

Abstract
CYP51 enzymes (sterol 14α-demethylases) are cytochromes P450 that catalyze multistep reactions. The CYP51 reaction occurs in all biological kingdoms and is essential in sterol biosynthesis. It removes the 14α-methyl group from cyclized sterol precursors by first forming an alcohol, then an aldehyde, and finally eliminating formic acid with the introduction of a Δ14–15 double bond in the sterol core. The first two steps are typical hydroxylations, mediated by an electrophilic compound I mechanism. The third step, C–C bond cleavage, has been proposed to involve either compound I (i.e. FeO3+) or, alternatively, a proton transfer-independent nucleophilic ferric peroxo anion (compound 0, i.e. Fe3+O2–). Here, using comparative crystallographic and biochemical analyses of WT human CYP51 (CYP51A1) and its D231A/H314A mutant, whose proton delivery network is destroyed (as evidenced in a 1.98-Å X-ray structure in complex with lanosterol), we demonstrate that deformylation of the 14α-carboxaldehyde intermediate requires an active proton relay network to drive the catalysis. These results indicate a unified, compound I-based mechanism for all three steps of the CYP51 reaction, as previously established for CYP11A1 and CYP19A1. We anticipate that our approach can be applied to mechanistic studies of other P450s that catalyze multistep reactions, such as C–C bond cleavage.
Keywords: cytochrome P450, enzyme mechanism, enzyme catalysis, X-ray crystallography, conformational change, C–C bond cleavage, compound I, CYP51, sterol 14α-demethylase, sterol biosynthesis, proton relay, cytochrome P450 mechanism, X-ray structure, deformylation, hydroxylation, structural biology, proton relay
Cytochrome P450 (CYP) enzymes are heme–thiolate coordinated monooxygenases that catalyze a vast variety of reactions involving xenobiotic and endogenous compounds. CYP51 (sterol 14α-demethylases) play a crucial role in the production of sterols in all phyla and are important drug targets (1–3). The CYP51 reaction includes three steps (Fig. 1) (i.e. three cytochrome P450 catalytic cycles [Fig. 2]); the first two steps are formal hydroxylations, and the third step is a C–C bond cleavage.
Figure 1.

The three steps of the CYP51 reaction. *, lanosterol is the natural CYP51 substrate in vertebrates and in some yeasts. The other CYP51 substrates are 24,25-dihydrolanosterol, 24-methylene-dihydrolanosterol, C4-norlanosterol, and obtusifoliol. **, 3β-hydroxy-4,4-dimethyl-cholesta-8,14,24-triene. Examples of other P450s catalyzing multistep reactions are shown in Fig. S1.
Figure 2.

The cytochrome P450 catalytic cycle. Binding of the substrate (1) displaces the water molecule from the sixth (distal) coordination site of the ferric heme iron, changing its spin state from hexa-coordinated low to penta-coordinated high, increasing its redox potential, and facilitating the first electron transfer from the protein redox partner (2). The ferrous iron complex binds molecular oxygen (3), which triggers acceptance of the second electron producing the nucleophilic ferric peroxo anion (4) (compound 0, or Fe3+–O2−). The ferric peroxo complex is then protonated to form the ferric hydroperoxo state (5), the second protonation of the distal oxygen atom causing the O-O bond scission, release of a water molecule, and generation of a highly reactive electrophilic Fe4+-oxo cation radical (FeO3+, or compound I) (6). The compound I mechanism requires two protons delivered from the protein surface to the iron-bound dioxygen via the active proton delivery network. In the compound 0 mechanism, the dioxygen abstracts protons from the substrate, forming a peroxo-hemiacetal intermediate and, thus, being independent of proton delivery.
Whereas it is now generally agreed that most P450 reactions use compound I (4) (step 6 in Fig. 2, FeO3+) as the active iron species, the C–C bond cleavage (lyase) mechanism in the multistep reactions is still under debate (5). Akhtar and his associates proposed that catalysis involves nucleophilic attack of a ferric peroxo anion (Fe3+O2–; compound 0, step 4 in Fig. 2) (6–10), but the reliability of the 18O isotope incorporation and MS data has been questioned because of technical concerns about ambient formic acid (and the difficulties in discerning 2HCO2H from endogenous H13CO2H) (5, 11). In more recent studies, a ferric peroxide mechanism has been supported by a kinetic solvent isotope effect (12) and resonance Raman spectroscopy (13, 14) for the substrate-induced lyase reaction of CYP17A1, although evidence for a compound I pathway has also been presented (15, 16). Compound I was identified as the active oxidant for the lyase step of CYP19A1 (steroid aromatase) using resonance Raman spectroscopy (17), kinetic solvent isotope effects (18), and 18O isotope labeling (11) and for CYP11A1 (P450scc) using electron paramagnetic resonance/electron nuclear double resonance spectroscopy (19), 18O isotope labeling (20), and resonance Raman spectroscopy (21). For the third step of the CYP51 reaction, theoretical studies have proposed the operation of a ferric peroxo anion (compound 0) (22), but definitive experimental evidence so far has been lacking.
A distinguishing difference between the two types of mechanisms is that the formation of compound I requires a specific proton relay network (to minimize the unwanted production of hydrogen peroxide via nonspecific proton donation to dioxygen in P450 enzymes) (23–26), whereas the ferric peroxo-anion abstracts hydrogens from substrates and does not depend on proton delivery (13). It has been generally accepted (going back to the 1980s [27–29]) that the proton delivery network involves a conserved P450 threonine in the I helix, a preceding charged residue, and a solvent-accessible residue(s) of opposite charge in the middle of helix F (3, 30, 31). The conserved threonine is Thr-315 in human CYP51 (also called CYP51A1) and corresponds to Thr-252 in P450cam (24). A preceding charged residue is always a His in CYP51 versus an acidic residue (Asp/Glu) in other P450s, and a solvent-accessible charged residue(s) in the helix F is always Asp/Glu in CYP51 versus Lys/Arg in other P450s.
In the ligand-free and inhibitor-bound CYP51 structures (which all display a very high overall similarity), this histidine and the carboxylate of an F helix acidic side chain are always salt bridged (e.g. His-294/Glu-205 in Trypanosoma cruzi and His-314/Asp-231 in human CYP51 orthologs). However, as we recently reported, binding of the substrate (obtusifoliol) to the T. cruzi CYP51 I105F mutant induces a large-scale conformational switch that causes the His-294/Glu-205 salt bridge opening, allowing the charged side chains to participate in proton delivery (32).
