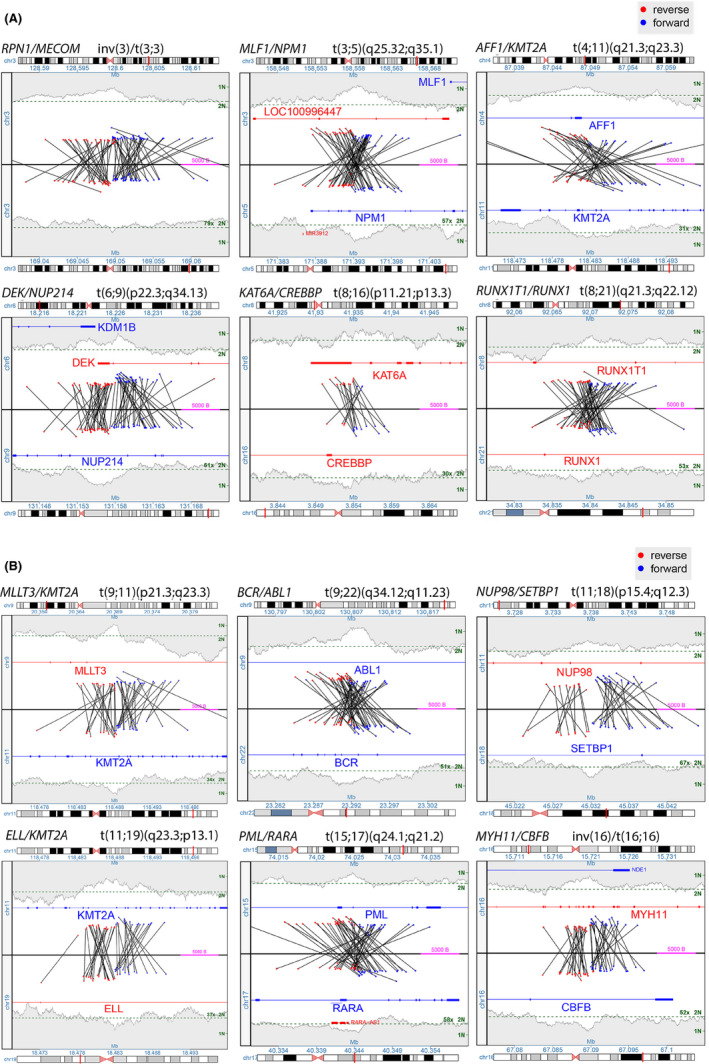
(panel A and B) Representative junction plots of the classic‐AML rearrangements to be detected by this assay. Each junction plot shows the lowest number chromosome on the top and the higher number on the bottom. Each black line represents a junction between two rearranged fragments with blue in the forward orientation and red in the reverse. Copy state is shown by the gray shaded area on the far top and bottom of the plot. All this data taken together allows for reconstruction of the abnormal region
3. RESULTS
To establish the MPseq assay's limits of detection, we evaluated the minimum requirements for (a) DNA input, (b) number of sequenced fragments, and (c) tumor percentage (Supplemental Data, Figures S1‐S5 and summarized in Table S2). In summary, eleven samples tested with decreasing amounts of input DNA indicated that the minimum amount yielding accurate results was 0.5 µg (Figure S3). In silico dilution studies of six samples with various rearrangements/CNVs determined that a minimum of 20 million sequenced fragments were required to ensure detection of all targeted abnormalities (Figure S4). Tumor content requirements were assessed by performing various dilutions of six samples with known rearrangements/CNVs, and established a minimum of 10% tumor for the detection of rearrangements and 25% tumor for the detection of CNVs (Figure S5).
3.1. Detection of primary cytogenetic abnormality using MPseq
We assessed the accuracy of MPseq results compared to results provided by clinical FISH or chromosome studies by evaluating DNA from 68 known abnormal samples extracted from either fresh bone marrow (n = 27), fresh peripheral blood (n = 25) or fixed cell pellets obtained from a conventional chromosome study (n = 16), as well as from 20 karyotypically‐normal fresh bone marrow (n = 10) and peripheral blood (n = 10) samples. For all samples, some known variants representing secondary abnormalities as evidenced by FISH studies were present, but below the resolution of the testing performed. The cutoffs of reporting MPseq results were established based on limit of detection data (Supplemental Data, Figures S1‐S5 and summarized in Table S2).
