

Abstract
G-protein–coupled receptors (GPCRs) are a ubiquitously expressed family of receptor proteins that regulate many physiological functions and other proteins. They act through two dissociable signaling pathways: the exchange of GDP to GTP by linked G-proteins and the recruitment of β-arrestins. GPCRs modulate several members of the transient receptor potential (TRP) channel family of nonselective cation channels. How TRP channels reciprocally regulate GPCR signaling is less well-explored. Here, using an array of biochemical approaches, including immunoprecipitation and fluorescence, calcium imaging, phosphate radiolabeling, and a β-arrestin–dependent luciferase assay, we characterize a GPCR–TRP channel pair, angiotensin II receptor type 1 (AT1R), and transient receptor potential vanilloid 4 (TRPV4), in primary murine choroid plexus epithelial cells and immortalized cell lines. We found that AT1R and TRPV4 are binding partners and that activation of AT1R by angiotensin II (ANGII) elicits β-arrestin–dependent inhibition and internalization of TRPV4. Activating TRPV4 with endogenous and synthetic agonists inhibited angiotensin II–mediated G-protein–associated second messenger accumulation, AT1R receptor phosphorylation, and β-arrestin recruitment. We also noted that TRPV4 inhibits AT1R phosphorylation by activating the calcium-activated phosphatase calcineurin in a Ca2+/calmodulin–dependent manner, preventing β-arrestin recruitment and receptor internalization. These findings suggest that when TRP channels and GPCRs are co-expressed in the same tissues, many of these channels can inhibit GPCR desensitization.
Keywords: transient receptor potential vanilloid 4 (TRPV4), angiotensin II receptor type 1 (AT1R), G-protein–coupled receptor (GPCR), β-arrestin, phosphorylation, desensitization, calcineurin, cell signaling, cell sensor, receptor desensitization, transient receptor potential channels (TRP channels), beta-arrestin
Canonical GPCR signaling comprises G-protein–mediated alterations in the production of the second messenger molecules cAMP, inositol (1,4,5)-trisphosphate (IP3), and diacylglycerol (1). GPCRs can also signal through the recruitment of β-arrestins (2). β-Arrestins translocate to the cell membrane and bind GPCRs following ligand-induced phosphorylation of the GPCR C terminus by G-protein–coupled receptor kinases (GRKs) (2, 3). β-Arrestin interactions hinder further G-protein binding, desensitize the GPCR, and also may lead to receptor internalization, recycling, or degradation (2, 3). β-Arrestin also elicits G-protein–independent signaling consequences through mitogen-activated protein kinases and serves as an adaptor protein linking GPCRs to other nearby membrane proteins (3). For example, ANGII stimulation of AT1R can direct chemotaxis of HEK293 cells in a G-protein–independent fashion, as well as induce β-arrestin–dependent ubiquitination of TRPV4 ion channels in rat vascular smooth muscle cells (4, 5). Thus, β-arrestin recruitment is a critical regulatory event that dictates the signaling of GPCRs, as well as other nearby proteins.
TRP channels are a large family of multimodal cation channels permeable to Ca2+, Na+, and Mg2+ (6, 7). TRP channels are located in the plasma membrane of most animal cells and mediate a wide range of sensory signaling including taste, mechanosensation, osmosensation, and thermosensation (7). Because of their ubiquity, TRP channels are regulated by a wide array of GPCRs. For example, AT1R activation induces catecholamine secretion from chromaffin cells by recruiting and activating transient receptor potential canonical 3 (TRPC3) (8); μ-opioid receptor (MOR1) activation inhibits cold sensation by internalizing transient receptor potential melastatin 8 (TRPM8) (9); and metabotropic glutamate receptor 1 can stimulate neuronal excitatory post synaptic conductance mediated by transient receptor potential C1 (TRPC1) channels in neurons (10). A less well-explored aspect of the GPCR–TRP channel interaction is the ability of TRP channels to reciprocally regulate GPCRs. We recently reported that transient receptor potential vanilloid 1 (TRPV1) can bind MOR1, inhibit MOR1 phosphorylation, and block β-arrestin recruitment (11). TRPV1 activity inhibits ligand-induced phosphorylation of MOR1 by GRK5 and can induce β-arrestin nuclear translocation preventing recruitment to MOR1 (11, 12).
We hypothesize that diverse GPCR–TRP channel-binding partners exhibit regulatory relationships. In particular, GPCRs and TRP channels that physically bind and have antagonistic signaling consequences, such as AT1R and TRPV4, may act as reciprocal regulators. AT1R and TRPV4 are each ubiquitously expressed, but are both particularly abundant in vascular endothelial/smooth muscle cells and the choroid plexus (5, 13–16). In these cell types, AT1R and TRPV4 display opposite cellular effects. In the vasculature, AT1R activation stimulates vasoconstriction, whereas TRPV4 activity induces vasodilation (13, 17). In the choroid plexus, AT1R and TRPV4 have similarly oppositional signaling consequences. AT1R activation stimulates the secretion of vasopressin from choroid plexus epithelial cells (CPECs), leading to diminished blood flow to the choroid plexus and decreased secretion of CSF. In contrast, TRPV4 activity increases blood flow to the choroid and increases the flux of Cl− and K+ ions into the cerebral ventricles increasing CSF production. Thus, TRPV4 and AT1R are ideally suited to regulate CSF homeostasis by regulating one another's activity.
In this study we examine whether TRPV4 and AT1R exhibit a similar regulatory relationship as TRPV1 and MOR1. We explore the ability of TRPV4 to regulate AT1R activity, phosphorylation, and β-arrestin recruitment in the choroid plexus and immortalized cells and investigate how this regulatory relationship generalizes to other pairs of GPCR and TRP channels.
Results
TRPV4 physically and functionally interacts with AT1R
TRPV4 and AT1R are co-expressed in a variety of cells types including vascular endothelial cells, vascular smooth muscle cells, and CPECs (5, 13, 15). Despite their co-expression in multiple tissues, TRPV4 and AT1R have only been shown to interact physically in rat vascular smooth muscle cells (5). In HEK293 cells, overexpressed HA–AT1R co-immunoprecipitates (co-IP) with FLAG–TRPV4 (Fig. 1A). Treating cells with the potent TRPV4 agonist GSK101 does not affect the binding of TRPV4 to AT1R (Fig. 1A). Cultured primary murine CPECs derived from WT mice at postnatal day 21 express endogenous TRPV4 and AT1R protein (Fig. 1B). Knockout of TRPV4 in CPECs cultured from TRPV4 null (TRPV4-KO) mice does not significantly alter AT1R expression in CPECs in vitro (Fig. 1B). TRPV4-KO eliminates the response of CPECs in vitro to 10 μm GSK101 by calcium imaging (Fig. S1A). WT CPECs express mRNA for both TRPV4 (probe Trpv4) and AT1R (probe Agt1ra) (Fig. 1C). IP AT1R from whole choroid plexus of WT and TRPV4-KO mice suggests that TRPV4 and AT1R bind in primary cells at endogenous expression levels (Fig. S1B). AT1R is predominantly a Gαq-coupled GPCR. Ligand binding to AT1R increases phospholipase C activity, which cleaves membranous phosphatidylinositol trisphosphate into the second messengers diacylglycerol and IP3 (8, 18). To monitor IP3 production, HEK293 cells overexpressing HA–AT1R and FLAG–TRPV4 were labeled with tritiated myo-inositol, and HPLC was used to separate the different phosphoinositol isoforms. ANGII stimulation of AT1R results in robust IP3 production, whereas pretreatment with 10 nm GSK101 for 5 min significantly inhibits IP3 production (Fig. 1D). HPLC analysis of the major phosphorylated inositol subtypes indicates that TRPV4 activation does not significantly alter levels of other phosphoinositol isoforms (Fig. S1C). Additionally, we did not find evidence that TRPV4 activation by GSK101 disrupts Gαq binding to AT1R (Fig. S1D).
Figure 1.

