

Abstract
Retinoid X receptor (RXR) plays a pivotal role as a transcriptional regulator and serves as an obligatory heterodimerization partner for at least 20 other nuclear receptors (NRs). Given a potentially limiting/sequestered pool of RXR and simultaneous expression of several RXR partners, we hypothesized that NRs compete for binding to RXR and that this competition is directed by specific agonist treatment. Here, we tested this hypothesis on three NRs: peroxisome proliferator-activated receptor gamma (PPARγ), vitamin D receptor (VDR), and retinoic acid receptor alpha (RARα). The evaluation of competition relied on a nuclear translocation assay applied in a three-color imaging model system by detecting changes in heterodimerization between RXRα and one of its partners (NR1) in the presence of another competing partner (NR2). Our results indicated dynamic competition between the NRs governed by two mechanisms. First, in the absence of agonist treatment, there is a hierarchy of affinities between RXRα and its partners in the following order: RARα > PPARγ > VDR. Second, upon agonist treatment, RXRα favors the liganded partner. We conclude that recruiting RXRα by the liganded NR not only facilitates a stimulus-specific cellular response but also might impede other NR pathways involving RXRα.
Keywords: nuclear receptors, nuclear transport, heterodimerization, retinoic acid receptor (RAR), retinoid X receptor (RXR), peroxisome proliferator‐activated receptor (PPAR), vitamin D receptor (VDR), transcription factor, confocal microscopy, nuclear receptor, dimerization, retinoid, vitamin D, transcription regulation, ChIP sequencing (ChIP-seq)
Retinoid X receptor (RXR) plays a pivotal role as a transcriptional regulator. It serves as an obligatory heterodimerization partner for many other nuclear receptors (NRs) (1). Activation of RXR heterodimers exerts transcriptional activity controlling a wide variety of important biological processes, such as development, cell differentiation, metabolism, and cell death (2). Consequently, dysfunction of these signaling pathways may result in proliferative and metabolic diseases, like cancer, obesity, and diabetes (3), and makes nuclear receptors promising therapeutic targets (4, 5).
Nuclear receptors share a common structure composed of several functional domains: a ligand-independent N-terminal transcription activation function domain (AF-1), a DNA-binding domain (DBD), a flexible hinge region, and a ligand-binding domain (LBD), also functioning as a ligand-dependent transcription activation function domain (AF-2) at the C terminus (6, 7). The main function of the DBD is to bind to a specific DNA sequence called a response element (RE) (8). RXR heterodimers recognize REs composed of two AGGTCA half-sites arranged as direct repeats (DRs) and separated by a spacer of zero to eight base pairs, termed DR0 to DR8 (9, 10). Moreover, the DBD also contributes to receptor heterodimerization and harbors between its two zinc finger motifs a nuclear localization signal, termed NLS1, mediating the nuclear localization of the receptors (11). The LBD accommodates the core of NR actions, because it contains the ligand-binding pocket and regions that mediate dimer formation, coregulator binding, and ligand-dependent transactivation (12).
The mechanism of activation, called the molecular switch, is also a common characteristic of these receptors (13). In the absence of the cognate ligand, the heterodimeric complex of RXR and its partner binds to DNA in its apo form and recruits corepressors, leading to a transcriptionally repressed state. The liganded receptors change their conformation to their holo form, which reduces their affinity for corepressors and leads to coactivator binding and transcription initiation (14). This similarity in structure, mechanism of activation, and sharing of coregulators suggest cross-talk between the members of this NR family and a competition for the common binding partners, i.e. for RXR, coactivator, or DNA binding sites (15).
Numerous fluorescence microscopy studies have analyzed the molecular mechanism of NR activation by observing protein-protein and protein-DNA interactions and dynamics (16–23). Homodimerization of RXR-LBD is enhanced upon 9-cis-retinoic acid treatment, as demonstrated by fluorescence fluctuation analysis (24). Previously we have shown by fluorescence correlation spectroscopy that specific agonists increase chromatin binding of RARα and RXRα in a coactivator-dependent manner (25, 26). We have shown by light-sheet microscopy-based FRET and fluorescence cross-correlation measurements (SPIM-FRET-FCCS) that dimerization of RARα and RXRα and chromatin binding of the dimer are enhanced upon agonist treatment (27).
Typically, multiple heterodimeric partners of RXR are present in certain cell types. In cells where there is a limiting or sequestered pool of RXR (13, 15, 28, 29) combined with the expression of several RXR heterodimerization partners, the mechanism by which RXR partner selection is mediated remains unclear. We hypothesized that there is a competition between RXR partners for binding to RXR and that binding of a specific agonist increases the affinity of a given receptor to RXR and favors their heterodimerization. RXRα and three NR partners were included in this study: PPARγ, VDR, and RARα.
The present work was undertaken to better understand RXRα partitioning between its different heterodimeric partners in the absence and presence of their cognate agonists. This may have pharmacological implications by improving the current usage of the present drugs in triggering specific signaling pathways and may explain how targeting one NR pathway can interfere with a seemingly distinct NR pathway.
Results
Subcellular distribution of WT and NLS mutant NRs: effect of agonist treatment and coexpression with RXRα
We hypothesized that competition for heterodimerization with RXR is dictated, at least in part, by the availability of the partner-specific agonist. To test this hypothesis, we developed a robust system relying on a nuclear translocation assay applied in a three-color imaging model system by detecting changes in heterodimerization between RXRα and one of its partners (NR1) in the presence of another competing partner (NR2). To this end, NR1 was present in a form showing homogeneous distribution when expressed alone and translocating into the nucleus when interacting with RXRα (for details, see Fig. 1 and Experimental procedures). In this system, fluorescent proteins attached N-terminally to the NRs: TagBFP, EGFP, and mCherry (indicated as prefixes B-, G-, and C-, respectively, to the NRs throughout the text).
Figure 1.

Schematic representation of the translocation assay in a three-color imaging model system. I, single expression of NRs; distribution of NR1, RXRα, and NR2 expressed alone in WT HEK293 cells. A, PPARγ/nlsm (NLS mutant form) as an example of NR1 having a homogeneous, nucleocytoplasmic distribution. B, WT RXRα having nuclear localization. C, WT RARα as an example of NR2 having nuclear localization. II, double expression; NR1 or NR2 expressed in HEK293B-RXRα. D, nuclear redistribution of NR1 in HEK293B-RXRα. E, nuclear distribution of NR2 in HEK293B-RXRα. III, triple expression of NR1 + RXRα + NR2 to detect the competition between NR1 and NR2 for heterodimerization with RXRα, in the absence or presence of specific ligands of NR1 and/or NR2. F, NR1 is expected to translocate to the nucleus if NR1 rather than NR2 is the preferred heterodimerization partner of RXRα. G, NR1 is expected to be redistributed homogeneously if NR2 is the preferred heterodimerization partner of RXRα rather than NR1.
To minimize the influence of endogenously expressed NRs on the observed results, we used wt HEK293 cells, which express the NRs involved in our study at low/medium levels. Using publicly available RNA-seq data from WT HEK-293T cells (30), we plotted the average gene expression values of the studied NRs (Fig. S1E). We also generated a stable cell line, HEKB-RXRα, overexpressing B-RXRα. According to our immunofluorescence and Western blot analyses, the endogenous RXR expression level in WT HEK293 is ca. one-fourth the level in HEKB-RXRα (Fig. S1A–D).
First, we expressed NRs tagged with EGFP and studied their changes of localization in response to the addition of ligands and coexpression of RXRα using the WT HEK293 and HEK293B-RXRα cell lines. G-PPARγ and G-RARα were localized mainly in the nucleus (Fig. 2A), even in the absence of exogenously added RXRα or agonist. To impede the nuclear transport of these NRs in the absence of RXRα, we induced a mutation in the nuclear localization signal, NLS1 (denoted /nlsm).
Figure 2.

Subcellular distribution of NRs used in this study. A, representative confocal images show the nuclear localization of G-PPARγ and G-RARα in HEK293 cells. B, representative confocal images of NR1s. The NLS mutants of G-PPARγ/nlsm and G-RARα/nlsm and WT G-VDR transiently transfected into WT HEK293 (top) or HEK293B-RXRα cells stably expressing B-RXRα (bottom) in the absence (left) or presence (right) of specific NR1 agonists (10−6 m RSG, 10−7 m AM580, and 10−7 m cal) is shown. C, Nuclear-to-cytoplasmic fluorescence intensity/pixel ratios (NCR) of NR1; ***, p < 0.0001 compared with NR1 distribution in WT HEK293; ns, not significant. D, representative confocal image shows the nuclear localization of B-RXRα forms, B-RXRα and the mutants B-RXRα/znm and B-RXRα/nlsm in WT HEK293. E, NCR of B-RXRα forms. F, effect of calcitriol on governing the nuclear localization of B-RXRα/nlsm cotransfected with G-VDR. Box-and-whiskers plots represent 10th, 25th, 50th, 75th, and 90th percentiles; +, mean value. G, EGFP; B, TagBFP; C, mCherry. Scale bar, 10 μm. ***, p < 0.0001.
We then observed the subcellular distribution of these NLS mutant receptors (G-PPARγ/nlsm and G-RARα/nlsm) in WT HEK293 cells and studied the effect of specific agonist treatment (rosiglitazone [RSG] and AM580, respectively) on their localization. As shown in Fig. 2C, the nuclear-to-cytoplasmic ratio (NCR) for G-PPARγ/nlsm and G-RARα/nlsm was around 1 irrespective of ligand treatment, i.e. these homogeneously distributed mutant receptors did not show considerable enrichment in the nucleus.
We next transfected the NLS mutant receptors into HEK293B-RXRα cells to see the effect of dimerization with RXRα on their localization (Fig. 2B and C). We detected a 6-fold enrichment in the nucleus for G-PPARγ/nlsm and 5-fold for G-RARα/nlsm. Specific agonist treatment caused a further increase in the nuclear accumulation of these receptors.
Contrary to both G-PPARγ and G-RARα, G-VDR (expressed alone in WT HEK293 cells in the absence of agonist) was distributed in the cell with an NCR of ∼2 (Fig. 2C). Treatment with a specific agonist, calcitriol (cal), or coexpression of RXRα (using HEK293B-RXRα cells) enhanced the translocation of G-VDR to the nucleus, resulting in an NCR value of ∼3. Expressing G-VDR in HEK293B-RXRα combined with calcitriol treatment led to an even higher NCR value of ∼6 (Fig. 2B and C). Because WT VDR showed an increased localization in the nucleus in the presence of RXRα, it could be used in its WT form to detect heterodimerization with RXRα.
The next question was whether DNA binding of RXRα plays a role in the RXRα-dependent nuclear enrichment of NR1 or depends merely on heterodimerization. Therefore, a similar experiment was carried out in which RXRα was expressed either as a zinc finger mutant, RXRα/znm, or as an NLS mutant, RXRα/nlsm. RXRα/znm had a nuclear localization similar to that of WT RXRα despite lacking a direct DNA binding capability (which is mediated through the two zinc finger motifs in the DBD), whereas the mutation in the NLS1 of RXRα resulted in a homogeneous distribution of RXRα/nlsm (Fig. 2D and E).
Interestingly, RXRα/znm boosted translocation of NR1 into the nucleus just as effectively as WT RXRα did, whereas RXRα/nlsm failed to do so (Fig. 2C). Thus, nuclear enrichment of NR1 in the presence of RXRα is because of heterodimerization rather than binding to DNA. As we have learned before from Fig. 2B and C, calcitriol treatment alone could cause nuclear enrichment of its cognate receptor, VDR, and, intriguingly, when VDR was coexpressed with RXRα/nlsm, both of these homogeneously distributed receptors were enriched in the nucleus upon calcitriol treatment (Fig. 2F).
