

Abstract
Circular yeast artificial chromosomes (YACs) provide significant advantages for cloning and manipulating large segments of genomic DNA in Saccharomyces cerevisiae. However, it has been difficult to exploit these advantages, because circular YACs are difficult to isolate and purify. Here we describe a method for purification of large circular YACs that is more reliable compared with previously described protocols. This method has been used to purify YACs up to 600 kb in size. The purified YAC DNA is suitable for restriction enzyme digestion, DNA sequencing and functional studies. For example, YACs carrying full-size genes can be purified from yeast and used for transfection into mammalian cells or for the construction of a synthetic genome that can be used to produce a synthetic cell. This method for isolating high-quality YAC DNA in microgram quantities should be valuable for functional and synthetic genomic studies. The entire protocol takes ~3 d to complete.
INTRODUCTION
Yeast artificial chromosomes (YACs) were first developed as linear molecules stabilized by terminal telomeres for cloning, propagating and manipulating large segments of DNA in yeast cells1. In an early application, traditional YAC cloning was critical to the success of the Human Genome Project. However, traditional YAC cloning has not been more widely used, at least in part because it is tedious and difficult to separate linear YACs from endogenous yeast chromosomes. A significant development in yeast cloning was achieved when human genomic libraries were created in circular YACs2,3, which are more easily separated from linear yeast chromosomes. In addition, circular YACs can be readily modified by homologous recombination (HR) in yeast and retrofitted into bacterial artificial chromosomes (BACs) for propagation in Escherichia coli, significantly simplifying subsequent analysis and manipulation of selected YAC clones4.
Circular YACs have several advantages over circular BACs and P1-derived artificial chromosomes (PACs)5,6. First, YACs can be used to clone megabase-size DNA fragments that are too large for BAC or PAC cloning, and YACs can be readily amplified7 and modified by HR in yeast cells. The high efficiency and specificity of HR in yeast is unique, offering a powerful tool for introducing point mutations, deletions and insertions or other DNA modifications into large DNA fragments with high accuracy and fidelity and usually without unwanted secondary rearrangements or mutations (reviewed in ref. 8). Second, DNA sequences that are inherently unstable when cloned as BACs in E. coli, such as imperfect palindromes and AT-rich regions, are often stable and can be propagated as YACs in yeast cells9,10. Third, transformation-associated recombination (TAR) cloning is a powerful method for isolating any chromosome regions (including genes and pathways) as circular YACs without constructing a genomic library of random clones11,12. TAR cloning is based on in vivo HR between a specific genome target and a linearized TAR cloning vector with terminal homology to the target. During cotransformation into yeast, the vector and target gene recombine to form a circular YAC, which can readily be propagated, segregated and selected for in yeast. Several dozen genes and genomic regions have been isolated by TAR cloning from human and mouse genomes for structural, evolutionary and functional studies (reviewed in ref. 12). Physical and functional analyses have demonstrated that TAR cloning is a relatively error-free, reliable and efficient method for generating circular YACs containing a desired chromosomal region or gene. So far, TAR cloning has been used to isolate genomic segments and genes from complex genomes up to 300 kb12 and whole bacterial genomes up to 600 kb in size13. With optimized conditions that avoid DNA shearing, it may be possible to isolate even larger DNA fragments by YAC/TAR cloning. The YAC/TAR cloning system can also be used to assemble overlapping DNA regions into large contigs. For example, long arrays of synthetic alphoid DNA can be constructed in circular YACs by TAR cloning and used to develop human artificial chromosomes (HACs)14. HACs are currently of great interest because of their potential applications in gene therapy15 and in the analysis of the regulation and expression of human genes in mammalian cells16.
Circular YAC cloning has also proven to be useful in the propagation of synthetic and natural bacterial genomes17–20. For example, a set of overlapping chemically synthesized fragments was assembled into a complete ~580 kb Mycoplasma genitalium genome using this circular YAC method17,18. More recently, a complete functional synthetic M. mycoides genome and its assembly intermediates were cloned in yeast as circular YACs20. For these and other similar experiments, including cloning of entire mammalian genes and loci or construction of whole microbial genomes, the large cloning capacity (>2 Mb)21 of circular YACs is indispensable. Importantly, these groundbreaking experiments and approaches not only point the way toward constructing new microbes for biotechnology but also promise to transform the fields of functional and synthetic genomics.
The above techniques require purification and manipulation of large amounts of highly purified large circular YACs, which has remained a problem. Circular YACs can, in principle, be separated from linear yeast chromosomes by pulse-field gel electrophoresis22, but this time-consuming, low-yield, low-throughput procedure cannot be used to produce large amounts of high-quality DNA suitable for downstream applications such as DNA sequencing, transfection into mammalian cells or further recombination into larger constructs. An alternative method for purifying circular YACs was reported by Devenish and Newlon23. These authors described purification of a ~200-kb circular derivative of yeast chromosome III by alkaline extraction. Although this method was used to construct the first yeast chromosome-specific library, the reproducibility of the method is poor and the quality of DNA is low23. Another method based on the alkaline extraction approach has since been published24. This more-streamlined protocol avoids the use of time-consuming equilibrium centrifugation in cesium chloride–ethidium bromide gradients, but has additional drawbacks, including low DNA yield and a size limit of 120 kb. Another approach used is retrofitting of large YAC clones for transfer and propagation in E. coli as BACs4, followed by purification of the clones from large-scale bacterial cultures and additional DNA characterization and manipulation. These additional steps of YAC retrofitting are time consuming and have several obvious limitations, including the ~300 kb limit on insert size in BACs and PACs, and the fact that YAC/BAC clones frequently undergo undesirable rearrangements during transformation into bacterial cells25.
Isolation of circular YAC DNA molecules has become crucial for analysis of synthetic and natural bacterial genomes17–20 because entire genomes, and their smaller assembly intermediates, cannot be introduced into and replicate in E. coli cells. Therefore, they must be directly isolated from yeast cells for downstream applications, such as the activation of these genomes in recipient cells. To address the need for an improved method for purifying large circular YACs directly from yeast cells, we developed a new method24 based on the alkaline extraction approach of Devenish and Newlon23. This simple and highly reproducible method has been optimized for large-scale purification of circular YACs up to 600 kb in size (Figs. 1 and 2) and has been used to purify large circular YACs carrying synthetic M. genitalium genomes17,18; however, the isolation of megabase-size circular DNA molecules may be inefficient because of shearing during the procedure. We have demonstrated that YAC DNA produced by this method is of very high quality, and can be used for DNA sequencing, structural and functional studies of TAR-cloned genes and chromosome regions, and purification of assembly intermediates that can be used to build a functional synthetic genome10,17–19. A detailed protocol for this method is provided below.