TRPV4 and AT1R are co-expressed in the same tissue, bind to one another, and interact physiologically. A, FLAG–TRPV4 and HA–AT1R co-IP in transiently transfected HEK293 cells left untreated or treated with 10 nm GSK101. GSK101 was applied for 10-min prelysis. B, Western blotting from CPECs from WT and TRPV4-KO. WT CPECs express both AT1R and TRPV4. TRPV4-KO CPECs express AT1R and no TRPV4. C, RT-qPCR of mRNA extracted from CPECs from WT mice. Error bars denote means ± S.D. D, left panel, IP3 production measured using HPLC from HEK293 cells expressing TRPV4 and AT1R. The cells were stimulated with combinations of 10 nm GSK101 for 10 min and 100 nm ANGII for 2 min. Right panel, total IP3 is measured by calculating the area under the curve for elution fractions 21–27 (n = 4). Error bars denote means ± S.D. *, p < 0.05. Pulldown Westerns are indicated by pulldown (PD). CPECs were cultured for 12 days in vitro prior to experimentation. No TX, no treatment.
TRPV4 inhibits AT1R phosphorylation
We next examined whether TRPV4 activity impacts ligand-induced phosphorylation of a GPCR-binding partner. AT1R phosphorylation, the most proximal signaling event that we can monitor, is required for AT1R-mediated inhibition of TRPV4 (5). HEK293 cells overexpressing HA–AT1R and FLAG–TRPV4 were incubated in [32P]orthophosphate and treated with combinations of 10 nm GSK101 and 100 nm ANGII. ANGII stimulates the phosphorylation of AT1R, but TRPV4 activation prior to ANGII treatment significantly inhibits the AT1R phosphorylation (Fig. 2A). Compared with baseline, there is also a nonsignificant decrease in phosphorylation of AT1R when cells are treated with GSK101 alone (Fig. 2A). We did not measure AT1R phosphorylation changes in response to GSK101 when no TRPV4 was expressed. After a GPCR is phosphorylated, β-arrestin is recruited to bind the GPCR C terminus. IP of AT1R from HEK293 cells expressing FLAG–TRPV4, HA–AT1R, and YFP–β-arrestin after treatment with ANGII pulls down β-arrestin, which is blocked by pretreatment with GSK101 (Fig. 2B).
Figure 2.

TRPV4 inhibits ligand-induced AT1R phosphorylation and subsequent β-arrestin recruitment. A, left panel, autoradiograph from IP for HA from HEK293 cells overexpressing HA–AT1R and FLAG–TRPV4. HEK293 cells were incubated in [32P]orthophosphate and stimulated with the indicated combinations of 100 nm ANGII for 10 min and 10 nm GSK101 for 5 min. Right panel, densitometry quantification of phosphorylated band normalized to total phosphorylated protein (n = 3). B, HEK293 cells overexpressing HA–AT1R, FLAG–TRPV4, and YFP–β-arrestin 1 were treated with indicated drug combinations of 10 nm GSK101 and 100 nm ANGII for 10 and 20 min, respectively. IP indicates immunoprecipitation with HA primary and blot for HA and YFP pulldown (PD). C–E, HTLA cells expressing FLAG–TRPV4 and AT1R-Tango stimulated with GSK101 and ANGII for 10 and 30 min, respectively. C, normalized luciferase production stimulated with indicated drug combinations (n = 6). D, normalized luciferase production stimulated by 100 nm ANGII and increasing doses of GSK101 (n = 3). E, normalized luciferase production in response to combinations of 100 nm ANGII and 5 nm GSK101 in the presence and absence of 5 μm HC067, a TRPV4 antagonist (n = 3). All error bars denote means ± S.D. All graphs analyzed by one-way ANOVA corrected for multiple comparisons with a post hoc Tukey test. *, p < 0.05; ***, p < 0.0005. Normalization is described under “Experimental procedures.” RLU, relative luminescence unit.
We employed the luciferase-based TANGO system to explore details of β-arrestin recruitment to AT1R. The TANGO system leverages an engineered HEK293 cell line (HTLA) that produces the luciferase enzyme following β-arrestin translocation to the cell membrane and binding to a GPCR (19). Activation of AT1R with 100 nm and 1 μm ANGII significantly increases β-arrestin recruitment to AT1R, which is blocked by TRPV4 activation with 10 nm GSK101 (Fig. 2C). The endogenous TRPV4 agonist 11,12-epoxyeicosatrienoic acid also inhibits β-arrestin recruitment to AT1R (Fig. S2A) (20). Interestingly, 10 nm GSK101 alone is sufficient to significantly decrease β-arrestin recruitment and luciferase production compared with baseline, which could indicate a reduction in ligand-independent phosphorylation (Fig. 2C). To determine whether our assay was measuring a reduction in ligand-independent phosphorylation, we treated HTLA cells with olmesartan, an inverse agonist of AT1R. Applying 5 nm olmesartan to HLTA cells expressing AT1R is sufficient to block β-arrestin recruitment but does not lower baseline luciferase production (Fig. S2B). Stimulating TRPV4 with increasing concentrations of GSK101 causes a dose-responsive decrease in the amount of β-arrestin recruitment to AT1R with an approximate IC50 between 0.5 and 1 nm (Fig. 2D). GSK101 has no effect when TRPV4 is not expressed (Fig. S2C). Preincubation of cells with the TRPV4 antagonist HC067 (Fig. 2E) and ruthenium red, another TRP channel antagonist, dose-dependently rescues ANGII-stimulated β-arrestin recruitment to AT1R (Fig. S2, D and E). Taken together, these data indicate that TRPV4 activation is necessary to block ligand-induced binding of β-arrestin to AT1R.
TRPV4 activates the phosphatase calcineurin to dephosphorylate AT1R
Protein phosphorylation can be regulated by two mechanisms: the initial phosphorylation by a protein kinase and the dephosphorylation by a protein phosphatase. TRPV1 inhibits MOR1 phosphorylation by stimulating the nuclear translocation of GRK5 (11). TRPV4 activation can also induce the translocation of GRK5 out of the plasma membrane (Fig. S3A). We performed an order-of-addition experiment to determine whether TRPV4 regulates AT1R phosphorylation by inhibiting a protein kinase, such as GRK5, or activates a protein phosphatase. If TRPV4 inhibits a kinase, then reversing the order in which TRPV4 and AT1R are activated will produce different results, because once a kinase has completed phosphorylation of a substrate, it does not matter if it is inhibited. Conversely, if TRPV4 activates a phosphatase, the order in which TRPV4 and AT1R are activated is irrelevant, because the phosphatase will immediately dephosphorylate its substrate. The order in which TRPV4 and AT1R are activated has no significant effect on TRPV4-mediated inhibition of β-arrestin recruitment (Fig. 3A). This suggests that TRPV4 inhibits AT1R phosphorylation by activating a protein phosphatase.
Figure 3.

TRPV4 activates calcineurin to dephosphorylate AT1R receptor in a Ca2+/calmodulin–dependent manner. A–C, HTLA cells expressing FLAG–TRPV4 and AT1R-Tango stimulated with GSK101 for 10 min and ANGII for 30 min. A, normalized luciferase production in response to activating AT1R before or after TRPV4 activation (n = 3–6). B, normalized luciferase production in response to AT1R and TRPV4 activation in the presence of 30 μm CAP (n = 3). C, normalized luciferase production in response to 0.5 nm GSK101 stimulation followed by 100 nm ANGII stimulation in the presence of increasing concentrations of CAP (n = 3). D, left panel, cell fractionation and immunoblotting to determine subcellular localization of p-NFATC1 and NFATC1 in HEK293 cells expressing FLAG–TRPV4. Cells were treated with the indicated doses of GSK101 for 10 min prior to lysis. Right panel, densitometry of cytoplasmic p-NFATC1 (top right panel) and nuclear NFATC1 (bottom right panel). GAPDH is a cytoplasmic protein loading control. Histone 2B is a nuclear protein loading control. E and F, HTLA cells expressing FLAG–TRPV4 and AT1R-Tango stimulated with GSK101 for 10 min and ANGII for 30 min. E, normalized luciferase production in response to activating TRPV4 with 0.5 nm GSK101 for 10 min followed by activating AT1R with 100 nm ANGII for 30 min in the presence or absence of 100 nm okadaic acid (n = 3). F, normalized luciferase production in response to 0.5 nm GSK101 stimulation followed by 100 nm ANGII stimulation in the presence of increasing concentrations of calmidazolium, calmodulin inhibitor (n = 3). G and H, HTLA cells expressing FLAG–TRPV1 and AT1R-Tango stimulated with 50 nm capsaicin for 10 min and 100 nm ANGII for 30 min. G, normalized luciferase production in response to TRPV1 and AT1R activation. H, normalized luciferase production in response to AT1R and TRPV1 activation in the presence of 30 μm CAP (n = 6). All error bars denote means ± S.D. All graphs were analyzed by one-way ANOVA corrected for multiple comparisons with a post hoc Tukey test. *, p < 0.05; ***, p < 0.005. RLU, relative luminescence unit; ns, not significant; No Tx, no treatment.