In summary, the mutation in NLS1 abolished the spontaneous nuclear accumulation of RARα and PPARγ but conserved their ability to heterodimerize with RXRα and to bind their ligands efficiently. Because G-PPARγ/nlsm, G-RARα/nlsm, and WT G-VDR had homogeneous distribution when expressed alone and moved to the nucleus when heterodimerizing with RXRα, irrespective of the presence or absence of ligand, they could serve as a suitable model system in our study (we refer to them later as NR1). Via a piggy-back mechanism (31), the NR1-RXRα heterodimeric complex translocated into the nucleus depending on the NLS of RXRα.
PPARγ heterodimerization with RXRα is abolished in the presence of RARα and restored by RSG
In all competition experiments, the combined expression level of NR1 and NR2 was higher than that of RXRα, (NR1 + NR2) > RXRα, resulting in a limiting pool of RXRα. This was assessed by comparing the relative fluorescence intensities of the fluorescent protein labels of the NRs to those of EGFP-mCherry and EGFP-TagBFP fusion proteins expressing the fluorescent proteins at a 1:1 ratio (see Experimental procedures).
Competition between PPARγ and RARα for binding to RXRα was assessed by tracing the distribution changes of G-PPARγ/nlsm (NR1) in HEK293B-RXRα in the presence of C-RARα (NR2) and cognate agonists. Nuclear accumulation of G-PPARγ/nlsm in HEK293B-RXRα was strikingly reduced when cells were additionally cotransfected with C-RARα; G-PPARγ/nlsm became homogeneously distributed with an NCR value of ∼1, similar to the case when expressed alone in WT HEK293. Treatment with RSG caused a 4-fold nuclear enrichment of G-PPARγ/nlsm, whereas AM580 or LG268 (RXR agonist) treatment kept G-PPARγ/nlsm homogeneously distributed (Fig. 3A and B). The challenge was to prove that dominance of RARα over PPARγ in competing for RXRα was not because of expressing mutant PPARγ and intact RARα; therefore, a complementary experiment was also carried out. Here, the changes in the distribution of G-RARα/nlsm (NR1) were traced in HEK293B-RXRα in the presence of C-PPARγ (NR2) and agonists. The results were consistent with our previous observation; C-PPARγ failed to inhibit binding of G-RARα/nlsm to B-RXRα. G-RARα/nlsm was still dominantly located in the nucleus with an NCR of ∼5.5, and this was further enhanced by AM580 treatment. On the other hand, RSG treatment facilitated the recovery of C-PPARγ binding to RXRα and led to a homogeneous redistribution of G-RARα/nlsm (Fig. 3C and D).
Figure 3.
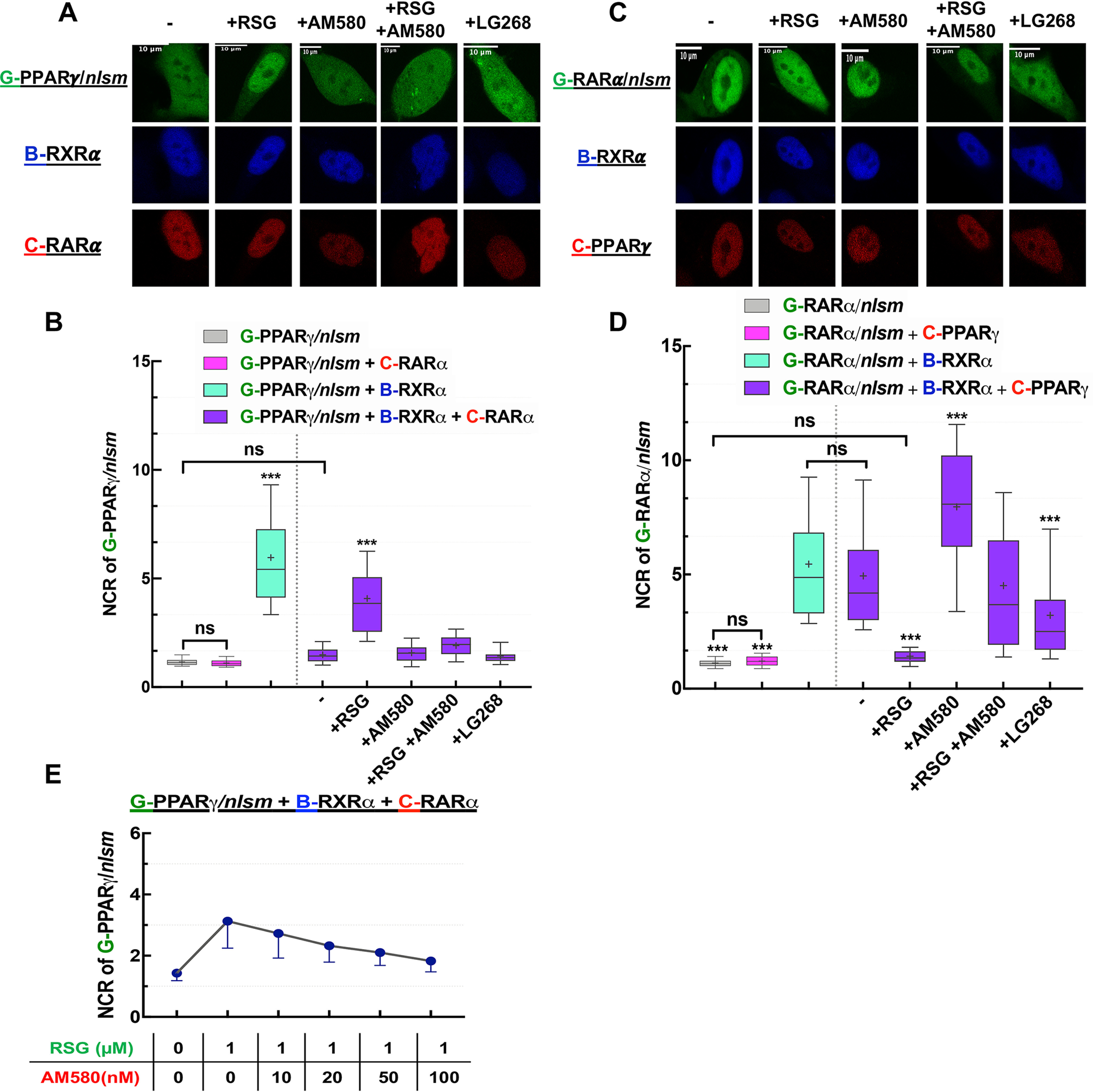
Competition between PPARγ and RARα: RXRα is more prone to heterodimerize with RARα than with PPARγ unless PPARγ gets liganded. A and C, representative confocal images show NR1 in green, RXRα in blue, and NR2 in red. Scale bar, 10 μm. A, changes in the distribution of G-PPARγ/nlsm in HEK293B-RXRα cotransfected with C-RARα were assessed in the absence or presence of agonists (10−6 m RSG, 10−7 m AM580, or 10−7 m LG268). C, analogously, changes in the distribution of G-RARα/nlsm were assessed in HEK293B-RXRα cotransfected with C-PPARγ. B and D, nuclear-to-cytoplasmic fluorescence intensity/pixel ratios (NCR) of NR1. ***, p < 0.0001 compared with NR1 distribution in HEK293B-RXRα cotransfected with NR2; −, nontreated sample; ns, not significant. E, dose-response curve showing that RXRα partner selection is dose dependent. Box-and-whiskers plots represent 10th, 25th, 50th, 75th, and 90th percentiles; +, mean value. G, EGFP; B, TagBFP; C, mCherry.
We were interested in how the affinities of liganded RARα and liganded PPARγ toward RXRα compare. To answer this question, we first treated our samples with saturating doses of RSG and AM580 simultaneously. G-RARα/nlsm and G-PPARγ/nlsm responded to the double treatment similarly to AM580 treatment alone, i.e. G-RARα/nlsm became nucleus localized, whereas G-PPARγ/nlsm remained homogeneous (Fig. 3). This implies that liganded RARα has a higher affinity toward RXRα than liganded PPARγ. We then cotreated the cells with RSG, the agonist of the weak partner, at its saturating dose (1 μm) and titrated AM580, the ligand of the dominant partner, from 0 to saturation, 100 nm. RSG-dependent nuclear enrichment of G-PPARγ/nlsm in HEK293B-RXRα cotransfected with C-RARα was abolished gradually with increasing doses of AM580 (Fig. 3E).
In summary, these data show that in the absence of an agonist, RARα has a higher binding affinity to RXRα than PPARγ, whereas treatment with a specific PPARγ agonist, RSG, tips the scale in favor of PPARγ.
RXRα is more likely to heterodimerize with RARα than with VDR unless calcitriol is present
We next investigated the competition between VDR and RARα in a similar way. The enrichment of G-RARα/nlsm in the nucleus of HEK293B-RXRα was reduced in the presence of C-VDR; the NCR decreased from 5.5 to 3.8, as shown in Fig. 4B. Treatment with agonists had a considerable effect on the redistribution of their cognate receptors. Whereas the nuclear accumulation of G-RARα/nlsm was augmented by AM580 treatment, it was abolished by calcitriol treatment, after which G-RARα/nlsm was distributed more homogeneously in HEK293B-RXRα, just as in WT HEK293. Simultaneous treatment with AM580 and calcitriol increased the NCR value to ∼6, even more than that of AM580 alone. LG268 treatment had no impact on this competition (Fig. 4A and B). Associated changes in the localization of C-VDR are shown in Fig. 4C; treatment with AM580 kept C-VDR in the cytoplasm, whereas calcitriol induced its nuclear translocation with an NCR of ∼5. Interestingly, double treatment with AM580 and calcitriol increased the NCR value to ∼6.6.
Figure 4.
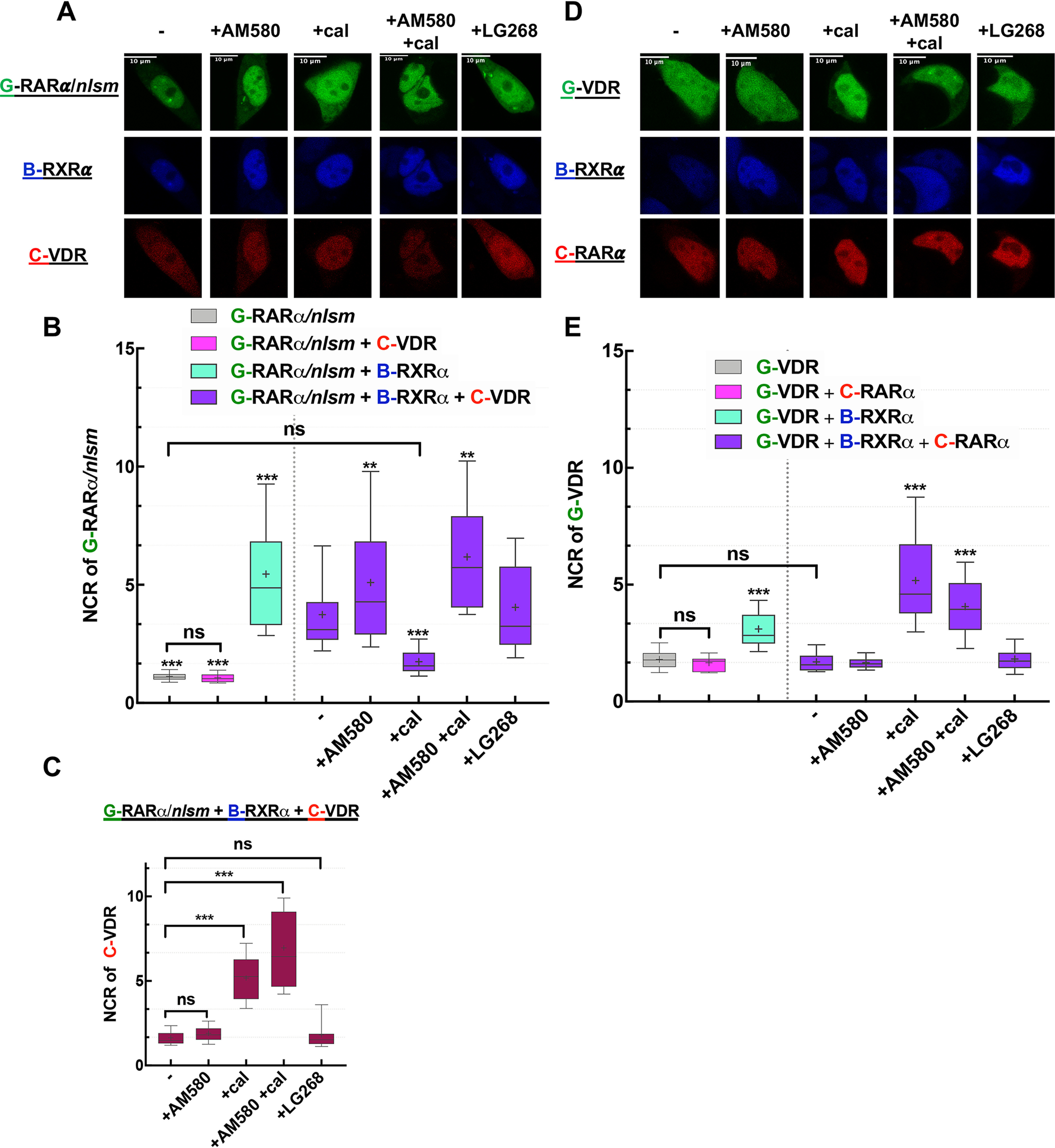
Competition between RARα and VDR: RXRα is more prone to heterodimerize with RARα than with VDR unless VDR gets liganded. A and D, representative confocal images show NR1 in green, RXRα in blue, and NR2 in red. Scale bar, 10 μm. A, changes in the distribution of G-RARα/nlsm in HEK293B-RXRα cotransfected with C-VDR were assessed in the absence or presence of treatment with agonists (10−7 m AM580, 10−7 m cal, or 10−7 m LG268). D, analogously, changes in the distribution of G-VDR were assessed in HEK293B-RXRα cotransfected with C-RARα. B and E, nuclear-to-cytoplasmic fluorescence intensity/pixel ratios (NCR) of NR1. ***, p < 0.0001; **, p < 0.01 compared with NR1 distribution in HEK293B-RXRα cotransfected with NR2; −, nontreated sample; ns, not significant. C, NCR values of NR2, C-VDR in HEK293B-RXRα cotransfected with G-RARα/nlsm. Box-and-whiskers plots represent 10th, 25th, 50th, 75th, and 90th percentiles; +, mean value. G, EGFP; B, TagBFP; C, mCherry.
The complementary experiment was also carried out as in the previous case: changes of G-VDR localization in HEK293B-RXRα were monitored in the presence of the competing partner, C-RARα. G-VDR failed to maintain its nuclear accumulation in HEK293B-RXRα when cells were cotransfected with C-RARα and redistributed homogeneously with NCR values similar to those in WT HEK293. Calcitriol treatment abolished the dominance of C-RARα over G-VDR in competing for RXRα and boosted the nuclear accumulation of VDR, resulting in an NCR value of ∼5, whereas calcitriol treatment in combination with AM580 resulted in an NCR of ∼4 (Fig. 4E).
In summary, RXRα is more likely to heterodimerize with RARα than with VDR, unless VDR gets liganded with its specific agonist, calcitriol.
PPARγ heterodimerization with RXRα is slightly reduced by VDR, abolished by calcitriol, and boosted by RSG
To investigate the competition between PPARγ and VDR, we first assessed changes of the NCR of G-PPARγ/nlsm in HEK293B-RXRα ensuing in the presence of C-VDR and specific agonists. As shown in Fig. 5A and B, without ligand, the nuclear accumulation of G-PPARγ/nlsm was diminished in cells coexpressing C-VDR (from an NCR of 6 to 3.7), although to a lesser extent than that because of C-RARα (Fig. 3B). Agonist treatment shifted competition between the NRs in favor of the liganded partner. RSG treatment induced G-PPARγ/nlsm enrichment in the nucleus of triply transfected cells, whereas calcitriol redistributed G-PPARγ/nlsm within HEK293B-RXRα cells with an NCR value equaling that measured in WT cells. Double treatment with RSG and calcitriol increased the NCR value of G-PPARγ/nlsm to ∼4.4, favoring again the binding of RXRα to G-PPARγ/nlsm and abolishing the effect of calcitriol. LG268 did not affect the competition: G-PPARγ/nlsm remained nucleus localized. Changes in the localization of C-VDR are shown in Fig. 5C. In these triply transfected cells, C-VDR was homogeneously distributed in the absence of ligand and upon RSG or LG268 treatment, whereas its NCR value increased to ∼4 after treatment with calcitriol alone and to ∼4.5 in combination with RSG.
Figure 5.
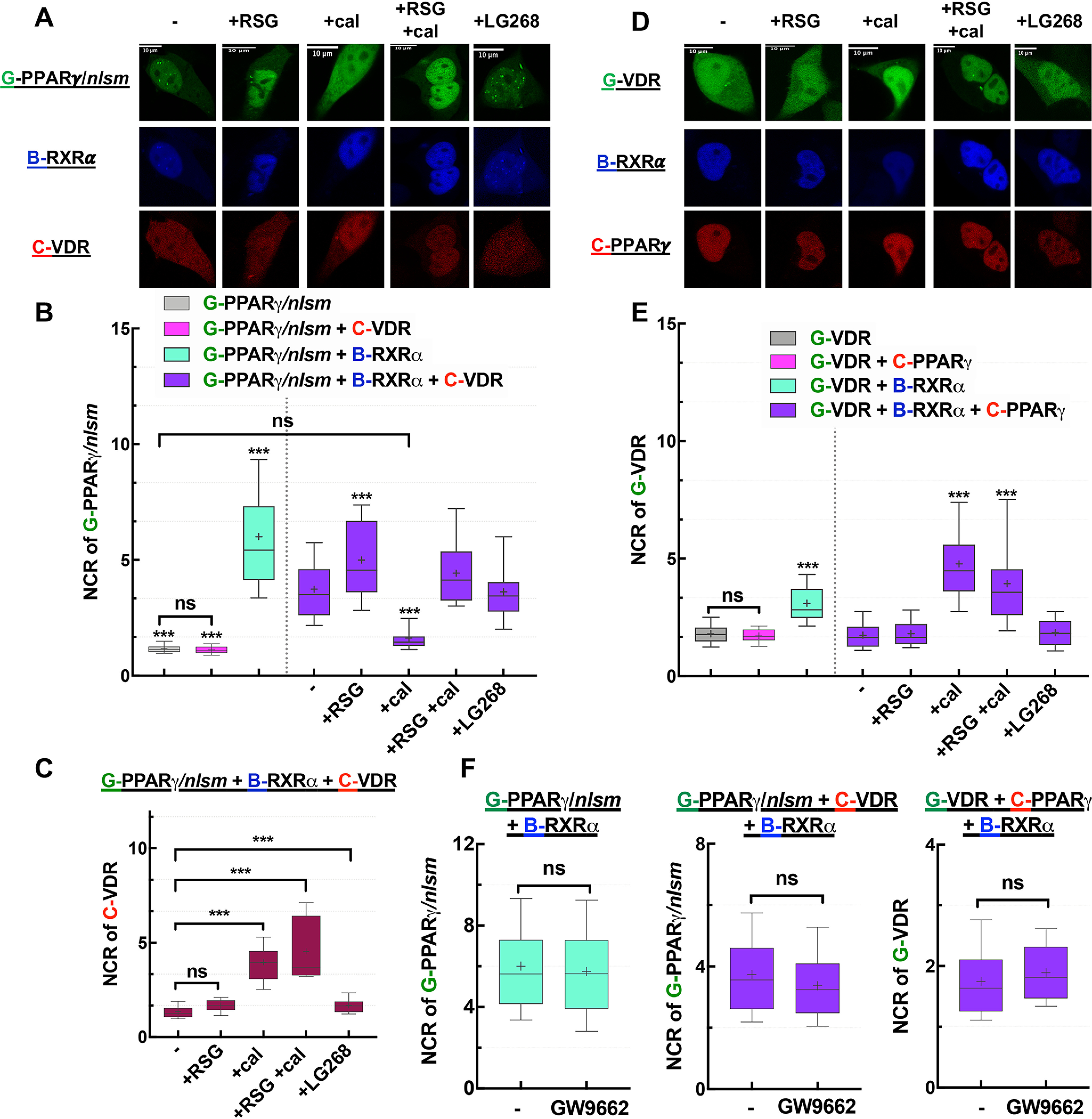
Competition between PPARγ and VDR: RXRα is more likely to heterodimerize with PPARγ than with VDR unless VDR gets liganded. A and D, representative confocal images show NR1 in green, RXRα in blue, and NR2 in red. Scale bar, 10 μm. A, changes in the distribution of G-PPARγ/nlsm in HEK293B-RXRα cotransfected with C-VDR were assessed in the absence or presence of treatment with agonists (10−6 m RSG, 10−7 m cal, or 10−7 m LG268). D, analogously, changes in the distribution of G-VDR were assessed in HEK293B-RXRα cotransfected with C-PPARγ. B and E, nuclear-to-cytoplasmic fluorescence intensity/pixel ratios (NCR) of NR1; ***, p < 0.0001 compared with NR1 distribution in HEK293B-RXRα cotransfected with NR2; −, nontreated sample; ns, not significant. C, NCR values of NR2, C-VDR in HEK293B-RXRα cotransfected with G-PPARγ/nlsm. F, PPARγ antagonist GW9662 (10−6 m) did not change the NCR of NR1 (compared with nontreated samples). Box-and-whiskers plots represent 10th, 25th, 50th, 75th, and 90th percentiles; +, mean value. G, EGFP; B, TagBFP; C, mCherry.
Second, in the complementary experiment, we traced localization changes of G-VDR in HEK293B-RXRα cells cotransfected with C-PPARγ and treated with the above-mentioned agonists (Fig. 5D and E). Without agonist treatment, the NCR of G-VDR was ∼2, similar to its value in WT HEK293. RSG or LG268 treatment kept the NCR of VDR low, impeding the interaction between VDR and RXRα, whereas calcitriol boosted the NCR of G-VDR to ∼4.8 when applied alone and to ∼4 when in combination with RSG.
To exclude the effect of endogenously produced PPARγ ligands on our results, we also used a PPARγ antagonist (GW9662) to displace endogenous ligands. This antagonist enhances corepressor binding and stabilizes PPARγ in its apo (nonliganded) conformation (32). The dominance of PPARγ over VDR was retained even when GW9662 was present (Fig. 5F), suggesting that this dominance was not because of binding endogenous agonists.
In summary, RSG treatment favors RXRα heterodimerization with PPARγ and calcitriol augments VDR binding to RXRα, whereas without agonists RXRα binds to PPARγ with a higher affinity.
Competition of VDR/nlsm, as NR1, with RARα or PPARγ
In contrast to NLS mutants of PPARγ and RARα, G-VDR/nlsm showed a cytoplasmic distribution in WT HEK293 cells (NCR of ∼0.6) and remained so even in HEK293B-RXRα cells (Fig. 6A and B). Intriguingly, even RXRα followed G-VDR/nlsm to the cytoplasm, indicating that G-VDR/nlsm was competent to heterodimerize with RXRα but the heterodimeric complex failed to translocate to the nucleus. Therefore, we also traced changes in the localization of B-RXRα to detect the presence or absence of heterodimerization with G-VDR/nlsm (Fig. 6C). Specific agonist treatment had an interesting effect. In WT HEK293, the NCR of G-VDR/nlsm was doubled upon treatment with calcitriol, reaching a value of ∼1.3 (Fig. 6B). Moreover, both G-VDR/nlsm and RXRα became enriched in the nucleus of HEK293B-RXRα cells upon calcitriol treatment; the NCR of G-VDR/nlsm increased to ∼3.3 (Fig. 6B), and that of B-RXRα was restored to ∼5, close to its normal value (Fig. 6C).
Figure 6.

EGFP-VDR/nlsm as NR1 in competition with RARα and PPARγ: both RARα and PPARγ dominate over VDR/nlsm in competing for dimerization with RXRα. A, representative confocal images of G-VDR/nlsm transiently transfected into WT HEK293 (left) or HEK293B-RXRα cells (right) in the absence (top) or presence (bottom) of specific NR1 agonists (10−7 M cal). Scale bar, 10 μm. B, nuclear-to-cytoplasmic fluorescence intensity/pixel ratios (NCR) of G-VDR/nlsm. C, NCR values of B-RXRα. D, NCR of G-VDR/nlsm in HEK293B-RXRα cotransfected with C-RARα were assessed in the absence or presence of agonists (10−7 m AM580, 10−7 m cal, or 10−7 m LG268). E, changes in the distribution of G-VDR/nlsm in HEK293B-RXRα cotransfected with C-PPARγ were assessed in the absence or presence of agonists (10−6 m RSG, 10−7 m cal, or 10−7 m LG268). Box-and-whiskers plots represent 10th, 25th, 50th, 75th, and 90th percentiles; +, mean value. G, EGFP; B, TagBFP; C, mCherry. *, p < 0.05; ***, p < 0.0001; ns, not significant.
In competition experiments between G-VDR/nlsm and C-RARα in HEK293B-RXRα cells, RXRα was enriched in the nucleus together with PPARγ rather than being sequestered in the cytoplasm with VDR/nlsm, suggesting the preference of RXRα for RARα over VDR (Fig. 6D).
Similarly, when G-VDR/nlsm and C-PPARγ were cotransfected in HEK293B-RXRα cells, RXR was trapped in the nucleus where PPARγ resided rather than colocalizing with VDR/nlsm in the cytoplasm. This also indicated the dominance of PPARγ over VDR (Fig. 6E).
In both experiments, G-VDR/nlsm became slightly enriched in the nucleus of HEK293B-RXRα after treatment with calcitriol (Fig. 6D and E).
In summary, results on VDR/nlsm without ligand treatment are in accordance with those gained with WT VDR, indicating the dominance of both RARα and PPARγ over VDR. The role of calcitriol in the competition is less obvious in the case of VDR/nlsm.
Overexpression of RXRα abrogates competition between its potential heterodimerization partners
We have shown that NRs compete for RXRα and revealed differences in the binding affinities between RXRα and its partners. Competition was observed in a situation where the combined expression levels of NR1 and NR2 were larger than that of RXRα alone; therefore, RXRα was limiting.
To assess whether the competition for RXRα is the primary cause for our observed results, experiments were repeated in cells expressing less NR1 and NR2. Thus, HEK293B-RXRα cells were transfected with half the amount of the plasmids of NR1 and NR2 used previously, and experiments were carried out 24 h rather than 48 h after transfection. In the new model system, RXRα is not limiting anymore: RXRα > (NR1 + NR2). The dominance of RARα over PPARγ was abolished because RXRα became sufficient for both partners (Fig. 7A). G-PPARγ/nlsm in HEK293B-RXRα was enriched in the nucleus with an NCR of ∼5, very close to the case where C-RARα was absent. This supports our previous conclusions. The homogeneous distribution of G-PPARγ/nlsm in HEK293B-RXRα cotransfected with C-RARα is because of monopolizing of RXRα by the dominant partner, RARα, when RXRα is limiting. In the same manner, we showed how the reduced amount of heteroassociation partners relative to RXRα abolished the dominance of RARα (Fig. 7B) and PPARγ (Fig. 7C) over VDR.
Figure 7.

Increased availability of RXRα abolishes the competition between NRs for RXRα heterodimerization. Distribution of NR1 in WT HEK293 is shown in gray, in HEK293B-RXRα cells in light green, and in HEK293B-RXRα cells cotransfected with NR2 in blue. In the first blue box, HEK293B-RXRα cells were transfected with 80 ng of each of NR1 and NR2, and experiments were carried out 48 h after transfection; these conditions result in a limiting pool of RXRα: (NR1 + NR2) > RXRα. In the second blue box, HEK293B-RXRα cells were transfected with 40 ng of each of NR1 and NR2; experiments were carried out 24 h after transfection, resulting in a larger RXRα pool relative to NR1 and NR2: RXRα > (NR1 + NR2). Box-and-whiskers plots represent 10th, 25th, 50th, 75th, and 90th percentiles; +, mean value. G, EGFP; B, TagBFP; C, mCherry.
In summary, in cells where there is a limiting or sequestered pool of RXR combined with the expression of several RXR heterodimerization partner NRs, a competition between NRs for their common partner, RXR, is most likely to occur.
Chromatin binding of VDR is dynamically regulated by calcitriol
Our finding that specific agonist binding increases the affinity of the studied NRs toward RXR brings up the question of whether it also affects their chromatin binding properties. We investigated this possibility in the case of VDR. Genomic binding sites (GBSs) of VDR were detected by chromatin immunoprecipitation sequencing (ChIP-seq) in THP-1 monocytes. The GBSs (n = 13,915) were divided into three groups: (i) n = 1866 VDREs containing a full vitamin D response element (a direct repeat of AGGTCA or similar sequence with a 3-nucleotide spacer, DR3), (ii) n = 1572 less specific NR half-sites (single AGGTCA sequences), and (iii) the rest, n = 10,477, where neither a VDRE nor an NR half-site sequence could be mapped, denoted as “None.” Upon calcitriol activation, VDRE-containing GBSs showed considerably higher occupancies on average than those in the control, vehicle-treated sample. A similar induction, but to a much lesser extent, was detected for the NR half-site-containing regions. In contrast, the None GBSs did not show any induction upon calcitriol treatment (Fig. 8A).
Figure 8.
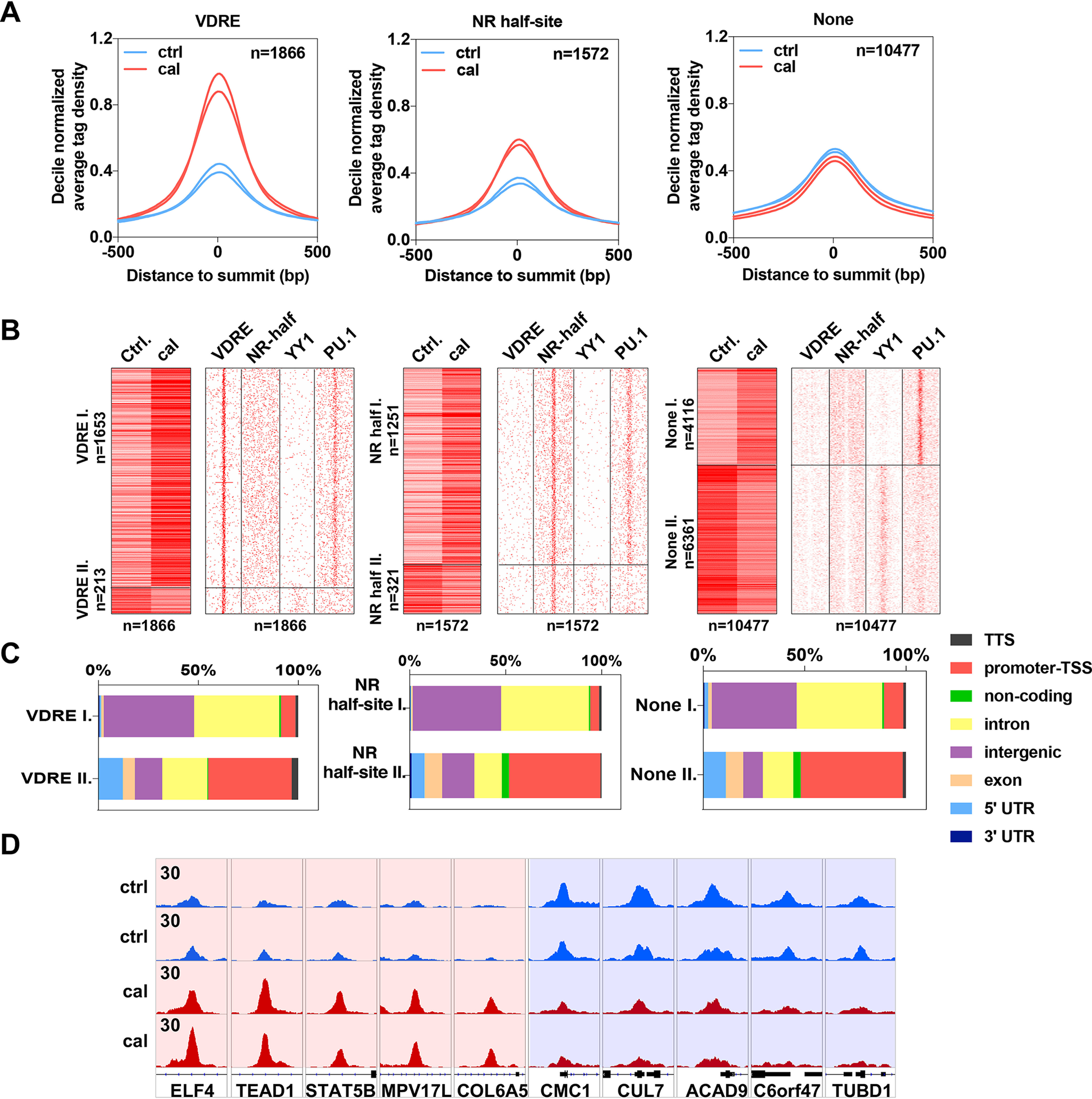
Chromatin binding of VDR is dynamically regulated by calcitriol. A, histograms showing the decile-normalized average tag density of the VDR binding sites that contain VDRE (n = 1866), NR half-site (n = 1572), or None (n = 10477) in the presence of cal or vehicle (Ctrl). B, left, heat maps representing the tag densities of VDR binding sites with VDRE, NR half-site, or None upon control (Ctrl) and calcitriol treatment. Regions were divided by k-means clustering (k = 2). Right, motif distribution heat maps showing the presence of VDRE, NR half-site, YY1, and PU.1 motifs in 2-kb frames around the VDR binding sites. C, bar charts showing the genomic distribution of VDR binding sites separately for the two clusters (I and II) of regions that contain VDRE, NR half-site, or None. D, an Integrative Genomics Viewer (IGV) snapshot of VDR coverage representing ten VDR binding sites upon control (Ctrl) or cal treatment from clusters I (left) and II (right). The interval scale is 30 in all cases.
To reveal more details of the stimulatory/inhibitory effect of calcitriol on VDR binding, we clustered the VDR GBSs in each group according to their tag densities and response types. We found that in the None group, a significant proportion (n = 6361) of the GBSs displayed a negative response, i.e. a decrease of tag densities upon calcitriol treatment, whereas only a few VDRE (n = 213)- and NR half-site (n = 321)-containing GBSs behaved this way (Fig. 8B, clusters denoted by II). To better understand the factors that are responsible for this pattern, we applied de novo motif enrichment analysis within each group. In addition to the expected motifs, the VDRE- and NR half-site-containing GBSs mostly showed the presence of the well-known PU.1, C/EBP (enhancer-specific pioneering factors purine-rich box 1 and CCAAT enhancer-binding protein), and AP-1 (activator protein 1) motifs, which occurred mainly in intergenic or intronic regions, also suggesting that they are enhancers. On the other hand, in the None group, the promoter-specific YY1 (Yin Yang 1 transcriptional repressor protein) motif was significantly enriched (Fig. S2). We observed that the YY1 motif was enriched at the negatively responding sites (cluster II of each group, Fig. 8B), and many of these GBSs were indeed located within promoter-transcription start site regions (Fig. 8C).
From the above results, we can conclude that direct DNA binding (to VDREs or NR half-sites) is enhanced by calcitriol, which, together with our microscopy data, suggests that heterodimerization with RXR and direct DNA binding are correlated events. On the other hand, binding at None GBS regions is characteristic for promoters and is diminished by calcitriol treatment. Figure 8D demonstrates the effect of calcitriol treatment on VDR accumulation on the GBSs of some of its target genes in either enhancer or promoter regions. There is an overlap between these two sets: in the case of 507 genes, binding both to the enhancer and the promoter of the same gene was found, suggesting chromatin looping (Fig. S2D).
Discussion
Nuclear receptors are widely used and are excellent targets for drug intervention in many diseases. However, in recent trials, unexpected side effects have been observed despite the high specificity of the drug-NR interaction. Here, we propose how the activation of one NR pathway can interfere indirectly with other NR pathways through competition for their common heterodimeric partner, RXRα. We show that there is a hierarchy in the affinities of NR partners toward RXRα, which is overridden by ligand binding. Our findings can help to explain the complex response observed in in vivo tests and may help design novel treatment strategies utilizing already FDA-approved drugs in an appropriate combination.
NLS1 mutants as a model system to study heterodimerization with RXR
The localization of NRs in the nucleus is mandatory for exerting their function as transcription factors; thus, their nuclear trafficking has been studied extensively. Like other nuclear proteins, nuclear localization of NRs is mediated through binding of carrier proteins, called importins, to the nuclear localization signal of the receptor (33), which is a specific sequence characterized by basic, positively charged amino acids. Changing these residues to uncharged, polar, or nonpolar amino acids abolishes receptor binding to importins and abrogates its nuclear accumulation (34, 35). Based on our observations, we can conclude that mutation of the NLS1 of RXR partners increases their presence in the cytoplasm but retains their ability to heterodimerize with RXR and to bind their ligands effectively. In line with our results, NLS mutants of pregnane xenobiotic receptor and constitutive androstane receptor (CAR) also showed RXR heterodimerization-dependent nuclear import (36). These findings allowed us to establish the NLS1 mutant receptors as a good model to detect the occurrence of heterodimerization with RXR by using a translocation assay.
Competition of NRs for their common partner, RXRα
In this paper, we addressed the questions of whether RXR has different affinities to its heterodimerization partners in the absence of ligand and if specific agonist treatment enhances these affinities. Our data demonstrated that in the absence of ligand, RXRα had the highest affinity toward RARα, intermediate for PPARγ, and the lowest for VDR, whereas agonist treatment always tipped the scale in favor of the liganded partner.
The dependence of RXR partner selection on the availability of the partner's specific agonist is consistent with the regulation of metabolism in which triggering a specific metabolic pathway is dictated by the availability of the endogenous substrate, e.g. NRs like liver X receptor (LXR) and PPARα are involved in lipid metabolism (storage versus degradation, respectively). These receptors serve as sensors for their endogenous ligands, free fatty acids (FFA), and oxysterol (37). In a fasting state where energy production is needed, FFA accumulates in the liver, reducing the expression of genes involved in fatty acid and cholesterol synthesis while activating genes promoting fatty acid catabolism. FFA increases the expression of PPARα and evokes its transcriptional activity, regulating genes involved in mitochondrial β-oxidation (38, 39). On the other hand, in a fed state with a high-fat diet, fatty acid delivery to the liver is decreased and oxysterol accumulates as a result of an increasing concentration of cholesterol (40). Oxysterol-dependent activation of LXR helps to eliminate the excess of cholesterol by increasing the expression of genes involved in bile acid synthesis, cholesterol absorption, transport, and excretion (41). This example of regulation of metabolism by two NRs implies how the liver can avoid competition between two partners of RXR mediating two interacting signaling pathways by providing only one partner's ligand at a time.
In HEK293B-RXRα cells, the originally nuclear localization of PPARγ/nlsm was massively reduced in the presence of RARα, whereas RARα/nlsm maintained its nuclear localization even in the presence of either PPARγ or VDR. Monopolization of RXRα by RARα indicates competition between the two partners (because of the limiting amount of RXRα expressed by the stable cell line) with an outcome favoring RARα. PPARγ/nlsm managed to maintain its nuclear localization in HEK293B-RXRα cells cotransfected with VDR; in addition, VDR was redistributed homogeneously, implying a dominance of PPARγ over VDR. The role of a potential endogenous ligand in this competition was excluded by applying a PPARγ antagonist, which did not change the distribution of the NRs. We also showed that if the RXRα pool is not limiting, competition between the heterodimerization partners is abolished; both NRs can bind to RXRα and become enriched in the nucleus. These experiments show a well-defined order of preference of RXRα for its heterodimerizing partners in the absence of agonist treatment, which is absolutely overridden by the effect of specific agonists (Fig. 9). Heterodimerization with RXRα of even the strongest partner, RARα/nlsm, persisted only until its competing partner, PPARγ or VDR, was liganded with its specific agonist, RSG or calcitriol.
Figure 9.

Summary of the results represented as chemical equations. Reactants, the two competing partners, and RXR are shown in black. For the products, the weak partner is shown in blue, and the dominant NR:RXR heterodimerization complex is shown in orange. 0, without ligand treatment.
In previous studies, PPARγ was shown to dominate the signaling pathway of both thyroid hormone receptor and VDR; it suppressed thyroid hormone receptor signaling in epiphyseal chondrocytes (42) and attenuated 1,25-dihydroxyvitamin-D3-mediated transactivation of VDR (43). PPAR/RXR binding to the response element of PPAR was shown to be reduced in the presence of LXRα or LXRβ (44). In the previously mentioned studies, the dominance of one RXR partner over the other was abolished when RXR was overexpressed and became abundantly available for both partners simultaneously. This might be the case in cells with a very large pool of uncommitted or sequestered RXR (13, 15, 28). Previously, we have shown that retinoic acid treatment of undifferentiated mouse embryonic stem cells decreases the expression of LXR target genes and decreases RXR binding at LXR response elements while inducing RXR redistribution to RAR target genes. This ligand-induced switch in RXR preference from LXR to RAR is critical for proper cellular differentiation (45).
There are other examples of ligand-directed competition for a shared subunit between different protein complexes (46). We also previously showed that interleukin-2 and -15 regulate the association of their membrane-localized receptor chains in a similar fashion. The β and γc chains of their heterotrimeric receptors are used by the two cytokines, whereas the IL-2Rα and IL-15Rα chains are cytokine specific. The βγc heterodimer forms a complex with the receptor α chain, the ligand of which is present (47).
Our or similar studies may have some limitations. We note that (i) we cannot entirely exclude the presence of endogenous ligands for the receptors; (ii) our approach may not mimic all possible physiological conditions; (iii) as for the limiting nature of RXR, antibody-based approaches are, at best, semiquantitative to determine endogenous NR concentrations; and finally, (iv) we used indirect measures to assess downstream gene expression events.
Similarities and differences in the localization of NRs
Despite the similar positioning of the NLS1 of different NRs, the studied NLS mutant NRs partitioned between the nucleus and the cytoplasm with different ratios and responded to RXRα heterodimerization with different localizations. PPARγ/nlsm and RARα/nlsm were distributed homogeneously in WT HEK293. Nuclear accumulation of PPARγ/nlsm and RARα/nlsm was observed in the presence of RXRα (in HEK293B-RXRα cells). The translocation of NLS mutant receptors into the nucleus can be explained by the piggy-back mechanism, which is mediated by protein-protein interaction with their shuttling partner, RXRα. This mechanism was described previously for other NRs, such as progesterone receptors (31).
Contrary to PPARγ/nlsm and RARα/nlsm, VDR/nlsm was more cytoplasmic in WT HEK293 cells. In HEK293B-RXRα, it not only failed to translocate into the nucleus but also induced a significant redistribution of RXRα. Translocation of RXRα to the cytoplasm implies that heterodimerization of RXRα with VDR/nlsm masks RXR's NLS1 and prevents its recognition by importins. Intriguingly, calcitriol treatment promoted the nuclear import of both VDR/nlsm and RXRα in HEK293B-RXRα. In line with this, when using RXRα/nlsm, calcitriol evoked the nuclear enrichment of both VDR and RXRα/nlsm, whereas both receptors were retained in the cytoplasm in the absence of calcitriol, similar to previous studies (48, 49). Another study also suggests that the translocation of the VDR-RXR heterodimer into the nucleus is facilitated by the NLS of VDR (50). Heterodimerization of GFP-VDR and RXR-BFP in the cytoplasm has also been affirmed by FRET (49), and the initiation of heterodimerization with RXR in the cytoplasm has been suggested for other NRs, such as xenobiotic receptors (pregnane xenobiotic receptor and CAR) (36).
Because VDR/nlsm did not translocate to the nucleus upon heterodimerization with RXRα in the absence of ligand, we did not rely on this mutant alone in the competition assay. WT VDR matched our needs better; it was distributed between the nucleus and the cytoplasm evenly in WT HEK293 cells and translocated into the nucleus when treated with calcitriol. It accumulated in the nucleus of HEK293B-RXRα cells and was further enriched there when treated with calcitriol. Thus, calcitriol increases the affinity of VDR to RXRα, which results in the accumulation of VDR in the nucleus, in line with previous findings showing that RXRα inhibits VDR export (48, 49). The effect of calcitriol to enhance the nuclear accumulation of VDR is because of increasing its affinity to importin (50). Nuclear enrichment of VDR/nlsm after calcitriol treatment may be explained by the presence of a further, ligand-responsive NLS in the hinge region between the DBD and the LBD (51, 52). A similar NLS, also in the hinge region, has been reported for other NRs like progesterone receptors (53).
In competition experiments for dimerization with RXRα, the application of a single agonist (of NR1 or NR2) always shifted the balance toward the liganded receptor. When ligands of NR1 and NR2 were both present, the outcome was different for the different pairs. RARα dominated over PPARγ when both NRs were liganded. In experiments involving WT VDR and another NR, both NRs accumulated in the nucleus. In these cases, RARα/nlsm or PPARγ/nlsm probably relied on the NLS of RXRα using the piggy-back mechanism, whereas calcitriol treatment may have exposed an alternative NLS of VDR facilitating its binding to importins.
Ligand-induced chromatin binding of VDR and heterodimerization with RXR are correlated
Calcitriol treatment had a triple effect on VDR: it enhanced its nuclear localization along with an augmented heterodimerization with RXRα, and it increased its binding to its response elements, stronger to VDREs binding the heterodimer and to a lesser extent to NR half-sites. This is in line with our finding that heterodimerization of RARα and RXRα was also positively correlated with chromatin binding as measured by comobility (27).
The GBSs of VDR were primarily enhancers, where the motifs of the enhancer-specific transcription factors PU.1 and C/EBP were also enriched. On the other hand, the majority of GBSs not containing a VDRE or a half-site were localized in promoter regions and were enriched in the motif of the promoter-specific YY1 transcription factor. In these regions, VDR binds indirectly. The latter GBSs became less occupied upon calcitriol treatment; thus, we can conclude that calcitriol induces a transition from promoters (and possibly from the non-DNA-bound pool of VDR) to enhancers. 507 genes were doubly assigned to an enhancer region (from the cluster of positively responding sites) as well as to a promoter region (from the negative responders), suggesting chromatin looping (54). A possible interpretation of the observed complex behavior is that calcitriol strengthens binding of VDR to enhancers and initiates transcription, causing the transcription machinery to move from the promoter toward the coding region of the regulated gene.
Conclusions
Tracing the dynamic distribution pattern of EGFP-VDR, EGFP-PPARγ/nlsm, and EGFP-RARα/nlsm (homogeneous in the absence of RXRα and nuclear enriched in response to RXRα binding) serves as a good model system for studying their competition for heterodimerization with RXRα. There is indeed dynamic competition between RXR partners, which is governed by two mechanisms. First, in the absence of agonist treatment, there is a hierarchy of affinities between RXRα and its partners in the following order: RARα > PPARγ > VDR. Second, in the presence of agonist treatment, RXRα partner selection is shifted toward the liganded partner.
These results may explain certain side effects of drugs targeting NRs. Competition for RXR could be responsible for the symptoms of vitamin D deficiency developed in a child upon receiving systemic retinoid treatment for ichthyosis (55) or the antagonistic effect of coadministered vitamin A on serum calcium response to vitamin D treatment (56).
Our observations regarding these three RXR partners, consistent with metabolism regulation by two other RXR partners (LXR and PPARα) and our similar previous findings on membrane-localized IL-2/15 receptors, encourage us to generalize the concept that specific ligand binding may often govern competition between different partners of a promiscuous receptor. Our findings are a proof of concept of a hierarchy of affinities between NRs and their common partner, RXRα. These studies can be extended to a larger number of receptors to uncover the network of hierarchies, and follow-up studies will also focus on testing this concept in a broader and physiological context.
Experimental procedures
Plasmid construction
Expression constructs for EGFP-PPARγ, EGFP-RARα, and EGFP-VDR were generated by PCR using PPARγ, RARα, and VDR human cDNA, splice variant (isoform) 1, as templates. As a replacement for the start codon ATG, TTT was inserted in the designed cloning primers. PCR products were subcloned into EGFP-C3 vector (Clontech Laboratories, Mountain View, CA) using the restriction sites XhoI/SalI, BglII/HindIII, and XhoI/HindIII, respectively. mCherry-PPARγ, mCherry-RARα, and mCherry-VDR constructs were generated from EGFP-tagged receptors by replacing EGFP with mCherry. Candidate NLSs in the DBDs of the involved receptors have been defined to be between amino acids 136 and 141 for PPARγ, 113 and 118 for RARα, 49 and 54 for VDR, and 160 and 165 for RXRα in a previous study (48). NLS mutant receptors were prepared by introducing a point mutation in the NLS1 into the coding sequence of EGFP-tagged receptors using site-directed mutagenesis via the overlap extension PCR method (57). Accordingly, the following point mutations were introduced: in EGFP-PPARγ/nlsm, R136Q, R137G, R140G, and L141Q; in EGFP-RARα/nlsm, R113Q, R114G, Q117G, and K118Q; in EGFP-VDR/nlsm, R49Q, R50G, K53G, and R54Q; in EGFP-RXRα/nlsm, K160Q, R161G, R164G, and K165Q. TagBFP-RXRα/nlsm was generated from EGFP-RXRα/nlsm by replacing EGFP with TagBFP using NheI and BglII restriction enzymes. The zinc finger mutant RXR, RXR/znm, was prepared by mutating all 8 cysteines within the two zinc finger motifs of the DBD to alanines using the overlap extension PCR method. Accordingly, the following point mutations were introduced: C135A, C138A, C152A, C155A, C171A, C177A, C190A, and C195A.
In all cases, the fluorescent proteins were fused to the N termini of the receptors. All plasmids were sequenced and are available upon request.
Control plasmid constructs EGFP-mCherry and EGFP-TagBFP are complemented with a CGC GAT CCA CCG GTA ATG linker sequence after the sequence of EGFP. In the proline-separated TagBFP-P30-EGFP construct, there is a GGT CCG GTC GCC ACC CGC GAT CCA CCG GTA ATG linker before the proline spacer (30 repeats, mixed from the 4 proline codons) after the sequence of TagBFP.
Generation of TagBFP-RXRα stable cell line
The adherent version of human embryonic kidney (HEK293; also called AD-293) cell line stably expressing TagBFP-RXRα, HEK293B-RXRα, was prepared using viral transduction. Virus particles harboring the TagBFP-RXRα gene were purchased from VectorBuilder (Santa Clara, CA, USA). Multiplicity of infection, which is the number of virus particles needed to transduce one cell (58), was optimized experimentally for HEK293. A multiplicity of infection of 12 gave 70% transduction efficiency and kept the cells in good viability conditions; thus, it was chosen to transduce the cells. G418 as a selection marker was applied in a dose of 500 µg/ml to get rid of nontransduced cells. Cells were further sorted with a FACSAria III (BD Bioscience, San Jose, CA, USA) cell sorter to collect a cell population expressing TagBFP-RXR within an expression range with a coefficient of variation of 50%.
Cell culture and transient transfection
Both HEK293 and HEK293B-RXRα cells were maintained in DMEM, supplemented with phenol red, 10% fetal calf serum (Sigma-Aldrich, Saint Louis, MO), 1× GlutaMAX (Fisher Scientific, Tokyo, Japan), and 50 mg/liter gentamycin (KARA, Novo Mesto, Slovenia). For microscopy experiments, HEK293 or HEK293B-RXRα cells were subcultured in 8-well chambered coverslips (ibidi, Munich, Germany) and maintained in phenol red-free DMEM because of the potential estrogenic effect of phenol red (59) and supplemented with 10% charcoal-stripped fetal calf serum (PAN-Biotech, Aidenbach Germany) to get rid of the endogenous NR ligands. 24 h after seeding, cells reached 50–60% confluency and were transiently transfected with 75–80 ng of either one RXR partner (NR1) or two competing partners (NR1 + NR2) using FuGENE® HD transfection reagent (Promega, MA, USA), as suggested by the manufacturer. The amounts of plasmids yielding equal expression levels of NR1 and NR2 were determined by comparing the red-to-green fluorescence intensity ratio of the receptors to that of the EGFP-mCherry fusion protein, expressing the fluorophores in equal amounts. Unless otherwise indicated, cells were used for microscopy within 48 h after transient transfection.
Ligand treatment
Ligands were purchased from BioVision (Milpitas, CA, USA) and were applied to the transfected cells 30–60 min before imaging at 37 °C, 5% CO2. The following ligands with the mentioned concentrations were used: RSG, a PPAR agonist (1 μm); AM580, an RAR agonist (100 nm); calcitriol, a VDR agonist (100 nm); and LG268, an RXR agonist (100 nm). PPARγ-specific antagonist (GW9662; 1 μm) was purchased from Promega, MA, USA. Stock solutions of ligands were prepared in DMSO. For use, ligand aliquots were prepared in 1:1 DMSO–ethanol solvent to prevent repetitive freezing and thawing. Ligands were stored at −20 °C.
Microscopy
Confocal images were recorded by a Zeiss LSM 880 (Carl Zeiss, Jena, Germany) confocal microscope using a 40×, 1.2 numeric aperture water immersion objective. A 405-nm laser was used to excite the blue fluorescence signal of TagBFP detected between 429 and 481 nm, the 488-nm line of an argon-ion laser for the green signal of EGFP detected between 499 and 562 nm, and a 543-nm HeNe laser for the red signal of mCherry measured between 561 and 735 nm. An incubator built around the microscope maintained the temperature at 37 °C during the measurements.
Analysis of microscopy data
The average intensity per pixel was measured in two separately, manually selected regions of interest, one contouring the entire cytoplasm and another contouring the entire nucleus, excluding the nucleoli. The values were corrected for background by subtracting the mean intensity calculated for nontransfected HEK293 cells. The intensities were determined using the open-source FIJI distribution of ImageJ (version 2.0.0-rc-69/1.52i).
Relative expression levels of the fluorescently tagged NRs were assessed as described previously (60). Briefly, the intensity of B-RXR in the blue channel and that of G-NR1 in the green channel were measured, and the ratio of these signals, IG/IB, was calculated. Similar IG/IB ratios from TagBFP-P30-EGFP (and EGFP-TagBFP) fusion proteins expressing the two fluorophores at a 1:1 ratio were determined and averaged, serving as a standard value, Q. The molecular expression ratio NG/NB of the NRs was then calculated as NG/NB = (IG/IB)/Q.
For determining the relative expression ratio of G-NR1 to C-NR2, NG/NC, we used the EGFP-mCherry fusion protein as a standard in a similar fashion, whereas the relative expression ratio of C-NR2 to B-RXRα was calculated as NG/NB divided by NG/NC. In our triple coexpression experiments, the combined expression of NR1 and NR2 was greater than that of RXR to ensure a limiting pool of RXR; thus, competition could be observed.
Data were presented as a ratio of nuclear-to-cytoplasmic fluorescence intensities (NCR), displayed as box-and-whiskers plots depicting the 10th, 25th, 50th, 75th, and 90th percentiles and the arithmetic means for at least 30 cells. Analysis of variance was used to compare the differences between the groups. Prism 8.4.0 was used for statistical analysis and graphs.
Translocation assay
In this study, we intended to understand how the promiscuous RXR molecule chooses its interaction partner in the presence of several potential heterodimeric partners and agonists. We evaluated competition between these NRs for their common partner, RXRα, by using a translocation assay in a three-color model imaging system as illustrated in Fig. 1. RXRα was tagged with TagBFP and stably expressed in HEK293 cells. NR1, called the “studied partner,” was labeled with EGFP and NR2, or the “competing partner,” with mCherry. Applying a translocation assay required a version of the studied partner present in both the cytoplasm and the nucleus when not interacting with RXRα and getting enriched in the nucleus when interacting with RXRα. Inducing a mutation in the NLS1 of PPARγ and RARα fulfilled these criteria, whereas VDR was more appropriate in its WT form; we call these the “homogeneous form of the receptor” and refer to it as NR1. A pilot study confirmed the homogeneous localization of EGFP-PPARγ/nlsm, EGFP-RARα/nlsm, and EGFP-VDR in WT HEK293 cells and their nuclear accumulation in HEK293B-RXRα. Next, competition between RXRα partners for binding to RXRα was evaluated by detecting changes in heterodimerization between RXRα and the homogeneous form of NR1 in the presence of WT NR2. Competition between any two selected NRs was tested in two complementary experiments, e.g. to study competition between RARα and PPARγ for binding to RXRα, we traced the distribution changes of both EGFP-PPARγ/nlsm in the presence of mCherry-RARα and that of EGFP-RARα/nlsm in the presence of mCherry-PPARγ in HEK293B-RXRα cells. The role of specific agonists in governing the competition was assessed by treating the samples with the specific agonists of the involved receptors selectively or in combination.
ChIP
Cell culture and ligand treatment
The human monocytic cell line THP-1 was maintained in Roswell Park Memorial Institute medium supplemented with 10% fetal calf serum (Sigma-Aldrich, Saint Louis, MO), 1× GlutaMAX, (Fisher Scientific, Tokyo, Japan), 50 mg/liter gentamycin (KARA, Novo Mesto, Slovenia). 60 min prior to ChIP assay, THP-1 cells were maintained in phenol red-free Roswell Park Memorial Institute medium supplemented with 10% charcoal-stripped fetal calf serum and treated with either vehicle, 1:1 DMSO–ethanol, or calcitriol (100 nm).
The ChIP experiment was performed as previously published (61) and optimized with minor modifications. A mouse monoclonal anti-VDR antibody was applied (sc-13133, Santa Cruz Biotechnology). TruSeq ChIP library systems (Illumina) were used for library preparation according to the manufacturer's instructions.
ChIP-seq data analysis and peak prediction
Raw ChIP-seq reads were aligned to the hg19 reference genome assembly with default parameters by using the BWA tool (v07.17), and BAM files were generated with SAMtools (v1.7) (62, 63). Peaks were predicted with the findPeaks program of the HOMER toolkit, and their widths were fixed to 200 bp relative to their summits (64). Artifacts were removed according to the blacklisted genomic regions of ENCODE (65). Peaks that were present in both replicates (separately for control and calcitriol-treated samples) and reached a minimum of 3 units (normalized tag densities as determined within ±25 bp relative to peak summits by HOMER's annotatePeaks; n = 8401 for the control and n = 10,927 for the calcitriol-treated samples) were merged (n = 13,915) and considered in further analyses. Genome coverage (bedgraph) files were generated by HOMER's makeUCSCfile program.
Classification and characterization of peaks
Peaks were divided into three groups based on the presence or absence of the VDRE or NR half-site. For this, VDRE and NR half-site motif matrices were downloaded from the HOMER Motif Database. Tag density of the central 50-bp region of the 13,915 peaks was determined for each sample with HOMER's annotatePeaks (-hist 50 and -ghist options). Tag density values were normalized with the per-sample upper decile value and were averaged per condition. For heat maps, the three peak groups (with VDRE, NR half, or None) were further divided by k-means clustering (k = 2), applying the centered correlation similarity metric using the Cluster 3.0 (v1.59) software. For histograms, average tag densities were generated with HOMER's annotatePeaks (-hist 10) and were normalized with the previously determined decile values. Genomic distribution and annotation of the peak groups also were defined by HOMER's annotatePeaks. The pathway terms were predicted using the KEGG database (66).