Mutation of each of the corresponding salt bridge-forming residues in human CYP51 strongly affects catalysis of lanosterol, with the double mutant (D231A/H314A) displaying a decrease in kcat of more than two orders of magnitude. There were no changes in the stability, spectral properties (absolute absorbance and CO complex formation upon chemical reduction), or apparent substrate binding affinity of the mutants, but the amplitude of the type I spectral response to lanosterol (low-to-high spin state transition in the heme iron) increased in the order WT < D231A < H314A < D231A/H314A (32). The D231A/H314A mutant is the first example of a CYP51 that, upon the addition of substrate, is found completely in the high-spin form (Fig. 3), although without structural evidence it has remained unclear whether it indeed forms a stoichiometric enzyme–substrate complex in solution or if the heme iron acquires the ability to exist in the high-spin (water-free pentacoordinated) form even without the substrate.
Figure 3.

The D231A/H314 mutation enhances spectrally detectable binding of substrate to human CYP51. A, absolute absorbance spectra before (blue) and after (black) titration with lanosterol. B, high-spin-form content in the protein samples (n = 2 ± S.D.).
In this report, we determined the 1.98-Å X-ray structure of the human CYP51 D231A/H314A mutant in complex with lanosterol and applied comparative crystallographic and biochemical analyses of this mutant and WT human enzymes to probe the CYP51 mechanism. We found that the activity of the mutant toward the 14α-aldehyde substrate is affected as much as its activity with lanosterol, leading to the conclusion that proton transfer is critical for the third step of the CYP51 reaction and compound I is the active oxidant.
Results and Discussion
Crystallographic analysis
The enzyme-substrate complex was crystallized in the orthorhombic C2221 space group, and the final structure was refined to 1.98-Å resolution with an Rfree of 22% and Rwork of 19% (Table 1). The atomic coordinates and structure factors have been deposited in the Protein Data Bank under accession code 6UEZ. The asymmetric unit consisted of two P450 molecules, each of them revealing a clear electron density for one molecule of lanosterol bound within the CYP51 active site (Fig. 4A and Fig. S2).
Table 1.
Data collection and refinement statistics
| Complex | D231A/H314A human CYP51, lanosterol |
|---|---|
| Data collection | |
| Space group | C2221 |
| Cell dimensions | |
| a, b, c (Å) | 91.180, 165.070, 154.030 |
| α, β, γ (°) | 90.00, 90.00, 90.00 |
| Wavelength, Å | 1.12713 |
| Resolution (outer shell), Å | 43.60–2.03 (2.03–1.98) |
| No. of molecules per asymmetric unit | 2 |
| Rmerge (outer shell) | 0.056 (0.934) |
| I/σ (outer shell) | 16.1 (1.1) |
| Completeness (outer shell), % | 89.6 (52.8) |
| Redundancy (outer shell) | 8.6 (5.7) |
| Refinement | |
| No. of reflections | |
| Total | 68,672 |
| Test set | 3792 |
| Rwork/Rfree | 0.191/0.222 |
| RMSD from ideal geometry | |
| Bond lengths, Å | 0.002 |
| Bond angles, ° | 1.27 |
| Ramachandran plot | |
| Residues in favorable/allowed regions, % | 98/100 |
| Outliers, % | 0 |
| Wilson B-factor | 45.1 |
| No. of atoms (mean B-factor, Å2) | 7790 (53.0) |
| No. of residues per molecule | A/B |
| Protein (mean B-factor, Å2) | 446/445 (58/55) |
| Heme (mean B-factor, Å2) | 1/1 (39/38) |
| Lanosterol (mean B-factor, Å2) | 1/1 (41/40) |
| Water (mean B-factor, Å2) | 225/290 (58) |
| PDB code | 6UEZ |
Figure 4.
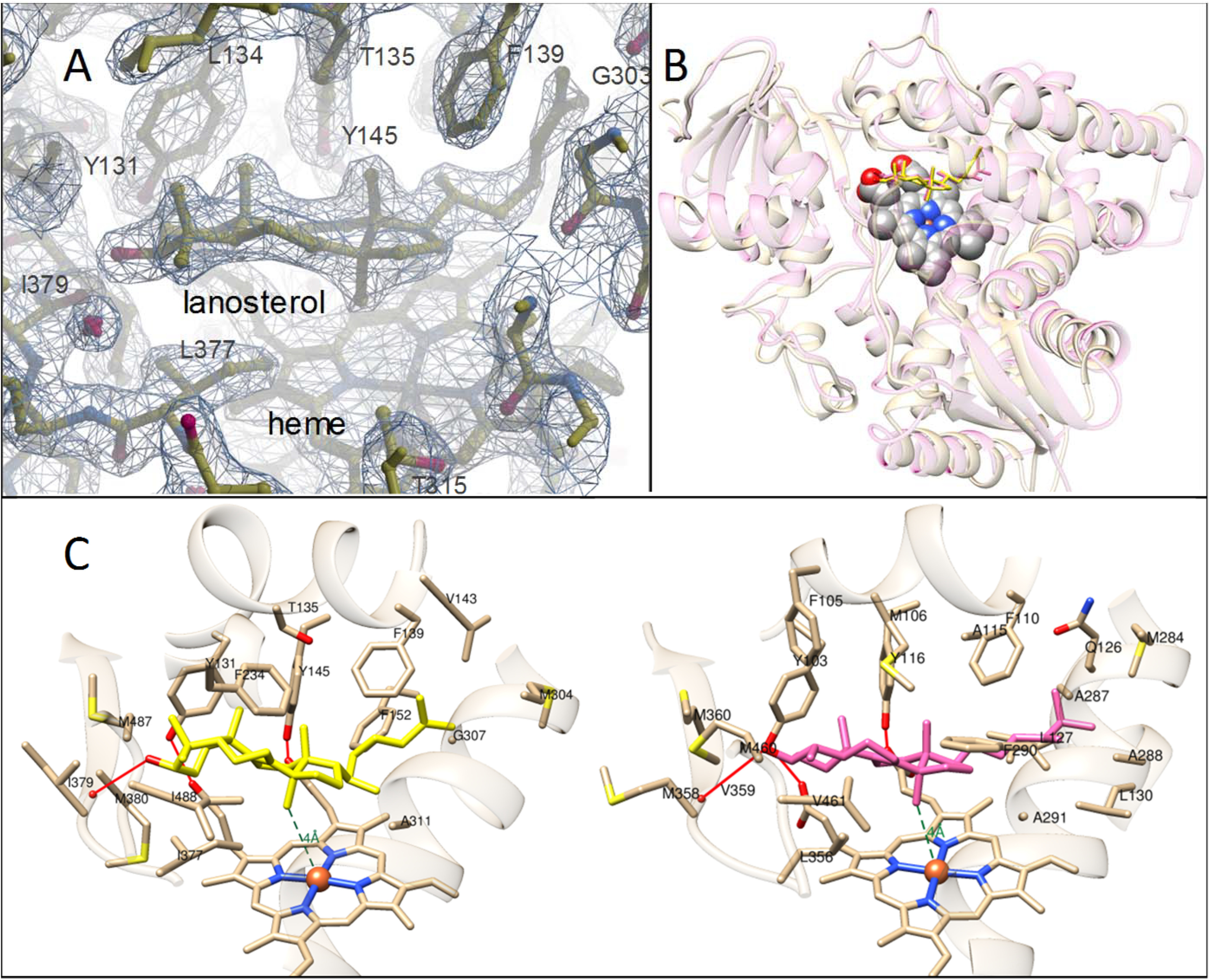
The D231A/H314 mutation does not affect the substrate binding mode in CYP51. A, the 2Fo-Fc electron density map (1.5 σ) for the active-site area of the D321A/H314A mutant of human CYP51. Lanosterol, heme, and some of the protein residues are marked. The snapshot was taken in Coot. B, superimposed structures of obtusifoliol-bound I105F T. cruzi CYP51 (pink, PDB code 6FMO) and lanosterol-bound D231A/H314A human CYP51 (yellow). Shown is the distal P450 face. The RMSD of Cα is 1.2 Å. C, substrate-contacting residues in human (left) and T. cruzi CYP51 (right). The active-site volumes are 1,200 Å3 and 1,300 Å3, respectively. The complete list of the residues located within 4.5 Å of the sterol molecules can be found in Table S1.
