

Abstract
Accurate DNA replication and segregation are critical for maintaining genome integrity and suppressing cancer. Metnase and EEPD1 are DNA damage response (DDR) proteins frequently dysregulated in cancer and implicated in cancer etiology and tumor response to genotoxic chemo- and radiotherapy. Here, we examine the DDR in human cell lines with CRISPR/Cas9 knockout of Metnase or EEPD1. The knockout cell lines exhibit slightly slower growth rates, significant hypersensitivity to replication stress, increased genome instability and distinct alterations in DDR signaling. Metnase and EEPD1 are structure-specific nucleases. EEPD1 is recruited to and cleaves stalled forks to initiate fork restart by homologous recombination. Here, we demonstrate that Metnase is also recruited to stalled forks where it appears to dimethylate histone H3 lysine 36 (H3K36me2), raising the possibility that H3K36me2 promotes DDR factor recruitment or limits nucleosome eviction to protect forks from nucleolytic attack. We show that stalled forks are cleaved normally in the absence of Metnase, an important and novel result because a prior study indicated that Metnase nuclease is important for timely fork restart. A double knockout was as sensitive to etoposide as either single knockout, suggesting a degree of epistasis between Metnase and EEPD1. We propose that EEPD1 initiates fork restart by cleaving stalled forks, and that Metnase may promote fork restart by processing homologous recombination intermediates and/or inducing H3K36me2 to recruit DDR factors. By accelerating fork restart, Metnase and EEPD1 reduce the chance that stalled replication forks will adopt toxic or genome-destabilizing structures, preventing genome instability and cancer. Metnase and EEPD1 are overexpressed in some cancers and thus may also promote resistance to genotoxic therapeutics.
INTRODUCTION
Accurate DNA replication and proper chromosome segregation to daughter cells are required to maintain genome stability and suppress cancer. DNA is under constant threat from endogenous and exogenous genotoxins that cause DNA lesions that block replication or reduce replication fidelity. DNA damage activates checkpoint signaling and DNA repair pathways, collectively termed the DNA damage response (DDR). The DDR promotes cell survival and suppresses cancer by promoting genome stability, and programmed cell death when damage is excessive. It is crucial to understand how DDR networks manage replication stress because DDR defects predispose to cancer, determine tumor response to chemo- and radiotherapy, and underlie several congenital conditions, including Seckel syndromes, neurological diseases, primordial dwarfism and premature aging syndromes (1–4). Thus, the DDR is an enticing target to augment cancer therapy (5–11). The DDR is a complex network of interacting/cross-talking pathways and changes in one pathway may lead to compensatory changes in other pathways that confer therapeutic resistance. A better understanding of DDR pathways can reveal synthetic lethal relationships that can be exploited to augment cancer therapy and devise personalized therapies (10,12–16).
DNA lesions, common fragile sites and repeated sequences impede replication, causing replication stress (17). Replication stress is also induced when DNA polymerase is inhibited, nucleotide pools are depleted [e.g. with hydroxyurea (HU)] and when MCM helicase decouples from DNA polymerase (18). Stressed replication forks can assume branched structures (‘chicken feet’) that resemble Holliday junctions of homologous recombination (HR) intermediates (19,20). Some branched DNA structures are toxic, or they may cause genome rearrangements, so a first line of defense is cleavage of stressed forks by structure-specific nucleases, creating double-strand breaks (DSBs). Broken forks are resected to single-stranded DNA (ssDNA) tails, which are first bound by RPA and then RAD51 to promote accurate fork repair/restart by HR (21–23). DDR and replication stress response proteins are subject to cell cycle regulation by cyclin-dependent kinase (CDK) that phosphorylates certain RPA residues before RPA is further phosphorylated by ATM, ATR, DNA-PK and Chk1/2 kinases, with final hyperphosphorylated RPA (pRPA) marked by phospho-Ser4/Ser8 (24–27). Thus, RPA bound to ssDNA and pRPA play critical roles in checkpoint activation and HR.
In human cells, several structure-specific nucleases have been implicated in replication fork restart in response to replication stress, including MUS81 with its EME2 binding partner, Metnase (SETMAR) and EEPD1 (23,28–31). Like many DDR proteins, MUS81 is ancient and conserved over >1 billion years from yeast to humans (32). MUS81 partners with EME2 to promote restart of stressed forks (31). EEPD1 arose later with the first homologs appearing ∼676 million years ago (Mya) in chordates, and is well conserved during vertebrate evolution (32). Metnase arose very recently (∼50 Mya) in monkeys, and is therefore unique to monkeys, apes and humans (33). Here, we focus on the two late-evolving nucleases and their roles in replication stress responses. EEPD1 cleaves forks stalled by HU and promotes Exo1-mediated resection to promote accurate fork repair/restart via HR (23,29,34). Metnase also promotes fork restart after HU stress in a nuclease-dependent manner (28,30), although its precise role is unclear. Metnase and EEPD1 are frequently up- or downregulated in cancer, and they may influence tumor response to therapy (23,35–41).
Nearly all genetic analyses of Metnase and EEPD1 have relied on gene knockdown or overexpression. One study of a Metnase knockout (KO) examined growth rate and etoposide sensitivity (42); no prior studies employed EEPD1 KO cells. siRNA-mediated knockdown is typically incomplete, and biological processes can be strongly and unpredictably affected by changes in gene/protein expression. For example, HR is stimulated by moderate overexpression of RAD51 (43,44), but HR is inhibited by high-level overexpression of both RAD51 and RAD52 (45). Here, we describe replication stress responses in complete CRISPR/Cas9 KOs of Metnase and EEPD1 in HeLa cells. The results confirm that Metnase and EEPD1 promote cell growth, genome stability, replication fork restart and resistance to replication stress. We present several novel results including differences in DDR signaling in each KO, recruitment of Metnase and dimethylation of histone H3 lysine 36 (H3K36me2) at stalled replication forks and a role for EEPD1, but not Metnase, in cleavage of stalled forks. A prior study showed that fork restart is significantly delayed in cells expressing nuclease-defective Metnase (30). Thus, rather than cleaving stalled forks, Metnase nuclease may act later by processing branched or flap intermediates that arise during HR-mediated fork restart. We further show that EEPD1/Metnase double-KO cells are no more sensitive to etoposide than either single KO. Together, these results indicate that Metnase and EEPD1 play distinct roles in a common replication stress response pathway.
MATERIALS AND METHODS
Cell culture, CRISPR/Cas9 genome editing and KO complementation
HeLa (CCL-2; ATCC) cells were grown in α-MEM (Thermo Fisher) supplemented with 10% (v/v) fetal bovine serum (Atlas Biotechnologies), 100 μg/ml penicillin/streptomycin (Thermo Fisher) and 1 mM sodium pyruvate (Thermo Fisher), at 37°C in 5% CO2. HEK293 cells expressing V5-tagged Metnase were constructed and cultured as described (46). CRISPR/Cas9 KO plasmids encoded spCas9, green fluorescent protein (GFP) and single-guide RNAs (sgRNAs) to target Metnase or EEPD1 (sc-417901 and sc-413405; Santa Cruz Biotechnology). For each gene, a three-plasmid mixture was used, each encoding an sgRNA targeting an exon sequence. Cells were transfected using Lipofectamine 3000 (Thermo Fisher). Briefly, 300 000 cells were seeded in each well of six-well dishes and incubated for 24 h. Before transfection, medium was replaced with Opti-MEM without antibiotics. Cells were transfected with 500 ng of CRISPR/Cas9 plasmid mixtures for 16 h, and transfection was confirmed by visualizing GFP by fluorescence microscopy. Cells were seeded to 96-well dishes (0.3 cells/well) 48 h after transfection, and clonal isolates were expanded and screened for Metnase or EEPD1 expression by western blot. KO mutations at target loci were identified by using the EnGen Mutation Detection Kit (New England Biolabs), and confirmed by sequencing target loci amplified by PCR. A double-KO cell line was constructed by transfecting EEPD1 KO #27 cells with pooled Metnase CRISPR vectors as above. Metnase KO cells were complemented by stable transfection with plasmid pFLAG-CMV2-Metnase (47), selected for 14 days in medium with 400 μg/ml G418. EEPD1 KO cells were complemented by stable transfection with a C-terminal FLAG-Myc-tagged EEPD1 plasmid derived from pCMV6-Entry (NM_030636; Origene, Rockville, MD), also selected with G418.
Replication stress agents and irradiation
HU and mitomycin C (MMC) were purchased from Sigma-Aldrich, etoposide was purchased from Tocris, and fresh solutions were prepared for each experiment. Cells were irradiated with a 5000 Ci γ-ray cesium source.