Figure 1 |.
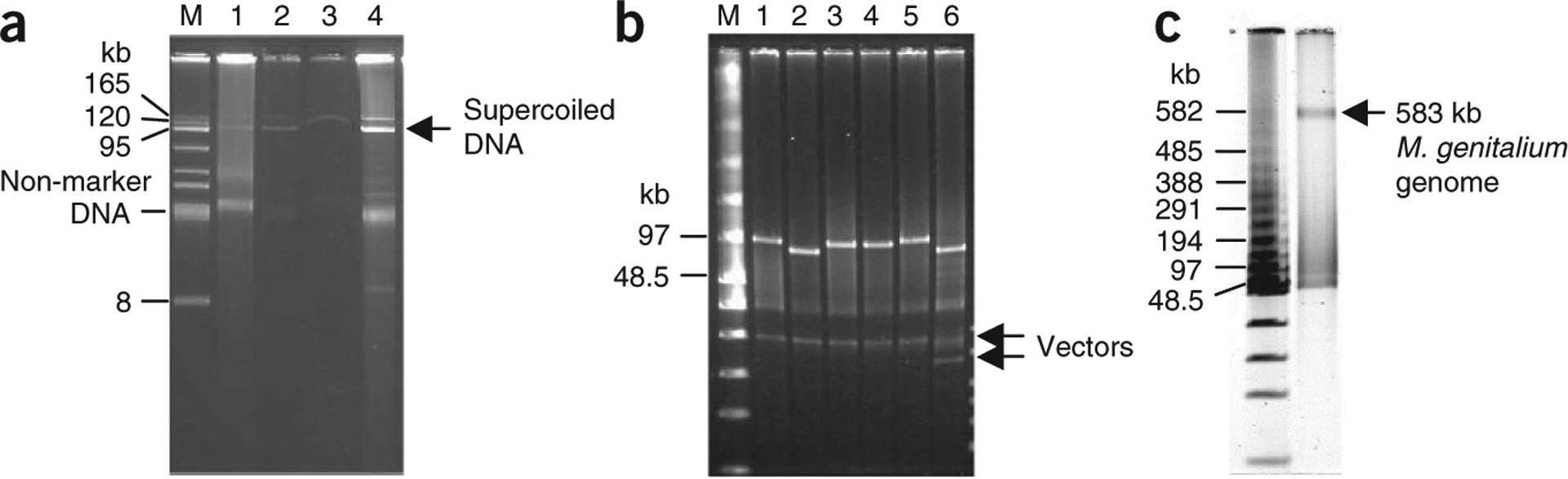
Purification of circular YAC from 400 ml of yeast culture. (a) Electrophoresis analysis of a 100-kb intermediate YAC of M. mycoides19, YAC 2a-100, recovered during purification. DNA was separated on a 1% agarose/1× TAE electrophoresis gel and stained as described in EQUIPMENT SETUP. M, BAC-tracker supercoiled DNA ladder; lane 1, 20 μl of DNA sample from Step 15 (before adding exonuclease); lane 2, 20 μl of DNA sample from Step 16 (after 35 min incubation); lane 3, 20 μl of the eluate (Step 21 before DNA precipitation); and lane 4, 1 μl of DNA sample from Step 25 (DNA pellet was dissolved in 150 μl of TE buffer). (b) NotI restriction enzyme digestion analysis of six YACs. A 0.75 μl sample of each YAC DNA was digested with NotI, which releases the insert DNA of YACs, and analyzed by FIGE. FIGE analysis was performed as described in EQUIPMENT SETUP. M, low-range PFG marker; lane 1, YAC 301–400 (~100 kb); lane 2, YAC 2a-100 (~96 kb); lane 3, YAC 101–200 (~100 kb); lane 4, YAC 401–500 (~100 kb); lane 5, YAC 501–600 (~100 kb); and lane 6, YAC 811a-900 (~88 kb). (c) NotI digestion analysis of ~600 kb YAC containing a synthetic M. genitalium genome17,18. A 0.75-μl DNA sample (from a total volume of 150 μl) was digested with NotI, which releases the M. genitalium genome (~580 kb) from the vector, and analyzed in a 1% (wt/vol) agarose gel by FIGE. The parameters were essentially as described in EQUIPMENT SETUP, with the following modifications: forward 90 V, initial switch 5.0 s, final switch 30 sec, with linear ramp.
Figure 2 |.