Calcineurin is a Ca2+/calmodulin–activated protein phosphatase that can be activated by TRPV4 (21, 22). Furthermore, calcineurin can inhibit the desensitization of 5-HT1c receptors, a GPCR expressed in the choroid plexus (23, 24). Incubating HTLA cells transiently transfected with TRPV4 and AT1R in 30 μm of a cell-permeable calcineurin autoinhibitory peptide (CAP) blocks TRPV4 inhibition of β-arrestin recruitment (Fig. 3B). CAP rescues β-arrestin binding in a dose-responsive manner with an approximate EC50 of ∼1.2 μm (Fig. 3C). Phosphorylated nuclear factor of activated T-cells (p-NFAT) is the canonical target of calcineurin dephosphorylation (25). Activated calcineurin dephosphorylates p-NFAT and initiates NFAT translocation from the cytoplasm to the nucleus (25). Activating TRPV4 in HEK293 cells with 10 nm GSK101 is sufficient to significantly dephosphorylate p-NFAT and reduce NFATC1 in the cytoplasm (Fig. 3D and Fig. S3B). There is no significant change of NFATC1 in the nucleus (Fig. 3D). Incubating HEK293 cells expressing TRPV4 in 30 μm CAP blocks TRPV4-mediated p-NFAT dephosphorylation (Fig. 3D). We found inhibiting calcineurin completely blocks the effects of TRPV4 on β-arrestin recruitment. We wondered whether other phosphatases are activated by TRPV4. Incubation of cells in 100 nm okadaic acid, a broad phosphatase inhibitor that does not block calcineurin, does not recapitulate the effects of inhibiting calcineurin (Fig. 3E) (26). Furthermore, calcineurin activation requires calmodulin (25, 27). Consequently, culturing HTLA cells in calmidazolium, a calmodulin inhibitor, similarly prevents TRPV4 from blocking β-arrestin recruitment to AT1R in a dose-dependent manner, with an EC50 of ∼3 μm (Fig. 3F). Taken together, the results demonstrate that TRPV4 specifically activates calcineurin in a Ca2+/calmodulin–dependent manner calcineurin autoinhibitory peptide is sufficient to dephosphorylate AT1R and block β-arrestin recruitment.
TRPV4 and TRPV1 both can inhibit phosphorylation of GPCRs that are in close proximity in a Ca2+/calmodulin-dependent manner. It is likely that many other TRP channels similarly regulate nearby GPCRs. Co-expressing and activating TRPV1 by capsaicin (Fig. 3G) and TRPA1 by iodoacetamide (Fig. S3C) with AT1R both block ANGII-stimulated β-arrestin recruitment. Similarly, when TRPV4 is co-expressed with another GPCR, mas-related G-protein–coupled receptor membrane A1 (MRGA1), GSK101 stimulation blocks β-arrestin recruitment to MRGA1 stimulated with Phe-Met-Arg-Phe-amide (Fig. S3D). Previous work demonstrated that TRPV1 inhibits phosphorylation of MOR1 by inhibiting a kinase (11). Incubating TRPV1- and AT1R-expressing cells with CAP does not rescue TRPV1-mediated inhibition of β-arrestin recruitment (Fig. 3H). Taken together, our findings reveal that diverse TRP channels inhibit GPCR phosphorylation but may do so via distinct molecular pathways for different TRP channels.
TRPV4 activity is regulated by AT1R activation
The choroid plexus is a tissue in which TRPV4 and AT1R are likely to regulate each other's activity. TRPV4 and AT1R have opposite roles in regulating blood flow to the choroid plexus and in the production of CSF (13, 15, 17, 28). To test whether AT1R activation can inhibit TRPV4 in cells other than vascular smooth muscle cells (5), we used CPECs as a model tissue for TRPV4–AT1R reciprocal regulation. Primary murine CPECs exposed to 100 nm ANGII in vitro have significantly reduced calcium inflows in response to GSK101 treatment (Fig. 4A). Calcium influx in CPECs induced by GSK101 is specific to TRPV4 activation and is completely blocked when CPECs are incubated in GSK205, a TRPV4-specific antagonist (Fig. 4A). AT1R inhibits TRPV4 by recruiting β-arrestin (5). We found that activating TRPV4 before AT1R blocks recruitment of β-arrestin to AT1R. Therefore, TRPV4 can prevent its own inhibition. Accordingly, calcium imaging of CPECs that were incubated in GSK101 before treating the cells with ANGII partially rescues TRPV4-mediated calcium influx in response to further GSK101 treatment (Fig. 4B).
Figure 4.
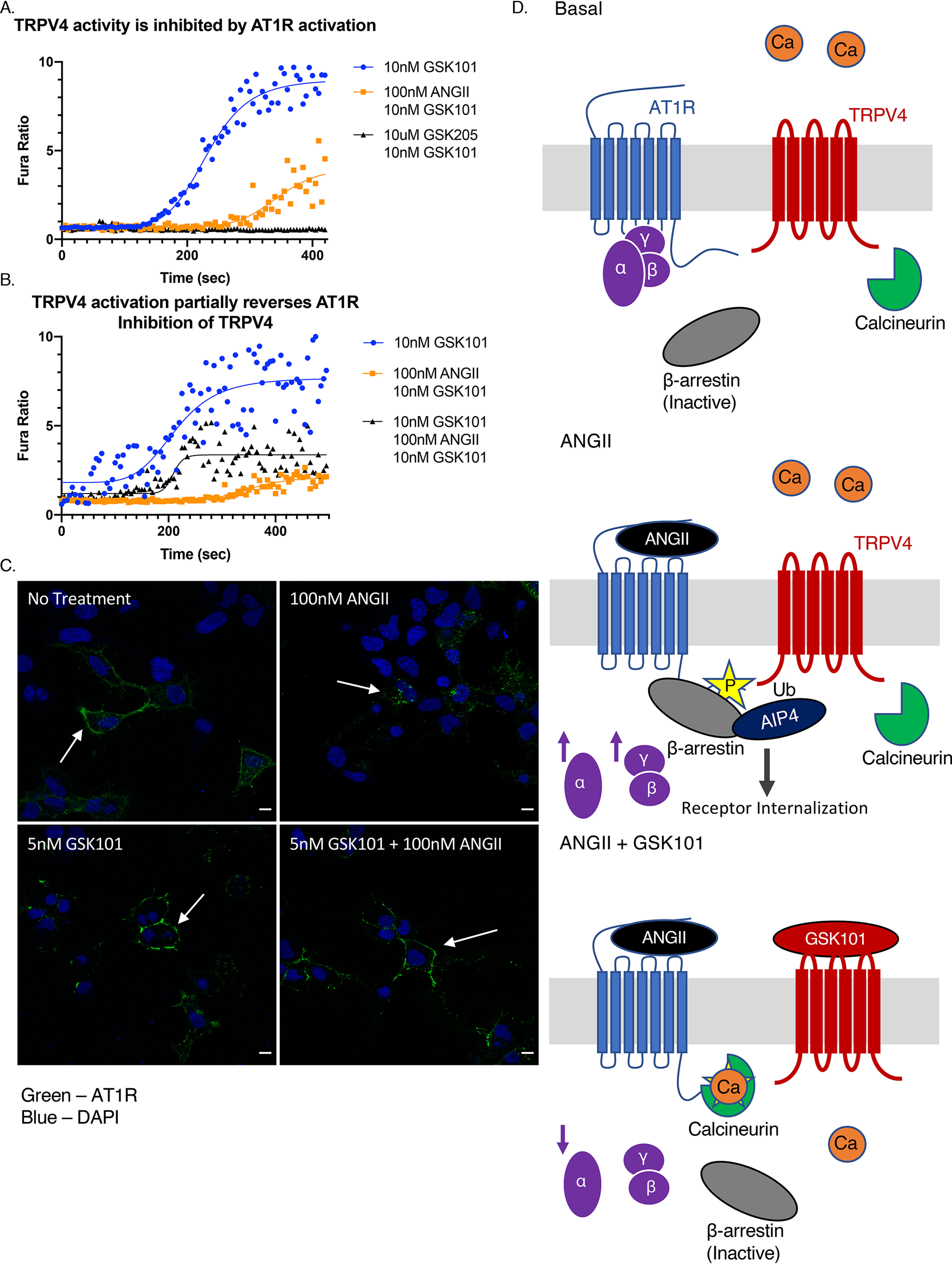
TRPV4 activity can be regulated by AT1R activation. A and B, calcium imaging of CPECs cultured for 12 days in vitro. The cells are imaged only for the last 10 nm GSK101 dose. A, CPECs stimulated with 10 nm GSK101 (blue) and 100 nm ANGII for 30 min followed by 10 nm GSK101 (orange), and the cells were incubated in 10 μm GSK205 followed by 10 nm GSK101 (black; n = 7–10). B, CPECs stimulated with 10 nm GSK101 (blue), 100 nm ANGII for 30 min followed by 10 nm GSK101 (orange), and 10 nm GSK101 for 10 min, followed by 100 nm ANGII for 30 min and then 10 nm GSK101 a second time (n = 10–23). C, IF of HEK293 cells expressing HA–AT1R and FLAG–TRPV4 treated with indicated combinations 10 nm GSK101 and 100 nm ANGII for 10 and 20 min, respectively. Primary antibody was added prior to drug applications. White arrows indicate specific locations on cells of interest. Green, HA–AT1R; blue, Hoechst. Scale bar, 10 μm. D, hypothetical schematic of the TRPV4 interaction with AT1R. Top panel, at baseline TRPV4 and AT1R are co-expressed in the same cell and may physically interact. Middle panel, ANGII-mediated activation of AT1R catalyzes the activation of Gαq and βγ signaling and recruits β-arrestin to bind its phosphorylated C terminus. β-Arrestin then mediates ubiquitination of TRPV4 by AIP4. Bottom panel, TRPV4 activation activates calcineurin in Ca2+/calmodulin–dependent manner, which dephosphorylates AT1R, inhibiting recruitment of β-arrestin and therefore inhibiting internalization. Separately, TRPV4 activation inhibits Gαq signaling by reducing IP3 accumulation. DAPI, 4[prime],6[prime]-diamino-2-phenylindole.