Motif analysis and mapping
De novo motif enrichment analyses were carried out with HOMER's findMotifsGenome and were performed on the central 200-bp regions of the peaks. The targeted motif lengths were 10, 12, 14, and 16 bp. p values were calculated by comparing the enrichment within the target regions and that of a random set of regions (background) generated by HOMER. For motif distribution plots, matrices of the enriched motifs were mapped by following the order of regions in the tag density heat maps by using the -mbed parameter of HOMER's annotatePeaks in 20-bp windows within 2-kb frames relative to the peak centers.
Visualization
Heat maps were visualized by Java TreeView (v1.1.6r4) (67). Histograms and bar charts were plotted by using GraphPad Prism (v8). An area-proportional Venn diagram was generated by BioVenn (68). Genome coverage (bedgraph) files were visualized by Integrative Genomics Viewer (v2.4.16) (69).
Data availability
ChIP-seq data have been submitted to Sequence Read Archive and Gene Expression Omnibus databases under accession numbers PRJNA632899 and GSE150652, respectively.
Supplementary Material
Acknowledgments
We thank Prof. John Schwabe (University of Leicester) and Dr. Péter Vilmos for helpful discussions and Edina Nagy for technical assistance. Microscopy was carried out in the Sándor Damjanovich Cell Analysis Core Facility of the University of Debrecen (Cellular BioImaging Hungary, a Euro-BioImaging Node).
This article contains supporting information.
Author contributions—L. F., D. B., G. N., and G. V. data curation; L. F., D. B., G. N., and G. V. formal analysis; L. F., J. V., Z. K., and É. H. investigation; L. F., B. R., J. V., and G. V. methodology; L. F., D. B., Z. K., G. N., and G. V. writing-original draft; L. F., Z. K., Z. S., G. S., K. T., L. N., and G. V. writing-review and editing; G. S., K. T., and G. V. supervision; L. N. and G. V. conceptualization; L. N. and G. V. funding acquisition; L. F., G. M., and Z. S. plasmid construction.
Funding and additional information—This work was supported by the Stipendium Hungaricum Scholarship by the Tempus Public Foundation (to L. F.), NR-NET FP7-PEOPLE-2013-ITN-606806 Marie Curie Initial Training Networks (ITN) (to L. N. and L. F.), the National Research, Development, and Innovation Office grants NN129371, KKP129909, K124298, GINOP-2.3.2-15-2016-00026, and GINOP-2.3.3-15-2016-00003 (to G. V. and L. N.), the German Academic Exchange Service, and the Tempus Public Foundation no. 273478 (to G. V. and K. T).
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- AP-1
- activator protein 1
- TagBFP
- blue fluorescent proteins
- mCherry
- red fluorescent protein
- cal, calcitriol
- VDR-specific agonist
- CEBPA
- CCAAT enhancer-binding protein alpha
- ChIP-seq
- chromatin immunoprecipitation sequencing
- DBD
- DNA-binding domain
- FFA
- free fatty acids
- FP
- fluorescent protein
- EGFP
- enhanced green fluorescent protein
- G418
- Geneticin
- GBSs
- genomic binding sites
- HEK293
- human embryonic kidney adherent cells
- HEK293B-RXRα
- HEK293 cells stably expressing TagBFP-RXRα
- HRE
- hormone response element
- LBD
- ligand-binding domain
- CLSM
- confocal laser scanning microcopy
- LXR
- liver X receptor
- NCR
- nuclear-to-cytoplasmic ratio
- NLS
- nuclear localization signal
- NLS1
- nuclear localization signal between the two zinc finger motifs
- nlsm
- mutation in the NLS
- NR
- nuclear receptor
- NR/nlsm
- nuclear receptor with a mutation in its NLS
- NR1
- homogeneous version of RXR partner or the studied partner
- NR2
- wild-type version of the RXR partner or the competing partner
- PPARγ
- peroxisome proliferator-activated receptor gamma
- PU.1
- purine-rich box 1
- RARα
- retinoid acid receptor alpha
- RE
- response element
- ROI
- region of interest
- RSG
- rosiglitazone
- RUNX1
- Runt-related transcription factor
- RXRα/znm
- RXR with a mutation in its two zinc fingers motifs
- RXRα
- retinoid X receptor alpha
- THP-1
- immortalized monocyte-like cell line
- VDR
- vitamin D receptor
- YY1
- Yin Yang 1 transcriptional repressor protein.
References
- 1. Giner X. C., Cotnoir-White D., Mader S., and Levesque D. (2015) Selective ligand activity at Nur/retinoid X receptor complexes revealed by dimer-specific bioluminescence resonance energy transfer-based sensors. FASEB J. 29, 4256–4267 10.1096/fj.14-259804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shulman A. I., and Mangelsdorf D. J. (2005) Retinoid X receptor heterodimers in the metabolic syndrome. N. Engl. J. Med. 353, 604–615 10.1056/NEJMra043590 [DOI] [PubMed] [Google Scholar]
- 3. Gronemeyer H., Gustafsson J. A., and Laudet V. (2004) Principles for modulation of the nuclear receptor superfamily. Nat. Rev. Drug Discov. 3, 950–964 10.1038/nrd1551 [DOI] [PubMed] [Google Scholar]
- 4. Burris T. P., Solt L. A., Wang Y., Crumbley C., Banerjee S., Griffett K., Lundasen T., Hughes T., and Kojetin D. J. (2013) Nuclear receptors and their selective pharmacologic modulators. Pharmacol. Rev. 65, 710–778 10.1124/pr.112.006833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Le Maire A., Alvarez S., Shankaranarayanan P., Lera A. R., Bourguet W., and Gronemeyer H. (2012) Retinoid receptors and therapeutic applications of RAR/RXR modulators. Curr. Top. Med. Chem. 12, 505–527 10.2174/156802612799436687 [DOI] [PubMed] [Google Scholar]
- 6. Pawlak M., Lefebvre P., and Staels B. (2012) General molecular biology and architecture of nuclear receptors. Curr. Top. Med. Chem. 12, 486–504 10.2174/156802612799436641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dawson M. I., and Xia Z. (2012) The retinoid X receptors and their ligands. Biochim. Biophys. Acta 1821, 21–56 10.1016/j.bbalip.2011.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rastinejad F., Wagner T., Zhao Q., and Khorasanizadeh S. (2000) Structure of the RXR-RAR DNA-binding complex on the retinoic acid response element DR1. EMBO J. 19, 1045–1054 10.1093/emboj/19.5.1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Khorasanizadeh S., and Rastinejad F. (2001) Nuclear-receptor interactions on DNA-response elements. Trends Biochem. Sci. 26, 384–390 10.1016/S0968-0004(01)01800-X [DOI] [PubMed] [Google Scholar]
- 10. Chatagnon A., Veber P., Morin V., Bedo J., Triqueneaux G., Sémon M., Laudet V., d'Alché-Buc F., and Benoit G. (2015) RAR/RXR binding dynamics distinguish pluripotency from differentiation associated cis-regulatory elements. Nucleic Acids Res. 43, 4833–4854 10.1093/nar/gkv370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vivat-Hannah V., Bourguet W., Gottardis M., and Gronemeyer H. (2003) Separation of retinoid X receptor homo- and heterodimerization functions. Mol. Cell. Biol. 23, 7678–7688 10.1128/mcb.23.21.7678-7688.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Huang P., Chandra V., and Rastinejad F. (2010) Structural overview of the nuclear receptor superfamily: insights into physiology and therapeutics. Annu. Rev. Physiol. 72, 247–272 10.1146/annurev-physiol-021909-135917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dong D., and Noy N. (1998) Heterodimer formation by retinoid X receptor: regulation by ligands and by the receptor's self-association properties. Biochemistry 37, 10691–10700 10.1021/bi980561r [DOI] [PubMed] [Google Scholar]
- 14. Nagy L., and Schwabe J. W. (2004) Mechanism of the nuclear receptor molecular switch. Trends Biochem. Sci. 29, 317–324 10.1016/j.tibs.2004.04.006 [DOI] [PubMed] [Google Scholar]
- 15. Chan L. S., and Wells R. A. (2009) Cross-talk between PPARs and the partners of RXR: a molecular perspective. PPAR Res. 2009, 925309 10.1155/2009/925309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Feige J. N., Gelman L., Tudor C., Engelborghs Y., Wahli W., and Desvergne B. (2005) Fluorescence imaging reveals the nuclear behavior of peroxisome proliferator-activated receptor/retinoid X receptor heterodimers in the absence and presence of ligand. J. Biol. Chem. 280, 17880–17890 10.1074/jbc.M500786200 [DOI] [PubMed] [Google Scholar]
- 17. van Royen M. E., van de Wijngaart D. J., Cunha S. M., Trapman J., and Houtsmuller A. B. (2013) A multi-parameter imaging assay identifies different stages of ligand-induced androgen receptor activation. Cytometry A 83, 806–817 10.1002/cyto.a.22284 [DOI] [PubMed] [Google Scholar]
- 18. Mikuni S., Yamamoto J., Horio T., and Kinjo M. (2017) Negative correlation between the diffusion coefficient and transcriptional activity of the glucocorticoid receptor. Int. J. Mol. Sci. 18, 1855 10.3390/ijms18091855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Maruvada P., Baumann C. T., Hager G. L., and Yen P. M. (2003) Dynamic shuttling and intranuclear mobility of nuclear hormone receptors. J. Biol. Chem. 278, 12425–12432 10.1074/jbc.M202752200 [DOI] [PubMed] [Google Scholar]
- 20. Jankevics H., Prummer M., Izewska P., Pick H., Leufgen K., and Vogel H. (2005) Diffusion-time distribution analysis reveals characteristic ligand-dependent interaction patterns of nuclear receptors in living cells. Biochemistry 44, 11676–11683 10.1021/bi050744v [DOI] [PubMed] [Google Scholar]
- 21. Lionnet T., Wu B., Grunwald D., Singer R. H., and Larson D. R. (2010) Nuclear physics: quantitative single-cell approaches to nuclear organization and gene expression. Cold Spring Harb. Symp. Quant. Biol. 75, 113–126 10.1101/sqb.2010.75.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Savatier J., Jalaguier S., Ferguson M. L., Cavaillès V., and Royer C. A. (2010) Estrogen receptor interactions and dynamics monitored in live cells by fluorescence cross-correlation spectroscopy. Biochemistry 49, 772–781 10.1021/bi9013006 [DOI] [PubMed] [Google Scholar]
- 23. Hendrix J., Gijsbers R., De Rijck J., Voet A., Hotta J., McNeely M., Hofkens J., Debyser Z., and Engelborghs Y. (2011) The transcriptional co-activator LEDGF/p75 displays a dynamic scan-and-lock mechanism for chromatin tethering. Nucleic Acids Res. 39, 1310–1325 10.1093/nar/gkq933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen Y., Wei L. N., and Muller J. D. (2003) Probing protein oligomerization in living cells with fluorescence fluctuation spectroscopy. Proc. Natl. Acad. Sci. U S A 100, 15492–15497 10.1073/pnas.2533045100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brazda P., Szekeres T., Bravics B., Toth K., Vamosi G., and Nagy L. (2011) Live-cell fluorescence correlation spectroscopy dissects the role of coregulator exchange and chromatin binding in retinoic acid receptor mobility. J. Cell Sci. 124, 3631–3642 10.1242/jcs.086082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Brazda P., Krieger J., Daniel B., Jonas D., Szekeres T., Langowski J., Toth K., Nagy L., and Vamosi G. (2014) Ligand binding shifts highly mobile retinoid X receptor to the chromatin-bound state in a coactivator-dependent manner, as revealed by single-cell imaging. Mol. Cell. Biol. 34, 1234–1245 10.1128/MCB.01097-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rehó B., Lau L., Mocsár G., Müller G., Fadel L., Brázda P., Nagy L., Tóth K., and Vámosi G. (2020) Simultaneous mapping of molecular proximity and comobility reveals agonist-enhanced dimerization and DNA binding of nuclear receptors. Anal. Chem. 92, 2207–2215 10.1021/acs.analchem.9b04902 [DOI] [PubMed] [Google Scholar]
