

Abstract
Receptor tyrosine kinases (RTKs) are single-pass membrane proteins that control vital cell processes such as cell growth, survival, and differentiation. There is a growing body of evidence that RTKs from different subfamilies can interact and that these diverse interactions can have important biological consequences. However, these heterointeractions are often ignored, and their strengths are unknown. In this work, we studied the heterointeractions of nine RTK pairs, epidermal growth factor receptor (EGFR)–EPH receptor A2 (EPHA2), EGFR–vascular endothelial growth factor receptor 2 (VEGFR2), EPHA2–VEGFR2, EPHA2–fibroblast growth factor receptor 1 (FGFR1), EPHA2–FGFR2, EPHA2–FGFR3, VEGFR2–FGFR1, VEGFR2–FGFR2, and VEGFR2–FGFR3, using a FRET-based method. Surprisingly, we found that RTK heterodimerization and homodimerization strengths can be similar, underscoring the significance of RTK heterointeractions in signaling. We discuss how these heterointeractions can contribute to the complexity of RTK signal transduction, and we highlight the utility of quantitative FRET for probing multiple interactions in the plasma membrane.
Keywords: protein-protein interaction, membrane protein, receptor tyrosine kinase (RTK), thermodynamics, cell signaling, epidermal growth factor receptor (EGFR), EPH receptor A2 (EPHA2), vascular endothelial growth factor receptor 2 (VEGFR2), fibroblast growth factor receptor (FGFR), fluorescence resonance energy transfer (FRET), receptor tyrosine kinase
The 58 human receptor tyrosine kinases (RTKs) are grouped into 20 subfamilies based on the homology of their N-terminal extracellular (EC) domains (1–4). They are single-pass membrane receptors, which are activated upon dimerization and phosphorylation of tyrosines in their C-terminal intracellular (IC) kinase domains (3, 5–8). Adaptor proteins bind to the phosphorylated tyrosines, leading to the initiation of downstream signaling cascades that control cellular processes such as growth, motility, survival, and differentiation (9–14). Moreover, RTKs are often dysregulated in many growth disorders and diseases, such as cancer (15–20).
Whereas RTKs are best known for forming signaling homodimers, they are also known to interact with many other binding partners, including other RTKs from the same subfamily, G protein–coupled receptors, and cell adhesion molecules (21–26). Furthermore, it is becoming clear that they are capable of interacting with RTKs from unrelated subfamilies. A recent review of the literature identified nearly 100 studies that support the idea that RTKs from different subfamilies can interact (27). Despite these studies, heterodimerization across subfamilies has largely been ignored, both in conceptual models of RTK signaling and in the interpretation of RTK signaling data. However, these heterodimers have been shown to play important and diverse roles, such as amplifying or inhibiting downstream signaling (28–34), increasing the signal diversity (35–38), or providing signaling back-up (27).
It is also becoming increasingly apparent that unliganded RTK dimers can form in physiological contexts and can have important effects, especially in disease (39–41). RTK overexpression has been linked to many cancers, as it promotes RTK interactions even in the absence of ligand (17, 18). Interestingly, some of these cancers are associated with not just increased RTK expression, but loss of ligand as well. It can be expected that such conditions (high RTK expression and no ligand) also favor the formation of heterodimers of unrelated RTKs, as these RTKs typically do not share ligands. However, it is currently not clear how likely cross-subfamily heterointeractions are to occur, and it is thus difficult to say how significant a role they play in RTK signaling. Indeed, heterointeraction strengths for RTKs from different subfamilies are unknown, as prior work has had limited quantification capabilities.
In this work, we study the heterointeractions of nine RTK pairs (EGFR–EPHA2, EGFR–VEGFR2, EPHA2–VEGFR2, EPHA2–FGFR1, EPHA2–FGFR2, EPHA2–FGFR3, VEGFR2–FGFR1, VEGFR2–FGFR2, and VEGFR2–FGFR3) using a fluorescence-based method: fully quantified spectral imaging Förster resonance energy transfer (FSI-FRET). The FSI-FRET method reports on the occurrence of specific interactions and yields the strength of the interactions (1). We advance this FRET methodology to also enable the determination of the oligomer size (e.g. dimer, trimer, tetramer) of the heterointeraction. For three of the RTK pairs, we demonstrate that they form dimers in the absence of ligand, and we quantify their heterodimerization strengths. Surprisingly, we find that the heterodimerization and homodimerization strengths are similar. This suggests that the heterodimers are likely to form under physiological and pathological conditions. Furthermore, we show that the presence of ligands modulates the abundance of the heterodimers. This work underscores the significance of heterointeractions in RTK signaling and highlights a method that can be used to study a multitude of interactions in the plasma membrane of live cells.
Results
Heterointeraction models
In most cases, RTKs are thought to signal as homodimers (2, 3). Accordingly, if two different RTKs, X and Y, were to interact, the obvious assumption is that they would form a heterodimer, XY. However, there is no experimental evidence that excludes the possibility of higher-order heterooligomers (e.g. trimers, tetramers) forming. For example, as each RTK explores a monomer-dimer equilibrium, it is possible that a dimer, YY, and a monomer, X, interact to form a trimer, XYY. It is also possible that two dimers interact to form a tetramer, XXYY. These cases are illustrated in Fig. 1. Here, we develop physical-chemical models for these cases, which can be used to identify the model that best describes experimental data.
Figure 1.

RTK models. Shown are cartoon representations of the different RTK interaction models (A–C) and RTK constructs used in the study (D) (not drawn to scale and not meant to represent a mechanism of dimerization (e.g. the TM domains might not be directly interacting)). A, heterodimer model. B, heterotrimer model. C, heterotetramer model. D, fluorescent proteins (a FRET pair) are attached to the C termini of the RTK constructs used in this work.
Heterodimers (Fig. 1A)
The heterodimer case has been considered previously by Del Piccolo et al. (42). Even in this simplest case, three coupled reactions are needed to describe the fact that both X and Y form homodimers, XX and YY, in addition to the heterodimer, XY.
Here, KX and KY are the homodimer association constants, and KXY is the heterodimer association constant. These three association constants can be written in terms of the concentrations of the monomers and dimers.
| (Eq. 1) |
| (Eq. 2) |
| (Eq. 3) |
Assuming that the total concentration of each RTK is constant—an assumption that is valid for the conditions under which the FRET experiments are performed (43)—then simple equations for mass conservation can be written as follows.
| (Eq. 4) |
| (Eq. 5) |
By substituting in the values of KX and KY from Equations 1–2, the total concentrations can be written in terms of the monomer and heterodimer concentrations as follows.
| (Eq. 6) |
| (Eq. 7) |
These are quadratic equations which can be solved for in terms of the monomer concentration.
| (Eq. 8) |
| (Eq. 9) |
Last, the concentration of the heterodimer can be written in terms of the monomer concentrations and the heterodimer association constant by rearranging Equation 3 as follows.
| (Eq. 10) |
By substituting the values of [X] and [Y] from Equations 8 and 9 into Equation 10, we arrive at an equation where KXY and [XY] are defined in terms of [Xtotal], [Ytotal], KX, and KY. As described previously (1, 6, 42, 44), quantitative FRET experiments can directly measure [Xtotal] and [Ytotal], and the FRET efficiency. FRET efficiency is a function of [XY], as shown in Equation 32 (see “Materials and methods”). Accordingly, if KX and KY have been measured previously, then FRET experiments can be used to determine the best-fit KXY. For a detailed description of this fitting process, see Ref. 42.
Heterotrimers (Fig. 1B)
There are two possible heterotrimers, XYY and XXY; however, these are equivalent, as the assignment of X and Y for the two RTKs can be switched. Accordingly, only the case of XYY will be considered here. We begin with defining the three coupled reactions needed to fully describe the formation of the homodimers, XX and YY, and the heterotrimer, XYY:
where KXYY is the heterotrimer association constant. These three association constants can be written in terms of the concentrations of the monomers, homodimers, and heterotrimers:
| (Eq. 11) |
| (Eq. 12) |
| (Eq. 13) |
The equations for mass conservation can be written as follows.
| (Eq. 14) |
| (Eq. 15) |
and these can be rewritten in terms of the association constants.
| (Eq. 16) |
| (Eq. 17) |
These are quadratic equations which can be solved for in terms of [X] and [Y].
| (Eq. 18) |
| (Eq. 19) |
Finally, the concentration of the heterotrimer can be written in terms of the monomer concentrations and the heterotrimer association constant by rearranging Equation 13.
| (Eq. 20) |
By substituting the values of [X] and [Y] from Equations 18 and 19 into Equation 20, we arrive at an equation where KXYY and [XYY] are defined in terms of [Xtotal], [Ytotal], KX, and KY, as in the case of the heterodimer. Analogously to the heterodimer case, the best-fit KXYY can be determined by fitting this model to the experimental FRET data.
Heterotetramers (Fig. 1C)
Here, we consider two dimers, XX and YY, forming a tetramer, XXYY. We begin with defining the three coupled reactions needed to fully describe the formation of the homodimers and the heterotetramer,
where KXXYY is the heterotetramer association constant. These three association constants can be written in terms of the concentrations of the monomers, dimers, and tetramers as follows.
| (Eq. 21) |
| (Eq. 22) |
| (Eq. 23) |
The equations for mass conservation are written as follows,
| (Eq. 24) |
| (Eq. 25) |
and these can be rewritten in terms of the association constants.
| (Eq. 26) |
| (Eq. 27) |
The monomer concentration can be determined by solving these quadratic equations.
| (Eq. 28) |
| (Eq. 29) |
Last, the concentration of the heterotetramer can be written in terms of the monomer concentrations and the heterotetramer association constant by rearranging Equation 23 as follows.
| (Eq. 30) |
By substituting the values of [X] and [Y] from Equations 28 and 29 into Equation 30, we arrive at an equation where KXXYY and [XXYY] are defined in terms of [Xtotal], [Ytotal], KX, and KY. As with the heterodimer and heterotrimer cases, FRET can be used to determine KXXYY and hence heterotetramer stability.
Experimental assessments of heterointeractions
The first RTK pair that we studied was EGFR and EPHA2. EGFR is a member of the ErbB family and is important for cell growth, division, migration, and survival (45–47). It is known to be overexpressed or mutated in many cancers (48, 49). EPHA2 is a member of the EPH family, and it plays a role in neuronal development, cell proliferation, and axon guidance (50–52). Overexpression and mutations of the EPH receptor are generally oncogenic (16, 53), but unliganded EPHA2 dimers have been shown to suppress tumorigenic signaling (54). Both EGFR and EPHA2 can be expressed in the same cells (55–57), and they have been reported to interact, based on fluorescence colocalization and coimmunoprecipitation (58). Moreover, there is a report that increased EPHA2 levels in colorectal cancer correlates with a poor response to the EGFR inhibitor cetuximab (59). Taken together, these studies indicate that the EGFR–EPHA2 interaction could be affecting signaling and could be a potential target in therapeutic strategies. Therefore, we asked whether we could detect the heterointeraction between EGFR and EPHA2 using FRET.
To study heterointeractions using FRET, one RTK must be labeled with a donor fluorophore, and the other RTK must be labeled with an acceptor fluorophore. If the receptors interact, the donor will be brought close to the acceptor, and FRET will occur. In these FRET experiments, HEK 293T cells were transiently co-transfected with plasmids that encode for full-length EGFR labeled with mTurquoise (MT, a FRET donor) and full-length EPHA2 labeled with eYFP (a FRET acceptor). The cells were grown for at least 24 h after transfection and were serum-starved for at least 12 h prior to imaging to ensure that no ligands were present.
FRET experiments were performed using the FSI method, which has been described in detail previously (1). In brief, the cells were imaged using a two-photon microscope. Two scans were taken of each cell: one scan to maximally excite the donor and one scan to maximally excite the acceptor. Fluorescence intensities were converted into concentrations using purified solutions of the fluorophores of known concentrations. The cells were analyzed using MATLAB to determine the 2D concentration of the donor, the 2D concentration of the acceptor, and the apparent FRET efficiency of selected regions of the plasma membrane.
We encountered difficulties in these experiments: whereas the co-transfection was performed with a 2:1 or 3:1 EGFR–MT/EPHA2–YFP ratio of pasmid DNA, all cells had very low EGFR expression. It appeared that EPHA2 expression supressed EGFR expression, such that only a small percentage of the cells that we imaged had quantifiable concentrations of both EGFR and EPHA2 in the plasma membrane. A representative image of such a cell that was selected for analysis is shown in Fig. 2A. For this cellular subset, the experimentally measured (i.e. apparent) FRET efficiencies (Eapp) for the EGFR–EPHA2 pair are shown in Fig. 2B as a function of acceptor concentration (EPHA2–YFP). Each black star represents the measurement for a 2–3-µm stretch of plasma membrane; two such membrane regions were selected per cell. The blue line in Fig. 2B is a previously modeled monomer proximity line (60, 61), which represents the maximum proximity FRET that can occur at a given acceptor concentration. Proximity FRET increases with increasing fluorophore concentration, as it occurs when a donor and acceptor are randomly close to each in the absence of specific interactions.
Figure 2.

