

SUMMARY
Nuclear restorer of fertility (Rf) genes suppress the effects of mitochondrial genes causing cytoplasmic male sterility (CMS), a condition in which plants fail to produce viable pollen. Rf genes, many of which encode RNA‐binding pentatricopeptide repeat (PPR) proteins, are applied in hybrid breeding to overcome CMS used to block self‐pollination of the seed parent. Here, we characterise the repertoire of restorer‐of‐fertility‐like (RFL) PPR genes in barley (Hordeum vulgare). We found 26 RFL genes in the reference genome (‘Morex’) and an additional 51 putative orthogroups (POGs) in a re‐sequencing data set from 262 barley genotypes and landraces. Whereas the sequences of some POGs are highly conserved across hundreds of barley accessions, the sequences of others are much more variable. High sequence variation strongly correlates with genomic location – the most variable genes are found in a cluster on chromosome 1H. A much higher likelihood of diversifying selection was found for genes within this cluster than for genes present as singlets. This work includes a comprehensive analysis of the patterns of intraspecific variation of RFL genes. The RFL sequences characterised in this study will be useful for the development of new markers for fertility restoration loci.
Keywords: Restorer‐of‐fertility‐like gene, mitochondria, pentatricopeptide repeat protein, cytoplasmic male sterility, hybrid breeding, Hordeum vulgare
Significance statement
Restorer of fertility genes are applied in plant breeding to overcome mitochondrially conditioned male sterility in hybrid crop production. Although these genes are known to vary between related species, intraspecific variation in the gene family has not been comprehensively studied. By systematic analysis of restorer of fertility‐like sequences across hundreds of barley accessions, we show that a huge diversity of these RNA‐binding proteins exists within a single species, with strong diversifying selection on the residues known to determine binding specificity.
INTRODUCTION
After wheat, maize and rice, barley is the fourth most important cereal crop in regard to production area, with a world output of 144 million tonnes in 2014 (FAOSTAT). As an abiotic‐stress‐resilient cereal, the seed yield of barley is more stable against seasonal variation than that of wheat and most other small grains. The possibilities of exploiting hybrid heterosis in barley have been explored for decades with the major goals of further increasing crop yield and stability, particularly in marginal environments. The potential heterosis of F1 hybrid varieties in barley has been estimated at about 10% yield gain compared with inbred parental lines (Longin et al., 2012; Muhleisen et al., 2013).
Three methods have been applied to block self‐pollination of crop plants in hybrid breeding. Manual emasculation of flowers is widely used in hybrid production in maize but, due to flower architecture, this method is not applicable on a commercial scale in barley or wheat. The efficiency of treatment with gametocidal chemicals is strongly influenced by weather conditions and it can also negatively impact yield. In comparison, the use of cytoplasmic male sterility (CMS) can be, once established in a given crop, a labour and cost‐effective way of large scale emasculation of the seed parents of hybrids (Chase, 2007; Chen and Liu, 2014). CMS is induced by mitochondrial genes, the effects of which can be overcome by nuclear restorer of fertility genes (Schnable and Wise, 1998; Hanson and Bentolila, 2004). The majority of known restorer genes belong to the family of pentatricopeptide repeat (PPR) proteins (Dahan and Mireau, 2013; Chen and Liu, 2014; Hu et al., 2014; Gaborieau et al., 2016). PPR proteins are targeted to mitochondria or chloroplasts where they participate in a plethora of RNA‐associated processes (Barkan and Small, 2014). The PPR family has expanded significantly in plants and, on average, ~500 PPR genes are present in diploid plant genomes (O'Toole et al., 2008; Fujii et al., 2011; Cheng et al., 2016; Sykes et al., 2016). Members of the PPR family are identified by the presence of tandem repeats of degenerate 31–36 amino acids, and can be divided into P‐ and PLS‐classes based on the PPR motif structure (Lurin et al., 2004; Cheng et al., 2016). The P subfamily consists of PPR proteins with canonical 35‐amino‐acid‐long PPR motifs, while the PLS subfamily includes proteins with additional S (for short) and L (for long) motif variants arranged into PLS triplets (Lurin et al., 2004; Cheng et al., 2016). P‐class PPRs are involved in RNA stabilisation and processing, including 5’ and 3’ RNA cleavage and intron splicing; they are also involved in the initiation of mRNA translation (Meierhoff et al., 2003; Raynaud et al., 2007; de Longevialle et al., 2008; Pfalz et al., 2009; Prikryl et al., 2011). In comparison, the main function assigned to PLS‐class PPRs is the C‐U editing of organellar transcripts (Okuda et al., 2007; Sosso et al., 2012; Chateigner‐Boutin et al., 2013). PPR proteins recognise their organellar RNA targets in a sequence‐specific manner with base specificity relying on specific amino acids in each PPR motif, with the strongest effect observed for residues at positions 5 and 35 (Fujii et al., 2011; Barkan et al., 2012; Takenaka et al., 2013; Yagi et al., 2013; Miranda et al., 2018).
Rf proteins are generally P‐class PPR proteins (Dahan and Mireau, 2013; Gaborieau et al., 2016) and, although plant genomes encode hundreds of P‐class PPR proteins, on average, only ~10% of them belong to the restorer‐of‐fertility‐like (RFL) clade (Fujii et al., 2011; Melonek et al., 2016; Sykes et al., 2016). Characteristic features that discriminate RFL genes from other P‐class PPR proteins include clustering at a small number of genomic locations, and their relatively high number of PPR motifs with close similarity to the PPR consensus (Fujii et al., 2011; Melonek et al., 2016). The most remarkable attribute of RFL proteins is their evolutionary plasticity, reflected in much higher evolution rates compared with any other group of PPR proteins, with diversifying selection acting particularly on amino acid residues involved in binding to RNA targets (Fujii et al., 2011). The role of RFL proteins in suppressing CMS by blocking expression of CMS‐associated ORFs has been proposed as a possible explanation for their diversity and unusual evolutionary behaviour (Chase, 2007). The co‐evolution of nuclear RFL genes with CMS‐inducing mitochondrial genes has been compared with the co‐evolution of pathogen effectors and resistance (R) genes in plant–pathogen interactions, described as an ‘evolutionary arms race’ between the mitochondrial and nuclear genomes (Touzet and Budar, 2004; Dahan and Mireau, 2013).
The development of a CMS‐based hybrid breeding system requires three types of breeding lines: a ‘female line’ or cytoplasmic male sterile line, which carries a mitochondrial gene that causes CMS, a maintainer line that is required for propagating the sterile line and a restorer line, which carries a nuclear Rf gene that restores male fertility by suppressing the action of the CMS‐causing gene. Two CMS cytoplasms, designated msm1 and msm2, respectively, were found as natural variants of the wild progenitor of barley Hordeum vulgare ssp. spontaneum (C. Koch) Thellung (Ahokas, 1979, 1982). So far, only a single dominant restorer gene Rfm1 (restorer of fertility in msm1) has been identified that restores the fertility of both msm1 and msm2 cytoplasm (Ahokas, 1980a,b, 1982), and the Rfm1‐CMS system has been used to develop the HYVIDO® family of high‐yielding 6‐row winter barley by Syngenta (Rizzolatti et al., 2017). As these hybrids were shown to display several advantages over non‐hybrid varieties, including consistency in the seed yield from year to year by better overcoming severe weather conditions and higher resistance to diseases, the prospective benefits of breeding hybrid varieties in barley based on CMS are promising (Muhleisen et al., 2013, 2014a,b). However, the application of Rfm1‐CMS in hybrid breeding in barley is limited due to its thermosensitivity, as spontaneous fertility restoration in the absence of the Rfm1 gene occurs during periods of higher temperatures around heading and flowering time (Bernhard et al., 2017). Therefore, the identification of new sources of CMS and alternative restorer genes, the most likely origin of which will be RFL genes from wild barley relatives, will be crucial for the development of new hybrid varieties.
In this study, the RFL family in the genus Hordeum was comprehensively characterised, and the conservation and sequence variation of identified RFLs across hundreds of barley accessions, landraces and wild relatives analysed.
RESULTS
Identification of RFL genes in the genus Hordeum
For H. vulgare cv. ‘Morex’ the newest barley reference genome Refseqv1.0 (Mascher et al., 2017) was used. For comparison, previously published draft whole‐genome shotgun (WGS) assemblies of H. vulgare cvs. ‘Morex’, ‘Barke’ and ‘Bowman’ (The International Barley Genome Sequencing Consortium, 2012) were included in the study (Table 1). The genomic sequences were analysed to identify RFL sequences as previously described for rice (Melonek et al., 2016). In total, 245 PLS‐class and 215 P‐class PPR genes were identified in the H. vulgare cv. ‘Morex’ reference genome (Table 1). These genes were found to be distributed across all seven barley chromosomes (Figure 1a). Analysis of PPR gene density across the genome revealed the presence of two PPR‐rich regions on chromosomes 1H and 2H showing higher gene density compared with other regions in the barley genome (Figure 1a).
Table 1.
Identified RFL sequences in barley ‘Morex’ Refseqv1.0 reference genome and WGS data sets of ‘Morex’, ‘Barke’ and ‘Bowman’
| # | Species | Genomes coded names | Data set type | References | Identification of ORFs containing PPR motifs | # of RFLs (10 or more PPR motifs) | |||
|---|---|---|---|---|---|---|---|---|---|
| # ORFs/6 frame translations (× 1000) | # ORFs with PPR repeats | # ORFs with PLS‐class PPR repeats (> 240)a | # ORFs with P‐class PPR repeats (> 100)a | ||||||
| 1. | Hordeum vulgare cv. ‘Morex’ | HvMo | WGA | (Mascher et al., 2017) | 10 908 | 953 | 245 | 215 | 26 |
| 2. | Hordeum vulgare cv. ‘Morex’ | HvM | WGS | (The International Barley Genome Sequencing Consortium, 2012) | 2824 | 683 | 222 | 116 | 12 |
| 3. | Hordeum vulgare cv. ‘Bowman’ | HvBo | WGS | (The International Barley Genome Sequencing Consortium, 2012) | 2988 | 659 | 209 | 115 | 13 |
| 4. | Hordeum vulgare cv. ‘Barke’ | HvBa | WGS | (The International Barley Genome Sequencing Consortium, 2012) | 2613 | 633 | 160 | 94 | 13 |
PPR protein scores as judged from hmmsearch.