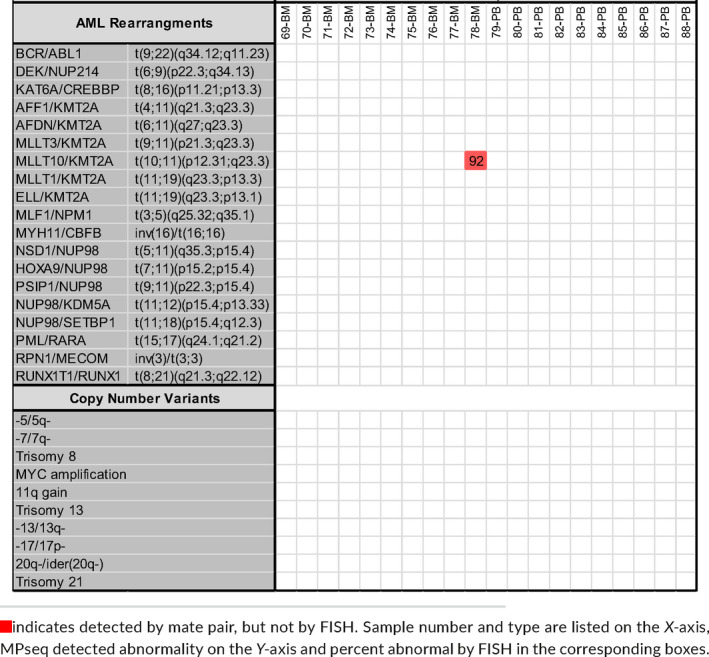
All 68 abnormal samples demonstrated concordance with known FISH studies when the abnormal FISH percentage was greater than 10% for rearrangements and greater than 25% for CNVs (Table S2). Three samples (2‐PB, 8‐BM and 40‐BM) were positive for trisomy 8 (each with FISH percentage less than 9%) but were not detected by MPseq (Table 1). Twenty apparently normal samples were also evaluated by MPseq and 19 of 20 were found to be concordant (95%) with no AML‐panel abnormalities identified (Table 2).
Table 1.
Accuracy results from 68 known abnormal samples extracted from either fresh bone marrow (n = 27), fresh peripheral blood (n = 25), or fixed cell pellets (n = 16)
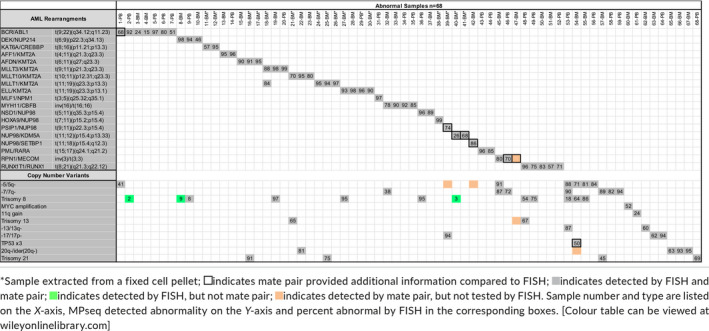
Table 2.
Accuracy results from 20 karyotypically normal fresh bone marrow (n = 10) and peripheral blood (n = 10) samples [Colour figure can be viewed at wileyonlinelibrary.com]
3.2. MPseq clarified gene fusions when fish was abnormal
MPseq was necessary to more clearly define the specific gene fusion in eight (12%) of the 68 abnormal cases when FISH either revealed a gene break‐apart pattern or three abnormal signals (Table 3). In one of these cases, FISH identified three BCR signals when using the BCR/ABL1 FISH probe (which may indicate BCR rearrangement or copy number gain of the BCR locus), while MPseq confirmed a BCR/FGFR1 fusion (1‐PB). Similarly, in another case, we identified three MECOM signals using the RPN1/MECOM FISH probe in the absence of an RPN1/MECOM fusion, while MPseq confirmed a MECOM/CDK6 gene fusion (46‐PB). Further, a TP53/IGL fusion was found by MPseq in a case with three TP53 FISH signals (54‐BM).
Table 3.