Heterointeractions between full-length EGFR and full-length EPHA2. A, representative image of a HEK 293T cell co-transfected with EGFR–MT and EPHA2–eYFP. Regions selected for analysis are shown with the red stars surrounded by blue boxes. B, measured FRET versus acceptor concentration in cells co-expressing EGFR–MT and EPHA2–eYFP. The cells were transfected with varying donor/acceptor ratios, with a total of 1–2 μg of EPHA2 DNA and 2–4 μg of EGFR DNA. Each black star represents the measurement for a 2–3 μm stretch of plasma membrane. Two regions were selected per cell, for a total of 48 data points. The solid blue line is the monomer FRET proximity as a function of acceptor concentration, which represents the maximum FRET that can occur at any given acceptor concentration in the absence of specific interactions (60, 61). Almost all points lie above the proximity line, demonstrating specific interactions. C and D, FRET data for LAT, a monomeric control. HEK 293T cells were transiently transfected with LAT-MT and LAT-eYFP in a 1:3 donor/acceptor ratio with a total of 1–4 μg of DNA. C, the apparent FRET is plotted as a function of LAT-eYFP concentration, along with the monomer FRET proximity (solid blue line). The FRET data (a total of 288 points) effectively straddle the proximity line, providing an experimental validation for the proximity FRET prediction. D, distribution of FRET efficiencies after the FRET has been corrected for proximity. Red line, Gaussian fit. The mean is −0.0009, and the S.E. is 0.005.
We sought to experimentally validate the predicted monomer proximity FRET using a monomer control protein. To do this, we performed FRET experiments with linker for activation of T cells (LAT). LAT is a single-pass membrane protein that plays a key role in T-cell receptor downstream signaling (62–64), and it has been used as a monomer control in the literature (65). HEK 293T cells were transfected with versions of LAT that have been labeled by either MT or eYFP on the C terminus via a flexible linker. As seen in the plot of apparent FRET versus acceptor concentration in Fig. 2C, the LAT FRET effectively straddles the monomer proximity line, confirming that the modeled monomer proximity line accurately describes the proximity FRET of a monomer. Further evidence is seen in Fig. 2D, where the FRET data in Fig. 2C have been corrected for the monomer proximity FRET contribution, histogrammed (blue bars), and fit to a Gaussian (red line). As expected, the result is effectively random noise (i.e. a Gaussian centered around zero).
As can be seen in Fig. 2A, almost all of the FRET data for the EGFR–EPHA2 pair lie above the modeled proximity line. This is indicative of a specific interaction occurring between EGFR and EPHA2, consistent with the literature (58, 59). Thus, we were able to observe the heterointeraction between full-length EGFR and EPHA2 using FRET. However, we were not able to calculate the heteroassociation constant, as a much larger data set is needed to fit the data with the models shown in Fig. 1 (A–C).
There is a published report that the deletion of amino acids in the EGFR post-kinase tail may improve EGFR expression (66). Accordingly, we created an EGFRΔTail construct where the post-kinase tail (∼220 amino acids) was deleted and replaced with a fluorescent protein (MT). We also used an EPHA2ΔSAM construct where the SAM domain was replaced with a fluorophore (eYFP), such that the fluorescent proteins were attached to the kinase domains in both constructs (see Fig. 1D, middle). This design should ensure that the FRET pair will be in close proximity if heterointeractions occur, allowing us to measure FRET.
The overall expression of EGFR was improved upon the deletion of the post-kinase tail, but most of it did not traffic to the plasma membrane, and thus the concentration of EGFR in the plasma membrane was still low. Only a small fraction of the imaged cells had a quantifiable amount of both receptors. For these cells, Fig. 3A shows the FRET efficiency versus the acceptor concentration for the EGFRΔTail–EPHA2ΔSAM pair. Again, almost all of the data lie above the modeled proximity line, indicating that a specific interaction is occurring between EGFR and EPHA2. Accordingly, the post-kinase tail of EGFR and the SAM domain of EPHA2 are not required for the interaction to occur.
Figure 3.

Heterointeractions between truncated EGFR and EPHA2 (A) and truncated VEGFR2 and EPHA2 (B). In the case of EPHA2, the SAM domain was replaced with eYFP, and in the case of EGFR and VEGFR2, the post-kinase tail was replaced with MT. The cells were transfected with varying donor/acceptor ratios, with a total of 0.5–3 μg of EPHA2–eYFP DNA and either 1–5 μg of EGFR–MT DNA (A) or 3–8 μg of VEGFR2–MT DNA (B). Each black star represents the measurement for a 2–3-μm stretch of plasma membrane in a cell. Two regions were selected per cell, for a total of 56 and 50 data points in A and B, respectively. The solid blue line is the modeled monomer FRET proximity as a function of acceptor concentration (60, 61). In both cases, almost all data points lie above the monomer proximity line, demonstrating specific interactions.
In Fig. 3B, we show FRET data reporting on the interactions between VEGFR2 and EPHA2. Specifically, we studied the interactions between VEGFR2ΔTail–MT and EPHA2ΔSAM–YFP, as the full-length VEGFR2 exhibited virtually no plasma membrane expression in HEK 293T cells. VEGFR2 is critically important for angiogenesis. It plays a role in organ development and wound healing and is overexpressed in many cancers (30, 67). We chose to study this interaction, as VEGFR2 is sometimes expressed in the same cells as EGFR and EPHA2 (55–57). Moreover, EPHA2 is also involved in angiogenesis, and its activity is at least partly dependent on VEGFR2 (68–70).
Nearly all of the FRET data in Fig. 3B lie above the proximity line, indicative of a specific interaction between VEGFR2 and EPHA2. Thus, these FRET experiments suggest that VEGFR2 and EPHA2 interact in the membrane. However, no quantitative information about the interaction could be determined, as a much larger data set is needed to differentiate betwen the different oligomerization models in Fig. 1 (A–C).
Finally, we wanted to see whether EGFR and VEGFR2 interact. There is substantial evidence of cross-talk between the EGFR and VEGFR2 signaling pathways, especially in cancer (71–73). In fact, the use of EGFR inhibitors to treat cancer is associated with increased VEGFR2 activity. Furthermore, resistance to VEGFR2 inhibitors is associated with increased EGFR activity (74–76). We attempted to study the EGFRΔTail–VEGFR2ΔTail pair, but we were unable to collect any data, as both receptors had poor expression on the plasma membrane.
In previous works, insights about RTK interactions and function were gained by using truncated RTK constructs where the IC domains were replaced by fluorophores (Fig. 1D, right) attached directly to the TM domains via flexible linkers; such constructs are referred to as “ECTM RTKs.” (1, 77, 78). In other words, an ECTM RTK is a construct with an EC and TM domain, but no IC domain. The fluorescent proteins in these ECTM constructs are in close enough proximity for FRET to occur if an interaction is occurring, and the proteins both express and traffic to the plasma membrane efficiently. Accordingly, we used these truncated versions of the RTKs of interest in the FRET experiments to gain quantitative insights into the interactions. Because RTK kinase domains have a high degree of homology (79, 80) and the kinase domains almost always have an attractive interaction (6, 78), we expect that the general conclusions for the truncated receptors will hold for the full-length receptors.
ECTM EGFR and ECTM EPHA2 form a heterodimer
We next performed FRET experiments with ECTM EGFR and ECTM EPHA2, following the same imaging protocol as in the full-length and post-kinase tail deletion cases. Fig. 4A shows the apparent FRET for the ECTM EGFR–ECTM EPHA2 pair as a function of acceptor concentration. These data combine the results of two types of experiments, one where EGFR was labeled with MT while EPHA2 was labeled with eYFP (orange) and one where EGFR was labeled with eYFP while EPHA2 was labeled with MT (green). Fig. 4B shows the concentration of EGFR versus the concentration of EPHA2 for the entire data set. We see that the FRET data in Fig. 4A lie above the proximity line, which indicates that a specific interaction is occurring between ECTM EGFR and ECTM EPHA2.
Figure 4.
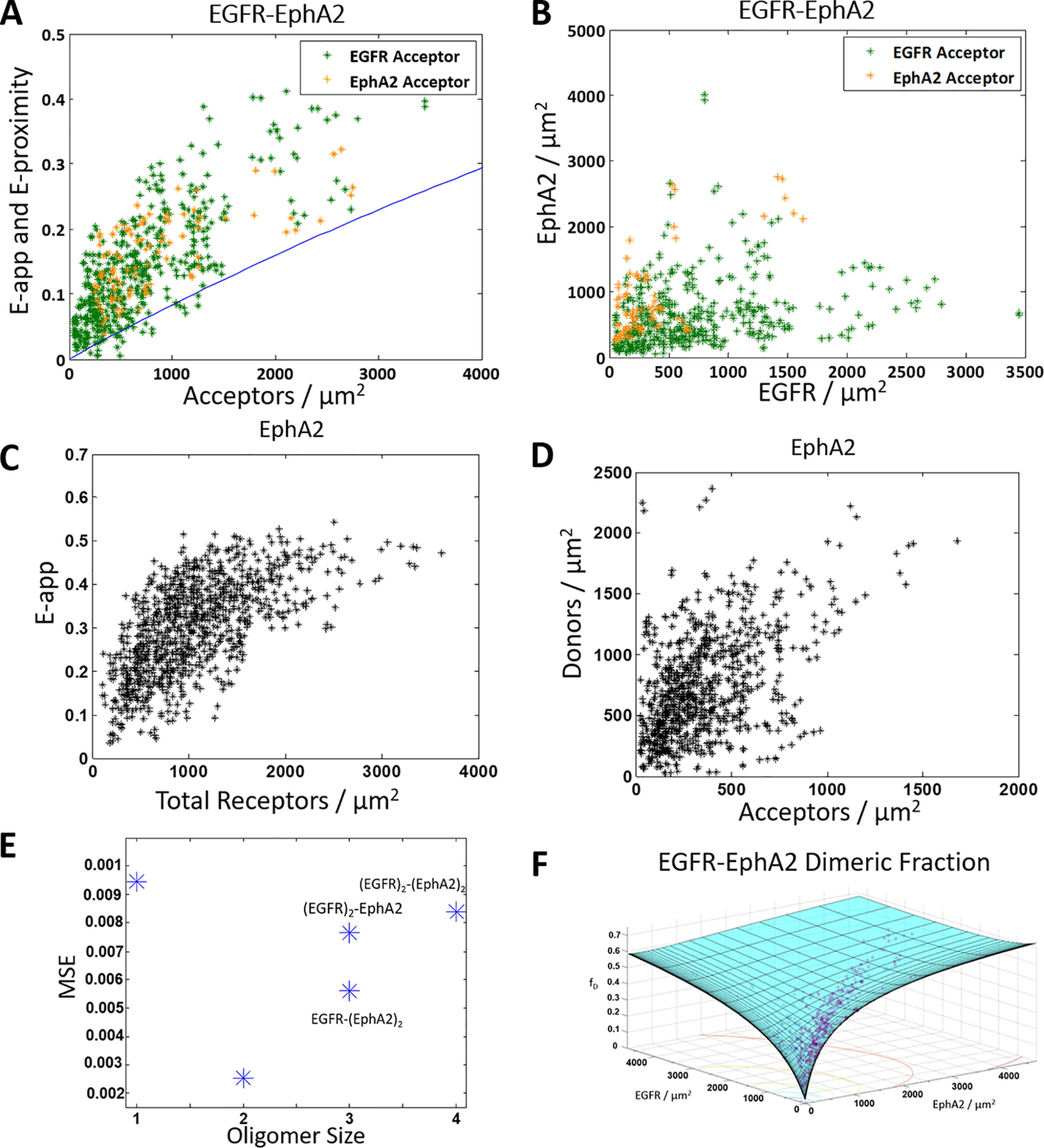
ECTM EGFR and ECTM EPHA2 form a heterodimer. A and B, HEK 293T cells were transiently co-transfected with versions of EGFR and EPHA2 where the IC domain has been replaced with a fluorophore: either MT as the donor or eYFP as the acceptor. Orange, ECTM EGFR–MT and ECTM EPHA2–eYFP. Green, ECTM EGFR–eYFP and ECTM EPHA2–MT. The cells were transfected with varying ratios of EGFR and EPHA2, with a total of 0.5–4 μg of EGFR DNA and 0.5–3 μg of EPHA2 DNA. A, apparent FRET versus acceptor concentration. Each data point represents the measurement for a 2–3 μm stretch of the plasma membrane of a cell. Two regions were selected per cell, for a total of 574 data points (480 with EGFR labeled with the acceptor and 94 with EPHA2 labeled with the acceptor). The solid blue line is the monomer FRET proximity as a function of acceptor concentration (60, 61). B, EGFR concentration versus EPHA2 concentration. C and D, HEK 293T cells were transiently co-transfected with ECTM EPHA2–MT and ECTM EPHA2–eYFP, with a 1:3 donor/acceptor ratio and a total of 0.5–3 μg of DNA. C, apparent FRET versus EPHA2 concentration (a total of 962 data points). D, donor concentration versus acceptor concentration. E, MSEs for the different interaction models in Fig. 1 (A–C). The heterodimer model yields the best fit. F, dimer fraction as a function of EGFR and EPHA2 concentrations. The purple symbols are the experimentally determined dimeric fractions, and the solid cyan surface is the best-fit surface for the heterodimer model.