PPR, pentatricopeptide repeat; RFL, restorer‐of‐fertility‐like.
Figure 1.
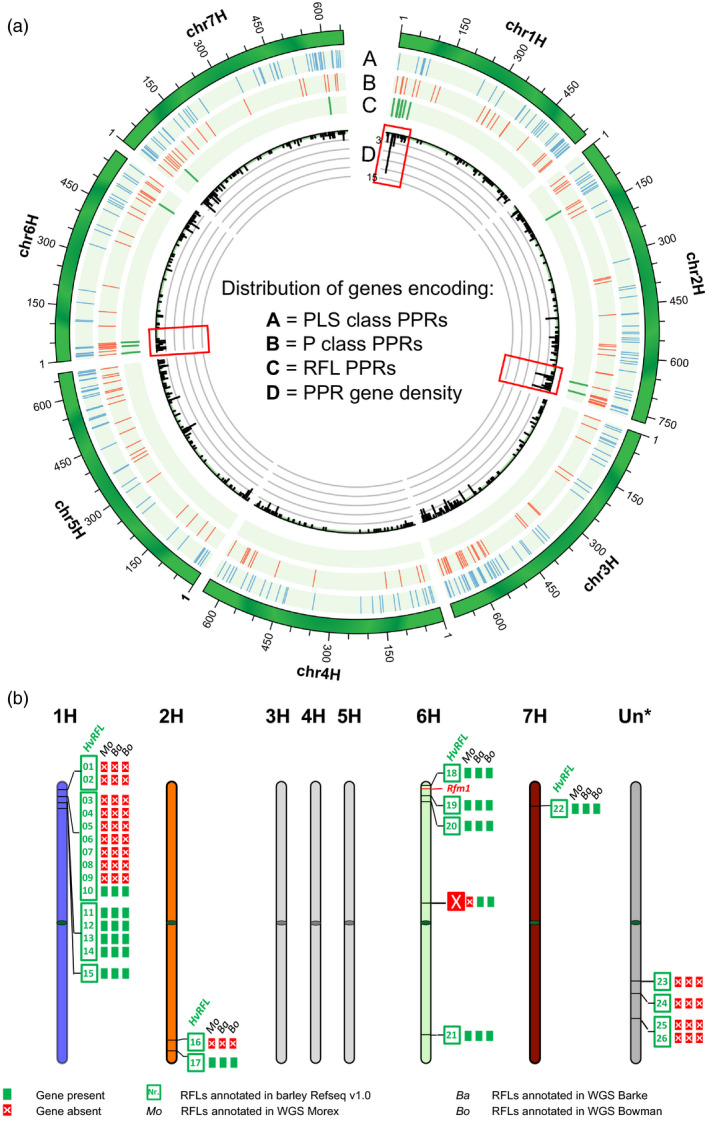
Genome‐wide distribution of the pentatricopeptide repeat (PPR) and restorer‐of‐fertility‐like (RFL) family members in the Hordeum vulgare cv. ‘Morex’ Refseqv1.0 reference genome. (a) Circos diagram illustrating the genome‐wide distribution of the PLS‐ and P‐class PPRs as well as RFL sequences in barley. The density of PPR genes per 2‐Mb windows along the chromosomes is shown. Red frames indicate the genomic regions showing unusually high RFL gene density.(b) Schematic drawing showing the locations of the identified 26 RFLs in the newest version of the H. vulgare cv. ‘Morex’ Refseqv1.0 genome (Mascher et al., 2017) as well as whole‐genome shotgun (WGS) data sets of cvs. ‘Morex’ (Mo), ‘Barke’ (Ba) and ‘Bowman’ (Bo) (The International Barley Genome Sequencing Consortium, 2012). The genomic location of the mapped position of the Rfm1 locus (Ui et al., 2015; Rizzolatti et al., 2017) is shown on the short arm of chromosome 6H.
RFL sequences were identified by two approaches: phylogenetic analysis; and by inferring orthologous groups with OrthoMCL‐DB (Chen et al., 2006; Table S1). In total, 55 RFL sequences with amino acid length ranging from 925 to 93 were identified (Table S1). Of these, 26 represent genes encoding putative full‐length PPR proteins with 10 or more PPR motifs (Tables 1 and S1).
The analysis of the WGS draft assemblies of H. vulgare cvs. ‘Morex’, ‘Barke’ and ‘Bowman’ identified 12 RFL genes in ‘Morex’ and 13 in the two latter accessions (Figure S1; Tables 1 and S2). The majority of the missing RFL sequences correspond to genes in the cluster on chromosome 1H in ‘Morex’ (Table 2). Detailed sequence analyses revealed that seven RFL genes were identical between all three cultivars (sequence identity > 99%), three genes were identical between ‘Bowman’ and ‘Barke’ (sequence identity 100%), and one gene was identical between ‘Bowman’ and ‘Morex’ (sequence identity 100%; Table S2). One gene, HvBo_RFL11 = HvBa_RFL11, was found only in the cvs. ‘Barke’ and ‘Bowman’ (Table S2). In regard to ‘Morex’, for all 12 RFL genes identified in the WGS assembly, a corresponding gene was found in the barley Refseqv1.0 reference genome (Figures 1 and S1; Table S2).
Table 2.
Overview of RFL sequences identified in the ‘Morex’ Refseqv1.0 reference genome and WGS data sets of ‘Morex’, ‘Barke’ and ‘Bowman’
| Data set | Chromosome H1 | 2H | 6H | Unknown | # of extra genes | Total | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sub‐cluster 1 | Sub‐cluster 2 | Sub‐cluster 3 | ||||||||||||||||||||||||||
| HvRFL01 | HvRFL02 | HvRFL03 | HvRFL04 | HvRFL05 | HvRFL06 | HvRFL07 | HvRFL08 | HvRFL09 | HvRFL10 | HvRFL11 | HvRFL12 | HvRFL13 | HvRFL14 | HvRFL15 | HvRFL16 | HvRFL17 | HvRFL18 | HvRFL19 | HvRFL20 | HvRFL21 | HvRFL22 | HvRFL23 | HvRFL24 | HvRFL25 | HvRFL26 | |||
| ‘Morex’ Refseqv1.0 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 26 |
| WGS ‘Morex’ | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 12 |
| WGS ‘Barke’ | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 1 | 13 |
| WGS ‘Bowman’ | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 1 | 13 |
Genome‐wide distribution of RFLs in Hordeum vulgare cv. ‘Morex’
Twenty‐two of the identified ‘Morex’ RFLs were located on four chromosomes (Figure 1b; Table 3), and four sequences were located on unanchored scaffolds (chrUn; Figure 1b; Table 3). The highest number of RFLs was present on chromosome 1H, where 13 RFLs were organised into three sub‐clusters and one gene (HvRFL15) was found as a singlet (Figure 1b; Table 3). The biggest sub‐cluster on chromosome 1H (sub‐cluster 2) spanned a region of ~2 Mbp and contained eight full‐length RFL sequences (Figure 1b; Table 3). Sequence clustering with CD‐HIT suggested that all four RFLs (RFL23–26) located on unanchored scaffolds most likely originate from chromosome 1H (Table S3). Including these genes, the total number of RFL genes in 1H sub‐cluster 2 may be as high as 12 (Table 3). Sub‐cluster 1 and sub‐cluster 3 on chromosome 1H were composed of two and four RFLs, respectively (Figure 1; Table 3). All four RFL genes located on chromosome 6H were separated by long DNA stretches ranging from 10 to 23 Mbp, and thus were classified as singlets (Figure 1b; Table 3). Sequence alignment of the 26 RFL sequences revealed that one of the genes identified on chromosome 6H (HvRFL21) shared 70% sequence identity to HvRFL14 and HvRFL11, both located on chromosome 1H, suggesting a possible misassembly of this gene on chromosome 6H or a recent chromosomal relocation of the gene within the barley genome (Figure 1b). A single RFL gene (HvRFL22) was found on chromosome 7H (Figure 1b). The genomic distances between the RFL genes located in the gene clusters on chromosome 1H and 2H were much shorter compared with the distances between RFLs present as singlets or any other type of PPR genes located anywhere else in the genome as visualised by the density plot (Figure 1a). The mapped Rfm1 interval (Ui et al., 2015) is indicated on the short arm of chromosome 6H (Figure 1b) and does not include any of the RFL sequences.
Table 3.
