

Abstract
Aim
Severe mental illnesses (SMI), such as bipolar disorder and schizophrenia, are highly heritable, and have a complex pattern of inheritance. Genome‐wide association studies detect a part of the heritability, which can be attributed to common genetic variation. Examination of rare variants with next‐generation sequencing may add to the understanding of the genetic architecture of SMI.
Methods
We analyzed 32 ill subjects from eight multiplex families and 33 healthy individuals using whole‐exome sequencing. Prioritized variants were selected by a three‐step filtering process, which included: deleteriousness by five in silico algorithms; sharing within families by affected individuals; rarity in South Asian sample estimated using the Exome Aggregation Consortium data; and complete absence of these variants in control individuals from the same gene pool.
Results
We identified 42 rare, non‐synonymous deleterious variants (~5 per pedigree) in this study. None of the variants were shared across families, indicating a ‘private’ mutational profile. Twenty (47.6%) of the variant harboring genes were previously reported to contribute to the risk of diverse neuropsychiatric syndromes, nine (21.4%) of which were of Mendelian inheritance. These included genes carrying novel deleterious variants, such as the GRM1 gene implicated in spinocerebellar ataxia 44 and the NIPBL gene implicated in Cornelia de Lange syndrome.
Conclusion
Next‐generation sequencing approaches in family‐based studies are useful to identify novel and rare variants in genes for complex disorders like SMI. The findings of the study suggest a potential phenotypic burden of rare variants in Mendelian disease genes, indicating pleiotropic effects in the etiology of SMI.
Keywords: Mendelian, polygenic, schizophrenia, bipolar disorder, rare variant
Bipolar disorder (BD) and schizophrenia (SCZ) are severe mental illness (SMI) syndromes with a median lifetime prevalence of 2.4 and 3.3 per thousand persons, respectively,1, 2 and an estimated heritability of 70–90%.3, 4 Evidence from family and molecular genetic studies suggests shared, perhaps overlapping, risk factors across these syndromes.5, 6 The outcomes from large‐scale genome‐wide association studies (GWAS) exploring the common disease–common variant (CDCV) hypothesis detect a proportion of the estimated genetic risk.7 In this context, next‐generation sequencing (NGS) technology, by evaluating rare genetic variants, has enabled a deeper examination of complex traits using alternate models of risk, such as the ‘oligogenic quasi‐Mendelian model’8 and the ‘omnigenic models’9 of inheritance. Several recent studies in autism, SCZ, BD, and depression have detected rare variants using NGS in case–control or family‐based designs, across different genes implicated to play a key role in critical biological pathways.10, 11 Findings from such studies have shown that the majority of the rare variants identified are private to a family (Table S1),12, 13, 14 indicating the underlying heterogeneity in the genetic architecture of SMI. Multiplex families may provide valuable insights into the genetic correlates of these syndromes15, 16 when tested using high throughput sequencing.
A cross‐nosology approach has been quite informative in identifying potential disease‐relevant pathways in SCZ and BD.17, 18 Single‐nucleotide polymorphisms (SNP) associated with these two syndromes show a high mutual correlation, among combinations of neuropsychiatric syndromes.7 Such overlaps have also been observed across diverse neuropsychiatric syndromes, for both common and rare genetic variations, as well as in gene expression profiles in the cerebral cortex.19, 20 These findings indicate an underlying shared molecular pathology in the pathobiology of SMI.
As part of a longitudinal study, ‘Accelerator Program for Discovery of Brain Disorders Using Stem Cells’ (ADBS),21 aimed at understanding the developmental trajectories and basic biology of SMI, we describe in this study the results of a variant discovery analysis using whole‐exome sequencing (WES) in eight multiplex pedigrees with SCZ and BD phenotypes from a well characterized Indian cohort. Such studies have been predominantly conducted in large cohorts of European origin,16 and representation from other populations is perhaps necessary to validate earlier findings, and identify population‐specific signatures underlying SMI. In the current study, we aimed to identify rare, damaging, exonic variants that co‐segregate with SMI in multiplex families, and to examine their relevance to the disease.
Methods
Sample selection
The families were recruited as part of the ADBS longitudinal study, which has been approved by the ethics committee of the National Institute of Mental Health and Neurosciences, Bengaluru, India. The details of screening, informed consent, recruitment, and phenotyping have been previously published.21 Some of the families in the cohort have been on follow up for longer than 10 years. We have previously noted evidence of linkage in psychosis at chromosome 18p11.2,22 and the sex‐specific association to the DISC1 gene using a case–control study design23 in samples taken from this cohort. For the current study, eight families (A through H) with high loading of SMI (SCZ, BD, and psychosis in the context of these eight pedigrees; Fig. 1a, Fig. S1) were assessed in detail. From these, 32 individuals (‘cases’; 16 females) with SMI were available for blood sampling and were subjected to WES. Two senior psychiatrists evaluated all patients and unaffected relatives independently. Diagnoses were made with the ICD‐10 Classification for Mental and Behavioural Disorders and were verified in the longitudinal course of follow ups. From five of the eight families, we could also sample eight unaffected individuals who had crossed the age at risk and are defined as a ‘family‐specific control’ for the respective pedigree henceforth in this report. An independent set of 25 individuals without a history of SMI were further sampled as population‐matched controls. Together this group constituted a total of 33 asymptomatic ‘controls.’
Figure 1.