Additional information obtained from MPseq testing on nine samples that were not apparent through conventional karyotyping or FISH studies
| Additional information from MPseq | Abnormal FISH pattern | Karyotype | |
|---|---|---|---|
| 1‐PB | identified the BCR partner as FGFR1 | (BCR)x3[341/500] | BM ‐ 46,XX,t(8;22)(p11.2;q11.2)[3] |
| 39‐BM[Link] | identified the NUP98 partner as PSIP1 | (NUP98x2)(5'NUP98 sep 3'NUP98x1)[148/200] | 45,XX,add(1)(p22),add(2)(p13),der(4)t(1;4)(p32;q21),‐5,inv(6)(p11.2q21),t(9;11)(p22;p15),‐12,add(12)(p13),add(15)(p15),add(16)(q22),add(17)(p11.2),add(20)(p13),+mar[cp20] |
| 40‐BM[Link] | identified the NUP98 partner as KDM5A | (NUP98x2)(3'NUP98 con 5'NUP98x1)[51/100] | 46,XY,del(13)(q12q22)[2]/48,idem,+6,+8[1]/46,XY[17] |
| 41‐BM[Link] | identified the NUP98 partner as KDM5A | (NUP98x2)(3'NUP98 sep 5'NUP98x1)[135/200] | 46,XX,+6,dic(14;19)(p13;p13.3)[14]/48,X,t(X;7)(q24;q11.2),+6,+7[5]/48,sl2,del(13)(q12q22)[1] |
| 42‐BM | identified the NUP98 partner as SETBP1 | (NUP98x2)(3'NUP98 con 5'NUP98x1)[171/200] | 46,XX,del(5)(q31q33),add(11)(p15),add(18)(q21)[16]/46,sl,add(6)(q21)[2]/46,XX[2] |
| 46‐PB | identified the MECOM partner as CDK6 | (RPN1x2,M ECOMx3)[348/500] | BM ‐ 46,XX,del(7)(q22q34)[6] |
| 47‐BM |
identified a complex MLF1/MECOM/RPN1 rearrangement |
no FISH | 46,XY,t(3;5)(q21;q31)[20] |
| 54‐BM | identified a TP53/IGL rearrangement | (TP53x3,D17Z1x2)[100/200] | 54,XX,+1,del(4)(q21q27),+5,del(5)(q13q33)x2,+8,t(10;22)(p15;q11.2),+der(10)t(10;22),+11,+1 5,add(17)(p13),+18,+21[15]/46,XX[5] |
| 78‐BM | identified a cryptic MLLT10/KMT2A rearrangement in an apparently normal sample | (MLLT10x3,MLLx2)(MLLT10 con MLLx1)[462/500] | 46,XX[20] |
Sample extracted from a fixed cell pellet.
In four cases, MPseq identified the NUP98 fusion partner (either PSIP1 or SETBP1 in one case each or KDM5A in two cases) when FISH identified a NUP98 break‐apart pattern (no knowledge of the fusion partner). Thus, MPseq was necessary to molecularly define a subset of abnormalities that would not have been elucidated using our current AML FISH panel.
3.3. MPseq detected cryptic rearrangements missed by conventional cytogenetic assays
In two cases, MPseq identified cryptic rearrangements that were not apparent using conventional cytogenetic tests. Sample (78‐BM) was initially classified as a normal sample for this verification (Table 2) because we reported a normal karyotype and normal AML FISH panel result. However, the mate pair data specifically demonstrated the insertion of exons 9‐24 of MLLT10 into intron 9 of KMT2A resulting from a cryptic, insertional rearrangement which was later confirmed using a MLLT10/KMT2A D‐FISH probe and not visible by karyotype or by the break‐apart KMT2A FISH probe (Figure 3 and Table 3). Therefore, this discordance was due to the limitation of the clinical FISH testing approach, as our current clinical AML FISH panel includes the KMT2A break‐apart FISH probe, and reflex D‐FISH KMT2A probes are only utilized when there is an abnormal KMT2A break‐apart result. In another case (47‐BM) (Table 3), an apparent t(3;5)(q21;q31) translocation was identified with breakpoints concerning for a NPM1/MLF1 fusion. MPseq identified a classic MECOM/RPN1 fusion as part of a more complex 3;5 translocation also involving the SLC22A5 gene on chromosome 5, and both the RPN1 locus and RSRC1 locus on chromosome 3.
Figure 3.
