

Abstract
Antibodies are vital proteins of the immune system that recognize potentially harmful molecules and initiate their removal. Mammals can efficiently create vast numbers of antibodies with different sequences capable of binding to any antigen with high affinity and specificity. Because they can be developed to bind to many disease agents, antibodies can be used as therapeutics. In an organism, after antigen exposure, antibodies specific to that antigen are enriched through clonal selection, expansion, and somatic hypermutation. The antibodies present in an organism therefore report on its immune status, describe its innate ability to deal with harmful substances, and reveal how it has previously responded. Next-generation sequencing technologies are being increasingly used to query the antibody, or B-cell receptor (BCR), sequence repertoire, and the amount of BCR data in public repositories is growing. The Observed Antibody Space database, for example, currently contains over a billion sequences from 68 different studies. Repertoires are available that represent both the naive state (i.e. antigen-inexperienced) and that after immunization. This wealth of data has created opportunities to learn more about our immune system. In this review, we discuss the many ways in which BCR repertoire data have been or could be exploited. We highlight its utility for providing insights into how the naive immune repertoire is generated and how it responds to antigens. We also consider how structural information can be used to enhance these data and may lead to more accurate depictions of the sequence space and to applications in the discovery of new therapeutics.
Keywords: antibody, bioinformatics, immunology, protein sequence, protein structure, adaptive immunity, B-cell receptor (BCR), observed antibody space database, structural annotation, next-generation sequencing, antibody
Antibodies are proteins that play a key role in the adaptive immune response. They are produced by B cells and are either secreted or membrane-bound (in the latter case, they are known as B-cell receptors, or BCRs). They are able to neutralize and initiate the removal of foreign entities (known as antigens) from the body by binding to them (1). The ability of the immune system to respond to a huge range of antigens originates in the diversity of the antibodies that can be generated—antibodies can be produced that bind to nearly every antigen, with both high specificity and affinity (2). This property has made antibodies highly successful as therapeutics; to date, 87 have been approved for use in the clinic across a number of disease areas, and many more are undergoing clinical trials (3, 4). Antibodies are currently the largest class of biotherapeutic (5).
It is estimated that the human antibody repertoire contains around 1013 unique sequences (6). This diversity is a result of how the proteins are encoded in the genome. Antibodies are composed of two types of protein chain, known as the heavy and light chains (Fig. 1). Each of these is encoded by multiple gene segments that are spliced together using a process called V(D)J recombination (7). The sequence for the light-chain variable region (Fv) is made up of two segments: the variable segment (V) and the joining segment (J). The heavy chain is encoded from variable, joining, and diversity (D) segments. There are many genes for each of the V, D, and J segments, which can be matched up in different combinations to produce a diverse range of antibody sequences. Further diversity is introduced through the insertion or deletion of nucleotides at the segment junctions (8) and somatic hypermutation (a process through which the number of random mutations that occur is increased) (9). The majority of the variation in sequence occurs in the complementarity-determining regions, or CDRs—there are three of these on each of the heavy and light chains. The most variable of these is the H3 loop (the third CDR on the heavy chain), because the DNA encoding it is found at the join between the V, D, and J segments. By creating a large, diverse repertoire of antibody sequences, an individual is able to react to almost any antigen it may encounter.
Figure 1.

A, antibody structure. An antibody is made up of four chains: two light (orange) and two heavy (blue). Each chain is made up of a series of domains—the variable domains of the light and heavy chains together are known as the Fv region (shown on the right; PDB entry 12E8). The Fv features six loops known as CDRs (shown in dark blue); these are mainly responsible for antigen binding. B, example sequences for the VH and VL, highlighting the CDR regions and the genetic composition.
The ability of an antibody to bind to its target antigen is governed by its three-dimensional structure. Knowledge of an antibody's structure therefore allows for a deeper understanding of its physicochemical properties than can be gained from sequence alone. The general structure of an antibody is depicted in Fig. 1 The heavy and light variable domains both adopt a β-sandwich structure known as the immunoglobulin fold. Framework (non-CDR) regions are very highly conserved between different antibodies; in accordance with the observed variability of antibody sequences, the structural diversity that allows binding to many different targets occurs mainly in the CDRs. These correspond to loops in the three-dimensional structure, which are responsible for most of the antigen-binding interactions (10). For five of the six CDRs (H1, H2, and L1–L3), structural diversity is limited—only a few different shapes have been observed, forming a set of discrete conformational classes known as canonical structures. However, as described above, the H3 loop is much more variable in sequence than the other CDRs and consequently is also more structurally diverse. It is thought that the H3 loop contributes the most to antigen-binding properties (11, 12).
Upon exposure to an antigen, antibodies that are able to bind to it do so and are thus selected from the repertoire (clonal selection) (13). Having a large repertoire of antibodies present in the body at any time increases the chance that at least one has the ability to bind to the antigen, even if only weakly, thereby allowing the initiation of an appropriate immune response. B cells producing binding antibodies undergo cycles of proliferation (clonal expansion) with simultaneous somatic hypermutation (9) to produce antibodies with higher affinity. The antibody repertoire is consequently enriched with antibodies that bind to the target antigen.
The antibodies present in an organism therefore describe both its current and past immune status; what it is able to respond to, and what it has previously dealt with. Whereas previously only a handful of sequences could be obtained at a time, technological advances mean that large snapshots of this repertoire can now be obtained using next-generation sequencing approaches. This technique of BCR repertoire sequencing was first described by Glanville et al. in 2009 (14), and since then the volume of data available has increased exponentially (Fig. 2). As it is the H3 loop that mostly determines binding properties, many studies have focused only on sequencing this region. However, BCR repertoires containing full-length sequences are increasingly being produced—commonly only the heavy chain (15), but some studies have focused only the light chain (e.g. Refs. 16 and 17), and some data sets include both (e.g. Refs. 18 and 19). Recent advances in sequencing technology have led to a small but growing number of repertoires that also include native pairing information (i.e. which heavy-chain sequences belong with which light-chain sequences).
Figure 2.

The cumulative growth of publicly available (redundant) antibody sequences over time (data from the Observed Antibody Space database (28)).
The largest repertoire sequencing study to date, by Briney et al. (20), alone resulted in a set of over 300 million heavy-chain sequences. In addition, many algorithms and pipelines have now been created that preprocess the generated data ready for analysis, performing tasks such as translation from nucleotides to amino acids, error estimation and correction, and sequence numbering (21). Recently, efforts have been made to create standardized, publicly available repositories for these sequencing data (e.g. iReceptor (22), VDJServer (23), ImmuneDB (24), and others (25–29)). This has provided researchers with easy access to a vast number of sequences and created opportunities for large-scale data mining. The Observed Antibody Space (OAS) database, for example, which collates full-length variable region sequences, currently contains over 1 billion sequences spanning 68 different studies (28).
The studies included in OAS cover many different repertoire characteristics. Sequences are available for six different species, with the majority (64%) being human. Diseased states are represented (i.e. repertoires from individuals who have been exposed to a specific antigen) as well as healthy ones (meaning the individual has not been exposed to the antigen of interest and also has not suffered from a disorder of the immune system). Repertoires from vaccination studies also feature (e.g. HIV, hepatitis B, flu, etc.), and in some cases, OAS has the repertoires of the same individual both pre- and post-immunization. Although the snapshots of the repertoire achieved through sequencing are actually small relative to the potential number of antibodies present in an organism (e.g. data sets in OAS contain between 20,000 and 300 million redundant sequences) and most studies feature only the heavy chain or have no pairing information, the data available still provides opportunities to investigate many different aspects of the immune response. In this review, we explore what can be done with the wealth of antibody sequence data stored in repositories such as OAS. We give examples of how this data has been used to give insights into the workings of the immune system, look at how it can be enhanced with structural information, explore how it offers new avenues for therapeutic antibody discovery and development, and consider what advances may be made in the future.
Biological insights from antibody repertoire data
Until the advent of BCR repertoire sequencing, antibody sequences were analyzed in much smaller numbers (normally a few hundred B cells per experiment (15)), only a tiny fraction of the estimated total repertoire. This approach can be useful when investigating a few key antibodies (e.g. those that bind to an antigen of interest (e.g. Refs. 30 and 31)) but cannot give an in-depth view of the repertoire as a whole (e.g. little can be learned about its diversity). Analysis of larger repertoire snapshots, on the other hand, gives a much more detailed picture and can provide valuable insights into how the immune system works. It can be used to explain how in its naive state (i.e. before exposure to a given antigen) it is capable of protecting against such diverse threats and can give a deeper understanding of the processes that produce higher-affinity antibodies after antigen exposure.
Sequencing data has been used to learn more about the underlying mechanisms that shape the repertoire, such as V(D)J recombination (32, 33). Increasing amounts of large-scale sequence data, along with the development of computational tools that annotate sequences with their V(D)J gene origins (34–37), have allowed trends in this process to be identified. It has been shown that the process is intrinsically biased; the available V, D, and J segments in the genome are not used with the same frequency, and therefore some combinations are observed more commonly than others (14, 38–41). Mathematical models of V(D)J recombination have been developed that reproduce the natural biases (42, 43). It has been proposed that this has the potential to aid in the discovery of new antibody therapeutics—replicating the underlying architecture of observed human repertoires should lead to the creation of more human-like (and hence less immunogenic) screening libraries (44).
During the proliferation of B cells in clonal selection, the rate of mutation is increased up to 106-fold (45) compared with normal cells, due to somatic hypermutation (as described earlier). Variations on the original antigen-binding antibody sequence are therefore generated, and higher-affinity antibodies are iteratively produced. Repertoire data has been used to analyze this process (46–50). This has increased our understanding of mutation frequencies, substitution bias, and the location of mutation hot spots and, hence, how the repertoire reacts to an antigenic stimulus. For example, researchers have demonstrated that memory cells of different isotypes experience different selection pressures (46) and that substitution profiles vary between V genes (47), are dependent on neighboring bases, and are conserved across individuals (48). As in the case of V(D)J recombination, these insights have enabled accurate models of somatic hypermutation to be established (49, 50). These models have led to the creation of software that simulates repertoires (51) and mean that more accurate B-cell lineages can be established (49). These phylogenies have the potential to be used in the identification of antibodies with high binding affinities (50).
Researchers have also investigated the interplay between all the processes that dictate repertoire diversity to ascertain how much is genetically predetermined and how much is antigen-driven; analysis indicates that both are important factors, but genetics are more influential (39). Further research has compared the repertoires of humans and other species (52, 53), revealing that immune system development is broadly similar across different mammals (53) and that mice BCR repertoires tend to be closer to germline sequences than those of humans (52). The effect of disease on the immune system has also been studied (54) and has indicated that repertoire analysis can have more practical applications; for example, it can be used to monitor the diversity of the repertoire before and after an organ transplant (55), and machine learning methods have been used to predict vaccination status or the presence of disease (56–58).
The overall architecture of the antibody repertoire can be investigated by inferring relationships between sequences (i.e. by predicting which ones originated from the same precursor antibody and hence which bind to the same antigen). One approach is to consider the repertoire as a network, with each sequence being a separate node and the presence of an edge between them indicating an evolutionary relationship (44). These relationships are normally defined based on sequence identity; for example, two sequences can be connected if they differ by one amino acid in their H3 region (44). Common network analysis metrics can then be used to explore the repertoire architecture—for example, the degree distribution (the degree of a node is the number of edges it is connected to) can reveal the presence or absence of clonal expansion (33), because highly connected nodes are likely to represent sequences derived from a common precursor during affinity maturation (Fig. 3).
Figure 3.