The repertoire of RFL proteins in Hordeum vulgare cv. ‘Morex’ Refseqv1.0 genome
| # | RFL ID | Gene name | Genomic location | Cluster/sensing | Physical position [start–end] (orientation) | Gene length (bp) | PPR motif structure | mTP yes [length amino acids) | Possible pseudogene |
|---|---|---|---|---|---|---|---|---|---|
| 1. | HvRFL01 | HORVU1Hr1G004750 | chr1H | Sub‐cluster 1 | [9851312–9848892] (REVERSE) | 2421 | 56‐P‐46‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐38 | Yes [111] | |
| 2. | HvRFL02 | Not annotated | chr1H | [9907009–9904493] (REVERSE) | 2517 | 56‐P‐46‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐315 | Yes [25] | Yes | |
| 3. | HvRFL03 | HORVU1Hr1G010890 | chr1H | Sub‐cluster 2 | [25542037–25539329] (REVERSE) | 2709 | 199‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐70 | Yes [18] | |
| 4. | HvRFL04 | HORVU1Hr1G010970 | chr1H | [25974315–25976849] (FORWARD) | 2535 | 58‐P‐48‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐70 | Yes [21] | ||
| 5. | HvRFL05 | HORVU1Hr1G011020 | chr1H | [26214086–26211741] (REVERSE) | 2346 | 149‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐70 | Yes [53] | ||
| 6. | HvRFL06 | Not annotated | chr1H | [26421244–26418548] (REVERSE) | 2697 | 195‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐35 | Yes [77] | ||
| 7. | HvRFL07 | Not annotated | chr1H | [26485385–26488081] (FORWARD) | 2697 | 195‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐35 | Yes [77] | ||
| 8. | HvRFL08 | HORVU1Hr1G011160 | chr1H | [26659630–26662332] (FORWARD) | 2703 | 197‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐70 | Yes [18] | ||
| 9. | HvRFL09 | HORVU1Hr1G011300 | chr1H | [27069847–27072621] (FORWARD) | 2775 | 221‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐70 | Yes [18] | ||
| 10. | HvRFL10 | HORVU1Hr1G011400 | chr1H | [27458351–27456489] (REVERSE) | 1863 | 68‐P‐48‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐35‐P‐8 | Yes [107] | ||
| 11. | HvRFL11 | HORVU1Hr1G013810 | chr1H | Sub‐cluster 3 | [37317192–37319720] (FORWARD) | 2529 | 140‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐70 | Yes [32] | |
| 12. | HvRFL12 | HORVU1Hr1G014030 | chr1H | [38711706–38709145] (REVERSE) | 2562 | 150‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐70 | Yes [19] | ||
| 13. | HvRFL13 | HORVU1Hr1G014040 | chr1H | [38766653–38769199] (FORWARD) | 2547 | 145‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐35‐P | Yes [46] | ||
| 14. | HvRFL14 | HORVU1Hr1G014060 | chr1H | [38811031–38813742] (FORWARD) | 2712 | 198‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐70 | Yes [33] | ||
| 15. | HvRFL15 | Not annotated | chr1H | Singlet | [48954576–48956174] (FORWARD) | 1599 | 5‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐37 | No | Yes |
| 16. | HvRFL16 | HORVU2Hr1G105460 | chr2H | Cluster 1 | [706791942–706790239] (REVERSE) | 1704 | 44‐P‐P‐P‐P‐P‐P‐P‐53‐P‐P‐P‐P‐P‐50 | No | Yes |
| 17. | HvRFL17 | HORVU2Hr1G116650 | chr2H | [738768328–738766658] (REVERSE) | 1671 | P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐86 | No | Yes | |
| 18. | HvRFL18 | HORVU6Hr1G002890 | chr6H | Singlet | [7033847–7031583] (REVERSE) | 2265 | 124‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐31‐P‐P‐P‐P‐36 | Yes [25] | |
| 19. | HvRFL19 | HORVU6Hr1G014310 | chr6H | Singlet | [30413113–30415251] (FORWARD) | 2139 | 103‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐11 | Yes [46] | Yes |
| 20. | HvRFL20 | HORVU6Hr1G014310 | chr6H | Singlet | [43353403–43354881] (FORWARD) | 1479 | 101‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐6 | No | Yes |
| 21. | HvRFL21 | HORVU6Hr1G076780 | chr6H | Singlet | [527185061–527182566] (REVERSE) | 2496 | 128‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐35 | Yes [10] | |
| 22. | HvRFL22 | HORVU7Hr1G029840 | chr7H | Singlet | [56755710–56753593] (REVERSE) | 2118 | 36‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐38 | No | Yes |
| 23. | HvRFL23 | Not annotated | Unknowna | Unknown | [177465399–177467981] (FORWARD) | 2583 | 157‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐70 | Yes [35] | |
| 24. | HvRFL24 | HORVU0Hr1G031920 | Unknowna | Unknown | [190422976–190420268] (REVERSE) | 2709 | 199‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐70 | Yes [18] | |
| 25. | HvRFL25 | Not annotated | Unknowna | Unknown | [210337547–210339712] (FORWARD) | 2166 | 126‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P | No | Yes |
| 26. | HvRFL26 | HORVU0Hr1G035310 | Unknowna | Unknown | [210357231–210359657] (FORWARD) | 2427 | 105‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐P‐70 | Yes [35] |
Genes most likely located in RFL sub‐cluster 2 on chromosome 1H (based on CD‐HIT clustering).
mTP, mitochondrial targeting sequence; PPR, pentatricopeptide repeat; RFL, restorer‐of‐fertility‐like.
Characterisation of identified RFL sequences: functional genes versus pseudogenes
The 26 identified HvRFL sequences contain between 11 and 19 PPR motifs (Table 3). For 16 of them a mitochondrial localisation (Small et al., 2004) was predicted using Predotar (Table 3), in agreement with the expected mitochondrial location of RFL proteins. Nine of the RFL sequences may represent pseudogenes as the encoded proteins either lack a predicted mitochondrial targeting sequence (mTP) or are truncated in the middle of a PPR motif (Table 3). In addition to these long RFL sequences encoding proteins composed of 10 or more PPR motifs, the genomic regions carrying RFL genes contain an additional 29 short RFL sequences (Table S1). Such partial RFL sequences may represent remnants of RFL genes disrupted by unequal crossing‐over reported to occur frequently during recombination events within RFL clusters (Melonek et al., 2016).
RFL gene variation across 262 barley accessions
Target enrichment re‐sequencing data from 262 barley accessions (Mascher et al., 2013; Russell et al., 2016) were analysed to estimate the sequence variation of RFL genes in the genus Hordeum. One‐hundred and sixty‐eight data sets were from barley cultivars and landraces, and 94 were from wild Hordeum spontaneum accessions (Table S4). De novo assembly of captured reads was performed with MaSuRCA (Zimin et al., 2013), and the obtained sequence scaffolds were screened for the presence of PPR ORFs of P‐ and PLS‐type, the number of which varied from 174 to 357 and 186 to 383, respectively (Table S5). About 20–40 RFL ORFs were found in the analysed accessions (Table S5) compared with the 55 RFL ORFs found in the ‘Morex’ Refseqv1.0 genome (Figure 2a), suggesting that only about half of the RFL sequences were captured and assembled. The captured RFL ORFs are, on average, shorter, as illustrated in Figure 2(b). Whereas the length distributions for PPR ORFs are relatively consistent between the different data sets, the exome capture (EC) and genome shotgun assemblies contain a dearth of long RFL sequences (over 800 amino acids) and a large excess of short RFL sequences (less than 200 amino acids).
Figure 2.
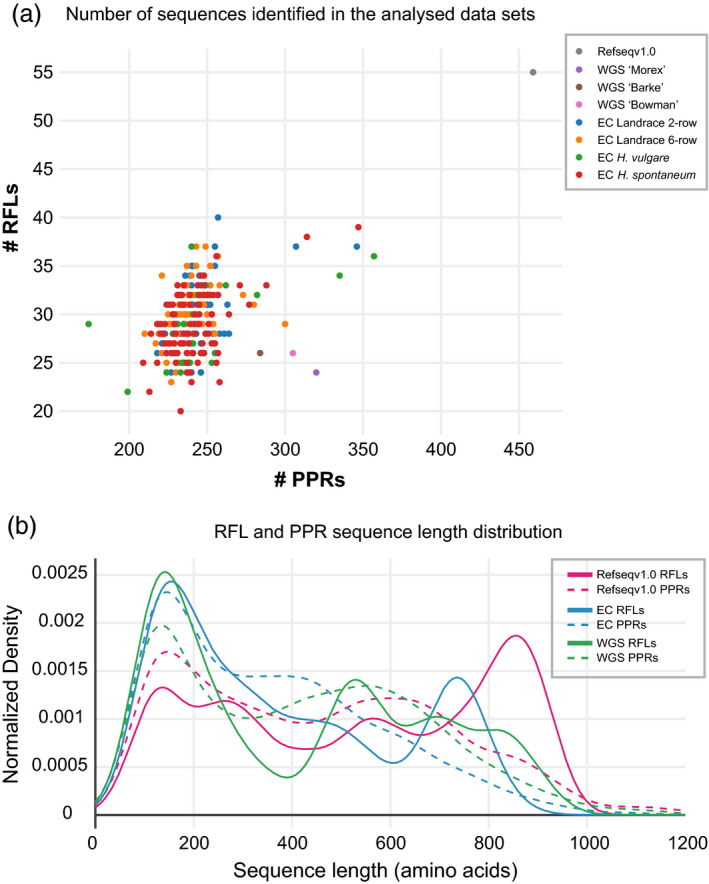
Characterisation of restorer‐of‐fertility‐like (RFL) sequences recovered from various barley sequence data sets.(a) Scatter plot illustrating the number of identified RFLs in each barley accession compared with the total number of pentatricopeptide repeat (PPR) sequences. The plot shows results from the analysis of Refseqv1.0 genome as well as from whole‐genome shotgun (WGS) assemblies and exome capture (EC) data sets.(b) Length distribution of PPR and RFL‐type ORFs in the Refseqv1.0 genome, EC data sets and WGS data sets. Gaussian kernel density estimates of the ORF length distributions were generated with the sklearn.neighbors.KernelDensity estimator version 0.18.1 (Pedregosa et al., 2011), and the data were visualised in an offline version of plotly (https://plot.ly/). PPR ORFs longer than 279 nucleotides (= 93 amino acids) were included in the analysis.