Substrate binding mode
The binding mode of lanosterol in the human structure was found to be essentially the same as the binding mode of obtusifoliol in the T. cruzi CYP51 I105F mutant (32) (Fig. 4, B and C). The C3-OH group of the sterol nucleus is directed toward the substrate access channel entrance approaching β-strand 1–4 and forming an H-bond with the main-chain oxygen of Ile-379 (corresponds to Met-358 in T. cruzi CYP51), the aliphatic arm occupies the deepest portion of the active site reaching helices C and I, and the C14α-methyl group is located 4 Å above the heme iron, indicative of a catalytically competent orientation.
Structural response to substrate binding
The active-site volumes of both protozoan and human substrate-bound CYP51 enzymes are very similar (1,200 and 1,300 Å3), and so is the large-scale conformational switch seen in the superimposed ligand-free/obtusifoliol-bound T. cruzi CYP51 (of the four P450 molecules in the asymmetric unit in that structure, three are obtusifoliol-bound and one is ligand-free) and inhibitor-bound/lanosterol-bound human CYP51 (Fig. 5). The rearrangements involve most of the structural segments of the CYP51 active site. The 5–6-Å inward movement of the HI arm and helix C changes the topology of the area to create a tighter interface with NADPH-CYP reductase (CPR), and the side-chain flip of Lys-156 out of the active site is analogous to that of Arg-124 in T. cruzi CYP51, the residue we have shown to directly participate in the electron transfer (32). In both, the H-bond with the heme propionate is lost and a positively charged group is now exposed above the proximal P450 surface (Fig. 5, upper, and Fig. S3). The 5–7-Å movement of the FG arm downstream of helix I (Fig. 5, lower, and Fig. 6) closes the entrance into the substrate access channel and increases the distance between the backbones of residues 231 and 314. In the human CYP51 mutant, where these two residues are now alanines, there are no charged side chains in the vicinity capable of enabling the transfer of protons from solvent to the iron-bound dioxygen required for the formation of compound I (formally Fe3+O).
Figure 5.

The substrate-induced conformational switch is the same in protozoan and human CYP51. Superimposed structures of obtusifoliol-bound (magenta) and ligand-free (blue) T. cruzi CYP51 (PDB code 6FMO), RMSD of Cα of 1.83 Å, maximal XYZ displacement of 7.55 Å (A), and lanosterol-bound (violet, PDB code 6UEZ) and VFV-bound (green, PDB code 4UHI) human CYP51, RMSD of Cα of 1.98 Å, maximal XYZ displacement of 9.44 Å (B). Middle, the overall distal P450 view. The arrows mark the areas of the molecules enlarged above (rearrangements for electron delivery) and below (rearrangements for proton delivery). The conserved salt bridges are shown as red dashes.
Figure 6.

Movement of the FG arm closes the substrate entry. Green, VFV-bound human CYP51 (PDB code 4UHI); violet, lanosterol-bound D231A/H314A human CYP51. A, ribbon representation. B, distal P450 face in surface representation. Four water molecules (green spheres), three within 4.5 Å of A231 and A314 and one that marks the location of the substrate entrance, are all positioned outside the protein globule. Similar to the structure of obtusifoliol-bound T. cruzi CYP51 (32), there are no open channels in the area. An enlarged fragment of the surface and a stick/ribbon representation of the corresponding protein segments in both lanosterol-bound human CYP51 molecules can be seen in Fig. S4.
Biochemical analysis
Electron transfer
Both WT human CYP51 and the D231A/H314A mutant were readily reduced chemically with sodium dithionite, regardless of the presence of the substrate, and the P450 concentrations determined from the difference spectra of the CO complexes corresponded to the concentrations calculated from the absolute absorbance spectra. The efficiency of enzymatic reduction with NADPH via CPR, however, was facilitated by the presence of the substrate, 100% in the mutant versus 65% in the WT sample, whereas without lanosterol the amount of detectable CO complexes did not exceed 15% in either case (Fig. 7A). These results support the conclusion that a crystallographically detectable substrate-induced large-scale conformational change does take place in solution, and the 35% lower enzymatic reduction of the WT sample is because of a lower lanosterol-bound fraction (33% versus 100% in the mutant).
Figure 7.
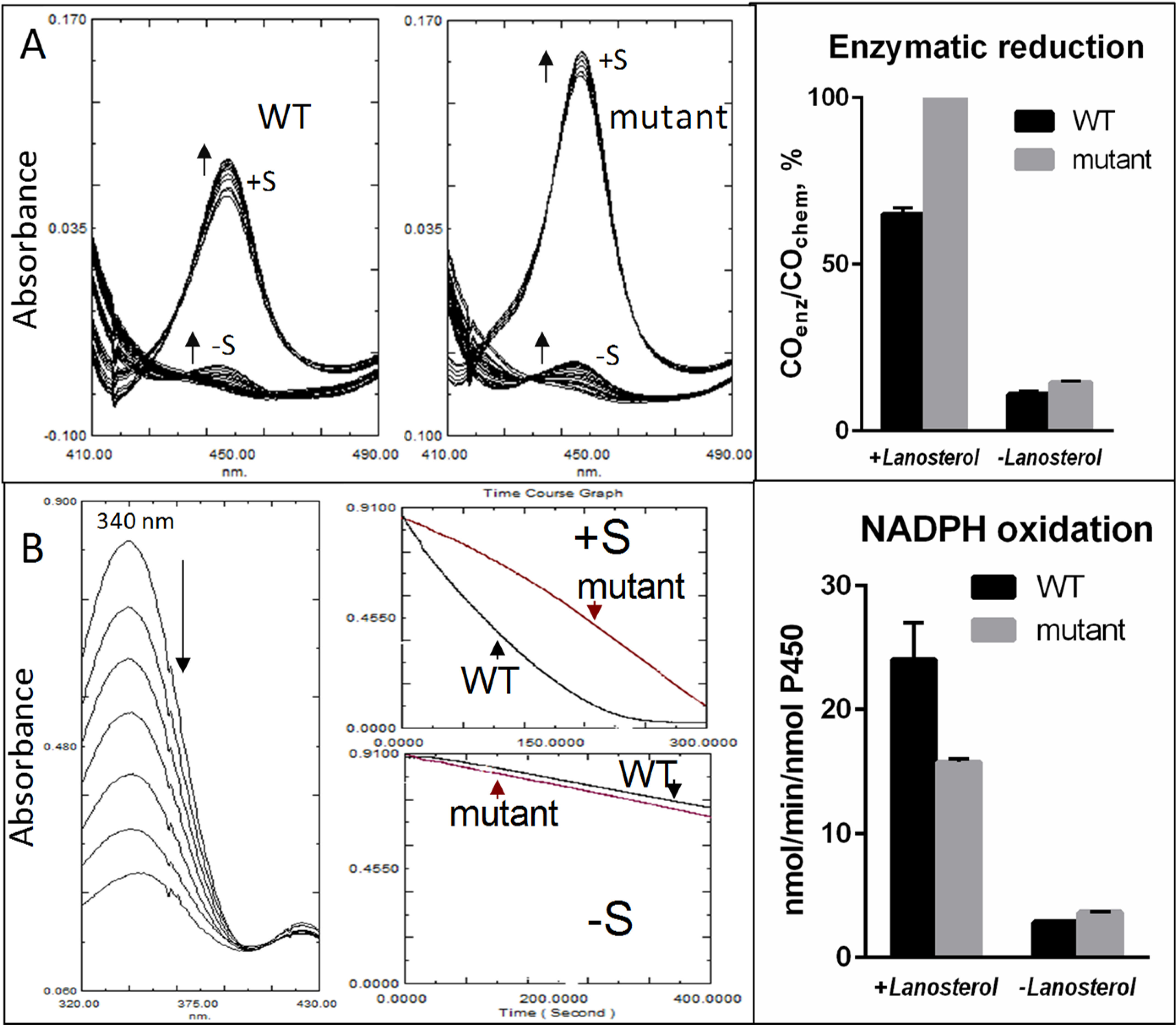
Electron transfer efficiency in the D231A/H314A mutant and WT human CYP51. A, enzymatic reduction with NADPH in the presence of CPR. Left, spectral changes introduced by binding of carbon monoxide to the ferrous CYP51. Right, efficiency of enzymatic reduction relative to the chemical reduction with sodium dithionite. B, NADPH oxidation. Left, decrease of the 340-nm absorbance upon NADPH consumption. Left, a sample of absorbance spectra; right, time course graphs. Right, rates of NADPH consumption. The mixture contained 2 μm CYP51, 4 μm CPR, 150 μm NADPH, and either 0 or 8 μm lanosterol (S); n = 2 ± S.D.
Slightly faster rates of NADPH oxidation by the WT enzyme (24 versus 16 min−1) (Fig. 7B) were observed, because WT CYP51 performed lanosterol 14α-demethylation while the mutant was catalytically inactive. In the absence of the substrate, the rates of NADPH consumption were comparable for WT CYP51 and the mutant, both one order of magnitude slower. Thus, the structural and biochemical data indicate that the D231A/H314A mutation did not affect (but rather enhanced) the ability of human CYP51 to bind substrate, interact with the electron donor protein CPR, and accept electrons. The lack of charged residues that enable the proton flow is the only observed reason for the dramatic loss of enzymatic activity.
Deformylation of lanosterol 14α-carboxaldehyde
The mutant provided a unique opportunity to elucidate the mechanism of the C–C bond cleavage because, if this step in CYP51 catalysis is performed by a nucleophilic peroxo oxidant, it should not require protons from the solvent, and the ability of the D231A/H314A mutant to catalyze deformylation of the 14α-carboxaldehyde intermediate of lanosterol (as opposed to its ability to hydroxylate lanosterol) should be comparable with or higher than that of the WT. If this reaction occurs via compound I, for which the formation transfer of protons from the solvent is critical (Fig. 2), the ability of the D231A/H314A mutant to catalyze deformylation of the 14α-carboxaldehyde intermediate should remain low.
We observed that catalytic turnover of the WT enzyme with the aldehyde intermediate as the substrate (kcat of 95 min−1) was twice as high as that with lanosterol (kcat of 46 min−1), which is expected in that the reaction occurs in one final step instead of three (Km of ∼5 μm in each case). The activity of the mutant was low, as in the case of lanosterol (kcat of 0.6 min−1) (Fig. 8A), revealing the same decreased reaction velocity, with Km values being too low to measure accurately (32) and longer reaction time required for detectable product to be formed. The data strongly suggest that all three steps of CYP51 catalysis have the same compound I-mediated mechanism.
Figure 8.
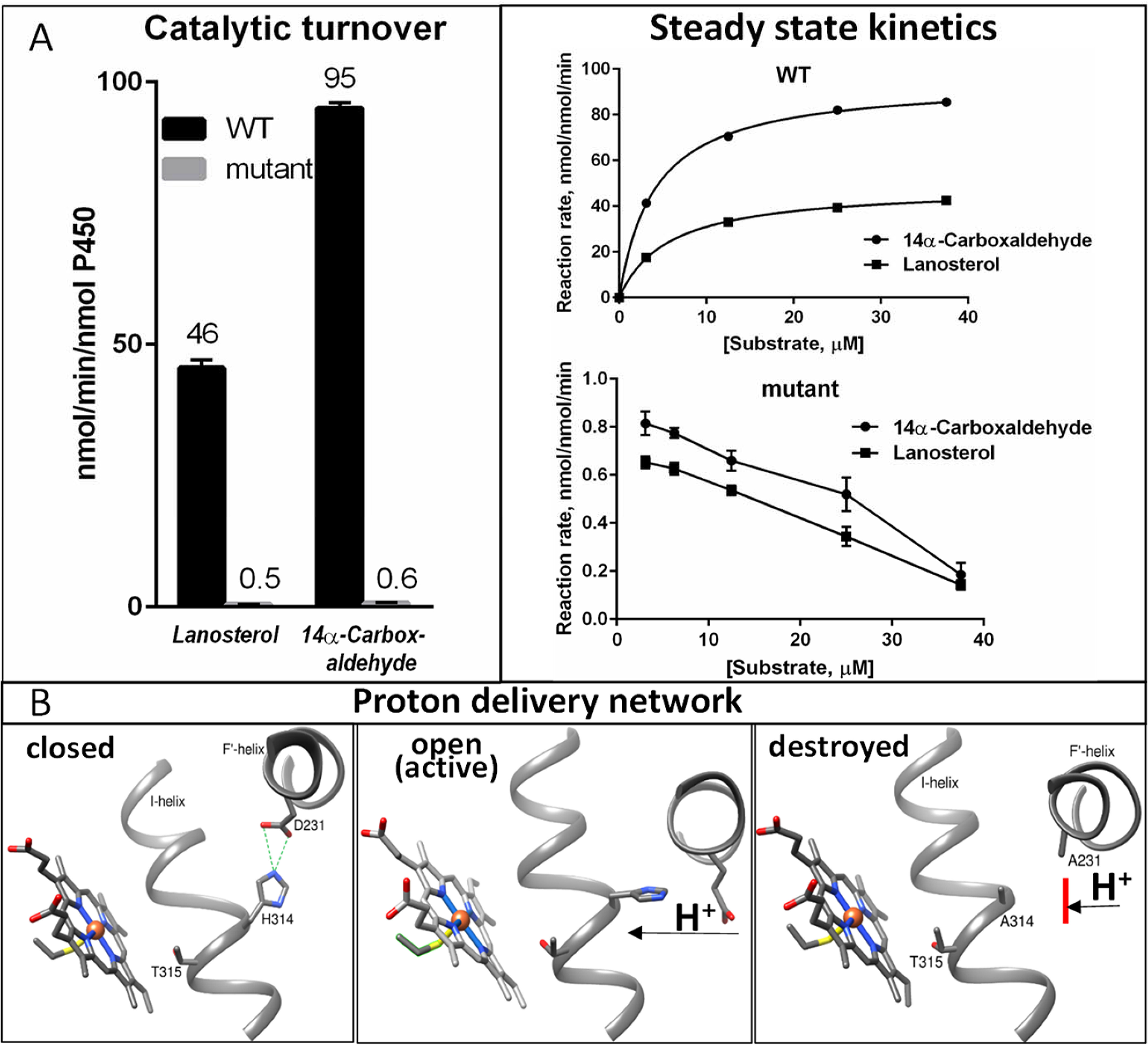
The third step of CYP51 reaction proceeds via the compound I mechanism. A, catalytic parameters of WT and D231A/H314A human CYP51 with lanosterol and its 14α-carboxaldehyde intermediate as substrates. Points are shown as means of two determinations ± S.D. Examples of HPLC profiles are shown in Fig. S6. B, proton relay network in CYP51 catalysis: closed in ligand-free and inhibitor-bound structures (4UHI), open (active) upon substrate binding (6FMO), and destroyed by mutagenesis (6UEZ).
It has been proposed that for a C–C bond cleavage reaction to proceed through the ferric peroxo oxidant instead of compound I, a P450 substrate must bear a hydroxyl group juxtaposed to the Fe-O-O– fragment, which was found to be the case for CYP17A1/17OH-pregnenolone but not for CYP19A1 (14, 17). This is also not the case for CYP51, as in the CYP51 substrates the only OH group (at the C3 carbon) is located 8 Å from the C14α-aldehyde group and H-bonded with the main chain carbonyl oxygen in the β 1-4 strand (Fig. 4C). Thus, the current evidence supports compound I mechanisms in three of the four major steroid C–C bond cleavage reactions (Fig. 1 and Fig. S1).
It should be noted that in CYP17A1, some of the evidence suggests the interaction of the substrate hydroxyl group with compound I (5, 16). However, in the case of CYP17A1, the 18O labeling work is not unambiguous in discerning between the two pathways (5, 15), and the reported spectral intermediates have not been shown to be catalytically competent (5, 13, 14, 17), so more experimental work on this enzyme is in order. Overall, the number of P450 reactions that can be attributed to compound 0 ferric peroxo anion C–C bond cleavage and other types of transformation has decreased (5, 33–35), although the possibility exists that some facile reactions do involve this species.
Conclusions
To clarify whether the third step of CYP51 catalysis can proceed through a proton-independent ferric peroxo anion-mediated mechanism, we applied a combined crystallographic and biochemical approach. The work was conceived based on our previous findings that substrate binding induces a large-scale conformational switch in the structure of CYP51 from the protozoan pathogen T. cruzi (32). Among the changes was a 4–6-Å movement of the FG arm that led to the opening of the His-294/Glu-205 salt bridge, a process expected for the activation of the CYP51 proton delivery network.
Mutation of the corresponding residues in human CYP51 (His-314 and Asp-231) to alanine, without changing the apparent lanosterol binding affinity, caused a decrease of two orders of magnitude in the rate of lanosterol 14α-demethylation yet increased the amplitude of the mutant spectral response to lanosterol binding. Cocrystallization of the mutant with lanosterol produced a 1.98-Å structure of the enzyme-substrate complex, providing convincing evidence that the large conformational switch accompanying substrate binding must be general for CYP51 across phylogeny, physiologically relevant, and required for catalysis.
The D231A/H314A mutation did not cause any apparent disturbances in substrate binding (including the spectral Kd values), protein folding, stability, conformational dynamics, or the ability to accept electrons from NADPH via its electron donor partner CPR, and the disruption of the proton delivery network (Fig. 8B) is judged to be the only observable reason for the loss of catalytic activity. Because proton delivery is a critical step in the activation of iron-linked molecular oxygen, required for splitting of the O–O bond and formation of compound I, the finding that the mutant catalytic rates are dramatically affected both for lanosterol (hydroxylation, the first step of the three step reaction) and for the 14α-carboxaldehyde intermediate (C–C bond cleavage, the third step [a proposed reaction scheme is shown in Fig. 9]) strongly supports the view that CYP51 enzymes follow the same compound I-mediated mechanism during all three steps of the reaction.
Figure 9.

Proposed compound I-mediated mechanism for the third step of CYP51 reaction (adapted from reference 5).
On the basis of our work here and previous studies with CYP11A1 and CYP19A1, we conclude that the C–C bond cleavage mechanisms involve compound I in at least three of the four major steroid P450s that perform multistep reactions The approach we used should be applicable in distinguishing between Fe3+-O2− and FeO3+ as catalytically active species for other P450 cytochromes that catalyze multistep reactions.
Experimental procedures
Protein expression and purification
All proteins were expressed in Escherichia coli (HMS-174, Novagen) in a pCW expression vector. The expression and purification of human CYP51 (full-length WT and the D231A/H314A mutant) was performed as described previously (32). For crystallization purposes, the 50-amino-acid membrane anchor sequence at the N terminus of D231A/H314A was replaced with an MAKKTSSKGKL fragment (36). The expression and purification of rat NADPH-cytochrome P450 reductase (CPR) and T. brucei CYP51 and CPR were performed as described previously (37, 38).
Spectroscopic characterization
UV-visible spectra were recorded at room temperature (23 °C) using a dual-beam Shimadzu UV-240IPC spectrophotometer. P450 concentrations were determined from the Soret band absorbance in the absolute spectrum, using an absolute molar extinction coefficient (ε417) of 117 mm−1 cm−1 for the low-spin oxidized form of the protein or a difference molar extinction coefficient (Δε446–490) of 91 mm−1 cm−1 for the reduced carbon monoxide complex in the difference spectra (Fe2+-CO versus Fe2+). The spin states of P450 samples were estimated from the absolute spectra as the ΔA393–470/ΔA418–470 ratio, with values of 0.4 and 2.1 corresponding to 100% low- and 100% high-spin iron, respectively (36, 39). NADPH oxidation rates were monitored at 340 nm (Δε340, 6.22 mm−1 cm−1).
Crystallization, structure determination, and analysis
The human CYP51 D231A/H314A mutant [5 μm in 20 mm potassium phosphate buffer (pH 7.4) containing 500 mm NaCl, 5.6 mm tris(2-carboxyethyl)phosphine, and 10% (v/v) glycerol] was gradually saturated with lanosterol (using a 1 mm stock solution in 45% [w/v] 2-hydroxypropyl-β-cyclodextrin [HPCD]), incubated for 20 min at room temperature, centrifuged to remove the precipitate, concentrated using an Amicon Ultra 50K (Millipore) to 500 μm, diluted 2-fold with 5 mm phosphate buffer (pH 7.4), and mixed with 23 mm cyclohexylpentanoyl-N-hydroxyethylglucamide (Ana-trace). Crystals were obtained at 26 °C by vapor diffusion in hanging drops using 2 μl protein mixture and 2 μl of mother liquid consisting of 100 mm calcium acetate (pH 7.3), 18% (w/v) PEG 3350, and 0.1 mm EDTA. The crystals were transferred in three steps of increasing glycerol concentration to a cryoprotectant solution containing 25% (v/v) glycerol in mother liquor and flash-cooled in liquid nitrogen. X-ray diffraction data were collected from a single crystal at the 21-ID-D beamline at the Advanced Photon Source, Argonne National Laboratory, at a wavelength of 1.12713 Å and using a Dectris Eiger 9M detector. The diffraction images were processed with HKL-2000, and the initial electron density map was obtained by molecular replacement using the coordinates of the VFV-bound human CYP51 (PDB entry 4UHI) as a search model in Phaser MR (CCP4 Program Suite [40]). The structure was built with Coot (41) and refined with Refmac5 (CCP4 Suite). Details of the data collection and refinement statistics are listed in Table 1. Structure superimposition and root mean square deviation (RMSD) calculation were performed in lsqcab (CCP4 Suite). Active-site volumes were calculated in BioVia Discovery Studio Visualizer 2019. Molecular graphics were rendered using Chimera.
Preparation of lanosterol 14α-carboxaldehyde
The 14α-carboxaldehyde intermediate of lanosterol (i.e. the substrate for the third step of the CYP51 reaction, the C–C bond cleavage) was prepared using a feature of T. brucei CYP51, i.e. that of producing a large amount of this intermediate if lanosterol is used as the substrate (38). The reaction mixture contained 10 μm T. brucei CYP51, 5 μm T. brucei CPR, and 50 μm lanosterol (for this purpose, 0.5 mm solutions of unlabeled and [3-3H]-labeled lanosterol [∼4000 dpm/nmol] were mixed in 45% HPCD at a 10:1 molar ratio). The radiolabeled lanosterol was added to be able to quantify the amount of aldehyde by its peak area. The reaction was started by the addition of 1 mm NADPH, conducted at 37 °C for 2 h, and the 14α-carboxaldehyde intermediate was separated by reverse-phase HPLC and collected as described previously (38), except that the time of the linear gradient (vide infra) was extended to 40 min (Fig. S5). The aldehyde fraction was dried under a nitrogen stream, dissolved in 45% (w/v) HPCD, and used as a substrate for comparative analysis of WT and D231A/H314A human CYP51 (Fig. S6).
Reconstitution of human CYP51 catalytic activity
The standard reaction mixture (500 µl) contained 0.25 μm human CYP51, 1.0 μm CPR, 100 μm l-α-1,2-dilauroyl-sn-glycero-3-phosphocholine, 0.4 mg/ml isocitrate dehydrogenase, and 25 mm sodium isocitrate in 50 mm potassium phosphate buffer (pH 7.2) containing 10% (v/v) glycerol (36). After addition of the radiolabeled [3-3H]-lanosterol (∼4,000 dpm/nmol) or lanosterol 14α-carboxaldehyde (∼400 dpm/nmol), the mixture was preincubated for 30 s at 37 °C in a shaking water bath, and the reaction was initiated by the addition of 100 μm NADPH and stopped by extraction of the sterols with 5 ml of ethyl acetate. The extracted sterols were dried, dissolved in CH3OH, and analyzed by a reverse-phase HPLC system (Waters) equipped with a β-RAM detector (INUS Systems) using a NovaPak octadecylsilane (C18) column (particle size, 4 μm, 3.9 mm × 150 mm) and a linear gradient from H2O:CH3CN:CH3OH (1.0:4.5:4.5, v/v/v) (solvent A) to CH3OH (solvent B), increasing from 0 to 100% B for 30 min at a flow rate of 1.0 ml/min. For steady-state kinetic analysis, the reactions wererun for 1 min (WT) and 30 min (D231A/H314A) at 37 °C, and the sterol concentration range was 3.1–38 μm. Michaelis-Menten parameters were calculated using hyperbolic fitting in Prism v.6 (GraphPad, La Jolla, CA), with the reaction rates (nmol product formed/nmol P450/min) plotted versus total substrate concentration.
Data availability
The atomic coordinates and structure factors (code 6UEZ) have been deposited in the Protein Data Bank (http://wwpdb.org/), and all other data are contained within the manuscript and the supporting information.
Supplementary Material
Acknowledgments
Vanderbilt University is a member institution of the Life Sciences Collaborative Access Team at Sector 21 of the Advanced Photon Source (Argonne, IL). Use of the Advanced Photon Source at Argonne National Laboratory was supported by the United States Department of Energy, Office of Science, Office of Basic Energy Sciences, under contract DE-AC02-06CH11357.
This article contains supporting information.
Author contributions—T. Y. H., F. P. G., and G. I. L. conceptualization; T. Y. H. and G. I. L. data curation; T. Y. H., Z. W., F. P. G., and G. I. L. formal analysis; T. Y. H. and G. I. L. supervision; T. Y. H. and G. I. L. funding acquisition; T. Y. H., F. P. G., and G. I. L. validation; T. Y. H., Z. W., F. P. G., and G. I. L. investigation; T. Y. H. and G. I. L. visualization; T. Y. H., F. P. G., and G. I. L. methodology; T. Y. H. and G. I. L. writing-original draft; T. Y. H. and G. I. L. project administration; T. Y. H., F. P. G., and G. I. L. writing-review and editing.
Funding and additional information—The study was supported by National Institutes of Health grants R01 GM067871 (G. I. L.) and R01 GM118122 (F. P. G.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- CYP or P450
- cytochrome P450
- CYP51
- sterol 14α-demethylase
- CPR
- NADPH-cytochrome P450 reductase
- RMSD
- root mean square deviation
- HPCD
- 2-hydroxypropyl-β-cyclodextrin
- DLPC
- l-α-1,2-dilauroyl-sn-glycero-3-phosphocholine.
References
- 1. Lepesheva G. I., Friggeri L., and Waterman M. R. (2018) CYP51 as drug targets for fungi and protozoan parasites: past, present and future. Parasitology 145, 1820–1836 10.1017/S0031182018000562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Friggeri L., Hargrove T. Y., Wawrzak Z., Guengerich F. P., and Lepesheva G. I. (2019) Validation of human sterol 14α-demethylase (CYP51) druggability: structure-guided design, synthesis, and evaluation of stoichiometric, functionally irreversible inhibitors. J. Med. Chem. 62, 10391–11040 10.1021/acs.jmedchem.9b01485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lepesheva G. I., and Waterman M. R. (2007) Sterol 14α-demethylase cytochrome P450 (CYP51), a P450 in all biological kingdoms. Biochim. Biophys. Acta 1770, 467–477 10.1016/j.bbagen.2006.07.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rittle J., and Green M. T. (2010) Cytochrome P450 compound I: capture, characterization, and C-H bond activation kinetics. Science 330, 933–937 10.1126/science.1193478 [DOI] [PubMed] [Google Scholar]
- 5. Guengerich F. P., and Yoshimoto F. K. (2018) Formation and cleavage of C-C bonds by enzymatic oxidation-reduction reactions. Chem. Rev. 118, 6573–6665 10.1021/acs.chemrev.8b00031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Akhtar M., Corina D., Miller S., Shyadehi A. Z., and Wright J. N. (1994) Mechanism of the acyl-carbon cleavage and related reactions catalyzed by multifunctional P-450s: studies on cytochrome P-450(17)α. Biochemistry 33, 4410–4418 10.1021/bi00180a039 [DOI] [PubMed] [Google Scholar]
- 7. Caspi E., Arunachalam T., and Nelson P. A. (1986) Biosynthesis of estrogens: aromatization of (19R)-, (19S)-, and (19S)-[19-3H,2H,1H]-3β-hydroxyandrost-5-en-17-ones by human placental aromatase. J. Am. Chem. Soc. 108, 1847–1852 10.1021/ja00268a023 [DOI] [Google Scholar]
- 8. Akhtar M., Calder M. R., Corina D. L., and Wright J. N. (1982) Mechanistic studies on C-19 demethylation in oestrogen biosynthesis. Biochem. J. 201, 569–580 10.1042/bj2010569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shyadehi A. Z., Lamb D. C., Kelly S. L., Kelly D. E., Schunck W.-H., Wright J. N., Corina D., and Akhtar M. (1996) The mechanism of the acyl-carbon bond cleavage reaction catalyzed by recombinant sterol 14α-demethylase of Candida albicans (other names are: lanosterol 14α-demethylase, P-45014DM, and CYP51). J. Biol. Chem. 271, 12445–12450 10.1074/jbc.271.21.12445 [DOI] [PubMed] [Google Scholar]
- 10. Akhtar M., Corina D., Pratt J., and Smith T. (1976) Studies on the removal of C-19 in oesterogen biosynthesis using 18O2. J. Chem. Soc. Chem. Commun. 21, 854–856 10.1039/c39760000854 [DOI] [Google Scholar]
- 11. Yoshimoto F. K., and Guengerich F. P. (2014) Mechanism of the third oxidative step in the conversion of androgens to estrogens by cytochrome P450 19A1 steroid aromatase. J. Am. Chem. Soc. 136, 15016–15025 10.1021/ja508185d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gregory M. C., Denisov I. G., Grinkova Y. V., Khatri Y., and Sligar S. G. (2013) Kinetic solvent isotope effect in human P450 CYP17A1-mediated androgen formation: evidence for a reactive peroxoanion intermediate. J. Am. Chem. Soc. 135, 16245–16247 10.1021/ja4086403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mak P. J., Gregory M. C., Denisov I. G., Sligar S. G., and Kincaid J. R. (2015) Unveiling the crucial intermediates in androgen production. Proc. Natl. Acad. Sci. U S A 112, 15856–15861 10.1073/pnas.1519376113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mak P. J., Duggal R., Denisov I. G., Gregory M. C., Sligar S. G., and Kincaid J. R. (2018) Human cytochrome CYP17A1: the structural basis for compromised lyase activity with 17-hydroxyprogesterone. J. Am. Chem. Soc. 140, 7324–7331 10.1021/jacs.8b03901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yoshimoto F. K., Gonzalez E., Auchus R. J., and Guengerich F. P. (2016) Mechanism of 17α,20-lyase and new hydroxylation reactions of human cytochrome P450 17A1: 18O labelling and oxygen surrogate evidance for a role of a perferryl oxygen. J. Biol. Chem. 291, 17143–17164 10.1074/jbc.M116.732966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gonzalez E., Johnson K. M., Pallan P. S., Phan T. T. N., Zhang W., Lei L., Wawrzak Z., Yoshimoto F. K., Egli M., and Guengerich F. P. (2018) Inherent steroid 17α,20-lyase activity in defunct cytochrome P450 17A enzymes. J. Biol. Chem. 293, 541–556 10.1074/jbc.RA117.000504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mak P. J., Luthra A., Sligar S. G., and Kincaid J. R. (2014) Resonance Raman spectroscopy of the oxygenated intermediates of human CYP19A1 implicates a compound I intermediate in the final lyase sep. J. Am. Chem. Soc. 136, 4825–4828 10.1021/ja500054c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Khatri Y., Luthra A., Duggal R., and Sligar S. G. (2014) Kinetic solvent isotope effect in steady-state turnover by CYP19A1 suggests involvement of compound I for both hydroxylation and aromatization steps. FEBS Lett. 588, 3117–3122 10.1016/j.febslet.2014.06.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Davydov R., Strushkevich N., Smil D., Yantsevich A., Gilep A., Usanov S., and Hoffman B. M. (2015) Evidence that compound I is the active species in both the hydroxylase and lyase steps by which P450scc converts cholesterol to pregnenolone: EPR/ENDOR/cryoreduction/annealing studies. Biochemistry 54, 7089–7097 10.1021/acs.biochem.5b00903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yoshimoto F. K., Jung I. J., Goyal S., Gonzalez E., and Guengerich F. P. (2016) Isotope-labeling studies support the electrophilic compound I iron active species, FeO3+, for the carbon-carbon bond cleavage reaction of the cholesterol side-chain cleavage enzyme, cytochrome P450 11A1. J. Am. Chem. Soc. 138, 12124–12141 10.1021/jacs.6b04437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhu Q., Mak P. J., Tuckey R. C., and Kincaid J. R. (2017) Active site structures of CYP11A1 in the presence of its physiological substrates and alterations upon binding of adrenodoxin. Biochemistry 56, 5786–5797 10.1021/acs.biochem.7b00766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sen K., and Hackett J. C. (2010) Peroxo-iron mediated deformylation in sterol 14α-demethylase catalysis. J. Am. Chem. Soc. 132, 10293–11030 10.1021/ja906192b [DOI] [PubMed] [Google Scholar]
- 23. Denisov I. G., Makris T. M., Sligar S. G., and Schlichting I. (2005) Structure and chemistry of cytochrome P450. Chem. Rev. 105, 2253–2277 10.1021/cr0307143 [DOI] [PubMed] [Google Scholar]
- 24. Tripathi S., Li H., and Poulos T. L. (2013) Structural basis for effector control and redox partner recognition in cytochrome P450. Science 340, 1227–1230 10.1126/science.1235797 [DOI] [PubMed] [Google Scholar]
- 25. Vidakovic M., Sligar S. G., Li H., and Poulos T. L. (1998) Understanding the role of the essential asp251 in cytochrome P450cam using site-directed mutagenesis, crystallography, and kinetic solvent isotope effect. Biochemistry 37, 9211–9219 10.1021/bi980189f [DOI] [PubMed] [Google Scholar]
- 26. Follmer A. H., Tripathi S., and Poulos T. L. (2019) Ligand and redox partner binding generates a new conformational state in cytochrome P450cam (CYP101A1). J. Am. Chem. Soc. 141, 2678–2683 10.1021/jacs.8b13079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Martinis S. A., Atkins W. M., Stayton P. S., and Sligar S. G. (1989) A conserved residue of cytochrome P-450 is involved in heme-oxygen stability and activation. J. Am. Chem. Soc. 111, 9252–9253 10.1021/ja00208a031 [DOI] [Google Scholar]
- 28. Vaz A. D. N., Pernecky S. J., Raner G. M., and Coon M. J. (1996) Peroxo-iron and oxenoid-iron species as alternative oxygenating agents in cytochrome P450-catalyzed reactions: switching by threonine-302 to alanine mutagenesis of cytochrome P450 2B4. Proc. Natl. Acad. Sci. U. S. A. 93, 4644–4648 10.1073/pnas.93.10.4644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Imai M., Shimada H., Watanabe Y., Matsushima-Hibiya Y., Makino R., Koga H., Horiuchi T., and Ishimura Y. (1989) Uncoupling of the cytochrome P-450cam monooxygenase reaction by a single mutation, threonine-252 to alanine or valine: a possible role of the hydroxy amino acid in oxygen activation. Proc. Natl. Acad. Sci. U S A 86, 7823–7827 10.1073/pnas.86.20.7823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lepesheva G. I., and Waterman M. R. (2011) Structural basis for conservation in the CYP51 family. Biochim. Biophys. Acta 1814, 88–93 10.1016/j.bbapap.2010.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sen K., and Hackett J. C. (2009) Molecular oxygen activation and proton transfer mechanisms in lanosterol 14α-demethylase catalysis. J. Phys. Chem. B 113, 8170–8182 10.1021/jp902932p [DOI] [PubMed] [Google Scholar]
- 32. Hargrove T. Y., Wawrzak Z., Fisher P. M., Child S. A., Nes W. D., Guengerich F. P., Waterman M. R., and Lepesheva G. I. (2018) Binding of a physiological substrate causes large-scale conformational reorganization in cytochrome P450 51. J. Biol. Chem. 293, 19344–19353 10.1074/jbc.RA118.005850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ogliaro F., de Visser S. P., Cohen S., Sharma P. K., and Shaik S. (2002) Searching for the second oxidant in the catalytic cycle of cytochrome P450: a theoretical investigation of the iron(III)-hydroperoxo species and its epoxidation pathways. J. Am. Chem. Soc. 124, 2806–2817 10.1021/ja0171963 [DOI] [PubMed] [Google Scholar]
- 34. Wang J. B., Huang Q., Peng W., Wu P., Yu D., Chen B., Wang B., and Reetz M. T. (2020) P450-BM3-catalyzed sulfoxidation versus hydroxylation: a common or two different catalytically active species? J. Am. Chem. Soc. 142, 2068–2073 10.1021/jacs.9b13061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Newcomb M., Aebisher D., Shen R., Chandrasena R. E., Hollenberg P. F., and Coon M. J. (2003) Kinetic isotope effects implicate two electrophilic oxidants in cytochrome P450-catalyzed hydroxylations. J. Am. Chem. Soc. 125, 6064–6065 10.1021/ja0343858 [DOI] [PubMed] [Google Scholar]
- 36. Hargrove T. Y., Friggeri L., Wawrzak Z., Sivakumaran S., Yazlovitskaya E. M., Hiebert S. W., Guengerich F. P., Waterman M. R., and Lepesheva G. I. (2016) Human sterol 14α-demethylase as a target for anticancer chemotherapy: towards structure-aided drug design. J. Lipid Res. 57, 1552–1563 10.1194/jlr.M069229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hanna I. H., Teiber J. F., Kokones K. L., and Hollenberg P. F. (1998) Role of the alanine at position 363 of cytochrome P450 2B2 in influencing the NADPH- and hydroperoxide-supported activities. Arch. Biochem. Biophys. 350, 324–332 10.1006/abbi.1997.0534 [DOI] [PubMed] [Google Scholar]
- 38. Lepesheva G. I., Nes W. D., Zhou W., Hill G. C., and Waterman M. R. (2004) CYP51 from Trypanosoma brucei is obtusifoliol-specific. Biochemistry 43, 10789–10799 10.1021/bi048967t [DOI] [PubMed] [Google Scholar]
- 39. Lepesheva G. I., Strushkevich N. V., and Usanov S. A. (1999) Conformational dynamics and molecular interaction reactions of recombinant cytochrome P450scc (CYP11A1) detected by fluorescence energy transfer. Biochim. Biophys. Acta 1434, 31–43 10.1016/S0167-4838(99)00156-9 [DOI] [PubMed] [Google Scholar]
- 40. Potterton E., Briggs P., Turkenburg M., and Dodson E. (2003) A graphical user interface to the CCP4 program suite. Acta Crystallogr. D 59, 1131–1137 10.1107/s0907444903008126 [DOI] [PubMed] [Google Scholar]
- 41. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The atomic coordinates and structure factors (code 6UEZ) have been deposited in the Protein Data Bank (http://wwpdb.org/), and all other data are contained within the manuscript and the supporting information.