Next, we asked which of the different interaction models (dimer, trimer, or tetramer) in Fig. 1 (A–C) (described above under “Heterointeraction models”) best describe these data. To answer this question and to determine the heteroassociation constant, we need to know the homodimer association constants, KX and KY, for both ECTM EGFR and ECTM EPHA2. The homodimer association constant KEGFR has been measured previously (77), but KEPHA2 has not been. To measure it, we performed FRET homodimerization experiments in HEK 293T cells that were co-transfected with plasmids for ECTM EPHA2–MT and ECTM EPHA2–eYFP, and we followed a well-established homodimer data analysis protocol (1, 81). Fig. 4C shows the apparent FRET efficiency versus the total concentration of receptors, and Fig. 4D shows the donor versus acceptor concentration. The ECTM EPHA2 homodimer association constant is determined by fitting a monomer-dimer model to the FRET data (1, 81). The best-fit value for KEPHA2 is 0.0012 ± 0.0001 μm2/receptor, and the corresponding dissociation constant (KEPHA2−1) is 860 ± 50 receptors/μm2. Using ΔG = −RTlnK—where R is the ideal gas constant and T is temperature—and a reference state of 1 nm2/receptor gives a ΔGEPHA2 of −4.0 ± 0.1 kcal/mol.
With KEGFR and KEPHA2 known, the heterointeraction FRET data in Fig. 4 (A and B) were fit to predictions derived from the three heterodimerization models in Fig. 1 (A–C) as well as a monomer model where the proximity line is regarded as the best fit (60, 61). For these four models, the best-fit theoretical FRET was compared with the experimental FRET, and mean squared errors (MSEs) were calculated for each of the four models, as shown in Fig. 4E. The lowest MSE in Fig. 4E is observed for the heterodimer, suggesting that EGFR and EPHA2 likely form a heterodimer and not higher-order oligomers. Fig. 4F shows the dimeric fractions, which depend on both EGFR and EPHA2 concentrations. The experimentally derived fractions are shown as purple circles, and the best-fit surface is shown in cyan. The best-fit KEGFR–EPHA2−1 for the heterodimer is 5000 ± 1000 receptors/μm2, corresponding to a heterodimer stability of ΔGEGFR–EPHA2 = −3.1 ± 0.2 kcal/mol. In Table 1, these values are compared with the previously determined homodimer dissociation constants and ΔG values for the EGFR and EPHA2 homodimers. We see that the heterointeraction strength is very similar to the homointeraction strengths.
Table 1.
Homo- and heterodimerization strengths for three of the studied RTKs
Shown are Kd and dimer stabilities (ΔG) for EGFR homodimers, EPHA2 homodimers, VEGFR2 homodimers, EGFR–EPHA2 heterodimers, EGFR–VEGFR2 heterodimers, and EPHA2–VEGFR2 heterodimers. Errors represent the 95% confidence interval from the fit. All values are for the ECTM versions of the receptors. Values for the EGFR and VEGFR2 homodimers have been measured previously (1, 77).
| Kd | ΔG | |
|---|---|---|
| receptors/μm2 | kcal/mol | |
| EGFR | 2800 ± 200 | −3.5 ± 0.1 |
| EPHA2 | 860 ± 50 | −4.0 ± 0.1 |
| VEGFR2 | 2700 ± 450 | −3.5 ± 0.1 |
| EGFR–EPHA2 | 5000 ± 1000 | −3.1 ± 0.2 |
| EGFR–VEGFR2 | 5200 ± 1200 | −3.1 ± 0.2 |
| EPHA2–VEGFR2 | 2900 ± 900 | −3.4 ± 0.2 |
ECTM EGFR and ECTM VEGFR2 form a heterodimer
Next, we studied the heterointeraction between ECTM EGFR and ECTM VEGFR2 using the same FRET imaging protocol. Fig. 5A shows the apparent FRET efficiencies versus acceptor concentration data for the ECTM EGFR–ECTM VEGFR2 pair. Again, experiments were performed with two different labeling schemes: ECTM EGFR–MT, ECTM VEGFR2–eYFP (orange) and ECTM EGFR–eYFP, ECTM VEGFR2–MT (green). Fig. 5B shows the expression of VEGFR2 versus EGFR expression. Virtually all of the data in Fig. 5A lie above the proximity line, which indicates that a specific interaction is occurring between ECTM EGFR and ECTM VEGFR2.
Figure 5.
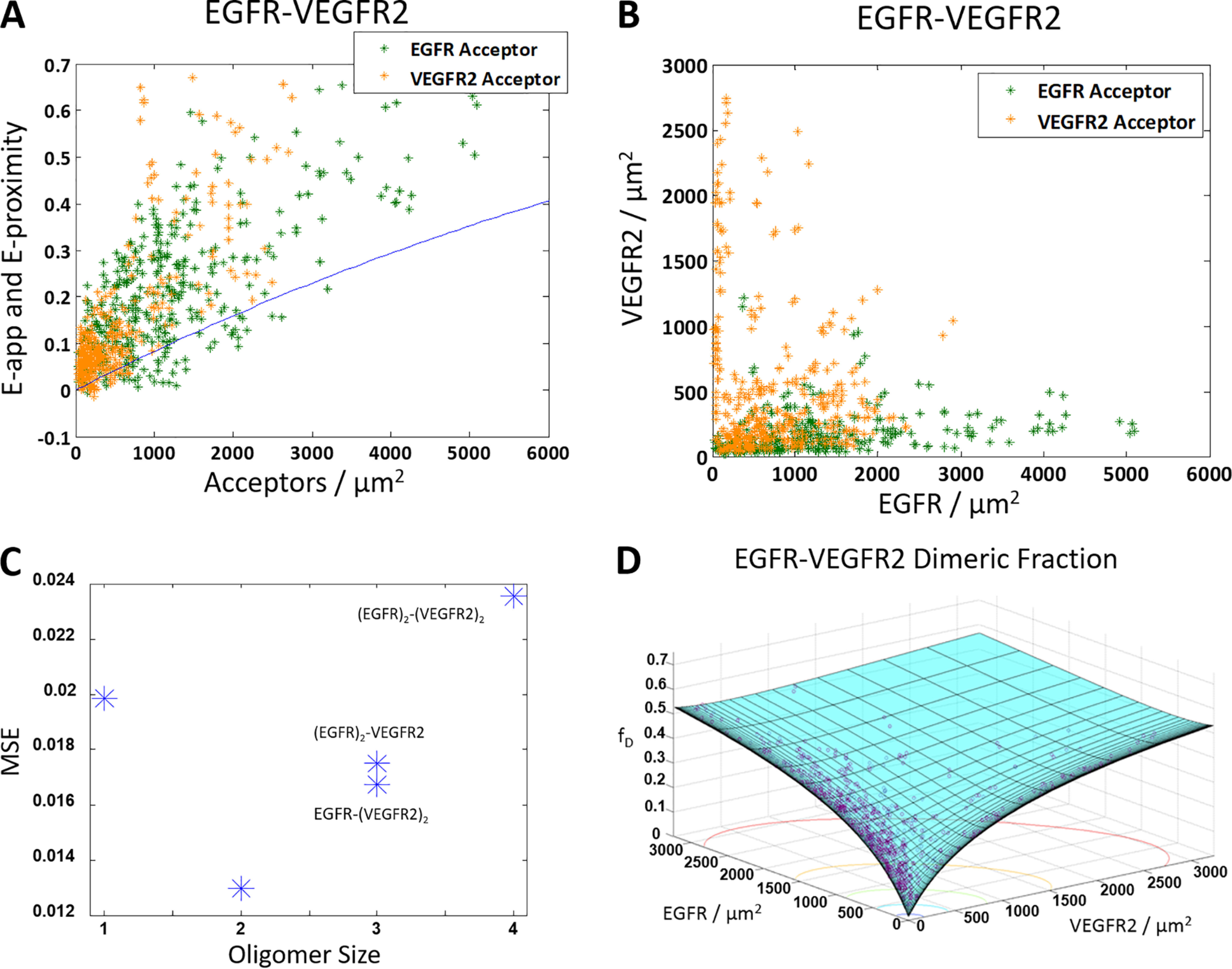
The truncated ECTM EGFR and ECTM VEGFR2 form a heterodimer. HEK 293T cells were transiently co-transfected with versions of EGFR and VEGFR2 where the IC domain has been replaced with a fluorophore: either MT for the donor or eYFP for the acceptor. The cells were transfected with varying ratios of EGFR and VEGFR2, with a total of 0.5–4 μg of EGFR DNA and 3–6 μg of VEGFR2 DNA. A, apparent FRET efficiency as a function of acceptor concentration. Shown are 844 data points (388 points for VEGFR2–MT and EGFR–eYFP (green) and 456 points for EGFR–MT and VEGFR2–eYFP (orange)). The solid blue line is the monomer FRET proximity (60, 61). B, VEGFR2 concentration versus EGFR concentration. C, the MSE plot, showing that the heterodimer model gives the best fit. D, dimer fraction as a function of EGFR and VEGFR2 concentration. The purple symbols are the experimentally determined dimeric fractions, and the solid cyan surface is the best-fit surface for the heterodimer model.
The data in Fig. 5 (A and B) were fit to the models shown in Fig. 1 (A–C) and the monomer model using the previously measured values of KEGFR (77) and KVEGFR2 (1). An MSE analysis for the different heterointeraction models is shown in Fig. 5C, and it suggests that EGFR and VEGFR2 form a heterodimer. Fig. 5D shows the dimeric fraction as a function of EGFR and VEGFR2 concentrations, where the best-fit surface is in cyan and the data are the purple circles. The best-fit KEGFR–VEGFR2−1 for the heterodimer is 5200 ± 1200 receptors/μm2, which corresponds to heterodimer stability of ΔGEGFR–VEGFR2 = −3.1 ± 0.2 kcal/mol. These values are comparable with the previously determined homodimerization constants and ΔG values for the ECTM EGFR and ECTM VEGFR2 homodimers, as shown in Table 1.
ECTM EPHA2 and ECTM VEGFR2 form a heterodimer
We also studied the heterointeractions between ECTM EPHA2 and ECTM VEGFR2. Fig. 6A shows the apparent FRET efficiencies versus acceptor concentration for the EPHA2–VEGFR2 pair, and Fig. 6B shows the expression of the two RTKs. As with the previous pairs, experiments were performed both with ECTM EPHA2–MT and ECTM VEGFR2–eYFP, shown in green, and ECTM EPHA2–eYFP and ECTM VEGFR2–MT, shown in orange. Nearly every data point in Fig. 6A lies above the proximity line, indicating the presence of specific interactions between ECTM EPHA2 and ECTM VEGFR2.
Figure 6.
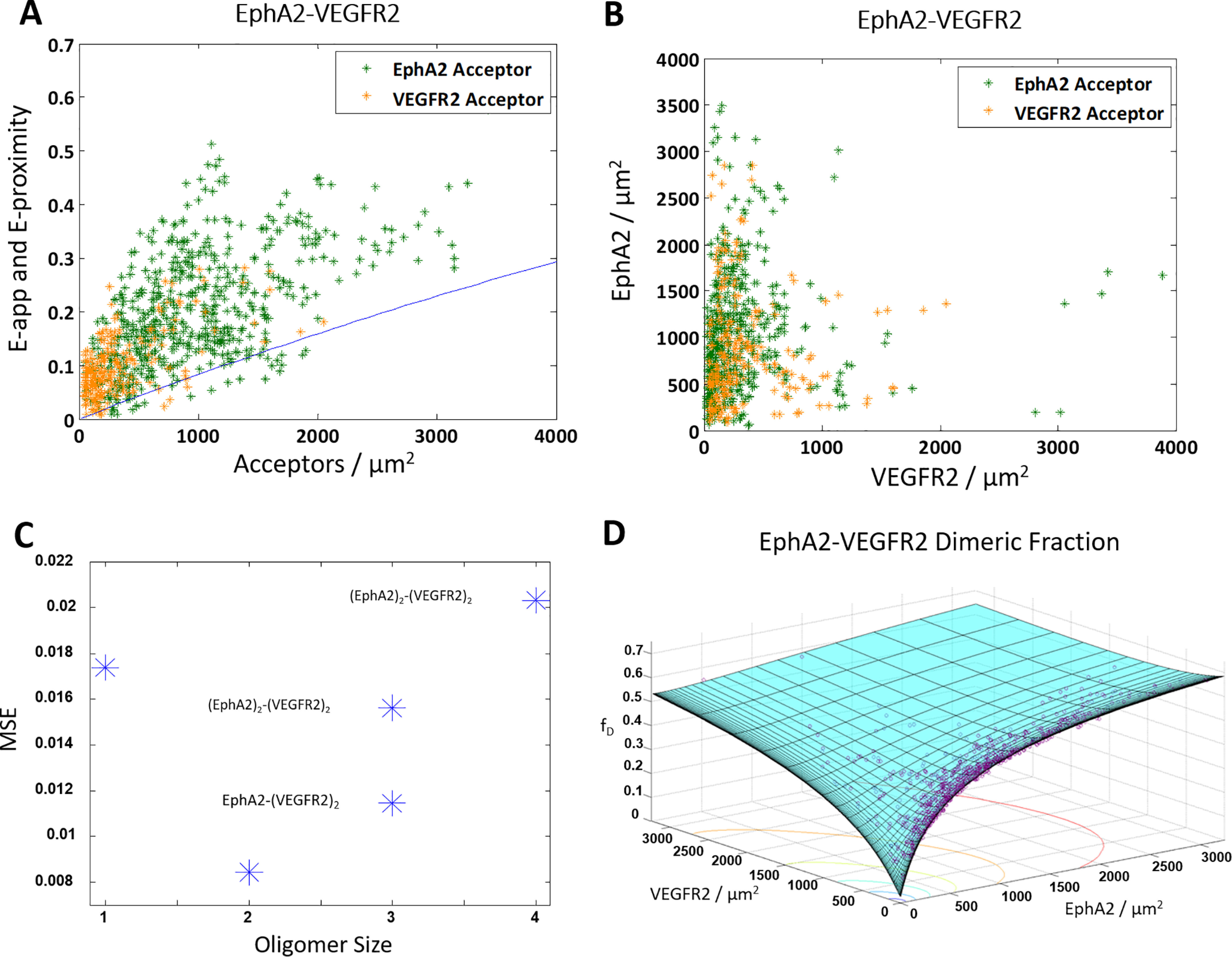
The truncated ECTM EPHA2 and ECTM VEGFR2 form a heterodimer. HEK 293T cells were transiently co-transfected with versions of EPHA2 and VEGFR2 where the IC domain has been replaced with a fluorophore: either MT for the donor or eYFP for the acceptor. The cells were transfected with varying ratios of EPHA2 and VEGFR2, with a total of 0.5–3 μg of EPHA2 DNA and 3–6 μg of VEGFR2 DNA. A, apparent FRET efficiency versus acceptor concentration. Shown are 856 data points (592 for VEGFR2–MT and EPHA2–eYFP (green) and 264 for EPHA2–MT and VEGFR2–eYFP (orange)). The solid blue line is the monomer proximity FRET (60, 61). B, EPHA2 concentration versus VEGFR2 concentration. C, an MSE plot, showing that the heterodimer model gives the best fit. D, dimeric fraction as a function of VEGFR2 and EPHA2. The purple symbols are the experimentally determined dimeric fractions, and the solid cyan surface is the best-fit surface for the heterodimer model.
The data in Fig. 6 (A and B) were fit to the models in Fig. 1 (A–C) and a monomer model. An MSE analysis of the different models is shown in Fig. 5C. The lowest MSE is attained for the heterodimer, suggesting that EPHA2 and VEGFR2 form a heterodimer. Fig. 6D shows the dimeric fraction versus the concentration of EPHA2 and VEGFR2, where the best-fit surface is in cyan and the data are shown as purple circles. The best-fit KEPHA2–VEGFR2−1 for the heterodimer is 2900 ± 900 receptors/μm2, corresponding to a heterodimer stability of ΔGEPHA2–VEGFR2 = −3.4 ± 0.2 kcal/mol. In Table 1, these values are compared with the previously determined homodimer dissociation constants and ΔG values for ECTM EPHA2 and ECTM VEGFR2, and again these values are very similar.
ECTM EPHA2 and ECTM VEGFR2 both engage in heterointeractions with ECTM FGFR1, ECTM FGFR2, and ECTM FGFR3
We performed FRET experiments with FGFR1, FGFR2, and FGFR3, with the goal to study their heterointeractions with EPHA2 and VEGFR2. The FGFRs are important for musculoskeletal development (82, 83), and their misfunction has been linked to many growth disorders (20, 84). These three FGFRs have been shown to interact with each other (42), and immunoprecipitation studies suggest that they can also interact with many RTKs from other subfamilies (27). To the best of our knowledge, no interactions have been reported for FGFRs and EPHA2 or for FGFRs and VEGFR2. However, all FGFRs have been shown to interact with EPHA4 (85, 86). We therefore hypothesized that there may be FGFR interactions, in particular FGFR-EPHA2 interactions, that have not yet been identified.
In the experiments, HEK 293T cells were co-transfected with either ECTM VEGFR2–MT or ECTM EPHA2–MT and one of ECTM FGFR1–eYFP, ECTM FGFR2–EYFP, or ECTM FGFR3–eYFP. The apparent FRET efficiencies for all six pairs are shown as a function of acceptor concentration in Fig. 7, with the EPHA2 data in Fig. 7A and the VEGFR2 data in Fig. 7B. In all cases, virtually all of the data lie above the proximity line, which indicates that EPHA2–FGFR1, EPHA2–FGFR2, EPHA2–FGFR3, VEGFR2–FGFR1, VEGFR2–FGFR2, and VEGFR2–FGFR3 all engage in specific heterointeractions.
Figure 7.
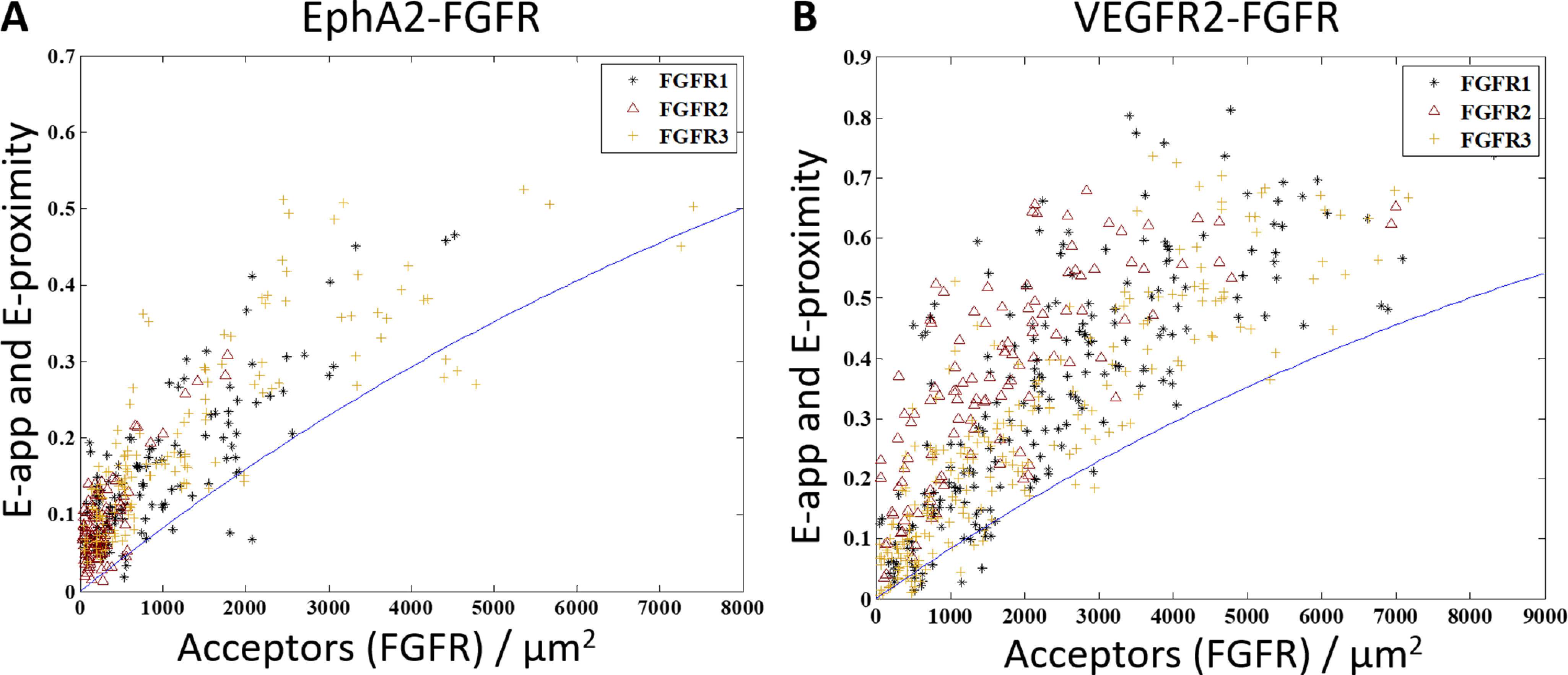
ECTM EPHA2 and ECTM VEGFR2 interact with ECTM FGFR1, ECTM FGFR2, and ECTM FGFR3. HEK 293T cells were transiently co-transfected with ECTM FGFR1, ECTM FGFR2, or ECTM FGFR3 and ECTM EPHA2 or ECTM VEGFR2. In each experiment, FGFR1, FGFR2, or FGFR3 was labeled with eYFP, and EPHA2 or VEGFR2 was labeled with MT. A, experiments with EPHA2. B, experiments with VEGFR2. The cells were co-transfected with both the donor and the acceptor in varying ratios, with a total of 1–3 μg of the DNA for the FGFR and 0.5–3 μg of EPHA2 DNA or 2–4 μg of VEGFR2 DNA. Shown is apparent FRET efficiency versus acceptor concentration. Black stars, FGFR1; maroon triangles, FGFR2; gold plus signs, FGFR3. There are 136 EPHA2–FGFR1 data points, 112 EPHA2–FGFR2 data points, 148 EPHA2–FGFR3 data points, 206 VEGFR2–FGFR1 data points, 114 VEGFR2–FGFR2 data points, and 228 VEGFR2–FGFR3 data points. The solid blue line is the modeled monomer FRET proximity as a function of acceptor concentration (60, 61). In all cases, almost all of the points lie above the FRET proximity line, indicative of specific interactions.
EGF and mEphrin-A1 Fc ligands decrease the heterointeractions
The data presented above suggest that RTK homo- and heterodimers are approximately equally likely to form in the absence of ligand; accordingly, their abundance will depend on the expression of the RTKs. If ligands are present, however, homodimers that are bound to their cognate ligand will be stabilized, and thus the concentration of monomers will decrease, which is expected to lead to a depletion of the heterodimer population. In other words, if the ligands do not bind the heterodimers, the heterodimer population will decrease as the ligand concentration is increased. Thermodynamic predictions have shown that when the ligand concentration substantially exceeds the ligand dissociation constant (27), the heterodimer population becomes near zero.
With these considerations in mind, we quantified the effect of the EGF ligand on the ECTM EGFR–ECTM VEGFR2 heterodimer population. We used 10 nm EGF, which is about an order of magnitude higher than the reported EGF-EGFR apparent dissociation constant (87–90). Cells were prepared identically to the EGFR–VEGFR2 experiments, except 0.1% (w/v) BSA was added to the starvation and imaging media to prevent ligand binding to the imaging dish, and the ligand was added 10 min prior to imaging. In Fig. 8A, the apparent FRET versus acceptor concentration is shown for cells in the presence of 10 nmol of EGF; EGF data are shown in red, and the no-ligand data (see Fig. 5A) are in black. We observe a marked decrease in the FRET efficiency in the presence of EGF, as the FRET efficiency now appears to be straddling the monomer proximity line. Thus, a substantial decrease in the heterointeraction occurred, as expected.
Figure 8.