This observed sequence fragmentation could be due to the repetitive nature of highly similar RFL sequences, which creates ambiguities in alignment and computational challenges during assembly (Table S6 lists examples of partial exon assemblies). These issues make it impossible to determine whether a sequence is truly absent in a given accession or whether it has simply escaped detection by the current approach. With that caveat in mind, a hierarchical cluster analysis (HCA) with CD‐HIT (Huang et al., 2010) was performed to assess gene conservation and look for new RFL variants within the 262 barley accessions. During this process, similar RFL sequences were iteratively grouped into clusters representing putative orthogroups (POGs). Ideally, we were aiming to assign sequences of a single RFL gene into one POG across all 262 accessions but, due to extremely high sequence similarity between some RFL genes, it is unavoidable that in some cases a POG will contain sequences from several distinct genes. For example, POG3 contains both HvRFL06 and HvRFL07 (Table S7) as the two sequences are identical. By using the described approach across all 262 EC data sets and the ‘Morex’ Refseqv1.0, 68 putative POGs were identified (Figure 3;Table S7). Fifteen POGs each correspond to a single gene found in the Refseqv1.0 genome, two contain multiple ‘Morex’ reference genes and 51 POGs contain no Refseqv1.0 genes (Table S7). Four POGs (POG67, POG15, POG66 and POG06) were found in more than 90% of the accessions and the WGS assemblies of ‘Barke’ and ‘Morex’ (Figure 3; Table S8). These correspond to ‘Morex’ HvRFL18, HvRFL19, HvRFL21 and HvRFL12, respectively (Table S8), which, apart from HvRFL12, were annotated on chromosome 6H as singlets (Table 3). Sequence alignments of these genes revealed high sequence conservation with very few amino acid substitutions (Figure S2a). In comparison, three genes located on chromosome 1H in ‘Morex’ POG68, POG05 and POG01 corresponding to HvRFL11, HvRFL09 and HvRFL01, respectively, were found in 70% of the accessions (Table S8) and show high sequence divergence (Figure S2b). The remaining 61 POGs were found in 50% or less of the accessions, and show different levels of sequence variability (Table S8).
Figure 3.
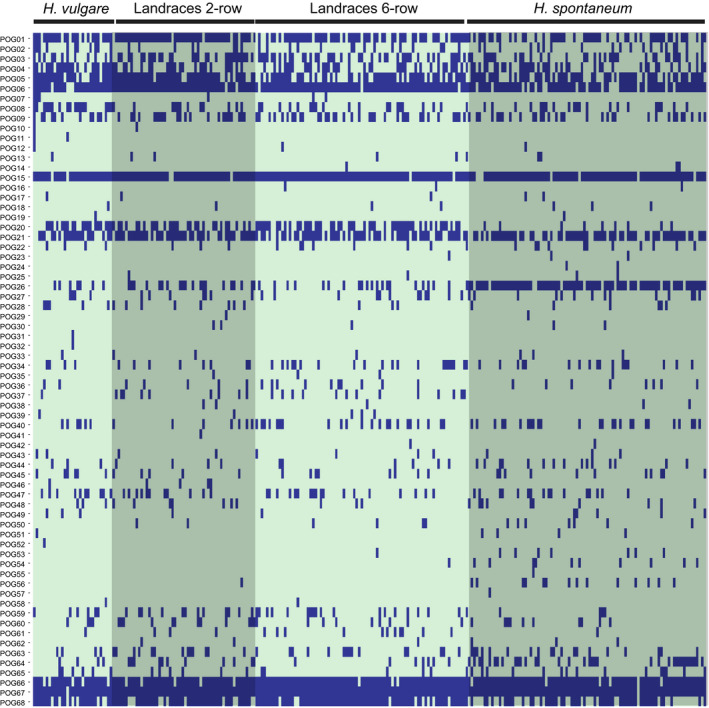
Identifying the repertoire of putative orthogroups (POGs) across 262 barley accessions.Matrix illustrating the presence (blue bar) of representative sequences of 68 reference POG sequences identified by hierarchical cluster analysis (HCA) of 7704 sequences identified in data sets from 262 barley accessions. The matrix was visualised in an offline version of plotly (https://plot.ly/).
Estimation of dN/dS rate ratio of barley RFL proteins to assess positive selection
To measure the strength and mode of natural selection acting on barley RFL genes, the ratio of non‐synonymous (dN) to synonymous (dS) substitutions (ω = dN/dS) was calculated (Table S9). First, we estimated the average ω values for each RFL gene by using the M0 model in the CODEML package (Yang, 2007), which averages the ω ratio across all sites in a protein (Jeffares et al., 2015). We observed that the average ω values for RFL genes correlate with their genomic location (Figure 4a). In particular, RFLs located within sub‐cluster 2 on chromosome 1H, which contains the highest number of RFLs among all clusters identified in the barley reference genome, show elevated ω values reaching 1 (Figure 4a; Table S9). The lowest ω values (ω = 0.22) were reported for two singlets, HvRFL15 and HvRFL21, located on chromosomes 1H and 6H, respectively (Figure 4a; Table S9). This indicates that purifying rather than diversifying selection is acting on them (Figure 4a).
Figure 4.
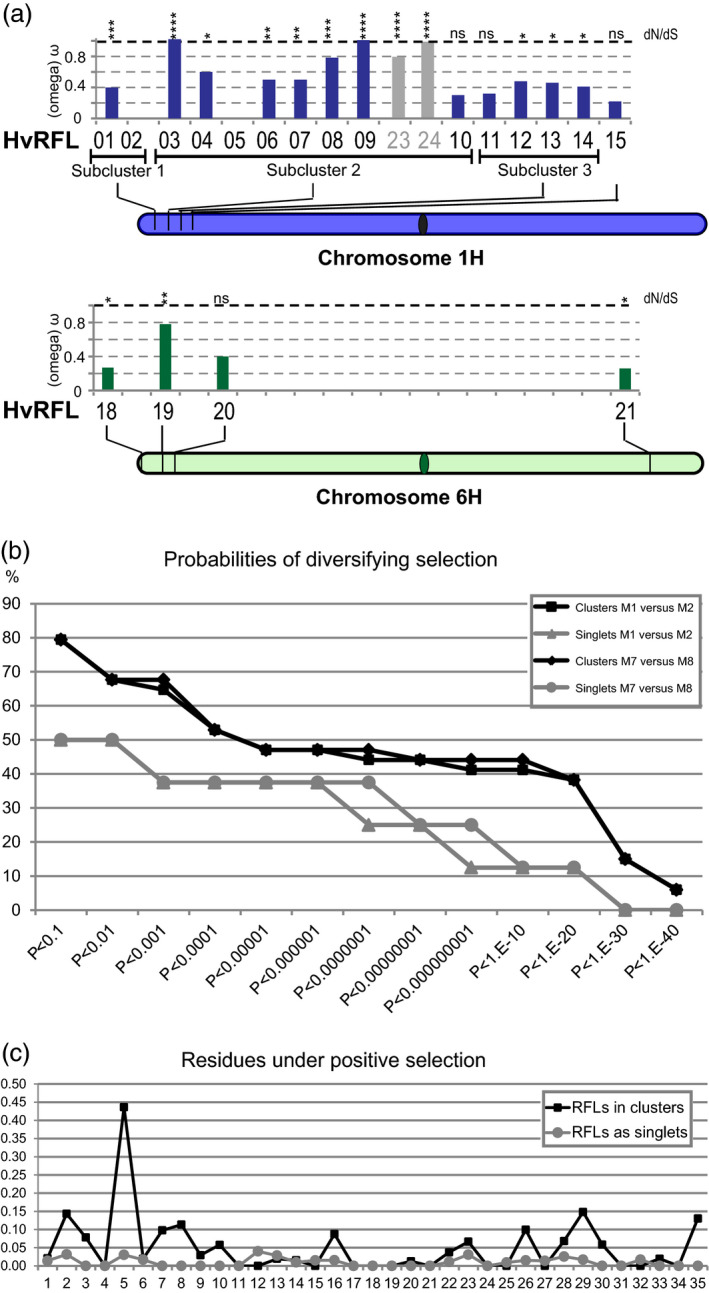
Diversifying selection acting on barley restorer‐of‐fertility‐like (RFL) genes.(a) Average ω values (Model 0) calculated for each HvRFL protein and plotted along the chromosomal positions of corresponding genes along with probabilities of diversifying selection calculated with CODEML. HvRFL23 and HvRFL24 are depicted within sub‐cluster 2 on chromosome 1H based on CD‐HIT clustering, but their position within the sub‐cluster is arbitrary. *P < 0.1, **P < 1.E‐5, ***P < 1.E‐10, ****P < 1.E‐20, ns – no probabilities of diversifying selection.(b) Cumulative proportions of genes that fit models allowing diversifying selection (M2 or M8) better than models assuming only purifying/neutral evolution (M1 or M7), respectively at different P‐values. (c) Bayes Empirical Bayes (BEB) probabilities of positive selection mapped onto positions of a pentatricopeptide repeat (PPR) motif. The line chart displays mean positive‐selection probabilities at each amino acid position within the PPR motif from either RFL genes organised in clusters or present in the genome as singlets.
The probabilities of diversifying selection (dN/dS ratios) for POGs identified by the HCA approach were calculated and compared with predictions from codon substitution models assuming only purifying/neutral evolution or also allowing diversifying selection. Nearly one‐third of analysed POGs showed very strong indications of positive selection (P < 1.E‐11 Table S10). In contrast, for 10 genes no evidence of diversifying selection was found (Table S10). Comparison of POGs located within a cluster or present as singlets revealed that genes located in clusters show much higher probabilities of diversifying selection compared with singlets (Figure 4a and b).
Amino acid residues within PPR motifs of RFL proteins that are under positive selection were predicted (Figure 4c) by using the Bayes Empirical Bayes (BEB) approach (Yang et al., 2005) implemented in CODEML. In particular, positions 5 and 35 but also 2, 7, 8 and 29 of each PPR motif were found to be under much stronger diversifying selection compared with other residues (Figure 4c). These probabilities of diversifying selection, acting on PPR residues reported to be involved directly in contact with the target RNA, i.e. positions 5 and 35 (Barkan et al., 2012), were much higher for POGs organised in clusters than as singlets (Figure 4c).