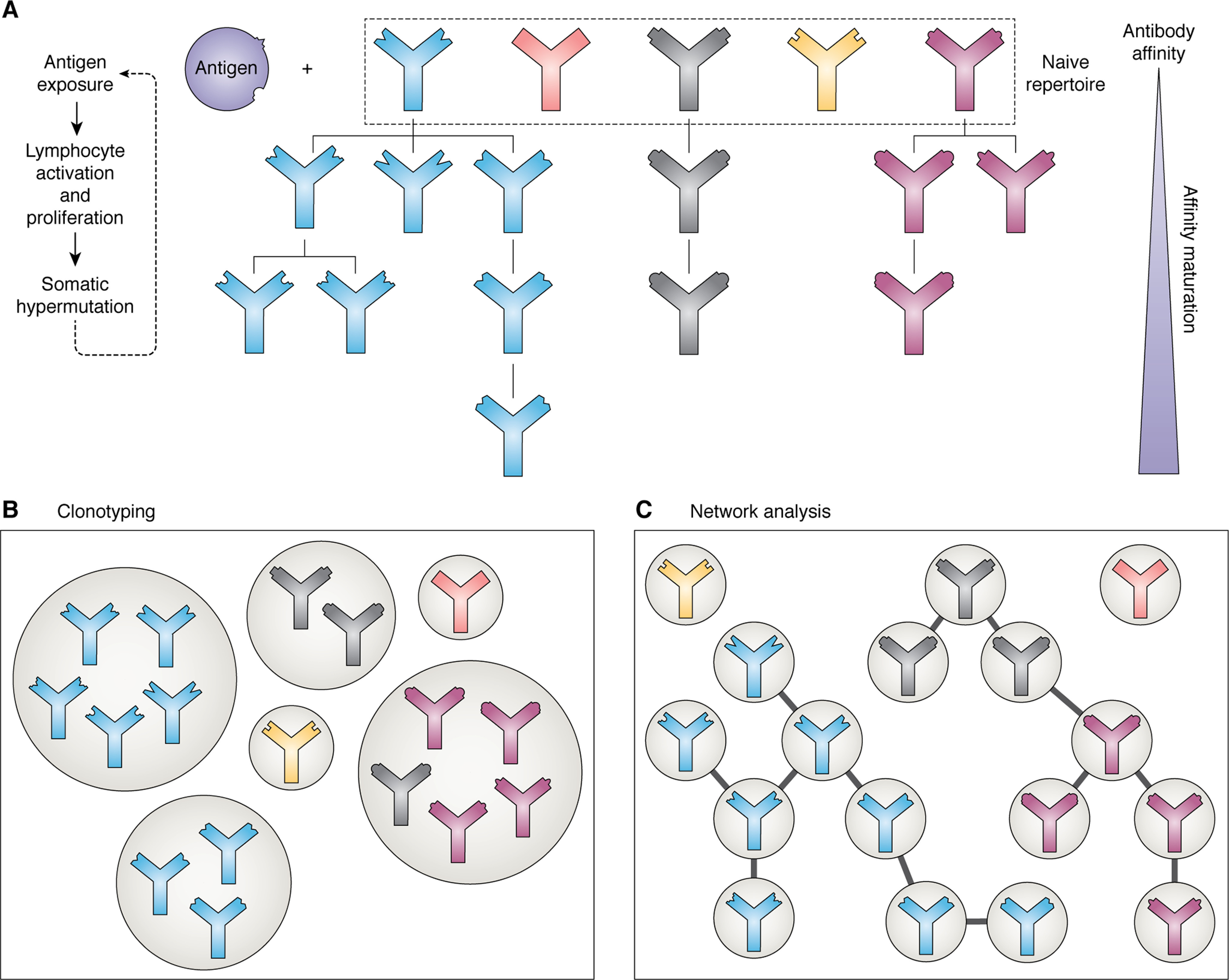
The process of affinity maturation and methods of analyzing the resulting antibody repertoires. A, upon exposure to an antigen, those antibodies present in the naive repertoire that are able to bind to it proliferate, undergoing somatic hypermutation to produce variations upon the initial binder. Successive rounds of this process produce antibodies with high affinity. B, clonotyping groups antibodies in the repertoire based on sequence similarity; normally they must originate from the same V and J genes and have an H3 sequence identity of 80–100%. Antibodies of the same clonotype are predicted to bind to the same epitope. C, network analysis of antibody repertoires, where each node is a different sequence and edges are present between them if they meet set sequence similarity criteria. The lineages of different antibodies can be inferred using this method.
Clonotyping is another related way of investigating the diversity of repertoires and, in particular, how they change upon antigen exposure. Similar antibody sequences are clustered into “clonotypes”; these are generally defined as sequences originating from the same V and J genes and with H3s that are the same length and similar in sequence (normally a sequence identity of 80–100%) (59–62), although alternative approaches have been used (63). Antibodies belonging to the same clonotype are assumed to share the same precursor sequence (i.e. they arose from the proliferation of the same B cell) and are therefore predicted to bind to the same epitope. This is therefore a method of monitoring the clonal selection and expansion that occurs after exposure to an antigen and can be used to identify the antibodies that bind to a particular target.
Because the repertoires of many individuals have now been sequenced, we can compare them to identify which characteristics of the repertoire are shared and which are unique to each organism. The idea of “public sequences” has recently been proposed—a set of sequences or clonotypes that are observed in the repertoires of two or more individuals (20, 44, 61, 64–66). One may expect that this is rare, due to the enormous potential number of sequences (estimated at 1013) and the relatively small proportion of those sequences sampled in current data sets (the largest samples from a single individual currently have on the order of 106 sequences). However, whereas repertoires are largely unique to the organism (67), it has been shown that individuals share more heavy-chain sequences than would be expected by coincidence. Briney et al. (20), in their recent large-scale study, showed that in the repertoires of 10 individuals, on average 0.95% of clonotypes were shared between at least two subjects, and 0.022% were common to all 10. The pool of subjects contained both men and women, individuals from both Caucasian and African American ethnic backgrounds, and a variety of blood types; the authors report that the repertoires did not cluster based on these factors. The work of Soto et al. (64) indicates that this public subrepertoire could be even larger, making up between 1 and 6% of the whole. Greiff et al. (68) have used machine learning techniques, trained on publicly available data sets such as those in OAS, to predict the public or private nature of a given sequence with 80% accuracy, hinting that this property is not random and that there are fundamental characteristics of the sequences that separate the two subsets. In their network-based analysis of antibody H3 sequences, where each node is a unique H3 sequence, Miho et al. (44) demonstrated that public clonotypes were among the most connected nodes (i.e. they are similar in sequence to many other nodes) and that most private clonotypes (74%) were connected to at least one public one. The removal of public clonotypes from the network therefore changed the underlying repertoire architecture; however, the system was robust to the removal of a large number of randomly selected clonotypes. This implies that public clonotypes are key in maintaining functional immunity against antigens, whereas the presence of other clonotypes is able to fluctuate over time.
Light chain data has also been analyzed; VL sequences are less diverse than their VH counterparts (52, 69, 70), so the percentage of the repertoire comprising public sequences is much larger. For instance, Soto et al., in a three-individual experiment, observed that 20–34% of light chains (of both κ and λ types) were shared by at least two people (64).
Overall, the presence of shared clonotypes across different individuals, although small, may signal the existence of a baseline common functionality of the immune system. This core subset of the repertoire may be responsible for an organism's response to common antigens (66), and it has been hypothesized that these public clonotypes are more likely to display low levels of immunogenicity and be more versatile binders and hence may be useful starting points in therapeutic development (71).1
Combining sequence with structure
Although much can be learned from sequences alone, it is the three-dimensional structure of the antibody that determines how it interacts with an antigen and therefore governs its binding properties (1, 72). It is known that CDRs belonging to the same canonical class (i.e. that have nearly identical structures) can have very different sequences, and conversely H3 loops with similar sequences can adopt different conformations (Fig. 4) (73). Therefore, by considering sequence alone (e.g. in clonotyping), antibodies may be grouped together that have structurally dissimilar binding sites, and vice versa (74). It is therefore crucial to consider structure as well as sequence to allow more accurate comparisons to be made and to properly understand antibody function.
Figure 4.

Sequence is not always a reliable indicator of structural similarity. A, L1 loops of the PDB entries 3PHO (red) and 3QUM (blue). The two loops differ in sequence at every position except one (sequence identity = 10%); however, they have very similar conformations (RMSD = 0.60 Å). B, H3 loops of the PDB entries 5I1G (red) and 5I1C (blue). These loops have very similar sequences (sequence identity = 92%) and therefore may be predicted to have similar structures; however, this is not the case (RMSD = 4.15 Å). RMSDs were calculated across all backbone atoms after superposition of the anchor residues (two residues on each side of the loop, shown in gray).
Antibody structures can be obtained experimentally, normally through X-ray crystallography or NMR. However, the sequence-structure gap is large—whereas OAS consists of over a billion sequences, SAbDab, a database of publicly available antibody structures (75), currently contains ∼4000 entries. This is because experimental structure determination is time-consuming and hence low-throughput; as such, it can be used to probe the chemistry of a select few sequences (76, 77), but it cannot yet be used to structurally characterize a BCR repertoire.
Computational modeling offers an alternative. It has been shown that the majority of antibody sequences from BCR repertoires can be mapped to known structures (74). A number of algorithms have been developed that predict the structure of an antibody's Fv region from its sequence (78–91). Due to the conserved nature of the antibody framework structure (see Fig. 1) and the existence of canonical classes, these tools generally rely on homology modeling (i.e. an existing structure with high sequence identity to a segment of or to the whole target is used as a template). Normally the structure is considered as separate regions, first the frameworks of the VH and VL and then the six CDRs. Separate templates may be chosen for the VH and VL; however, if a single template is available with high sequence identity to both chains, only one is required (78). In this case, the orientation of the two chains can be directly copied from the chosen template; otherwise, a further template that is similar in sequence to both chains is required, or the orientation between the chains must be predicted (92). The framework can be modeled with very high accuracy, typically with a root mean square deviation (RMSD) of below 1 Å. In the second Antibody Modelling Assessment (AMA-II), a blind test of prediction accuracy, VH and VL were modeled with an average backbone-atom RMSD of 0.65 and 0.50 Å, respectively (87, 88, 90, 93–96). Prediction of the orientation of the two domains was more challenging, however, with predicted tilt angles differing from the true angle by 5–12∘ (93).
Once a framework template has been selected, CDR structures can then be predicted, again using templates through knowledge-based loop modeling algorithms. As mentioned previously, in the majority of cases, CDRs L1–L3, H1, and H2 adopt a limited number of known conformations known as canonical classes (97–99). As a result, they can be predicted accurately and quickly using this technique. Templates are selected from a database of known CDR structures based on sequence identity and the geometry of the anchor residues (the residues on either side of the CDR). The database of CDR structures can either include all known structures or be limited to the known conformations for the predicted canonical class of the target (78, 80). Average RMSDs achieved during AMA-II ranged from 0.50 Å for L2 to 1.6 Å for L3 (93).
H3 can also be modeled using this method; however, its sequence and structural diversity compared with the other CDRs makes prediction more challenging (100). The H3 loop has also been shown to be structurally distinct from typical protein loops (101); researchers have therefore developed specialized software to model H3 loops more accurately (102–105). Ab initio techniques, which create potential loop conformations without knowledge of templates, are often used here, either in isolation or in combination with knowledge-based strategies as a hybrid algorithm (102). Despite the existence of H3-specific prediction algorithms, H3 modeling remains challenging, achieving RMSDs normally in the region of 2-3 Å (74, 93). In addition, ab initio methods typically require much longer run times than knowledge-based methods, and therefore H3 prediction is currently the main bottleneck for accurate modeling of BCR repertoires. Attempts have been made to circumvent this issue, either by imposing an H3 length cutoff (long loops are modeled less accurately due to the absence of experimental data) (107) or by only considering those H3 sequences that can be confidently modeled using a knowledge-based algorithm (74, 107).1 Whereas this may introduce some biases into the analysis—for example, long H3 loop structures will be underrepresented in model libraries—it increases the confidence we have in the models that are considered and subsequently in the conclusions that are drawn.
Several studies have used antibody modeling to enhance the information given by BCR repertoires. DeKosky et al. (108) modeled 2,000 VH/VL pairs using RosettaAntibody (82, 83), limiting their sequences to those with high-identity templates available. They analyzed the physico-chemical properties of the antibodies, such as solvent-accessible surface area and hydrophobicity, and were able to demonstrate how these properties change with antigen experience and link their observations to germline usage. Raybould et al. (106) used ABodyBuilder (78) to predict the structures of a large subset of a BCR repertoire (∼19,000 sequences) and compared these models with those of a set of therapeutics to deduce which properties are required to reduce developability issues. Because antibody properties can be predicted with greater accuracy with the inclusion of structural data (109), models representing the repertoire have the potential to improve strategies such as directed design by using them as inputs to other computational tools (e.g. predictors of the sets of residues on the antibody and antigen that are involved in binding (known as the epitope and paratope respectively) and developability predictors).
One problem with modeling the antibody sequences obtained through repertoire sequencing is that they are normally not paired (i.e. we do not know which VH belongs with which VL). Native pairings are important in creating accurate models that represent the repertoire and will affect the properties of the antibody, such as its folding, stability, expression, and binding. Pairing is currently thought to be mostly random (20, 65), meaning that most VH chains are capable of associating with most VLs. Prediction of true pairings is therefore difficult. Techniques currently used to propose likely pairings include comparing all of the potential interfaces with those observed in known structures (106),2 pairing based on the relative frequency of the sequences (110), or by constructing phylogenetic trees (111). Recently, experimental methods for immunoglobulin sequencing that preserve native pairings have been developed (112); as these techniques become more widespread, the amount of paired data will increase, and these approximations will no longer be required.
Producing complete models of the antibody variable region can be time-consuming; for example, in the study by DeKosky et al. (108), RosettaAntibody took 570,000 CPU hours to produce 2,000 models. Even for algorithms that are considered to be fast, execution times would be prohibitive—ABodyBuilder, for example, takes on average 567 CPU hours per 1,000 sequences (78). An alternative, faster method of characterizing a repertoire is the structural annotation of sequences. Instead of running a complete modeling protocol, sequences can be quickly matched up to their predicted templates using sequence identity. The conformations of the CDRs can be assigned by either exploiting a knowledge-based loop modeling algorithm (74) or a canonical class predictor (for the non-H3 CDRs) (99, 107). Sequences can therefore be structurally annotated in much greater numbers than could be done using modeling tools. It has been shown that the majority of sequences can be mapped to an existing structure in this way (74).
SAAB (Structural Annotation of Antibodies) (74) and its successor SAAB+ (107) are algorithms that have been used to annotate millions of sequences with their proposed template structures, allowing thorough analysis of repertoire-wide structural properties. For example, Kovaltsuk et al. (107) investigated structural changes that occur with B-cell differentiation. Clustering based on their proposed H3 templates resulted in the separation of antibodies from different stages of the immune response, indicating that there are structural changes that occur as the response progresses. The effect of aging on the repertoire has also been studied in this way, revealing that older individuals have a higher number of antibodies that are structurally distinct from the germline (113).
The idea of public sequences has been extended to that of public structures. Instead of searching for sequences that are observed in the repertoires of multiple individuals, we can look instead for antibodies with shared backbone conformations, which may be a greater indicator of common functionality. Sequence-only analyses have shown that the shared space is present but only makes up a small percentage of the overall repertoire (20); however, by incorporating structure it can be seen that the public repertoire is likely to be much larger (107).1
BCR repertoire sequencing and therapeutic discovery
Discovering antibodies specific to an antigen of interest
Currently, potential therapeutic antibodies are commonly discovered in two ways: through the immunization of an animal, such as a mouse, with the target antigen and subsequent extraction of the antibodies it produces and through phage display, where viruses displaying antibodies on their surface are screened against the target antigen. High-throughput sequencing of the antibody repertoire has been used successfully to enhance both approaches. For example, researchers have genetically engineered mice such that they contain human antibody genes—the antibodies produced by these mice are therefore less likely to be immunogenic. The “humanness” of the repertoire was validated through sequencing of the mouse BCR repertoire (114). Sequencing techniques have been used to characterize phage display libraries, to monitor their diversity and hence evaluate their capability of isolating antibodies that bind to different antigens (115). Screening libraries can also be designed using BCR repertoire data—Zhai et al. (116) and Prassler et al. (117) have shown how this is possible, by reproducing the observed amino acid usages at each sequence position. Both groups found that the antibodies in their libraries exhibited better expression levels than other synthetic libraries, with high genetic diversity, and they were able to isolate high-affinity antibodies for a range of different antigens.
It is now becoming possible to identify binders directly from BCR repertoire data. If an antibody that binds to the target antigen is already known, approaches such as clonotyping can be used to identify more potential binders with closely related sequences, expanding the pool of candidates that can be taken forward for further study. Known binders are not essential, however. The immunization of an organism with an antigen, as explained previously, leads to the enrichment of the repertoire with antibodies that bind to that antigen. Therefore, by analyzing how often a given sequence or clonotype appears in the repertoire after antigen exposure, specific antibodies can be identified. This approach can be used either to find antibodies that might work as therapeutics or to monitor the immune response during the development of vaccines (66, 118–122). The repertoires of multiple individuals who have been exposed to the same antigen can be investigated to find potential binders, by identifying common features that hint at shared functionality (e.g. identical H3 sequences) (123). The volume of data produced also means that deep learning techniques can be used effectively; for example Mason et al. (124) have generated neural networks that classify antibodies as HER2 binders or nonbinders based on sequence and thereby successfully identified 30 antigen-specific antibodies. BCR repertoire sequencing experiments have been carried out to discover binders for a wide range of antigens, including HIV (71, 111, 125, 126), Ebola (127), hepatitis B (66, 128), and many others (77, 110, 116, 118, 120, 123, 128–133).
Following the isolation of binders in this way, a small number can be taken forward as starting points for further development (77), or a larger number can be employed as a targeted screening library (110). A comparison between repertoire mining and phage display has demonstrated that the antibodies isolated by each method are not necessarily the same, and therefore it could be beneficial to use the two techniques together (129).
Much of the data from these experiments has been deposited in public sequence repertoires (28), meaning it can be exploited by other researchers in their therapeutic discovery pipelines, for example to provide new lead molecules. It has recently been shown that there is a close sequence match to many known therapeutic antibodies in the OAS database (134). Of 242 antibodies that are either currently used as therapeutics or undergoing clinical trials (Phase II or later), sequences with over 90% identity were available for 90 H chains and 158 light chains. Notably, for H3, which is thought to contribute the most to an antibody's binding properties, 54 perfect matches were found. Given the huge number of potential sequences, this is significantly more than would be expected by chance alone in a sequence database of this size (around 1 billion sequences) and implies that artificially developed sequences are not dissimilar from their natural counterparts. It therefore follows that natural sequence repertoires could potentially be mined for new therapeutic leads, perhaps removing the need for large-scale screening experiments at the beginning of an antibody discovery project.
Structural annotations and modeling can also be applied to discover antigen-specific antibodies. Krawczyk et al. (74) annotated ∼3.4 million sequences from individuals who had been exposed to the influenza virus with their proposed templates and therefore whose repertoires were enriched with influenza-specific binders. They discovered that many of the templates assigned came from known influenza-binding antibodies. They therefore propose that sharing of a similar structural template could be an indication of similar specificity. Assuming that a structure of an antibody specific to a given antigen or epitope is known, antibodies can be selected from a repertoire if they are predicted to have a high degree of structural similarity to it. Other computational tools can also be exploited to find potential therapeutics: a large set of models generated from repertoire data can be used as an in silico screening library (135)1 in conjunction with epitope predictors (109, 136–144), paratope predictors (72, 145–150), and docking algorithms (83, 151–164). As computational methods continue to improve and become faster, this approach will become more accurate and more feasible, potentially making an entirely in silico antibody discovery platform a reality.
However, issues arise due to most sequencing experiments focusing on only the heavy chain and unknown native pairings even when both the heavy and light chains are sequenced. Antibodies with high affinity and specificity are identified more often when the true VH/VL pairings are known (165); however, this is not achievable with most of the available data. As previously stated, single-cell approaches that retain pair information have been developed (112); however, the method is not as high-throughput as other sequencing techniques, so less data is currently available. In the future, this is likely to change, but for now, other approaches must be applied. For experiments resulting in both heavy- and light-chain sequences, pairings can be exhaustively tested for plausibility (135)1 or by observing relative frequencies (110). Alternatively, especially when light chains have not been sequenced, it may be possible to use an artificial light chain with the ability to associate with a range of heavy chains (166). The concept of public sequences may also help here; a subset of the public light chain sequences could be used as a pairing library, as these sequences are clearly widely used and are therefore more likely to form successful pairings. In general, known public sequences may be a good place to start when attempting to discover a new therapeutic (e.g. in the design of a screening library), because they are likely to have low immunogenicity and be of high importance in the immune response to many common antigens.
Using BCR repertoire data to identify undesirable properties during therapeutic development
Binding affinity is not the only feature of a potential therapeutic that needs to be optimized. In addition to being biologically active, it must be safe to administer to humans and be able to withstand the stresses of the production process (i.e. the antibody should have good “developability”) (167). Antibodies discovered through the immunization of an organism (such as a mouse) against the target antigen cannot be used directly as therapeutics, because they would be identified as nonnative by the human immune system and would therefore cause an unwanted response themselves (168). Changes made to potential therapeutics during the development process can also introduce nonhuman-like characteristics. It is therefore desirable to be able to quantify the similarity of a sequence to those from natural human repertoires (its “humanness”) and to propose changes that could be made to a sequence to make it more human and hence less likely to be rejected by a patient. This “humanization” process can be guided through comparisons with human BCR repertoires, because they are natural and represent what is “allowed” and what is safe in an organism (see Fig. 5). Previous work has used small sets of reference sequences (such as known germline sequences) to infer humanness (169–171), but the growth of BCR repertoire sequencing has created new opportunities. The amount of data now available allows not only the identification of which amino acids are allowed at which positions, but also the investigation of residue couplings and covariation (172). Recently, Wollacott et al. (172) described a machine learning-based humanization method, trained on large sets of sequence data, and demonstrated that it outperformed other methods at evaluating the humanness of antibodies from sequence.
Figure 5.

WebLogo representations for the second framework region (residues 39–55 in the IMGT numbering scheme) for known human and mouse antibody sequences. Hydrophobic amino acids are shown in red, hydrophilic in blue, and neutral in gray. Data were extracted from OAS; we only considered repertoires from individuals with no disease and no vaccine recorded. Whereas amino acid usage is the same at many positions along the sequence, it can be seen that there are differences that could potentially be used to measure “humanness” and guide the humanization process. For example, it is rare to observe lysine at position 43 in human antibodies, but this is common in mice. Changing a lysine to an arginine at this position in a potential therapeutic may therefore reduce immunogenicity.
The chemical properties of a potential therapeutic can also cause problems, such as instability, self-association, high viscosity, polyspecificity, and poor expression (167). These characteristics can be determined experimentally; however, this is time-consuming and hence low-throughput, meaning the examination of thousands or millions of sequences from a BCR repertoire is not feasible. However, some of these properties can be predicted from the amino acid sequence of the antibody. For example, a number of sequence motifs have been identified that indicate sites of potential post-translational modification (78, 173); hydrophobic residues in the CDRs are thought to lead to high aggregation, viscosity, and polyspecificity (167, 174–179); patches of electrostatic charge on the antibody surface have been linked to high clearance rates and poor expression (180, 181); and asymmetric charges of the heavy and light variable domains result in self-association and high viscosity (175, 182). A number of computational tools have therefore been developed that predict these risk factors (e.g. Refs. 175–179 and 183). Whereas some of these attempt to predict solely from sequence, the majority require structural knowledge—for instance, it is important to know which residues are located on the antibody surface (176, 177). The tools can be exploited during the identification of binders as described above to minimize issues further along the therapeutic development pipeline.
The properties described above can also be examined by calculating repertoire-wide distributions. As a simple example, consider the lengths of the CDRs. Using sequence repertoires, the distribution of observed lengths can be obtained. If a given length falls outside the range of this distribution, it can be assumed that this property is “unnatural,” and therefore the antibody is more likely to have undesirable characteristics in vivo. Raybould et al. (106) used this approach, alongside the generation of antibody model libraries, to contextualize known therapeutic sequences against human repertoires. They were therefore able to define five developability guidelines that predict whether a given antibody will be successful as a therapeutic, based on total CDR length, patches of hydrophobicity, patches of positive and negative charge, and the overall surface charge of VH and VL domains. Testing the guidelines on sequences from two antibody discovery projects showed that this approach successfully highlighted candidates with known developability issues.