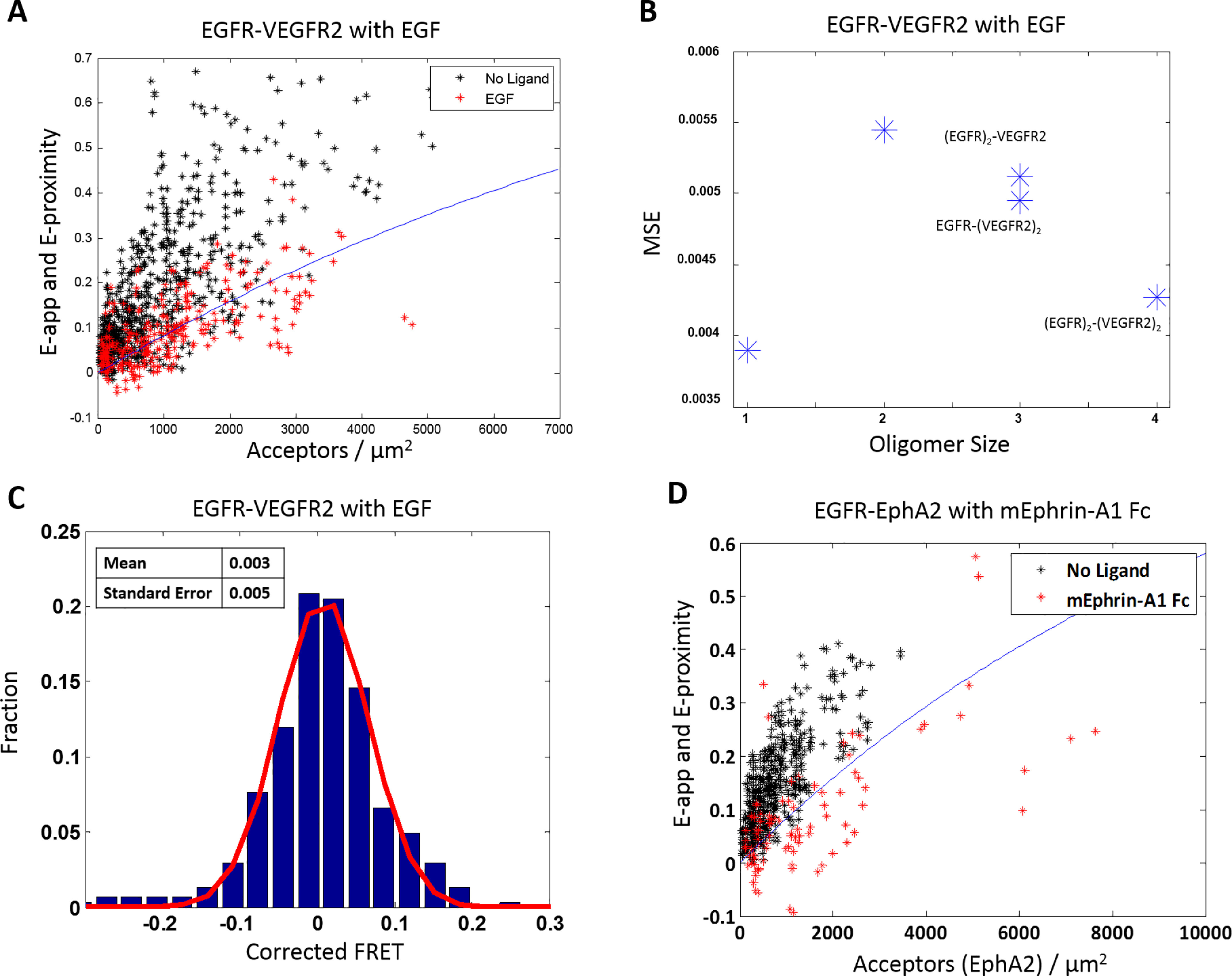
Ligands decrease the heterointeractions. A, FRET efficiency versus acceptor concentration for ECTM EGFR and ECTM VEGFR2, when two different labeling schemes are used as in Fig. 5A. The cells were co-transfected with varying ratios of EGFR and VEGFR2, with a total of 0.5–4 μg of EGFR DNA and 3–6 μg of VEGFR2 DNA. The black stars are the data in the absence of ligand (also shown in Fig. 5A), and the red stars are in the presence of 10 nmol of EGF. There is a total of 302 data points in the presence of ligand (156 with EGFR as the acceptor and 146 with VEGFR2 as the acceptor). The solid blue line is the proximity FRET as a function of acceptor concentration (60, 61). The presence of ligand decreases the apparent FRET efficiency. B, mean square error as a function of oligomer size. The monomer model yields the best fit. C, distribution of the FRET efficiencies in the presence of ligand, after correction for proximity FRET. The distribution is well-described by a Gaussian (red line). The mean is 0.003, and the S.E. is 0.005. D, FRET versus acceptor concentration for ECTM EGFR–MT and ECTM EPHA2–YFP. Black symbols, no-ligand data (also shown in Fig. 4A). Red stars, data in the presence of 50 nm mEphrin-A1 Fc (92 points total). Red stars fall on or below the FRET proximity line, showing that the ligand abolishes the heterointeractions.
Under the conditions of this experiment, over 90% of the EGFR present is expected to be ligand-bound. We therefore considered these data in the context of the models in Fig. 1 (A–C), where one receptor is EGF-bound EGFR, and the other receptor is unliganded VEGFR2. The homodimerization constant for EGF-bound EGFR has been measured previously as 0.053 μm2/receptor (77). The MSE analysis, shown in Fig. 8B, reveals that the monomer model exhibits the lowest MSE, supporting the fact that EGF largely abolished the heterointeraction.
We further analyzed the FRET data by first correcting for the monomer proximity FRET contribution and then histogramming the corrected FRET data. As seen in Fig. 8C, the data for EGFR–VEGFR2 in the presence of EGF is effectively a Gaussian centered around zero, similar to the case of the monomer control LAT (Fig. 2D) but very different from the case of EGFR–VEGFR2 in the absence of ligand. In agreement with this assessment, an analysis of variance indicates that the LAT and EGF data are not significantly different, but the no-ligand data and EGF data are very significantly different (p < 0.0001). These results provide more evidence that EGF substantially decreases the EGFR–VEGFR heterointeraction, consistent with expectations.
Next, we performed a similar experiment, adding the ligand ephrin-A1-Fc to ECTM EGFR–ECTM EPHA2 heterodimers. This is a ligand with subnanomolar affinity for EPHA2, which induces EPHA2 cluster formation (91, 92). It is dimeric, as it is fused to the Fc fragment of an antibody that dimerizes constitutively. As shown in Fig. 8D, the presence of 50 nm ephrin-A1-Fc caused a substantial decrease in FRET. Of note, the majority of the measured FRET efficiencies are very low and now lie below the monomer FRET proximity line. This can be explained by the fact that the liganded EPHA2 forms clusters, and thus the monomer proximity FRET model is now inaccurate. Indeed, it has been shown previously that the contribution of proximity FRET decreases as the cluster size grows (60, 61). Thus, the data in Fig. 8D are consistent with the idea that ephrin-A1-Fc sequesters EPHA2 into clusters and decreases its heterointeractions with EGFR.
Discussion
FRET as a tool to study heterointeractions
Here, we demonstrate that RTKs from unrelated subfamilies can interact with each other in the absence of ligand using a quantitative FRET technique that reports on the heterointeraction strength in live cells. We expand the previously published methodology (42) to determine the type of heterooligomer that forms when two different RTKs interact. In particular, we develop models that describe heterodimers, heterotrimers, and heterotetramers, and then we fit these models to FRET data to determine which of these models best describe the data. This method requires the acquisition of FRET binding curves over a broad range of concentrations. We were able to collect such binding curves for ECTM versions of several RTKs and to determine the oligomer size and the stability of the oligomer. Constructs that incorporated the kinase domains had poor plasma membrane expression, such that only a small fraction of the imaged cells had both receptors in their plasma membranes. Thus, whereas we showed that these RTKs interact, we were not able to quantitatively assess the interactions. In all cases where we were able to determine the oligomer size, we showed that the RTKs interact as heterodimers.
Although such heterointeractions have been studied previously, almost all of the research has been done with qualitative techniques. The most common method used in the literature to assess heterointeractions is co-immunoprecipitation (co-IP), followed by Western blotting (93, 94). One of the RTKs is immunoprecipitated, and the other RTK is blotted against. In this case, some concerns arise as the interactions in the native plasma membrane and in the immunoprecipitate may be different. Whereas the two-dimensional, hydrophobic membrane imposes structural constraints on RTKs, these may be relieved or altogether absent in the aqueous immunoprecipitate. Furthermore, weak interactions can easily fall apart during the lysing and precipitation process, meaning that such interactions are likely to be missed. Co-IP alters the concentrations of the interacting partners, and it thus cannot yield binding curves and association constants.
Another method that has been used to assess propensities for interaction is chemical cross-linking, where a chemical cross-linking agent is added before Western blotting is performed under nondenaturing conditions (44, 95, 96). This helps to stabilize weak or environment-dependent interactions that are often lost in co-IP. However, in addition to having similar issues as co-IP and not being able to provide interaction free energies, it has other shortcomings. First, the cross-linker is nonspecific, meaning that it cross-links all proteins in close proximity. As a result, bands of cross-linked proteins on gels are smeared and generally cannot be accurately quantified. Second, the cross-linking propensities depend not only on close proximity, but also on structure. Third, the cross-linker itself is expected to drive the equilibrium toward the oligomeric state, although this effect has not been investigated in detail.
Although less common than the previous two methods, another method that is used to study heterointeractions relatively frequently is the proximity ligation assay (97–100). In this technique, DNA probes are attached to primary or secondary antibodies against the proteins of interest. When the probes come in close proximity, a rolling circular amplification reaction occurs, and this DNA product can be hybridized with fluorescently labeled oligonucleotides. This assay is performed in cells rather than lysates, preserving the native environment, although fixation and permeabilization are generally required. However, this assay is not able to provide information about interaction energies or stoichiometries, as it simply reports on the close proximity of the proteins of interest.
Whereas these methods will continue to have utility in the future, the FRET method that we use here can be used in conjunction with these methods to yield a more comprehensive description of the interactions. We are thus hopeful that this method will have a broad applicability in RTK research.
Strength of heterointeractions
Here we find that in the absence of ligand, EPHA2 interacts with FGFR1, FGFR2, and FGFR3; VEGFR2 interacts with FGFR1, FGFR2, and FGFR3. We further show that EGFR–VEGFR2, EGFR–EPHA2, and EPHA2–VEGFR2 all form heterodimers. Surprisingly, we find that the stabilities of the heterodimers are quite similar to the stabilities of the homodimers in the absence of ligand. This is an unexpected result, and it suggests that these heterointeractions could have a significant effect in regulating RTK function. As the strengths of interaction are similar, both homodimers and heterodimers are expected to form under physiological conditions and to depend primarily on the relative RTK expression levels, in accordance with the law of mass action. In cancer, RTKs are often overexpressed, and ligand levels can be reduced (17, 18). Under these conditions, unliganded RTK heterodimers are expected to form with high probabilities.
Open structural and mechanistic questions
An open question is what physical contacts between the RTKs are enabling these heterointeractions. One possibility is that the TM domains play a key role, as it has been argued in the literature that the interactions between RTK TM domains are promiscuous, forming many weak, nonspecific interactions (15, 101, 102). It is also possible that interactions between the kinase domains contribute, as the kinase domain is highly conserved across families (79, 80). Previous FRET studies of RTK homodimers have shown that the deletion of their intracellular domains usually leads to dimer destabilization, both in the presence and absence of ligand (6, 78). However, the intracellular domain is not required, as the ECTM versions of all RTKs that were studied here engaged in heterointeractions. It is also possible that there is no universal mechanism for the heterointeraction, but rather, each pair interacts differently.
High-resolution structural information about RTK heterodimers is currently not available, and the steps leading to their phosphorylation and signaling are unknown. The mechanism of activation of the different RTK homodimers is believed to be different. For instance, EGFR is believed to be activated through a distinct allosteric mechanism that involves the formation of asymmetric dimers (103). Questions therefore arise as to how the EGFR kinase and the other kinases will function within heterodimers. Further work is needed to understand the mechanistic details of RTK heterodimer formation and activation.
Interactions with ligands and the RTK interactome
We investigated the effect of ligand on heterointeractions by adding the cognate ligands of EGFR, EGF, and EPHA2, ephrinA1-Fc. We showed that the presence of saturating quantities of these ligands abolished the EGFR–VEGFR2 and EGFR–EPHA2 interactions, respectively. This means that the presence of ligand does not just shift the population of RTKs from monomers to dimers, but also decreases the concentration of heterodimers. One study found that an interaction between ROR1 and ErbB3 resulted in a unique tyrosine on ErbB3 being phosphorylated, and this resulted in a novel modulation of the Hippo-YAP pathway (38). Thus, the ligand can act as a signaling modulator that alters the extent to which a signaling pathway is being activated, by altering the equilibrium between heterodimers and homodimers.
It is possible that all RTKs interact with each other and form an interaction network, which we called the “RTK interactome” (27). In this view, each RTK can influence numerous interconnected signaling pathways via direct interactions with RTK partners in the plasma membrane. There exists a complex equilibrium between multiple RTKs and their ligands, which is controlled by the expression of all interaction partners and the association constants that govern every interaction. This greatly increases the degree of signaling complexity that RTKs are able to achieve and makes the signaling highly responsive to environmental changes that alter RTK and ligand concentrations. The data presented here provide experimental support for this view and add a quantitative perspective to our understanding of the RTK interactome.
Implications
The RTK interactome concept may provide an explanation for differences in experimental data acquired for an RTK in different cell lines (27). Typically, the expression of unrelated RTKs is not taken into account, and the possibility of RTK heterodimer formation is ignored. However, it is conceivable that the presence of these heterodimers greatly influences the cell signaling outcomes. The data in this study are consistent with this view.
Heterointeractions, such as the ones quantified here, need to be considered in the design of RTK inhibitors for cancers and other diseases or disorders (27). Even if a drug successfully inhibits the signaling of an RTK homodimer, the drug will not have the desired effect if signaling-competent heterodimers are also present. Therefore, the RTK expression pattern in a cell type may be a critical factor in determining the performance of the inhibitor. Because an inhibitor can affect the functions of both homodimers and heterodimers—or possibly even affect the entire RTK interactome—it can lead to many unanticipated consequences. Accordingly, the understanding of RTK heterointeractions may help in understanding drug resistance in cancer therapy and in designing more effective treatments. Significant work is still needed to quantify all homo- and heterointeractions and to arrive at a comprehensive understanding of RTK signaling and its biological functions. The use of quantitative experimental methodologies and a theoretical framework will help guide such investigations.