Interspecific RFL sequence conservation and synteny
To study RFL gene conservation in other Hordeum species, RFL sequences were identified in draft WGS assemblies of Hordeum pubiflorum, also known as Antarctic barley, native to South America, and Hordeum bulbosum, another wild relative of cultivated barley. These were compared with RFL sequences found in the ‘Morex’ Refseqv1.0 reference genome. Eight of the nine RFLs found in the H. bulbosum genome show 78–93% identity with RFLs from H. vulgare ‘Morex’ (Figure S1; Table S11). Out of 22 sequences identified in H. pubiflorum, only 10 share 64–100% identity with RFL sequences from ‘Morex’ (Table S11), and the remaining 10 sequences group together and show low sequence similarity to H. vulgare RFL sequences (Table S11). This result might reflect the greater genetic distance between H. vulgare and H. pubiflorum than between H. vulgare and H. bulbosum.
For a broader interspecific comparison, the set of barley Refseqv1.0 reference RFLs was compared with RFLs identified in three other grass species: Sorghum bicolor (Fujii et al., 2011); Brachypodium distachyon (Fujii et al., 2011); and Oryza sativa ssp. indica (Melonek et al., 2016; Figure 5). Arabidopsis RFLs were included as an outgroup (Figure 5). Clusters of RFLs from one species are grouped with whole clusters or larger groups of RFL sequences from other species (Figure 5). Within a species, RFL sequences located on the same chromosome show higher sequence similarity to each other than to RFL sequences located on other chromosomes, for example, all sorghum RFL sequences located on chromosome 5 form one group and sequences located on chromosome 2 form another group (Figure 5). Interestingly, HvRFL18 and HvRFL19 located on chromosome 6H and identified as highly conserved sequences in barley show high sequence similarity to four sorghum RFLs located on chromosome 2 (Figure 5). Moreover, the majority of RFLs from chromosome 1H cluster in barley are grouped with RFLs located on chromosome 2 in Brachypodium (Figure 5). This indicates that these genes might have originated from a common ancestor cluster.
Figure 5.
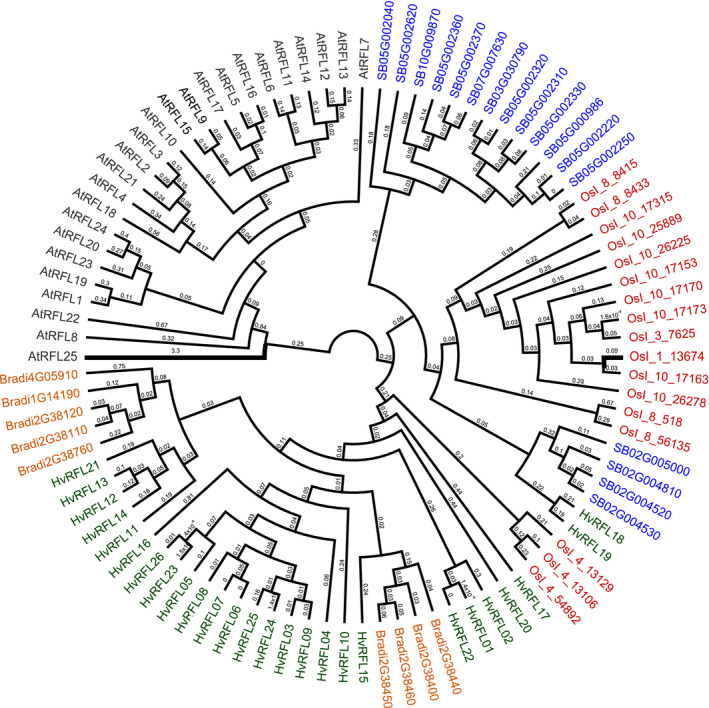
Phylogenetic relationships between restorer‐of‐fertility‐like (RFLs) from Hordeum vulgare cv. ‘Morex’ (green), Sorghum bicolor (blue), Oryza sativa ssp. indica (red) and Brachypodium distachyon (orange).Arabidopsis RFLs were added as an outgroup (black). Protein sequences were aligned with Muscle v.3.8.31 (Edgar, 2004), and the cladogram was built with FastTree (Price et al., 2009) and visualised in Geneious (www.geneious.com).
In addition to different levels of sequence divergence, differences in level of synteny between RFL clusters were also observed among the analysed species and H. vulgare ‘Morex’ (Figure 6). Whereas genomic locations of RFL clusters in H. vulgare, B. distachyon and O. sativa ssp. indica partially overlap, no synteny between RFL clusters identified in H. vulgare and S. bicolor was observed (Figure 6).
Figure 6.
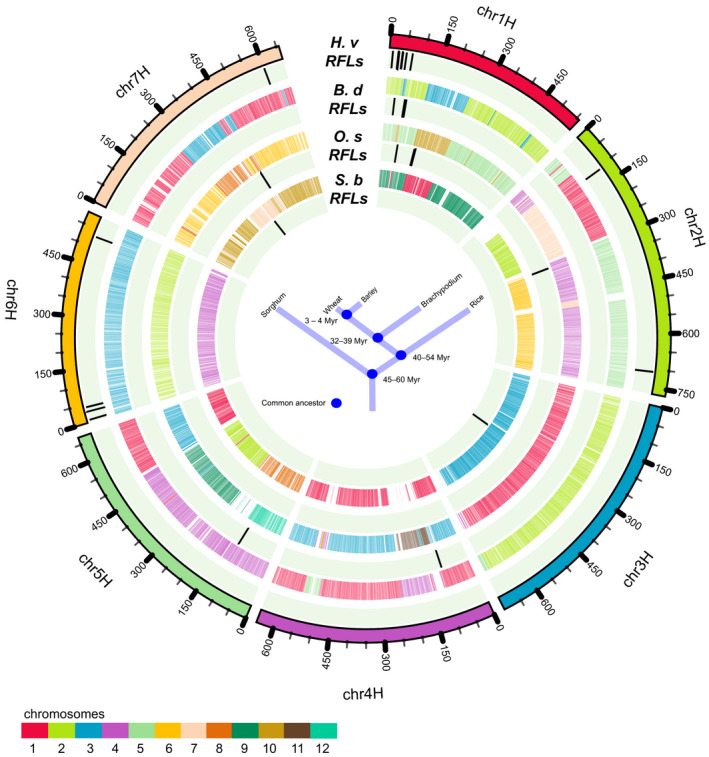
Circos diagram showing synteny between Hordeum vulgare, Brachypodium distachyon, Oryza sativa ssp. indica and Sorghum bicolor chromosomes as reported earlier (The International Barley Genome Sequencing Consortium, 2012). Genomic locations of restorer‐of‐fertility‐like (RFL) gene clusters are shown as black vertical bars.
DISCUSSION
Over the last few years, as a result of rapid advances in sequencing technologies and computational techniques, increasing numbers of high‐quality plant genome sequences have become available. Among them is the first high‐quality reference (Refseqv1.0) genome of barley (Mascher et al., 2017). It is now possible in cereals to characterise large gene families that have long been known and extensively studied in model plants such as Arabidopsis or rice. Some of these gene families are of agronomic importance, and thus the knowledge gains are expected to advance the breeding of new varieties with higher yield and better tolerance to changing environments. One such family is the PPR family, in particular a subclade of it referred to as RFL proteins. Rf genes have played a crucial role in the success of hybrid rice varieties (Huang et al., 2014). Therefore, the identification of restorer lines carrying strong Rf genes is likely to be helpful for further development of commercial CMS‐based restoration systems in barley.
We initially focused on the analysis of the PPR and RFL families in cultivated barley, represented by H. vulgare cv. ‘Morex’. The pool of 460 PPR proteins identified in the barley Refseqv1.0 reference genome is similar to the number of PPRs identified in other diploid plant genomes such as Arabidopsis and rice (Cheng et al., 2016). The number of identified RFL proteins (26) is also similar to that observed in Arabidopsis and rice (Fujii et al., 2011; Melonek et al., 2016), but is fewer than the ~30–50 RFL proteins comprised of 10 or more PPR motifs that were found to be encoded by each of the three bread wheat (Triticum aestivum Chinese Spring CS42) sub‐genomes (The International Wheat Genome Sequencing Consortium, 2018).
As reported previously for rice and bread wheat, where ~90% of the RFL genes were found in clusters (Melonek et al., 2016; The International Wheat Genome Sequencing Consortium, 2018), barley RFL genes are organised in two clusters, with sub‐cluster 2 on chromosome 1H being by far the largest. Within each cluster the genes show a close relationship, such that RFL sequences originating from the same genomic region show higher sequence similarity to each other than to sequences located on other chromosomes. This feature, typical of RFL genes, distinguishes them from other PPR genes that do not show such clustering. Regions of the barley genome carrying RFL clusters show higher PPR gene density than regions with other types of PPR genes. The origin of such RFL‐rich regions can be explained by the proposed mechanism of RFL gene expansion by tandem duplications and unequal crossover (Dahan and Mireau, 2013; Melonek et al., 2016), which generate sequence variation that fuels the ‘molecular arms‐race’ between the nuclear and mitochondrial genomes also known as nucleocytoplasmic conflict (Touzet and Budar, 2004).
Analysis of conservation of RFL genes across the genus Hordeum gave striking insight into the RFL sequence retention and variability across hundreds of individual accessions and landraces, despite the fact that only about half of ‘Morex’ reference RFLs were found in the draft WGS assemblies (The International Barley Genome Sequencing Consortium, 2012) and 262 barley EC data sets (Mascher et al., 2013). Two main factors could have contributed to this result: (i) non‐exhaustive coverage of the sequence capture experiment; and (ii) limitations of the de novo assembly of RFL sequences from short reads. Based on the comparison with the barley draft genome assembly, regions covered by the capture targets were estimated to encompass ~78% of high‐confidence exonic sequence and ~41% of low‐confidence exon sequence (Mascher et al., 2013). Due to the repetitive nature of RFL genes, one capture probe could hybridise to several paralogous RFL regions, therefore it is rather unlikely that the EC approach is the sole cause of the low recovery of RFL sequences. Most likely, the high similarity of RFL sequences, often originating from duplications, created ambiguities in short read alignments and assemblies. This, in turn, generated shorter (partial) sequence scaffolds or chimeric sequences formed from several highly similar RFL paralogues being merged into a single sequence. These assembly issues are likely to be particularly prevalent for the larger RFL clusters containing multiple similar genes, and probably explain why, for example, the sequences missing from the draft WGS assemblies predominantly correspond to the largest cluster on chromosome 1H. Taking into account these considerations, the total number of 68 RFL POGs determined by HCA of ~7700 RFL sequences is probably an underestimate. More POGs are expected to be identified in high‐quality whole‐genome sequences obtained, ideally, in a hybrid assembly approach with long and short reads, a method that has recently been shown to improve discovery of gene family expansions in plants (Miller et al., 2017) and was successfully applied in the assembly of several plant genomes, including the large and highly repetitive genome of Aegilops tauschii (Zimin et al., 2017). Resolving variation in RFL gene clusters will be crucial for identifying new Rf gene variants and will help in understanding the evolution of this complex gene family.
For some POGs, high retention (a representative sequence present in more than ~50% of surveyed samples) across hundreds of barley accessions was observed. Four of these ‘core’ POGs show extremely high sequence conservation and, based on similarity with ‘Morex’ genes, they most likely represent singlets. On the other hand, a few of the ‘core’ RFLs show much higher nucleotide polymorphism across accessions, and their genomic locations in ‘Morex’ coincide with RFL clusters. These genes show much higher average ω values than other POGs. RFL clusters have been proposed as sites in the nuclear genome where novel Rf gene variants are created and selected for their ability to target novel RNAs causing plant sterility created by recombination events in the mitochondrial genome (Fujii et al., 2011). Previous studies have shown rapid RFL sequence divergence in interspecific comparisons (Fujii et al., 2011; Melonek et al., 2016). In this study, a much larger set of RFL sequences from closely related barley accessions and landraces was analysed, allowing for the first systematic intraspecific analyses of RFL diversity to be carried out. The large sample size means that the calculated probabilities for diversifying selection are much higher than those obtained in previous studies (Geddy and Brown, 2007; Fujii et al., 2011). Diversifying selection was previously detected on particular amino acid residues within PPR motifs (Fujii et al., 2011). The amino acid residues at positions 5 and 35, which are in direct contact with target RNAs (Shen et al., 2016), were reported to be under strong diversifying selection in interspecific comparisons (Fujii et al., 2011). In our study, residue 5 (which helps distinguish between purine and pyrimidine nucleotides; Barkan et al., 2012; Shen et al., 2016) is the major target of diversifying selection. Elevated probabilities of diversifying selection could be used as markers for detecting active Rf loci (i.e. those under natural selection) among the many RFL sequences that can be identified in complex genomic data sets. Rapid copy number variation of RFL sequences accompanied by equally rapid selective sequence changes contribute to the overall high sequence plasticity of the RFL family members, making it necessary to sequence every prospective restorer line – working with the reference genome alone only gives a very partial view of the diversity of RFL sequences within the gene pool.
Studies on CMS and fertility restoration in barley are still very limited and, to date, only Rfm1 has been reported as a locus controlling fertility restoration in barley (Ui et al., 2015; Rizzolatti et al., 2017). The genomic location of the mapped Rfm1 region in the barley ‘Morex’ reference genome does not coincide with either of the two RFL clusters on chromosome 1H and 2H, or any of the single RFL genes identified in this study. Recently, sequencing of BAC libraries developed from the barley restorer line Re08 allowed the probable identification of Rfm1 as a PLS‐class PPR gene (Rizzolatti et al., 2017). So far, the only other PLS‐class Rf candidate was reported from sorghum (Klein et al., 2005). The restoring capability of these two PLS‐class genes remains to be proven and the molecular mechanism underlying the mode of action of PLS‐type Rf genes investigated.
As the majority of Rf genes identified to date in plant species belong to the RFL subclade, the RFL clusters on chromosome 1H and 2H in barley are expected to coincide with the location of genomic intervals carrying putative yet to be identified Rf restorer genes in H. spontaneum, the cytoplasm donor of msm1 and msm2 cytoplasms. Of particular interest is the sub‐cluster 2 on chromosome 1H, as the RFL sequences located within show high copy number and sequence variation as well as elevated probabilities for diversifying selection. It was shown in rice that several Rf restorer genes including Rf1a, Rf1b, Rf4 and Rf5 are all located within the same RFL cluster located on chromosome 10, the largest RFL cluster in the rice genome (Kazama and Toriyama, 2003; Akagi et al., 2004; Hu et al., 2012). The combination of the H. spontaneum mitochondrial genome sequence obtained recently (Hisano et al., 2016) with the RFL sequence data obtained once the H. spontaneum reference genome becomes available will bring new insights into the mechanisms underlying sterility and fertility restoration in barley.
Our analysis represents the most comprehensive characterisation of the PPR and RFL gene families in the genus Hordeum. The sequence data obtained in this study are a valuable resource that can be used in the design of sequence baits destined for capture‐based target enrichment of samples prior to next‐generation sequencing (NGS). The development of high‐throughput cost‐effective NGS‐based methods will allow screening of hundreds of elite lines and wild barley accessions, and will enable a more in‐depth analysis of sequence and structural variation of RFL family in the barley pan‐genome. The obtained sequence knowledge has the potential to accelerate genomic‐based improvement of barley elite lines and will be beneficial for the development of hybrid breeding systems based on CMS.
EXPERIMENTAL PROCEDURES
Identification of RFL sequences in genomic sequence data
The barley ‘Morex’ Refseqv1.0 genome was downloaded from the Plant Genomics and Phenomics Research Data Repository https://doi.org/10.5447/ipk/2016/34 (Mascher et al., 2017). The WGS assemblies of H. pubiflorum and H. bulbosum were accessed from The National Center for Biotechnology (NCBI) (https://www.ncbi.nlm.nih.gov/bioproject/) project reference number: PRJEB3404 and PRJEB3403, respectively. The PPR sequences in the genomic sequence data were identified as published recently (Cheng et al., 2016). Only P‐ and PLS‐class ORFs with scores above 100 and 240 (as judged by hmmsearch scores), respectively, were chosen for further analyses. The identification of RFL sequences was performed as described earlier (Melonek et al., 2016), and was based on inferring orthologous sequences using phylogenetic approaches and OrthoMCL (http://www.orthomcl.org/orthomcl/) (Li et al., 2003). In addition, previously identified RFL sequences from S. bicolor and B. distachyon (Fujii et al., 2011) and O. sativa indica (Melonek et al., 2016) were included in the study. The Circos diagrams were drawn with Circos software (Krzywinski et al., 2009).
Analysis of 262 barley exome capture data sets
The barley EC data sets were downloaded from the Sequence Read Archive (SRA) (https://www.ncbi.nlm.nih.gov/sra) accession number PRJEB1810 (Mascher et al., 2013). The sequencing reads’ insert sizes were estimated by aligning reads with ‘Morex’ Refseqv1.0 using BWA (Li and Durbin, 2009) and subsequently were assembled with MaSuRCA v3.2.2 (Zimin et al., 2013). The obtained sequence scaffolds were screened for the presence of RFL genes as described above. HCA (Huang et al., 2010) was applied to assign the identified RFL sequences into POGs. Three iterated runs of CD‐HIT clustering with identity thresholds (‐c) of 98, 96 and 93%, respectively, were performed. The parameters of single CD‐HIT run were as follows: ‐c 0.98 ‐n 5 ‐g 1 ‐G 0 ‐aS 0.99 ‐d 0. The presence/absence matrix of POGs in the barley accessions was generated with an offline version of plotly (https://plot.ly/python/) in Jupyter notebook (http://jupyter.org/).
Calculation of probabilities of positive/diversifying selection with CODEML
To detect positive selection, the NSites test implemented in the CODEML program from the Phylogenetic Analysis by Maximum Likelihood (PAML) package version 4.9 (Yang, 2007) was used. Neutral models were compared with alternative models allowing positive selection and performing likelihood ratio tests of the following PAML models: M1 versus M2 and M7 versus M8. For the analysis we used only RFL genes for which representatives in more than four accessions were identified. First, sequences assigned to each POG by HCA longer than 400 amino acids were aligned with Muscle (Edgar, 2004). The number of sequences included in each sequence alignment is given in Table S10. The sequence alignments were used to construct a maximum‐likelihood tree based on the JTT matrix‐based model (Jones et al., 1992) in MEGA software version 7.0 (Kumar et al., 2016). Initial trees for the heuristic search were obtained automatically by applying Neighbor‐Join and BioNJ algorithms to a matrix of pairwise distances estimated using a JTT model, and then selecting the topology with superior log likelihood value. The tree with the highest log likelihood was exported into Newick standard format that was directly used by CODEML. Sequence alignments along with the topology trees generated for each POG were deposited in the UWA Research Repository (https://doi.org/10.26182/5ba4695d7ff38) (Melonek et al., 2018). PAL2NAL (Suyama et al., 2006) was used to generate codon‐protein alignments. CODEML was run with the following settings: runmode = 0, CodonFreq = 2:F3x4, model = 0, Nsites = 0 1 2 7 8. The BEB approach implemented in CODEML (Yang et al., 2005) was used to identify sites potentially under positive selection.
Sequence homology and synteny analysis
The analysis of sequence homology was performed on a set of RFLs identified earlier in sorghum and Arabidopsis (Fujii et al., 2011), as well as rice and Brachypodium (Melonek et al., 2016). The sequences were aligned with Muscle v.3.8.31 (Edgar, 2004) and the alignment was used to generate a tree with FastTree (Price et al., 2009). The tree branches were coloured in Geneious (http://www.geneious.com/). To study the conservation of genomic locations of RFL regions between barley and three other cereal species, a data set with chromosomal synteny reported earlier (The International Barley Genome Sequencing Consortium, 2012) was used. The figure was generated with Circos (Krzywinski et al., 2009).
