

Abstract
Small GTPases of the RAS and RHO families are related signaling proteins that, when activated by growth factors or by mutation, drive oncogenic processes. While activating mutations in KRAS, NRAS, and HRAS genes have long been recognized and occur in many kinds of cancers, similar mutations in RHO family genes such as RAC1 and RHOA have only recently been detected as the result of extensive cancer genome sequencing efforts and are linked to a restricted set of malignancies. In this review, we focus on the role of RAC1 signaling in malignant melanoma, emphasizing recent advances that describe how this oncoprotein alters melanocyte proliferation and motility and how these findings might lead to new therapeutics in RAC1-mutant tumors.
Keywords: malignant melanoma, driver mutations, small GTPases, fast-cycling, signal transduction, effectors, cancer therapeutics
RAC1 pathway activation in cancer
RAC1 is a well-studied and highly conserved member of the RHO family of small GTPases, a family that includes two other RAC isoforms (RAC2 and RAC2), CDC42 and RHO subgroups, and several other less-studied related proteins [1]. Like other enzymes in this class, RAC1 acts as a molecular switch, active when bound to GTP and inactive when bound to GDP. RAC1 is ubiquitously expressed and its downstream effectors are known to play key roles in numerous cellular processes such as proliferation, survival, differentiation, apical-basal polarity, actin dynamics, reactive oxygen species (ROS) production, and inflammatory responses [2–4]. The signaling activity of RAC1 is tightly regulated by activators, including guanine-nucleotide exchange factors (GEFs), and inhibitors, including GTPase activating proteins (GAPs) and guanine-nucleotide disassociation inhibitors (GDIs). In addition, RAC1 is regulated by modifications at it C-terminus, including carboxylmethylation, geranylgeranylation, and palmitoylation, as well as numerous additional posttranslational modifications that influence its localization, quantity, activity, and ability to bind effectors [5, 6]. The RAC1 protein has long been recognized as a central signaling hub that is required for transformation by many oncogenes, but the recent discovery of activating mutations in the RAC1 gene in melanoma and other malignancies suggest a previously unappreciated driver role in cancer and that targeting RAC1 and/or its effectors could be beneficial in this setting. Here, we discuss the role of RAC1 in cancer, advances in our understanding of key signaling pathways altered by activated RAC1, as revealed by new cell and animal models, and the potential therapeutic implications of these findings.
RAC GEFs in Cancer
In the absence of RAC1 mutations, upstream elements of the RAC1 signaling pathway, in particular its GEFs, are often overexpressed and/or activated by mutation in a variety of cancers and may influence drug resistance [6–8]. For example, the phosphatidylinositol-3,4,5-triphosphate-dependent RAC exchangers PREX1 and −2 are commonly overexpressed in cancer, and promote invasion and colony formation in melanoma cells in vitro [9]. High levels of PREX1 protein drive a more migratory, invasive, and adhesive phenotype in ER+ breast cancer cells [10], and breast tumor tissue that expresses PREX1 has a higher recurrence and metastasis rate [11]. The related GEF PREX2 can be activated by overexpression or by missense and/or nonsense mutations that result in truncation of its C-terminal autoinhibitory element [7]. Such mutations in the PREX2 gene result in constitutive activation of the GEF, and are seen in approximately 14% of melanomas [7]. Inducible transgenic mice containing a common cancer-associated Prex2 mutation, when combined with a concurrent NrasQ61K mutation, showed accelerated melanoma development [12]. In this setting, tumor cells displayed increased phospho-AKT, which was abrogated by the RAC1 inhibitor EHT1864. Genes encoding additional RAC1 GEFs such as TIAM1, VAV, ECT2 and DOCK are also frequently altered in human cancers [13], highlighting the ubiquity of RAC pathway activation in malignant disease.