Materials and methods
Plasmids
Plasmids encoding for different constructs of EGFR, EPHA2, VEGFR2, FGFR1, FGFR2, FGFR3, and LAT were used. In almost all cases, the plasmids was in the pcDNA3.1+ vector, and the fluorescent protein (either MT or eYFP, which together form a FRET pair) was positioned at the C terminus of the RTK constructs, attached via a flexible (GGS)5 linker to enable free rotation (104). The one exception is the full-length EGFR, which is in the pSSX vector and has a GGS linker. The plasmids encoding for ECTM EGFR, ECTM VEGFR2, ECTM FGFR1, ECTM FGFR2, ECTM FGFR3, EPHA2ΔSAM, full-length EGFR, and full-length EPHA2 have been described in detail in previous studies (1, 77, 78, 81, 92, 105, 106). The plasmids encoding for ECTM EPHA2, EGFRΔTail, VEGFR2ΔTail, and LAT were cloned using Gibson assembly. In all four cases, the receptor constructs were cloned into the pcDNA3.1+ vector using the NEBuilder HiFi DNA assembly kit (New England Biolabs, E5520S), and the primers were designed using the online NEBuilder assembly tool. For ECTM EPHA2, the last amino acid in the TM domain before the hydrophilic flexible (GGS)5 linker was Ile-558; for EGFRΔTail, the last amino acid was Leu-979; and for VEGFR2ΔTail, the last amino acid was Asn-1162; numbering is based on the sequences for isoform 1 in UniProt.
Cell culture and transient transfection
HEK 293T cells were purchased from ATCC. The cells were cultured in Dulbecco's modified Eagle's medium (Thermo Fisher Scientific, 31600034) supplemented with 3.5 g/liter glucose, 1.5 g/liter sodium bicarbonate, and 10% fetal bovine serum (Hylone, SH30070.03). The cells were maintained at 37 °C with 5% CO2 and passed every third day.
For the FRET experiments, HEK 293T cells were seeded at a density of 2.0 × 105 cells/dish in collagen-coated, glass-bottom Petri dishes (MatTek, P35GCOL-1.5-14-C). Approximately 24 h later, the cells were transfected with the desired plasmids using Lipofectamine 3000 (Invitrogen, L3000008) according to the manufacturer's protocol. Twelve hours after transfection, cells were serum-starved by replacing the medium with fetal bovine serum–free Dulbecco's modified Eagle's medium that also lacked phenol red (MilliporeSigma, D2902) to ensure that endogenous ligands were not present during imaging. To remove all traces of phenol red, the imaging dishes were washed twice with the phenol red–free medium. When using the VEGFR2 plasmids, 10 nm sodium butyrate was added 6 h after transfection and after serum starvation to increase expression. In the case of ligand experiments, 0.1% (w/v) BSA was added to the imaging dish during starvation to prevent ligands from binding to the imaging dish.
FRET imaging and data analysis
FRET experiments were performed using the FSI method described previously (1). The medium in the imaging dish was replaced with hypoosmotic swelling medium (1:9 serum-free media/distilled H2O, 25 mm HEPES), as this reversibly “unwrinkled” the plasma membrane (107), enabling accurate concentration measurements. If ligands—either 10 nm EGF (Cell Signaling Technology, 8916SC) or 50 nm dimeric Ephrin-A1 Fc (R&D Systems, 602-A1-200)—were used, 0.1% (w/v) BSA was added to the swelling medium, and then the ligands were added. Each dish was imaged for no more than 2 h. Imaging was done using a two-photon microscope equipped with the OptiMiS True Line Spectral Imaging system (Aurora Spectral Technologies, Shorewood, WI, USA) (108, 109). A Mai Tai laser (Spectra-Physics, Santa Clara, CA, USA) was used to generate femtosecond mode-locked pulses, and two images were acquired per cell: one at 840 nm to primarily excite the donor and one at 960 nm to primarily excite the acceptor. Cells singly transfected with a construct labeled with MT as well as cells singly transfected with a construct labeled with eYFP were also imaged to obtain the fluorescent spectra of the fluorophores. Pixel-level fluorescent intensities were converted into 2D concentrations using purified MT and eYFP solutions (110) to create calibration curves as described (1).
Cells were analyzed in MATLAB as described previously (1, 42). For each cell, two 2-3-µm stretches of plasma membrane were analyzed. The 2D concentration of the RTK labeled with the donor ([donor]), the 2D concentration of the RTK labeled with the acceptor ([acceptor]), and the apparent FRET efficiency (Eapp) were determined in each membrane region. Proximity FRET (Eprox) (111, 60) was corrected for using the following,
| (Eq. 31) |
where Eoligo is the FRET due to sequence-specific interactions (61).
Furthermore,
| (Eq. 32) |
where [heterooligomer] is the concentration of either heterodimers, heterotrimers, or heterotetramers and E˜ is the intrinsic FRET, a structural parameter depending on the distance between the two fluorophores and their orientation (112). By combining this with the equations under “Heterointeraction models,” a two-parameter fit for the heteroassociation constant, Khetero (e.g. KXY, KXYY), and E˜ was performed using the experimentally measured [donor], [acceptor], and Eapp. Data fitting was performed as described previously using MATLAB (1, 42). Statistical significance was determined using one-way analysis of variance in PRISM.
Acknowledgments
We thank Joana Ye for help with data analysis. We thank Dr. Marek Cebecauer for providing the LAT plasmid used to construct LAT-MT and LAT-eYFP.
Data availability
All raw FRET data are available free of charge upon request from Kalina Hristova (kh@jhu.edu). All other data are contained within the article.
Acknowledgments
We thank Joana Ye for help with data analysis. We thank Dr. Marek Cebecauer for providing us with the LAT plasmid used to construct LAT-MT and LAT-eYFP.
Author contributions—M. D. P. and K. H. conceptualization; M. D. P. data curation; M. D. P. software; M. D. P. formal analysis; M. D. P. and H. N. G. investigation; M. D. P. visualization; M. D. P. writing-original draft; M. D. P., H. N. G., and K. H. writing-review and editing; K. H. supervision; K. H. funding acquisition; K. H. project administration.
Funding and additional information—This work was supported by National Institutes of Health Grant GM068619 and National Science Foundation Grant MCB 1712740. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- RTK
- receptor tyrosine kinase
- FSI
- fully quantified spectral imaging
- MT
- mTurquoise
- 2D
- two-dimensional
- EC
- extracellular
- TM
- transmembrane
- IC
- intracellular
- EGF
- epidermal growth factor
- EGFR
- EGF receptor
- VEGFR2
- vascular endothelial growth factor receptor 2
- FGFR
- fibroblast growth factor receptor
- EPHA2
- EPH receptor A2
- YFP
- yellow fluorescent protein
- eYFP
- enhanced YFP
- LAT
- linker for activation of T cells
- MSE
- mean squared error
- co-IP
- co-immunoprecipitation.
References
- 1. King C., Stoneman M., Raicu V., and Hristova K. (2016) Fully quantified spectral imaging reveals in vivo membrane protein interactions. Integr. Biol. (Camb.) 8, 216–229 10.1039/c5ib00202h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fantl W. J., Johnson D. E., and Williams L. T. (1993) Signaling by receptor tyrosine kinases. Annu. Rev. Biochem. 62, 453–481 10.1146/annurev.bi.62.070193.002321 [DOI] [PubMed] [Google Scholar]
- 3. Lemmon M. A., and Schlessinger J. (2010) Cell Signaling by Receptor Tyrosine Kinases. Cell 141, 1117–1134 10.1016/j.cell.2010.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lawrence M. C., and Ward C. W. (2015) Structural features of the receptor tyrosine kinase ectodomains. Receptor Tyrosine Kinases: Structure, Functions and Role in Human Disease (Wheeler D. L., and Yarden Y., eds) pp. 163–193, Humana Press, Totowa, NJ [Google Scholar]
- 5. Chung I., Akita R., Vandlen R., Toomre D., Schlessinger J., and Mellman I. (2010) Spatial control of EGF receptor activation by reversible dimerization on living cells. Nature 464, 783–U163 10.1038/nature08827 [DOI] [PubMed] [Google Scholar]
- 6. Sarabipour S., Ballmer-Hofer K., and Hristova K. (2016) VEGFR-2 conformational switch in response to ligand binding. Elife 5, e13876 10.7554/eLife.13876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dietz M. S., Haße D., Ferraris D. M., Göhler A., Niemann H. H., and Heilemann M. (2013) Single-molecule photobleaching reveals increased MET receptor dimerization upon ligand binding in intact cells. BMC Biophys. 6, 6 10.1186/2046-1682-6-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Paul M. D., and Hristova K. (2019) The transition model of RTK activation: a quantitative framework for understanding RTK signaling and RTK modulator activity. Cytokine Growth Factor Rev. 49, 23–31 10.1016/j.cytogfr.2019.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schlessinger J. (2003) Autoinhibition control. Science 300, 750–752 10.1126/science.1082024 [DOI] [PubMed] [Google Scholar]
- 10. Lemmon M. A., Schlessinger J., and Ferguson K. M. (2014) The EGFR family: not so prototypical receptor tyrosine kinases. Cold Spring Harb. Perspect. Biol. 6, a020768 10.1101/cshperspect.a020768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schlessinger J. (2014) Receptor tyrosine kinases: legacy of the first two decades. Cold Spring Harb. Perspect. Biol. 6, a008912 10.1101/cshperspect.a008912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Arteaga C. L., and Engelman J. A. (2014) ERBB receptors: from oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell 25, 282–303 10.1016/j.ccr.2014.02.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Belov A. A., and Mohammadi M. (2013) Molecular mechanisms of fibroblast growth factor signaling in physiology and pathology. Cold Spring Harb. Perspect. Biol. 5, a015958 10.1101/cshperspect.a015958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wagner M. J., Stacey M. M., Liu B. A., and Pawson T. (2013) Molecular mechanisms of SH2- and PTB-domain-containing proteins in receptor tyrosine kinase signaling. Cold Spring Harb. Perspect. Biol. 5, a008987 10.1101/cshperspect.a008987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li E., and Hristova K. (2006) Role of receptor tyrosine kinase transmembrane domains in cell signaling and human pathologies. Biochemistry 45, 6241–6251 10.1021/bi060609y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pasquale E. B. (2010) Eph receptors and ephrins in cancer: bidirectional signalling and beyond. Nat. Rev. Cancer 10, 165–180 10.1038/nrc2806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Barker F. G., Simmons M. L., Chang S. M., Prados M. D., Larson D. A., Sneed P. K., Wara W. M., Berger M. S., Chen P. C., Israel M. A., and Aldape K. D. (2001) EGFR overexpression and radiation response in glioblastoma multiforme. Int. J. Radiat. Oncol. Biol. Phys. 51, 410–418 10.1016/S0360-3016(01)01609-1 [DOI] [PubMed] [Google Scholar]
- 18. Kornmann M., Beger H. G., and Korc M. (1998) Role of fibroblast growth factors and their receptors in pancreatic cancer and chronic pancreatitis. Pancreas 17, 169–175 10.1097/00006676-199808000-00010 [DOI] [PubMed] [Google Scholar]
- 19. Krejci P. (2014) The paradox of FGFR3 signaling in skeletal dysplasia: why chondrocytes growth arrest while other cells over proliferate. Mutat. Res. Rev. Mutat. Res. 759, 40–48 10.1016/j.mrrev.2013.11.001 [DOI] [PubMed] [Google Scholar]
- 20. Foldynova-Trantirkova S., Wilcox W. R., and Krejci P. (2012) Sixteen years and counting: the current understanding of fibroblast growth factor receptor 3 (FGFR3) signaling in skeletal dysplasias. Hum. Mutat. 33, 29–41 10.1002/humu.21636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Coon B. G., Baeyens N., Han J., Budatha M., Ross T. D., Fang J. S., Yun S., Thomas J. L., and Schwartz M. A. (2015) Intramembrane binding of VE-cadherin to VEGFR2 and VEGFR3 assembles the endothelial mechanosensory complex. J. Cell Biol. 208, 975–986 10.1083/jcb.201408103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Somanath P. R., Ciocea A., and Byzova T. V. (2009) Integrin and growth factor receptor alliance in angiogenesis. Cell Biochem. Biophys. 53, 53–64 10.1007/s12013-008-9040-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schneller M., Vuori K., and Ruoslahti E. (1997) αvβ3 integrin associates with activated insulin and PDGFβ receptors and potentiates the biological activity of PDGF. EMBO J. 16, 5600–5607 10.1093/emboj/16.18.5600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gao B., Song H., Bishop K., Elliot G., Garrett L., English M. A., Andre P., Robinson J., Sood R., Minami Y., Economides A. N., and Yang Y. (2011) Wnt signaling gradients establish planar cell polarity by inducing Vangl2 phosphorylation through Ror2. Dev. Cell 20, 163–176 10.1016/j.devcel.2011.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Grumolato L., Liu G., Mong P., Mudbhary R., Biswas R., Arroyave R., Vijayakumar S., Economides A. N., and Aaronson S. A. (2010) Canonical and noncanonical Wnts use a common mechanism to activate completely unrelated coreceptors. Genes Dev. 24, 2517–2530 10.1101/gad.1957710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bin-Nun N., Lichtig H., Malyarova A., Levy M., Elias S., and Frank D. (2014) PTK7 modulates Wnt signaling activity via LRP6. Development 141, 410–421 10.1242/dev.095984 [DOI] [PubMed] [Google Scholar]