Activation of GPCRs results in β-arrestin–mediated receptor internalization (2, 3). We performed nonpermeabilized IF of HEK293 cells transiently transfected with HA–AT1R and FLAG–TRPV4 to examine changes in AT1R localization following stimulation with ANGII and GSK101. At baseline, AT1R is largely restricted to the cell membrane (Fig. 4C, top left panel), but when treated with ANGII, it undergoes robust internalization (Fig. 4C, top right panel). GSK101 treatment alone does not influence membrane localization of AT1R (Fig. 4C, bottom left panel). Treating cells with GSK101 prior to the application of ANGII largely prevents AT1R internalization (Fig. 4C, bottom right panel). This result implies that β-arrestin is only recruited to the membrane when ANGII is given alone. IF shows that β-arrestin is only recruited to the cell membrane (Fig. S4) when HEK293 cells expressing AT1R and TRPV4 are treated with ANGII and not when cells are treated with the combination of GSK101 and ANGII (Fig. S4). In summary, TRPV4 and AT1R can both regulate one another's activity/signaling, as well as their membrane localization (a summary of the interaction is shown in Fig. 4D).
Discussion
Our study reveals that TRPV4 and AT1R reciprocally interact in the choroid plexus and that TRPV4 activity can inhibit AT1R signaling. Activation of TRPV4 stimulates the protein phosphatase calcineurin, which dephosphorylates AT1R, inhibiting the β-arrestin–mediated internalization of AT1R. TRPV4 activation also appears to decrease AT1R Gαq signaling by reducing ANGII-stimulated IP3 production. Notably, the regulatory mechanism we describe here likely generalizes to other pairs of TRP channels and GPCRs. Our study expands on the underappreciated role that TRP channel activity has in reciprocally regulating the activity, localization, and sensitization of GPCRs.
Co-IP studies in HEK293 cells and primary CPECs suggest that TRPV4 and AT1R are binding partners or interact in a macromolecular complex (Fig. 1A and Fig. S1B). This reinforces prior evidence that endogenous TRPV4 and AT1R bind in rat vascular smooth muscle cells (5). Shukla et al. (5) showed that AT1R stimulation by ANGII causes β-arrestin–dependent ubiquitination and functional inhibition of TRPV4 by AIP4 (an E3-ubiquitin ligase) (Fig. 4C, middle panel). Analogously, in our study exposure of CPECs to ANGII inhibits TRPV4 responses to GSK101 (Fig. 4A). Interestingly there are other proteins that are activated by ANGII such as Mas1 and angiotensin receptor type 2 that may also be able to interact with TRPV4 in the choroid plexus. GPCRs can regulate TRP channels through a variety of mechanisms including direct G-protein–mediated activation (8, 29), allosteric sensitization (30), inhibition by internalization (5), removal of inhibition (31), and inhibition of sensitization (32). The potential for TRP channels to regulate GPCRs has been examined far less.
Our results provide converging physiologic and pharmacologic evidence that TRP channels are important regulators of GPCR signaling, phosphorylation, β-arrestin recruitment, and GPCR internalization. The exact mechanism by which TRPV4 inhibits Gαq signaling is not known. We found no compelling evidence that TRPV4 activity interrupts G-protein GPCR coupling (Fig. S1D). We did, however, notice in our HPLC experiments that TRPV4 activation seemed to increase the accumulation of IP1 (Fig. S1C). The enzyme responsible for degrading IP3 to IP1 is polyphosphate 5-phosphatase. The rate of polyphosphate 5-phosphatase conversion of IP3 to IP1 is increased in the presence of increasing Ca2+ and elevated protein kinase C activity (33). Therefore, TRPV4 may not be directly inhibiting Gαq signaling but rather decreasing the accumulation of the second messenger IP3. The ability of TRPV4 to inhibit AT1R Gαq signaling contrasts with our work with TRPV1 and MOR1, in which TRPV1 had no influence on MOR1 mediated Gαi signaling (11). A limitation of this experiment is we did not assay the production of IP3 in the absence of TRPV4 when cells were still treated with GSK101. Therefore, we did not control for potential off-target effects GSK101 may have on downstream AT1R signaling. However, given that GSK101 does not affect calcium signaling in TRPV4 knockout cells (Fig. S1B) and has no effect on β-arrestin recruitment to AT1R in untransfected HEK293 (Fig. S2C), we do not believe those off-target effects significantly affect our conclusions.
TRP channels can inhibit GPCR phosphorylation and regulate β-arrestin localization (11, 12). In contrast to TRPV1, which inhibits the phosphorylation of MOR1 by GRK5, TRPV4 activates calcineurin to dephosphorylate AT1R. TRPV4 activates calcineurin in a Ca2+/calmodulin–dependent manner in osteoclasts, airway smooth muscles cells, and skeletal muscle (21, 22, 34). Our results indicate that TRPV4 can stimulate calcineurin activity in a wide range of tissues including the choroid plexus. Calcineurin canonically signals by stimulating the nuclear translocation of NFAT, which induces cell type–specific changes in gene transcription (25). Calcineurin can also inhibit the desensitization of GPCRs (23, 35). Thus, TRPV4 not only directs calcineurin to dephosphorylate AT1R but may also drive NFAT-dependent transcriptional changes in many tissues. An important caveat to this is that our results did not show a significant increase of NFAT in the nucleus, but only decreased pNFAT in the cytoplasm (Fig. 3D).
Our findings suggest that many TRP channels can regulate GPCR phosphorylation, although not necessarily by activating calcineurin. When TRPA1 and TRPV1 were artificially expressed with AT1R, both proteins could inhibit ligand-induced receptor phosphorylation. Additionally, when TRPV4 was transiently expressed with MRGA1, it was similarly able to inhibit MRGA1 phosphorylation. It is possible that we randomly selected special GPCR–TRP pairs but more likely that many pairs of GPCR–TRP channels exhibit an equivalent regulatory relationship. The specificity of the regulation is likely determined by the cell type being studied and whether the TRP channel and GPCR of interest physically interact. Interestingly, although activating TRPV1 and TRPV4 lead to an equivalent functional outcome on AT1R phosphorylation, the result is achieved through molecularly distinct signaling mechanisms (Fig. 3, B and H).