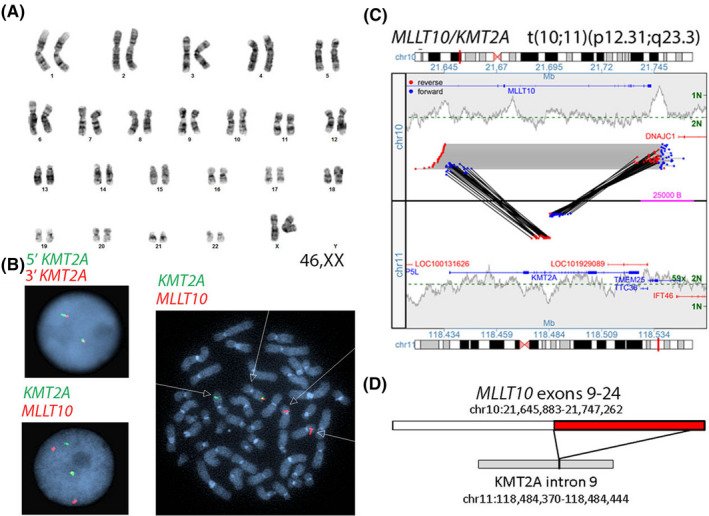
Sample 78‐BM classified as a normal sample for this verification because we reported a normal karyotype shown in A and normal KMT2A break‐apart FISH B, top figure. MPseq junction plot confirming an insertion of exons 9‐24 of MLLT10 (NM_004641) into intron 9 of KMT2A (NM_005933) in C and confirmed using an MLLT10/KMT2A D‐FISH probe B, bottom figure
4. DISCUSSION
Here, we describe an NGS‐based technology, MPseq, to detect diagnostic and prognostic chromosomal rearrangements and copy number changes in patients with AML. Following an extensive familiarization and pipeline development stage involving >200 samples with detectable cytogenetic rearrangements (data not shown), we tested 88 samples; 68 samples with known recurrent AML rearrangements identified by FISH and/or conventional chromosomes and 20 karyotypically normal samples from patients with a known diagnosis of AML to validate MPseq for AML. When the established minimum limit of detection metrics were achieved (Table S2), the MPseq results from all 88 samples were 98.9% concordant with the existing clinical FISH assay. Importantly, we demonstrate the added clinical value of MPseq, particularly in detecting chromosomal rearrangements that may be cryptic by FISH or when a fusion‐specific FISH probe is not available to further characterize an abnormal FISH result. Although these cases were considered concordant by our definition because an abnormality was identified by either FISH or chromosomes, MPseq was necessary to molecularly define the precise gene fusion. This was particularly evident for case 47‐BM, which was found to have a t(3;5)(q21;q31) translocation identified by chromosomes concerning for a NPM1/MLF1 fusion translocation. Because our clinical NPM1/MLF1 fusion D‐FISH probe5 had not yet been developed, this case was interpreted as having a known, recurrent chromosome abnormality with intermediate prognosis found in various myeloid malignancies particularly MDS/AML.10 An NPM1/MLF1 fusion was not identified by MPseq; instead, MPseq revealed a MECOM/RPN1 fusion associated with aggressive MDS or AML.10, 11, 12 While this case was considered concordant in this validation study, we identified a “cryptic” MECOM/RPN1 fusion later confirmed by FISH.
MPseq provided further value in cases where we identified additional FISH signals or a FISH break‐apart result without the availability of a reflex FISH probe to further define a suspected rearrangement. One example (case 1‐PB) is the identification of an FGFR1/BCR fusion in a case with three BCR FISH signals. The identification of an FGFR1 rearrangement provides further classification of this patient's AML in the WHO category of "Myeloid/lymphoid neoplasms with FGFR1 rearrangements"3 and could provide targeted therapeutic treatment options including FGFR inhibitors such as ponatinib.13, 14, 15, 16, 17 In addition, MPseq identified a MECOM/CDK6 fusion in AML case 46‐PB with three MECOM FISH signals and a deletion 7q. MECOM/CDK6 fusions have been described as recurrent “cryptic” abnormalities associated with increased MECOM/EVI expression and unfavorable prognosis.18, 19 Finally, while MPseq identified a TP53/IGL fusion in patient 54‐BM with a RFR of rule out AML and with a FISH pattern indicating three TP53 signals, the significance of this fusion is unknown. This patient (67 years old) is known to have AML with approximately 30%‐40% blasts and a moderately hypercellular bone marrow (70%). The lymphocytes are not increased, they have unremarkable morphology and there is no evidence of lymphoma. To our knowledge, immunoglobulin translocations involving TP53 have not been reported20 in AML.