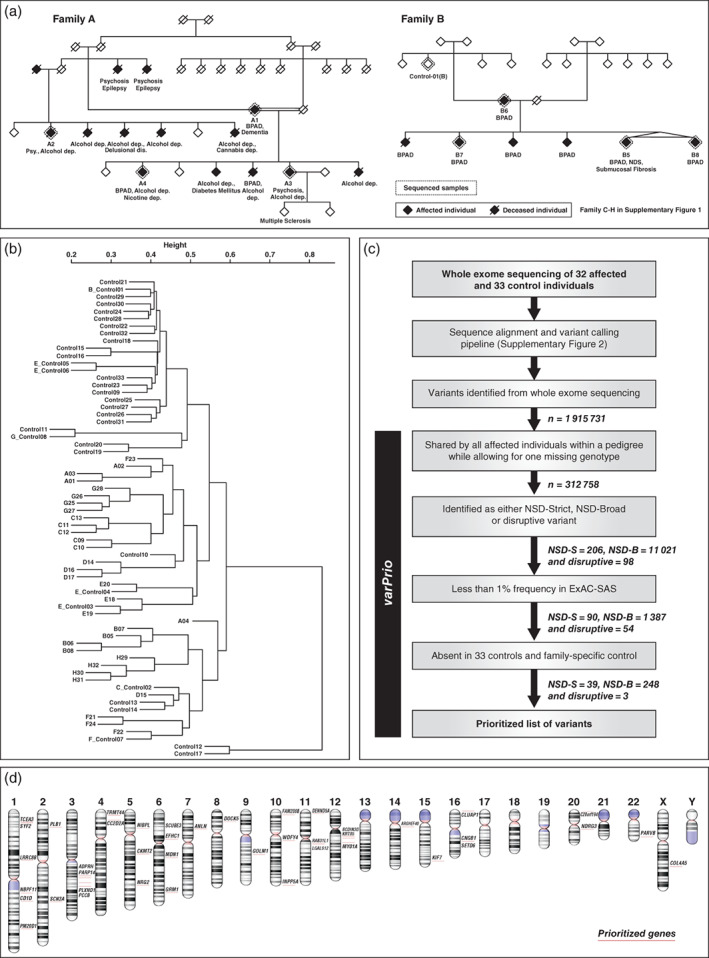
(a) Two representative pedigrees analyzed with exome sequencing (Families A and B). (b) Cluster dendrogram created with a distance matrix based on the degree of variant sharing between pairs of cases and controls analyzed in the study. (c) ‘varPrio’ – variant prioritization pipeline with numbers indicating the reduction in the total number of variants in each prioritization step. (d) Ideogram representing the 42 genes that harbored variants prioritized by non‐synonymous damaging strict (NSD‐S) and disruptive definition generated with NCBI genome decoration page.
Exome sequencing and analysis
Sequencing was carried out on the Illumina Hiseq NGS platform with libraries prepared using Illumina exome kits. Reads were aligned with reference human genome GRCh37 using the Burrows–Wheeler algorithm tool.24 Variants were called from realigned BAM files using Varscan2 with the standard criteria (min coverage = 8, MAF ≥ 0.25, and P ≤ 0.001).25 Standard quality control protocols were employed at sequencing, alignment, and variant calling (Fig.S2). The resulting variant called files were annotated with ANNOVAR.26
Pedigree‐based analysis
All variant segregation analysis was performed at the level of individual pedigrees. To ascertain the degree of variance (between pedigrees) and relatedness within family structures, we generated a dendrogram with hierarchical clustering analysis using an allele‐sharing matrix of the exonic variants (Appendix S1).
Variant prioritization
Variants were prioritized if:
the variant was found to be shared by all affected individuals within the pedigree while allowing for one missing genotype, a method shown to be useful in an earlier study of familial BD12;
the variant fell into any of the following deleterious categories – the non‐synonymous damaging strict (NSD‐S) set predicted to be damaging by five prediction algorithms (SIFT,27 Polyphen‐2 HDIV,28 Mutation taster 2,29 Mutation assessor,30 and LRT31[Appendix S1]), the Disruptive set predicted to result in protein truncation (splice site, stop gain, or stop loss variants), or the non‐synonymous damaging broad (NSD‐B) set predicted to be damaging by one or more of the five prediction algorithms; and
the variant was rare <1% in Exome Aggregation Consortium – South Asian sample (ExAC‐SAS)32 and completely absent from a control cohort of 33 individuals from the same gene pool (http://indexdb.ncbs.res.in).
The above variant prioritization was carried out using an in‐house automated pipeline ‘varPrio’. Details of the pipeline and the resulting variant enrichment are summarized in Figure1c. To rule out any false positive calls at the final variant list, a representative set of prioritized variants (n=10) was independently confirmed by Sanger sequencing and we noted a 100% concordance.
Functional annotation
We adapted two approaches for evaluating functional impact to the prioritized variants:
We reviewed the literature on individual genes identified in the NSD‐S and the disruptive set carrying rare variants of highest priority (all five in silico predictors) for prior evidence of disease association in neuropsychiatric phenotype.
For the NSD‐B set carrying rare variants of plausible disease relevance (1–5 in silico predictors), we tested for enrichment of the aggregate list using DAVID functional annotation tool 6.8.33, 34 To test the enrichment on the categories of biological process, molecular function, protein domain, protein–protein interaction, and tissue expression we selected the sources as –‘GOTERM_BP_DIRECT’, ‘GOTERM_MF_DIRECT’, ‘INTERPRO’, ‘KEGG_PATHWAY’ and‘UP_TISSUE’ in this in silico approach. Modified Fisher's exact test with Benjamini–Hochberg correction built‐in to this algorithm was used to infer enrichment.
Results
Sample characteristics
Of the 32 cases sequenced in the study, 26 were diagnosed with BD, four with SCZ, and one each with SCZ‐like psychosis and schizoaffective disorder. They had been ill for a mean (SD) duration of 23.7 (11.1) years, and the mean (SD) age at onset was 23.1 (7.9) years. In most of the pedigrees, there was heterogeneity in the age of onset, illness severity, global outcomes, and segregation of suicidality and psychosis (in BD) with the primary phenotype. Substance use disorder was a common comorbidity, followed by hypothyroidism, seizure disorder, and dementia (Appendix S2, Table S2).
In the analysis of relatedness using the cluster dendrogram, ‘cases’ and ‘controls’ formed a single cluster possibly resulting from sharing of a large number of common and/or benign exonic variants. As expected, members from each pedigree clustered together due to the relatively larger magnitude of variant sharing (Fig. 1b, Appendix S1).