Whether a GPCR–TRP channel pair are physiologically important regulators likely depends on their tissue expression profiles. TRPV4 and AT1R are co-expressed in many tissues, but in the vasculature and the choroid plexus, they elicit opposite signaling outcomes, making them potential reciprocal regulators. In the vasculature AT1R is a vasoconstrictor and TRPV4 is a vasodilator. Cross-talk between the two proteins may be essential for maintenance of appropriate vascular tone (5, 13). Under physiologic conditions, AT1R and TRPV4 interact in the choroid plexus to maintain homeostatic rates of CSF production on a minute to minute time scale. CPECs produce ∼80% of CSF and express all the molecular components of the renin–angiotensin–aldosterone system (28, 36). When intraventricular ANGII binds AT1R, CPECs secrete arginine vasopressin, which results in reduced blood flow to the choroid plexus and diminished Cl− efflux into the cerebral ventricles (28). The net effect of AT1R signaling is decreased CSF production. Conversely, activating TRPV4, which functions as an osmolarity sensor in the choroid plexus, increases Cl− and K+ efflux through calcium-activated chloride/potassium channels, resulting in enhanced CSF production (15, 37). TRPV4 and AT1R are uniquely situated as a GPCR–TRP channel pair to regulate each other's activity and localization and maintain homeostatic CSF production.
In summary, we have demonstrated a novel mechanism for TRPV4 to inhibit AT1R receptor phosphorylation and activity. Calcium influx through TRPV4 leads to the Ca2+/calmodulin–dependent activation of calcineurin and subsequent dephosphorylation of AT1R. This interaction is not only important for the regulation of AT1R activity but also relieves AT1R mediated inhibition of TRPV4. Our evidence indicates that this regulatory pathway is likely conserved across other GPCR–TRP channel pairs. Our model suggests that other pairs, such as TRPC1–dopamine receptor D2 and TRPM8–serotonin receptor 1B, would be good targets to examine and expand our understanding this regulatory pathway because they are already known to interact physically and physiologically (30, 38). Given the broad expression of TRP channels and the medical importance of GPCRs, it is possible that TRP channel agonists will have therapeutic potential as modulators of GPCR activity, desensitization, and localization.
Experimental procedures
Drugs/chemicals
The chemicals used to generate buffers were purchased from Sigma unless otherwise indicated. The concentrations of drugs used are indicated in the text and figure legends: GSK1016790A (G0798), angiotensin II (A9525), EGTA (03777), calcineurin autoinhibitory peptide (Millipore, 207001), ruthenium red (R2751), HC067047 (SML0143), 11,12-epoxyeicosatrienoic acid (Cayman Chemical, 50511), calmidazolium chloride (C3930), okadaic acid (Abcam, ab120375), iodoacetamide (I1149), Phe-Met-Arg-Phe-amide (N3637), PhosSTOP (4906845001), and protease inhibitor mixture (P8340).
Cell culture and transfection
HEK293 cells were grown in regular Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (Sigma), 2 mm glutamine (Invitrogen), and 1000 units of penicillin/streptomycin (Invitrogen). Where serum-free medium is indicated, no fetal bovine serum was supplemented to DMEM. Primary choroid plexus epithelial cells were cultured in regular DMEM supplemented with 100 ng/ml epidermal growth factor (Sigma) and AraC 20 μm (Sigma). HTLA cells were maintained in regular DMEM supplemented with 1 μg/ml hygromycin (Invivogen) and puromycin 3 ug/ml (Gibco) and were generously donated by Bryan Roth (19).
The cells were transfected using Lipofectamine 3000 (Invitrogen) according to company provided protocol. WT HA–AT1R was generated by site directed mutagenesis using QuikChange/Stratagene techniques from pcDNA3.1 AT1R DRY-AAY (rat), which was a gift from Laszlo Hunyady and Robert Lefkowitz (Addgene plasmid 45759) (39). FLAG–TRPV4 (human) construct has been previously described. TRPV4 cDNA was provided by Miguel Angel Valverde (40). The MRGA1 construct has been previously described (41) and was cloned into the Tango vector (a gift from Bryan Roth) using standard techniques. AT1R-Tango (human) construct was a gift from Bryan Roth (Addgene plasmid 66222) (19). YFP–β-arrestin 1 (rat) was a gift from Robert Lefkowitz (Addgene plasmid 36916) (42). GFP-Gαq (mouse) was a gift from Catherine Berlot (Addgene plasmid 66080) (43). TRPV1 cDNA and TRPA1 cDNA (gifts from Michael Caterina) and pWZL Neo Myr FLAG GRK5 (human; a gift from William Hahn and Jean Zhao; Addgene plasmid 20495) (44) were inserted into p3×FLAG–CMV-7 (Sigma) by standard cloning methods.
Western blotting
The cells were lysed in lysis buffer (20 mm Tris-HCl, pH 7.4, 140 mm NaCl, 1% Ipegal 630) supplemented with a complete protease inhibitor mixture (Roche) and phosphatase inhibitor tablet (Roche) on ice, unless otherwise indicated. Lysate protein content was determined using Bradford reagent (Sigma). The lysates were boiled in 1× NuPAGE lithium dodecyl sulfate sample buffer (Invitrogen) and subjected to SDS-PAGE using the Novex system (Invitrogen) following the manufacturer's instructions, transferred to polyvinylidene difluoride membranes (Millipore), blocked in 3% BSA (Sigma) in TBS, 0.1% Tween 20 for 1 h at room temperature, and incubated with primary antibodies overnight at 4 °C. In figures containing multiple Western blotting panels, triplicate gels were run containing equivalent samples.
The antibodies used were Na+/K+ ATPase (DSHB, a6f) 1:1000, FLAG M2 (Sigma, F3165) 1:2000, HA (Roche, 11867423001) 1:1000, 12CA5 HA antibody (Roche, 11583816001), TRPV4 (Abcam, ab39260) 1:1000, AT1R1 (Abcam, ab18801) 1:1000, phospho-NFATC1 (R&D, MAB5640) 1:1000, NFAT1 (CST, 4389S) 1:1000, GFP (Abcam, ab290) 1:1000, myc 9E10 (Roche, 11667203001) 1:1000, GRK5 (Santa Cruz, sc-565) 1:1000, Amersham Biosciences ECL mouse IgG HRP-linked (GE, NA931) 1:10,000, and Amersham Biosciences ECL rabbit IgG HRP-linked (GE, NA934) 1:10,000, rat IgG HRP-linked (CST, 7077S) 1:1000.
Immunoprecipitation
Co-immunoprecipitation experiments were performed using EZview Red FLAG-agarose (Sigma) for FLAG-tagged constructs and protein A/G–agarose (EMD Millipore) for HA-tagged and endogenous protein pulldowns. The ratio of primary antibody:agarose beads was 5 μg:40 μl. Lysis buffer for immunoprecipitation experiments was the same as for Western experiments unless otherwise noted. Protease inhibitor mixture (Sigma) and PhosphoSTOP tablets (Roche) were added on the day of cell lysis. Protein content was determined using Bradford reagent (Sigma). Equal amounts of protein were added to beads, and 25 μg of protein was kept for input gels. Lysates incubated with beads overnight at 4 °C and were washed 3× in lysis buffer before protein was eluted and run on SDS-page gel. Lysis buffer for β-arrestin immunoprecipitation experiments consisted of 5 mm HEPES, 0.5% Nonidet P-40, 250 mm NaCl, 2 mm EDTA, 10% (v/v) glycerol.
HPLC and inositol triphosphate measurement
HEK293 cells were plated in 6-well plates and transfected with HA–AT1R and FLAG–TRPV4. The medium was supplemented with 40 μCi of [3H]d-myo-inositol (PerkinElmer) 6 h after transfection. After 48 h, the medium was exchange for serum-free medium, and the cells were incubated at 37 °C for 2 h. The cells were washed with Hanks' balanced salt solution (HBSS, Gibco) and then allowed to incubate in HBSS supplemented with 20 mm LiCl for 20 min. Combinations of 10 nm GSK101 and 100 nm ANGII were added for 5 and 2 min, respectively. The cells were lysed with 200 μl of lysis solution, 0.6 m perchloric acid (Sigma), 0.2 mg/ml phytic acid (Sigma), 2 mm EDTA. Cell plates were then allowed to sit at −80 °C for 10 min. The lysates were neutralized with 30 μl of 1 m K2CO3. The lysates were collected and shaken overnight at 4 °C. Supernatants were run over a PARTiSPHERE SAX 5-μm HPLC column and collected in ninety 1-min fractions. Ultimo-Flo AP scintillation fluid was added to each fraction, and the radioactivity was measured using a LS 6500 multipurpose scintillation counter (Beckman Coulter).