MPseq characterized the fusion partner of each of the four cases with a NUP98 break‐apart FISH result. Nucleoporin 98 (NUP98) located at 11p15 is a structural component of the nuclear pore complex and has been identified as a gene partner involved in numerous gene fusions.21 NUP98 rearrangements are most common in myeloid neoplasms, especially therapy‐related AML, but can also be found in T‐ALL.21 AML patients with NUP98 translocations are typically younger and have unfavorable prognosis compared to other AML subtypes.22 Although the most common NUP98 partner gene is HOXA9, in our validation cohort, we identified rarer fusions including NUP98/SETBP1 (case 42‐BM) in a young adult with AML, a NUP98/PSIP1 (case 39‐BM) in an adult with therapy‐related AML23 and NUP98/KDM5A (cases 40‐BM and 41‐BM) in two pediatric patients with acute megakaryoblastic leukemia (AMKL) representing a rare subtype of AML.24, 25, 26 Since fusion of NUP98 with KDM5A is a recurrent rearrangement in pediatric non‐Down syndrome AMKL,24, 25, 26 the MPseq result would have provided a specific, molecularly defined diagnosis with risk group stratification.
Importantly, a limitation of the MPseq assay is the inability to reliably detect clonal aberrations present at a very low level. MPseq cannot reliably detect structural rearrangements below 10%, while D‐FISH strategies can detect specific rearrangements as low as 0.6%.27 MPseq also cannot reliably detect copy number changes below 25%, while FISH can detect copy number changes as low as 1.5%‐2.0% for homozygous deletions and trisomies and 4.5%‐9.5% for heterozygous deletions and monosomies (data from our internal laboratory validation studies). In the accuracy cohort, MPseq was unable to reliably identify an apparent secondary trisomy 8 in three samples with abnormal FISH results at 2%, 3%, and 9%. Future improvements in the limit of detection of MPseq could be achieved by increasing the depth of sequencing and by improving the CNV detection algorithm. These limitations, however, are common to recently developed NGS‐based technologies. A recent study by McKerrell et al28 demonstrated an NGS‐based tool for the diagnosis of myeloid malignancies; however, it was unable to detect some CNVs at 35% and was limited to detecting only four types of rearrangements. Another tool, Archer technology,29, 30 is also utilized for the detection of fusions common in AML but requires RNA for the detection of fusions, which is not an ideal specimen type compared to DNA since RNA is less stable and therefore requires additional measures taken by the laboratory to ensure RNA stability. Finally, the MPseq assay is not designed to detect point mutations; therefore, those subtypes of AML with recurrent genetic abnormalities based on genetic mutations, such as CEBPA, FLT3 or NPM1, would not be detected by this MPseq assay.
In conclusion, we evaluated the performance of an NGS‐based whole‐genome MPseq technology with a targeted analysis approach to detect recurrent diagnostic and prognostic chromosomal rearrangements and copy number changes in patients with AML. We demonstrate the clinical utility of MPseq as a potential replacement assay for conventional FISH on diagnostic AML samples and highlight the resulting increased diagnostic yield and clarity in comparison to other testing methodologies (Table S4). MPseq provided important clinical value in cases in which there was a “cryptic” rearrangement not detected by FISH or chromosomes, or for those cases with additional uncharacterized FISH signals, or when a FISH break‐apart result was identified without the availability of a reflex FISH probe to further define a suspected rearrangement. Due to the limitations in detecting very low‐level abnormalities, MPseq would not be recommended for follow‐up post therapy or minimal residual disease testing; utilization of FISH, RT‐PCR or a custom fusion‐qPCR strategy could be considered in those cases. It is important to note that the additional information identified in the MPseq data (Table 3) was filtered to reveal only the abnormalities on our predefined AML‐panel filter. Future studies are underway to identify novel abnormalities throughout the genome associated with myeloid malignancies. As novel rearrangements and copy number alterations important in AML are uncovered in the future, the clinical MPseq panel can be easily expanded to include additional targets. The studies presented here demonstrate the value of MPseq as a novel NGS‐based technology that has the potential to revolutionize the diagnosis of hematologic malignancies and provide an opportunity to advance precision medicine.
CONFLICT OF INTEREST
Algorithms described in this manuscript are licensed to WholeGenome LLC owned by George Vasmatzis.