In summary, by representing the allowed antibody sequence space, BCR repertoires can be used to guide the antibody discovery and development process toward more successful therapeutic candidates. Using developability or humanness prediction algorithms in conjunction with in silico screening of BCR repertoires should be of great benefit to the therapeutic development community, and as sequence repositories continue to grow and computational techniques become more sophisticated, we can expect more advances to be made.
Conclusions
Advances in next-generation sequencing and its increasing use in characterizing the immune system has led to the exponential growth of the number of known antibody sequences. Subsequently, there is now a wealth of information, which has increased opportunities for large-scale data mining. The amount of data presents its challenges, however. Curated, publicly available sequence repositories such as the OAS are addressing the problem of storage and accessibility, but changes may have to be made as we learn more about the needs of researchers wishing to use the data. The increase in the amount of data will also create computational obstacles; we must continue to develop methods that can analyze huge numbers of sequences in a time- and resource-efficient manner.
Repertoire data can be used to gain a deeper understanding of human immune system, including the mechanisms that drive repertoire diversity and its response to antigen exposure. Comparisons between individuals have detected the presence of a core set of shared sequences or clonotypes known as the public repertoire, potentially of great importance in protecting against common antigens.
The antigen-binding properties of antibodies are governed by their structures. Sequence-similar antibodies may adopt different structures, and vice versa; by using sequence alone, these subtleties are not discerned. The incorporation of structural information into repertoire analyses, through annotation or modeling, therefore allows more accurate comparisons to be made and hence provides a better representation of the repertoire space. Ongoing improvements in modeling algorithms, in particular increased speed and accuracy of H3 structure prediction, will mean that larger subsets of the repertoire can be analyzed in this manner and with more reliability. An increase in the number of available templates would also improve structural modeling—repertoire data itself may be used in this process, to highlight areas of sequence space for which structures are currently lacking.
Large-scale sequencing data can also be of great benefit during the discovery of antibodies for therapeutic use. Clonal selection and expansion leads to the enrichment of the repertoire with antigen binders post-exposure; these can be identified and used as starting points for further development. The presence of sequence-similar antibodies to known therapeutics in OAS (74) indicates that it should be possible to mine these repositories for new therapeutic leads without performing specific experiments. For example, in silico screening libraries could be developed, by combining BCR repertoire data with modeling protocols and other computational tools (e.g. docking algorithms) to select likely binders.
Currently, it is possible for the computational approaches such as those described in this review to be used in tandem with experimental work. For example, after a potential binder is identified experimentally, clonotyping can be used to select similar antibodies from a repertoire, thereby expanding the pool of candidates for further study. In the long term, however, the objective of many researchers is to make the discovery of new therapeutic antibodies completely computational, with little or no human input. Consolidating all of the knowledge gained from large-scale repertoire analysis may enable the creation of an in silico immune system, or at the least a completely human-like synthetic repertoire that can be screened to identify potential therapeutics. Although it is too soon to say whether an entirely in silico protocol would produce better results than an experimental one, it would remove the need for expensive and time-consuming experimental work and would mean the immunization of animals is no longer required. There are many obstacles to achieve this, perhaps most importantly in the initial selection of antibodies that bind to a specific antigen of interest—improvements in structural modeling, docking, and binding affinity prediction in particular will help this.
Even though there is a large quantity of data already available, there is a vast amount of the antibody sequence space that remains unknown. For example, at around one billion sequences (including redundant sequences), the Observed Antibody Space database represents less than 0.01% of the potential total number (predicted to be around 1013 nonredundant sequences). Efforts should also be made to sequence repertoires with different attributes (e.g. ethnic background)—currently, this is not routinely disclosed, making analysis of its effect on the repertoire difficult. The continued growth of available sequence information should mean that currently unknown parts of sequence space are investigated, and therefore we should be able to analyze the workings of the immune system and predict antibody/repertoire properties more accurately. Importantly, with the development of experimental techniques that preserve the native VH-VL pairings, we will no longer have to rely on approximations and exhaustive combinatorics to achieve an accurate view of what binding sites are present. Overall, access to large-scale sequencing data has provided many opportunities to deepen our understanding of the immune system and improve our ability to design biotherapeutics and will surely continue to do so.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
M. I. J. Raybould, C. Marks, A. Kovaltsuk, A. P. Lewis, J. Shi, and C. M. Deane, manuscript in preparation.
- BCR
- B-cell receptor
- Fv
- variable region
- V
- variable segment
- J
- joining segment
- D
- diversity segment
- CDR
- complementarity-determining region
- OAS
- Observed Antibody Space
- PDB
- Protein Data Bank
- RMSD
- root mean square deviation.
References
- 1. Sela-Culang I., Kunik V., and Ofran Y. (2013) The structural basis of antibody-antigen recognition. Front. Immunol. 4, 302 10.3389/fimmu.2013.00302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Saper C. B. (2009) A guide to the perplexed on the specificity of antibodies. J. Histochem. Cytochem. 57, 1–5 10.1369/jhc.2008.952770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ecker D. M., Jones S. D., and Levine H. L. (2015) The therapeutic monoclonal antibody market. mAbs 7, 9–14 10.4161/19420862.2015.989042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Raybould M. I. J., Marks C., Lewis A. P., Shi J., Bujotzek A., Taddese B., and Deane C. M. (2020) Thera-SAbDab: the therapeutic structural antibody database. Nucleic Acids Res. 48, D383–D388 10.1093/nar/gkz827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kaplon H., and Reichert J. M. (2019) Antibodies to watch in 2019. mAbs 11, 219–238 10.1080/19420862.2018.1556465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Greiff V., Miho E., Menzel U., and Reddy S. T. (2015) Bioinformatic and statistical analysis of adaptive immune repertoires. Trends Immunol. 36, 738–749 10.1016/j.it.2015.09.006 [DOI] [PubMed] [Google Scholar]
- 7. Tonegawa S. (1983) Somatic generation of antibody diversity. Nature 302, 575–581 10.1038/302575a0 [DOI] [PubMed] [Google Scholar]
- 8. Jeske D. J., Jarvis J., Milstein C., and Capra J. D. (1984) Junctional diversity. J. Immunol. 133, 1090–1092 [PubMed] [Google Scholar]
- 9. Schramm C. A., and Douek D. C. (2018) Beyond hot spots: biases in antibody somatic hypermutation and implications for vaccine design. Front. Immunol. 9, 1876 10.3389/fimmu.2018.01876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Collis A. V., Brouwer A. P., and Martin A. C. (2003) Analysis of the antigen combining site: correlations between length and sequence composition of the hypervariable loops and the nature of the antigen. J. Mol. Biol. 325, 337–354 10.1016/S0022-2836(02)01222-6 [DOI] [PubMed] [Google Scholar]
- 11. Xu J. L., and Davis M. M. (2000) Diversity in the CDR3 region of V. Immunity 13, 37–45 10.1016/S1074-7613(00)00006-6 [DOI] [PubMed] [Google Scholar]
- 12. Kuroda D., Shirai H., Jacobson M. P., and Nakamura H. (2012) Computer-aided antibody design. Protein Eng. Des. Sel. 25, 507–521 10.1093/protein/gzs024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Burnet F. M. (1960) Theories of immunity. Perspect. Biol. Med. 3, 447–458 10.1353/pbm.1960.0034 [DOI] [PubMed] [Google Scholar]
- 14. Glanville J., Zhai W., Berka J., Telman D., Huerta G., Mehta G. R., Ni I., Mei L., Sundar P. D., Day G. M., Cox D., Rajpal A., and Pons J. (2009) Precise determination of the diversity of a combinatorial antibody library gives insight into the human immunoglobulin repertoire. Proc. Natl. Acad. Sci. U.S.A. 106, 20216–20221 10.1073/pnas.0909775106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Georgiou G., Ippolito G. C., Beausang J., Busse C. E., Wardemann H., and Quake S. R. (2014) The promise and challenge of high-throughput sequencing of the antibody repertoire. Nat. Biotechnol. 32, 158–168 10.1038/nbt.2782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ota M., Duong B. H., Torkamani A., Doyle C. M., Gavin A. L., Ota T., and Nemazee D. (2010) Regulation of the B cell receptor repertoire and self-reactivity by BAFF. J. Immunol. 185, 4128–4136 10.4049/jimmunol.1002176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhou T., Zhu J., Wu X., Moquin S., Zhang B., Acharya P., Georgiev I. S., Altae-Tran H. R., Chuang G.-Y., Joyce M. G., Kwon Y. D., Longo N. S., Louder M. K., Luongo T., McKee K., et al. (2013) Multidonor analysis reveals structural elements, genetic determinants, and maturation pathway for HIV-1 neutralization by VRC01-class antibodies. Immunity 39, 245–258 10.1016/j.immuni.2013.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vander Heiden J. A., Stathopoulos P., Zhou J. Q., Chen L., Gilbert T. J., Bolen C. R., Barohn R. J., Dimachkie M. M., Ciafaloni E., Broering T. J., Vigneault F., Nowak R. J., Kleinstein S. H., and O'Connor K. C. (2017) Dysregulation of B cell repertoire formation in myasthenia gravis patients revealed through deep sequencing. J. Immunol. 198, 1460–1473 10.4049/jimmunol.1601415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gidoni M., Snir O., Peres A., Polak P., Lindeman I., Mikocziova I., Sarna V. K., Lundin K. E. A., Clouser C., Vigneault F., Collins A. M., Sollid L. M., and Yaari G. (2019) Mosaic deletion patterns of the human antibody heavy chain gene locus shown by Bayesian haplotyping. Nat. Commun. 10, 628 10.1038/s41467-019-08489-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Briney B., Inderbitzin A., Joyce C., and Burton D. R. (2019) Commonality despite exceptional diversity in the baseline human antibody repertoire. Nature 566, 393–397 10.1038/s41586-019-0879-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. López-Santibáñez-Jácome L., Avendaño-Vázquez S. E., and Flores-Jasso C. F. (2019) The pipeline repertoire for Ig-Seq analysis. Front. Immunol. 10, 899 10.3389/fimmu.2019.00899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Corrie B. D., Marthandan N., Zimonja B., Jaglale J., Zhou Y., Barr E., Knoetze N., Breden F. M. W., Christley S., Scott J. K., Cowell L. G., and Breden F. (2018) iReceptor: a platform for querying and analyzing antibody/B-cell and T-cell receptor repertoire data across federated repositories. Immunol. Rev. 284, 24–41 10.1111/imr.12666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Christley S., Scarborough W., Salinas E., Rounds W. H., Toby I. T., Fonner J. M., Levin M. K., Kim M., Mock S. A., Jordan C., Ostmeyer J., Buntzman A., Rubelt F., Davila M. L., Monson N. L., et al. (2018) VDJServer: A cloud-based analysis portal and data commons for immune repertoire sequences and rearrangements. Front. Immunol. 9, 976 10.3389/fimmu.2018.00976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rosenfeld A. M., Meng W., Luning Prak E. T., and Hershberg U. (2018) ImmuneDB, a novel tool for the analysis, storage, and dissemination of immune repertoire sequencing data. Front. Immunol. 9, 2107 10.3389/fimmu.2018.02107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chailyan A., Tramontano A., and Marcatili P. (2012) A database of immunoglobulins with integrated tools: DIGIT. Nucleic Acids Res. 40, 1230–1234 10.1093/nar/gkr806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Swindells M. B., Porter C. T., Couch M., Hurst J., Abhinandan K. R., Nielsen J. H., Macindoe G., Hetherington J., and Martin A. C. (2017) abYsis: integrated antibody sequence and structure-management, analysis, and prediction. J. Mol. Biol. 429, 356–364 10.1016/j.jmb.2016.08.019 [DOI] [PubMed] [Google Scholar]