Tango–Luciferase
The Tango–luciferase system has been described in detail previously (19). Briefly, the cells were plated in a 24-well plate transfected with AT1R-Tango and FLAG–TRPV4. After 48 h, the medium was exchanged for serum-free medium, and the cells were allowed to incubate for 4 h. The cells were then incubated with drug combinations mentioned in the text. TRP channel agonist were applied for 10 min, and GPCR agonists were applied for 30 min After drug treatments, the medium was exchanged for serum-free DMEM, and the cells were incubated at 37 °C for 6 h. The cells were lysed with 100 μl passive lysis buffer (Promega, E194A) containing protease inhibitor mixture. The lysates were spun down, and then 10 μl of each supernatant was combined with 10 μl of Bright-Glo (Promega E2610). Luminescence was measured and integrated of 10 s with a TD-20/20 luminometer (Turner Designs).
Animal protocol and primary cell collection
Unless otherwise indicated, 3–4-week-old C57BL/6 male/female mice (Jackson Laboratory) were used for all the animal-based experiments. TRPV4-KO mice have been previously described and were obtained through Makoto Suzuki (45). Animal breeding and procedures were conducted in strict accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The animal experiments were approved by the Johns Hopkins University Animal Care and Use Committee. The animals were kept on a 12-h light/dark cycle and were provided food and water ad libitum.
Extensive protocols for culture of primary mouse CPECs have been published (24, 46). Briefly, the mice were euthanized by cervical dislocation and rapidly decapitated, and the brain was placed in HBSS. The whole choroid plexus was dissected from both the lateral and the fourth ventricles. The choroid plexus from multiple mice of the same genotype were pooled and digested using 0.25% trypsin for 20 min Following digestion, the cells were plated on 35-mm glass culture dishes (MatTek) precoated with 0.1% collagen. The cells were cultured for 12 days with medium exchanges every 3 days. The medium contained 10 ng/ml epidermal growth factor (Sigma) and cytosine arabinoside 20 μm (Sigma).
Calcium imaging
CPEC cultured for 12 days in vitro on 0.1% collagen precoated MatTek 35-mm dishes. 24 h prior to imaging, regular medium was exchanged for serum-free medium. The cells were treated with 5 μl of Fura-2 AM in 95% DMSO, 5% pluronic acid for 40 min prior to imaging. The cells were imaged every 5 s with 40–160 ms of 340-nm exposure and 200–800 ms of 380-nm exposure. The final ratiometric data were background subtracted and analyzed for drawn regions of interest that covered the entire cell.
Immunofluorescence/whole mount
HEK293 cells were plated on 35-mm MatTek dishes precoated with poly-l-ornithine (Sigma), and transiently transfected with FLAG–TRPV4 and HA–AT1R. After 24 h, the medium was exchanged for serum-free medium, and the cells were incubated for 24 h more. For AT1R internalization experiments, primary antibody (HA Roche 11867423001, 1:100) was added prior to permeabilization and drug treatments. For β-arrestin staining, primary antibody (GFP Abcam ab290, 100) was added after permeabilization and drug treatments. The cells were incubated in blocking buffer, 0.1% BSA in PBS for 30 min at 4°C and then incubated with primary antibody for 1 h at 4°C. HEK293 cells were then fixed with 4% paraformaldehyde in PBS (Electron Microscopy Sciences) for 15 min. The cells were washed twice in PBS and permeabilized in 0.1% Triton X-100 in PBS for 15 min. The cells were washed three times in PBS and then blocked for 45 min at room temperature in 0.1% BSA, 0.3% milk in Tris-buffered saline, pH 7.4. The cells were then then incubated in secondary antibody (1:1000) for 1 h at room temperature. Hoechst (Sigma) added for the last 3 min of secondary antibody incubation. The cells were washed three times and then sealed with Vectashield HardSet mounting medium (Vectorlabs).
Cell fractionation
Cell fractionation and cytoplasm and crude membrane cell compartments were isolated per our published protocol (47). Briefly, HEK293 cells expressing proteins of interest were collected from 60-mm culture dishes, pelleted cells, and snap-frozen in buffer 1 (10 mm HEPES, pH 7.4, 1 mm EGTA, 1 mm DTT, 10% sucrose, protease, and phosphatase inhibitors (Roche)). The pellets were centrifuged at 1000 × g for 10 min; the supernatant was saved; the pellet was rehomogenized in buffer 1 and spun at 1000 × g for 10 min at 4 °C; and then we saved and combined the supernatants. Supernatants contain crude membrane and cytoplasm; the nuclear proteins have been removed. Centrifuge combined supernatants at 11,000 × g for 20 min at 4 °C. Supernatant contains cytoplasmic proteins, the pellet contains crude membrane. The pellet was resuspended in buffer 2 (10 mm HEPES, pH 7.4, 1 mm EGTA, 1 mm DTT, 1 mm MgCl2) and centrifuged at 21,000 × g for 20 min at 4 °C. The final pellet was resuspended in 40 μl of buffer 2. The proteins were separated by SDS-Page and immunoblotted for proteins of interest.
Radioactive phosphate labeling
A detailed protocol on radiolabeling GPCRs with hot phosphate has been previously described (48). Briefly, HEK293 cells were transfect in with HA–AT1R and FLAG–TRPV4 as indicated above. The cells were cultured overnight in serum-free medium and then incubated in phosphate-free medium supplemented with [32P]orthophosphate (PerkinElmer). The cells were treated with combinations of 10 nm GSK101 for 5 min and 100 nm ANGII for 10 min. The cells were lysed with 300 μl of lysis buffer containing 50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 4 mg/ml n-dodecyl m-maltoside (Sigma) and 0.5 mg/ml cholesteryl hemisuccinate (Sigma), protease inhibitor mixture (Sigma), 10 mm sodium fluoride (Sigma), 4.5 mg/ml sodium pyrophosphate, 0.5 μm okadaic acid (Abcam). Protein was pulled down on protein A–agarose (Roche) with 12CA5 anti-HA antibody (Roche). The beads were washed three times in various salt solutions. 30 μl0 of sample was added per lane. SDS-PAGE separation was performed as indicated above. The gels were dried on vacuum gel drier for 1 h. Dried gel was exposed to a storage phosphor screen (Molecular Dynamics) for 1 h and imaged on a Typhoon FLA9500 (GE) with a 635-nm laser, 800V PMT, and 100 μm pixel scanning. Lane intensity was measured in the ImageJ (National Institutes of Health) software package.
RT-qPCR
TaqMan RNA-to-CT one-step (Applied Biosystems, 4392938) was used for all mRNA measurement. RNA was collected using the RNeasy mini kit (Qiagen, 74104). RT-qPCR was performed with a Step One Plus real-time PCR system (Applied Biosystems) according to the company guidelines. All primers were obtained from Thermo Fisher Scientific: Trpv4 (Mm00499025), Agt1ra (Mm00507771), Hprt (Mm00446968), and ActB (Mm00607939). All probes are exon-spanning.
Statistics and image analysis
Western blotting image analysis/densitometry was performed in ImageJ (National Institutes of Health). Immunofluorescence images were contrast enhanced to improve image visibility, all contrast changes were performed uniformly across the image, and no quantitative analysis was performed on any contrast modified image. Densitometry measurements were normalized to loading proteins for each cellular compartment: GAPDH for cytoplasmic proteins and histone 2B for nuclear compartment. The no-treatment group was used to standardize across blots and set to a density of 1. The graphs and statistics were performed in the statistics and graphing program Prism 8. Dose-response curve lines of fit were calculated by normalizing the data to a dose of zero and curve fitting with a four-variable nonlinear regression. Estimated EC50 and IC50 values were calculated by the best line of fit. All luciferase production experiments were normalized to total protein/sample (luminescence/[total protein]). When graphed luciferase experiments were normalized to the no-treatment condition so that control = 100%. Unless otherwise indicated, a one-way ANOVA corrected for multiple comparisons and a post hoc Tukey test were performed to judge statistical significance. A significant result was considered p < 0.05.
Data availability
All data used in generation of this article are presented in the main text and supporting information. Requests for more information can be directed to the corresponding author: Dr. Solomon Snyder, ssnyder@jhmi.edu.