Protein detection by western blot
Whole cell lysates were prepared in M-PER buffer with 1× protease inhibitor and protein phosphatase inhibitor cocktail (Thermo Fisher). Soluble fractions of lysates were collected and protein was quantified by using the bicinchoninic acid assay. Total protein (30 μg) was resolved on a 10% Nu-PAGE Bis-Tris gel (Thermo Fisher) in 1× MOPS buffer and transferred onto 0.45-μm PVDF-FL membranes (IPFL07810; Sigma) in 1× Tris-glycine buffer with 20% methanol (v/v). Milk (5%) in TBST was used as a blocking buffer for all antibodies using horseradish peroxidase (HRP) detection; blots processed for Li-COR near-IR fluorescent imaging were blocked in 5% bovine serum albumin in TBST, or commercial blocking buffer (927-50000; LI-COR Biosciences). Primary antibodies were purchased from Santa Cruz Biotechnology and used at 1:1000 dilution except as noted: anti-EEPD1 rabbit monoclonal antibody (C3; produced by the Hromas lab); anti-SETMAR (ab-129455 or ab-3823; Abcam); anti-MUS81 (sc-53382), anti-GAPDH (sc-8432; 1:2000), anti-β-actin (sc-47724; 1:5000), anti-RPA32 (sc-58770; 1:2000), anti-pRPA pS4/8 (A300-2454; Bethyl Laboratories; 1:2500), anti-Chk1 (13303) and anti-phospho-Chk1 pS345 (26105; Cell Signaling Technology Inc.; 1:750), anti-histone H3 (sc-8654; 1:5000), anti-γ-H2AX phospho-S139 (05-636; Millipore; 1:4000) and anti-V5 (R960-25, Invitrogen). Secondary HRP conjugated antibodies were anti-goat/rabbit/mouse (Jackson ImmunoResearch Laboratories; 1:10 000), and secondary near-IR fluorescent anti-rabbit 800 nm or anti-mouse 680 nm (925-32211 and 925-68070; LI-COR Biosciences; 1:25 000).
Cell plating efficiency, proliferation and survival assays
Plating efficiency was determined by seeding appropriate numbers of cells in 10-cm dishes, incubating for 10–12 days and counting colonies stained with 0.5% crystal violet in 70% methanol. Cell proliferation rates were determined by seeding 104 cells in each well of 96-well dishes in growth medium, and percent confluence was captured hourly using an IncuCyte system (Essen BioScience). Cell survival after etoposide or MMC treatment was determined by seeding appropriate numbers of cells in fresh media with drugs. Cells were either chronically exposed to MMC or acutely exposed to etoposide for 90 min followed by two washes with phosphate-buffered saline (PBS), re-seeding at appropriate densities. Cells were incubated for 11 days and colonies were stained with crystal violet and counted.
Nuclear morphology and γ-H2AX foci assays
Nuclear abnormalities reflecting genome instability and mitotic catastrophe were determined as described (23). Induction and repair of DSBs was scored as γ-H2AX foci in cells on chamber slides after γ-irradiation as described (23). Images were acquired using a 63× oil objective and a Zeiss Axio Imager Z2 microscope equipped with Zen Blue software (Carl Zeiss Microscopy). Images were obtained as Z-stack sections of 0.2 μm per section containing 18 Z-stacks per channel. Image processing for foci quantification involved separating channels and producing a maximum projection file to identify and count both the number of foci and the number of cells using a combination of ImageJ (http://rsb.info.nih.gov/ij/) and Cell Profiler (http://www.cellprofiler.org/) software packages with the following settings: minimum size 5; maximum size: 100; rolling ball: 5. Approximately 200–300 cells were analyzed per condition, and nuclei with >5 foci were counted as positive.
Cell cycle phase-specific analysis of DNA damage responses by immunofluorescence flow cytometry
DDR responses (pRPA, γ-H2AX and pChk1) in specific cell cycle phases were determined following etoposide treatment by multiparameter flow cytometry. Cells (5 × 105) were seeded into T-25 flasks, incubated for 24 h and treated for 90 min with 20 μM etoposide or mock treated. Cells were washed with PBS (37°C), fresh medium was added and cells were incubated for indicated recovery times, trypsinized, harvested by centrifuging at 560 × g for 5 min, washed once with PBS, fixed with 4% paraformaldehyde in PBS for 10 min, washed once with PBS and stored at 4°C in PBS until further processing. Cells were permeabilized with 0.5% Triton X-100 in PBS, washed with PBS, incubated with primary antibodies overnight at 4°C, washed twice with PBS, incubated with secondary antibodies for 1 h at room temperature, washed twice with PBS and suspended in PBS containing 0.25 μl/ml SYTOX AADvanced (Thermo Fisher) and 80 μg/ml RNAse A. Cells were stored at 4°C in the dark until analyzed with a three-laser CyAn flow cytometer and FlowJo software.
DNA replication analysis by EdU incorporation and DNA fiber assays
Replication recovery after HU arrest was determined by 5-ethynyl-2′-deoxyuridine (EdU) incorporation as follows. Cells (5 × 105) were seeded into T-25 flasks, incubated for 24 h, treated with 10 mM HU for 1 h or mock treated, washed three times with PBS (37°C), fresh growth medium was added and 10 μM EdU was added for 30 min at indicated times during recovery from HU. After the EdU pulse, cells were trypsinized, harvested, fixed with gradual addition of 70% chilled ethanol and stored at 4°C until further processing. Cells were processed using the Click-iT Cell Reaction Kit (Thermo Fisher) using manufacturer’s instructions, suspended in PBS containing 0.25 μl/ml SYTOX AADvanced and 80 μg/ml RNase A, incubated at 4°C overnight in the dark to allow the cell cycle dye to saturate and analyzed by flow cytometry as above. Single-molecule analysis of replication fork restart was performed by DNA fiber analysis as described (23,28).
Single cell electrophoresis (comet) and iPOND assays
Comet assays were performed according to manufacturer’s instructions (Trevigen). Briefly, 50 000 cells were seeded into each well in six-well dishes, incubated for 24 h and treated with 10 mM HU for 18 h. Cells were washed and harvested in the dark and stored in cold PBS. Cells were counted and 105 cells/ml were combined with molten LMA-agarose at a ratio of 1:10, and 50 μl aliquots were transferred to each well of two-well comet slides. Slides were incubated in the dark at 4°C for 10 min, and then immersed in cold lysis solution for 30–60 min. Excess buffer was drained and slides were incubated for 20 min in freshly prepared alkaline denaturation buffer (200 mM NaOH, 1 mM EDTA, pH >13). Electrophoresis was performed in alkaline denaturation buffer at 25 V for 30 min. Slides were washed twice with dH2O, and once with 70% ethanol. Slides were dried at 37°C for 20 min, stained with 50 μl of SYBR Gold for 30 min and washed briefly with dH2O. Images were acquired by epifluorescence microscopy and lengths of comet tails (48) were measured using ImageJ software. iPOND was performed as described (23,49).
RESULTS
Construction and initial characterization of Metnase and EEPD1 KO cell lines
Pools of CRISPR/Cas9 vectors targeting either Metnase or EEPD1 were transfected into HeLa cells and ∼40 candidate subclones were isolated from each set. HeLa cells are pseudo-triploid, with most chromosomes present in three to four copies, including three copies each of chromosomes 3 and 7 (50,51) that harbor Metnase and EEPD1 genes, respectively. Western blot analysis indicated partial or complete KO of all copies of either Metnase or EEPD1 in ∼30% of candidates. Two complete KOs from each set were identified by western blot, and neither KO altered Chk1 or MUS81 expression (Figure 1A). KO mutations were confirmed by T7 endonuclease assay, and insertion/deletion mutations near CRISPR targets were confirmed by sequencing PCR products from genomic DNA (Supplementary Figure S1). Knockdown of Metnase or EEPD1 in HEK293 and A549 cells reduces cell growth (23,28), and overexpression of Metnase increases cell growth (28). Plating efficiencies of parent HeLa cells and Metnase or EEPD1 KOs were indistinguishable (Figure 1B), but cell growth rates showed slight, statistically significant reductions in both sets of KO cell lines (Figure 1C). The slight growth rate reduction by Metnase KO in HeLa cells mirrors that observed in DLD-1 colon cancer cells (42). Knockdown of Metnase or EEPD1 sensitizes cells to a variety of genotoxins (23,28), and we confirmed that both sets of KO mutants were hypersensitive to the TopoIIα poison etoposide and the cross-linking agent MMC (Figure 1D). DLD-1 Metnase KO cells are similarly hypersensitive to etoposide (42). To rule out off-target CRISPR effects, Metnase and EEPD1 KO cells were transfected with FLAG-tagged wild-type (WT) Metnase or FLAG- and Myc-tagged WT EEPD1, respectively. Derivatives expressing complementing proteins at levels similar to WT parent cell lines were isolated, and HU resistance was almost fully restored in complemented cell lines (Supplementary Figure S2), indicating that KO sensitivities are not affected by off-target mutations.
Figure 1.

Characterization of Metnase and EEPD1 KO cell lines. (A) Western blot analysis of WT HeLa cells and derivatives with either Metnase or EEPD1 KO mutations. Two KO candidates each for Metnase (#25 and #39) and EEPD1 (#22 and #27) are shown. Blots were probed with antibodies to Metnase and EEPD1, and to MUS81, Chk1 and β-actin as controls. (B) Plating efficiency was calculated as the ratio of colonies formed per cell plated. Values are averages ± standard deviation (SD) (n = 6). (C) Cell proliferation rates determined as percent confluence in an IncuCyte device. Values are averages ± SD (n = 3); statistical analysis of slopes was conducted using linear regression (GraphPad Prism). (D) Sensitivities to etoposide and MMC. (Left) Cells were treated with etoposide for 18 h, harvested and appropriate numbers of cells were seeded in 10-cm dishes, incubated for 12 days before colonies were counted. Values are averages (+SD); n = 3 or 6 determinations per condition. (Right) Cells were incubated in medium with MMC for 14 days and colonies were counted. Values are averages (+SD); n = 4 for treated cells and n = 2 for non-treated cells. Statistics were calculated using unpaired t-tests. In this and subsequent figures, P values are indicated as *P < 0.05, **P < 0.01 and ***P < 0.0001.
KO of Metnase or EEPD1 increases genome instability, suppresses DSB repair and alters DDR signaling
Etoposide causes replication stress and inhibits chromosome decatenation (52), causing genome instability and mitotic catastrophe revealed as micronuclei, fragmented nuclei (‘blebs’) and anaphase bridges (53–57). Compared to WT, Metnase and EEPD1 KO cells showed significantly increased abnormal nuclei after treatment with etoposide (Figure 2A and B); slight increases were observed in untreated cells, but these differences were not statistically significant. Metnase and EEPD1 are implicated in DSB repair by non-homologous end joining (NHEJ) and HR, respectively (23,58–60). We tested the ability of KO cells to signal DSB induction and repair DSBs induced by γ-rays by measuring the kinetics of γ-H2AX foci formation and resolution. Metnase and EEPD1 KO cells showed similar γ-H2AX induction to WT but repair was significantly delayed (Figure 2C and D).
Figure 2.

Metnase and EEPD1 KO cells display enhanced genome instability and delayed DSB repair after IR. (A) Representative images of normal and abnormal DAPI-stained nuclei; arrows indicate abnormalities associated with genome instability. (B) Percent abnormal nuclei in cells treated with 20 μM etoposide for 90 min, 48 h recovery or mock treated. A total of 258–410 cells were scored per condition (average = 374). Statistics were calculated by Fisher exact tests. (C) Representative images of γ-H2AX foci in DAPI-stained nuclei after 4 Gy γ-ray and mock irradiated controls. (D) Percentage of cells with >5 γ-H2AX foci. An average of 240 cells were scored per condition (range = 195–315). Statistics were calculated by Fisher exact tests.
EEPD1 knockdown reduces pRPA in response to HU-induced replication stress (23). To further explore Metnase and EEPD1 roles in DDR signaling, we first measured pRPA induction and resolution by western blot in response to stress induced by etoposide and HU. WT cells showed marked pRPA induction after 1.5 h treatment with 20 μM etoposide and this largely resolved after a 48 h recovery period, but pRPA persisted in Metnase KO cells (Figure 3A). We next examined pRPA in response to a 3 h treatment with 3 mM HU. WT cells showed strong pRPA induction after HU treatment, and this signal resolved during a 9 h recovery (Figure 3B). EEPD1 KO cells showed reduced pRPA immediately after HU treatment, consistent with reduced fork cleavage and resection with EEPD1 knockdown (23). After the 9 h recovery, EEDP1 KO cells showed a robust pRPA signal, perhaps due to eventual fork collapse and defective repair. Similar to WT, pRPA was strongly induced by HU in Metnase KO, but instead of declining during recovery, pRPA persisted in the mutant (Figure 3B); thus, Metnase KO also displays a repair defect. The different initial pRPA signals are consistent with comet assay results (below). Although both KOs display repair defects, these results indicate that Metnase and EEDP1 have distinct effects on replication stress responses.
Figure 3.
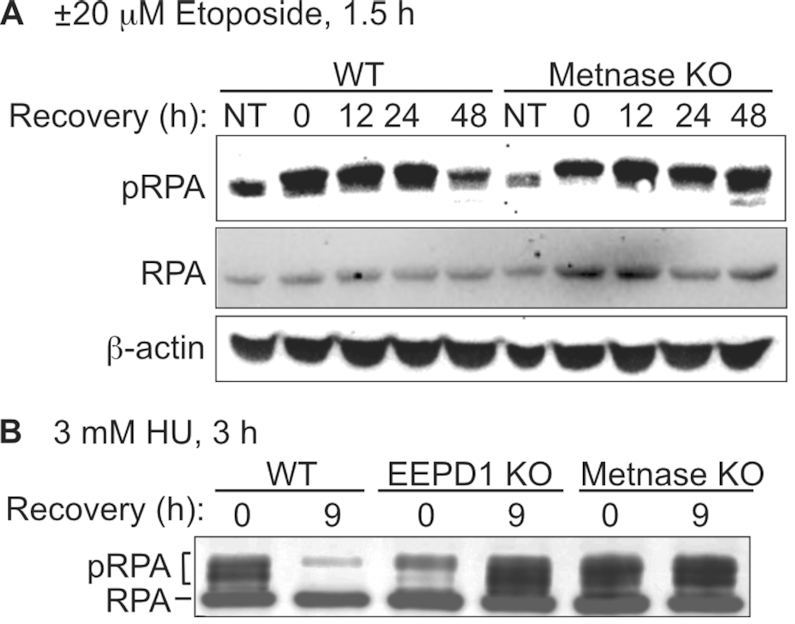
Metnase and EEPD1 differentially regulate DDR signaling. (A) WT and Metnase KO cells were treated with 0 or 20 μM etoposide for 1.5 h, and allowed to recover for indicated times. Blots probed with antibodies to RPA32 phospho-S4/S8 and β-actin as a loading control. (B) WT, Metnase KO and EEPD1 KO cells were treated with 3 mM HU for 3 h, harvested immediately or after a 9 h recovery and blots were probed with antibodies to RPA32 to detect both native and phosphorylated forms.
Western blots reveal DDR signaling within an entire cell population. Stress responses can vary with cell cycle phase, such as increased γ-H2AX in S phase reflecting broken replication forks (61) and suppression of resection in G1 phase due to limited CDK activity (62,63). To explore Metnase and EEPD1 roles in DDR signaling during the cell cycle, we used multiparameter flow cytometry to measure DNA content and pRPA, pChk1 or γ-H2AX signals in response to a 1.5 h treatment with 20 μM etoposide. We chose etoposide for these experiments because preliminary tests with HU showed insufficient sensitivity to detect signals in single cells by flow cytometry. Cells were analyzed before treatment, immediately after treatment (time = 0) and after a 48 h recovery period. This approach also revealed DDR signaling in cells with sub-G1 DNA content (dying/apoptotic cells) or greater than G2 DNA content (hyper-G2) reflecting over-replication, re-replication or other mitotic abnormalities (52). Cell cycle distributions were similar among untreated WT, Metnase KO and EEPD1 KO cells, and these distributions were not significantly altered during the 1.5 h etoposide treatment (Supplementary Figure S3). After 48 h recovery, there were significant increases in both sub-G1 and hyper-G2 populations in WT and KO cells, and EEPD1 KO cells in particular had reduced S and G2 fractions, and significantly increased sub-G1 fractions (Supplementary Figure S3).
The percentages of pRPA+ cells before and after etoposide treatment are shown in Figure 4A and Supplementary Figure S4. Among untreated WT and KO cells, pRPA+ cells were rare among G1, S and G2 cells, but common (35–50%) in hyper-G2 cells and slightly increased in sub-G1 cells, consistent with significant stress in these (rare) populations. After 1.5 h etoposide treatment, pRPA signals increased dramatically in all cell cycle phases except G1, consistent with limited resection in G1 phase (62,63). However, 48 h after etoposide treatment, pRPA+ cells comprised ∼35% of G1 cells in WT and Metnase KO, and nearly 80% in EEPD1 KO. During this 48 h recovery period, pRPA+ fractions in S- and G2-phase cells declined in WT and Metnase KO, but remained elevated in EEPD1 KO. The similar pRPA+ values in WT and Metnase KO after 48 h (Figure 4A) do not match higher pRPA+ in Metnase KO by western blot (Figure 3A), probably because western blots score the entire cell population (dead/dying cells, doublets, aggregates, cell debris), whereas flow cytometry scores only intact singlet cells. The distinct pRPA signaling in Metnase and EEPD1 KO cells further indicates their distinct roles in replication stress responses.
Figure 4.

Cell cycle phase-specific DDR signaling in WT, Metnase KO and EEPD1 KO cells. Cells were incubated with or without 20 μM etoposide for 1.5 h and processed for flow cytometric detection of DNA content and pRPA, γ-H2AX or pChk1. (A) Average percentages (+SD) of pRPA+ cells in G1, S and G2/M cell cycle phases for two determinations, and in cells with sub-G1 (<G1) or hyper-G2 (>G2) DNA content. Total percentage of pRPA+ cells in all phases (including sub-G1 and hyper-G2) is shown to the right. A representative scatter plot of the pRPA flow cytometry data is shown in Supplementary Figure S4. (B) As described for panel (A) but probed for γ-H2AX; representative scatter plot is shown in Supplementary Figure S5. (C) As described for panel (A) but probed for pChk1; representative scatter plot is shown in Supplementary Figure S6. Statistics were calculated using t-tests with Welch’s correction.
Etoposide causes replication stress and induces DSBs during all phases of the cell cycle, although effects are enhanced in S phase (64,65). DSBs marked by γ-H2AX were more prevalent in sub-G1 and hyper-G2 cells in untreated WT or KO populations, and etoposide greatly increased γ-H2AX+ fractions in all cell cycle phases (Figure 4B, Supplementary Figure S5). Metnase KO and EEPD1 KO cells showed significantly higher γ-H2AX+ fractions than WT in G1, S and G2 phases after etoposide treatment, and this was generally true after a 48 h recovery (Figure 4B). The percentage of γ-H2AX+ G2-phase cells was significantly higher in Metnase KO than WT or EEPD1 KO after 48 h recovery, which may reflect DSB repair deficiency and/or additional DSBs due to the absence of Metnase enhancement of TopoIIα-mediated chromosome decatenation (40,41,66). Chk1 is phosphorylated and activated in response to DSBs, RPA-ssDNA and ATR activation, leading to cell cycle arrest (22,24,25,67). Although pRPA and γ-H2AX responses to etoposide differed in WT and KO cells, etoposide induction of pChk1 was strong in all cell cycle phases (including sub-G1 and hyper-G2 cells), and was largely unaffected by the absence of Metnase or EEPD1 (Figure 4C, Supplementary Figure S6). This suggests that Chk1 is activated in response to etoposide independently of Metnase and EEPD1.
Replication fork restart is delayed in Metnase KO and EEPD1 KO cells
Knockdown of Metnase or EEPD1 delays restart of HU-stalled replication forks (28,30,68). We compared fork restart in WT and KO cell lines by measuring EdU incorporation into replicated DNA following a 30 min EdU pulse at intervals following release from HU (Figure 5A). In unstressed controls, EdU incorporation was indistinguishable between WT and Metnase KO cells (Figure 5B). EdU incorporation was sharply reduced after release from HU, but Metnase KO cells showed greater, statistically significant reductions in EdU incorporation 0–2 h after release. EdU incorporation almost fully recovered 2 h after HU release in WT cells, but Metnase KO cells did not recover until 4 h after release (Figure 5B). EEPD1 KO cells responded to HU stress similarly to Metnase KO, with lower EdU incorporation after HU and delayed recovery to pre-stress levels (Figure 5C). However, unstressed EEPD1 KO cells showed lower EdU incorporation than WT and Metnase KO (Figure 5C), perhaps reflecting delayed or failed restart of forks stalled at spontaneous DNA lesions and natural pause sites. These results indicate that Metnase and EEPD1 both enhance replication recovery after stress.
Figure 5.

Delayed fork restart in Metnase KO and EEPD1 KO cells. (A) Replication restart after HU treatment was assayed by EdU incorporation using flow cytometry as diagrammed. (B) Plots of EdU incorporation (mean fluorescence intensity; MFI) as a function of time during HU recovery in WT and Metnase KO cells. Values are averages ± SD for two determinations (all error bars are smaller than symbols). Values for control cells not treated with HU are indicated by NT; EdU was added to cultures for 30 min immediately after HU treatment (time = 0), or at indicated recovery times. Statistics were calculated using t-tests. (C) Data for WT and EEPD1 KO plotted as in panel (B). (D) Experimental scheme and examples of DNA fibers showing IdU (red) and CldU (green) incorporated before or after HU treatment, respectively. (E) Percentages of stopped forks in HU-treated or untreated cells; 87–330 fibers were scored per condition (average = 117). Statistics were calculated using Fisher exact tests. (F) Ratios of IdU:CldU lengths among restarted forks (stopped forks excluded). Plotted are average ratios (±SEM) for three determinations per condition; for each determination, 25–84 fibers were scored (average = 41). IdU:CldU ratios of ∼1.0 indicate rapid fork restart after HU release; greater ratios indicate delayed restart. Top graph shows data from untreated cultures, bottom from HU-treated cultures. (G) Percentages of restarted forks that showed significant delay (IdU:CldU ratios >1.5). Data were pooled from the three determinations described in panel (F). Statistics were calculated by Fisher exact tests for 76–251 restarted fibers per condition (average = 124).
We then assessed replication fork restart and fork progression by DNA fiber analysis, with 20 min pulses of 5-iodo-2′-deoxyuridine (IdU) and 5-chloro-2′-deoxyuridine (CldU) flanking a 60 min HU treatment (Figure 5D). Compared to WT, the percentage of forks that failed to restart (‘stopped forks’) was significantly higher in EEDP1 KO, but not in Metnase KO (Figure 5E). Although this might suggest that Metnase has no role in fork restart, measurements of pre- and post-stress fiber lengths indicate otherwise. Ratios of pre- (IdU) and post-stress (CldU) fiber lengths were increased in both Metnase KO and EEDP1 KO cells (Figure 5F and G). Increased IdU:CldU ratios indicate reduced CldU incorporation after release from stress, i.e. delayed fork restart and/or slower progression (69). IdU is incorporated prior to HU treatment, so we pooled IdU fiber length measurements from treated and untreated cultures and found that replication speed is not reduced in Metnase or EEPD1 KO cells (Supplementary Figure S7). In WT and mutant cells, CldU lengths were significantly shorter in HU-treated than control cells, but greater reductions were observed in Metnase KO and EEDP1 KO (∼2.2-fold) compared to WT (1.45-fold), another measure of delayed restart in the mutants. Thus, Metnase KO cells were able to restart most stalled forks during the 20 min recovery period similar to WT, but restart was delayed. In EEDP1 KO cells, restart was less frequent, and when forks did restart it was delayed compared to WT cells.
Distinct roles for Metnase and EEPD1 nucleases in processing stressed replication forks
Metnase and EEPD1 are structure-specific endonucleases capable of cleaving branched structures in vitro, such as ‘chicken feet’ that may arise during replication stress (23,30). EEPD1 knockdown studies indicate that EEPD1 plays a direct role in cleaving stalled replication forks (23,29,34), but the role of the Metnase nuclease domain in DNA repair and fork restart is unclear (30,59). We investigated this by using comet assays (Figure 6). EEPD1 KO cells displayed a marked defect in HU-induced DNA breaks, confirming knockdown results (23), but such breaks were induced to the same extent in Metnase KO and WT cells. Thus, unlike EEPD1, our results rule out Metnase contributions to DNA breaks after an 18 h HU treatment. Other potential roles for Metnase nuclease, and its protein methylase, in promoting fork restart are discussed below.
Figure 6.

Metnase does not cleave stalled replication forks. (A) Representative images of alkaline comet assays in untreated or HU-treated WT or Metnase KO cells. (B) Average comet lengths (±SD) of 10 determinations per condition; statistics were calculated by t-tests.
Metnase and H3K36 dimethylation at stalled replication forks
EEPD1 is recruited to and cleaves replication forks stalled by HU (23), but fork cleavage occurs normally in Metnase KO cells (Figure 6). Metnase is recruited to frank DSBs induced by ionizing radiation and I-SceI nuclease (60), it promotes restart of stalled forks (28) (Figure 5) and fork restart is delayed in cells expressing nuclease-defective Metnase (30). We therefore tested whether Metnase is recruited to stalled forks using the iPOND assay. Consistent with prior results (23,70), proliferating cell nuclear antigen (PCNA) is rapidly evicted from HU-stalled replication forks (Figure 7A). V5-tagged Metnase is recruited to HU-stalled forks within 30 min (Figure 7A). Endogenous (untagged) Metnase is also recruited to HU-stalled forks (Figure 7B and C). Thus, both Metnase and EEPD1 are recruited to stalled forks, but only EEPD1 is involved in fork cleavage, suggesting that Metnase nuclease acts later. Metnase dimethylates H3K36 at DSBs (60), so we explored whether H3K36me2 was present at stalled replication forks. Nucleosomes, comprising histones H2A, H2B, H3 and H4, are evicted ahead of replication forks and reassembled on daughter DNA strands, accounting for the delayed appearance of H3 on nascent DNA upon HU treatment (Figure 7A), as reported previously (70). We detected H3K36me2 at HU-stalled forks with similar kinetics to that of H3 and Metnase (Figure 7A). Although suggestive, further studies with SET-defective Metnase are required to conclusively demonstrate that Metnase directly dimethylates H3K36 at stalled forks (discussed below).
Figure 7.

Metnase is recruited to stalled replication forks. (A) iPOND analysis of V5-tagged Metnase, PCNA and H3K36me2 in HEK293 cells. (B) iPOND analysis of endogenous Metnase in HeLa cells. For iPOND, cells were incubated for 10 min in medium with 10 μM EdU to label nascent DNA, and then treated with 3 mM HU for 0.5–4 h to stall replication forks, or not treated (0 h, NT) as control. Histone H3 served as a loading control. Results are representative of at least two independent experiments. (C) Quantitation of iPOND capture of endogenous Metnase relative to histone H3 by scanning densitometry.
Evidence for epistasis between Metnase and EEPD1
Metnase and EEPD1 KO cells show comparable sensitivities to replication stress agents (Figure 1D). To explore whether this reflects epistasis, Metnase was knocked out in EEPD1 KO cells, producing a double-KO mutant (Figure 8A). As shown in Figure 8B, Metnase/EEPD1 double-KO cells are no more sensitive to etoposide than either single KO. This suggests that, at least for etoposide-induced stress, Metnase and EEPD1 function in the same stress response pathway (epistatic).
Figure 8.
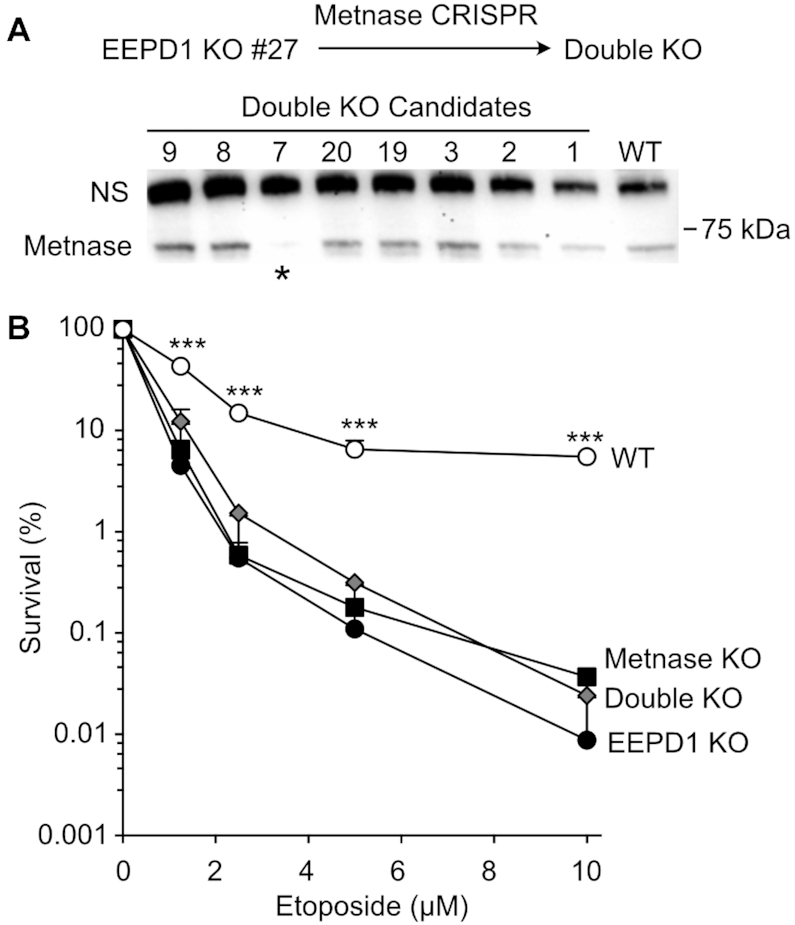
Epistasis of Metnase and EEPD1. (A) KO scheme and western blot of Metnase/EEPD1 double-KO candidates. (B) Etoposide sensitivity of WT, single KO and double KO (candidate #7, panel A). Values are averages (+SD) for three to six determinations.
DISCUSSION
Metnase and EEPD1 are two relatively late-evolving structure-specific nucleases that confer resistance to genotoxins, and promote restart of replication forks and genome stability. We present here several novel findings, including demonstration of their distinct effects on DDR signaling, distinct roles in stalled fork processing, recruitment of Metnase to stalled forks, evidence of H3K36me2 at stalled forks, and epistasis between Metnase and EEPD1. The growth and damage sensitivity phenotypes of Metnase and EEPD1 KO cells generally mirror those of knockdowns, confirming prior knockdown results and allaying concerns related to residual expression in knockdown cells. As DNA repair and replication stress factors, these proteins evolved relatively late and should thus be regarded as repair/replication augmentation factors. Hence, it is not surprising that neither protein is essential for cell viability.
Metnase and EEPD1 enhance cell growth (23,28,42,66) (Figure 1). Metnase promotes NHEJ (all cell cycle phases), replication fork restart (S phase) and chromosome decatenation (G2/M) (58,60), so Metnase defects could slow progression throughout the cell cycle. EdU incorporation during unstressed growth, a combined measure of DNA replication speed and the number of active replication forks, was unaffected in Metnase KO cells but reduced in EEPD1 KO cells (Figure 5B and C). Neither Metnase nor EEPD1 affects replication speed (Supplementary Figure S7), suggesting that reduced EdU incorporation in EEPD1 KO cells is due to fewer active replication forks and/or delayed fork restart when forks encounter spontaneous DNA damage or natural pause sites, such as common fragile sites (71,72). This would delay S-phase progression and increase the fraction of S-phase cells, as observed in unstressed EEPD1 KO cells (Supplementary Figure S3). Indirect evidence suggests that EEPD1 is important for replication of common fragile sites, as EEPD1 is required for RAD52-mediated repair of stressed forks in BRCA1-mutant cells (73), and RAD52 is critical for fragile site replication (74).
EEPD1 cleaves stalled replication forks, interacts with Exo1 and thereby promotes Exo1-mediated resection of broken forks (23,29,69). This produces ssDNA required for RPA binding/phosphorylation and HR-dependent fork restart. Metnase also enhances Exo1-dependent resection of stalled replication forks (75). Here, pRPA serves as a proxy for resection/ssDNA at DSBs, and induction/resolution of pRPA and γ-H2AX are proxies for DSB induction/repair. Unlike HU treatment, where EEPD1 (but not Metnase) is required to induce DSBs (Figure 6) (23), neither Metnase nor EEPD1 is required for DSB induction by etoposide (Figure 4B, Supplementary Figure S5). In addition, etoposide induces DSBs in all cell cycle phases, as we observed (Figure 4B, Supplementary Figure S5), although it is more effective (and more cytotoxic) in S phase (65). This is consistent with findings that etoposide-poisoned TopoII cleavage complexes are processed to DSBs by proteolysis and TTRAP/TDP2, by collisions with transcription machinery and indirectly through etoposide production of reactive oxygen species (65,76,77). In contrast, HU, camptothecin and aphidicolin only induce DSBs in S phase (52). Both Metnase and EEPD1 KOs show persistent γ-H2AX in G1/S cells, and Metnase KO shows persistent γ-H2AX in G2 cells (Figure 4B, Supplementary Figure S5), indicating that both proteins contribute to repair of etoposide DSBs. pRPA is prevalent in S- and G2-phase cells after etoposide, but not in G1 cells (Figure 4A, Supplementary Figure S4), consistent with suppressed resection in G1 (62,63). Nonetheless, 48 h after release from etoposide, significant pRPA signals are seen in G1 cells in WT and both mutants, potentially reflecting delayed resection in G1 and/or progression of S/G2 cells through mitosis due to failed G2 checkpoint arrest (78). The strong pRPA signals in EEPD1 KO cells after etoposide treatment (Figure 4A, Supplementary Figure S4) contrast with reduced pRPA after HU in EEPD1 knockdown cells (23). The dependence of pRPA on EEPD1 after HU is a downstream consequence of the requirement for EEPD1 cleavage of HU-stalled forks (23); here, we show that both induction and resection of etoposide DSBs are independent of EEPD1 and Metnase (Figure 4A and B, Supplementary Figures S4 and S5). Since Metnase and EEPD1 both interact with Exo1 (29,69,75), they may function redundantly to promote Exo1 resection and pRPA signaling. The greater persistence of pRPA in Metnase KO versus EEPD1 KO cells treated with etoposide (Figure 4A, Supplementary Figure S4) may reflect differential effects on resection and/or repair. It will be interesting to determine whether resection and pRPA signaling are suppressed in Metnase/EEPD1 double-KO cells.
Chk1 is activated in response to ssDNA; hence, pChk1 signals after etoposide treatment largely paralleled the pRPA results, i.e. low pChk1 in G1 cells despite significant G1 DSBs (γ-H2AX). After a 48 h recovery from etoposide, both pRPA and pChk1 are elevated in all cell cycle phases. Despite differences in pRPA and γ-H2AX among WT, EEPD1 KO and Metnase KO cells (Figure 4A and B, Supplementary Figures S4 and S5), Chk1+ fractions were similar among the three cell types (Figure 4C, Supplementary Figure S6). This may reflect persistent Chk1 activation independent of repair kinetics during the assay period. As noted above, EEPD1 knockdown suppresses events downstream of fork cleavage in response to HU, and this includes pChk1 suppression (23), contrasting with the strong pChk1 induction by etoposide in EEPD1 KO cells.
EEPD1 is a structure-specific nuclease that promotes DSB repair and genome stability, is recruited to and promotes HR-mediated restart of replication forks by cleaving stalled forks, and promotes Exo1-dependent resection (23,69). Metnase is a structure-specific nuclease that cleaves branched DNA structures and flaps (29,79), promotes DSB repair by NHEJ (30,58–60,79,80) and is recruited to and promotes fork restart (Figures 5 and 7) (28,30,68,75). Importantly, fork restart is delayed in cells expressing nuclease-defective Metnase (30). There are several, not mutually exclusive ways that Metnase could promote fork restart. Metnase promotes Exo1-dependent resection by mediating Exo1 recruitment to stalled forks (75). Metnase does not cleave HU-stalled forks (Figure 6), yet its nuclease is important for timely fork restart (30). Thus, Metnase may act later, processing branched or flap structures that arise during HR-mediated fork restart (30,58,59,79,80), analogous to FEN1, a related 5′ flap nuclease (81). Metnase is a SET protein methylase, and the SET domain is also important for fork restart (68). The Metnase SET domain enhances its nuclease, particularly cleavage of branched DNA structures (68). Metnase dimethylates H3K36 at DSBs to promote NHEJ by enhancing recruitment of Ku70 and NBS1 to DSBs (60). Although H3K36me2 appears at stalled forks with similar kinetics as H3 and Metnase (Figure 7A), confirmation of direct role for Metnase in this modification awaits further tests in cells expressing SET-defective Metnase. Nucleosomes are reassembled on new daughter duplexes with equal proportions of parental and naïve histones, and parental H3 and H4 histones retain their modifications during reassembly into chromatin to affect epigenetic memory (82), raising the question whether H3K36me2 is present ahead of the fork and then recycled, or produced de novo in replicated chromatin in response to stress. This is important because chromatin modifications regulate many aspects of DNA dynamics, including replication stress responses and DSB repair (83–85). If Metnase dimethylates H3K36 at stalled forks, this could underlie Metnase promotion of fork restart by regulating DDR factor recruitment to stalled forks, as it does at I-SceI DSBs (60), or by limiting nucleosome eviction (86) to protect ssDNA or branched structures from adverse nucleolytic attack. Another SET protein, G9a/KMT1C, methylates histone H3K56 to promote PCNA docking to chromatin in G1 that is important for (unstressed) DNA replication (87). Current evidence suggests that Metnase and EEPD1 operate epistatically to promote replication fork restart (Figure 8B), but do so by distinct mechanisms (Figures 3, 4 and 6). Future studies comparing fork restart and other stress responses of single- and double-KO cells will further clarify epistatic relationships of these proteins. We propose a model in which restart of HU-stalled forks is promoted by EEPD1 cleavage to initiate HR, and that Metnase may have roles in processing HR intermediates and/or DDR recruitment via H3K36 dimethylation, distinct from the MUS81 pathway (Supplementary Figure S8).
Defects and/or altered expression of DDR factors are common in cancer. Although there is no evidence of functional or driver mutations in Metnase or EEPD1 in the COSMIC or IntOGen cancer databases (88–90), altered expression of these proteins is common in many cancers. Each of the following represent >2-fold expression changes (P < 0.0001): Metnase is overexpressed in colon carcinoma and adenoma (91), and underexpressed in renal cell carcinoma (92) and melanoma (93). Metnase overexpression increases resistance to TopoIIα inhibitors in breast cancer and leukemia cells (40,41). EEPD1 is overexpressed in colorectal (23,94), brain (95) and colon cancer (96), and underexpressed in breast carcinomas (including male breast cancer) (97) (The Cancer Genome Atlas: https://cancergenome.nih.gov), ovarian adenocarcinoma (98) and acute myeloid leukemia (99). As replication stress response factors, underexpression of Metnase and EEPD1 may drive genome instability that often precedes cancer, and overexpression may help cancer cells manage oncogenic replication stress and increase their resistance to genotoxic cancer therapeutics (15,23,35–41,100–103).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Lynn Taylor and Dr Claudia Wiese for helpful comments and Noelia Altina for assistance with γ-H2AX experiments. The authors acknowledge the CSU Flow Cytometry and Cell Sorting Facility for support in method development, optimization and sample analysis.
Contributor Information
Neelam Sharma, Department of Environmental and Radiological Health Sciences, Colorado State University, 1618 Campus Delivery, Fort Collins, CO 80523-1618, USA.
Michael C Speed, Department of Environmental and Radiological Health Sciences, Colorado State University, 1618 Campus Delivery, Fort Collins, CO 80523-1618, USA.
Christopher P Allen, Department of Microbiology, Immunology, and Pathology, Colorado State University, 1601Campus Delivery, Fort Collins, CO 80523-1601, USA.
David G Maranon, Department of Environmental and Radiological Health Sciences, Colorado State University, 1618 Campus Delivery, Fort Collins, CO 80523-1618, USA.
Elizabeth Williamson, Division of Hematology and Medical Oncology, Department of Medicine, University of Texas HealthScience Center, 7703 Floyd Curl Drive, San Antonio, TX 78229, USA.
Sudha Singh, Division of Hematology and Medical Oncology, Department of Medicine, University of Texas HealthScience Center, 7703 Floyd Curl Drive, San Antonio, TX 78229, USA.
Robert Hromas, Division of Hematology and Medical Oncology, Department of Medicine, University of Texas HealthScience Center, 7703 Floyd Curl Drive, San Antonio, TX 78229, USA.
Jac A Nickoloff, Department of Environmental and Radiological Health Sciences, Colorado State University, 1618 Campus Delivery, Fort Collins, CO 80523-1618, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Cancer Online.
FUNDING
National Institutes of Health [R01 GM084020 to J.A.N. and R.H.]; College of Veterinary Medicine and Biomedical Sciences, Colorado State University [to J.A.N.]. Publication costs were partially covered by the Colorado State University Libraries Open Access Research and Scholarship Fund.
Conflict of Interest statement. None declared.
REFERENCES
- 1. Shaheen R., Faqeih E., Ansari S., Abdel-Salam G., Al-Hassnan Z.N., Al-Shidi T., Alomar R., Sogaty S., Alkuraya F.S. Genomic analysis of primordial dwarfism reveals novel disease genes. Genome Res. 2014; 24:291–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Croteau D.L., Popuri V., Opresko P.L., Bohr V.A. Human RecQ helicases in DNA repair, recombination, and replication. Annu. Rev. Biochem. 2014; 83:519–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Casper A.M., Durkin S.G., Arlt M.F., Glover T.W. Chromosomal instability at common fragile sites in Seckel syndrome. Am. J. Hum. Genet. 2004; 75:654–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. McKinnon P.J. DNA repair deficiency and neurological disease. Nat. Rev. Neurosci. 2009; 10:100–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Basu B., Yap T.A., Molife L.R., de Bono J.S. Targeting the DNA damage response in oncology: past, present and future perspectives. Curr. Opin. Oncol. 2012; 24:316–324. [DOI] [PubMed] [Google Scholar]
- 6. Curtin N.J. DNA repair dysregulation from cancer driver to therapeutic target. Nat. Rev. Cancer. 2012; 12:801–817. [DOI] [PubMed] [Google Scholar]
- 7. Gavande N.S., VanderVere-Carozza P.S., Hinshaw H.D., Jalal S.I., Sears C.R., Pawelczak K.S., Turchi J.J. DNA repair targeted therapy: the past or future of cancer treatment?. Pharmacol. Ther. 2016; 160:65–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. O’Connor M.J. Targeting the DNA damage response in cancer. Mol. Cell. 2015; 60:547–560. [DOI] [PubMed] [Google Scholar]
- 9. Pearl L.H., Schierz A.C., Ward S.E., Al-Lazikani B., Pearl F.M. Therapeutic opportunities within the DNA damage response. Nat. Rev. Cancer. 2015; 15:166–180. [DOI] [PubMed] [Google Scholar]
- 10. Nickoloff J.A. Wondrak G.T. DNA repair dysregulation in cancer: from molecular mechanisms to synthetic lethal opportunities. Stress Response Pathways in Cancer: From Molecular Targets to Novel Therapeutics. 2014; NY: Springer; 7–28. [Google Scholar]
- 11. Desai A., Yan Y., Gerson S.L. Advances in therapeutic targeting of the DNA damage response in cancer. DNA Repair. 2018; 66–67:24–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Helleday T. The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Mol. Oncol. 2011; 5:387–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Killock D. Targeted therapies: DNA polymerase theta—a new target for synthetic lethality?. Nat. Rev. Clin. Oncol. 2015; 12:125. [DOI] [PubMed] [Google Scholar]
- 14. Rehman F.L., Lord C.J., Ashworth A. Synthetic lethal approaches to breast cancer therapy. Nat. Rev. Clin. Oncol. 2010; 7:718–724. [DOI] [PubMed] [Google Scholar]
- 15. Shaheen M., Allen C., Nickoloff J.A., Hromas R. Synthetic lethality: exploiting the addiction of cancer to DNA repair. Blood. 2011; 117:6074–6082. [DOI] [PubMed] [Google Scholar]
- 16. Nickoloff J.A., Jones D., Lee S.-H., Williamson E.A., Hromas R. Drugging the cancers addicted to DNA repair. J. Natl. Cancer Inst. 2017; 109:djx059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Errico A., Costanzo V. Mechanisms of replication fork protection: a safeguard for genome stability. Crit. Rev. Biochem. Mol. Biol. 2012; 47:222–235. [DOI] [PubMed] [Google Scholar]
- 18. Cortez D. Unwind and slow down: checkpoint activation by helicase and polymerase uncoupling. Genes Dev. 2005; 19:1007–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rass U. Resolving branched DNA intermediates with structure-specific nucleases during replication in eukaryotes. Chromosoma. 2013; 122:499–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Branzei D., Foiani M. Maintaining genome stability at the replication fork. Nat. Rev. Mol. Cell Biol. 2010; 11:208–219. [DOI] [PubMed] [Google Scholar]
- 21. Allen C., Ashley A.K., Hromas R., Nickoloff J.A. More forks on the road to replication stress recovery. J. Mol. Cell Biol. 2011; 3:4–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Budzowska M., Kanaar R. Mechanisms of dealing with DNA damage-induced replication problems. Cell Biochem. Biophys. 2009; 53:17–31. [DOI] [PubMed] [Google Scholar]
- 23. Wu Y., Lee S.H., Williamson E.A., Reinert B.L., Cho J.H., Xia F., Jaiswal A.S., Srinivasan G., Patel B., Brantley A.et al. EEPD1 rescues stressed replication forks and maintains genome stability by promoting end resection and homologous recombination repair. PLoS Genet. 2015; 11:e1005675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ashley A.K., Shrivastav M., Nie J., Amerin C., Troksa K., Glanzer J.G., Liu S., Opiyo S.O., Dimitrova D.D., Le P.et al. DNA-PK phosphorylation of RPA32 Ser4/Ser8 regulates replication stress checkpoint activation, fork restart, homologous recombination and mitotic catastrophe. DNA Repair. 2014; 21:131–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu S., Opiyo S.O., Manthey K., Glanzer J.G., Ashley A.K., Troksa K., Shrivastav M., Nickoloff J.A., Oakley G.G. Distinct roles for DNA-PK, ATM, and ATR in RPA phosphorylation and checkpoint activation in response to replication stress. Nucleic Acids Res. 2012; 40:10780–10794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Anantha R.W., Vassin V.M., Borowiec J.A. Sequential and synergistic modification of human RPA stimulates chromosomal DNA repair. J. Biol. Chem. 2007; 282:35910–35923. [DOI] [PubMed] [Google Scholar]
- 27. Block W.D., Yu Y., Lees-Miller S.P. Phosphatidyl inositol 3-kinase-like serine/threonine protein kinases (PIKKs) are required for DNA damage-induced phosphorylation of the 32 kDa subunit of replication protein A at threonine 21. Nucleic Acids Res. 2004; 32:997–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. De Haro L.P., Wray J., Williamson E.A., Durant S.T., Corwin L., Gentry A.C., Osheroff N., Lee S.H., Hromas R., Nickoloff J.A. Metnase promotes restart and repair of stalled and collapsed replication forks. Nucleic Acids Res. 2010; 38:5681–5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kim H.S., Nickoloff J.A., Wu Y., Williamson E.A., Sidhu G.S., Reinert B.L., Jaiswal A.S., Srinivasan G., Patel B., Kong K.et al. Endonuclease EEPD1 is a gatekeeper for repair of stressed replication forks. J. Biol. Chem. 2017; 292:2795–2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kim H.-S., Chen Q., Kim S.-K., Nickoloff J.A., Hromas R., Georgiadis M.M., Lee S.-K. The DDN catalytic motif is required for Metnase functions in NHEJ repair and replication restart. J. Biol. Chem. 2014; 289:10930–10938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pepe A., West S.C. MUS81–EME2 promotes replication fork restart. Cell Rep. 2014; 7:1048–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zerbino D.R., Achuthan P., Akanni W., Amode M.R., Barrell D., Bhai J., Billis K., Cummins C., Gall A., Giron C.G.et al. Ensembl 2018. Nucleic Acids Res. 2018; 46:D754–D761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cordaux R., Udit S., Batzer M.A., Feschotte C. Birth of a chimeric primate gene by capture of the transposase gene from a mobile element. Proc. Natl Acad. Sci. U.S.A. 2006; 103:8101–8106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu Y. EEPD1: breaking and rescuing the replication fork. PLoS Genet. 2016; 12:e1005742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dussaussois-Montagne A., Jaillet J., Babin L., Verrelle P., Karayan-Tapon L., Renault S., Rousselot-Denis C., Zemmoura I., Auge-Gouillou C. SETMAR isoforms in glioblastoma: a matter of protein stability. Oncotarget. 2017; 8:9835–9848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Apostolou P., Toloudi M., Kourtidou E., Mimikakou G., Vlachou I., Chatziioannou M., Kipourou V., Papasotiriou I. Potential role for the Metnase transposase fusion gene in colon cancer through the regulation of key genes. PLoS One. 2014; 9:e109741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kuci V., Nordstrom L., Conrotto P., Ek S. SOX11 and HIG-2 are cross-regulated and affect growth in mantle cell lymphoma. Leuk. Lymphoma. 2016; 57:1883–1892. [DOI] [PubMed] [Google Scholar]
- 38. Wang X., Bjorklund S., Wasik A.M., Grandien A., Andersson P., Kimby E., Dahlman-Wright K., Zhao C., Christensson B., Sander B. Gene expression profiling and chromatin immunoprecipitation identify DBN1, SETMAR and HIG2 as direct targets of SOX11 in mantle cell lymphoma. PLoS One. 2010; 5:e14085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Williamson E.A., Damiani L., Leitao A., Hu C., Hathaway H., Oprea T., Sklar L., Shaheen M., Bauman J., Wang W.et al. Targeting the transposase domain of the DNA repair component Metnase to enhance chemotherapy. Cancer Res. 2012; 72:6200–6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wray J., Williamson E.A., Fnu S., Lee S.-H., Libby E., Willman C.L., Nickoloff J.A., Hromas R. Metnase mediates chromosome decatenation in acute leukemia cells. Blood. 2009; 114:1852–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wray J., Williamson E.A., Royce M., Shaheen M., Beck B.D., Lee S.H., Nickoloff J.A., Hromas R. Metnase mediates resistance to topoisomerase II inhibitors in breast cancer cells. PLoS One. 2009; 4:e5323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Carlson S.M., Moore K.E., Sankaran S.M., Reynoird N., Elias J.E., Gozani O. A proteomic strategy identifies lysine methylation of splicing factor snRNP70 by the SETMAR enzyme. J. Biol. Chem. 2015; 290:12040–12047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lambert S., Lopez B.S. Characterization of mammalian RAD51 double strand break repair using non-lethal dominant-negative forms. EMBO J. 2000; 19:3090–3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yanez R.J., Porter A.C.G. Gene targeting is enhanced in human cells overexpressing hRAD51. Gene Ther. 1999; 6:1282–1290. [DOI] [PubMed] [Google Scholar]
- 45. Kim P.M., Allen C., Wagener B.M., Shen Z., Nickoloff J.A. Overexpression of human RAD51 and RAD52 reduces double-strand break-induced homologous recombination in mammalian cells. Nucleic Acids Res. 2001; 29:4352–4360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hromas R., Williamson E., Fnu S., Lee Y.-J., Park S.-J., Beck B.D., You J.-S., Laitao A., Nickoloff J.A., Lee S.-H. Chk1 phosphorylation of Metnase enhances DNA repair but inhibits replication fork restart. Oncogene. 2012; 31:4245–4254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Roman Y., Oshige M., Lee Y.-J., Goodwin K., Georgiadis M.M., Hromas R.A., Lee S.-H. Biochemical characterization of a SET and transposase fusion protein, Metnase (SETMAR) for its DNA binding and DNA cleavage activity. Biochemistry. 2007; 46:11369–11376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Singh N.P., McCoy M.T., Tice R.R., Schneider E.L. A simple technique for quantitation of low levels of DNA damage in individual cells. Exp. Cell Res. 1988; 175:184–191. [DOI] [PubMed] [Google Scholar]
- 49. Sirbu B.M., Couch F.B., Cortez D. Monitoring the spatiotemporal dynamics of proteins at replication forks and in assembled chromatin using isolation of proteins on nascent DNA. Nat. Protoc. 2012; 7:594–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Landry J.J.M., Pyl P.T., Rausch T., Zichner T., Tekkedil M.M., Stütz A.M., Jauch A., Aiyar R.S., Pau G., Delhomme N.et al. The genomic and transcriptomic landscape of a HeLa cell line. G3 (Bethesda). 2013; 3:1213–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Macville M., Schrock E., Padilla-Nash H., Keck C., Ghadimi B.M., Zimonjic D., Popescu N., Ried T. Comprehensive and definitive molecular cytogenetic characterization of HeLa cells by spectral karyotyping. Cancer Res. 1999; 59:141–150. [PubMed] [Google Scholar]
- 52. Vesela E., Chroma K., Turi Z., Mistrik M. Common chemical inductors of replication stress: focus on cell-based studies. Biomolecules. 2017; 7:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Aguilera A., Gomez-Gonzalez B. Genome instability: a mechanistic view of its causes and consequences. Nat. Rev. Genet. 2008; 9:204–217. [DOI] [PubMed] [Google Scholar]
- 54. Bouwman P., Aly A., Escandell J.M., Pieterse M., Bartkova J., van der Gulden H., Hiddingh S., Thanasoula M., Kulkarni A., Yang Q.et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Struct. Mol. Biol. 2010; 17:688–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bunting S.F., Callen E., Wong N., Chen H.T., Polato F., Gunn A., Bothmer A., Feldhahn N., Fernandez-Capetillo O., Cao L.et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010; 141:243–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Feng L., Fong K.W., Wang J., Wang W., Chen J. RIF1 counteracts BRCA1-mediated end resection during DNA repair. J. Biol. Chem. 2013; 288:11135–11143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Fenech M., Kirsch-Volders M., Natarajan A.T., Surralles J., Crott J.W., Parry J., Norppa H., Eastmond D.A., Tucker J.D., Thomas P. Molecular mechanisms of micronucleus, nucleoplasmic bridge and nuclear bud formation in mammalian and human cells. Mutagenesis. 2011; 26:125–132. [DOI] [PubMed] [Google Scholar]
- 58. Lee S.H., Oshige M., Durant S.T., Rasila K.K., Williamson E.A., Ramsey H., Kwan L., Nickoloff J.A., Hromas R. The SET domain protein Metnase mediates foreign DNA integration and links integration to nonhomologous end-joining repair. Proc. Natl Acad. Sci. U.S.A. 2005; 102:18075–18080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tellier M., Chalmers R. The roles of the human SETMAR (Metnase) protein in illegitimate DNA recombination and non-homologous end joining repair. DNA Repair. 2019; 80:26–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Fnu S., Williamson E.A., De Haro L.P., Brenneman M., Wray J., Shaheen M., Radhakrishnan K., Lee S.H., Nickoloff J.A., Hromas R. Methylation of histone H3 lysine 36 enhances DNA repair by nonhomologous end-joining. Proc. Natl Acad. Sci. U.S.A. 2011; 108:540–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lobrich M., Shibata A., Beucher A., Fisher A., Ensminger M., Goodarzi A.A., Barton O., Jeggo P.A. γH2AX foci analysis for monitoring DNA double-strand break repair: strengths, limitations and optimization. Cell Cycle. 2010; 9:662–669. [DOI] [PubMed] [Google Scholar]
- 62. Ferretti L.P., Lafranchi L., Sartori A.A. Controlling DNA-end resection: a new task for CDKs. Front. Genet. 2013; 4:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Symington L.S. Mechanism and regulation of DNA end resection in eukaryotes. Crit. Rev. Biochem. Mol. Biol. 2016; 51:195–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Terasawa M., Shinohara A., Shinohara M. Canonical non-homologous end joining in mitosis induces genome instability and is suppressed by M-phase-specific phosphorylation of XRCC4. PLoS Genet. 2014; 10:e1004563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zhao H., Rybak P., Dobrucki J., Traganos F., Darzynkiewicz Z. Relationship of DNA damage signaling to DNA replication following treatment with DNA topoisomerase inhibitors camptothecin/topotecan, mitoxantrone, or etoposide. Cytometry A. 2012; 81:45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Williamson E.A., Rasila K.K., Corwin L.K., Wray J., Beck B.D., Severns V., Mobarak C., Lee S.H., Nickoloff J.A., Hromas R. The SET and transposase domain protein Metnase enhances chromosome decatenation: regulation by automethylation. Nucleic Acids Res. 2008; 36:5822–5831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Doksani Y., Bermejo R., Fiorani S., Haber J.E., Foiani M. Replicon dynamics, dormant origin firing, and terminal fork integrity after double-strand break formation. Cell. 2009; 137:247–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kim H.S., Kim S.K., Hromas R., Lee S.H. The SET domain is essential for Metnase functions in replication restart and the 5′ end of SS-overhang cleavage. PLoS One. 2015; 10:e0139418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Chun C., Wu Y., Lee S.H., Williamson E.A., Reinert B.L., Jaiswal A.S., Nickoloff J.A., Hromas R.A. The homologous recombination component EEPD1 is required for genome stability in response to developmental stress of vertebrate embryogenesis. Cell Cycle. 2016; 15:957–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Sirbu B.M., Couch F.B., Feigerle J.T., Bhaskara S., Hiebert S.W., Cortez D. Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev. 2011; 25:1320–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kaushal S., Freudenreich C.H. The role of fork stalling and DNA structures in causing chromosome fragility. Genes Chromosomes Cancer. 2019; 58:270–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Mirkin E.V., Mirkin S.M. Replication fork stalling at natural impediments. Microbiol. Mol. Biol. Rev. 2007; 71:13–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hromas R., Kim H.S., Sidhu G., Williamson E.A., Jaiswal A., Totterdale T.A., Nole J., Lee S.-H., Nickoloff J.A., Kong K.Y. The endonuclease EEPD1 mediates synthetic lethality in RAD52-depleted BRCA1-mutant breast cancer cells. Breast Cancer Res. 2017; 19:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wang H., Li S., Oaks J., Ren J., Li L., Wu X. The concerted roles of FANCM and Rad52 in the protection of common fragile sites. Nat. Commun. 2018; 9:2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kim H.S., Williamson E.A., Nickoloff J.A., Hromas R.A., Lee S.H. Metnase mediates loading of exonuclease 1 onto single-strand overhang DNA for end resection at stalled replication forks. J. Biol. Chem. 2016; 292:1414–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Montecucco A., Zanetta F., Biamonti G. Molecular mechanisms of etoposide. EXCLI J. 2015; 14:95–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Tanaka T., Halicka H.D., Traganos F., Seiter K., Darzynkiewicz Z. Induction of ATM activation, histone H2AX phosphorylation and apoptosis by etoposide: relation to cell cycle phase. Cell Cycle. 2007; 6:371–376. [DOI] [PubMed] [Google Scholar]
- 78. Nakada S., Katsuki Y., Imoto I., Yokoyama T., Nagasawa M., Inazawa J., Mizutani S. Early G2/M checkpoint failure as a molecular mechanism underlying etoposide-induced chromosomal aberrations. J. Clin. Invest. 2006; 116:80–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Beck B.D., Lee S.S., Williamson E., Hromas R.A., Lee S.H. Biochemical characterization of Metnase’s endonuclease activity and its role in NHEJ repair. Biochemistry. 2011; 50:4360–4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Hromas R., Wray J., Lee S.H., Martinez L., Farrington J., Corwin L.K., Ramsey H., Nickoloff J.A., Williamson E.A. The human set and transposase domain protein Metnase interacts with DNA ligase IV and enhances the efficiency and accuracy of non-homologous end-joining. DNA Repair. 2008; 7:1927–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ward T.A., McHugh P.J., Durant S.T. Small molecule inhibitors uncover synthetic genetic interactions of human flap endonuclease 1 (FEN1) with DNA damage response genes. PLoS One. 2017; 12:e0179278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Stewart-Morgan K.R., Petryk N., Groth A. Chromatin replication and epigenetic cell memory. Nat. Cell Biol. 2020; 22:361–371. [DOI] [PubMed] [Google Scholar]
- 83. Gong F., Miller K.M. Histone methylation and the DNA damage response. Mutat. Res. 2019; 780:37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Fournier L.A., Kumar A., Stirling P.C. Chromatin as a platform for modulating the replication stress response. Genes. 2018; 9:622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Rondinelli B., Gogola E., Yucel H., Duarte A.A., van de Ven M., van der Sluijs R., Konstantinopoulos P.A., Jonkers J., Ceccaldi R., Rottenberg S.et al. EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nat. Cell Biol. 2017; 19:1371–1378. [DOI] [PubMed] [Google Scholar]
- 86. Smolle M., Workman J.L., Venkatesh S. reSETting chromatin during transcription elongation. Epigenetics. 2013; 8:10–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Yu Y., Song C., Zhang Q., DiMaggio P.A., Garcia B.A., York A., Carey M.F., Grunstein M. Histone H3 lysine 56 methylation regulates DNA replication through its interaction with PCNA. Mol. Cell. 2012; 46:7–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Forbes S.A., Beare D., Gunasekaran P., Leung K., Bindal N., Boutselakis H., Ding M., Bamford S., Cole C., Ward S.et al. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015; 43:D805–D811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Rubio-Perez C., Tamborero D., Schroeder M.P., Antolin A.A., Deu-Pons J., Perez-Llamas C., Mestres J., Gonzalez-Perez A., Lopez-Bigas N. In silico prescription of anticancer drugs to cohorts of 28 tumor types reveals targeting opportunities. Cancer Cell. 2015; 27:382–396. [DOI] [PubMed] [Google Scholar]
- 90. Gonzalez-Perez A., Perez-Llamas C., Deu-Pons J., Tamborero D., Schroeder M.P., Jene-Sanz A., Santos A., Lopez-Bigas N. IntOGen-mutations identifies cancer drivers across tumor types. Nat. Methods. 2013; 10:1081–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Skrzypczak M., Goryca K., Rubel T., Paziewska A., Mikula M., Jarosz D., Pachlewski J., Oledzki J., Ostrowski J. Modeling oncogenic signaling in colon tumors by multidirectional analyses of microarray data directed for maximization of analytical reliability. PLoS One. 2010; 5:e13091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Beroukhim R., Brunet J.P., Di Napoli A., Mertz K.D., Seeley A., Pires M.M., Linhart D., Worrell R.A., Moch H., Rubin M.A.et al. Patterns of gene expression and copy-number alterations in Von-Hippel Lindau disease-associated and sporadic clear cell carcinoma of the kidney. Cancer Res. 2009; 69:4674–4681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Talantov D., Mazumder A., Yu J.X., Briggs T., Jiang Y., Backus J., Atkins D., Wang Y. Novel genes associated with malignant melanoma but not benign melanocytic lesions. Clin. Cancer Res. 2005; 11:7234–7242. [DOI] [PubMed] [Google Scholar]
- 94. Gaedcke J., Grade M., Jung K., Camps J., Jo P., Emons G., Gehoff A., Sax U., Schirmer M., Becker H.et al. Mutated KRAS results in overexpression of DUSP4, a MAP-kinase phosphatase, and SMYD3, a histone methyltransferase, in rectal carcinomas. Genes Chromosomes Cancer. 2010; 49:1024–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Sun L., Hui A.M., Su Q., Vortmeyer A., Kotliarov Y., Pastorino S., Passaniti A., Menon J., Walling J., Bailey R.et al. Neuronal and glioma-derived stem cell factor induces angiogenesis within the brain. Cancer Cell. 2006; 9:287–300. [DOI] [PubMed] [Google Scholar]
- 96. Sabates-Bellver J., Van der Flier L.G., de Palo M., Cattaneo E., Maake C., Rehrauer H., Laczko E., Kurowski M.A., Bujnicki J.M., Menigatti M.et al. Transcriptome profile of human colorectal adenomas. Mol. Cancer Res. 2007; 5:1263–1275. [DOI] [PubMed] [Google Scholar]
- 97. Curtis C., Shah S.P., Chin S.F., Turashvili G., Rueda O.M., Dunning M.J., Speed D., Lynch A.G., Samarajiwa S., Yuan Y.et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012; 486:346–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Yoshihara K., Tajima A., Komata D., Yamamoto T., Kodama S., Fujiwara H., Suzuki M., Onishi Y., Hatae M., Sueyoshi K.et al. Gene expression profiling of advanced-stage serous ovarian cancers distinguishes novel subclasses and implicates ZEB2 in tumor progression and prognosis. Cancer Sci. 2009; 100:1421–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Andersson A., Ritz C., Lindgren D., Eden P., Lassen C., Heldrup J., Olofsson T., Rade J., Fontes M., Porwit-Macdonald A.et al. Microarray-based classification of a consecutive series of 121 childhood acute leukemias: prediction of leukemic and genetic subtype as well as of minimal residual disease status. Leukemia. 2007; 21:1198–1203. [DOI] [PubMed] [Google Scholar]
- 100. Bartkova J., Horejsi Z., Koed K., Kramer A., Tort F., Zieger K., Guldberg P., Sehested M., Nesland J.M., Lukas C.et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005; 434:864–870. [DOI] [PubMed] [Google Scholar]
- 101. Weaver B.A., Silk A.D., Montagna C., Verdier-Pinard P., Cleveland D.W. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell. 2007; 11:25–36. [DOI] [PubMed] [Google Scholar]
- 102. Gaillard H., Garcia-Muse T., Aguilera A. Replication stress and cancer. Nat. Rev. Cancer. 2015; 15:276–289. [DOI] [PubMed] [Google Scholar]
- 103. Hills S.A., Diffley J.F. DNA replication and oncogene-induced replicative stress. Curr. Biol. 2014; 24:R435–R444. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.