- 27. Zhang W., Wang L., Liu K., Wei X., Yang K., Du W., Wang S., Guo N., Ma C., Luo L., Wu J., Lin L., Yang F., Gao F., Wang X., et al. (2019) PIRD: Pan Immune Repertoire Database. Bioinformatics 36, 897–903 10.1093/bioinformatics/btz614 [DOI] [PubMed] [Google Scholar]
- 28. Kovaltsuk A., Leem J., Kelm S., Snowden J., Deane C. M., and Krawczyk K. (2018) Observed antibody space: a resource for data mining next-generation sequencing of antibody repertoires. J. Immunol. 201, 2502–2509 10.4049/jimmunol.1800708 [DOI] [PubMed] [Google Scholar]
- 29. DeWitt W. S., Lindau P., Snyder T. M., Sherwood A. M., Vignali M., Carlson C. S., Greenberg P. D., Duerkopp N., Emerson R. O., and Robins H. S. (2016) A public database of memory and naive B-cell receptor sequences. PLoS ONE 11, e0160853 10.1371/journal.pone.0160853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wrammert J., Smith K., Miller J., Langley W. A., Kokko K., Larsen C., Zheng N. Y., Mays I., Garman L., Helms C., James J., Air G. M., Capra J. D., Ahmed R., and Wilson P. C. (2008) Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature 453, 667–671 10.1038/nature06890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yu X., Tsibane T., McGraw P. A., House F. S., Keefer C. J., Hicar M. D., Tumpey T. M., Pappas C., Perrone L. A., Martinez O., Stevens J., Wilson I. A., Aguilar P. V., Altschuler E. L., Basler C. F., and Crowe J. E. Jr. (2008) Neutralizing antibodies derived from the B cells of 1918 influenza pandemic survivors. Nature 455, 532–536 10.1038/nature07231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Frost S. D., Murrell B., Hossain A. S. M., Silverman G. J., and Pond S. L. (2015) Assigning and visualizing germline genes in antibody repertoires. Phil. Trans. R. Soc. B 370, 20140240 10.1098/rstb.2014.0240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Miho E., Yermanos A., Weber C. R., Berger C. T., Reddy S. T., and Greiff V. (2018) Computational strategies for dissecting the high-dimensional complexity of adaptive immune repertoires. Front. Immunol. 9, 224 10.3389/fimmu.2018.00224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gadala-Maria D., Yaari G., Uduman M., and Kleinstein S. H. (2015) Automated analysis of high-throughput B-cell sequencing data reveals a high frequency of novel immunoglobulin V gene segment alleles. Proc. Natl. Acad. Sci. U.S.A. 112, E862–E870 10.1073/pnas.1417683112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gupta N. T., Vander Heiden J. A., Uduman M., Gadala-Maria D., Yaari G., and Kleinstein S. H. (2015) Change-O: a toolkit for analyzing large-scale B cell immunoglobulin repertoire sequencing data. Bioinformatics 31, 3356–3358 10.1093/bioinformatics/btv359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Corcoran M. M., Phad G. E., Vázquez Bernat N. V., Stahl-Hennig C., Sumida N., Persson M. A., Martin M., and Karlsson Hedestam G. B. (2016) Production of individualized v gene databases reveals high levels of immunoglobulin genetic diversity. Nat. Commun. 7, 13642 10.1038/ncomms13642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Marcou Q., Mora T., and Walczak A. M. (2018) High-throughput immune repertoire analysis with IGoR. Nat. Commun. 9, 561 10.1038/s41467-018-02832-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Feeney A. J., Tang A., and Ogwaro K. M. (2000) B-cell repertoire formation: role of the recombination signal sequence in non-random V segment utilization. Immunol. Rev. 175, 59–69 10.1111/j.1600-065X.2000.imr017508.x [DOI] [PubMed] [Google Scholar]
- 39. Greiff V., Menzel U., Miho E., Weber C., Riedel R., Cook S., Valai A., Lopes T., Radbruch A., Winkler T. H., and Reddy S. T. (2017) Systems analysis reveals high genetic and antigen-driven predetermination of antibody repertoires throughout B cell development. Cell Rep. 19, 1467–1478 10.1016/j.celrep.2017.04.054 [DOI] [PubMed] [Google Scholar]
- 40. Weinstein J. A., Jiang N., White R. A. 3rd, Fisher D. S., and Quake S. R. (2009) High-throughput sequencing of the zebrafish antibody repertoire. Science 324, 807–810 10.1126/science.1170020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Glanville J., Kuo T. C., von Büdingen H. C., Guey L., Berka J., Sundar P. D., Huerta G., Mehta G. R., Oksenberg J. R., Hauser S. L., Cox D. R., Rajpal A., and Pons J. (2011) Naive antibody gene-segment frequencies are heritable and unaltered by chronic lymphocyte ablation. Proc. Natl. Acad. Sci. U.S.A. 108, 20066–20071 10.1073/pnas.1107498108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Elhanati Y., Sethna Z., Marcou Q., Callan C. G. Jr., Mora T., and Walczak A. M. (2015) Inferring processes underlying B-cell repertoire diversity. Phil. Trans. R. Soc. B 370, 20140243 10.1098/rstb.2014.0243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Elhanati Y., Marcou Q., Mora T., and Walczak A. M. (2016) RepgenHMM: A dynamic programming tool to infer the rules of immune receptor generation from sequence data. Bioinformatics 32, 1943–1951 10.1093/bioinformatics/btw112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Miho E., Roškar R., Greiff V., and Reddy S. T. (2019) Large-scale network analysis reveals the sequence space architecture of antibody repertoires. Nat. Commun. 10, 1321 10.1038/s41467-019-09278-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Odegard V. H., and Schatz D. G. (2006) Targeting of somatic hypermutation. Nat. Rev. Immunol. 6, 573–583 10.1038/nri1896 [DOI] [PubMed] [Google Scholar]
- 46. Yaari G., Uduman M., and Kleinstein S. H. (2012) Quantifying selection in high-throughput immunoglobulin sequencing data sets. Nucleic Acids Res. 40, 10–12 10.1093/nar/gks457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sheng Z., Schramm C. A., Kong R., Mullikin J. C., Mascola J. R., Kwong P. D., Shapiro L., and NISC Comparative Sequencing Program (2017) Gene-specific substitution profiles describe the types and frequencies of amino acid changes during antibody somatic hypermutation. Front. Immunol. 8, 537 10.3389/fimmu.2017.00537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yaari G., Vander Heiden J. A., Uduman M., Gadala-Maria D., Gupta N., Stern J. N. H., O'Connor K. C., Hafler D. A., Laserson U., Vigneault F., and Kleinstein S. H. (2013) Models of somatic hypermutation targeting and substitution based on synonymous mutations from high-throughput immunoglobulin sequencing data. Front. Immunol. 4, 358 10.3389/fimmu.2013.00358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hoehn K. B., Lunter G., and Pybus O. G. (2017) A phylogenetic codon substitution model for antibody lineages. Genetics 206, 417–427 10.1534/genetics.116.196303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Horns F., Vollmers C., Dekker C. L., and Quake S. R. (2019) Signatures of selection in the human antibody repertoire: selective sweeps, competing subclones, and neutral drift. Proc. Natl. Acad. Sci. U.S.A. 116, 1261–1266 10.1073/pnas.1814213116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yermanos A., Greiff V., Krautler N. J., Menzel U., Dounas A., Miho E., Oxenius A., Stadler T., and Reddy S. T. (2017) Comparison of methods for phylogenetic B-cell lineage inference using time-resolved antibody repertoire simulations (AbSim). Bioinformatics 33, 3938–3946 10.1093/bioinformatics/btx533 [DOI] [PubMed] [Google Scholar]
- 52. Collins A. M., and Jackson K. J. (2018) On being the right size: antibody repertoire formation in the mouse and human. Immunogenetics 70, 143–158 10.1007/s00251-017-1049-8 [DOI] [PubMed] [Google Scholar]
- 53. Skaggs H., Chellman G. J., Collinge M., Enright B., Fuller C. L., Krayer J., Sivaraman L., and Weinbauer G. F. (2019) Comparison of immune system development in nonclinical species and humans: closing information gaps for immunotoxicity testing and human translatability. Reprod. Toxicol. 89, 178–188 10.1016/j.reprotox.2019.06.005 [DOI] [PubMed] [Google Scholar]
- 54. Bashford-Rogers R. J. M., Bergamaschi L., McKinney E. F., Pombal D. C., Mescia F., Lee J. C., Thomas D. C., Flint S. M., Kellam P., Jayne D. R. W., Lyons P. A., and Smith K. G. C. (2019) Analysis of the B cell receptor repertoire in six immune-mediated diseases. Nature 574, 122–126 10.1038/s41586-019-1595-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lai L., Zhou X., Chen H., Luo Y., Sui W., Zhang J., Tang D., Yan Q., and Dai Y. (2019) Composition and diversity analysis of the B-cell receptor immunoglobulin heavy chain complementarity determining region 3 repertoire in patients with acute rejection after kidney transplantation using high-throughput sequencing. Exp. Ther. Med. 17, 2206–2220 10.3892/etm.2019.7183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Greiff V., Bhat P., Cook S. C., Menzel U., Kang W., and Reddy S. T. (2015) A bioinformatic framework for immune repertoire diversity profiling enables detection of immunological status. Genome Med. 7, 3–5 10.1186/s13073-015-0169-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ostmeyer J., Christley S., Rounds W. H., Toby I., Greenberg B. M., Monson N. L., and Cowell L. G. (2017) Statistical classifiers for diagnosing disease from immune repertoires: a case study using multiple sclerosis. BMC Bioinformatics 18, 401 10.1186/s12859-017-1814-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Arora R., Kapllinsky J., Li A., and Arnaout R. (2019) Repertoire-based diagnostics using statistical biophysics. bioRxiv 10.1101/519108 [DOI]
- 59. Jiang N., He J., Weinstein J. A., Penland L., Sasaki S., He X. S., Dekker C. L., Zheng N. Y., Huang M., Sullivan M., Wilson P. C., Greenberg H. B., Davis M. M., Fisher D. S., and Quake S. R. (2013) Lineage structure of the human antibody repertoire in response to influenza vaccination. Sci. Transl. Med. 5, 171ra19 10.1126/scitranslmed.3004794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lindner C., Thomsen I., Wahl B., Ugur M., Sethi M. K., Friedrichsen M., Smoczek A., Ott S., Baumann U., Suerbaum S., Schreiber S., Bleich A., Gaboriau-Routhiau V., Cerf-Bensussan N., Hazanov H., et al. (2015) Diversification of memory B cells drives the continuous adaptation of secretory antibodies to gut microbiota. Nat. Immunol. 16, 880–888 10.1038/ni.3213 [DOI] [PubMed] [Google Scholar]