- 28. Chen Z. P., Iyer J., Bourguet W., Held P., Mioskowski C., Lebeau L., Noy N., Chambon P., and Gronemeyer H. (1998) Ligand- and DNA-induced dissociation of RXR tetramers. J. Mol. Biol. 275, 55–65 10.1006/jmbi.1997.1413 [DOI] [PubMed] [Google Scholar]
- 29. Wood R. J. (2008) Vitamin D and adipogenesis: new molecular insights. Nutr. Rev. 66, 40–46 10.1111/j.1753-4887.2007.00004.x [DOI] [PubMed] [Google Scholar]
- 30. Philippe L., van den Elzen A. M. G., Watson M. J., and Thoreen C. C. (2020) Global analysis of LARP1 translation targets reveals tunable and dynamic features of 5' TOP motifs. Proc. Natl. Acad. Sci. U S A 117, 5319–5328 10.1073/pnas.1912864117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tyagi R. K., Amazit L., Lescop P., Milgrom E., and Guiochon-Mantel A. (1998) Mechanisms of progesterone receptor export from nuclei: role of nuclear localization signal, nuclear export signal, and ran guanosine triphosphate. Mol. Endocrinol. 12, 1684–1695 10.1210/mend.12.11.0197 [DOI] [PubMed] [Google Scholar]
- 32. Daniel B., Nagy G., Czimmerer Z., Horvath A., Hammers D. W., Cuaranta-Monroy I., Poliska S., Tzerpos P., Kolostyak Z., Hays T. T., Patsalos A., Houtman R., Sauer S., Francois-Deleuze J., Rastinejad F., et al. (2018) The nuclear receptor PPARgamma controls progressive macrophage polarization as a ligand-insensitive epigenomic ratchet of transcriptional memory. Immunity 49, 615–626 10.1016/j.immuni.2018.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sessler R. J., and Noy N. (2005) A ligand-activated nuclear localization signal in cellular retinoic acid binding protein-II. Mol. Cell 18, 343–353 10.1016/j.molcel.2005.03.026 [DOI] [PubMed] [Google Scholar]
- 34. Kumar S., Saradhi M., Chaturvedi N. K., and Tyagi R. K. (2006) Intracellular localization and nucleocytoplasmic trafficking of steroid receptors: an overview. Mol. Cell Endocrinol. 246, 147–156 10.1016/j.mce.2005.11.028 [DOI] [PubMed] [Google Scholar]
- 35. Wagstaff K. M., and Jans D. A. (2009) Importins and beyond: non-conventional nuclear transport mechanisms. Traffic 10, 1188–1198 10.1111/j.1600-0854.2009.00937.x [DOI] [PubMed] [Google Scholar]
- 36. Dash A. K., Yende A. S., Jaiswal B., and Tyagi R. K. (2017) Heterodimerization of retinoid X receptor with xenobiotic receptor partners occurs in the cytoplasmic compartment: mechanistic insights of events in living cells. Exp. Cell Res. 360, 337–346 10.1016/j.yexcr.2017.09.024 [DOI] [PubMed] [Google Scholar]
- 37. Alaynick W. A. (2008) Nuclear receptors, mitochondria and lipid metabolism. Mitochondrion 8, 329–337 10.1016/j.mito.2008.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Georgiadi A., and Kersten S. (2012) Mechanisms of gene regulation by fatty acids. Adv. Nutr. 3, 127–134 10.3945/an.111.001602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kersten S., Seydoux J., Peters J. M., Gonzalez F. J., Desvergne B., and Wahli W. (1999) Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J. Clin. Invest. 103, 1489–1498 10.1172/JCI6223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhao C., and Dahlman-Wright K. (2010) Liver X receptor in cholesterol metabolism. J. Endocrinol. 204, 233–240 10.1677/JOE-09-0271 [DOI] [PubMed] [Google Scholar]
- 41. Wang B., and Tontonoz P. (2018) Liver X receptors in lipid signalling and membrane homeostasis. Nat. Rev. Endocrinol. 14, 452–463 10.1038/s41574-018-0037-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hunter J., Kassam A., Winrow C. J., Rachubinski R. A., and Capone J. P. (1996) Crosstalk between the thyroid hormone and peroxisome proliferator-activated receptors in regulating peroxisome proliferator-responsive genes. Mol. Cell Endocrinol. 116, 213–221 10.1016/0303-7207(95)03717-9 [DOI] [PubMed] [Google Scholar]
- 43. Alimirah F., Peng X., Yuan L., Mehta R. R., von Knethen A., Choubey D., and Mehta R. G. (2012) Crosstalk between the peroxisome proliferator-activated receptor gamma (PPARgamma) and the vitamin D receptor (VDR) in human breast cancer cells: PPARgamma binds to VDR and inhibits 1alpha,25-dihydroxyvitamin D3 mediated transactivation. Exp. Cell Res. 318, 2490–2497 10.1016/j.yexcr.2012.07.020 [DOI] [PubMed] [Google Scholar]
- 44. Ide T., Shimano H., Yoshikawa T., Yahagi N., Amemiya-Kudo M., Matsuzaka T., Nakakuki M., Yatoh S., Iizuka Y., Tomita S., Ohashi K., Takahashi A., Sone H., Gotoda T., Osuga J., et al. (2003) Cross-talk between peroxisome proliferator-activated receptor (PPAR) alpha and liver X receptor (LXR) in nutritional regulation of fatty acid metabolism. II. LXRs suppress lipid degradation gene promoters through inhibition of PPAR signaling. Mol. Endocrinol. 17, 1255–1267 10.1210/me.2002-0191 [DOI] [PubMed] [Google Scholar]
- 45. Simandi Z., Horvath A., Cuaranta-Monroy I., Sauer S., Deleuze J. F., and Nagy L. (2018) RXR heterodimers orchestrate transcriptional control of neurogenesis and cell fate specification. Mol. Cell Endocrinol. 471, 51–62 10.1016/j.mce.2017.07.033 [DOI] [PubMed] [Google Scholar]
- 46. Gonnord P., Angermann B. R., Sadtler K., Gombos E., Chappert P., Meier-Schellersheim M., and Varma R. (2018) A hierarchy of affinities between cytokine receptors and the common gamma chain leads to pathway cross-talk. Sci. Signal. 11, eaal1253 10.1126/scisignal.aal1253 [DOI] [PubMed] [Google Scholar]
- 47. Vamosi G., Bodnar A., Vereb G., Jenei A., Goldman C. K., Langowski J., Toth K., Matyus L., Szollosi J., Waldmann T. A., and Damjanovich S. (2004) IL-2 and IL-15 receptor alpha-subunits are coexpressed in a supramolecular receptor cluster in lipid rafts of T cells. Proc. Natl. Acad. Sci. U S A 101, 11082–11087 10.1073/pnas.0403916101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Prufer K., and Barsony J. (2002) Retinoid X receptor dominates the nuclear import and export of the unliganded vitamin D receptor. Mol. Endocrinol. 16, 1738–1751 10.1210/me.2001-0345 [DOI] [PubMed] [Google Scholar]
- 49. Prufer K., Racz A., Lin G. C., and Barsony J. (2000) Dimerization with retinoid X receptors promotes nuclear localization and subnuclear targeting of vitamin D receptors. J. Biol. Chem. 275, 41114–41123 10.1074/jbc.M003791200 [DOI] [PubMed] [Google Scholar]
- 50. Yasmin R., Williams R. M., Xu M., and Noy N. (2005) Nuclear import of the retinoid X receptor, the vitamin D receptor, and their mutual heterodimer. J. Biol. Chem. 280, 40152–40160 10.1074/jbc.M507708200 [DOI] [PubMed] [Google Scholar]
- 51. Michigami T., Suga A., Yamazaki M., Shimizu C., Cai G., Okada S., and Ozono K. (1999) Identification of amino acid sequence in the hinge region of human vitamin D receptor that transfers a cytosolic protein to the nucleus. J. Biol. Chem. 274, 33531–33538 10.1074/jbc.274.47.33531 [DOI] [PubMed] [Google Scholar]
- 52. Gocek E., Baurska H., Marchwicka A., and Marcinkowska E. (2012) Regulation of leukemic cell differentiation through the vitamin D receptor at the levels of intracellular signal transduction, gene transcription, and protein trafficking and stability. Leuk. Res. Treatment 2012, 713243 10.1155/2012/713243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Guiochon-Mantel A., Lescop P., Christin-Maitre S., Loosfelt H., Perrot-Applanat M., and Milgrom E. (1991) Nucleocytoplasmic shuttling of the progesterone receptor. EMBO J. 10, 3851–3859 10.1002/j.1460-2075.1991.tb04954.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hanel A., Malmberg H. R., and Carlberg C. (2020) Genome-wide effects of chromatin on vitamin D signaling. J. Mol. Endocrinol. 64, R45–R56 10.1530/JME-19-0246 [DOI] [PubMed] [Google Scholar]
- 55. Neema S., Mukherjee S., Vasudevan B., Verma R., Moorchung N., and Chatterjee M. (2015) Vitamin D deficiency after oral retinoid therapy for ichthyosis. Pediatr. Dermatol. 32, e151–e155 10.1111/pde.12614 [DOI] [PubMed] [Google Scholar]
- 56. Johansson S., and Melhus H. (2001) Vitamin A antagonizes calcium response to vitamin D in man. J. Bone Miner. Res. 16, 1899–1905 10.1359/jbmr.2001.16.10.1899 [DOI] [PubMed] [Google Scholar]
- 57. Barik S. (1993) Site-directed mutagenesis by double polymerase chain reaction: megaprimer method. Methods Mol. Biol. 15, 277–286 10.1385/0-89603-244-2:277 [DOI] [PubMed] [Google Scholar]
- 58. Sinha S., Grewal R. K., and Roy S. (2018) Modeling bacteria-phage interactions and its implications for phage therapy. Adv. Appl. Microbiol. 103, 103–141 10.1016/bs.aambs.2018.01.005 [DOI] [PubMed] [Google Scholar]
- 59. Welshons W. V., Wolf M. F., Murphy C. S., and Jordan V. C. (1988) Estrogenic activity of phenol red. Mol. Cell Endocrinol. 57, 169–178 10.1016/0303-7207(88)90072-X [DOI] [PubMed] [Google Scholar]
- 60. Vamosi G., Baudendistel N., von der Lieth C. W., Szaloki N., Mocsar G., Muller G., Brazda P., Waldeck W., Damjanovich S., Langowski J., and Toth K. (2008) Conformation of the c-Fos/c-Jun complex in vivo: a combined FRET, FCCS, and MD-modeling study. Biophys. J. 94, 2859–2868 10.1529/biophysj.107.120766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Nielsen R., and Mandrup S. (2014) Genome-wide profiling of transcription factor binding and epigenetic marks in adipocytes by ChIP-seq. Methods Enzymol. 537, 261–279 10.1016/B978-0-12-411619-1.00014-8 [DOI] [PubMed] [Google Scholar]
- 62. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., and 1000 Genome Project Data Processing Subgroup. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Li H., and Durbin R. (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Heinz S., Benner C., Spann N., Bertolino E., Lin Y. C., Laslo P., Cheng J. X., Murre C., Singh H., and Glass C. K. (2010) Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 10.1016/j.molcel.2010.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. The ENCODE Project Consortium. (2012) An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 10.1038/nature11247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kanehisa M., and Goto S. (2000) KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 10.1093/nar/28.1.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Saldanha A. J. (2004) Java Treeview–extensible visualization of microarray data. Bioinformatics 20, 3246–3248 10.1093/bioinformatics/bth349 [DOI] [PubMed] [Google Scholar]
- 68. Hulsen T., de Vlieg J., and Alkema W. (2008) BioVenn—a web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genomics 9, 488 10.1186/1471-2164-9-488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Thorvaldsdottir H., Robinson J. T., and Mesirov J. P. (2013) Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 14, 178–192 10.1093/bib/bbs017 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
ChIP-seq data have been submitted to Sequence Read Archive and Gene Expression Omnibus databases under accession numbers PRJNA632899 and GSE150652, respectively.