- 27. Paul M. D., and Hristova K. (2019) The RTK interactome: overview and perspective on RTK heterointeractions. Chem. Rev. 119, 5881–5921 10.1021/acs.chemrev.8b00467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lenferink A. E., Pinkas‐Kramarski R., van de Poll M. L., van Vugt M. J., Klapper L. N., Tzahar E., Waterman H., Sela M., van Zoelen E. J., and Yarden Y. (1998) Differential endocytic routing of homo‐and hetero‐dimeric ErbB tyrosine kinases confers signaling superiority to receptor heterodimers. EMBO J. 17, 3385–3397 10.1093/emboj/17.12.3385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Goyette M. A., Duhamel S., Aubert L., Pelletier A., Savage P., Thibault M. P., Johnson R. M., Carmeliet P., Basik M., Gaboury L., Muller W. J., Park M., Roux P. P., Gratton J. P., and Côté J. F. (2018) The receptor tyrosine kinase AXL is required at multiple steps of the metastatic cascade during HER2-positive breast cancer progression. Cell Rep. 23, 1476–1490 10.1016/j.celrep.2018.04.019 [DOI] [PubMed] [Google Scholar]
- 30. Olsson A. K., Dimberg A., Kreuger J., and Claesson-Welsh L. (2006) VEGF receptor signalling—in control of vascular function. Nat. Rev. Mol. Cell Biol. 7, 359–371 10.1038/nrm1911 [DOI] [PubMed] [Google Scholar]
- 31. Rahimi N., Dayanir V., and Lashkari K. (2000) Receptor chimeras indicate that the vascular endothelial growth factor receptor-1 (VEGFR-1) modulates mitogenic activity of VEGFR-2 in endothelial cells. J. Biol. Chem. 275, 16986–16992 10.1074/jbc.M000528200 [DOI] [PubMed] [Google Scholar]
- 32. Lu K. V., Chang J. P., Parachoniak C. A., Pandika M. M., Aghi M. K., Meyronet D., Isachenko N., Fouse S. D., Phillips J. J., Cheresh D. A., Park M., and Bergers G. (2012) VEGF inhibits tumor cell invasion and mesenchymal transition through a MET/VEGFR2 complex. Cancer Cell 22, 21–35 10.1016/j.ccr.2012.05.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Greenberg J. I., Shields D. J., Barillas S. G., Acevedo L. M., Murphy E., Huang J., Scheppke L., Stockmann C., Johnson R. S., Angle N., and Cheresh D. A. (2008) A role for VEGF as a negative regulator of pericyte function and vessel maturation. Nature 456, 809–813 10.1038/nature07424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cheng C., Haasdijk R. A., Tempel D., den Dekker W. K., Chrifi I., Blonden L. A. J., van de Kamp E. H. M., de Boer M., Bürgisser P. E., Noorderloos A., Rens J. A. P., ten Hagen T. L. M., and Duckers H. J. (2012) PDGF-induced migration of vascular smooth muscle cells is inhibited by heme oxygenase-1 via VEGFR2 upregulation and subsequent assembly of inactive VEGFR2/PDGFRβ heterodimers. Arterioscler. Thromb. Vasc. Biol. 32, 1289–1298 10.1161/ATVBAHA.112.245530 [DOI] [PubMed] [Google Scholar]
- 35. Rupp E., Siegbahn A., Rönnstrand L., Wernstedt C., Claesson‐Welsh L., and Heldin C. H. (1994) A unique autophosphorylation site in the platelet‐derived growth factor α receptor from a heterodimeric receptor complex. FEBS J. 225, 29–41 10.1111/j.1432-1033.1994.00029.x [DOI] [PubMed] [Google Scholar]
- 36. Wu E., Palmer N., Tian Z., Moseman A. P., Galdzicki M., Wang X., Berger B., Zhang H., and Kohane I. S. (2008) Comprehensive dissection of PDGF-PDGFR signaling pathways in PDGFR genetically defined cells. PLoS ONE 3, e3794 10.1371/journal.pone.0003794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Huang K., Andersson C., Roomans G. M., Ito N., and Claesson-Welsh L. (2001) Signaling properties of VEGF receptor-1 and-2 homo-and heterodimers. Int. J. Biochem. Cell Biol. 33, 315–324 10.1016/S1357-2725(01)00019-X [DOI] [PubMed] [Google Scholar]
- 38. Li C., Wang S., Xing Z., Lin A., Liang K., Song J., Hu Q., Yao J., Chen Z., Park P. K., Hawke D. H., Zhou J., Zhou Y., Zhang S., Liang H., et al. (2017) A ROR1-HER3-lncRNA signalling axis modulates the Hippo-YAP pathway to regulate bone metastasis. Nat. Cell Biol. 19, 106–119 10.1038/ncb3464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Turner N., Pearson A., Sharpe R., Lambros M., Geyer F., Lopez-Garcia M. A., Natrajan R., Marchio C., Iorns E., Mackay A., Gillett C., Grigoriadis A., Tutt A., Reis J. S., and Ashworth A. (2010) FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res. 70, 2085–2094 10.1158/0008-5472.CAN-09-3746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hafner C., Schmitz G., Meyer S., Bataille F., Hau P., Langmann T., Dietmaier W., Landthaler M., and Vogt T. (2004) Differential gene expression of Eph receptors and ephrins in benign human tissues and cancers. Clin. Chem. 50, 490–499 10.1373/clinchem.2003.026849 [DOI] [PubMed] [Google Scholar]
- 41. He L., Horton W. A., and Hristova K. (2010) The physical basis behind achondroplasia, the most common form of human dwarfism. J. Biol. Chem. 285, 30103–30114 10.1074/jbc.M109.094086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Del Piccolo N., Sarabipour S., and Hristova K. (2017) A new method to study heterodimerization of membrane proteins and its application to fibroblast growth factor receptors. J. Biol. Chem. 292, 1288–1301 10.1074/jbc.M116.755777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rauch C., and Farge E. (2000) Endocytosis switch controlled by transmembrane osmotic pressure and phospholipid number asymmetry. Biophys. J. 78, 3036–3047 10.1016/S0006-3495(00)76842-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ahmed F., and Hristova K. (2018) Dimerization of the Trk receptors in the plasma membrane: effects of their cognate ligands. Biochem. J. 475, 3669–3685 10.1042/BCJ20180637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Real F. X., Rettig W. J., Chesa P. G., Melamed M. R., Old L. J., and Mendelsohn J. (1986) Expression of epidermal growth factor receptor in human cultured cells and tissues: relationship to cell lineage and stage of differentiation. Cancer Res. 46, 4726–4731 [PubMed] [Google Scholar]
- 46. Shilo B.-Z. (2003) Signaling by the Drosophila epidermal growth factor receptor pathway during development. The EGF Receptor Family (Carpenter G., ed) pp. 147–156, Academic Press, Inc., Cambridge, MA: [DOI] [PubMed] [Google Scholar]
- 47. Singh B., Carpenter G., and Coffey R. J. (2016) EGF receptor ligands: recent advances. F1000Res 5, 2270 10.12688/f1000research.9025.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nicholson R., Gee J., and Harper M. (2001) EGFR and cancer prognosis. Eur. J. Cancer 37, 9–15 10.1016/S0959-8049(01)00231-3 [DOI] [PubMed] [Google Scholar]
- 49. Appert-Collin A., Hubert P., Cremel G., and Bennasroune A. (2015) Role of ErbB receptors in cancer cell migration and invasion. Front. Pharmacol. 6, 283 10.3389/fphar.2015.00283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kania A., and Klein R. (2016) Mechanisms of ephrin-Eph signalling in development, physiology and disease. Nat. Rev. Mol. Cell Biol. 17, 240–256 10.1038/nrm.2015.16 [DOI] [PubMed] [Google Scholar]
- 51. Wilkinson D. G. (2001) Multiple roles of EPH receptors and ephrins in neural development. Nat. Rev. Neurosci. 2, 155–164 10.1038/35058515 [DOI] [PubMed] [Google Scholar]
- 52. Conover J. C., Doetsch F., Garcia-Verdugo J.-M., Gale N. W., Yancopoulos G. D., and Alvarez-Buylla A. (2000) Disruption of Eph/ephrin signaling affects migration and proliferation in the adult subventricular zone. Nat. Neurosci. 3, 1091–1097 10.1038/80606 [DOI] [PubMed] [Google Scholar]
- 53. Boyd A. W., Bartlett P. F., and Lackmann M. (2014) Therapeutic targeting of EPH receptors and their ligands. Nat. Rev. Drug Discov. 13, 39–62 10.1038/nrd4175 [DOI] [PubMed] [Google Scholar]
- 54. Singh D. R., Ahmed F., King C., Gupta N., Salotto M., Pasquale E. B., and Hristova K. (2015) EphA2 receptor unliganded dimers suppress EphA2 pro-tumorigenic signaling. J. Biol. Chem. 290, 27271–27279 10.1074/jbc.M115.676866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chan B., and Sukhatme V. P. (2009) Receptor tyrosine kinase EphA2 mediates thrombin-induced upregulation of ICAM-1 in endothelial cells in vitro. Thromb. Res. 123, 745–752 10.1016/j.thromres.2008.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. LaRusch G. A., Mahdi F., Shariat-Madar Z., Adams G., Sitrin R. G., Zhang W. M., McCrae K. R., and Schmaier A. H. (2010) Factor XII stimulates ERK1/2 and Akt through uPAR, integrins, and the EGFR to initiate angiogenesis. Blood 115, 5111–5120 10.1182/blood-2009-08-236430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Maretzky T., Evers A., Zhou W., Swendeman S. L., Wong P.-M., Rafii S., Reiss K., and Blobel C. P. (2011) Migration of FGF7-stimulated epithelial cells and VEGF-A-stimulated HUVECs depends on EGFR transactivation by ADAM17. Nat. Commun. 2, 229 10.1038/ncomms1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Larsen A. B., Pedersen M. W., Stockhausen M. T., Grandal M. V., van Deurs B., and Poulsen H. S. (2007) Activation of the EGFR gene target EphA2 inhibits epidermal growth factor-induced cancer cell motility. Mol. Cancer Res. 5, 283–293 10.1158/1541-7786.MCR-06-0321 [DOI] [PubMed] [Google Scholar]
- 59. De Robertis M., Loiacono L., Fusilli C., Poeta M. L., Mazza T., Sanchez M., Marchionni L., Signori E., Lamorte G., Vescovi A. L., Garcia-Foncillas J., and Fazio V. M. (2017) Dysregulation of EGFR pathway in EphA2 cell subpopulation significantly associates with poor prognosis in colorectal cancer. Clin. Cancer Res. 23, 159–170 10.1158/1078-0432.CCR-16-0709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. King C., Sarabipour S., Byrne P., Leahy D. J., and Hristova K. (2014) The FRET signatures of noninteracting proteins in membranes: simulations and experiments. Biophys. J. 106, 1309–1317 10.1016/j.bpj.2014.01.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. King C., Raicu V., and Hristova K. (2017) Understanding the FRET signatures of interacting membrane proteins. J. Biol. Chem. 292, 5291–5310 10.1074/jbc.M116.764282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Weber J. R., Ørstavik S., Torgersen K. M., Danbolt N. C., Berg S. F., Ryan J. C., Taskén K., Imboden J. B., and Vaage J. T. (1998) Molecular cloning of the cDNA encoding pp36, a tyrosine-phosphorylated adaptor protein selectively expressed by T cells and natural killer cells. J. Exp. Med. 187, 1157–1161 10.1084/jem.187.7.1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zhang W., Sloan-Lancaster J., Kitchen J., Trible R. P., and Samelson L. E. (1998) LAT: the ZAP-70 tyrosine kinase substrate that links T cell receptor to cellular activation. Cell 92, 83–92 10.1016/S0092-8674(00)80901-0 [DOI] [PubMed] [Google Scholar]
- 64. Bunnell S. C., Kapoor V., Trible R. P., Zhang W., and Samelson L. E. (2001) Dynamic actin polymerization drives T cell receptor–induced spreading: a role for the signal transduction adaptor LAT. Immunity 14, 315–329 10.1016/S1074-7613(01)00112-1 [DOI] [PubMed] [Google Scholar]
- 65. Adámková L., Kvíčalová Z., Rozbeský D., Kukačka Z., Adámek D., Cebecauer M., and Novák P. (2019) Oligomeric architecture of mouse activating Nkrp1 receptors on living cells. Int. J. Mol. Sci. 20, 1884 10.3390/ijms20081884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Qiu C., Tarrant M. K., Boronina T., Longo P. A., Kavran J. M., Cole R. N., Cole P. A., and Leahy D. J. (2009) In vitro enzymatic characterization of near full length EGFR in activated and inhibited states. Biochemistry 48, 6624–6632 10.1021/bi900755n [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ferrara N., Gerber H.-P., and LeCouter J. (2003) The biology of VEGF and its receptors. Nat. Med. 9, 669–676 10.1038/nm0603-669 [DOI] [PubMed] [Google Scholar]