Rare deleterious variants in Mendelian genes segregate within SMI families
Familywise prioritization identified a total of 39 NSD‐S, three disruptive, and 248 NSD‐B variants. The NSD‐S and disruptive sets of variants (Table 1, Fig. 1d) spanning 42 genes were private to individual pedigrees (~5 variants per pedigree). Twelve of these were novel (not reported in dbSNP or other published databases) and the remaining were noted in very low frequencies (<1e −07 to 7.8e‐03) in ExAC‐SAS. None of the variants prioritized were present in 33 healthy Indian control samples (http://indexdb.ncbs.res.in). Nine (21.4%) of the 42 variants were found in genes that have been reported in Mendelian syndromes with early onset neurodevelopmental features, such as infantile epilepsy, intellectual disability, and structural brain abnormalities. Seven (16.67%) of these gene‐phenotype relationships were reported in the Online Mendelian Inheritance in Man (OMIM)35 and the remaining two were noted in the MedGen (NCBI) and ClinVar36 databases. This was significantly higher in comparison to a background list of 1310 out of 15 857 (8.26%) such genes (Appendix S1) listed in the OMIM database (P = 0.039, odds ratio [OR] = 2.423, confidence interval [CI] = 1.07–5.513, Fisher's exact test) while not accounting for potential gene length bias. Some of these variants were observed in close proximity to reported ‘pathogenic’ mutation of the relevant Mendelian syndrome and/or in highly conserved regions (Table 2(a)). Two of these nine variants, one each on the GRM1 gene (chr6:146351218, GRCh37) and NIPBL gene (chr5:37010263, GRCh37), were novel. Pathogenic mutations on the GRM1 gene, coding for metabotropic glutamate receptor 1 (mGluR1) result in autosomal dominant (type 44) (OMIM:617691) and recessive (type 13) (OMIM:614831) forms of spinocerebellar ataxia, both of which are characterized by early age of onset and associated intellectual disability. Missense variants have been identified spanning the entire exome of this gene in individuals and families with SCZ and other neuropsychiatric syndromes.37 Mutations in the NIPBL gene, coding for Cohesin Loading Factor involved cortical neuronal migration,38 cause Cornelia de Lange syndrome 1. The novel missense variant identified in the pedigree G (chr5:37010263), segregating with BD would result in substitution of polar amino acid glutamine by a hydrophobic amino acid proline. A non‐sense mutation at the same codon (rs797045760) is reported to be pathogenic of Cornelia de Lange syndrome 1 (ClinVarSCV000248215.1).
Table 1.
List of novel or rare variants prioritized by non‐synonymous damaging strict and disruptive definition
| Gene symbol | rsID/novel | chr:location | Transcript | Exon | Variant | Amino acid change | ExAC_SAS |
|---|---|---|---|---|---|---|---|
| LRRC8B | NOVEL | chr1:90049348 | NM_015350 | Exon5 | c.A1139C | p.Y380S | |
| GRM1 | NOVEL | chr6:146351218 | NM_001278064 | Exon1 | c.A565G | p.S189G | |
| SETD6 | NOVEL | chr16:58552094 | NM_001160305 | Exon6 | c.C932G | p.A311G | |
| SYF2 | NOVEL | chr1:25555567 | NM_015484 | Exon3 | c.A180C | p.K60N | |
| RAB3IL1 | NOVEL | chr11:61675047 | NM_001271686 | Exon3 | c.G491A | p.S164N | |
| BCDIN3D | NOVEL | chr12:50236792 | NM_181708 | Exon1 | c.G79A | p.G27S | |
| NDRG3 | NOVEL | chr20:35317139 | NM_022477 | Exon3 | c.G106T | p.G36C | |
| PARP14 | NOVEL | chr3:122423522 | NM_017554 | Exon8 | c.G3467A | p.S1156N | |
| NIPBL | NOVEL | chr5:37010263 | NM_015384 | Exon21 | c.A4496C | p.Q1499P | |
| SCUBE3 | NOVEL | chr6:35211460 | NM_001303136 | Exon16 | c.C1996T | p.L666F | |
| NBPF11 | NOVEL | chr1:147599423 | Splicing | ||||
| CKMT2 | NOVEL | chr5:80550306 | Splicing | ||||
| KRT85 | rs112554450 | chr12:52758810 | NM_002283 | Exon2 | c.G565A | p.D189N | 6.000E‐03 |
| NRG2 | rs148371256 | chr5:139231286 | NM_001184935 | Exon7 | c.C1477T | p.R493W | 5.000E‐04 |
| MDN1 | rs148868949 | chr6:90397121 | NM_014611 | Exon68 | c.C11392T | p.R3798W | 3.000E‐03 |
| MYO1A | rs151269703 | chr12:57431355 | NM_005379 | Exon19 | c.A2032T | p.I678F | 5.500E‐03 |
| EFHC1 | rs1570624 | chr6:52319050 | NM_018100 | Exon5 | c.G881A | p.R294H | 5.100E‐03 |
| CNGB1 | rs192843629 | chr16:57950041 | NM_001286130 | Exon22 | c.C2191T | p.R731C | 7.000E‐04 |
| PCCB | rs371155999 | chr3:136002730 | NM_000532 | Exon6 | c.C595T | p.P199S | 7.100E‐03 |
| TRMT44 | rs373816157 | chr4:8467199 | NM_152544 | Exon8 | c.C1405T | p.R469W | 0.000E+00 |
| CLUAP1 | rs531380218 | chr16:3558347 | NM_015041 | Exon4 | c.C278T | p.A93V | 9.000E‐04 |
| GOLM1 | rs534059912 | chr9:88661389 | NM_016548 | Exon5 | c.G463A | p.D155N | 3.700E‐03 |
| LGALS12 | rs534811017 | chr11:63277314 | NM_001142537 | Exon3 | c.C320T | p.T107M | 2.200E‐03 |
| ADPRH | rs547308034 | chr3:119301144 | NM_001291949 | Exon2 | c.T128C | p.L43S | 7.800E‐03 |
| FAM208B | rs548531206 | chr10:5789582 | NM_017782 | Exon15 | c.T4198C | p.S1400P | 4.000E‐03 |
| PM20D1 | rs553380022 | chr1:205809408 | NM_152491 | Exon10 | c.G1088A | p.R363Q | 2.700E‐03 |
| CD1D | rs569233577 | chr1:158152752 | NM_001766 | Exon5 | c.C692G | p.P231R | 1.400E‐03 |
| PLXND1 | rs569306898 | chr3:129279222 | NM_015103 | Exon31 | c.G5084C | p.R1695P | 6.124E‐05 |
| WDFY4 | rs571808731 | chr10:50030541 | NM_020945 | Exon35 | c.C5941A | p.P1981T | 3.100E‐03 |
| CC2D2A | rs574421639 | chr4:15559035 | NM_001080522 | Exon22 | c.A2734G | p.R912G | 1.300E‐03 |
| ANLN | rs575071809 | chr7:36435984 | NM_001284301 | Exon2 | c.C128T | p.P43L | 1.800E‐03 |
| PARVB | rs575240566 | chr22:44528830 | NM_001243385 | Exon6 | c.C463A | p.H155N | 1.000E‐04 |
| DOCK5 | rs61732769 | chr8:25174610 | NM_024940 | Exon14 | c.C1406T | p.T469M | 4.300E‐03 |
| KIF7 | rs749711306 | chr15:90176400 | NM_198525 | Exon13 | c.G2690C | p.G897A | 6.478E‐05 |
| C20orf194 | rs750188084 | chr20:3251118 | NM_001009984 | Exon30 | c.A2741G | p.N914S | 6.063E‐05 |
| TCEA3 | rs753347636 | chr1:23720470 | NM_003196 | Exon8 | c.C721T | p.R241C | 6.058E‐05 |
| ARHGEF40 | rs756016433 | chr14:21553914 | NM_001278529 | Exon19 | c.C1885T | p.R629W | 0.000E+00 |
| PLB1 | rs760022335 | chr2:28814039 | Splicing | 0.0004 | |||
| SCN3A | rs775711350 | chr2:166032822 | NM_001081676 | Exon3 | c.G83A | p.R28H | 0.000E+00 |
| INPP5A | rs775793924 | chr10:134521844 | NM_005539 | Exon7 | c.C502T | p.R168W | 6.083E‐05 |
| DENND5A | rs779817963 | chr11:9171664 | NM_001243254 | Exon15 | c.A2699G | p.H900R | 6.132E‐05 |
| COL4A5 | rs78972735 | chrX:107865996 | NM_000495 | Exon33 | c.G2858T | p.G953V | 6.900E‐03 |
Chr:location (chromosomal location); ExAC_SAS (variant allele frequency in ExAC south Asian sample).
Table 2.
Disease relevance of the genes harboring prioritized variants
| (a) Genes implicated in a Mendelian syndrome | |||
|---|---|---|---|
| Gene symbol | Name | Mendelian disease | Selected gene functions |
| GRM1 † | Glutamate metabotropic receptor 1 | Spinocerebellar ataxia AR 13 (MIM:617691) and SCA 44 (MIM:614831) | GO:0007216~G‐protein coupled glutamate receptor signaling pathway; GO:0007268~chemical synaptic transmission |
| EFHC1 | EF‐hand domain containing 1 | Myoclonic epilepsy, juvenile, susceptibility to, 1 (MIM:254770) | GO:0021795~cerebral cortex cell migration |
| DENND5A | DENN domain containing 5A | Epileptic encephalopathy, early infantile, 49 (MIM:617281) | GO:0043547~positive regulation of GTPase activity; GO:0070588~calcium ion transmembrane transport |
| KIF7 | Kinesin family member 7 | Acrocallosal syndrome, Joubert syndrome 12 (MIM:200990) | GO:0007018~microtubule‐based movement; GO:0045879~negative regulation of smoothened signaling pathway |
| SCN3A | Sodium voltage‐gated channel alpha subunit 3 | Cryptogeneicpaediatric partial epilepsy (Medgen CN240377) | GO:0019228~neuronal action potential; GO:0060078~regulation of postsynaptic membrane potential |
| PCCB | Propionyl‐CoA carboxylase beta subunit | Propionicacidemia (MIM:606054) | GO:0006633~fatty acid biosynthetic process |
| NIPBL † | NIPBL, cohesin loading factor | Cornelia de Lange syndrome 1(MIM:122470) | GO:0007420~brain development; GO:0045995~regulation of embryonic development |
| CLUAP1 | Clusterin‐associated protein 1 | Oculoectodermal syndrome, Joubert syndrome (ClinVar) | GO:0001843~neural tube closure; GO:0021508~floor plate formation |
| CC2D2A | Coiled‐coil and C2 domain containing 2A | COACH syndrome (MIM:216360), Joubert syndrome 9 (MIM:612285), Meckel syndrome 6 (MIM:612284) | GO:1990403~embryonic brain development; GO:0001843~neural tube closure |
| (b) Genes implicated in a human polygenic phenotype | |||
|---|---|---|---|
| Gene symbol | Name | Phenotypes and evidence | Selected gene functions |
| NRG2 | Neuregulin 2 | Schizophrenia gamma band oscillation – GWAS (suggestive)39 | GO:0038128~ERBB2 signaling pathway; GO:0014066~regulation of phosphatidylinositol 3‐kinase signaling |
| GOLM1 | Golgi membrane protein 1 | Alzheimer's dementia – GWAS43 | GO:0006997~nucleus organization |
| INPP5A | Inositol polyphosphate‐5‐phosphatase A | Cognitive function in older adults – EWAS46; ataxia and cerebellar degeneration – animal model51 | GO:0048016~inositol phosphate‐mediated signaling |
| MDN1 | Midasin AAA ATPase 1 | Bipolar disorder – Exome sequencing44 | GO:0000027~ribosomal large subunit assembly |
| DOCK5 | Dedicator of Cytokinesis 5 | Familial Parkinson's disease – CNV analysis; DOCK family proteins in multiple neuropsychiatric phenotypes50 | GO:0007264~small GTPase mediated signal transduction; GO:1900026~positive regulation of substrate adhesion‐dependent cell spreading |
| PARP14 | Poly polymerase family member 14 | PTSD, ADHD, MDD – Genome wide transcriptome42 | GO:0006355~regulation of transcription |
| TRMT44 | tRNA methyltransferase 44 homolog | Familial epilepsy – resequencing of linkage region45 | GO:0030488~tRNA methylation |
| PM20D1 | Peptidase M20 domain containing 1 | Parkinson's disease – GWAS49 | GO:1901215~negative regulation of neuron death |
| WDFY4 | WDFY family member 4 | Bipolar Disorder – GWAS (nominal)47 | GO:0016021~integral component of membrane |
| PARVB | Parvin beta | Schizophrenia – microRNA interaction48 | GO:0007155~cell adhesion; GO:0031532~actin cytoskeleton reorganization |
| NBPF11 | Neuroblastoma breakpoint family member 11 | Schizophrenia – CNV analysis case–control52 | No BP annotation |
| (c) Genes putatively significant to neurobiology | |||
|---|---|---|---|
| Gene symbol | Name | Evidence in brain biology or expression (including only animal model evidence) | Selected gene functions |
| PLXND1 | Plexin D1 | Neocortical synapse formation58 | GO:0007416~synapse assembly; GO:0048841~regulation of axon extension involved in axon guidance |
| SETD6 | SET domain containing 6 | Preliminary evidence in memory consolidation through epigenetic regulation56 | GO:0048863~stem cell differentiation; GO:0032088~negative regulation of NF‐kappaB transcription factor activity |
| NDRG3 | NDRG family member 3 | Highest tissue expression in cerebral cortex and cerebellum54 | GO:0007165~signal transduction; GO:0030154~cell differentiation |
| SCUBE3 | Signal peptide, CUB domain and EGF like domain containing 3 | Mouse knock out model – neurological behavioral phenotype55 | GO:0051260~protein homooligomerization; GO:0051291~protein heterooligomerization |
| CNGB1 | Cyclic nucleotide gated channel beta 1 | Upregulated in rat models for cognitive deficits53 | GO:0051480~regulation of cytosolic calcium ion concentration; GO:0033365~protein localization to organelle |
| LRRC8B | Leucine rich repeat containing 8 family member B | Involved in transport of Glutamate, GABA and D‐Serine57; highest tissue expression in brain54 | GO:0098656~anion transmembrane transport |
| C20orf194 | Chromosome 20 open reading frame 194 | Highest tissue expression in brain54 | |
| ANLN | Anillin actin binding protein | Highest tissue expression in brain54 | GO:0098609~cell–cell adhesion |
Variant in close proximity to a pathogenic mutation for a Mendelian syndrome.
CNV, copy number variation; EWAS, epigenome wide association study; GO, gene ontology; GWAS, genome‐wide association studies; MIM, Mendelian Inheritance in Man.
Ten other genes that harbored prioritized variants have been implicated in neuropsychiatric syndromes. We identified a variant (rs148371256) in the NRG2 gene (Neuregulin 2) that was earlier reported to be associated with gamma band oscillations in SCZ with suggestive genome‐wide significance.39 The encoded protein neuregulin‐2 has been shown to be critical for the formation and maturation of GABAergic synapses40 and its ablation results in dopamine dysregulation.41 Another novel variant (chr3:122423522, GRCh37) was identified in the PARP14 gene (Poly ADP ribose polymerase 14), and the gene has been implicated in post‐traumatic stress disorder (PTSD), major depressive disorder (MDD), and attention deficit hyperactivity disorder (ADHD).42 We also noted a variant (rs534059912) in the GOLM1 gene (Golgi membrane protein 1), which was earlier reported in sporadic Alzheimer's dementia (AD) to influence the pre‐frontal cortical volume.43 A list of these 10 genes, evidence for disease association, and gene ontology descriptions are presented in Table 2(b).44, 45, 46, 47, 48, 49, 50, 51, 52
Of the remaining genes, there were several with a plausible role in the biology of SMI, but not thus far implicated in any disease phenotype. These genes, with the ontology descriptions and plausible biological implications, are provided in Table 2(c).53, 54, 55, 56, 57, 58
Enrichment of coding variants with plausible functional role in SMI
The NSD‐B set consisted of 248 variants; of these, except for rs570064523 in the PCSK1 gene, which was identified in cases from two families (G and H), no other overlap at the level of family was noted for the remaining 247 variants (Appendix S2, Table S3). In the ‘protein domains’ category tested using the Interpro database as the source, the term ‘epidermal growth factor like domain’ showed a nominally significant enrichment with P = 0.0013, Benjamini–Hochberg false discovery rate corrected P = 0.073. Twelve genes that were enriched for this domain included the NRG2 and SCUBE3 genes, which were also categorized in the NSD‐S set along with the NOTCH1, JAG1 and WIF1 genes, which form critical nodes in the notch signaling pathway implicated in neurodevelopment and embryogenesis (Appendix S2, Table S4).59 There was no statistically significant enrichment in any of the remaining categories tested with this in silico approach.
Discussion
The results of our study highlight the usefulness of WES in multiplex families with SMI to identify rare and novel variants that may contribute to the susceptibility to common polygenic syndromes. Many of these variants prioritized by NSD‐S, and presumed to be disruptive, map to genes that have been previously reported in GWAS, candidate gene association, post‐mortem expression, or animal model studies of SMI. In addition, consistent with the WES approach, we identify variants in genes hitherto not reported in the context of an SMI, but that could potentially contribute to disease biology.
The segregation of rare and deleterious variants in Mendelian disease genes with a neuropsychiatric phenotype is in keeping with some recent observations. Studies have shown that heterozygous carriers of Mendelian disease mutations are at increased risk for specific common diseases.60 While Mendelian forms of common, complex traits, such as Alzheimer's disease, hypertension, hypercholesterolemia, and hypertriglyceridemia, have long been attributed to rare causal variants in single genes, population‐based GWAS in these traits have often implicated genes that also cause single gene disorders.60 More recently, using electronic health record data, the disease‐relevant phenotypic burden of rare variants in Mendelian genes, thus far not characterized as ‘pathogenic,’ has been demonstrated across diverse phenotypes.61
We explored the clinical significance of nine variants in Mendelian genes in the ClinVar database, a publicly available archive of human phenotype‐variation relations.36 None of these variants was annotated as ‘pathogenic’ or ‘likely pathogenic’ in the database for the corresponding Mendelian phenotype. As a corollary, none of the families had any identified or suspected case of a severe neurodevelopmental syndrome. However, the predicted deleteriousness by in silico algorithms, a very low prevalence in the population, physical proximity to known pathogenic mutations, and the reported physiological gene function suggest a plausible role for these variants in the etiology of SMI. The impact of these variants in cellular and/or animal models needs to be examined to validate these observations and to establish their causal role in SMI. Interestingly, an earlier WES study in families with BD also reported variants in genes of monogenic syndromes: holoprosencephaly and progressive myoclonic epilepsy.13
We detected rare variants in 10 additional genes that have been noted in earlier studies to contribute to the risk for polygenic syndromes, such as SCZ, BD, autism, MDD, ADHD, PTSD, AD, and Parkinson's disease. This finding is congruent with the evolving concept of shared molecular neuropathology across SMI.19 These, along with other identified genes known to be involved in neurodevelopmental processes (e.g., PLXND1) or known to have manifold higher brain expression (e.g., ANLN, LRRC8B) are potential targets to be examined in future studies of SMI. Lastly, of the 12 genes encoding highly conserved epidermal growth factor‐like domains and showing nominally significant enrichment to this domain, many encode for proteins that play critical roles during embryogenesis and neurodevelopment.59
Certain limitations are to be considered while interpreting the results of this study. The relatively small control set sequenced in our study precluded statistical association testing at the level of a variant or a gene. It has been estimated that rare variant association testing at gene level using case–control samples would require sample sizes greater than 20 000 individuals.62 As an alternative, we considered the minor allele frequency of the variant in ExAC South Asian samples in the prioritization approach, and many of the identified variants were noted to be extremely rare. Second, although we sampled a nearly equal number of affected persons from each family, the relationships within pedigrees were not uniform, potentially adding heterogeneity to the number of identified variants. Thus, we prioritized variants with complete sharing allowing for one missing genotype. This resulted in identification of some variants that were not fully penetrant. Third, like the previous studies of WES in SCZ and BD, we have relied on in silico predictions to infer the deleteriousness of a variant and have considered those predicted by five algorithms as the primary variants of interest. Supporting this approach, a recent analysis noted that the strength of disease association for a non‐synonymous variant increased with the greater number of deleterious predictions in silico. 63 Fourth, inherent to the prioritization criteria of rarity, deleteriousness and segregation, the NSD‐S and disruptive variant set presented above would explain only a part of an individual's liability to disease. The results of this analysis represent the shared familial risk for SMI, private to each pedigree, determined by variants of possible major/moderate effect. Lastly, we have not been able to sample all affected individuals from each multiplex pedigree. Among the unaffected individuals, we have been able to sample one to two representative individuals from five of the pedigrees. Thus, the prioritized variants might represent only a part of the shared genetic risk within each pedigree.
Using WES data in multiplex families with SMI, we find evidence that suggests intersections in the molecular pathways leading to the expression of polygenic SMI and Mendelian neuropsychiatric syndromes. The patient‐derived neural stem cell lines being developed as part of the program21 will be useful to explore the functional significance of the identified variants accounting for ‘modifier genetic background’,64 and to characterize mechanisms that underlie the observed genotype–phenotype correlates.
Conclusions
NGS approaches in a family‐based study design are useful to identify novel and rare variants in genes potentially relevant to complex disorders, such as SMI. The study further provides an independent validation for the phenotypic burden of rare deleterious variants in Mendelian disease genes that segregate privately in multiplex pedigrees with SCZ and BD. Our findings support the role of heterogeneity and pleiotropy in the genetic architecture of SMI encompassing a spectrum of neurodevelopmental and degenerative phenotypes.
Disclosure statement
The authors declare that they have no conflicts of interest.
Author contributions
S.G. analyzed the data and wrote the manuscript; H.A.P. built the VarPrio algorithm and performed variant prioritization and secondary analysis; R.K.N. and M.S. performed the detailed clinical assessments of the study participants under the supervision of B.V.; R.P.M. performed whole‐exome sequence data mining; O.M. supervised sequencing data generation, analysis of the results, and manuscript preparation. S.J. and M.S. provided vital inputs to data analysis and manuscript preparation. The study was conceived by the ADBS Consortium. All authors took part in editing the manuscript and approved the final version.
Supporting information
Appendix S1. Supplemental material for Mendelian disease genes in familial SMI
Appendix S2. Microsoft Excel file containing supplementary tables
Acknowledgments
The authors are grateful to all the patients, their family members, and healthy volunteers who participated in the study. Financial support for the study was provided by: the Department of Biotechnology funded grants, “Targeted generation and interrogation of cellular models and networks in neuro‐psychiatric disorders using candidate genes” (BT/01/CEIB/11/VI/11/2012) and “Accelerating program for discovery in brain disorders using stem cells” (BT/PR17316/MED/31/326/2015) (ADBS); Pratiksha Trust; and the Institute of Stem Cells and Regenerative Medicine (InStem), Bengaluru, India. The authors would like to thank the sequencing core facility at the Institute of Genomics and Integrative Biology (IGIB), Delhi (Dr Faruq Mohammed) and the National Centre for Biological Sciences (NCBS), Bengaluru (Dr Awadhesh Pandit) for sample processing and WES data generation. The authors would like to thank Manasa K.P., Anand Ganapathy Subramaniam, Soham Deepak Jagtap, Geetanjali Murari, Srividya Shetty, and Surya Prakash M. from NIMHANS and Vidhya Varadharajan, Priyanka Bhatia, Shubhra Acharya, and Batul Yusuf from InStem for their technical support during initial raw data curation. The authors would also like to thank all investigators of the ADBS Consortia for providing valuable inputs to the manuscript and having final approval of the manuscript. Suhas Ganesh is currently affiliated with and supported by the Schizophrenia Neuropharmacology Research Group at Yale University.
Contributor Information
Odity Mukherjee, Email: omukherjee@ncbs.res.in.
The ADBS Consortium:
Biju Viswanath, Naren P. Rao, Janardhanan C. Narayanaswamy, Palanimuthu T Sivakumar, Arun Kandaswamy, Muralidharan Kesavan, UrvakhshMeherwan Mehta, Ganesan Venkatasubramanian, John P. John, Meera Purushottam, Ramakrishnan Kannan, Bhupesh Mehta, Thennarasu Kandavel, B Binukumar, Jitender Saini, Deepak Jayarajan, A Shyamsundar, Jagadisha Thirthalli, Prabha S. Chandra, Bangalore N. Gangadhar, Pratima Murthy, Vivek Benegal, Mathew Varghese, Janardhan YC Reddy, Sanjeev Jain, Mitradas M. Panicker, Upinder S Bhalla, Padinjat Raghu, Odity Mukherjee, Sumantra Chattarji, and Mahendra Rao
References
- 1. Merikangas KR, Jin R, He J‐P et al Prevalence and correlates of bipolar spectrum disorder in the World Mental Health Survey Initiative. Arch. Gen. Psychiatry 2011; 68: 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Saha S, Chant D, Welham J, McGrath J. A systematic review of the prevalence of schizophrenia. PLoS Med. 2005; 2: e141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sullivan PF, Kendler KS, Neale MC. Schizophrenia as a complex trait: Evidence from a meta‐analysis of twin studies. Arch. Gen. Psychiatry 2003; 60: 1187–1192. [DOI] [PubMed] [Google Scholar]
- 4. Barnett JH, Smoller JW. The genetics of bipolar disorder. Neuroscience 2009; 164: 331–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lichtenstein P, Yip BH, Björk C et al Common genetic determinants of schizophrenia and bipolar disorder in Swedish families: A population‐based study. Lancet 2009; 373: 234–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lin PI, Mitchell BD. Approaches for unraveling the joint genetic determinants of schizophrenia and bipolar disorder. Schizophr. Bull. 2007; 34: 791–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cross‐Disorder Group of the Psychiatric Genomics Consortium , Lee SH, Ripke S et al Genetic relationship between five psychiatric disorders estimated from genome‐wide SNPs. Nat. Genet. 2013; 45: 984–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kerner B. Toward a deeper understanding of the genetics of bipolar disorder. Front. Psych. 2015; 6: 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Boyle EA, Li YI, Pritchard JK. An expanded view of complex traits: From polygenic to omnigenic. Cell 2017; 169: 1177–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chapman NH, Nato AQ, Bernier R et al Whole exome sequencing in extended families with autism spectrum disorder implicates four candidate genes. Hum. Genet. 2015; 134: 1055–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kato T. Whole genome/exome sequencing in mood and psychotic disorders: Whole genome/exome in mental disorders. Psychiatry Clin. Neurosci. 2015; 69: 65–76. [DOI] [PubMed] [Google Scholar]
- 12. Goes FS, Pirooznia M, Parla JS et al Exome sequencing of familial bipolar disorder. JAMA Psychiatry 2016; 73: 590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rao AR, Yourshaw M, Christensen B, Nelson SF, Kerner B. Rare deleterious mutations are associated with disease in bipolar disorder families. Mol. Psychiatry 2017; 22: 1009–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Timms AE, Dorschner MO, Wechsler J et al Support for the N‐methyl‐D‐aspartate receptor hypofunction hypothesis of schizophrenia from exome sequencing in multiplex families. JAMA Psychiatry 2013; 70: 582. [DOI] [PubMed] [Google Scholar]
- 15. DeLisi LE. A case for returning to multiplex families for further understanding the heritability of schizophrenia: A psychiatrist's perspective. Mol. Neuropsychiatry 2016; 2: 15–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Glahn DC, Nimgaonkar VL, Raventos H et al Rediscovering the value of families for psychiatric genetics research. Mol. Psychiatry 2018. 10.1038/s41380-018-0073-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cristino AS, Williams SM, Hawi Z et al Neurodevelopmental and neuropsychiatric disorders represent an interconnected molecular system. Mol. Psychiatry 2014; 19: 294–301. [DOI] [PubMed] [Google Scholar]
- 18. O'Dushlaine C, Rossin L, Lee PH et al Psychiatric genome‐wide association study analyses implicate neuronal, immune and histone pathways. Nat. Neurosci. 2015; 18: 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gandal MJ, Haney JR, Parikshak NN et al Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science 2018; 359: 693–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brainstorm Consortium , Anttila V, Bulik‐Sullivan B et al Analysis of shared heritability in common disorders of the brain. Science 2018; 360: eaap8757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Viswanath B, Rao NP, Narayanaswamy JC et al Discovery biology of neuropsychiatric syndromes (DBNS): A center for integrating clinical medicine and basic science. BMC Psychiatry 2018; 18: 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mukherjee O, Meera P, Ghosh S et al Evidence of linkage and association on 18p11.2 for psychosis. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2006; 141B: 868–873. [DOI] [PubMed] [Google Scholar]
- 23. Ram Murthy A, Purushottam M, Kiran Kumar HBG et al Gender‐specific association of TSNAX/DISC1 locus for schizophrenia and bipolar affective disorder in south Indian population. J. Hum. Genet. 2012; 57: 523–530. [DOI] [PubMed] [Google Scholar]
- 24. Li H, Durbin R. Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics 2009; 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Koboldt DC, Larson DE, Wilson RK. Using VarScan 2 for germline variant calling and somatic mutation detection In: Bateman A, Pearson WR, Stein LD, Stormo GD, Yates JR. (eds). Current Protocols in Bioinformatics. John Wiley & Sons, Hoboken, NJ, 2013; 15.4.1–15.4.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang K, Li M, Hakonarson H. ANNOVAR: Functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Res. 2010; 38: e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non‐synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009; 4: 1073–1081. [DOI] [PubMed] [Google Scholar]
- 28. Adzhubei IA, Schmidt S, Peshkin L et al A method and server for predicting damaging missense mutations. Nat. Methods 2010; 7: 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: Mutation prediction for the deep‐sequencing age. Nat. Methods 2014; 11: 361–362. [DOI] [PubMed] [Google Scholar]
- 30. Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011; 39: e118–e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chun S, Fay JC. Identification of deleterious mutations within three human genomes. Genome Res. 2009; 19: 1553–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lek M, Karczewski KJ, Minikel EV et al Analysis of protein‐coding genetic variation in 60,706 humans. Nature 2016; 536: 285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009; 4: 44–57. [DOI] [PubMed] [Google Scholar]