Supplementary Material
Acknowledgments
We thank Dr. Jeremy Nathans (Johns Hopkins University School of Medicine) for access to the luminometer. We thank Dr. Rachel Green (Johns Hopkins University School of Medicine) for access to her Typhoon phosphorimaging system. We also thank Roxanne Barrow (Johns Hopkins University) and Adele Snowman (Johns Hopkins University) for assistance with the molecular biology and construct cloning. We lastly thank Chirag Vasavda, Jeremy Sullivan, William Aisenberg, and Daniel Ramos (Johns Hopkins University School of Medicine) for helpful discussions.
This article contains supporting information.
Author contributions—N. W. Z., C. J. S., and S. H. S. conceptualization; N. W. Z., C. J. S., and S. H. S. data curation; N. W. Z., C. J. S., and S. H. S. formal analysis; N. W. Z. investigation; N. W. Z. visualization; N. W. Z. methodology; N. W. Z. writing-original draft; N. W. Z., C. J. S., and S. H. S. writing-review and editing; C. J. S. and S. H. S. resources; C. J. S. and S. H. S. supervision; S. H. S. funding acquisition.
Funding and additional information—This work was supported by National Institutes of Health Grants DA044123-1A1 (to S. H. S.), MH018501-48 (to S. H. S.), MH100024-05 (to S. H. S.), NS062869 (to C. J. S.), NS087579 (to C. J. S.), OD016374 (to S. Kuo), and GM007309 (to N. W. Z.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- GPCR
- G-protein coupled receptor
- TRP
- transient receptor potential
- TRPV
- TRP vanilloid
- AT1R
- angiotensin II receptor type 1
- ANGII
- angiotensin II
- IP3
- inositol trisphosphate
- GRK
- G-protein receptor kinase
- MOR1
- μ-opioid receptor 1
- TRPC
- TRP canonical
- TRPM
- TRP melanostatin
- CPEC
- choroid plexus epithelial cell
- CSF
- cerebrospinal fluid
- NFAT
- nuclear factor of activated T-cells
- IP
- immunoprecipitate
- KO
- knockout
- IF
- immunofluorescence
- CAP
- calcineurin autoinhibitory peptide
- DMEM
- Dulbecco's modified Eagle's medium
- HRP
- horseradish peroxidase
- HBSS
- Hanks' balanced salt solution
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- ANOVA
- analysis of variance.
References
- 1. Gilman A. G. (1987) G proteins: transducers of receptor-generated signals. Annu. Rev. Biochem. 56, 615–649 10.1146/annurev.bi.56.070187.003151 [DOI] [PubMed] [Google Scholar]
- 2. Hilger D., Masureel M., and Kobilka B. K. (2018) Structure and dynamics of GPCR signaling complexes. Nat. Struct. Mol. Biol. 25, 4–12 10.1038/s41594-017-0011-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wisler J. W., Xiao K., Thomsen A. R., and Lefkowitz R. J. (2014) Recent developments in biased agonism. Curr. Opin. Cell Biol. 27, 18–24 10.1016/j.ceb.2013.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hunton D. L., Barnes W. G., Kim J., Ren X.-R., Violin J. D., Reiter E., Milligan G., Patel D. D., and Lefkowitz R. J. (2005) Arrestin 2–dependent angiotensin II type 1A receptor–mediated pathway of chemotaxis. Mol. Pharmacol. 67, 1229–1236 10.1124/mol.104.006270 [DOI] [PubMed] [Google Scholar]
- 5. Shukla A. K., Kim J., Ahn S., Xiao K., Shenoy S. K., Liedtke W., and Lefkowitz R. J. (2010) Arresting a transient receptor potential (TRP) channel. J. Biol. Chem. 285, 30115–30125 10.1074/jbc.M110.141549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zheng J. (2013) Molecular mechanism of TRP channels. Compr. Physiol. 3, 221–242 10.1002/cphy.c120001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Luft F. C. (2015) Tripping out on TRPV4. J. Mol. Med. 93, 1283–1285 10.1007/s00109-015-1347-2 [DOI] [PubMed] [Google Scholar]
- 8. Liu C.-H., Gong Z., Liang Z.-L., Liu Z.-X., Yang F., Sun Y.-J., Ma M.-L., Wang Y.-J., Ji C.-R., Wang Y.-H., Wang M.-J., Cui F.-A., Lin A., Zheng W.-S., He D.-F., et al. (2017) Arrestin-biased AT1R agonism induces acute catecholamine secretion through TRPC3 coupling. Nat. Commun. 8, 14335 10.1038/ncomms14335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shapovalov G., Gkika D., Devilliers M., Kondratskyi A., Gordienko D., Busserolles J., Bokhobza A., Eschalier A., Skryma R., and Prevarskaya N. (2013) Opiates modulate thermosensation by internalizing cold receptor TRPM8. Cell Rep. 4, 504–515 10.1016/j.celrep.2013.07.002 [DOI] [PubMed] [Google Scholar]
- 10. Kim S. J., Kim Y. S., Yuan J. P., Petralia R. S., Worley P. F., and Linden D. J. (2003) Activation of the TRPC1 cation channel by metabotropic glutamate receptor mGluR1. Nature 426, 285–291 10.1038/nature02162 [DOI] [PubMed] [Google Scholar]
- 11. Scherer P. C., Zaccor N. W., Neumann N. M., Vasavda C., Barrow R., Ewald A. J., Rao F., Sumner C. J., and Snyder S. H. (2017) TRPV1 is a physiological regulator of μ-opioid receptors. Proc. Natl. Acad. Sci. U.S.A. 114, 13561–13566 10.1073/pnas.1717005114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Basso L., Aboushousha R., Fan C. Y., Iftinca M., Melo H., Flynn R., Agosti F., Hollenberg M., Thompson R. J., Bourinet E., and Trang T. (2018) TRPV1 regulates opioid analgesia during inflammation. bioRxiv 10.1101/274233 [DOI] [PubMed]
- 13. Filosa J. A., Yao X., and Rath G. (2013) TRPV4 and the regulation of vascular tone. J. Cardiovasc. Pharmacol. 61, 113–119 10.1097/FJC.0b013e318279ba42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Millar I. D., Bruce J. I., and Brown P. D. (2007) Ion channel diversity, channel expression and function in the choroid plexuses. Cerebrospinal Fluid Res. 4, 8 10.1186/1743-8454-4-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Takayama Y., Shibasaki K., Suzuki Y., Yamanaka A., and Tominaga M. (2014) Modulation of water efflux through functional interaction between TRPV4 and TMEM16A/anoctamin 1. FASEB J. 28, 2238–2248 10.1096/fj.13-243436 [DOI] [PubMed] [Google Scholar]
- 16. Mendoza S. A., Fang J., Gutterman D. D., Wilcox D. A., Bubolz A. H., Li R., Suzuki M., and Zhang D. X. (2010) TRPV4-mediated endothelial Ca 2+ influx and vasodilation in response to shear stress. Am. J. Physiol. Circ. Physiol. 298, H466–H476 10.1152/ajpheart.00854.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Matsusaka T., and Ichikawa I. (1997) Biological functions of angiotensin and its receptors. Annu. Rev. Physiol. 59, 395–412 10.1146/annurev.physiol.59.1.395 [DOI] [PubMed] [Google Scholar]
- 18. Mizuno N., and Itoh H. (2009) Functions and regulatory mechanisms of Gq-signaling pathways. Neurosignals 17, 42–54 10.1159/000186689 [DOI] [PubMed] [Google Scholar]
- 19. Kroeze W. K., Sassano M. F., Huang X.-P., Lansu K., McCorvy J. D., Giguère P. M., Sciaky N., and Roth B. L. (2015) PRESTO-Tango as an open-source resource for interrogation of the druggable human GPCRome. Nat. Struct. Mol. Biol. 22, 362–369 10.1038/nsmb.3014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bihzad S. M., and Yousif M. H. M. (2017) 11,12-Epoxyeicosatrienoic acid induces vasodilator response in the rat perfused mesenteric vasculature. Auton. Autacoid Pharmacol. 37, 3–12 10.1111/aap.12052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhao L., Sullivan M. N., Chase M., Gonzales A. L., and Earley S. (2014) Calcineurin/nuclear factor of activated T cells: coupled vanilliod transient receptor potential channel 4 Ca2+ sparklets stimulate airway smooth muscle cell proliferation. Am. J. Respir. Cell Mol. Biol. 50, 1064–1075 10.1165/rcmb.2013-0416OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Masuyama R., Vriens J., Voets T., Karashima Y., Owsianik G., Vennekens R., Lieben L., Torrekens S., Moermans K., Vanden Bosch A., Bouillon R., Nilius B., and Carmeliet G. (2008) TRPV4-mediated calcium influx regulates terminal differentiation of osteoclasts. Cell Metab. 8, 257–265 10.1016/j.cmet.2008.08.002 [DOI] [PubMed] [Google Scholar]