- 68. Chen J., Hicks D., Brantley-Sieders D., Cheng N., McCollum G. W., Qi-Werdich X., and Penn J. (2006) Inhibition of retinal neovascularization by soluble EphA2 receptor. Exp. Eye Res. 82, 664–673 10.1016/j.exer.2005.09.004 [DOI] [PubMed] [Google Scholar]
- 69. Hess A. R., Margaryan N. V., Seftor E. A., and Hendrix M. J. (2007) Deciphering the signaling events that promote melanoma tumor cell vasculogenic mimicry and their link to embryonic vasculogenesis: role of the Eph receptors. Dev. Dyn. 236, 3283–3296 10.1002/dvdy.21190 [DOI] [PubMed] [Google Scholar]
- 70. Baharuddin W. N. A., Yusoff A. A. M., Abdullah J. M., Osman Z. F., and Ahmad F. (2018) Roles of EphA2 receptor in angiogenesis signaling pathway of glioblastoma multiforme. Malays. J. Med. Sci. 25, 22–27 10.21315/mjms2018.25.6.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Larsen A. K., Ouaret D., El Ouadrani K., and Petitprez A. (2011) Targeting EGFR and VEGF(R) pathway cross-talk in tumor survival and angiogenesis. Pharmacol. Ther. 131, 80–90 10.1016/j.pharmthera.2011.03.012 [DOI] [PubMed] [Google Scholar]
- 72. Goldman C. K., Kim J., Wong W., King V., Brock T., and Gillespie G. (1993) Epidermal growth factor stimulates vascular endothelial growth factor production by human malignant glioma cells: a model of glioblastoma multiforme pathophysiology. Mol. Biol. Cell 4, 121–133 10.1091/mbc.4.1.121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Pore N., Jiang Z., Gupta A., Cerniglia G., Kao G. D., and Maity A. (2006) EGFR tyrosine kinase inhibitors decrease VEGF expression by both hypoxia-inducible factor (HIF)-1–independent and HIF-1–dependent mechanisms. Cancer Res. 66, 3197–3204 10.1158/0008-5472.CAN-05-3090 [DOI] [PubMed] [Google Scholar]
- 74. Amin D. N., Bielenberg D. R., Lifshits E., Heymach J. V., and Klagsbrun M. (2008) Targeting EGFR activity in blood vessels is sufficient to inhibit tumor growth and is accompanied by an increase in VEGFR-2 dependence in tumor endothelial cells. Microvasc. Res. 76, 15–22 10.1016/j.mvr.2008.01.002 [DOI] [PubMed] [Google Scholar]
- 75. Naumov G. N., Nilsson M. B., Cascone T., Briggs A., Straume O., Akslen L. A., Lifshits E., Byers L. A., Xu L., Wu H.-K., Jänne P., Kobayashi S., Halmos B., Tenen D., Tang X. M., et al. (2009) Combined vascular endothelial growth factor receptor and epidermal growth factor receptor (EGFR) blockade inhibits tumor growth in xenograft models of EGFR inhibitor resistance. Clin. Cancer Res. 15, 3484–3494 10.1158/1078-0432.CCR-08-2904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Cascone T., Herynk M. H., Xu L., Du Z., Kadara H., Nilsson M. B., Oborn C. J., Park Y.-Y., Erez B., Jacoby J. J., Lee J.-S., Lin H. Y., Ciardiello F., Herbst R. S., Langley R. R., et al. (2011) Upregulated stromal EGFR and vascular remodeling in mouse xenograft models of angiogenesis inhibitor–resistant human lung adenocarcinoma. J. Clin. Invest. 121, 1313–1328 10.1172/JCI42405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Singh D. R., King C., Salotto M., and Hristova K. (2019) Revisiting a controversy: the effect of EGF on EGFR dimer stability. Biochim. Biophys. Acta Biomembr. 1862, 183015 10.1016/j.bbamem.2019.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Sarabipour S., and Hristova K. (2016) Mechanism of FGF receptor dimerization and activation. Nat. Commun. 7, 10262 10.1038/ncomms10262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Süveges D., and Jura N. (2015) Structural features of the kinase domain. Receptor Tyrosine Kinases: Structure, Functions and Role in Human Disease (Wheeler D. L., and Yarden Y., eds) pp. 195–223, Humana Press, Totowa, NJ [Google Scholar]
- 80. Yarden Y., and Ullrich A. (1988) Growth factor receptor tyrosine kinases. Annu. Rev. Biochem. 57, 443–478 10.1146/annurev.bi.57.070188.002303 [DOI] [PubMed] [Google Scholar]
- 81. Singh D. R., Ahmed F., Paul M. D., Gedam M., Pasquale E. B., and Hristova K. (2017) The SAM domain inhibits EphA2 interactions in the plasma membrane. Biochim. Biophys. Acta 1864, 31–38 10.1016/j.bbamcr.2016.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Eswarakumar V. P., Lax I., and Schlessinger J. (2005) Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 16, 139–149 10.1016/j.cytogfr.2005.01.001 [DOI] [PubMed] [Google Scholar]
- 83. Ornitz D. M., and Itoh N. (2015) The fibroblast growth factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 4, 215–266 10.1002/wdev.176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Muenke M., and Schell U. (1995) Fibroblast-growth-factor receptor mutations in human skeletal disorders. Trends Genet. 11, 308–313 10.1016/S0168-9525(00)89088-5 [DOI] [PubMed] [Google Scholar]
- 85. Yokote H., Fujita K., Jing X., Sawada T., Liang S., Yao L., Yan X., Zhang Y., Schlessinger J., and Sakaguchi K. (2005) Trans-activation of EphA4 and FGF receptors mediated by direct interactions between their cytoplasmic domains. Proc. Natl. Acad. Sci. U.S.A. 102, 18866–18871 10.1073/pnas.0509741102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Sawada T., Jing X., Zhang Y., Shimada E., Yokote H., Miyajima M., and Sakaguchi K. (2010) Ternary complex formation of EphA4, FGFR and FRS2α plays an important role in the proliferation of embryonic neural stem/progenitor cells. Genes Cells 15, 297–311 10.1111/j.1365-2443.2010.01391.x [DOI] [PubMed] [Google Scholar]
- 87. Schlessinger J. (1986) Allosteric regulation of the epidermal growth factor receptor kinase. J. Cell Biol. 103, 2067–2072 10.1083/jcb.103.6.2067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Schlessinger J. (1988) The epidermal growth factor receptor as a multifunctional allosteric protein. Biochemistry 27, 3119–3123 10.1021/bi00409a002 [DOI] [PubMed] [Google Scholar]
- 89. Klein P., Mattoon D., Lemmon M. A., and Schlessinger J. (2004) A structure-based model for ligand binding and dimerization of EGF receptors. Proc. Natl. Acad. Sci. U.S.A. 101, 929–934 10.1073/pnas.0307285101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Macdonald J. L., and Pike L. J. (2008) Heterogeneity in EGF-binding affinities arises from negative cooperativity in an aggregating system. Proc. Natl. Acad. Sci. U.S.A. 105, 112–117 10.1073/pnas.0707080105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Miao H., Burnett E., Kinch M., Simon E., and Wang B. (2000) Activation of EphA2 kinase suppresses integrin function and causes focal-adhesion-kinase dephosphorylation. Nat. Cell Biol. 2, 62–69 10.1038/35000008 [DOI] [PubMed] [Google Scholar]
- 92. Singh D. R., Kanvinde P., King C., Pasquale E. B., and Hristova K. (2018) The EphA2 receptor is activated through induction of distinct, ligand-dependent oligomeric structures. Commun. Biol. 1, 15 10.1038/s42003-018-0017-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Phizicky E. M., and Fields S. (1995) Protein-protein interactions: methods for detection and analysis. Microbiol. Rev. 59, 94–123 10.1128/MMBR.59.1.94-123.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Bonifacino J. S., Dell'Angelica E. C., and Springer T. A. (2001) Immunoprecipitation. Curr. Protoc. Immunol. 41, 8.3.1–8.3.28 10.1002/0471142735.im0803s41 [DOI] [PubMed] [Google Scholar]
- 95. Lundqvist G., Yücel-Lindberg T., and Morgenstern R. (1992) The oligomeric structure of rat liver microsomal glutathione transferase studied by chemical cross-linking. Biochim. Biophys. Acta 1159, 103–108 10.1016/0167-4838(92)90081-N [DOI] [PubMed] [Google Scholar]
- 96. Kluger R., and Alagic A. (2004) Chemical cross-linking and protein–protein interactions—a review with illustrative protocols. Bioorg. Chem. 32, 451–472 10.1016/j.bioorg.2004.08.002 [DOI] [PubMed] [Google Scholar]
- 97. Fredriksson S., Gullberg M., Jarvius J., Olsson C., Pietras K., Gústafsdóttir S. M., Östman A., and Landegren U. (2002) Protein detection using proximity-dependent DNA ligation assays. Nat. Biotechnol. 20, 473–477 10.1038/nbt0502-473 [DOI] [PubMed] [Google Scholar]
- 98. Gullberg M., Gústafsdóttir S. M., Schallmeiner E., Jarvius J., Bjarnegård M., Betsholtz C., Landegren U., and Fredriksson S. (2004) Cytokine detection by antibody-based proximity ligation. Proc. Natl. Acad. Sci. U.S.A. 101, 8420–8424 10.1073/pnas.0400552101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Söderberg O., Gullberg M., Jarvius M., Ridderstrale K., Leuchowius K. J., Jarvius J., Wester K., Hydbring P., Bahram F., Larsson L. G., and Landegren U. (2006) Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods 3, 995–1000 10.1038/nmeth947 [DOI] [PubMed] [Google Scholar]
- 100. Söderberg O., Leuchowius K. J., Gullberg M., Jarvius M., Weibrecht I., Larsson L. G., and Landegren U. (2008) Characterizing proteins and their interactions in cells and tissues using the in situ proximity ligation assay. Methods 45, 227–232 10.1016/j.ymeth.2008.06.014 [DOI] [PubMed] [Google Scholar]
- 101. Lemmon M. A., and Engelman D. M. (1994) Specificity and promiscuity in membrane helix interactions. Q. Rev. Biophys. 27, 157–218 10.1017/s0033583500004522 [DOI] [PubMed] [Google Scholar]
- 102. Li E., and Hristova K. (2010) Receptor tyrosine kinase transmembrane domains: function, dimer structure, and dimerization energetics. Cell Adh. Migr. 4, 249–254 10.4161/cam.4.2.10725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Zhang X., Gureasko J., Shen K., Cole P. A., and Kuriyan J. (2006) An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 125, 1137–1149 10.1016/j.cell.2006.05.013 [DOI] [PubMed] [Google Scholar]
- 104. Evers T. H., van Dongen E. M., Faesen A. C., Meijer E., and Merkx M. (2006) Quantitative understanding of the energy transfer between fluorescent proteins connected via flexible peptide linkers. Biochemistry 45, 13183–13192 10.1021/bi061288t [DOI] [PubMed] [Google Scholar]
- 105. Chen L., Placone J., Novicky L., and Hristova K. (2010) The extracellular domain of fibroblast growth factor receptor 3 inhibits ligand-independent dimerization. Sci. Signal. 3, ra86 10.1126/scisignal.2001195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Del Piccolo N., and Hristova K. (2017) Quantifying the interaction between EGFR dimers and Grb2 in live cells. Biophys. J. 113, 1353–1364 10.1016/j.bpj.2017.06.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Sinha B., Köster D., Ruez R., Gonnord P., Bastiani M., Abankwa D., Stan R. V., Butler-Browne G., Vedie B., Johannes L., Morone N., Parton R. G., Raposo G., Sens P., Lamaze C., et al. (2011) Cells respond to mechanical stress by rapid disassembly of caveolae. Cell 144, 402–413 10.1016/j.cell.2010.12.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Raicu V., Stoneman M. R., Fung R., Melnichuk M., Jansma D. B., Pisterzi L. F., Rath S., Fox M., Wells J. W., and Saldin D. K. (2009) Determination of supramolecular structure and spatial distribution of protein complexes in living cells. Nat. Photonics 3, 107–113 10.1038/nphoton.2008.291 [DOI] [Google Scholar]
- 109. Biener G., Stoneman M. R., Acbas G., Holz J. D., Orlova M., Komarova L., Kuchin S., and Raicu V. (2013) Development and experimental testing of an optical micro-spectroscopic technique incorporating true line-scan excitation. Int. J. Mol. Sci. 15, 261–276 10.3390/ijms15010261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Sarabipour S., King C., and Hristova K. (2014) Uninduced high-yield bacterial expression of fluorescent proteins. Anal. Biochem. 449, 155–157 10.1016/j.ab.2013.12.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Wolber P., and Hudson B. (1979) An analytic solution to the Förster energy transfer problem in two dimensions. Biophys. J. 28, 197–210 10.1016/S0006-3495(79)85171-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Chen L. R., Novicky L., Merzlyakov M., Hristov T., and Hristova K. (2010) Measuring the energetics of membrane protein dimerization in mammalian membranes. J. Am. Chem. Soc. 132, 3628–3635 10.1021/ja910692u [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All raw FRET data are available free of charge upon request from Kalina Hristova (kh@jhu.edu). All other data are contained within the article.