- 61. Galson J. D., Trück J., Fowler A., Münz M., Cerundolo V., Pollard A. J., Lunter G., and Kelly D. F. (2015) In-depth assessment of within-individual and inter-individual variation in the B cell receptor repertoire. Front. Immunol. 6, 531 10.3389/fimmu.2015.00531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Galson J. D., Clutterbuck E. A., Trück J., Ramasamy M. N., Münz M., Fowler A., Cerundolo V., Pollard A. J., Lunter G., and Kelly D. F. (2015) BCR repertoire sequencing: different patterns of B-cell activation after two Mningococcal vaccines. Immunol. Cell Biol. 93, 885–895 10.1038/icb.2015.57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gupta N. T., Adams K. D., Briggs A. W., Timberlake S. C., Vigneault F., and Kleinstein S. H. (2017) Hierarchical clustering can identify B cell clones with high confidence in Ig repertoire sequencing data. J. Immunol. 198, 2489–2499 10.4049/jimmunol.1601850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Soto C., Bombardi R. G., Branchizio A., Kose N., Matta P., Sevy A. M., Sinkovits R. S., Gilchuk P., Finn J. A., and Crowe J. E. (2019) High frequency of shared clonotypes in human B cell receptor repertoires. Nature 566, 398–402 10.1038/s41586-019-0934-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. DeKosky B. J., Kojima T., Rodin A., Charab W., Ippolito G. C., Ellington A. D., and Georgiou G. (2015) In-depth determination and analysis of the human paired heavy- and light-chain antibody repertoire. Nat. Med. 21, 86–91 10.1038/nm.3743 [DOI] [PubMed] [Google Scholar]
- 66. Galson J. D., Trück J., Fowler A., Clutterbuck E. A., Münz M., Cerundolo V., Reinhard C., van der Most R., Pollard A. J., Lunter G., and Kelly D. F. (2015) Analysis of B cell repertoire dynamics following hepatitis B vaccination in humans, and enrichment of vaccine-specific antibody sequences. EBioMedicine 2, 2070–2079 10.1016/j.ebiom.2015.11.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wang C., Liu Y., Cavanagh M. M., Le Saux S., Qi Q., Roskin K. M., Looney T. J., Lee J. Y., Dixit V., Dekker C. L., Swan G. E., Goronzy J. J., and Boyd S. D. (2015) B-cell repertoire responses to varicella-zoster vaccination in human identical twins. Proc. Natl. Acad. Sci. U.S.A. 112, 500–505 10.1073/pnas.1415875112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Greiff V., Weber C. R., Palme J., Bodenhofer U., Miho E., Menzel U., and Reddy S. T. (2017) Learning the high-dimensional immunogenomic features that predict public and private antibody repertoires. J. Immunol. 199, 2985–2997 10.4049/jimmunol.1700594 [DOI] [PubMed] [Google Scholar]
- 69. Jackson K. J. L., Wang Y., Gaeta B. A., Pomat W., Siba P., Rimmer J., Sewell W. A., and Collins A. M. (2012) Divergent human populations show extensive shared IGK rearrangements in peripheral blood B cells. Immunogenetics 64, 3–14 10.1007/s00251-011-0559-z [DOI] [PubMed] [Google Scholar]
- 70. Hoi K. H., and Ippolito G. C. (2013) Intrinsic bias and public rearrangements in the human immunoglobulin Vλ light chain repertoire. Genes Immun. 14, 271–276 10.1038/gene.2013.10 [DOI] [PubMed] [Google Scholar]
- 71. Setliff I., McDonnell W. J., Raju N., Bombardi R. G., Murji A. A., Scheepers C., Ziki R., Mynhardt C., Shepherd B. E., Mamchak A. A., Garrett N., Karim S. A., Mallal S. A., Crowe J. E. Jr., Morris L., et al. (2018) Multi-donor longitudinal antibody repertoire sequencing reveals the existence of public antibody clonotypes in HIV-1 infection. Cell Host Microbe 23, 845–854.e6 10.1016/j.chom.2018.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Peng H. P., Lee K. H., Jian J. W., and Yang A. S. (2014) Origins of specificity and affinity in antibody-protein interactions. Proc. Natl. Acad. Sci. U.S.A. 111, E2656–E2665 10.1073/pnas.1401131111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kovaltsuk A., Krawczyk K., Galson J. D., Kelly D. F., Deane C. M., and Trück J. (2017) How B-cell receptor repertoire sequencing can be enriched with structural antibody data. Front. Immunol. 8, 1753 10.3389/fimmu.2017.01753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Krawczyk K., Kelm S., Kovaltsuk A., Galson J. D., Kelly D., Trück J., Regep C., Leem J., Wong W. K., Nowak J., Snowden J., Wright M., Starkie L., Scott-Tucker A., Shi J., et al. (2018) Structurally mapping antibody repertoires. Front. Immunol. 9, 1698 10.3389/fimmu.2018.01698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Dunbar J., Krawczyk K., Leem J., Baker T., Fuchs A., Georges G., Shi J., and Deane C. M. (2014) SAbDab: the structural antibody database. Nucleic Acids Res. 42, D1140–D1146 10.1093/nar/gkt1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Li Y., Li H., Yang F., Smith-Gill S. J., and Mariuzza R. A. (2003) X-ray snapshots of the maturation of an antibody response to a protein antigen. Nat. Struct. Biol. 10, 482–488 10.1038/nsb930 [DOI] [PubMed] [Google Scholar]
- 77. Huang K. A., Rijal P., Jiang H., Wang B., Schimanski L., Dong T., Liu Y. M., Chang P., Iqbal M., Wang M. C., Chen Z., Song R., Huang C. C., Yang J. H., Qi J., et al. (2019) Structure-function analysis of neutralizing antibodies to H7N9 influenza from naturally infected humans. Nat. Microbiol. 4, 306–315 10.1038/s41564-018-0303-7 [DOI] [PubMed] [Google Scholar]
- 78. Leem J., Dunbar J., Georges G., Shi J., and Deane C. M. (2016) ABodyBuilder: automated antibody structure prediction with data-driven accuracy estimation. mAbs 8, 1259–1268 10.1080/19420862.2016.1205773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Klausen M. S., Anderson M. V., Jespersen M. C., Nielsen M., and Marcatili P. (2015) LYRA, a webserver for lymphocyte receptor structural modeling. Nucleic Acids Res. 43, W349–W355 10.1093/nar/gkv535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Marcatili P., Rosi A., and Tramontano A. (2008) PIGS: automatic prediction of antibody structures. Bioinformatics 24, 1953–1954 10.1093/bioinformatics/btn341 [DOI] [PubMed] [Google Scholar]
- 81. Yamashita K., Ikeda K., Amada K., Liang S., Tsuchiya Y., Nakamura H., Shirai H., and Standley D. M. (2014) Kotai antibody builder: automated high-resolution structural modeling of antibodies. Bioinformatics 30, 3279–3280 10.1093/bioinformatics/btu510 [DOI] [PubMed] [Google Scholar]
- 82. Sivasubramanian A., Sircar A., Chaudhury S., and Gray J. J. (2009) Toward high-resolution homology modeling of antibody Fv regions and application to antibody-antigen docking. Proteins 74, 497–514 10.1002/prot.22309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Weitzner B. D., Jeliazkov J. R., Lyskov S., Marze N., Kuroda D., Frick R., Adolf-Bryfogle J., Biswas N., Dunbrack R. L. Jr., and Gray J. J. (2017) Modeling and docking of antibody structures with Rosetta. Nat. Protoc. 12, 401–416 10.1038/nprot.2016.180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Kemmish H., Fasnacht M., and Yan L. (2017) Fully automated antibody structure prediction using BIOVIA tools: validation study. PLoS ONE 12, e0177923–26 10.1371/journal.pone.0177923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Bujotzek A., Fuchs A., Qu C., Benz J., Klostermann S., Antes I., and Georges G. (2015) MoFvAb: modeling the Fv region of antibodies. mAbs 7, 838–852 10.1080/19420862.2015.1068492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Whitelegg N. R. J., and Rees A. R. (2000) WAM: an improved algorithm for modelling antibodies on the WEB. Protein Eng. Des. Sel. 13, 819–824 10.1093/protein/13.12.819 [DOI] [PubMed] [Google Scholar]
- 87. Zhu K., Day T., Warshaviak D., Murrett C., Friesner R., and Pearlman D. (2014) Antibody structure determination using a combination of homology modeling, energy-based refinement and loop prediction. Proteins 82, 1646–1655 10.1002/prot.24551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Maier J. K. X., and Labute P. (2014) Assessment of fully automated antibody homology modeling protocols in molecular operating environment. Proteins 82, 1599–1610 10.1002/prot.24576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Mandal C., Kingery B. D., Anchin J. M., Subramaniam S., and Linthicum D. S. (1996) ABGEN: a knowledge-based automated approach for antibody structure modeling. Nat. Biotechnol. 14, 323–328 10.1038/nbt0396-323 [DOI] [PubMed] [Google Scholar]
- 90. Berrondo M., Kaufmann S., and Berrondo M. (2014) Automated Aufbau of antibody structures from given sequences using Macromoltek's SmrtMolAntibody. Proteins 82, 1636–1645 10.1002/prot.24595 [DOI] [PubMed] [Google Scholar]
- 91. Lapidoth G., Parker J., Prilusky J., and Fleishman S. J. (2019) AbPredict 2: A server for accurate and unstrained structure prediction of antibody variable domains. Bioinformatics 35, 1591–1593 10.1093/bioinformatics/bty822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Bujotzek A., Dunbar J., Lipsmeier F., Schäfer W., Antes I., Deane C. M., and Georges G. (2015) Prediction of VH-VL domain orientation for antibody variable domain modeling. Proteins 83, 681–695 10.1002/prot.24756 [DOI] [PubMed] [Google Scholar]
- 93. Teplyakov A., Luo J., Obmolova G., Malia T. J., Sweet R., Stanfield R. L., Kodangattil S., Almagro J. C., and Gilliland G. L. (2014) Antibody modeling assessment II. Structures and models. Proteins 82, 1563–1582 10.1002/prot.24554 [DOI] [PubMed] [Google Scholar]
- 94. Fasnacht M., Butenhof K., Goupil-Lamy A., Hernandez-Guzman F., Huang H., and Yan L. (2014) Automated antibody structure prediction using Accelrys tools: results and best practices. Proteins 82, 1583–1598 10.1002/prot.24604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Shirai H., Ikeda K., Yamashita K., Tsuchiya Y., Sarmiento J., Liang S., Morokata T., Mizuguchi K., Higo J., Standley D. M., and Nakamura H. (2014) High-resolution modeling of antibody structures by a combination of bioinformatics, expert knowledge, and molecular simulations. Proteins 82, 1624–1635 10.1002/prot.24591 [DOI] [PubMed] [Google Scholar]
- 96. Weitzner B. D., Kuroda D., Marze N., Xu J., and Gray J. J. (2014) Blind prediction performance of RosettaAntibody 3.0: grafting, relaxation, kinematic loop modeling, and full CDR optimization. Proteins 82, 1611–1623 10.1002/prot.24534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Chothia C., and Lesk A. M. (1987) Canonical structures for the hypervariable regions of immunoglobulins. J. Mol. Biol. 196, 901–917 10.1016/0022-2836(87)90412-8 [DOI] [PubMed] [Google Scholar]
- 98. North B., Lehmann A., and Dunbrack R. L. Jr. (2011) A new clustering of antibody CDR loop conformations. J. Mol. Biol. 406, 228–256 10.1016/j.jmb.2010.10.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Nowak J., Baker T., Georges G., Kelm S., Klostermann S., Shi J., Sridharan S., and Deane C. M. (2016) Length-independent structural similarities enrich the antibody CDR canonical class model. mAbs 8, 751–760 10.1080/19420862.2016.1158370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Marks C., and Deane C. M. (2017) Antibody H3 structure prediction. Comput. Struct. Biotechnol. J. 15, 222–231 10.1016/j.csbj.2017.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Regep C., Georges G., Shi J., Popovic B., and Deane C. M. (2017) The H3 loop of antibodies shows unique structural characteristics. Proteins 85, 1311–1318 10.1002/prot.25291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Marks C., Nowak J., Klostermann S., Georges G., Dunbar J., Shi J., Kelm S., and Deane C. M. (2017) Sphinx: merging knowledge-based and ab initio approaches to improve protein loop prediction. Bioinformatics 33, 1346–1353 10.1093/bioinformatics/btw823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Messih M. A., Lepore R., Marcatili P., and Tramontano A. (2014) Improving the accuracy of the structure prediction of the third hypervariable loop of the heavy chains of antibodies. Bioinformatics 30, 2733–2740 10.1093/bioinformatics/btu194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Choi Y., and Deane C. M. (2011) Predicting antibody complementarity determining region structures without classification. Mol. Biosyst. 7, 3327–3334 10.1039/c1mb05223c [DOI] [PubMed] [Google Scholar]
- 105. Zhu K., and Day T. (2013) Ab initio structure prediction of the antibody hypervariable H3 loop. Proteins 81, 1081–1089 10.1002/prot.24240 [DOI] [PubMed] [Google Scholar]
- 106. Raybould M. I. J., Marks C., Krawczyk K., Taddese B., Nowak J., Lewis A. P., Bujotzek A., Shi J., and Deane C. M. (2019) Five computational developability guidelines for therapeutic antibody profiling. Proc. Natl. Acad. Sci. U.S.A. 116, 4025–4030 10.1073/pnas.1810576116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Kovaltsuk A., Raybould M. I. J., Wong W. K., Marks C., Kelm S., Snowden J., Trück J., and Deane C. M. (2019) Structural diversity of B-cell receptor repertoires along the B-cell differentiation axis in humans and mice. bioRxiv 10.1101/762880 [DOI] [PMC free article] [PubMed]
- 108. DeKosky B. J., Lungu O. I., Park D., Johnson E. L., Charab W., Chrysostomou C., Kuroda D., Ellington A. D., Ippolito G. C., Gray J. J., and Georgiou G. (2016) Large-scale sequence and structural comparisons of human naive and antigen-experienced antibody repertoires. Proc. Natl. Acad. Sci. U.S.A. 113, E2636–E2645 10.1073/pnas.1525510113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Krawczyk K., Liu X., Baker T., Shi J., and Deane C. M. (2014) Improving B-cell epitope prediction and its application to global antibody-antigen docking. Bioinformatics 30, 2288–2294 10.1093/bioinformatics/btu190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Reddy S. T., Ge X., Miklos A. E., Hughes R. A., Kang S. H., Hoi K. H., Chrysostomou C., Hunicke-Smith S. P., Iverson B. L., Tucker P. W., Ellington A. D., and Georgiou G. (2010) Monoclonal antibodies isolated without screening by analyzing the variable-gene repertoire of plasma cells. Nat. Biotechnol. 28, 965–969 10.1038/nbt.1673 [DOI] [PubMed] [Google Scholar]
- 111. Zhu J., Ofek G., Yang Y., Zhang B., Louder M. K., Lu G., McKee K., Pancera M., Skinner J., Zhang Z., Parks R., Eudailey J., Lloyd K. E., Blinn J., Alam S. M., et al. (2013) Mining the antibodyome for HIV-1-neutralizing antibodies with next-generation sequencing and phylogenetic pairing of heavy/light chains. Proc. Natl. Acad. Sci. U.S.A. 110, 6470–6475 10.1073/pnas.1219320110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. DeKosky B. J., Ippolito G. C., Deschner R. P., Lavinder J. J., Wine Y., Rawlings B. M., Varadarajan N., Giesecke C., Dörner T., Andrews S. F., Wilson P. C., Hunicke-Smith S. P., Willson C. G., Ellington A. D., and Georgiou G. (2013) High-throughput sequencing of the paired human immunoglobulin heavy and light chain repertoire. Nat. Biotechnol. 31, 166–169 10.1038/nbt.2492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Ghraichy M., Galson J. D., Kovaltsuk A., Niederhäusern V. V., and Trück J. (2019) Maturation of naïve and antigen-experienced B-cell receptor repertoires with age. bioRxiv 10.1101/609651 [DOI]
- 114. Lee E. C., Liang Q., Ali H., Bayliss L., Beasley A., Bloomfield-Gerdes T., Bonoli L., Brown R., Campbell J., Carpenter A., Chalk S., Davis A., England N., Fane-Dremucheva A., Franz B., et al. (2014) Complete humanization of the mouse immunoglobulin loci enables efficient therapeutic antibody discovery. Nat. Biotechnol. 32, 356–363 10.1038/nbt.2825 [DOI] [PubMed] [Google Scholar]
- 115. Lim C. C., Choong Y. S., and Lim T. S. (2019) Cognizance of molecular methods for the generation of mutagenic phage display antibody libraries for affinity maturation. Int. J. Mol. Sci. 20, 1861 10.3390/ijms20081861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Zhai W., Glanville J., Fuhrmann M., Mei L., Ni I., Sundar P. D., Van Blarcom T., Abdiche Y., Lindquist K., Strohner R., Telman D., Cappuccilli G., Finlay W. J., Van den Brulle J., Cox D. R., et al. (2011) Synthetic antibodies designed on natural sequence landscapes. J. Mol. Biol. 412, 55–71 10.1016/j.jmb.2011.07.018 [DOI] [PubMed] [Google Scholar]
- 117. Prassler J., Thiel S., Pracht C., Polzer A., Peters S., Bauer M., Nörenberg S., Stark Y., Kölln J., Popp A., Urlinger S., and Enzelberger M. (2011) HuCAL PLATINUM, a synthetic fab library optimized for sequence diversity and superior performance in mammalian expression systems. J. Mol. Biol. 413, 261–278 10.1016/j.jmb.2011.08.012 [DOI] [PubMed] [Google Scholar]
- 118. Fowler A., Galson J. D., Trück J., Kelly D. F., and Lunter G. (2018) Inferring B cell specificity for vaccines using a mixture model. bioRxiv 10.1101/464792 [DOI] [PMC free article] [PubMed]
- 119. Galson J. D., Pollard A. J., Trück J., and Kelly D. F. (2014) Studying the antibody repertoire after vaccination: practical applications. Trends Immunol. 35, 319–331 10.1016/j.it.2014.04.005 [DOI] [PubMed] [Google Scholar]
- 120. Devarajan P., and Swain S. L. (2019) Original antigenic sin: friend or foe in developing a broadly cross-reactive vaccine to influenza? Cell Host Microbe 25, 354–355 10.1016/j.chom.2019.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Lee J., Paparoditis P., Horton A. P., Frühwirth A., McDaniel J. R., Jung J., Boutz D. R., Hussein D. A., Tanno Y., Pappas L., Ippolito G. C., Corti D., Lanzavecchia A., and Georgiou G. (2019) Persistent antibody clonotypes dominate the serum response to influenza over multiple years and repeated vaccinations. Cell Host Microbe 25, 367–376.e5 10.1016/j.chom.2019.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Henry C., Zheng N. Y., Huang M., Cabanov A., Rojas K. T., Kaur K., Andrews S. F., Palm A.-K. E., Chen Y. Q., Li Y., Hoskova K., Utset H. A., Vieira M. C., Wrammert J., Ahmed R., et al. (2019) Influenza virus vaccination elicits poorly adapted B cell responses in elderly individuals. Cell Host Microbe 25, 357–366.e6 10.1016/j.chom.2019.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Trück J., Ramasamy M. N., Galson J. D., Rance R., Parkhill J., Lunter G., Pollard A. J., and Kelly D. F. (2015) Identification of antigen-specific B cell receptor sequences using public repertoire analysis. J. Immunol. 194, 252–261 10.4049/jimmunol.1401405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Mason D. M., Friedensohn S., Weber C. R., Jordi C., Wagner B., Meng S., Gainza P., Correia B., and Reddy S. T. (2019) Deep learning enables therapeutic antibody optimization in mammalian cells by deciphering high-dimensional protein sequence space. bioRxiv 10.1101/617860 [DOI]
- 125. Zhu J., Wu X., Zhang B., McKee K., O'Dell S., Soto C., Zhou T., Casazza J. P., NISC Comparative Sequencing Program, Mullikin J. C., Kwong P. D., Mascola J. R., Shapiro L., Becker J., Benjamin B., and Blakesley R. (2013) De novo identification of VRC01 class HIV-1-neutralizing antibodies by next-generation sequencing of B-cell transcripts. Proc. Natl. Acad. Sci. U.S.A. 110, E4088–E4097 10.1073/pnas.1306262110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Walker L. M., Huber M., Doores K. J., Falkowska E., Pejchal R., Julien J. P., Wang S. K., Ramos A., Chan-Hui P. Y., Moyle M., Mitcham J. L., Hammond P. W., Olsen O. A., Phung P., Fling S., et al. (2011) Broad neutralization coverage of HIV by multiple highly potent antibodies. Nature 477, 466–470 10.1038/nature10373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Wang B., Kluwe C. A., Lungu O. I., DeKosky B. J., Kerr S. A., Johnson E. L., Tanno H., Lee C.-H., Jung J., Rezigh A. B., Carroll S. M., Reyes A. N., Bentz J. R., Villanueva I., Altman A. L., et al. (2015) Facile discovery of a diverse panel of anti-Ebola virus antibodies by immune repertoire mining. Sci. Rep. 5, 13926 10.1038/srep13926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Sato S., Beausoleil S. A., Popova L., Beaudet J. G., Ramenani R. K., Zhang X., Wieler J. S., Schieferl S. M., Cheung W. C., and Polakiewicz R. D. (2012) Proteomics-directed cloning of circulating antiviral human monoclonal antibodies. Nat. Biotechnol. 30, 1039–1043 10.1038/nbt.2406 [DOI] [PubMed] [Google Scholar]
- 129. Saggy I., Wine Y., Shefet-Carasso L., Nahary L., Georgiou G., and Benhar I. (2012) Antibody isolation from immunized animals: comparison of phage display and antibody discovery via v gene repertoire mining. Protein Eng. Des. Sel. 25, 539–549 10.1093/protein/gzs060 [DOI] [PubMed] [Google Scholar]
- 130. Cheung W. C., Beausoleil S. A., Zhang X., Sato S., Schieferl S. M., Wieler J. S., Beaudet J. G., Ramenani R. K., Popova L., Comb M. J., Rush J., and Polakiewicz R. D. (2012) A proteomics approach for the identification and cloning of monoclonal antibodies from serum. Nat. Biotechnol. 30, 447–452 10.1038/nbt.2167 [DOI] [PubMed] [Google Scholar]
- 131. Ravn U., Gueneau F., Baerlocher L., Osteras M., Desmurs M., Malinge P., Magistrelli G., Farinelli L., Kosco-Vilbois M. H., and Fischer N. (2010) By-passing in vitro screening—next generation sequencing technologies applied to antibody display and in silico candidate selection. Nucleic Acids Res. 38, e193 10.1093/nar/gkq789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Wang B., DeKosky B. J., Timm M. R., Lee J., Normandin E., Misasi J., Kong R., McDaniel J. R., Delidakis G., Leigh K. E., Niezold T., Choi C. W., Viox E. G., Fahad A., Cagigi A., et al. (2018) Functional interrogation and mining of natively paired human v H:V L antibody repertoires. Nat. Biotechnol. 36, 152–155 10.1038/nbt.4052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Wang B., Lee C. H., Johnson E. L., Kluwe C. A., Cunningham J. C., Tanno H., Crooks R. M., Georgiou G., and Ellington A. D. (2016) Discovery of high affinity anti-ricin antibodies by B cell receptor sequencing and by yeast display of combinatorial VH:VL libraries from immunized animals. mAbs 8, 1035–1044 10.1080/19420862.2016.1190059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Krawczyk K., Raybould M. I. J., Kovaltsuk A., and Deane C. M. (2019) Looking for therapeutic antibodies in next-generation sequencing repositories. mAbs 11, 1197–1205 10.1080/19420862.2019.1633884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Raybould M. I. J., Wong W. K., and Deane C. M. (2019) Antibody-antigen complex modelling in the era of immunoglobulin repertoire sequencing. Mol. Syst. Des. Eng. 4, 679–688 10.1039/C9ME00034H [DOI] [Google Scholar]
- 136. Rapberger R., Lukas A., and Mayer B. (2007) Identification of discontinuous antigenic determinants on proteins based on shape complementarities. J. Mol. Recognit. 20, 113–121 10.1002/jmr.819 [DOI] [PubMed] [Google Scholar]
- 137. Sela-Culang I., Benhnia M. R.-E.-I., Matho M. H., Kaever T., Maybeno M., Schlossman A., Nimrod G., Li S., Xiang Y., Zajonc D., Crotty S., Ofran Y., and Peters B. (2014) Using a combined computational-experimental approach to predict antibody-specific B cell epitopes. Structure 22, 646–657 10.1016/j.str.2014.02.003 [DOI] [PubMed] [Google Scholar]
- 138. Sela-Culang I., Ashkenazi S., Peters B., and Ofran Y. (2015) PEASE: predicting B-cell epitopes utilizing antibody sequence. Bioinformatics 31, 1313–1315 10.1093/bioinformatics/btu790 [DOI] [PubMed] [Google Scholar]
- 139. Jespersen M. C., Mahajan S., Peters B., Nielsen M., and Marcatili P. (2019) Antibody specific B-cell epitope predictions: leveraging information from antibody-antigen protein complexes. Front. Immunol. 10, 298 10.3389/fimmu.2019.00298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Hua C. K., Gacerez A. T., Sentman C. L., Ackerman M. E., Choi Y., and Bailey-Kellogg C. (2017) Computationally-driven identification of antibody epitopes. eLife 6, e29023 10.7554/eLife.29023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Bourquard T., Musnier A., Puard V., Tahir S., Ayoub M. A., Jullian Y., Boulo T., Gallay N., Watier H., Bruneau G., Reiter E., Crépieux P., and Poupon A. (2018) MAbTope: a method for improved epitope mapping. J. Immunol. 201, 3096–3105 10.4049/jimmunol.1701722 [DOI] [PubMed] [Google Scholar]