AUTHOR CONTRIBUTIONS
UA, SAS, BAP, KEP, ECT, RBJ, HMK, NLH, and LBB designed validation study; UA, SAS, BAP, and LBB wrote paper with help from SHJ, JBS, JFP, KBG, DLV, ECT, RBJ, RPK, PTG, HMK, and NLH; UA, SAS, BAP, ECT, RBJ, HMK, NLH, and LBB analyzed and interpreted data; RMZ, GV, SHJ, JBS, HMK, and NLH provided critical analytical tools; SAS and BAP collected data and PTG and RPK provided samples highlighted in manuscript.
Supporting information
ACKNOWLEDGMENTS
We thank Saranya Sankaranarayanan, MS for bioinformatics help. This study was supported by development funds provided by the Department of Laboratory Medicine and Pathology, Mayo Clinic and partially funded by the Center for Individualized Medicine, Mayo Clinic.
Aypar U, Smoley SA, Pitel BA, et al. Mate pair sequencing improves detection of genomic abnormalities in acute myeloid leukemia. Eur J Haematol. 2019;102:87–96. 10.1111/ejh.13179
REFERENCES
- 1. Grimwade D, Ivey A, Huntly BJ. Molecular landscape of acute myeloid leukemia in younger adults and its clinical relevance. Blood. 2016;127(1):29‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. De Kouchkovsky I, Abdul‐Hay M. Acute myeloid leukemia: a comprehensive review and 2016 update. Blood Cancer J. 2016;6(7):e441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391‐2405. [DOI] [PubMed] [Google Scholar]
- 4. Leonard JP, Martin P, Roboz GJ. Practical implications of the 2016 revision of the World Health Organization classification of lymphoid and myeloid neoplasms and acute leukemia. J Clin Oncol. 2017;35(23):2708‐2715. [DOI] [PubMed] [Google Scholar]
- 5. Aypar U, Knudson RA, Pearce KE, Wiktor AE, Ketterling RP. Development of an NPM1/MLF1 D‐FISH probe set for the detection of t(3;5)(q25;q35) identified in patients with acute myeloid leukemia. J Mol Diagn. 2014;16(5):527‐532. [DOI] [PubMed] [Google Scholar]
- 6. Keefe JG, Sukov WR, Knudson RA, et al. Development of five dual‐color, double‐fusion fluorescence in situ hybridization assays for the detection of common MLL translocation partners. J Mol Diagn. 2010;12(4):441‐452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Johnson SH, Smadbeck JB, Smoley SA, et al. SVAtools for junction detection of genome‐wide chromosomal rearrangements by mate‐pair sequencing (MPseq). Cancer Genet. 2018;221:1‐18. [DOI] [PubMed] [Google Scholar]
- 8. Smadbeck JB, Johnson SH, Smoley SA, et al. Copy number variant analysis using genome‐wide mate‐pair sequencing. Genes Chromosomes Cancer. 2018;57:459‐470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Drucker TM, Johnson SH, Murphy SJ, Cradic KW, Therneau TM, Vasmatzis G. BIMA V3: an aligner customized for mate pair library sequencing. Bioinformatics. 2014;30(11):1627‐1629. [DOI] [PubMed] [Google Scholar]
- 10. Grimwade D, Hills RK, Moorman AV, et al. Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood. 2010;116(3):354‐365. [DOI] [PubMed] [Google Scholar]
- 11. Harris NL, Jaffe ES, Diebold J, et al. World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues: report of the Clinical Advisory Committee meeting‐Airlie House, Virginia, November 1997. J Clin Oncol. 1999;17(12):3835‐3849. [DOI] [PubMed] [Google Scholar]
- 12. Dohner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424‐447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Demiroglu A, Steer EJ, Heath C, et al. The t(8;22) in chronic myeloid leukemia fuses BCR to FGFR1: transforming activity and specific inhibition of FGFR1 fusion proteins. Blood. 2001;98(13):3778‐3783. [DOI] [PubMed] [Google Scholar]
- 14. Fioretos T, Panagopoulos I, Lassen C, et al. Fusion of the BCR and the fibroblast growth factor receptor‐1 (FGFR1) genes as a result of t(8;22)(p11;q11) in a myeloproliferative disorder: the first fusion gene involving BCR but not ABL. Genes Chromosomes Cancer. 2001;32(4):302‐310. [DOI] [PubMed] [Google Scholar]