- 23. Boddeke H. W., Hoffman B. J., Palacios J. M., and Hoyer D. (1993) Calcineurin inhibits desensitization of cloned rat 5-HT1C receptors. Naunyn. Schmiedebergs. Arch. Pharmacol. 348, 221–224 10.1007/BF00169147 [DOI] [PubMed] [Google Scholar]
- 24. Sanders-Bush E., and Breeding M. (1991) Choroid plexus epithelial cells in primary culture: a model of 5HT1C receptor activation by hallucinogenic drugs. Psychopharmacology (Berl.) 105, 340–346 10.1007/BF02244428 [DOI] [PubMed] [Google Scholar]
- 25. Crabtree G. R. (2001) Calcium, calcineurin, and the control of transcription. J. Biol. Chem. 276, 2313–2316 10.1074/jbc.R000024200 [DOI] [PubMed] [Google Scholar]
- 26. Swingle M., Ni L., and Honkanen R. E. (2007) Small-molecule inhibitors of Ser/Thr protein phosphatases: specificity, use and common forms of abuse. Methods Mol. Biol. 365, 23–38 10.1385/1-59745-267-X:23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Klee C. B., Crouch T. H., and Krinks M. H. (1979) Calcineurin: a calcium- and calmodulin-binding protein of the nervous system. Proc. Natl. Acad. Sci. U. S. A. 76, 6270–6273 10.1073/pnas.76.12.6270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Damkier H. H., Brown P. D., and Praetorius J. (2013) Cerebrospinal fluid secretion by the choroid plexus. Physiol. Rev. 93, 1847–1892 10.1152/physrev.00004.2013 [DOI] [PubMed] [Google Scholar]
- 29. Orlandi C., Cao Y., and Martemyanov K. A. (2013) Orphan receptor GPR179 forms macromolecular complexes with components of metabotropic signaling cascade in retina ON-bipolar neurons. Invest. Ophthalmol. Vis. Sci. 54, 7153–7161 10.1167/iovs.13-12907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vinuela-Fernandez I., Sun L., Jerina H., Curtis J., Allchorne A., Gooding H., Rosie R., Holland P., Tas B., Mitchell R., and Fleetwood-Walker S. (2014) The TRPM8 channel forms a complex with the 5-HT1B receptor and phospholipase D that amplifies its reversal of pain hypersensitivity. Neuropharmacology 79, 136–151 10.1016/j.neuropharm.2013.11.006 [DOI] [PubMed] [Google Scholar]
- 31. Chen J., and Hackos D. H. (2015) TRPA1 as a drug target: promise and challenges. Naunyn. Schmiedebergs. Arch. Pharmacol. 388, 451–463 10.1007/s00210-015-1088-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vetter I., Wyse B. D., Monteith G. R., Roberts-Thomson S. J., and Cabot P. J. (2006) The mu opioid agonist morphine modulates potentiation of capsaicin-evoked TRPV1 responses through a cyclic AMP-dependent protein kinase A pathway. Mol. Pain 2, 22 10.1186/1744-8069-2-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Majerus P. W. (1992) Inositol phosphate biochemistry. Annu. Rev. Biochem. 61, 225–250 10.1146/annurev.bi.61.070192.001301 [DOI] [PubMed] [Google Scholar]
- 34. Kusudo T., Wang Z., Mizuno A., Suzuki M., and Yamashita H. (2012) TRPV4 deficiency increases skeletal muscle metabolic capacity and resistance against diet-induced obesity. J. Appl. Physiol. 112, 1223–1232 10.1152/japplphysiol.01070.2011 [DOI] [PubMed] [Google Scholar]
- 35. Bahouth S. W., Gokmen-Polar Y., Coronel E. C., and Fain J. N. (1996) Enhanced desensitization and phosphorylation of the β1-adrenergic receptor in rat adipocytes by peroxovanadate. Mol. Pharmacol. 49, 10479–1057 [PubMed] [Google Scholar]
- 36. Segal M. B. (2000) The choroid plexuses and the barriers between the blood and the cerebrospinal fluid. Cell. Mol. Neurobiol. 20, 183–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Preston D., Simpson S., Halm D., Hochstetler A., Schwerk C., Schroten H., and Blazer-Yost B. L. (2018) Activation of TRPV4 stimulates transepithelial ion flux in a porcine choroid plexus cell line. Am. J. Physiol. Physiol. 315, C357–C366 10.1152/ajpcell.00312.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hannan M. A., Kabbani N., Paspalas C. D., and Levenson R. (2008) Interaction with dopamine D2 receptor enhances expression of transient receptor potential channel 1 at the cell surface. Biochim. Biophys. Acta 1778, 974–982 10.1016/j.bbamem.2008.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gáborik Z., Jagadeesh G., Zhang M., Spät A., Catt K. J., and Hunyady L. (2003) The role of a conserved region of the second intracellular loop in AT1 angiotensin receptor activation and signaling. Endocrinology 144, 2220–2228 10.1210/en.2002-0135 [DOI] [PubMed] [Google Scholar]
- 40. Arniges M., Fernández-Fernández J. M., Albrecht N., Schaefer M., and Valverde M. A. (2006) Human TRPV4 channel splice variants revealed a key role of ankyrin domains in multimerization and trafficking. J. Biol. Chem. 281, 1580–1586 10.1074/jbc.M511456200 [DOI] [PubMed] [Google Scholar]
- 41. Meixiong J., Vasavda C., Green D., Zheng Q., Qi L., Kwatra S. G., Hamilton J. P., Snyder S. H., and Dong X. (2019) Identification of a bilirubin receptor that may mediate a component of cholestatic itch. Elife 8, e44116 10.7554/eLife.44116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Violin J. D., Ren X.-R., and Lefkowitz R. J. (2006) G-protein–coupled receptor kinase specificity for β-arrestin recruitment to the β2-adrenergic receptor revealed by fluorescence resonance energy transfer. J. Biol. Chem. 281, 20577–20588 10.1074/jbc.M513605200 [DOI] [PubMed] [Google Scholar]
- 43. Hughes T. E., Zhang H., Logothetis D. E., and Berlot C. H. (2001) Visualization of a functional Gαq–green fluorescent protein fusion in living cells. J. Biol. Chem. 276, 4227–4235 10.1074/jbc.M007608200 [DOI] [PubMed] [Google Scholar]
- 44. Boehm J. S., Zhao J. J., Yao J., Kim S. Y., Firestein R., Dunn I. F., Sjostrom S. K., Garraway L. A., Weremowicz S., Richardson A. L., Greulich H., Stewart C. J., Mulvey L. A., Shen R. R., Ambrogio L., et al. (2007) Integrative genomic approaches identify IKBKE as a breast cancer oncogene. Cell 129, 1065–1079 10.1016/j.cell.2007.03.052 [DOI] [PubMed] [Google Scholar]
- 45. Zhang D. X., Mendoza S. A., Bubolz A. H., Mizuno A., Ge Z.-D., Li R., Warltier D. C., Suzuki M., and Gutterman D. D. (2009) Transient receptor potential vanilloid type 4-deficient mice exhibit impaired endothelium-dependent relaxation induced by acetylcholine in vitro and in vivo. Hypertension 53, 532–538 10.1161/HYPERTENSIONAHA.108.127100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Monnot A. D., and Zheng W. (2013) Culture of choroid plexus epithelial cells and in vitro model of blood–CSF barrier. Methods Mol. Biol. 945, 13–29 10.1007/978-1-62703-125-7_2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Vasavda C., Zaccor N. W., Scherer P. C., Sumner C. J., and Snyder S. H. (2017) Measuring g-protein-coupled receptor signaling via radio-labeled GTP binding. J. Vis. Exp. 2017, 55561 10.3791/55561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Thomas W. G. (2009) G Protein–coupled Receptors in Drug Discovery (Leifert W. R., ed) Humana Press, Totowa, NJ [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data used in generation of this article are presented in the main text and supporting information. Requests for more information can be directed to the corresponding author: Dr. Solomon Snyder, ssnyder@jhmi.edu.