- 142. Soga S., Kuroda D., Shirai H., Kobori M., and Hirayama N. (2010) Use of amino acid composition to predict epitope residues of individual antibodies. Protein Eng. Des. Sel. 23, 441–448 10.1093/protein/gzq014 [DOI] [PubMed] [Google Scholar]
- 143. Zhao L., and Li J. (2010) Mining for the antibody-antigen interacting associations that predict the B cell epitopes. BMC Struct. Biol. 10, S6 10.1186/1472-6807-10-S1-S6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Zhao L., Wong L., and Li J. (2011) Antibody-specified B-cell epitope prediction in line with the principle of context-awareness. IEEE/ACM Trans. Comput. Biol. Bioinform. 8, 1483–1494 10.1109/TCBB.2011.49 [DOI] [PubMed] [Google Scholar]
- 145. Krawczyk K., Baker T., Shi J., and Deane C. M. (2013) Antibody i-Patch prediction of the antibody binding site improves rigid local antibody-antigen docking. Protein Eng. Des. Sel. 26, 621–629 10.1093/protein/gzt043 [DOI] [PubMed] [Google Scholar]
- 146. Kunik V., Ashkenazi S., and Ofran Y. (2012) Paratome: an online tool for systematic identification of antigen-binding regions in antibodies based on sequence or structure. Nucleic Acids Res. 40, 521–524 10.1093/nar/gks480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Olimpieri P. P., Chailyan A., Tramontano A., and Marcatili P. (2013) Prediction of site-specific interactions in antibody-antigen complexes: the proABC method and server. Bioinformatics 29, 2285–2291 10.1093/bioinformatics/btt369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Liberis E., Velickovic P., Sormanni P., Vendruscolo M., and Liò P. (2018) Parapred: antibody paratope prediction using convolutional and recurrent neural networks. Bioinformatics 34, 2944–2950 10.1093/bioinformatics/bty305 [DOI] [PubMed] [Google Scholar]
- 149. Daberdaku S., and Ferrari C. (2019) Antibody interface prediction with 3D Zernike descriptors and SVM. Bioinformatics 35, 1870–1876 10.1093/bioinformatics/bty918 [DOI] [PubMed] [Google Scholar]
- 150. Deac A., Veličković P., and Sormanni P. (2019) Attentive cross-modal paratope prediction. J. Comput. Biol. 26, 536–545 10.1089/cmb.2018.0175 [DOI] [PubMed] [Google Scholar]
- 151. Brenke R., Hall D. R., Chuang G. Y., Comeau S. R., Bohnuud T., Beglov D., Schueler-Furman O., Vajda S., and Kozakov D. (2012) Application of asymmetric statistical potentials to antibody-protein docking. Bioinformatics 28, 2608–2614 10.1093/bioinformatics/bts493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Kozakov D., Hall D. R., Xia B., Porter K. A., Padhorny D., Yueh C., Beglov D., and Vajda S. (2017) The ClusPro web server for protein-protein docking. Nat. Protoc. 12, 255–278 10.1038/nprot.2016.169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Shimba N., Kamiya N., and Nakamura H. (2016) Model building of antibody-antigen complex structures using GBSA scores. J. Chem. Inf. Model. 56, 2005–2012 10.1021/acs.jcim.6b00066 [DOI] [PubMed] [Google Scholar]
- 154. Sircar A., and Gray J. J. (2010) SnugDock: paratope structural optimization during antibody-antigen docking compensates for errors in antibody homology models. PLoS Comput. Biol. 6, e1000644 10.1371/journal.pcbi.1000644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Ramírez-Aportela E., López-Blanco J. R., and Chacón P. (2016) FRODOCK 2.0: fast protein-protein docking server. Bioinformatics 32, 2386–2388 10.1093/bioinformatics/btw141 [DOI] [PubMed] [Google Scholar]
- 156. Macindoe G., Mavridis L., Venkatraman V., Devignes M. D., and Ritchie D. W. (2010) HexServer: an FFT-based protein docking server powered by graphics processors. Nucleic Acids Res. 38, 445–449 10.1093/nar/gkq311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Chen R., Li L., and Weng Z. (2003) ZDOCK: an initial-stage protein-docking algorithm. Proteins 52, 80–87 10.1002/prot.10389 [DOI] [PubMed] [Google Scholar]
- 158. Dominguez C., Boelens R., and Bonvin A. M. (2003) HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 125, 1731–1737 10.1021/ja026939x [DOI] [PubMed] [Google Scholar]
- 159. De Vries S. J., van Dijk A. D. J., Krzeminski M., van Dijk M., Thureau A., Hsu V., Wassenaar T., and Bonvin A. M. (2007) HADDOCK versus HADDOCK: new features and performance of HADDOCK2.0 on the CAPRI targets. Proteins 69, 726–733 10.1002/prot.21723 [DOI] [PubMed] [Google Scholar]
- 160. de Vries S. J., Schindler C. E., Chauvot de Beauchêne I., and Zacharias M. (2015) A web interface for easy flexible protein-protein docking with ATTRACT. Biophys. J. 108, 462–465 10.1016/j.bpj.2014.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Tovchigrechko A., and Vakser I. A. (2006) GRAMM-X public web server for protein-protein docking. Nucleic Acids Res. 34, 310–314 10.1093/nar/gkl206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Jiménez-García B., Pons C., and Fernández-Recio J. (2013) pyDockWEB: a web server for rigid-body protein-protein docking using electrostatics and desolvation scoring. Bioinformatics 29, 1698–1699 10.1093/bioinformatics/btt262 [DOI] [PubMed] [Google Scholar]
- 163. Torchala M., Moal I. H., Chaleil R. A., Fernandez-Recio J., and Bates P. A. (2013) SwarmDock: a server for flexible protein-protein docking. Bioinformatics 29, 807–809 10.1093/bioinformatics/btt038 [DOI] [PubMed] [Google Scholar]
- 164. Schneidman-Duhovny D., Inbar Y., Nussinov R., and Wolfson H. J. (2005) PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res. 33, 363–367 10.1093/nar/gki481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Adler A. S., Bedinger D., Adams M. S., Asensio M. A., Edgar R. C., Leong R., Leong J., Mizrahi R. A., Spindler M. J., Bandi S. R., Huang H., Tawde P., Brams P., and Johnson D. S. (2018) A natively paired antibody library yields drug leads with higher sensitivity and specificity than a randomly paired antibody library. mAbs 10, 431–443 10.1080/19420862.2018.1426422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Xue H., Sun L., Fujimoto H., Suzuki T., Takahashi Y., and Ohnishi K. (2019) Artificial immunoglobulin light chain with potential to associate with a wide variety of immunoglobulin heavy chains. Biochem. Biophys. Res. Commun. 515, 481–486 10.1016/j.bbrc.2019.05.149 [DOI] [PubMed] [Google Scholar]
- 167. Jarasch A., Koll H., Regula J. T., Bader M., Papadimitriou A., and Kettenberger H. (2015) Developability assessment during the selection of novel therapeutic antibodies. J. Pharm. Sci. 104, 1885–1898 10.1002/jps.24430 [DOI] [PubMed] [Google Scholar]
- 168. Safdari Y., Farajnia S., Asgharzadeh M., and Khalili M. (2013) Antibody humanization methods—a review and update. Biotechnol. Genet. Eng. 29, 175–186 10.1080/02648725.2013.801235 [DOI] [PubMed] [Google Scholar]
- 169. Abhinandan K. R., and Martin A. C. R. (2007) Analyzing the “degree of humanness” of antibody sequences. J. Mol. Biol. 369, 852–862 10.1016/j.jmb.2007.02.100 [DOI] [PubMed] [Google Scholar]
- 170. Lazar G. A., Desjarlais J. R., Jacinto J., Karki S., and Hammond P. W. (2007) A molecular immunology approach to antibody humanization and functional optimization. Mol. Immunol. 44, 1986–1998 10.1016/j.molimm.2006.09.029 [DOI] [PubMed] [Google Scholar]
- 171. Gao S. H., Huang K., Tu H., and Adler A. S. (2013) Monoclonal antibody humanness score and its applications. BMC Biotechnol. 13, 55 10.1186/1472-6750-13-55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Wollacott A. M., Xue C., Qin Q., Hua J., Bohnuud T., Viswanathan K., and Kolachalama V. B. (2019) Quantifying the nativeness of antibody sequences using long short-term memory networks. Protein Eng. Des. Sel. 32, 347–354 10.1093/protein/gzz031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Haberger M., Bomans K., Diepold K., Hook M., Gassner J., Schlothauer T., Zwick A., Spick C., Kepert J. F., Hienz B., Wiedmann M., Beck H., Metzger P., Mølhøj M., Knoblich C., et al. (2014) Assessment of chemical modifications of sites in the CDRs of recombinant antibodies. mAbs 6, 327–339 10.4161/mabs.27876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Xu Y., Roach W., Sun T., Jain T., Prinz B., Yu T. Y., Torrey J., Thomas J., Bobrowicz P., Vásquez M., Wittrup K. D., and Krauland E. (2013) Addressing polyspecificity of antibodies selected from an in vitro yeast presentation system: a FACS-based, high-throughput selection and analytical tool. Protein Eng. Des. Sel. 26, 663–670 10.1093/protein/gzt047 [DOI] [PubMed] [Google Scholar]
- 175. Sharma V. K., Patapoff T. W., Kabakoff B., Pai S., Hilario E., Zhang B., Li C., Borisov O., Kelley R. F., Chorny I., Zhou J. Z., Dill K. A., and Swartz T. E. (2014) In silico selection of therapeutic antibodies for development: viscosity, clearance, and chemical stability. Proc. Natl. Acad. Sci. U.S.A. 111, 18601–18606 10.1073/pnas.1421779112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Chennamsetty N., Voynov V., Kayser V., Helk B., and Trout B. L. (2009) Enhanced stability. Proc. Natl. Acad. Sci. U.S.A. 106, 11937–11942 10.1073/pnas.0904191106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Lauer T. M., Agrawal N. J., Chennamsetty N., Egodage K., Helk B., and Trout B. L. (2012) Developability index: a rapid in silico tool for the screening of antibody aggregation propensity. J. Pharm. Sci. 101, 102–115 10.1002/jps.22758 [DOI] [PubMed] [Google Scholar]
- 178. Jain T., Boland T., Lilov A., Burnina I., Brown M., Xu Y., and Vásquez M. (2017) Prediction of delayed retention of antibodies in hydrophobic interaction chromatography from sequence using machine learning. Bioinformatics 33, 3758–3766 10.1093/bioinformatics/btx519 [DOI] [PubMed] [Google Scholar]
- 179. Obrezanova O., Arnell A., de la Cuesta R. G., Berthelot M. E., Gallagher T. R., Zurdo J., and Stallwood Y. (2015) Aggregation risk prediction for antibodies and its application to biotherapeutic development. mAbs 7, 352–363 10.1080/19420862.2015.1007828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Datta-Mannan A., Thangaraju A., Leung D., Tang Y., Witcher D. R., Lu J., and Wroblewski V. J. (2015) Balancing charge in the complementarity-determining regions of humanized mAbs without affecting pl reduces non-specific binding and improves the pharmacokinetics. mAbs 7, 483–493 10.1080/19420862.2015.1016696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Popovic B., Gibson S., Senussi T., Carmen S., Kidd S., Slidel T., Strickland I., Xu J., Spooner J., Lewis A., Hudson N., Mackenzie L., Keen J., Kemp B., Hardman C., et al. (2017) Engineering the expression of an anti-interleukin-13 antibody through rational design and mutagenesis. Protein Eng. Des. Sel. 30, 303–311 10.1093/protein/gzx001 [DOI] [PubMed] [Google Scholar]
- 182. Yadav S., Laue T. M., Kalonia D. S., Singh S. N., and Shire S. J. (2012) The influence of charge distribution on self-association and viscosity behavior of monoclonal antibody solutions. Mol. Pharmaceut. 9, 791–802 10.1021/mp200566k [DOI] [PubMed] [Google Scholar]
- 183. Sydow J. F., Lipsmeier F., Larraillet V., Hilger M., Mautz B., Mølhøj M., Kuentzer J., Klostermann S., Schoch J., Voelger H. R., Regula J. T., Cramer P., Papadimitriou A., and Kettenberger H. (2014) Structure-based prediction of asparagine and aspartate degradation sites in antibody variable regions. PLoS ONE 9, e100736 10.1371/journal.pone.0100736 [DOI] [PMC free article] [PubMed] [Google Scholar]