- 15. Patterer V, Schnittger S, Kern W, Haferlach T, Haferlach C. Hematologic malignancies with PCM1‐JAK2 gene fusion share characteristics with myeloid and lymphoid neoplasms with eosinophilia and abnormalities of PDGFRA, PDGFRB, and FGFR1. Ann Hematol. 2013;92(6):759‐769. [DOI] [PubMed] [Google Scholar]
- 16. Chase A, Bryant C, Score J, Cross NC. Ponatinib as targeted therapy for FGFR1 fusions associated with the 8p11 myeloproliferative syndrome. Haematologica. 2013;98(1):103‐106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Khodadoust MS, Luo B, Medeiros BC, et al. Clinical activity of ponatinib in a patient with FGFR1‐rearranged mixed‐phenotype acute leukemia. Leukemia. 2016;30(4):947‐950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Haferlach C, Bacher U, Grossmann V, et al. Three novel cytogenetically cryptic EVI1 rearrangements associated with increased EVI1 expression and poor prognosis identified in 27 acute myeloid leukemia cases. Genes Chromosomes Cancer. 2012;51(12):1079‐1085. [DOI] [PubMed] [Google Scholar]
- 19. Capela de Matos RR, Othman M, Ferreira GM, et al. Molecular approaches identify a cryptic MECOM rearrangement in a child with a rapidly progressive myeloid neoplasm. Cancer Genet. 2018;221:25‐30. [DOI] [PubMed] [Google Scholar]
- 20. Xu‐Monette ZY, Medeiros LJ, Li Y, et al. Dysfunction of the TP53 tumor suppressor gene in lymphoid malignancies. Blood. 2012;119(16):3668‐3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gough SM, Slape CI, Aplan PD. NUP98 gene fusions and hematopoietic malignancies: common themes and new biologic insights. Blood. 2011;118(24):6247‐6257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chou WC, Chen CY, Hou HA, et al. Acute myeloid leukemia bearing t(7;11)(p15;p15) is a distinct cytogenetic entity with poor outcome and a distinct mutation profile: comparative analysis of 493 adult patients. Leukemia. 2009;23(7):1303‐1310. [DOI] [PubMed] [Google Scholar]
- 23. Yamamoto K, Nakamachi Y, Yakushijin K, et al. Expression of the novel NUP98/PSIP1 fusion transcripts in myelodysplastic syndrome with t(9;11)(p22;p15). Eur J Haematol. 2012;88(3):244‐248. [DOI] [PubMed] [Google Scholar]
- 24. de Rooij JD, Branstetter C, Ma J, et al. Pediatric non‐Down syndrome acute megakaryoblastic leukemia is characterized by distinct genomic subsets with varying outcomes. Nat Genet. 2017;49(3):451‐456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. de Rooij JD, Hollink IH, Arentsen‐Peters ST, et al. NUP98/JARID1A is a novel recurrent abnormality in pediatric acute megakaryoblastic leukemia with a distinct HOX gene expression pattern. Leukemia. 2013;27(12):2280‐2288. [DOI] [PubMed] [Google Scholar]
- 26. de Rooij JD, Masetti R, van den Heuvel‐Eibrink MM, et al. Recurrent abnormalities can be used for risk group stratification in pediatric AMKL: a retrospective intergroup study. Blood. 2016;127(26):3424‐3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shearer BM, Sukov WR, Flynn HC, Knudson RA, Ketterling RP. Development of a dual‐color, double fusion FISH assay to detect RPN1/EVI1 gene fusion associated with inv(3), t(3;3), and ins(3;3) in patients with myelodysplasia and acute myeloid leukemia. Am J Hematol. 2010;85(8):569‐574. [DOI] [PubMed] [Google Scholar]
- 28. McKerrell T, Moreno T, Ponstingl H, et al. Development and validation of a comprehensive genomic diagnostic tool for myeloid malignancies. Blood. 2016;128(1):e1–e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zheng Z, Liebers M, Zhelyazkova B, et al. Anchored multiplex PCR for targeted next‐generation sequencing. Nat Med. 2014;20(12):1479‐1484. [DOI] [PubMed] [Google Scholar]
- 30. Badar T, Johnson L, Trifilo K, et al. Detection of novel t(12;17)(p12;p13) in relapsed refractory acute myeloid leukemia by anchored multiplex PCR(AMP)‐based next‐generation sequencing. Appl Immunohistochem Mol Morphol. 2017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials