

Abstract
Several turn-on RNA aptamers that activate small molecule fluorophores have been selected in vitro. Among these, the ~30 nucleotide Mango-III is notable because it binds the thiazole orange derivative TO1-Biotin with high affinity and fluoresces brightly (quantum yield 0.55). Uniquely among related aptamers, Mango-III exhibits biphasic thermal melting, characteristic of molecules with tertiary structure. We report crystal structures of TO1-Biotin complexes of Mango-III, a structure-guided mutant Mango-III(A10U), and a functionally reselected mutant iMango-III. The structures reveal a globular architecture arising from an unprecedented pseudoknot-like connectivity between a G-quadruplex and an embedded non-canonical duplex. The fluorophore is restrained into a planar conformation by the G-quadruplex, a lone, long-range trans-Watson-Crick pair (whose A10U mutation increases quantum yield to 0.66), and a pyrimidine perpendicular to the nucleobase planes of those motifs. The improved iMango-III and Mango-III(A10U) fluoresce ~50% brighter than enhanced green fluorescent protein, making them suitable tags for live cell RNA visualization.
Introduction
Several RNA aptamers that strongly induce the fluorescence of their cognate small molecule fluorophores have recently been selected in vitro and used as molecular tags to image RNAs in vivo1–5. Such aptamers have the potential to transform the study of cellular RNAs, analogous to how fluorescent proteins revolutionized the study of the proteome6. For imaging applications, two key properties are the affinity with which the RNAs bind their cognate fluorophores, and the magnitude of fluorescence enhancement resulting from RNA binding. An in vitro selection experiment that explicitly sought3 to isolate RNA aptamers that bind with high affinity to the thiazole orange (TO) derivative TO1-Biotin yielded ‘RNA Mango’. This ~30 nucleotide (nt) aptamer binds TO1-Biotin, and the related TO3-Biotin (Supplementary Fig. 1) with high affinity (dissociation constant, Kd, of 3.0 and 8.0 nM, respectively). Crystallographic structure determination of the RNA Mango-TO1-Biotin complex revealed an RNA comprised of a three-tiered G-quadruplex linked to an A-form duplex through a flexible junction7. Consistent with the modest quantum yield (0.14) of this complex, the structure revealed that the methylquinoline (MQ) and benzothiazole (BzT) heterocycles of TO1-Biotin bind making a 45° angle relative to each other. Since a planar conformation should improve quantum yield8,9, it was hypothesized that RNA Mango variants may exist that induce brighter fluorescence from the same fluorophores10.
To discover brighter variants that also bind to TO1-Biotin with high affinity, the final pool from which RNA Mango (hereafter, Mango-I) was originally selected was subjected to a microfluidic, droplet-based functional selection, in which sequence variants were explicitly enriched for both brighter fluorescence and high affinity5. Three aptamers, Mango-II, Mango-III, and Mango-IV were isolated from these experiments and their properties characterized in vitro and in vivo. Of these, Mango-III is noteworthy for having the most intense fluorescence when bound to TO1-Biotin, being 40% brighter than enhanced green fluorescent protein (EGFP). This fluorescent tag may already be sufficiently bright for in vivo imaging techniques11.
To elucidate the molecular basis for the induction of bright fluorescence by Mango-III, and as a starting point for structure-guided optimization of this tool for live-cell imaging of RNAs, we now determined the co-crystal structure of Mango-III. Structure-guided mutation produced an 18% brighter mutant, Mango-III(A10U), and functional reselection produced iMango-III, a variant 13% brighter than Mango-III. Structures of these three RNAs revealed an unusually complex molecular architecture for a small RNA, including a pseudoknot-like base pairing interaction between nucleotides internal and adjacent to a two-tiered G-quadruplex. Mutational, photophysical, and reselection experiments demonstrated that nucleotides distant from the fluorophore were found to modulate properties of the aptamer-fluorophore complex.
Results
Overall Structure of Mango-III bound to TO1-Biotin
A 38-nt construct comprising the conserved 28-nt Mango-III core flanked by nucleotides presumed to form a 5-base pair Watson-Crick duplex was co-crystalized with TO1-Biotin. The structure was solved by the single-wavelength anomalous dispersion (SAD) method using data from an iridium derivative (Supplementary Table 1, Methods). The crystallographic asymmetric unit (A.U.) contains two similar RNA-fluorophore complexes (RMSD = 0.15 Å for all non-hydrogen atoms). The two RNAs in the AU each make three crystal contacts involving end-to-end stacking of duplexes (Supplementary Fig. 2). Analytical ultracentrifugation and dynamic light scattering (Supplementary Fig. 3) however, indicate that Mango-III is monomeric in solution.
The structure of Mango-III (Fig. 1a,b) is organized around a two-tiered G-quadruplex with all-parallel connectivity, except for G18 of the top tier (T2), which is antiparallel. This quadruplex, which coordinates a canonical axial K+ ion (MA, Supplementary Fig. 4), stacks on a base triple, which in turn stacks coaxially on an A-form duplex (paired element P1). Two nucleotides from the loop connecting G14 and G18 of T2, and four nucleotides from the loop that follows G20 (the last of the eight guanines of the G-quadruplex) form the three base pairs of a second paired element (P2) that is juxtaposed with the G-quadruplex. U17, which is extruded from the middle of P2, forms a trans Watson-Crick pair with A10 from the propeller loop that connects G9 to G13, on the diagonally opposite side of the T2 G-quartet. The BzT and MQ of the bound TO1-Biotin are sandwiched between the A10•U17 tertiary base pair and T2 of the G-quadruplex. Reflecting the high structural complexity of this RNA, only two nucleobases (A6, U11) of the 28-nt conserved core of Mango-III do not share at least a hydrogen bond with another nucleobase. In comparison, six nucleobases of the 22-nt core of Mango-I are fully unpaired (Fig. 1c).
Figure 1. Overall structure of the Mango-III-TO1-Biotin complex.

(a) Secondary structure of Mango-III. Thin lines with arrowheads and Leontis-Westhof symbols41 denote connectivity and base pairs, respectively. (b) Cartoon representation of the three-dimensional structure of the complex colored as in (a). Arrows indicate 5′ to 3′ chain direction, and purple and green spheres represent K+ and Na+, respectively. (c) Sequence alignment of Mango-I and Mango-III aptamer RNA cores colored as in (a). Lines depict base pairing, except for the G-quadruplex (black lines). Terminal co-varying nucleotides shaded gray.
A coaxial duplex-triplex-quadruplex stack
Mango-I and the closely related Mango-II are each comprised7,12 of a three-tiered G-quadruplex flexibly linked to an A-form helix through a GAAÂ-tetraloop-like junction (quadruplex insertion site, ^). The residues of Mango-III corresponding to those junctions (Fig. 1c) instead fold into P1 by Watson-Crick base-pairing with complementary nucleotides at the 3’ end of the aptamer (Fig. 1c). This is consistent with covariation analysis (ref. 5 and Supplementary Table 2). A25 of Mango-III (which corresponds in sequence to the last tetraloop residue of Mango-I) forms a base triple with A7 (which is part of the propeller loop between G5 and G8 of the Mango-III quadruplex) and U19 (which connects G18 an G20 of the Mango-III quadruplex) (Fig. 1a, 2a). The A7•U19•A25 triple is unusual because only two direct hydrogen bonds connect the three nucleobases, but each base also hydrogen bonds through its Watson-Crick face to a shared water molecule (W1) that lies near the center of the triple (Fig 2a).
Figure 2. Structural features of Mango-III.
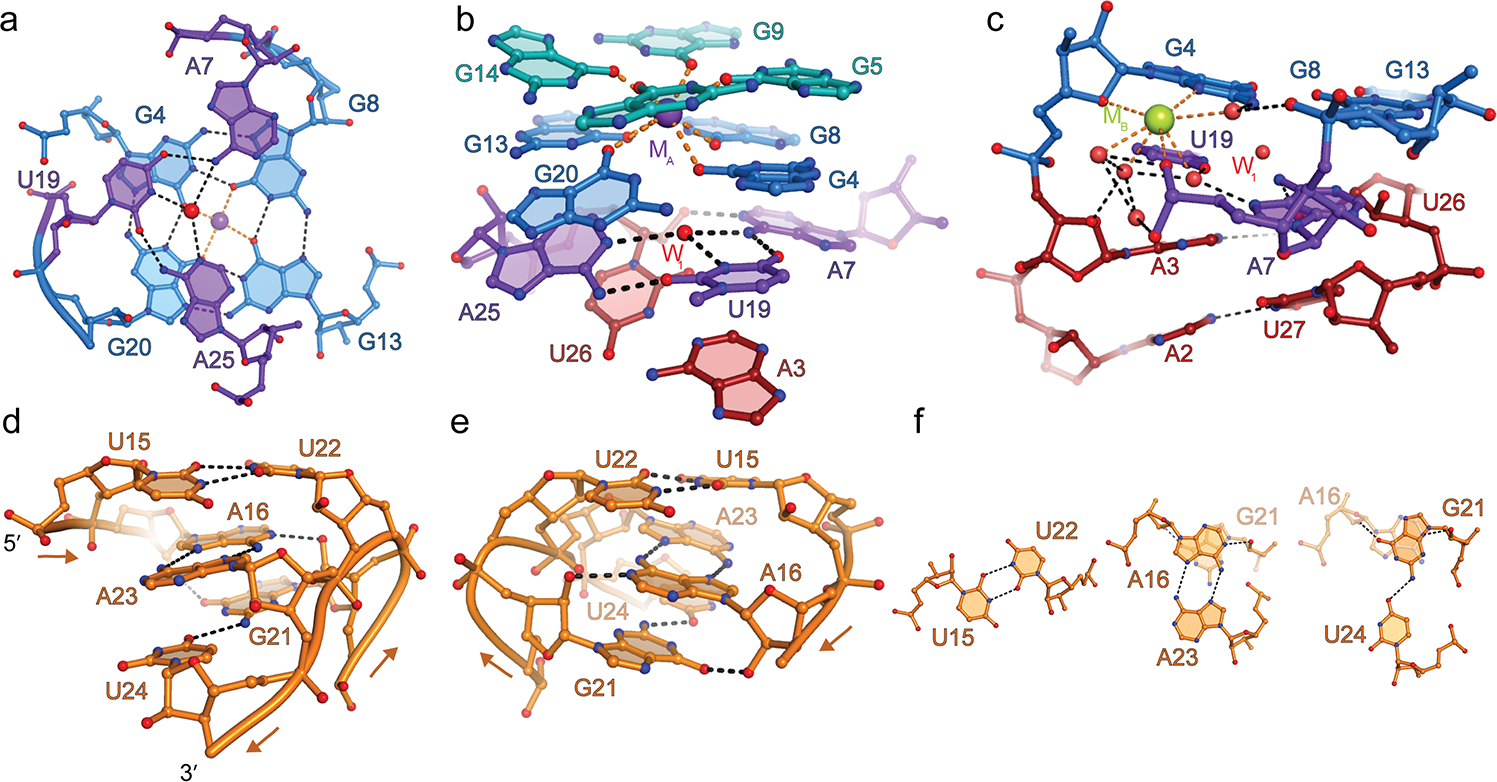
(a) Hydrogen bonding interactions of the A7•U19•A25 base triple (purple) and the G-quadruplex T1(blue). Red and purple spheres, water molecule at the center of the triple (W1), and K+ at the center of the G-quadruplex (MA), respectively. (b) The base triple stacks under the G-quadruplex and above the duplex P1. (c) Hydrated metal ion modeled as Na+ (green sphere) bridging the coaxial junction. (d) View of P2 from the direction of the G21-U24 strand. Arrows denote 5’ to 3’ chain direction. (e) 180° rotation along the vertical axis. (f) The three non-canonical base pairs of P2.
The A7•U19•A25 base triple links the P1 duplex with the quadruplex of Mango-III through three sets of interactions. First, the base triple stacks below the T1 of the quadruplex and above the A3•U26 Watson-Crick base pair that closes P1 (Fig. 2a,b). Second, the 2’-OH of U26 donates a hydrogen bond to the N1 of A7 of the triple, in effect expanding it to a pseudo-tetrad. Third, a hydrated cation (MB, modeled as Na+ due to lack of anomalous signal with 1.495 Å X-rays and coordination13 by N3 of a purine) and a network of ordered water molecules connects the phosphate of A7 to the minor groove faces of both, A3 and G4, thereby spanning the duplex-quadruplex junction (Fig. 2c). These interactions likely produce a rigid connection between P1 and the G-quadruplex moieties of Mango-III.
A partially parallel non-canonical duplex
The P2 duplex of Mango-III is unique because of the predominantly parallel orientation of its strands, and because its three base pairs are non-canonical (Fig. 2d,e,f). The backbones of the trans Watson-Crick U15•U22 and trans Hoogsteen A16•A23 base pairs are parallel. The third base pair is formed between U24 and G21, whose backbones are locally anti-parallel, being on opposite ends of the same loop connecting G20 of T1 (the last guanine of the G-quadruplex) to A25 of the base triple. The G21•U24 pair is formed in cis, and comprises a single hydrogen bond. The unusual structure of the P2 duplex is reflected in C1’-C1’ distances (8.5 Å, 11.4 Å and 12.6 Å, respectively, for the three pairs), different from that of canonical antiparallel A-form RNA duplexes (10.7 Å), and appears to be stabilized by a cross-strand stacking of A16 on G21. This latter interaction gives rise to reciprocal hydrogen bonds between N1 of A16 and 2’-OH of G21, and O6 of G21 and 2’-OH of A16.
The P2 helix contributes to the overall structure of Mango-III and the precise conformation of its fluorophore binding pocket in three ways. First, because P2 is followed immediately by A25, the duplex anchors the two-tiered G-quadruplex onto the base triple. Second, G18 and G20 form the only non-contiguous and anti-parallel guanine stack in the Mango-III G-quadruplex, and P2 anchors the backbones of these two nucleotides next to each other. This may be further facilitated by a hydrogen bond between the pro-Rp non-bridging phosphate oxygen (NBPO) of U15 and the 2’-OH of G14 from the adjacent guanine stack (Fig. 3a). Third, P2 helps position U17 of the tertiary base pair in the fluorophore binding pocket of Mango-III. Mutational analysis of P2 indicates that positions 16, 21, 23, 24 are sensitive to base identity while position 15 is less so (Supplementary Table 3).
Figure 3. Fluorophore binding by Mango-III.
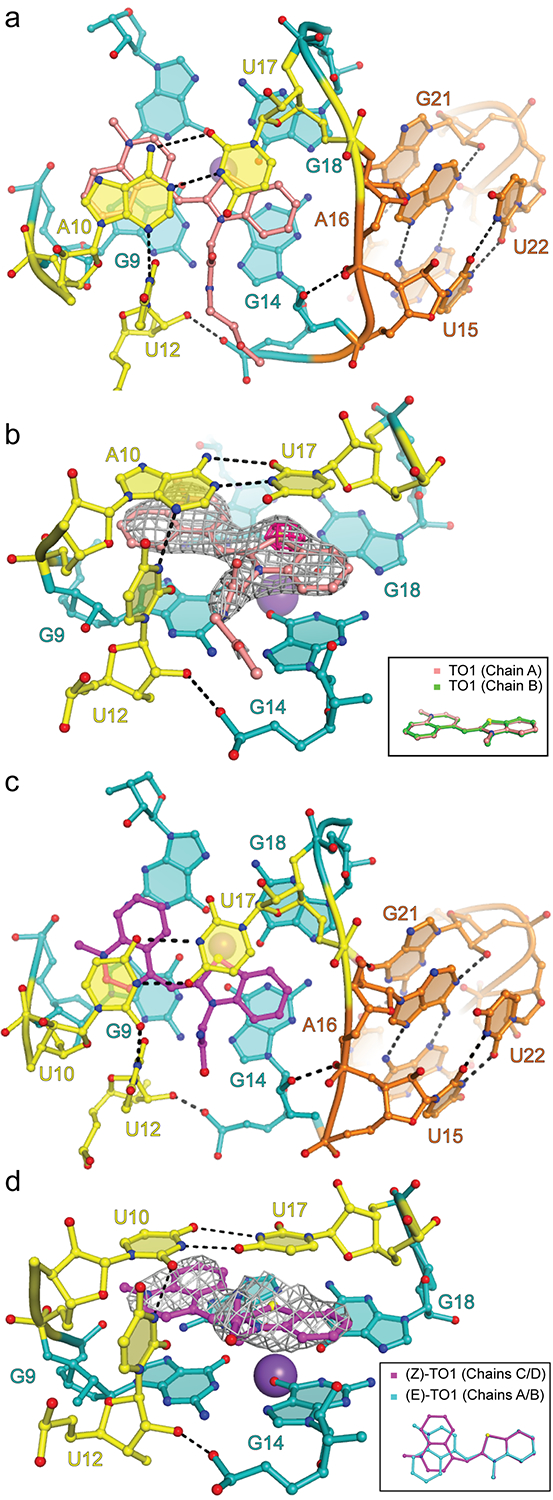
(a) Top view of the Mango-III-TO1-Biotin binding-pocket and the flanking P2. (b) Ligand binding pocket superimposed on the |Fo| - |Fc| electron density map prior to addition of fluorophore to the crystallographic model (grey mesh, 1.5 s.d.) Fuchsia surface, native anomalous difference Fourier synthesis (4 s.d.) Inset, all-atom superposition of the two TO1-Biotin fluorophores in the asymmetric unit. (c) Top view of the Mango-III(A10U)-TO1-Biotin binding-pocket. (d) Ligand binding pocket of Mango-III(A10U)-TO1-Biotin complex superimposed on the |Fo| - |Fc| electron density map prior to addition of fluorophore to the crystallographic model (grey mesh, 1.2 s.d.). Inset, structural superposition of the TO1-Biotin fluorophores from chains A and C of the Mango-III(A10U)-TO1-Biotin structure.
The TO1-Biotin-binding site of Mango-III
Unbiased residual electron density corresponding to the BzT and MQ, as well as the four proximal atoms of the PEG linker of TO1-Biotin unambiguously located the TO headgroup between T2 of the G-quadruplex and the tertiary A10•U17 trans Watson-Crick base pair. A strong anomalous difference Fourier feature corroborated the location of the sulfur of BzT (Fig. 3b). In its Mango-I complex, the planes of BzT and MQ of the fluorophore subtend a 45° angle, and its biotin moiety packs against MQ and the G-quadruplex of the aptamer. In contrast, when bound to Mango-III, the BzT and MQ rings are coplanar in both copies of the AU (Fig. 3b), and the biotin projects out into solvent, and lacks electron density.
To test the functional significance of the tertiary A10•U17 base pair of Mango-III, we generated the fifteen additional combinations of natural nucleotides at these two positions, as well as substitutions of A10 with 2-aminopurine and 2,6-diaminopurine, and examined their effect on TO1-Biotin binding and fluorescence enhancement (Supplementary Table 4). With one exception, these changes resulted in >10-fold decrease in binding affinity and >24% loss in fluorescence enhancement. The exception was A10U, which not only exhibited TO1-Biotin binding affinity indistinguishable from that of the wild-type, but also a 18% higher fluorescence enhancement. Thus, the tertiary base pair of Mango-III is important for fluorescence enhancement, of the sixteen possible nucleotide pairs, U10•U17 yields the brightest fluorescence.
Binding of TO1-Biotin to Mango-III appears to be further stabilized by U12. Its nucleobase is perpendicular to the planes of T2 and the A10•U17 pair, and it hydrogen bonds through its N3 to the N3 of A10, and through its 2’-OH to the pro-Sp NBPO of G14 (Fig. 3a,b). The nucleobase of U12 is also in van der Waals contact with the proximal atoms of the PEG linker of TO1-Biotin. Previous characterization of Mango-III showed that the U12C mutant was 40% less fluorescent5. We prepared U12A and abasic residue 12 variants and found that they exhibited >30-fold loss of affinity and >50% loss in fluorescence enhancement (Supplementary Table 4). Thus, residue 12 is important for Mango-III function, and a uracil at this position appears to be optimal for TO1-Biotin activation (the A10U, U17A double mutant of Mango-III improves fluorescence enhancement of TO3-Biotin by 30%, Supplementary Table 4).
Structure of the enhanced Mango-III(A10U) mutant
To determine the structural basis of the enhanced fluorescence of the A10U mutant, we solved its crystal structure in complex with TO1-Biotin at 2.9Å resolution (Supplementary Table 1, Methods, and Fig 3c, d). This mutant crystallized with four independent aptamer-fluorophore complexes in the A.U. The four complexes have similar overall structures; nonetheless, two of them (chains B and D) have P2 helices in which the cross-strand-stacked A16 and G21 are ordered, but U15, A23 and U24 are substantially disordered (Supplementary Fig. 5). In all four chains, the U10 base resulting from the mutation engages in a trans Watson-Crick base pair with U17. The C1’-C1’ distance of the mutant U10•U17 tertiary base pair (11.1 Å) is largely unchanged from that of the parental A10•U17 base pair (11.2 Å). However, the smaller pyrimidine at position 10 of the mutant reduces solvent occlusion of MQ of the fluorophore while retaining the H-bonding to U12. This and the partial disorder of P2 are reflected in a reduced shape complementarity14 between the A10U mutant and the fluorophore headgroup, compared to wild-type (S.C. scores of 0.837 ± 0.03 and 0.854 ± 0.01, respectively; mean ± r.m.s.d.) Consistent with the looser packing, the TO headgroups of the bound TO1-Biotin fluorophores adopt the (E) conformation, as seen in Mango-III, in two of the complexes in the A10U mutant crystal structure, while the other two are in the (Z) conformation (Fig. 3d).
Structure-guided reselection of Mango-III
The improved fluorescence enhancement of Mango-III(A10U) led us to hypothesize that other improved variants of Mango-III may exist. To test this, we subjected the aptamer to structure-guided functional reselection (Fig. 4). A starting pool of 4.1 × 106 sequences was generated by mixing two RNA pools in equal proportion, with pool 1 randomized at ten positions of the conserved Mango-III core (residues 3, 7, 10, 12, 17, 19, 21, 24, 25, 26), and pool 2 randomized at the same positions as pool 1 with an additional random insertion 3′ to position 26. We reasoned the latter could support a variant base quadruple (Fig. 4a). Two rounds of SELEX to enrich for high-affinity aptamers15,16 were followed by four rounds of droplet-based screening17 for bright aptamers. The selection converged on four dominant sequences with frequencies of 58%, 12%, 8.5%, and 1.2%. The four dominant reselected Mango-III variants were tested for TO1-Biotin binding and fluorescence enhancement. They exhibited dissociation constants of 3.6 – 6.4 nM and fluorescence enhancements (relative to wild-type) of −2% to +15% (Supplementary Table 5, 6). The most populated variant in the selection (iMango-III) exhibited fluorescence enhancement 13% higher than that of wild-type.
Figure 4. Structure guided re-selection.
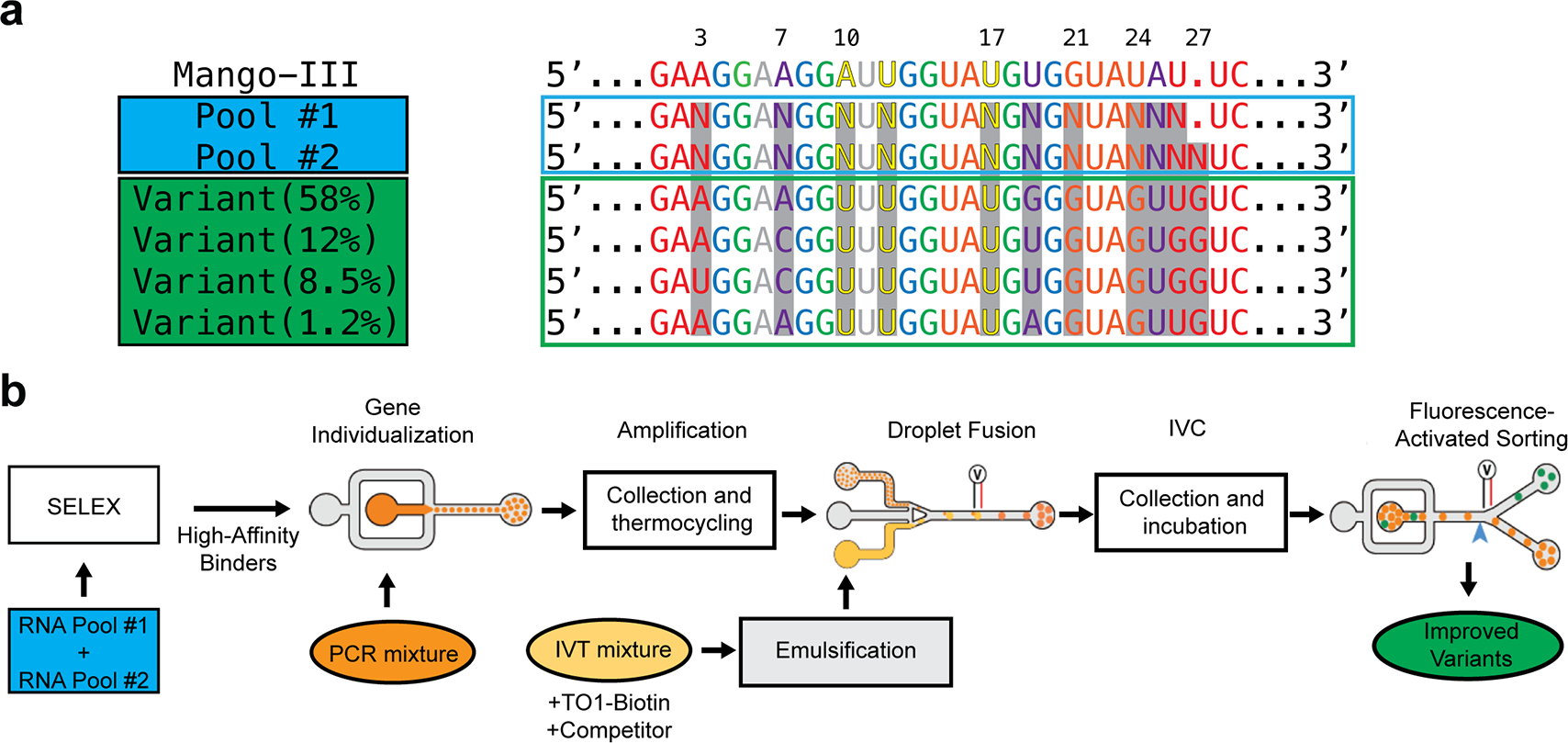
(a) Sequences of Mango-III, two structure-guided randomized pools (N, randomized positions), and four functionally reselected variants colored according to Mango-III structural elements (top). iMango-III is “Variant 58%”. (b) Reselection workflow. Equally mixed, randomized pools were first selected for high affinity binders through two rounds of SELEX. Resulting sequences underwent seven rounds of microfluidic droplet-based selection by individualizing each sequence in droplets (orange), thermocycling off chip, re-injected into a second chip to fuse with in vitro transcription mixture (yellow) followed by collection and incubation at 37˚C to amplify RNA, and transfer to a third chip for sorting based on fluorescence intensity (green).
All four functionally reselected sequences share uridines at positions 10 and 17, indicating that the tertiary base pair of Mango-III(A10U) is already optimal. All reselected sequences retain the insertion after residue 26 (hereafter residue 26:1) and in all cases this insertion is a guanine. The reselected sequences also differ from wild-type and Mango-III(A10U) in their duplex-triplex-quadruplex junction and in P2. Residue 7, which as an adenine participates in the junctional base triple in the wild-type, reselected as either adenine or cytosine, residue 19 (as a uracil, part of the wild-type triple) reselected as guanine, uracil or adenine, and residue 25 (which as an adenine completes the wild-type triple), reselected as an uracil. All four sequences harbor a guanine at position 24, which in the wild-type is occupied by a uracil that makes the G21•U24 closing base pair of P2. Collectively, these changes suggest that the structure of the junction region of the aptamer was altered.
Structure of the functionally reselected iMango-III
To determine the molecular consequences of functional reselection, we determined the structure of iMango-III in complex with TO1-Biotin at 1.55 Å resolution (Table 1, Online Methods). The structure reveals three sets of interrelated changes that propagate from G26:1, which forms a cis Watson-Crick pair with A3, thereby closing the G-quadruplex-proximal end of P1. U26, rather than pairing with A3 as in Mango-III and Mango-III(A10U), forms a base triple comprised of the A7•U26 Watson-Crick pair, and G19 (Fig. 5a). The four reselected variants of Mango-III can all form this triple (Supplementary Fig. 6). The repositioning of residue 26 into the triple, made possible by the presence of G26:1, extrudes residue 25 from the duplex-triplex-quadruplex junction (in wild-type, A25 is part of the junctional base triple) (Fig. 5b). Residue 25, a uridine in iMango-III, stacks below P2, and anchors it to the quadruplex by making three hydrogen bonds with G13 of T1 while the residue at position 24, a guanine in iMango-III, forms a cis Hoogsteen base pair with G21, which in turn stacks below the trans Watson-Crick A16•A23 pair (Fig 5c). As in the wild-type Mango-III, A16 and G21 make a cross-strand stack in P2 of iMango-III, but in the latter RNA, the two homopurine pairs (G21•G24 and A16•A23) are severely propeller-twisted compared to the near planar pairs in P2 of the wild-type. This twisting and a 28° rearrangement of the P2 and G-quadruplex helical axes (Supplementary Fig. 7), possibly abetted by the anchoring of the bottom of P2 by U25, allows G21 and G24 to make three direct and three water-mediated hydrogen bonds with the adjacent groove of T2 of the G-quadruplex (Fig 5d). None of these interactions are present in wild-type. P2 of iMango-III lacks the closing U15•U22 base pair. Instead, U22 is extruded into solvent and its nucleobase disordered, and U15 packs against the distal portion of the PEG linker of the bound TO1-Biotin, which in this crystal structure has defined electron density nearly in its entirety (Fig 5e).
Figure 5. Structure of iMango-III.
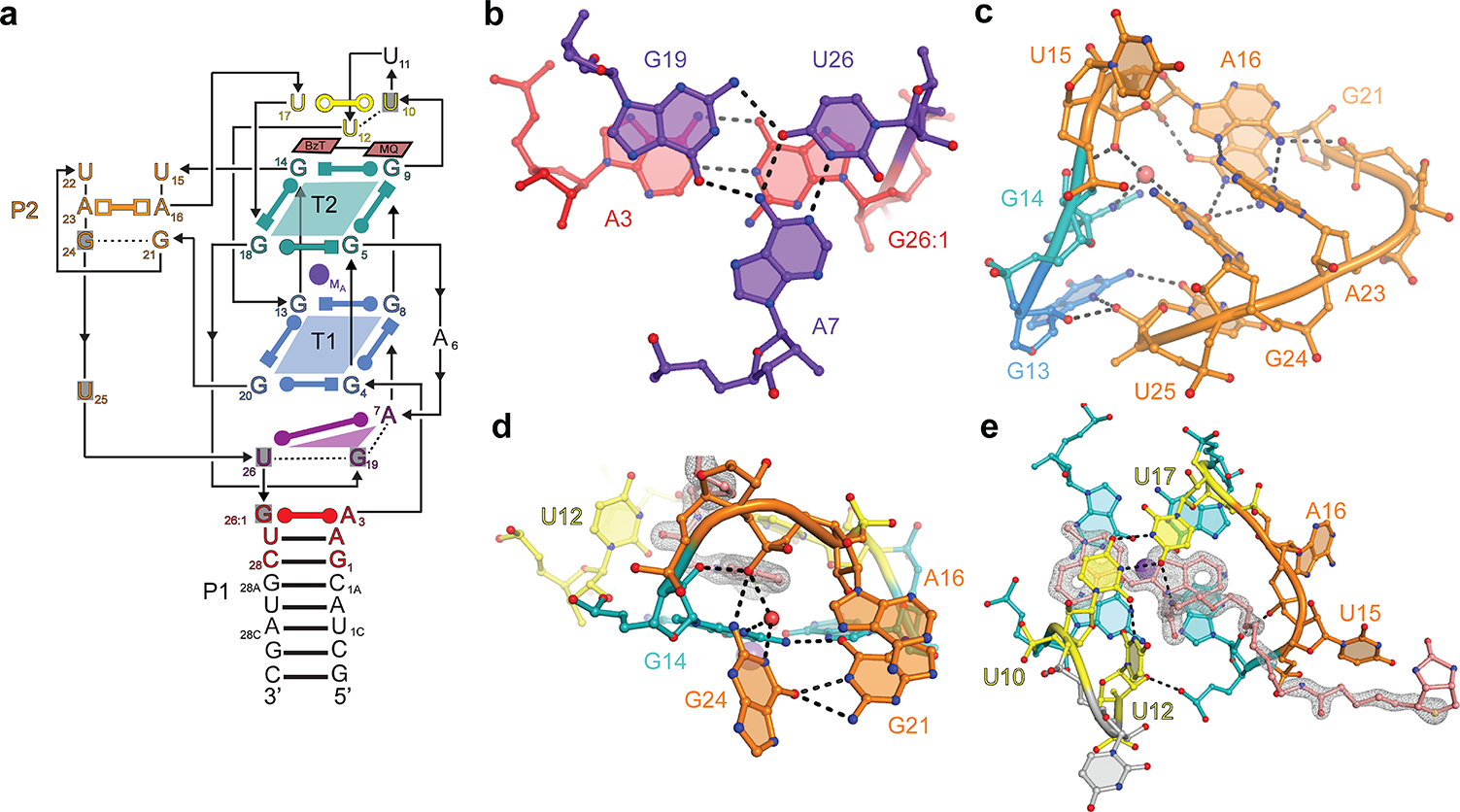
(a) Schematic secondary structure of iMango-III. Residues that differ from the Mango-III sequence are shaded gray. (b) Structure of the rearranged junctional base triple. (c) Structure of the rearranged P2. (d) Interaction of the G21•G24 base pair with T2 of the G-quadruplex. (e) TO-1-Biotin recognition by iMango-III. Gray mesh, |Fo| - |Fc| electron density prior to building the fluorophore (2.5 s.d.)
We tested the functional significance of iMango-III structural features by mutating P2 and the junctional triple. All P2 mutations reduced binding affinities to TO1-Biotin (Kd’s from 32 nM to 338 nM) and fluorescence enhancements (by 12% – 40%). Mutations to the junctional triple that were expected to maintain stability all had a marginal loss of fluorescence enhancement and affinity for TO1-Biotin, while mutations expected to disrupt the base triple resulted in Kd’s ranging from 137 nM – 408 nM, and reduced fluorescence enhancement by up to 20% (Supplementary Table 7).
Photophysical properties of Mango-III and variants
Despite the 18% and 13% increase in fluorescence enhancement by Mango-III(A10U) and iMango-III, respectively, relative to wild-type, as well as more than 20% of the core nucleotides being different in the functionally re-selected aptamer, Mango-III and its two variants exhibit indistinguishable affinities for TO1-Biotin (Supplementary Tables 4, 5). Consistent with this, there is no difference in thermal stability between them, and all three have the same profile for the first (tertiary) melting transition (Supplementary Fig. 8). The absorbance and fluorescence maxima of iMango-III are blue-shifted by 4 nm and 7 nm, respectively (Fig. 6a,b). To test whether the blue-shift of iMango-III is dependent on the U•U tertiary base pair, we generated an iMango-III(U10A) mutant in which the A•U tertiary base pair of the wild-type is restored. This mutant retains the spectrum of iMango-III while the spectrum of Mango-III(A10U) is identical to that of Mango-III (Supplementary Fig. 9), indicating that the iMango-III blue-shift results from its reselected core. Further photophysical analysis shows that the three RNA-fluorophore complexes have identical extinction coefficients, and the improved fluorescence enhancement by Mango-III(A10U) and iMango-III results from higher quantum yields [0.55, 0.66 and 0.64 for the TO1-Biotin complexes of Mango-III, Mango-III(A10U) and iMango-III, respectively; Supplementary Table 6].
Figure 6. Photophysical properties of Mango-III and variants.
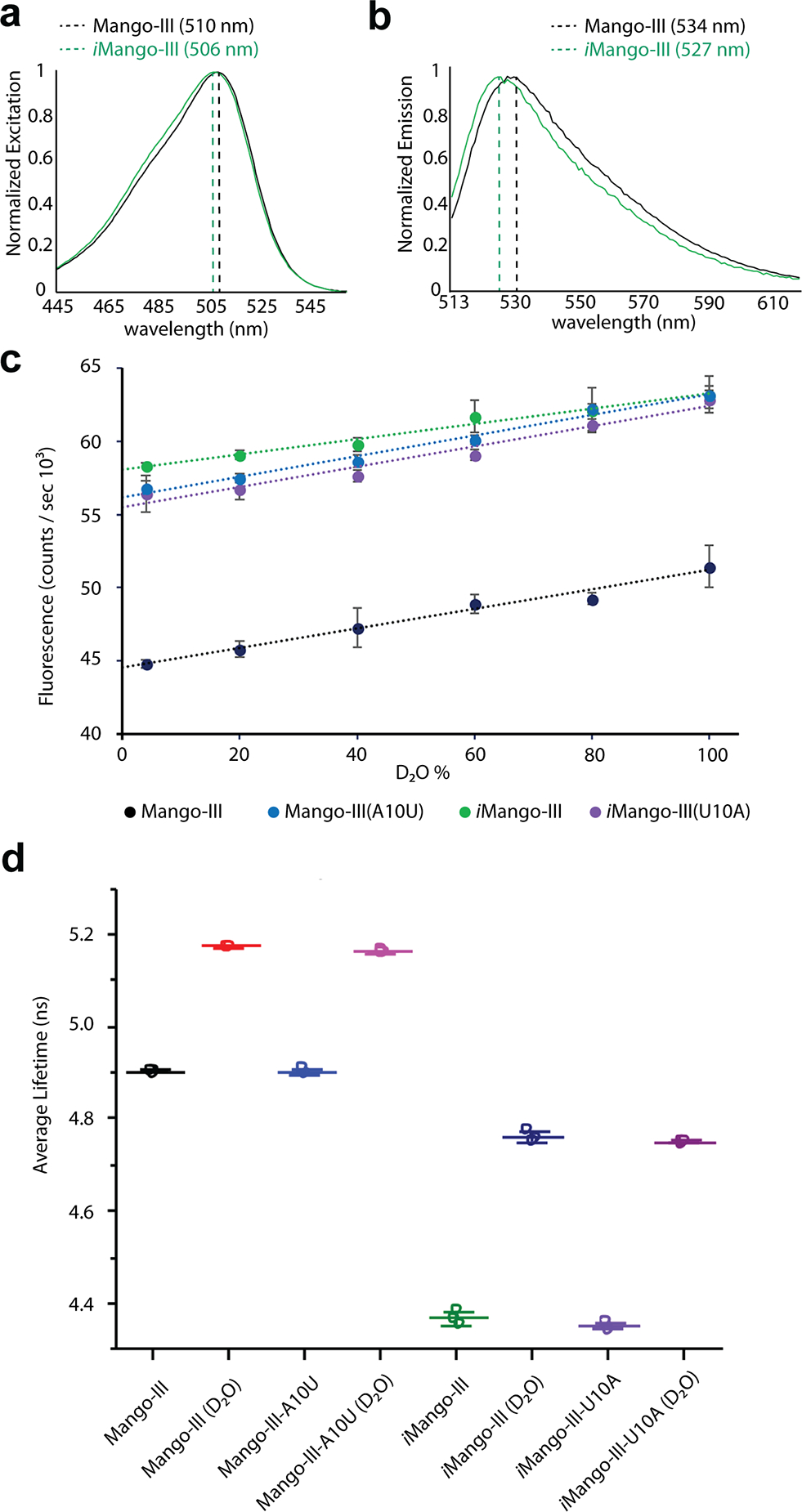
(a) Normalized absorbance spectra of the TO1-Biotin complexes of Mango-III and iMango-III (black and green, respectively). (b) Normalized fluorescence emission spectra of the TO1-Biotin complexes of Mango-III and iMango-III (black and green, respectively). Mango-III(A10U) and iMango-III(U10A) exhibit excitation and emission spectra indistinguishable from those of Mango-III and iMango-III, respectively (Supplementary Figure 9). (c) Fluorescence intensity of four aptamer-TO1-Biotin complexes as a function of D2O percentage. (d) Fluorescence lifetime of four aptamer-TO1-Biotin complexes in 100% water or 96.6% D2O. Individual data points, mean of three replicates, and standard deviations are shown (Supplementary Table 7).
In principle, higher quantum yield should be accompanied by longer fluorescence lifetime18. This, however, is not the case for Mango-III and variants. While the wild-type exhibits a lifetime of ~4.9 ns, Mango-III(A10U), iMango-III, and iMango-III(U10A) all had equal or shorter lifetimes (Supplementary Table 8). The lack of proportionality between fluorescence lifetimes and quantum yields could be due to contributions from ultra-fast decays (<100 ps)19–21 undetectable due to instrumental limitations (Online Methods). In an attempt to slow down potential molecular movements of the fluorophore, we performed lifetime measurements in solutions with increased viscosity. Reducing molecular motions has the potential to lessen ultra-fast decays, and in this case, help rationalize the discrepancy between quantum yields and lifetimes. Addition of either sucrose or glycerol, however, resulted in shorter average lifetimes and reduced brightness (Supplementary Fig. 10), suggesting that the structures of the RNAs are negatively perturbed by these solutes.
D2O can affect fluorescence lifetimes through a kinetic isotope effect on transfer of protons (deuterons)22 or through changes in viscosity. Since the protons of TO1-Biotin do not readily exchange, effects of D2O should arise from the latter under constant pH (or pD). To detect additional lifetime decays through increased viscosity, we performed D2O titrations on the Mango-III-TO1-Biotin complexes. To control for differences in direct solvent interaction with the fluorophore, we performed D2O titrations for iSpinach and Corn, two aptamers with demonstrated fluorophore-H2O hydrogen bonding23,24, and the DIR-Pro aptamer, a promiscuous aptamer with demonstrated fluorescence enhancement of TO1-Biotin25,26. Titration with D2O resulted in a linear increase in fluorescence intensity for Mango-III and iMango-III (Fig. 6c) and the control aptamers, Corn, iSpinach, and DIR-Pro (Supplementary Fig. 11). At 99.6% D2O, the fluorescence lifetimes of Mango-III and Mango-III(A10U) increased by 5.3% and those of iMango-III and iMango-III(U10A) by 9.2% (Fig. 6d, Supplementary Fig. 12), but additional lifetimes could not be resolved (Supplementary Table 8, Supplementary Fig. 13). The control aptamers exhibited longer lifetimes in D2O with Corn (14.0%)>DIR-Pro (6.1%)>iSpinach (1.3%) (Supplementary Table 9). Thus, no pattern was apparent between fluorophore-solvent hydrogen bonding and lifetimes.
Discussion
Previous Mango-I and Mango-II co-crystal structures revealed a shared molecular architecture comprised of a G-quadruplex and an A-form duplex (P1) flexibly linked through a tetraloop-like junction7,12. Despite its comparable size, the 3D structure of Mango-III is considerably more elaborate, incorporating a second helical element (P2), a long-range trans-Watson-Crick base pair, as well as a coaxially stacked junction between P1 and the G-quadruplex (Fig. 1). The formation of P2 by non-canonical base pairing between two nucleotides in a lateral loop of the G-quadruplex and four nucleotides immediately 3’ to it gives rise to a molecular connectivity that is analogous to that of a pseudoknot. A canonical pseudoknot arises from Watson-Crick base pairing between nucleotides in the loop of a stem-loop and those in an adjacent single-stranded segment27. The pseudoknot-like G-quadruplex fold of Mango-III is unprecedented. The complex tertiary structure of Mango-III explains why this aptamer, unlike Mango-I or Mango-II, exhibits a biphasic thermal melting profile5. As with proteins28, the requirement of structural complexity29,30 sufficient to make multidirectional interactions with the fluorophore in order to achieve bright fluorescence may set a lower bound on the size of fluorogenic RNA aptamers. At ~30 nt, almost all core nucleobases of Mango-III and iMango-III engage in hydrogen bonding. Perhaps not coincidentally, 30 nt is the lower limit in size of riboswitch aptamer domains31–33 and may be close to the minimal size for an RNA to possess tertiary structure and selectively bind a small molecule.
Most of the structurally characterized fluorescence turn-on aptamers incorporate G-quadruplex motifs within their functional cores (exceptions being34,25,35 malachite green, DIR, and Riboglow). G-quadruplexes can provide a large surface for multiple heterocycles to bind in-plane, thus maximizing fluorescence enhancement. However, in vivo, G-quadruplexes may suffer from off-target interactions with helicases36. Recent structural characterization37 of the substrate-bound mammalian helicase DHX36 demonstrated that it recognizes both, the guanine bases and the phosphodiester backbone of a simple, all-parallel G-quadruplex. By comprising flanking A-form P1 and irregular P2 helices, as well as a trans Watson-Crick base pair around a mixed-connectivity G-quadruplex core, Mango-III may exhibit reduced interactions with cellular G-quadruplex-binding proteins that may destabilize simpler G-quadruplex-containing RNAs.
Functional reselection based on the Mango-III cocrystal structure failed to produce a mutant brighter than Mango-III(A10U) demonstrating that U10•U17 pair is optimal for fluorescence enhancement. This is surprising because the smaller surface area of the homopyrimidine pair would constrain the TO headgroup of the fluorophore less than the A•U pair. Consistent with the lesser constraint is the presence of a mixture of (E) and (Z) stereoisomers of the fluorophore in the co-crystal structure of Mango-III(A10U). Mango-III-TO1-Biotin and Mango-III(A10U)-TO1-Biotin both exhibit the same number of decay pathways, on the ~150 ps – 10 ns timescale, despite Mango-III(A10U) stabilizing both (E) and (Z) stereoisomers in the crystal structure.
An unexpected consequence of the mutations that distinguish iMango-III from Mango-III is that the absorption and emission spectra of the functionally reselected aptamer are blue-shifted (Fig. 6a,b). Site-directed point mutations and DNA shuffling between the fluorescent proteins mScarlet and mCardinal demonstrated that mutations distant from the chromophore often result in blue-shifted emission and that emission maxima is anticorrelated with average lifetime38. Since Mango-III and Mango-III(U10A) have indistinguishable spectra (Supplementary Fig. 9), mutations distant from the binding pocket are responsible for these spectral shifts (as well as the shorter fluorescence lifetimes of the iMango-III and its U10A mutant). For Spinach, it was shown that mutation of the base triple that directly stacks on its DFHBI fluorophore resulted in excitation and emission blue shifts39. The blue shift of iMango-III relative to Mango-III is more surprising since the causative mutations are >10 Å from the fluorophore. These results demonstrate that the brightness and also the spectra of cyanine-dyes, such as TO1-Biotin, can be tuned by manipulating fluorophore-distal residues of Mango-III, and are reminiscent of the effects of distant mutations on the active sites of globular proteins40. The randomized sequence pool leading to iMango-III contained only a single point insertion; selections from pools containing additional insertions may lead to brighter and or spectrally distinct aptamers.
Online Methods
RNA preparation
RNA preparation and constructs used in this study are listed in Supplementary Note 1. Except where noted, RNAs were stored and analyzed in 20 mM MOPS-KOH pH 7.0, 150 mM KCl, and 10 μM EDTA.
Crystallization and diffraction data collection
Mango-III-TO1-Biotin (RNA 1) in 20 mM MOPS-KOH pH 7.0, 150 mM KCl, 10 μM EDTA was heated to 95˚C for 3 minutes, incubated at RT for 10 minutes, mixed with equimolar TO1-Bitoin, then incubated at RT for 30 minutes. For crystallization, 0.2 μl of RNA solution (300 μM) and 0.2 μl reservoir solution (0.04 M Na Cacodylate pH 6.5, 0.08 M NaCl, 0.012 M KCl, 0.02 M MgCl2, 0.02 M SrCl2, 0.012 M Spermine · 4 Cl, 5.5% Sucrose, 31% MPD) were mixed and equilibrated at 294 K by sitting drop vapor diffusion. Strongly fluorescent (500 nm illumination), tetragonal pyramidal crystals grew in 1–3 days to maximum dimensions of 300 × 100 × 100 μm3. Additional cryoprotection was not necessary prior to mounting the crystal in a nylon loop and vitrifying by plunging into liquid nitrogen. Data were collected at 100 K at APS 22-BM using 0.977 Å X-radiation, as well as APS 24-ID-C with 1.495 Å X-radiation. Data were reduced in HKL2000 (ref 42) with 10% of reflections flagged for RFree calculation. Mango-III-A10U-TO1-Biotin was crystallized using the sitting drop vapor diffusion method by mixing 0.2 μl of RNA 2 solution (300 μM, prepared similarly to above with addition of TO1-Biotin) and 0.2 μl reservoir solution (1.6 M Ammonium Citrate, 3.0% glycerol and 4% Acetone). Strongly fluorescent, triangular prism shaped crystals, grew in 1–2 days at 294 K with dimensions of 80 × 20 × 20 μm3. Crystals were mounted directly from mother liquor in a nylon loop and vitrified by plunging into liquid nitrogen. Data were collected at 100K ALS beamline 502 using 1.105 Å X-radiation, and reduced using DIALS (ref 43) with 5% of reflections being flagged for Rfree calculation. iMango-III was crystallized by sitting drop vapor diffusion by mixing 0.2 μl of RNA 3 (200 μM, prepared similarly to above with addition of TO1-Biotin) and 0.2 μl of reservoir solution (0.1 M Na Cacodylate pH 7.1, 16% PEG3350 and 0.25 M Magnesium acetate). Cryoprotection was performed by adding 1 μl of reservoir solution supplemented with 15% PEG400 to the mother liquor. Crystals were mounted in a nylon loop and vitrified by plunging into liquid nitrogen. Data were collected at 100 K at APS beamline 24-ID-C using 0.9792 Å X-radiation, and reduced using HKL2000 (ref 42) with 10% of reflections flagged for Rfree calculation. Data collection statistics are summarized in Supplementary Table 1.
Structure determination and refinement
Mango-III-TO1-Biotin crystals were soaked in drop conditions above supplemented with 10 mM Iridium hexamine for 90 minutes and then vitrifying by plunging into liquid nitrogen. Data sets of iridium-soaked crystals were collected at 100 K at APS 22-ID using 0.957 Å X-radiation. SHELXC reported significant anomalous signal extending to 3.5 Å from a single crystal diffracting to 3.2 Å. Two heavy atom sites were located by SHELXD (ref 44) with density modification performed in SHELXE (ref 44) resulting in an uninterpretable map. Heavy atom sites from SHELXD were refined using AutoSol (ref 45,46) yielding a mean overall figure of merit of 0.22. Density modification using Resolve (ref 45) resulted in an electron density map into which 16 nucleotides could be built manually using Coot (ref 39). This model was then refined using Phenix.refine (ref 47) and placed into the native data set (2.35 Å resolution) collected at APS 22-BM using the program Phaser (ref 48). The top solution had a TFZ score of 12.1. Manual building and refinement was performed in Coot (ref 39) and Phenix.refine (ref 47), respectively. Phase probability distributions from the Iridium dataset were part of the refinement target function. The Mango-III(A10U) structure was solved by molecular replacement using the program Phaser (ref 48) with the Mango-III-TO1 structure with residues 1, 2, 15, 22, 37, 38 and the TO1 ligand deleted as a search model. The top solution had a TFZ score of 15.2. Multiple rounds of manual building and refinement were performed in Coot and Phenix.refine respectively. Simulated annealing was performed every 6 refinement cycles at 5000 K until an RFree of 0.25 was reached. A new set of RFree flags composing 10% of reflections was selected and building and refinement continued with 4 rounds of simulated annealing. The structure of iMango-III was solved by molecular replacement using the program Phaser with coordinates from the Mango-III-TO1 complex structure with residues 1, 2, 15, 22, 37, 38 and the TO1 ligand deleted as a search model. The top solution had a TFZ score of 22.3. Multiple rounds of building and refinement were performed in Coot and Phenix.refine, respectively. Refinement statistics are summarized in Supplementary Table 1.
Analytical ultracentrifugation
Analytical Ultracentrifugation was performed on RNA 1 in the presence and absence of TO1-Biotin. Cells contained Mango-III at 5 μM concentration. 500 scans were collected and averaged on a Beckman XLI analytical ultracentrifuge. Absorbance was measured at 295 nm, under constant velocity with a run speed of 60,000 r.p.m. at 20˚C. The viscosity (0.01015 P) and density (1.0068 g ml−1) of the buffer were estimated with the Sednterp server (http://rasmb.org/sednterp)).
Dynamic light scattering
Dynamic light scattering was performed with a Dynapro Nanostar instrument. Samples were prepared by heating 300 μM Mango-III to 95˚C for 3 minutes then incubating at RT for 5 minutes. One molar equivalent of TO1-Biotin was added to the solution and mixed. Samples were then passed through a Millipore 0.1 μm spin filter immediately prior to data collection. Ten readings were taken per sample with maximum collection of 1s.
Fluorescence binding experiments
Fluorescence readings were recorded using a Varian Cary Eclipse spectrophotometer at 510 nm excitation and 532 nm emission. Fluorescence titrations were performed in Mango selection buffer (WB: 140 mM KCl, 1 mM MgCl2, 10 mM NaH2PO4 pH 7.2, 0.05% Tween-20). To determine binding affinity and Fmax, curves were fitted using least squares (Kaleidagraph 4.5) using the following equation for TO1-Biotin:
Or to the following equation for TO3-Biotin:
Structure-guided reselection
Two Mango 3-derived libraries (Fig. 4a) were designed based on the Mango-III crystal structure and were pooled together prior to being subjected to the workflow summarized in Fig. 4b. Pooled libraries contained a theoretical diversity of ~5,000,000 variants that exceeds the upper limit that can be analyzed by μIVC screening. Two rounds of SELEX were performed first to reduce the library diversity and to enrich it in high affinity variants. This first step was expected to reduce the sequence diversity to make possible covering the whole residual diversity with a μIVC screening procedure. A microfluidic-assisted screening similar to that used for the isolation5 of Mango III was then used to screen the SELEX-enriched library. Modifications from the original procedure were as follows. First, a unique barcode (Unique Droplet Identifier, UDI) was appended to each gene prior to starting each round of screening by mixing template DNA with 0.2 μM of forward barcode-containing primer (5’-TCG TCG GCA GCG TCA GAT GTG TAT AAG AGA CAG AAN NNN NNN NNN NNN NNN NNN NAA CAG CTA ATA CGA CTC ACT ATA GGA AGA ATG-3’) and of reverse primers (5’-CAA ACA TGG GAA GAA TGA-3’), 0.2 mM of each dNTP, and 0.02 U/μl Q5 DNA polymerase (New England Biolabs), and the supplied buffer (proprietary to NEB) at the recommended concentrations. The mixture was thermocycled starting with an initial step of denaturation of 2 min at 98°C followed by ~15 cycles of: 10 sec at 98°C, 30 sec at 55°C and 30 sec at 72°C, and PCR products were purified by Wizard SV Gel and PCR Clean-Up System (Promega). Next, barcoded DNA molecules were diluted into a 200 μg/ml yeast total RNA solution (Ambion) to reach the desired average number of DNA molecule per droplet (λ value) and introduced into a PCR mixture containing 0.2 μM of forward primer (5’- TCG TCG GCA GCG TCA GAT GTG TAT AAG AGA CAG-3’) and reverse primer (5’-CAA ACA TGG GAA GAA TGA-3’), 0.2 mM of each dNTP, 0.67 mg/mL Dextran-Texas Red 70 kDa (Molecular Probes), 0.1% Pluronic F68, 0.02 U μl−1 Q5 DNA polymerase (New England Biolabs) and the supplied buffer at the recommended concentrations. The mixture was then dispersed into 2.5 pL droplets as described previously5. Upon collection, the emulsion was thermocycled with an initial step of denaturation of 2 min at 98°C followed by 30 cycles of: 10 sec at 98°C, 30 sec at 55°C and 30 sec at 72°C. PCR droplets were then reinjected into a droplet fusion device where each droplet was fused to a 16 pL droplet containing an in vitro expression mixture (2.2 mM of each NTP, 24 mM MgCl2, 44 mM Tris-HCl pH 8.0, 50 mM KCl, 5 mM DTT, 1 mM Spermidine, 35 μg mL−1 of Dextran-Texas Red 70 kDa (Molecular Probes), 0.1% Pluronic F68, 100 nM TO1-Biotin, 70 U T7 RNA polymerase (New England Biolabs), 5 ng μl−1 inorganic pyrophosphatase (Roche) and supplemented optionally with appropriate TO3-Biotin concentration (Supplementary Table 10) as before5. Finally, upon incubation, droplets were re-injected into an analysis and sorting microfluidic device mounted onto a Thermo plate (Tokai Hit) held at the desired temperature (Supplementary Table 10). The most fluorescent droplets were sorted, broken and the DNA recovered. Then, a DNA aliquot was used either to prime a new round of screening (starting with the introduction of a new UDI barcode) or to prepare an indexed library for high-throughput sequencing analysis.
High-throughput sequencing analysis
DNA recovered from broken emulsions was subjected to a tandem of PCR amplification. First, sequencing adaptors were appended to both ends of DNA by introducing an aliquot of DNA into a PCR mixture containing 0.1 μM of forward (5’- TCG TCG GCA GCG TCA GAT GTG TAT AAG AGA CAG-3’) and reverse primers (5’-GTC TCG TGG GCT CGG AGA TGT GTA TAA GAG ACA GCA AAC ATG GGA AGA ATG A-3’), 0.2 mM of each dNTP, 0.02 U μl−1 Q5 DNA polymerase (NEB) and the supplied buffer. The mixture was then subjected to an initial step of 2 min at 98°C followed by 30 cycles of: 10 sec at 98°C, 30 sec at 55°C and 30 sec at 72°C. Then, each library was indexed using Nextera DNA library preparation kit (Illumina) as recommended. Indexed DNA libraries were purified on a 1 % agarose gel and the DNA extracted using the Monarch kit (New England Biolabs). DNA quality was validated on a Bioanalyzer (Agilent) platform and quantified using a Quant-iT dsDNA high sensitivity assay and QuBit fluorometer (Invitrogen). Finally, DNA libraries were sequenced on a MiSeq instrument using a MiSeq Reagent kit V3 150 cycles cartridge (Illumina). Upon sequencing, fastq files were analyzed using a custom Python bioinformatic pipeline. Briefly, sequences displaying the wrong length and/or unwanted mutations were filtered-out. Next, sequences sharing the same UDI barcode were clustered together. UDI displaying a low occurrence (i.e. represented by less than 50 reads) were considered spurious (e.g., point mutants resulting from PCR mutations or sequencing errors) and discarded. Finally, counting the number of different UDIs associated to the same aptamer-coding sequence allowed establishing the occurrence frequency of each sequence in the analysed library. Finally, sequences were ranked according to their occurrence frequency and the RNA corresponding to those the most represented in the final pool were further characterized. The overall success of the screening process was confirmed by the gradual reduction of the sequence diversity along the screening process (Supplementary Table 11 and Supplementary Dataset 1).
Fluorescence lifetime measurements
Lifetime measurements were performed using an in-house modified 2-photon fluorescence microscope based on a Zeiss LSM 510 unit capable of time-correlated single-photon counting (TCSPC) measurements. A diode-pumped solid-state laser (Millenia Prime, Spectra Physics, Newport, CA) was used to pump a Ti:Sapphire Tsunami oscillator (Model 3960C, Spectra Physics, Newport, CA), operating in femtosecond mode, at a repetition rate of 80 MHz, and tunable in the near infrared region. The output beam of the Ti:Sapphire oscillator was directed into a pulse-picker unit (Model 3980, Spectra Physics, Newport, CA) and the repetition rate of the laser was decreased to 4 MHz to allow for a broader time window for recording of the large fluorescence lifetime values observed for Mango3-derived aptamers. The beam was attenuated by using a variable neutral density filter before being directed onto a 700/488 dichroic mirror and reflected into the back aperture of a Plan Apochromat 63x, NA 1.4, oil immersion objective, and expanded to fill the back pupil. The fluorescence from the sample was collected from the same objective, intercepted by a 733LP dichroic mirror, sent toward the back port of the microscope, filtered through an ET750SP-2P and an ET680SP-2P filters (Chroma Technologies, Bellows Falls, VT) to remove residual scattering from the laser, and further passed through a 540/550 nm bandpass filter (Semrock, Rochester, NY). The fluorescence was finally focused onto the photocathode of a H7422P-40 GaAsP photomultiplier detector (Hamamatsu, Japan). The large NA of the objective contributes to a certain degree of depolarization of the incoming excitation beam, as well as of the detected fluorescence. According to Fisz (ref 49), this would allow one to omit an analyzer (polarizer) in front of our detector, because magic angle conditions are achieved by using a 45° angle detection (i.e., unpolarized) configuration. The signal from the detector and the synchronization signal from the laser were directed into an SPC150 photon counting card (Becker&Hickl, Germany), controlled by the SPCM64 proprietary software from the manufacturer (Becker&Hickl, Germany). The data were collected with a 50 ns time window in TCSPC histogramming mode with 1024 channels, with a width of 48.8 ps each. For the experiments the laser was tuned at 780 nm, and the power adjusted to ~1–1.5 mW before the objective lens, depending on the sample. RNA samples were diluted to a concentration of ~3 μM in buffer. For the preparation of sample chambers, a #1.5 coverslip was cleaned with deionized water, ethanol, ultra-pure water, and finally dried with a flow of Nitrogen gas. A CoverWell perfusion chamber (#PC8R-0.5, Grace Bio-Labs, Bend, OR) was adhered onto the clean coverslip, and the chamber was incubated for at least 1 hour with a 10 mg/ml solution of Bovine Serum Albumin (BSA) in water. After incubation, the chamber was thoroughly rinsed with ultrapure water to remove excess of BSA and allowed to dry before use. For measuring the samples in deuterated buffer, the chamber, after drying was pre-wetted with the deuterated buffer before loading the RNA-containing sample. The excitation was focused ~10 μm above the upper surface of the coverslip to reduce scatter and to avoid collecting signal from molecules immobilized on the glass surface. Data were collected until the maximum count in the peak channel of the TCSPC histogram reached 104 photons. Experiments were in triplicate. An instrument response function (IRF) was collected on every day of measurement by recording the second harmonic generation (SHG) signal from urea crystals deposited onto a glass coverslip, in this case the bandpass filter before the detector was substituted with a 440 nm short pass filter to collect the 390 nm SHG.
Fluorescence lifetime data analysis
Data were converted into ASCII files and further analyzed by using a MATLAB fitting routine based on the algorithm described (ref 50), and kindly provided by Dr. Joerg Enderlein (University of Goettingen, Germany). The fluorescence decay curves were fit using exponential functions of the form:
Where n is at most equal to 3 in our software. The data fitting routine uses a least-square algorithm where the exponential function is first reconvoluted with the measured IRF and then fit to the data. The color-shift of the IRF for each dataset was included as a free parameter during fitting. The number of exponentials used to fit the data was adapted in order to minimize the χ2 value. An F-test was performed on the ratio of χ2 values obtained from fitting the data with a different number of exponentials, to determine whether the addition of a component was statistically significant to the decrease of χ2. All the data presented in this manuscript were reasonably fit with 2-component exponential functions and fits with χ2<1.2.
Fluorescence D2O Titrations
RNA samples were taken up in 20 mM MOPS pH 7.0, 150 mM KCl, 10 μM EDTA in 99.6% D2O. Two solutions of 2 μM TO1-Biotin were made, one with pure H2O and the other with 99.6% D2O. The solutions were mixed to vary D2O with 20% increments while maintaining constant TO1-Biotin concentration. RNA samples were taken up in 20 mM MOPS pH 7.0, 150 mM KCl, 10 μM EDTA in 99.6% D2O. RNA was refolded as described above. RNA was added to a final concentration of 5μM. Fluorescence intensity was measured on a Photon Technologies International/ 820 Photomultiplier Detection System with an excitation and emission wavelength centered at 509 nm and 530 nm, respectively.
Statistics
Each dataset reported for X-ray crystallography was collected from a single crystal (Supplementary Table 1). Analytical Ultracentrifugation was performed on three independent samples with 50,000 traces acquired for each sample. Error is reported as the standard deviation of the resulting S values. Fluorescence binding titrations were performed on three independent samples. The Kd values reported are the mean of three experiments ± standard deviation. Fluorescence lifetime data are reported as the mean of three replicates (independently prepared samples), reported with ± standard deviation.
Data Availability
All data generated or analyzed during this study are included in this published article (and its supplementary information files). Atomic coordinates and structure factor amplitudes for the Mango-III-TO1-Biotin, Mango-III(A10U)-TO1-Biotin, and iMango-III-TO1-Biotin co-crystal structures have been deposited in the Protein Data Bank under accession codes 6E8S, 6E8T, and 6E8U, respectively.
Supplementary Material
Acknowledgments
We thank the staff of beamlines 5.0.1 and 5.0.2 of the Advanced Light Source, Lawrence Berkeley National Laboratory (ALS), and 22-BM, 22-ID, and 24-ID-C of the Advanced Photon Source, Argonne National Laboratory (APS) for crystallographic data collection; G. Piszczek of the Biophysics Core of the National Heart, Lung and Blood Institute (NHLBI) for analytical ultracentrifugation and dynamic light scattering; D.-Y. Lee of the Biochemistry Core of NHLBI and R. Levine for high-performance liquid chromatography; and members of the Ferré-D’Amaré, Ryckelynck and Unrau laboratories for discussions. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P30 GM124165). The Pilatus 6M detector on 24-ID-C beam line is funded by a NIH-ORIP HEI grant (S10 RR029205). Data were collected at Southeast Regional Collaborative Access Team (SER-CAT) 22-ID (or 22-BM) beamline at the Advanced Photon Source, Argonne National Laboratory. SER-CAT is supported by its member institutions (see www.ser-cat.org/members.html), and equipment grants (S10_RR25528 and S10_RR028976) from the National Institutes of Health. We also thank S. Koechler and A. Alioua from the IBMP Gene Expression Analysis facility (funded as part of the LabEx NetRNA) for technical assistance with high-throughput sequencing. This work was partially supported by an NSERC (Canada) operating grant (P.J.U.), the LabEx NetRNA (ANR-10-LABX-0036) (M.R.), ‘Agence Nationale pour la Recherche’ (ANR-16-CE11-0010-01) (M.R.), and the Intramural Program of the NHLBI, NIH.
Footnotes
Competing Financial Interests
The authors declare no competing financial interests.
References
- 1.Paige JS, Wu KY & Jaffrey SR RNA mimics of green fluorescent protein. Science 333, 642–646 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Paige JS, Nguyen-Duc T, Song WJ & Jaffrey SR Fluorescence imaging of cellular metabolites with RNA. Science 335, 1194–1194 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dolgosheina EV et al. RNA Mango aptamer-fluorophore: A bright, high-affinity complex for RNA labeling and tracking. ACS Chem. Biol 9, 2412–2420 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Song W et al. Imaging RNA polymerase III transcription using a photostable RNA-fluorophore complex. Nat. Chem. Biol 13, 1187–1194 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Autour A et al. Fluorogenic RNA Mango aptamers for imaging small non-coding RNAs in mammalian cells. Nat. Comm 9, 656 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rodriguez EA et al. The growing and glowing toolbox of fluorescent and photoactive proteins. Trends Biochem. Sci 42, 111–129 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Trachman RJ et al. Structural basis for high-affinity fluorophore binding and activation by RNA Mango. Nat. Chem. Biol 13, 807–813 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nygren J, Svanvik N & Kubista M The interactions between the fluorescent dye thiazole orange and DNA. Biopolymers 46, 39–51 (1998). [DOI] [PubMed] [Google Scholar]
- 9.Carreon JR, Mahon KP & Kelley SO Thiazole orange-peptide conjugates: Sensitivity of DNA binding to chemical structure. Organic Lett. 6, 517–519 (2004). [DOI] [PubMed] [Google Scholar]
- 10.Trachman RJ, Truong L & Ferré-D’Amaré AR Structural principles of fluorescent RNA aptamers. Trends Pharmacological Sci. 38, 928–939 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ishikawa-Ankerhold HC, Ankerhold R & Drummen GP Advanced fluorescence microscopy techniques--FRAP, FLIP, FLAP, FRET and FLIM. Molecules 17, 4047–132 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trachman RJ et al. Crystal structures of the Mango-II RNA aptamer reveal heterogeneous fluorophore binding and guide engineering of variants with improved selectivity and brightness. Biochemistry 57, 3544–3548 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leonarski F, D’Ascenzo L & Auffinger P Mg2+ ions: do they bind to nucleobase nitrogens? Nucleic Acids Res. 45, 987–1004 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lawrence MC & Colman PM Shape complementarity at protein/protein interfaces. J. Mol. Biol 234, 946–50 (1993). [DOI] [PubMed] [Google Scholar]
- 15.Tuerk C & Gold L Systematic evolution of ligands by exponential enrichment - RNA ligands to bacteriophage-T4 DNA-polymerase. Science 249, 505–510 (1990). [DOI] [PubMed] [Google Scholar]
- 16.Ellington AD & Szostak JW In vitro selection of RNA molecules that bind specific ligands. Nature 346, 818–22 (1990). [DOI] [PubMed] [Google Scholar]
- 17.Ryckelynck M et al. Using droplet-based microfluidics to improve the catalytic properties of RNA under multiple-turnover conditions. RNA 21, 458–469 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lakowicz JR Principles of Fluorescence Spectroscopy, 452 (Klewer Academic / Plenum Publishers, New York, 1999). [Google Scholar]
- 19.Chen RF, Knutson JR, Ziffer H & Porter D Fluorescence of tryptophan dipeptides: correlations with the rotamer model. Biochemistry 30, 5184–95 (1991). [DOI] [PubMed] [Google Scholar]
- 20.Xu J & Knutson JR Quasi-static self-quenching of Trp-X and X-Trp dipeptides in water: ultrafast fluorescence decay. J Phys. Chem. B 113, 12084–9 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jarikote DV, Krebs N, Tannert S, Röder B & Seitz O Exploring base-pair-specific optical properties of the DNA stain thiazole orange. Chemistry 13, 300–10 (2007). [DOI] [PubMed] [Google Scholar]
- 22.Toh KC, Stojkovic EA, van Stokkum IH, Moffat K & Kennis JT Proton-transfer and hydrogen-bond interactions determine fluorescence quantum yield and photochemical efficiency of bacteriophytochrome. Proc. Natl Acad. Sci. USA 107, 9170–9175 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Warner KD et al. A homodimer interface without base pairs in an RNA mimic of red fluorescent protein. Nat. Chem. Biol 13, 1195–1201 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fernandez-Millan P, Autour A, Ennifar E, Westhof E & Ryckelynck M Crystal structure and fluorescence properties of the iSpinach aptamer in complex with DFHBI. RNA 23, 1788–1795 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tan XH et al. Fluoromodules Consisting of a Promiscuous RNA Aptamer and Red or Blue Fluorogenic Cyanine Dyes: Selection, Characterization, and Bioimaging. J. Am. Chem. Soc 139, 9001–9009 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Shelke SA et al. Structural basis for activation of fluorogenic dyes by an RNA aptamer lacking a G-quadruplex motif. Nat. Comm 9, 4542 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pleij CW, Rietveld K & Bosch L A new principle of RNA folding based on pseudoknotting. Nucleic Acids Res. 13, 1717–31 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de Gennes PG Simple Views on Condensed Matter, 138–148 (Cambridge University Press, Cambridge, 1990). [Google Scholar]
- 29.Carothers JM, Oestreich SC, Davis JH & Szostak JW Informational complexity and functional activity of RNA structures. J. Am. Chem. Soc 126, 5130–7 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Szostak JW Functional information: Molecular messages. Nature 423, 689 (2003). [DOI] [PubMed] [Google Scholar]
- 31.Klein DJ, Edwards TE & Ferré-D’Amaré AR Cocrystal structure of a class I preQ1 riboswitch reveals a pseudoknot recognizing an essential hypermodified nucleobase. Nat. Struct. Mol. Biol 16, 343–344 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang L, Wang J & Lilley DMJ The structure of the guanidine-II riboswitch. Cell Chem. Biol 24, 695–702.e2 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reiss CW & Strobel SA Structural basis for ligand binding to the guanidine-II riboswitch. RNA 23, 1338–1343 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grate D & Wilson C Laser-mediated, site-specific inactivation of RNA transcripts. Proc. Natl Acad. Sci. USA 96, 6131–6136 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Braselmann E et al. A multicolor riboswitch-based platform for imaging of RNA in live mammalian cells. Nat. Chem. Biol 14, 964–971 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sauer M & Paeschke K G-quadruplex unwinding helicases and their function. Biochem. Soc. Trans 45, 1173–1182 (2017). [DOI] [PubMed] [Google Scholar]
- 37.Chen MC et al. Structural basis of G-quadruplex unfolding by the DEAH/RHA helicase DHX36. Nature 558, 465–469 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Canty L, Hariharan S, Liu Q, Haney SA & Andrews DW Peak emission wavelength and fluorescence lifetime are coupled in far-red, GFP-like fluorescent proteins. PLoS One 13, e0208075 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Warner KD et al. Structural basis for activity of highly efficient RNA mimics of green fluorescent protein. Nat. Struct. Mol. Biol 21, 658–663 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee J & Goodey NM Catalytic contributions from remote regions of enzyme structure. Chem. Rev 111, 7595–624 (2011). [DOI] [PubMed] [Google Scholar]
- 41.Leontis NB & Westhof E Geometric nomenclature and classification of RNA base pairs. RNA 7, 499–512 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Otwinowski Z & Minor W Processing of X-ray diffraction data collected in oscillation mode. Meth. Enzymol 276, 307–26 (1997). [DOI] [PubMed] [Google Scholar]
- 43.Parkhurst JM et al. Robust background modelling in. J Appl. Cryst 49, 1912–1921 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sheldrick G A short history of SHELX. Acta Cryst. D 64, 112–122 (2008). [DOI] [PubMed] [Google Scholar]
- 45.Terwilliger TC Maximum-likelihood density modification. Acta Cryst. D 56, 965–72 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Terwilliger T et al. Decision-making in structure solution using Bayesian estimates of map quality: the PHENIX AutoSol wizard. Acta Cryst. D 65, 582–601 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Afonine P et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Cryst. D 68, 352–367 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McCoy AJ et al. Phaser crystallographic software. J. Appl. Cryst 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fisz JJ Another look at magic-angle-detected fluorescence and emission anisotropy decays in fluorescence microscopy. J. Phys. Chem. A 111, 12867–70 (2007). [DOI] [PubMed] [Google Scholar]
- 50.Enderlein J & Erdmann R Fast fitting of multi-exponential decay curves. Optics Comm. 134, 371–378 (1997). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article (and its supplementary information files). Atomic coordinates and structure factor amplitudes for the Mango-III-TO1-Biotin, Mango-III(A10U)-TO1-Biotin, and iMango-III-TO1-Biotin co-crystal structures have been deposited in the Protein Data Bank under accession codes 6E8S, 6E8T, and 6E8U, respectively.