- 34. Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009; 37: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. OMIM . Online Mendelian Inheritance in Man. McKusick‐Nathans Institute of Genetic Medicine, Johns Hopkins University, Baltimore, MD, 2018. [Google Scholar]
- 36. Landrum MJ, Lee JM, Benson M et al ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018; 46: D1062–D1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ayoub MA, Angelicheva D, Vile D et al Deleterious GRM1 mutations in schizophrenia. PLoS ONE 2012; 7: e32849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. van den Berg DLC, Azzarelli R, Oishi K et al Nipbl interacts with Zfp609 and the integrator complex to regulate cortical neuron migration. Neuron 2017; 93: 348–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Konte B, Leicht G, Giegling I et al A genome‐wide association study of early gamma‐band response in a schizophrenia case–control sample. World J. Biol. Psychiatry 2017. 10.1080/15622975.2017.1366054 [DOI] [PubMed] [Google Scholar]
- 40. Lee K‐H, Lee H, Yang CH et al Bidirectional signaling of neuregulin‐2 mediates formation of GABAergic synapses and maturation of glutamatergic synapses in newborn granule cells of postnatal hippocampus. J. Neurosci. 2015; 35: 16479–16493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yan L, Shamir A, Skirzewski M et al Neuregulin‐2 ablation results in dopamine dysregulation and severe behavioral phenotypes relevant to psychiatric disorders. Mol. Psychiatry 2018; 23: 1233–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. de Jong S, Newhouse SJ, Patel H et al Immune signatures and disorder‐specific patterns in a cross‐disorder gene expression analysis. Br. J. Psychiatry 2016; 209: 202–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Inkster B, Rao AW, Ridler K et al Genetic variation in GOLM1 and prefrontal cortical volume in Alzheimer's disease. Neurobiol. Aging 2012; 33: 457–465. [DOI] [PubMed] [Google Scholar]
- 44. Kataoka M, Matoba N, Sawada T et al Exome sequencing for bipolar disorder points to roles of de novo loss‐of‐function and protein‐altering mutations. Mol. Psychiatry 2016; 21: 885–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Leschziner GD, Coffey AJ, Andrew T et al Q8IYL2 is a candidate gene for the familial epilepsy syndrome of partial epilepsy with pericentral spikes (PEPS). Epilepsy Res. 2011; 96: 109–115. [DOI] [PubMed] [Google Scholar]
- 46. Marioni RE, McRae AF, Bressler J et al Meta‐analysis of epigenome‐wide association studies of cognitive abilities. Mol. Psychiatry 2018. 10.1038/s41380-017-0008-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nurnberger JI Jr, Koller DL, Jung J et al Identification of pathways for bipolar disorder: A meta‐analysis. JAMA Psychiatry 2014; 71: 657–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Olde Loohuis NF, Nadif Kasri N, Glennon JC et al The schizophrenia risk gene MIR137 acts as a hippocampal gene network node orchestrating the expression of genes relevant to nervous system development and function. Prog. Neuropsychopharmacol. Biol. Psychiatry 2017; 73: 109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Satake W, Nakabayashi Y, Mizuta I et al Genome‐wide association study identifies common variants at four loci as genetic risk factors for Parkinson's disease. Nat. Genet. 2009; 41: 1303–1307. [DOI] [PubMed] [Google Scholar]
- 50. Shi L. Dock protein family in brain development and neurological disease. Commun. Integrative Biol. 2013; 6: e26839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yang AW, Sachs AJ, Nystuen AM. Deletion of Inpp5a causes ataxia and cerebellar degeneration in mice. Neurogenetics 2015; 16: 277–285. [DOI] [PubMed] [Google Scholar]
- 52. Sikela JM, Searles Quick VB. Genomic trade‐offs: Are autism and schizophrenia the steep price of the human brain? Hum. Genet. 2018; 137: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Buechel HM, Popovic J, Searcy JL, Porter NM, Thibault O, Blalock EM. Deep sleep and parietal cortex gene expression changes are related to cognitive deficits with age. PLoS ONE 2011; 6: e18387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Fagerberg L, Hallstrom BM, Oksvold P et al Analysis of the human tissue‐specific expression by genome‐wide integration of transcriptomics and antibody‐based proteomics. Mol. Cell. Proteomics 2014; 13: 397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Fuchs H, Sabrautzki S, Przemeck GK et al The first Scube3 mutant mouse line with pleiotropic phenotypic alterations. G3 (Bethesda) 2016; 6: 4035–4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Levy D, Kuo AJ, Chang Y et al Lysine methylation of the NF‐κB subunit RelA by SETD6 couples activity of the histone methyltransferase GLP at chromatin to tonic repression of NF‐κB signaling. Nat. Immunol. 2010; 12: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lutter D, Ullrich F, Lueck JC, Kempa S, Jentsch TJ. Selective transport of neurotransmitters and modulators by distinct volume‐regulated LRRC8 anion channels. J. Cell Sci. 2017; 130: 1122–1133. [DOI] [PubMed] [Google Scholar]
- 58. Wang F, Eagleson KL, Levitt P. Positive regulation of neocortical synapse formation by the Plexin‐D1 receptor. Brain Res. 2015; 1616: 157–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lasky JL, Wu H. Notch signaling, brain development, and human disease. Pediatr. Res. 2005; 57: 104R–109R. [DOI] [PubMed] [Google Scholar]
- 60. Lupski James R, Belmont JW, Boerwinkle E, Gibbs RA. Clan genomics and the complex architecture of human disease. Cell 2011; 147: 32–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bastarache L, Hughey JJ, Hebbring S et al Phenotype risk scores identify patients with unrecognized Mendelian disease patterns. Science 2018; 359: 1233–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sanders SJ, Neale BM, Huang H et al Whole genome sequencing in psychiatric disorders: The WGSPD Consortium. Nat. Neurosci. 2017; 20: 1661–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ganna A, Satterstrom FK, Zekavat SM et al Quantifying the impact of rare and ultra‐rare coding variation across the phenotypic spectrum. Am. J. Hum. Genet. 2018; 102: 1204–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Riordan JD, Nadeau JH. From peas to disease: Modifier genes, network resilience, and the genetics of health. Am. J. Human Genetics 2017; 101: 177–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Supplemental material for Mendelian disease genes in familial SMI
Appendix S2. Microsoft Excel file containing supplementary tables