Conflict of interest
This work has been partially funded by Groupe Limagrain.
Supporting information
Figure S1. Identification of RFL sequences in the genomic data sets of H. vulgare cvs. ‘Morex’, ‘Barke’ and ‘Bowman’.
Figure S2. Sequence alignment of POG15 and POG01 representative sequences.
Table S1 Summary of RFL genes identified in the barley cv. ‘Morex’ Refseqv1.0 genome.
Table S2 Summary of RFL genes identified in the WGS assemblies of barley cvs. ‘Morex’, ‘Barke’ and ‘Bowman’ compared with RFLs identified in the ‘Morex’ Refseqv1.0.
Table S3 CD‐HIT clustering of ‘unanchored’ barley RFLs.
Table S4 List of EC data sets (Mascher et al., 2013) used in this study.
Table S5 Number of PPR, P‐class, PLS‐class and RFL sequences identified in the EC data sets.
Table S6 Overlapping START and END between RFL ORFs and scaffolds.
Table S7 Representatives of 68 POG sequences identified across 262 barley accessions.
Table S8 Frequency of 68 POGs identified across 262 accessions.
Table S9 ω values calculated for HvRFLs with CODEML (Model 0).
Table S10 Comparison of codon substitution models M2 versus M1 and M8 versus M7 across POGs.
Table S11 Sequence conservation among RFL sequences identified in H. pubiflorum, H. bulbosum and H. vulgare.
ACKNOWLEDGEMENTS
I.S. and J.M. benefitted from support by the Australian Research Council Centre of Excellence (ARC CoE) in Plant Energy Biology (CE140100008) and Groupe Limagrain. P.E.B. acknowledges the support of the Forrest Research Foundation. The Australia–Germany Joint Research Cooperation Scheme (UA–DAAD: PPP Australien 1j 16, Projekt‐ID 57217869) is thanked for supporting collaborative exchange between the ARC CoE in Plant Energy Biology in Perth, and The Leibniz Institute of Plant Genetics and Crop Plant Research (IPK) in Gatersleben.
Contributor Information
Nils Stein, Email: stein@ipk-gatersleben.de.
Ian Small, Email: ian.small@uwa.edu.au.
REFERENCES
- Ahokas, H. (1979) Cytoplasmic male‐sterility in barley. Acta Agric. Scand. 29, 219–224. [Google Scholar]
- Ahokas, H. (1980a) Cytoplasmic male‐sterility in barley. 5. Physiological characterization of the Msm1‐Rfm1 a system. Physiol. Plant. 48, 231–238. [Google Scholar]
- Ahokas, H. (1980b) Cytoplasmic male‐sterility in barley. 7. Nuclear genes for restoration. Theor. Appl. Genet. 57, 193–202. [DOI] [PubMed] [Google Scholar]
- Ahokas, H. (1982) Cytoplasmic male‐sterility in barley. 11. The Msm2 cytoplasm. Genetics 102, 285–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akagi, H. , Nakamura, A. , Yokozeki‐Misono, Y. , Inagaki, A. , Takahashi, H. , Mori, K. and Fujimura, T. (2004) Positional cloning of the rice Rf‐1 gene, a restorer of BT‐type cytoplasmic male sterility that encodes a mitochondria‐targeting PPR protein. Theor. Appl. Genet. 108, 1449–1457. [DOI] [PubMed] [Google Scholar]
- Barkan, A. and Small, I. (2014) Pentatricopeptide repeat proteins in plants. Annu. Rev. Plant Biol. 65, 415–442. [DOI] [PubMed] [Google Scholar]
- Barkan, A. , Rojas, M. , Fujii, S. , Yap, A. , Chong, Y.S. , Bond, C.S. and Small, I. (2012) A combinatorial amino acid code for RNA recognition by pentatricopeptide repeat proteins. PLoS Genet. 8, e1002910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhard, T. , Friedt, R. , Snowdon, R.J. and Wittkop, B. (2017) New insights into genotypic thermodependency of cytoplasmic male sterility for hybrid barley breeding. Plant Breed. 136, 8–17. [Google Scholar]
- Chase, C.D. (2007) Cytoplasmic male sterility: a window to the world of plant mitochondrial‐nuclear interactions. Trends Genet. 23, 81–90. [DOI] [PubMed] [Google Scholar]
- Chateigner‐Boutin, A.L. , des Francs‐Small, C.C. , Fujii, S. , Okuda, K. , Tanz, S.K. and Small, I. (2013) The E domains of pentatricopeptide repeat proteins from different organelles are not functionally equivalent for RNA editing. Plant J. 74, 935–945. [DOI] [PubMed] [Google Scholar]
- Chen, L. and Liu, Y.G. (2014) Male sterility and fertility restoration in crops. Annu. Rev. Plant Biol. 65, 579–606. [DOI] [PubMed] [Google Scholar]
- Chen, F. , Mackey, A.J. , Stoeckert, C.J. and Roos, D.S. (2006) OrthoMCL‐DB: querying a comprehensive multi‐species collection of ortholog groups. Nucleic Acids Res. 34, D363–D368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, S.F. , Gutmann, B. , Zhong, X. et al (2016) Redefining the structural motifs that determine RNA binding and RNA editing by pentatricopeptide repeat proteins in land plants. Plant J. 85, 532–547. [DOI] [PubMed] [Google Scholar]
- Dahan, J. and Mireau, H. (2013) The Rf and Rf‐like PPR in higher plants, a fast‐evolving subclass of PPR genes. RNA Biol. 10, 1469–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar, R.C. (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii, S. , Bond, C.S. and Small, I.D. (2011) Selection patterns on restorer‐like genes reveal a conflict between nuclear and mitochondrial genomes throughout angiosperm evolution. Proc. Natl Acad. Sci. USA 108, 1723–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaborieau, L. , Brown, G.G. and Mireau, H. (2016) The propensity of pentatricopeptide repeat genes to evolve into restorers of cytoplasmic male sterility. Front. Plant Sci. 7, 1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geddy, R. and Brown, G.G. (2007) Genes encoding pentatricopeptide repeat (PPR) proteins are not conserved in location in plant genomes and may be subject to diversifying selection. BMC Genom. 8, 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson, M.R. and Bentolila, S. (2004) Interactions of mitochondrial and nuclear genes that affect male gametophyte development. Plant Cell 16, S154–S169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hisano, H. , Tsujimura, M. , Yoshida, H. , Terachi, T. and Sato, K. (2016) Mitochondrial genome sequences from wild and cultivated barley (Hordeum vulgare). BMC Genom. 17, 824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, J. , Wang, K. , Huang, W. et al (2012) The rice pentatricopeptide repeat protein RF5 restores fertility in Hong‐Lian cytoplasmic male‐sterile lines via a complex with the glycine‐rich protein GRP162. Plant Cell 24, 109–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, J. , Huang, W.C. , Huang, Q. , Qin, X.J. , Yu, C.C. , Wang, L.L. , Li, S.Q. , Zhu, R.S. and Zhu, Y.G. (2014) Mitochondria and cytoplasmic male sterility in plants. Mitochondrion 19, 282–288. [DOI] [PubMed] [Google Scholar]
- Huang, Y. , Niu, B.F. , Gao, Y. , Fu, L.M. and Li, W.Z. (2010) CD‐HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics 26, 680–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, J.Z. , E, Z.G. , Zhang, H.L. and Shu, Q.Y. (2014) Workable male sterility systems for hybrid rice: genetics, biochemistry, molecular biology, and utilization. Rice (N Y) 7, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffares, D.C. , Tomiczek, B. , Sojo, V. and dos Reis, M. (2015) A beginners guide to estimating the non‐synonymous to synonymous rate ratio of all protein‐coding genes in a genome. Methods Mol. Biol. 1201, 65–90. [DOI] [PubMed] [Google Scholar]
- Jones, D.T. , Taylor, W.R. and Thornton, J.M. (1992) The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 8, 275–282. [DOI] [PubMed] [Google Scholar]
- Kazama, T. and Toriyama, K. (2003) A pentatricopeptide repeat‐containing gene that promotes the processing of aberrant atp6 RNA of cytoplasmic male‐sterile rice. FEBS Lett. 544, 99–102. [DOI] [PubMed] [Google Scholar]
- Klein, R.R. , Klein, P.E. , Mullet, J.E. , Minx, P. , Rooney, W.L. and Schertz, K.F. (2005) Fertility restorer locus Rf1 [corrected] of sorghum (Sorghum bicolor L.) encodes a pentatricopeptide repeat protein not present in the colinear region of rice chromosome 12. Theor. Appl. Genet. 111, 994–1012. [DOI] [PubMed] [Google Scholar]
- Krzywinski, M. , Schein, J. , Birol, I. , Connors, J. , Gascoyne, R. , Horsman, D. , Jones, S.J. and Marra, M.A. (2009) Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, S. , Stecher, G. and Tamura, K. (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. and Durbin, R. (2009) Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, L. , Stoeckert, C.J. Jr and Roos, D.S. (2003) OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Longevialle, A.F. , Hendrickson, L. , Taylor, N.L. , Delannoy, E. , Lurin, C. , Badger, M. , Millar, A.H. and Small, I. (2008) The pentatricopeptide repeat gene OTP51 with two LAGLIDADG motifs is required for the cis‐splicing of plastid ycf3 intron 2 in Arabidopsis thaliana. Plant J. 56, 157–168. [DOI] [PubMed] [Google Scholar]
- Longin, C.F.H. , Muhleisen, J. , Maurer, H.P. , Zhang, H.L. , Gowda, M. and Reif, J.C. (2012) Hybrid breeding in autogamous cereals. Theor. Appl. Genet. 125, 1087–1096. [DOI] [PubMed] [Google Scholar]
- Lurin, C. , Andres, C. , Aubourg, S. et al (2004) Genome‐wide analysis of Arabidopsis pentatricopeptide repeat proteins reveals their essential role in organelle biogenesis. Plant Cell 16, 2089–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascher, M. , Richmond, T.A. , Gerhardt, D.J. et al (2013) Barley whole exome capture: a tool for genomic research in the genus Hordeum and beyond. Plant J. 76, 494–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascher, M. , Gundlach, H. , Himmelbach, A. et al (2017) A chromosome conformation capture ordered sequence of the barley genome. Nature 544, 427–433. [DOI] [PubMed] [Google Scholar]
- Meierhoff, K. , Felder, S. , Nakamura, T. , Bechtold, N. and Schuster, G. (2003) HCF152, an Arabidopsis RNA binding pentatricopeptide repeat protein involved in the processing of chloroplast psbB‐psbT‐psbH‐petB‐petD RNAs. Plant Cell 15, 1480–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melonek, J. , Stone, J.D. and Small, I. (2016) Evolutionary plasticity of restorer‐of‐fertility‐like proteins in rice. Sci. Rep. 6, 35 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melonek, J. , Zhou, R. , Bayer, P.E. , Edwards, D. , Stein, N. and Small, I. (2018) High intraspecific diversity of restorer‐of‐fertility‐like genes in barley, supplementary data set, The University of Western Australia Research Repository, 10.26182/5ba4695d7ff38. [accessed on 21 September 2018] [DOI]
- Miller, J.R. , Zhou, P. , Mudge, J. et al (2017) Hybrid assembly with long and short reads improves discovery of gene family expansions. BMC Genom. 18, 541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda, R.G. , McDermott, J.J. and Barkan, A. (2018) RNA‐binding specificity landscapes of designer pentatricopeptide repeat proteins elucidate principles of PPR‐RNA interactions. Nucleic Acids Res. 46, 2613–2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhleisen, J. , Maurer, H.P. , Stiewe, G. , Bury, P. and Reif, J.C. (2013) Hybrid breeding in barley. Crop Sci. 53, 819–824. [Google Scholar]
- Muhleisen, J. , Piepho, H.P. , Maurer, H.P. , Longin, C.F.H. and Reif, J.C. (2014a) Yield stability of hybrids versus lines in wheat, barley, and triticale. Theor. Appl. Genet. 127, 309–316. [DOI] [PubMed] [Google Scholar]
- Muhleisen, J. , Piepho, H.P. , Maurer, H.P. , Zhao, Y.S. and Reif, J.C. (2014b) Exploitation of yield stability in barley. Theor. Appl. Genet. 127, 1949–1962. [DOI] [PubMed] [Google Scholar]
- Okuda, K. , Myouga, F. , Motohashi, R. , Shinozaki, K. and Shikanai, T. (2007) Conserved domain structure of pentatricopeptide repeat proteins involved in chloroplast RNA editing. Proc. Natl Acad. Sci. USA 104, 8178–8183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Toole, N. , Hattori, M. , Andres, C. , Iida, K. , Lurin, C. , Schmitz‐Linneweber, C. , Sugita, M. and Small, I. (2008) On the expansion of the pentatricopeptide repeat gene family in plants. Mol. Biol. Evol. 25, 1120–1128. [DOI] [PubMed] [Google Scholar]
- Pedregosa, F. , Varoquaux, G. , Gramfort, A. et al (2011) Scikit‐learn: machine learning in python. J. Mach. Learn. Res. 12, 2825–2830. [Google Scholar]
- Pfalz, J. , Bayraktar, O.A. , Prikryl, J. and Barkan, A. (2009) Site‐specific binding of a PPR protein defines and stabilizes 5′ and 3′ mRNA termini in chloroplasts. EMBO J. 28, 2042–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price, M.N. , Dehal, P.S. and Arkin, A.P. (2009) FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 26, 1641–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prikryl, J. , Rojas, M. , Schuster, G. and Barkan, A. (2011) Mechanism of RNA stabilization and translational activation by a pentatricopeptide repeat protein. Proc. Natl Acad. Sci. USA 108, 415–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raynaud, C. , Loiselay, C. , Wostrikoff, K. , Kuras, R. , Girard‐Bascou, J. , Wollman, F.A. and Choquet, Y. (2007) Evidence for regulatory function of nucleus‐encoded factors on mRNA stabilization and translation in the chloroplast. Proc. Natl Acad. Sci. USA 104, 9093–9098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzolatti, C. , Bury, P. , Tatara, E. , Pin, P.A. , Rodde, N. , Berges, H. , Budar, F. , Mireau, H. and Gielen, J.J.L. (2017) Map‐based cloning of the fertility restoration locus Rfm1 in cultivated barley (Hordeum vulgare). Euphytica 213, 276. [Google Scholar]
- Russell, J. , Mascher, M. , Dawson, I.K. et al (2016) Exome sequencing of geographically diverse barley landraces and wild relatives gives insights into environmental adaptation. Nat. Genet. 48, 1024–1030. [DOI] [PubMed] [Google Scholar]
- Schnable, P.S. and Wise, R.P. (1998) The molecular basis of cytoplasmic male sterility and fertility restoration. Trends Plant Sci. 3, 175–180. [Google Scholar]
- Shen, C. , Zhang, D. , Guan, Z. et al (2016) Structural basis for specific single‐stranded RNA recognition by designer pentatricopeptide repeat proteins. Nat. Commun. 7, 11 285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small, I. , Peeters, N. , Legeai, F. and Lurin, C. (2004) Predotar: a tool for rapidly screening proteomes for N‐terminal targeting sequences. Proteomics 4, 1581–1590. [DOI] [PubMed] [Google Scholar]
- Sosso, D. , Mbelo, S. , Vernoud, V. et al (2012) PPR2263, a DYW‐subgroup pentatricopeptide repeat protein, is required for mitochondrial nad5 and cob transcript editing, mitochondrion biogenesis, and maize growth. Plant Cell 24, 676–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suyama, M. , Torrents, D. and Bork, P. (2006) PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 34, W609–W612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykes, T. , Yates, S. , Nagy, I. , Asp, T. , Small, I. and Studer, B. (2016) In‐silico identification of candidate genes for fertility restoration in cytoplasmic male sterile perennial ryegrass (Lolium perenne L.). Genome Biol. Evol. 9, 351–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takenaka, M. , Zehrmann, A. , Brennicke, A. and Graichen, K. (2013) Improved computational target site prediction for pentatricopeptide repeat RNA editing factors. PLoS ONE 8, e65343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The International Barley Genome Sequencing Consortium . (2012) A physical, genetic and functional sequence assembly of the barley genome. Nature 491, 711–716. [DOI] [PubMed] [Google Scholar]
- The International Wheat Genome Sequencing Consortium . (2018) Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 361, eaar7191. [DOI] [PubMed] [Google Scholar]
- Touzet, P. and Budar, F. (2004) Unveiling the molecular arms race between two conflicting genomes in cytoplasmic male sterility? Trends Plant Sci. 9, 568–570. [DOI] [PubMed] [Google Scholar]
- Ui, H. , Sameri, M. , Pourkheirandish, M. , Chang, M.C. , Shimada, H. , Stein, N. , Komatsuda, T. and Handa, H. (2015) High‐resolution genetic mapping and physical map construction for the fertility restorer Rfm1 locus in barley. Theor. Appl. Genet. 128, 283–290. [DOI] [PubMed] [Google Scholar]
- Yagi, Y. , Hayashi, S. , Kobayashi, K. , Hirayama, T. and Nakamura, T. (2013) Elucidation of the RNA recognition code for pentatricopeptide repeat proteins involved in organelle RNA editing in plants. PLoS ONE 8, e57286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Z. (2007) PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. [DOI] [PubMed] [Google Scholar]
- Yang, Z.H. , Wong, W.S.W. and Nielsen, R. (2005) Bayes empirical Bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 22, 1107–1118. [DOI] [PubMed] [Google Scholar]
- Zimin, A.V. , Marcais, G. , Puiu, D. , Roberts, M. , Salzberg, S.L. and Yorke, J.A. (2013) The MaSuRCA genome assembler. Bioinformatics 29, 2669–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimin, A.V. , Puiu, D. , Luo, M.C. , Zhu, T.T. , Koren, S. , Marcais, G. , Yorke, J.A. , Dvorak, J. and Salzberg, S.L. (2017) Hybrid assembly of the large and highly repetitive genome of Aegilops tauschii, a progenitor of bread wheat, with the MaSuRCA mega‐reads algorithm. Genome Res. 27, 787–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Identification of RFL sequences in the genomic data sets of H. vulgare cvs. ‘Morex’, ‘Barke’ and ‘Bowman’.
Figure S2. Sequence alignment of POG15 and POG01 representative sequences.
Table S1 Summary of RFL genes identified in the barley cv. ‘Morex’ Refseqv1.0 genome.
Table S2 Summary of RFL genes identified in the WGS assemblies of barley cvs. ‘Morex’, ‘Barke’ and ‘Bowman’ compared with RFLs identified in the ‘Morex’ Refseqv1.0.
Table S3 CD‐HIT clustering of ‘unanchored’ barley RFLs.
Table S4 List of EC data sets (Mascher et al., 2013) used in this study.
Table S5 Number of PPR, P‐class, PLS‐class and RFL sequences identified in the EC data sets.
Table S6 Overlapping START and END between RFL ORFs and scaffolds.
Table S7 Representatives of 68 POG sequences identified across 262 barley accessions.
Table S8 Frequency of 68 POGs identified across 262 accessions.
Table S9 ω values calculated for HvRFLs with CODEML (Model 0).
Table S10 Comparison of codon substitution models M2 versus M1 and M8 versus M7 across POGs.
Table S11 Sequence conservation among RFL sequences identified in H. pubiflorum, H. bulbosum and H. vulgare.