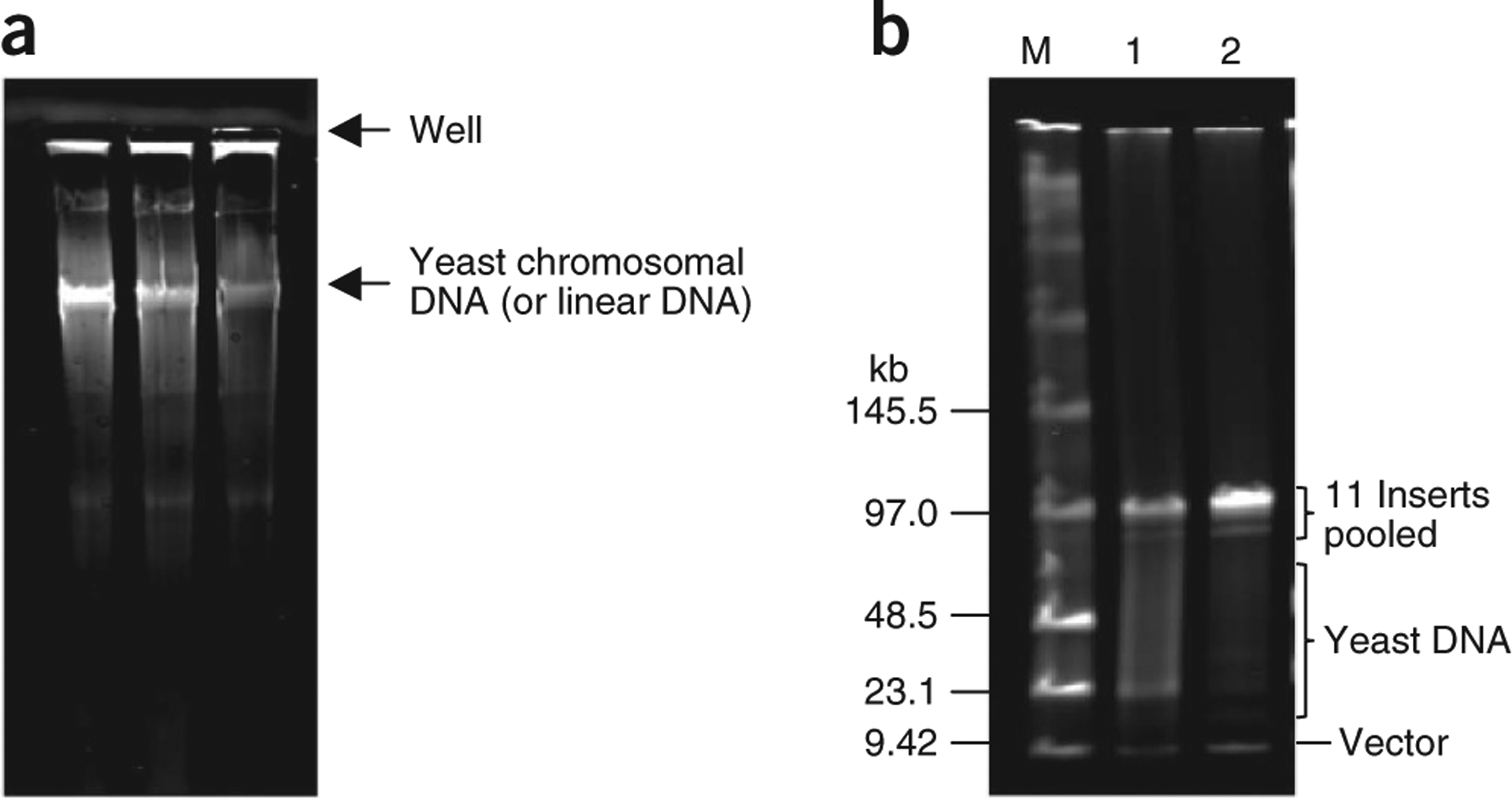
Topological trapping of circular YACs in agarose plugs. (a) Separation of yeast chromosomes. Agarose plugs containing 100-kb intermediates of circular YAC DNA of M. mycoides with contaminating yeast linear DNA were inserted into the wells of a 1% agarose/1× TAE gel. Electrophoresis was performed at 4.5 V cm−1 for 2 h. After the plugs were recovered, the gel was stained with SYBR Gold, as described in EQUIPMENT SETUP. During electrophoresis, linear yeast DNA entered the gel but circular DNAs were trapped in the plugs because they were topologically linked with agarose fibers. (b) YAC DNA purity after topological trapping. Samples (20 μl) of each of 11 intermediates of M. mycoides YAC with the insert DNA ranging in size from 88 to 104 kb were pooled together (final volume of 220 μl) and then mixed with 220 μl of 2% LMP agarose. Prepared plugs were subjected to the topological trapping procedure. At the final step of the procedure, the DNA pellet was resuspended in 30 μl of TE buffer. A measure of 5 μl of DNA sample before (lane 1) and after (lane 2) topological trapping were digested with NotI and analyzed by FIGE (parameters as described in EQUIPMENT SETUP). M, low-range PFG marker.
Experimental design
Cell number.
This protocol has been optimized for processing ~1.2 × 1010 cells from early stationary phase cultures grown in complete synthetic medium. The amount of buffers or reagents, such as Zymolyase-20T and DNA exonuclease, needs to be optimized accordingly if you use different amounts of cells, different phases of cell culture or different yeast strains.
Optimizing spheroplast preparation.
Although there are no limitations in the choice of a yeast host strain, the time of the Zymolyase treatment and/or the concentration of Zymolyase should be experimentally determined for each new batch of Zymolyase and for each strain, because different strains may exhibit a different sensitivity to the enzyme. The current protocol has been optimized for the strain VL6–48 and its derivatives. To check the level of spheroplasting, follow the protocol described in Box 1.
BOX 1 |. EFFICACY OF SPHEROPLASTING.
Prepare 10–20 samples of 1.8 μl of 2% (wt/vol) SDS and 1.8 μl of 1 M sorbitol solutions in 15-ml conical tubes and store them at RT.
Add 200 μl of yeast cell suspension treated by Zymolyase to the 1.8 μl of 2% (wt/vol) SDS and 1.8 μl of 1 M sorbitol. Take aliquots of the cell suspension at regular intervals (every 10 min) after 60 min of Zymolase treatment; see PROCEDURE Step 7.
Measure OD600 readings in SDS and sorbitol dilutions for each pair of samples.
The spheroplasts are determined to be ready when the difference between the two OD600 readings is four- to fivefold.
YAC confirmation.
Before starting a preparative YAC DNA isolation, a control experiment should be carried out to confirm that the YAC is maintained as a circular molecule in yeast cells. For this purpose, a standard separation of yeast chromosomes by field-inversion gel electrophoresis (FIGE) is recommended. Only bands corresponding to the 16 yeast chromosomes should be observed if the YAC is circular—large circular DNA molecules do not migrate during a FIGE run.
Topological trapping of circular DNA in agarose plugs.
Topological trapping (Steps 27–37) is optional, but is required when contamination by linear DNA molecules (predominantly yeast chromosomal DNA) should be minimized, e.g., for genome assembly. In this case, the major fraction of YAC molecules is represented by open circle DNA. These steps may be omitted when the YAC DNA is used for transfection into an alternative host. Without topological trapping, most of the purified YAC DNA molecules are in the supercoiled form, which is more resistant to shearing and thus more suitable for transfection.
MATERIALS
REAGENTS
Yeast strain containing YAC of interest. We have previously used derivatives of Saccharomyces cerevisiae strain VL6–48 (MATá, his3-Δ200, trp1-Δ1, ura3–52, lys2, ade2–101, met14, psi + cir0) carrying different size circular YACs for DNA isolation17,19. The VL6–48 strain is available from the American Type Culture Collection (ATCC, cat. no. MYA-3666).
SYBR Gold (Invitrogen, cat. no. S-11494)
Low-range PFG marker (New England Biolabs, cat. no. N0350S)
Agarose plug mold (Bio-Rad, cat. no. 1703706)
BAC-Tracker supercoiled DNA ladder (Epicentre, cat. no. BT010950)
Large-construct kit (Qiagen, cat. no. 12462) ▲ CRITICAL DNA purification is carried out as described in the manufacturer’s instructions, with the exception of an additional step (Step 19 of PROCEDURE) to remove any undissolved material before loading DNA on the column.
Zymolyase 20T (ICN Biochemicals, cat. no. 320921; see REAGENT SETUP)
Exonuclease (Qiagen, cat. no. 12462)
Glycerol (MP Biochemicals, cat. no. 8000688)
Sodium phosphate dibasic heptahydrate (Mallinckrodt, cat. no. 7892)
Sodium phosphate monobasic (Mallinckrodt, cat. no. 7914)
Phenol:chloroform:isoamyl alcohol (Invitrogen, cat. no. 15593–049)
Buffer-saturated phenol (Invitrogen, cat. no. 15513–039) ! CAUTION Phenol is a hazardous chemical: it is toxic and corrosive.
Sorbitol (Sigma, cat. no. S1876; see REAGENT SETUP)
EDTA (0.5 M, pH 7.5; Teknova, cat. no. E0375)
β-Mercaptoethanol (14 M; Sigma, cat. no. M3148) ! CAUTION It is highly toxic on contact with skin and is harmful if inhaled or swallowed.
Tris-HCl (1 M, pH 8; Invitrogen, cat. no. 15568–025)
Tris-HCl (1 M, pH 7.5; Invitrogen, cat. no. 15567–027)
SDS (Fluka, cat. no. 71725)
Sodium acetate (Sigma, cat. no. S7545)
Isopropanol (Fisher Scientific, cat. no. CA451–1) ! CAUTION It can cause eye and skin irritation.
GlycoBlue (Ambion, cat. no. AM9515)
Total yeast tRNA (Sigma, cat. no. R5636)
ATP powder (Sigma, cat. no. A3377; see REAGENT SETUP)
Bacto Agar (Difco, cat. no. 214010)
Agarose, low melting point (LMP agarose; Invitrogen, cat. no. 16520–100)
Agarose (1% (wt/vol))/1× TAE electrophoresis gel (Bio-Rad, cat. no. 161–3015)
β-Agarase (New England Biolabs, cat. no. M0392L)
Tris-acetate-EDTA buffer (TAE, 10×; cat. no. BP 1335–20)
Ethanol
Distilled, deionized water (ddH2O)
Yeast medium (see REAGENT SETUP). Because circular YACs may exhibit mitotic instability, we recommend using selective medium for culturing yeast cells. An appropriate selective liquid medium should be chosen based on the selective marker present in the YAC (HIS3, URA3 or TRP1). Medium can be prepared in house or purchased commercially (CM glucose broth minus HIS (Teknova, cat. no. C7112); CM glucose broth minus URA, (Teknova, cat. no. C1175); CM glucose broth minus TRP, (Teknova, cat. no. C1174)).
d-Glucose (Sigma-Aldrich, cat. no G8270)
Ammonium sulfate (NH4)2SO4 (Sigma-Aldrich, cat. no A6387)
Adenine sulfate (Sigma-Aldrich, cat. no 145815)
Uracil (Sigma-Aldrich, cat. no U075)
l-Arginine-HCl (Sigma-Aldrich, cat. no. A4881)
Yeast nitrogen base (Difco, cat. no. 239210)
l-Aspartic acid (Sigma-Aldrich, cat. no. A93100)
l-Glutamic acid (Sigma-Aldrich, cat. no. 128430)
l-Histidine-HCl (Sigma-Aldrich, cat. no. 1515668)
l-Isoleucine (Sigma-Aldrich, cat. no. 151718)
l-Leucine (Sigma-Aldrich, cat. no. L8000)
l-Lysine-HCl (Sigma-Aldrich, cat. no. L5501)
l-Methionine (Sigma-Aldrich, cat. no. M9625)
l-Phenylalanine (Sigma-Aldrich, cat. no. P2126)
l-Serine (Sigma-Aldrich, cat. no S4500)
l-Threonine (Sigma-Aldrich, cat. no T8625)
l-Tryptophan (Sigma-Aldrich, cat. no T0254)
l-Tyrosine (Sigma-Aldrich, cat. no T3754)
l-Valine (Sigma-Aldrich, cat. no V0500)
EQUIPMENT
Pyrex disposable serological pipette (10 ml; Corning, cat. no. 7078D-10)
Conical tube (225 ml; BD Falcon, cat. no. 352075)
Conical tube (50 ml; BD Falcon, cat. no. 352070)
Filter unit (0.22-μm pore, 1,000 ml; Corning, cat. no. 430517)
Plastic serological pipette (5 ml; Corning, cat. no. 4487)
Large orifice pipette tips (1 ml; USA Scientific, cat. no. 1011-9510)
Swing bucket rotor SH3000 (Thermo Scientific, cat. no. 11796)
Field-inversion gel electrophoresis system (FIGE; Bio-Rad, cat. no. 161-3016)
Typhoon 9410 imager (GE Healthcare, cat. no. 63-0055-80)
Rotator (Thermo Scientific, cat. no. T415110Q)
Gel-Doc 2000 (Bio-Rad, cat. no. 170-8126)
Digital thermoblock (VWR, cat. no.12621-084)
SLA-1500 rotor (Thermo Scientific, cat. no. 096-062034)
Eppendorf centrifuge 5417C (Eppendorf, cat. no. 22-62-170-0)
Eppendorf centrifuge fixed-angle rotor (Eppendorf, cat. no. 22-63-605-7)
Agarose plug molds (Bio-Rad)
REAGENT SETUP
Zymolyase solution (10 mg ml−1 of Zymolyase 20T in 25% (wt/vol) glycerol)
Add 200 mg of Zymolyase 20T and 1 ml of Tris-HCl (pH 7.5) to 9 ml of ddH2O. Stir until dissolved. Add 10 ml of 50% (wt/vol) glycerol. Mix well. Transfer 500-μl aliquots to 1.7-ml Eppendorf tubes and store at − 20 °C for up to 6 months.
SPE solution (1 M sorbitol, 0.01 M sodium phosphate, 0.01 M Na2EDTA (pH 7.5))
Add 182 g of sorbitol, 2.08 g of Na2HPO4·7H2O, 0.32 g of NaH2PO4·H2O and 20 ml of 0.5 M EDTA (pH 7.5) to ~800 ml of ddH2O in a 1-liter beaker. Stir until dissolved. Make up the volume to 1,000 ml in a 1,000-ml measuring cylinder and mix thoroughly. Transfer the solution to a glass storage bottle and autoclave for 15 min. Alternatively, the solution can be filter sterilized. SPE solution can be stored for up to 6 months at room temperature (RT, 25 °C).
Sorbitol solution (1 M)
Add 182 g of sorbitol to ~700 ml of ddH2O in a 1,000-ml beaker. Stir until dissolved. Make up the volume to 1,000 ml in a 1,000-ml measuring cylinder and mix thoroughly. Transfer the solution to a glass storage bottle and autoclave for 15 min. Alternatively, the solution can be filter sterilized. Sorbitol solution can be stored for up to 6 months at RT.
Lysis buffer (1 liter; 0.05 M Tris-HCl, 0.02 M EDTA, 1% SDS (pH 12.8))
Add 10 g SDS to 700 ml ddH2O. Stir until dissolved. Add 50 ml of 1 M Tris-HCl (pH 7.5) and 40 ml of 0.5 M EDTA (pH 8). Adjust pH with NaOH (6 N) to pH 12.8. Bring the final volume to 1 liter. Filter-sterilize the solution; store for up to 2–3 months at RT.
RNase A (10 mg ml−1)
Make 10× dilution from 100 mg ml−1 RNase A stock (included in the Qiagen large-construct kit) with sterile water. The solution can be stored for up to 6 months at RT.
ATP (100 mM; 20 ml)
Dissolve 1.1 g of ATP powder in 14 ml of sterilized ddH2O and then add 6 ml of 1 M Tris-HCl (pH 8). Mix the solution and transfer 1-ml aliquots of 100 mM ATP to 1.7-ml Eppendorf tubes. Store the solution at − 20 °C for up to 6 months.
TAE–3 M sodium acetate (10×)
Dissolve 40.8 g of sodium acetate trihydrate in 80 ml of 10× TAE buffer. Adjust pH to 8.0 with glacial acetic acid. Adjust volume to 100 ml with ddH2O and filter sterilize. The solution can be stored for 1 year at RT.
Tris-HCl/EDTA buffer (TE; 1×, pH 8)
Mix 1 ml of 1 M Tris-HCl (pH 8), 0.2 ml of 0.5 M EDTA (pH 8) and 98.8 ml of ddH2O. Filter-sterilize the solution and store for up to 1 year at RT.
CM glucose broth minus HIS
Combine 2% (wt/vol) D-glucose, 0.17% (wt/vol) yeast nitrogen base, 0.5% (wt/vol) (NH4)2SO4, 0.006% (wt/vol) adenine sulfate, 0.006% (wt/vol) uracil, 0.005% (wt/vol) l-arginine-HCl, 0.008% (wt/vol) l-aspartic acid, 0.01% (wt/vol) l-glutamic acid, 0.005% (wt/vol) l-isoleucine, 0.01% (wt/vol) l-leucine, 0.012% (wt/vol) l-lysine-HCl, 0.002% (wt/vol) l-methionine, 0.005% (wt/vol) l-phenylalanine, 0.0375% wt/vol l-serine, 0.01% (wt/vol) l-threonine, 0.005% (wt/vol) l-tryptophan, 0.005% (wt/vol) l-tyrosine, 0.006% (wt/vol) uracil and 0.015% (wt/vol) l-valine.
CM glucose broth minus URA
Combine 2% (wt/vol) d-glucose, 0.17% (wt/vol) yeast nitrogen base, 0.5% (wt/vol) (NH4)2SO4, 0.006% (wt/vol) adenine sulfate, 0.004% (wt/vol) histidine-HCl, 0.005% (wt/vol) l-arginine-HCl, 0.008% (wt/vol) l-aspartic acid, 0.01% (wt/vol) l-glutamic acid, 0.005% (wt/vol) l-isoleucine, 0.01% (wt/vol) l-leucine, 0.012% (wt/vol) l-lysine-HCl, 0.002% (wt/vol) l-methionine, 0.005% (wt/vol) l-phenylalanine, 0.0375% (wt/vol) l-serine, 0.01% (wt/vol) l-threonine, 0.005% (wt/vol) l-tryptophan, 0.005% (wt/vol) l-tyrosine, 0.006% (wt/vol) uracil and 0.015% (wt/vol) l-valine.
CM glucose broth minus TRP
Combine 2% (wt/vol) d-glucose, 0.17% (wt/vol) yeast nitrogen base, 0.5% (wt/vol) (NH4)2SO4, 0.006% (wt/vol) adenine sulfate, 0.006% (wt/vol) uracil, 0.004% (wt/vol) histidine-HCl, 0.005% (wt/vol) l-arginine-HCl, 0.008% (wt/vol) l-aspartic acid, 0.01% (wt/vol) l-glutamic acid, 0.005% (wt/vol) l-isoleucine, 0.01% (wt/vol) l-leucine, 0.012% (wt/vol) l-lysine-HCl, 0.002% (wt/vol) l-methionine, 0.005% (wt/vol) l-phenylalanine, 0.0375% (wt/vol) l-serine, 0.01% (wt/vol) l-threonine, 0.005% (wt/vol) l-tyrosine, 0.006% (wt/vol) uracil and 0.015% (wt/vol) l-valine.
EQUIPMENT SETUP
Agarose gel electrophoresis
DNA is separated on a 1% agarose/1× TAE electrophoresis gel by applying 4.5 V cm−1 for 3 h. After electrophoresis, the gel is stained with SYBR Gold (1/10,000 dilution) for 15–30 min and detected by the Gel Doc System.
FIGE
FIGE analysis is performed in a 1% (wt/vol) agarose gel with 1× TAE buffer without circulation. Typical forward parameters are as follows: 90 V, initial switch 0.1 s, and final switch 10 s, with linear ramp. Typical reverse parameters are: 60 V, initial switch 5.0 s, final switch 30 s, with linear ramp for 14 h. After electrophoresis, the gel is stained with SYBR Gold (1/10,000 dilution) for 15–30 min and detected by the Gel-Doc System.
PROCEDURE
Preparation of yeast cultures ● TIMING ~48 h (two overnight growth steps)
Grow yeast cells, harboring circular YAC, in 10 ml of appropriate selective medium in a BD Falcon 50-ml polypropylene conical tube (loosen the cap to allow aeration and secure it with tape). Incubate overnight (14–20 h) at 30 °C with vigorous shaking (225 r.p.m.).
Transfer 10 ml of overnight culture to a 2-liter Erlenmeyer flask containing 400 ml of appropriate selective medium and continue to grow at 30 °C for 14–20 h at 225 r.p.m.
Measure the optical density at 600 nm (OD600) at regular intervals (every 30 min) after 14 h of growth.
When the OD at 600 nm reaches 1.5, harvest cells by centrifugation in the SLA-1500 rotor at 1,860g, in two 225-ml tubes, for 5 min at RT.
-
Resuspend cell pellets with 100 ml distilled water and transfer cell suspension to two Falcon 50-ml polypropylene conical tubes. Centrifuge again at 1,860g for 5 min and discard the supernatant.
■ PAUSE POINT The cell pellets can be stored at − 20 °C for several weeks.
Cell lysis and DNA isolation ● TIMING 3 h
-
Resuspend each cell pellet with 40 ml of SPE solution.
▲ CRITICAL STEP This protocol has been optimized for processing ~1.2 × 1010 cells from the early stationary-phase culture grown in synthetic dropout medium. The amount of buffers or reagents, such as Zymolyase 20T (Step 7) and DNA exonuclease (Step 16), must be optimized accordingly if you use different amounts of cells, different phases of cell culture or different yeast strains.
-
Add 250 μl of Zymolyase 20T and 40 μl of β-mercaptoethanol. Incubate at 37 °C for 60 min. Measure efficacy of spheroplasting as described in Box 1.
! CAUTION β-Mercaptoethanol is highly toxic. It is toxic to the skin on contact and is harmful if inhaled or swallowed.
▲ CRITICAL STEP When preparing spheroplasts from frozen cells, the amount of Zymolyase should be reduced; the amount required can be determined as described in Box 1.
-
Harvest yeast spheroplasts by centrifugation at 1,294g for 5 min at 4 °C. Discard the supernatant. Resuspend cell pellets in 1 ml of 1 M sorbitol (using a 5-ml pipette).
▲ CRITICAL STEP The cell pellets may be very loose if cells were overdigested. Carefully remove the supernatant to avoid losing the cell pellet. It becomes more difficult to obtain a homogeneous cell suspension when cells have been overdigested.
-
Add 20 ml of lysis buffer to break open the yeast cells. Invert the tube ten times. Incubate at 37 °C for 30 min.
▲ CRITICAL STEP Handle the lysate carefully to avoid shearing of the YAC DNA.
-
Add 20 ml of phenol:chloroform:isoamyl alcohol (equilibrated to RT) and gently invert 20 times; this step removes protein and cell debris. Centrifuge at 4,194g for 20 min at RT.
! CAUTION Phenol is a hazardous chemical and is a toxic and corrosive solution.
-
Collect aqueous phase from each tube (~20 ml of the upper layer) into separate 50-ml Falcon tubes. Add 2 ml of 3 M sodium acetate and 20 ml of isopropanol to each tube to precipitate the DNA. Gently invert the tubes several times. Centrifuge at 4,194g for 30 min at 4 °C.
! CAUTION Isopropanol causes eye and skin irritation.
▲ CRITICAL STEP Avoid collecting the interphase material, which makes the pellets more difficult to dissolve (see Step 15).
-
Discard the supernatant and rinse the pellets with 10 ml of 70% (vol/vol) ethanol; this helps to remove excess salt that may interfere with downstream applications.
■ PAUSE POINT Precipitated pellets can be kept in 70% (vol/vol) ethanol and stored at − 20 °C for several weeks.
Centrifuge at 4,194g for 5 min and remove the supernatant, and then spin again at the same speed for 30 s. Remove the remaining liquid.
Qiagen large-construct kit purification ● TIMING 3 h
-
Add 1 ml of TE (pH 8.0) and 3 μl of RNase A (10 mg ml−1) to the pellets to degrade any cellular RNA. Mix without pipetting. Incubate at 37 °C for 30 min.
▲ CRITICAL STEP Pipetting may cause shearing of the YAC DNA.
-
Gently resuspend DNA pellet using 1-ml large orifice pipette tips. At this point, combine the two DNA samples.
▲ CRITICAL STEP Remove a 20-μl sample and set it aside for DNA analysis at Step 26 (see Fig. 1a). Keep the sample on ice until DNA analysis.
-
Add 10 ml of Buffer EX (included in large-construct kit), 200 μl ATP-dependent exonuclease and 300 μl 100 mM ATP. Invert the tube several times and incubate at 37 °C for 30–35 min. Linear DNA molecules are degraded in this step.
▲ CRITICAL STEP Remove a 20-μl sample for DNA analysis at Step 26 (see Fig. 1a). Keep the sample on ice until DNA analysis.
Equilibrate a Qiagen-tip 500 column by applying 10 ml of Buffer QBT (included in large-construct kit), and then allow the column to empty by gravity flow. This should be done immediately before applying the sample.
Add 12 ml of Buffer QS (included in large-construct kit) to DNA sample from Step 16, and gently invert tubes several times to adjust DNA sample to the buffer, equilibrating the column.
-
Centrifuge at 4,194g for 10 min at 4 °C. Carefully pour the whole sample onto the equilibrated column (Step 17) and allow it to run through the column by gravity flow.
▲ CRITICAL STEP The purpose of centrifugation is to remove any undissolved material, which will reduce the flow rate during column purification.
Wash the column with 30 ml of Buffer QC (included in large-construct kit) and allow it run through the column by gravity flow.
-
Elute DNA with 12 ml of Buffer QF (included in large-construct kit; equilibrated to 55 °C). Collect the eluate in a BD Falcon 50-ml tube.
▲ CRITICAL STEP Remove a 20-μl sample for DNA analysis at Step 26 (see Fig. 1a). Keep the sample on ice until DNA analysis.
-
Add 15 μl of GlycoBlue (15 mg ml−1), 15 μl of tRNA (~10 mg ml−1), 1.2 ml 3 M sodium acetate (pH 5.2) and 12 ml of isopropanol to precipitate the DNA.
▲ CRITICAL STEP Alternatively, DNA can be precipitated by adding two volumes of ethanol in the presence of 15 μl of GlycoBlue (15 mg ml−1), 15 μl of tRNA (~10 mg ml−1) and 1.2 ml of 3 M sodium acetate (pH 5.2), and it can be stored at − 20 °C for several weeks.
Centrifuge at 4,194g for 50 min at 4 °C. Discard the supernatant.
Rinse the pellets with 5 ml of 70% (vol/vol) ethanol and centrifuge at 4,194g for 5 min. Discard the supernatant and centrifuge at the same speed for 30 s. Carefully remove the remaining liquid.
-
Air-dry the pellets for 2 min. Dissolve the pellets in an appropriate volume of TE buffer (i.e., 150 μl when YAC DNA is purified from 400 ml of culture).
▲ CRITICAL STEP Remove two 1-μl samples for DNA analysis at Step 26 and, if required, at Step 36 (see Fig. 1). Keep the samples on ice until DNA analysis.
■ PAUSE POINT At this point, DNA can be stored at 4 °C for 1–2 d.
-
Analyze enrichment of the fraction of supercoiled YAC DNA during the procedure by 1% (wt/vol) agarose gel electrophoresis of the samples taken at Steps 15, 16, 21 and 25 (see Fig. 1a). Large supercoiled DNA molecules migrate according to their size during electrophoresis and the degree of enrichment indicates the quality of YAC DNA purification. Alternatively, quality of YAC DNA can be tested by the digestion of a DNA sample from Step 25 with an appropriate endonuclease, followed by FIGE analysis (see EQUIPMENT SETUP) as shown in Figure 1b,c.
? TROUBLESHOOTING
Topological trapping of circular DNA in agarose plugs (optional) ● TIMING 6 h
Place a microfuge tube containing 50 μl of DNA solution in a digital thermoblock set at 50 °C for 5 min.
Add 50 μl of 2% (wt/vol) LMP agarose, equilibrated to 50 °C, to the DNA sample. Mix gently by stirring with a pipette tip.
Transfer the mixture to a Bio-Rad agarose plug mold. Place on ice for 30 min.
-
Insert solidified plug into the well of a 1% agarose/1× TAE electrophoresis gel. Transfer the gel to an electrophoresis chamber and add enough 1× TAE buffer to cover the plug. Perform constant voltage electrophoresis at 4.5 V cm−1 for 2 h.
▲ CRITICAL STEP Agarose gel and electrophoresis buffer should not contain ethidium bromide because it can cause DNA nicks.
Recover the plug from the agarose gel and transfer it to a microfuge tube. Add 10 μl of 10× TAE/3 M sodium acetate buffer. Keep at 42 °C for 30 min.
-
Transfer the tube to a digital thermoblock set at 68 °C. Keep it there until the agarose is melted.
▲ CRITICAL STEP The time of agarose melting should not exceed 7 min because high temperature can affect DNA integrity.
Equilibrate the melted agarose plug to 42 °C for 15 min. Add 2 μl of β-agarase and mix by pipetting. Continue incubation at 42 °C for 1 h to degrade agarose.
-
Add 100 μl of phenol (buffer saturated), equilibrated to RT before use, to purify DNA from agarose fragments. Place it on a low-speed rotator (8–10 r.p.m.) for 10 min.
! CAUTION Phenol is hazardous; it is toxic and corrosive.
Spin at 16,100g in the Eppendorf centrifuge at RT for 10 min. Transfer the aqueous phase to another microfuge tube.
Add 1 μl of GlycoBlue and 100 μl of isopropanol to precipitate the YAC DNA. Gently invert the tube several times. Centrifuge at 16,100g at RT for 20 min. Remove the supernatant.
-
Wash the pellet with 70% (vol/vol) ethanol and then allow it to air-dry at RT for 5 min. Gently resuspend in appropriate volume of TE buffer (i.e., 30 μl when YAC DNA is purified from 400 ml of culture). Remove a 5-μl sample for DNA analysis. Check DNA quality by restriction digestion followed by FIGE analysis (see EQUIPMENT SETUP). Compare purity of the YAC DNA samples taken before (Step 25) and after topological trapping (Step 37; see Fig. 2b).
■ PAUSE POINT At this point, DNA can be stored at 4°C for 2–3 weeks.
? TROUBLESHOOTING
? TROUBLESHOOTING
Troubleshooting advice can be found in Table 1.
TABLE 1 |.
Troubleshooting table.
| Steps | Problem | Possible reason | Solution |
|---|---|---|---|
| 26 | No (or low yield) supercoiled DNA | Yeast cells grown in wrong medium for selecting YAC (Steps 1–5) | Ensure that you are using the correct growth medium |
| No (or low yield) supercoiled DNA | The pH of the lysis buffer is too high (Step 9) | Make sure the pH is 12.5–12.8 | |
| No (or low yield) supercoiled DNA | Exonuclease overdigestion (Step 16) | Optimize the conditions for exonuclease digestion by titrating the amount of exonuclease or adjusting the reaction time | |
| 26, 37 | No (or low yield) supercoiled DNA | Nicked DNA caused by oxidized phenol (Steps 10 and 35) | Check the quality of phenol:chloroform:isoamyl alcohol |
| No (or low yield) supercoiled DNA | Mechanical DNA shearing (Steps 8–37) | Handle DNA samples carefully in each step of the purification, especially for larger DNA (> 300 kb) Use wide-bore pipette tips to resuspend DNA pellets Mix the DNA solution by stirring with the pipette tip instead of tapping the tube |
|
| 37 | No (or low yield) supercoiled DNA | Agarose digestion is not complete (Step 33) | To ensure complete agarose digestion, incubate β-agarase-digested solution at 4 °C for 20 min. If only partial liquefaction is observed, incubate the reaction for an additional 1 h at 42 °C or add more p-agarase in Step 33 |
Note that all these problems can be visualized by gel electrophoresis analysis with aliquots of DNA samples taken from Steps 15, 16, 21, 26 and 37.
● TIMING
Steps 1–5, Growth of yeast culture: ~48 h (two overnight steps)
Steps 6–13, Cell lysis and DNA isolation: 3 h
Steps 14–25, Circular DNA enrichment: 3 h
Step 26, DNA Supercoiled DNA analysis: 4 h
Steps 27–37, Topological DNA trapping: 6 h
Step 37, Analysis of DNA quality by restriction digestion and FIGE analysis: 20 h
ANTICIPATED RESULTS
Overall, the procedure, from Steps 1–26, produces ~1 μg of 100 kb circular YAC DNA from 1.2 × 1010 yeast cells (Fig. 1). When a higher purity is needed, such as for isolation of DNA for genome assembly19, we recommend including the topological trapping procedure (Steps 27–37; Fig. 2). However, this will reduce DNA recovery to ~0.5 μg, because during this procedure only open circle DNA remains in the agarose plug; the supercoiled DNA is likely to run out of the agarose plug together with the contaminating linear DNA. Nevertheless, the reduction in DNA quantity is an acceptable consequence of the necessity for more highly purified DNA. The size of the YAC insert also affects the yield of YAC DNA. For example, in our laboratory, the yield of the synthetic M. genitalium JCVI genome (~600 kb circular DNA) was ~0.5 μg (from Steps 1 to 26). This may be mainly a result of mechanical shearing of DNA during the purification procedure. Thus, careful handing is essential for obtaining higher quality DNA.
ACKNOWLEDGMENTS
The research reported in this article was supported by Synthetic Genomics (SGI) (V.N.N., R.-Y.C. and D.G.G.) and by the intramural research program of the National Institutes of Health National Cancer Institute, Center for Cancer Research (V.L. and N.K.).
Footnotes
COMPETING FINANCIAL INTERESTS The authors declare no competing financial interests.
References
- 1.Burke T et al. Cloning of large segments of exogeneous DNA into yeast by means of artificial chromosome vectors. Science 236, 806–812 (1987). [DOI] [PubMed] [Google Scholar]
- 2.McGonidal T et al. Construction of a human DNA library in a circular centromere-based yeast plasmid. Gene 155, 267–271 (1995). [DOI] [PubMed] [Google Scholar]
- 3.Zeng C et al. New BAC/YAC libraries allowing to selectively re-isolate a desired genomic region by in vivo recombination in yeast. Genomics 77, 27–34 (2001). [DOI] [PubMed] [Google Scholar]
- 4.Kouprina N et al. Functional copies of a human gene can be directly isolated by TAR cloning with a small 3′ end target sequence. Proc. Natl. Acad. Sci. USA 95, 4469–4474 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shizuya H et al. Cloning and stable maintenance of 300-kilobase-pair fragments of human DNA in Escherichia coli using an F-factor-based vector. Proc. Natl. Acad. Sci. USA 89, 8794–8797 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ioannou PA A new bacteriophage P1-derived vector for the propagation of large human DNA fragments. Nat. Genet 6, 84–89 (1994). [DOI] [PubMed] [Google Scholar]
- 7.Smith DR et al. Amplification of large artificial chromosomes. Proc. Natl. Acad. Sci. USA 87, 8242–8246 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kouprina N & Larionov V Exploiting the yeast Saccharomyces cerevisiae for the study of the organization of complex genomes. FEMS Microbiol. Rev 27, 629–649 (2003). [DOI] [PubMed] [Google Scholar]
- 9.Kouprina N et al. Segments missing from the draft human genome sequence can be isolated by TAR cloning in yeast. EMBO Rep. 4, 257–262 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leem SH et al. Closing the gaps on human chromosome 19 revealed genes with a high density of repetitive tandemly arrayed elements. Genome Res. 14, 239–246 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Larionov V et al. Direct isolation of human BRCA2 gene by transformation-associated recombination in yeast. Proc. Natl. Acad. Sci. USA 94, 7384–7387 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kouprina N & Larionov V TAR cloning: insights into gene function, long-range haplotypes and genome structure and evolution. Nat. Rev. Genet 7, 805–812 (2006). [DOI] [PubMed] [Google Scholar]
- 13.Benders GA et al. Cloning whole bacterial genomes in yeast. Nucleic Acids Res. 38, 2558–2569 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ebersole T et al. Rapid generation of long synthetic tandem repeats and its application for analysis in human artificial chromosome formation. Nucleic Acids Res. 33, e130 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ayabe F et al. A novel expression system for genomic DNA loci using a human artificial chromosome vector with transformation-associated recombination cloning. J. Hum. Genet 50, 592–599 (2005). [DOI] [PubMed] [Google Scholar]
- 16.Iida Y et al. Human artificial chromosome with a conditional centromere for gene delivery and gene expression. DNA Res. 17, 293–301 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gibson DG et al. Complete chemical synthesis, assembly, and cloning of a Mycoplasma genitalium genome. Science 319, 1215–1220 (2008). [DOI] [PubMed] [Google Scholar]
- 18.Gibson DG et al. One-step assembly in yeast of 25 overlapping DNA fragments to form a complete synthetic Mycoplasma genitalium genome. Proc. Natl. Acad. Sci. USA 105, 20404–20409 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gibson DG et al. Creation of a bacterial cell controlled by a chemically synthesized genome. Science 329, 52–56 (2010). [DOI] [PubMed] [Google Scholar]
- 20.Lartigue C et al. Creating bacterial strains from genomes that have been cloned and engineered in yeast. Science 325, 1693–1696 (2009). [DOI] [PubMed] [Google Scholar]
- 21.Den Dunnen JT et al. Reconstruction of the 2.4 Mb human DMD-gene by homologous YAC recombination. Hum. Mol. Genet 1, 19–28 (1992). [DOI] [PubMed] [Google Scholar]
- 22.Larionov V et al. Highly selective isolation of human DNAs from rodent-human hybrid cells as circular YACs by TAR cloning. Proc. Natl. Acad. Sci. USA 93, 13925–13930 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Devenish RJ & Newlon C Isolation and characterization of yeast ring chromosome III by a method applicable to other circular DNAs. Gene 18, 277–288 (1982). [DOI] [PubMed] [Google Scholar]
- 24.Leem SH et al. Purification of circular YACs from yeast for DNA sequencing. Genome 51, 155–158 (2008). [DOI] [PubMed] [Google Scholar]
- 25.Kouprina N et al. Segments missing from the draft human genome sequence can be isolated by TAR cloning in yeast. EMBO Rep. 4, 257–262 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]