Overexpression of RAC1 and its splice variants
Many lines of evidence have established that RAC1 signaling is important in transformation by other oncogenes. Experiments in the 1990s showed that expression of a dominant negative form of RAC1 blocked transformation of NIH-3T3 cells by HRASG12V [4] (possibly by preventing RAS-induced ruffling and macropinocytosis [14, 15]), and overexpression of an engineered GTPase-deficient form of RAC1 was able to transform these cells. Later experiments showed that tissue-specific deletion of Rac1 prevented tumor formation in murine Kras/Tp53-driven models of lung cancer and pancreatic cancer, respectively [16, 17], and in a DMBA/TPA-induced (Kras mutant) model of skin cancer [18], indicating that Rac1 is essential for transformation by Kras in vivo. These studies established RAC1 as an essential, cell-autonomous element in transformation by the KRAS oncoprotein, but did not address whether activated RAC itself is a bona-fide oncoprotein. At the time of these early studies, “canonical” RAC1 mutations (i.e., those analogous to exon 2 or 4 mutations in RAS) had never been encountered in human tumors or cell lines, so the clinical relevance of these experiments with GTPase-deficient forms was unclear. On the other hand, overexpression of wild-type RAC1 has long been reported in many cancers, including melanoma, lung adenocarcinoma, glioma, pancreatic, prostate, colorectal, gastric, cervical, breast head and neck cancer as well as in various leukemias [17, 19–29]. RAC1 overexpression is correlated with poor prognosis in nearly all cancers [30] and has been shown to confer resistance to both targeted therapy [23, 31] and chemotherapy [32–34]. Also, overexpression of a RAC1 splice variant, termed RAC1b, has been described in breast, lung, and colorectal cancer, and is associated with chemoresistance in the latter [29, 33, 35, 36]. This variant contains an addition of 19 amino acids directly following the switch II domain. This addition impairs the intrinsic GTPase hydrolysis ability of the enzyme [36], and is associated with NF-κB activation, increased activity of matrix metalloproteinase-3, epithelial-mesenchymal transition, and genomic instability [37–40]. These reports indicate that wild-type RAC1 signaling plays a vital role in several oncogenic processes.
RAC1 mutations in cancer
When intentionally mutated and overexpressed, activated RAC1 genes (e.g., RAC1G12V or RAC1Q61L) can transform cells and induce tumor formation in transgenic mice [41], and can cooperate with HRASG12V in tumorigenesis in zebrafish [42], but these forms of RAC1 have only recently been found in a few types of human tumors. Instead, when exome sequencing efforts first revealed the existence of activating mutations in RAC1 in malignant melanoma, the vast majority of these mutations were found to affect codon 29, changing the wild-type proline to either serine or, less commonly, to leucine. Such mutations, occurring at a dipyrimidine (CCT -> TCT) site, represent a typical UV signature and occur in up to 9% of sun-exposed, cutaneous melanomas [23, 43]. Unlike common RAS oncogenic mutations that impair or abolish intrinsic GTP hydrolysis ability and render the protein constitutively active in terms of signaling, the RAC1P29S mutant protein retains the ability to hydrolyze GTP and GDP. Instead, this mutation activates RAC1 by conferring “fast-cycling” properties, in which GDP is exchanged more quickly for GTP [24, 44] (Box 1). The biochemical basis for the increased exchange rate is thought to be related to hydrogen bonding between the backbone carbonyl of Ser29 and the ribose hydroxyl group of GTP, favoring the GTP bound form (Fig. 1). These factors suggest that RAC1P29S may also retain regulation by modulators such as GEFs or GAPs, which in turn may be important when considering therapeutic strategies. This view is supported by the finding that deletion of the gene encoding the RAC-GEF DOCK1 reduces invasion and macropinocytosis in RAC1P29S melanoma and breast cancer cells [45].
Box 1. Fast Cycling GTPase Mutants.
Mutations that alter the intrinsic GTP on-off rate, as opposed to the hydrolysis rate, were first encountered during structure-function studies of HRAS [94]. Such mutants were found to be more potent transforming agents than classical GTPase deficient mutants, leading to the idea that GTPase cycling itself may be required for or may contribute to oncogenic signaling. Alternatively, it is possible that completely GTPase deficient versions are too toxic to most cells, as has been proposed for the fast cycling RHOY42C mutant found in diffuse gastric cancer [95]. Mutants such as RAC1P29S may hit a “sweet-spot” of activity: enough to support transformation, but not so active as to induce senescence or apoptosis.
Figure 1. Structure and Biochemistry of Wild-type RAC1, the Constitutively Active RAC1Q61L mutant, and the Fast Cycling RAC1P29S Mutant.
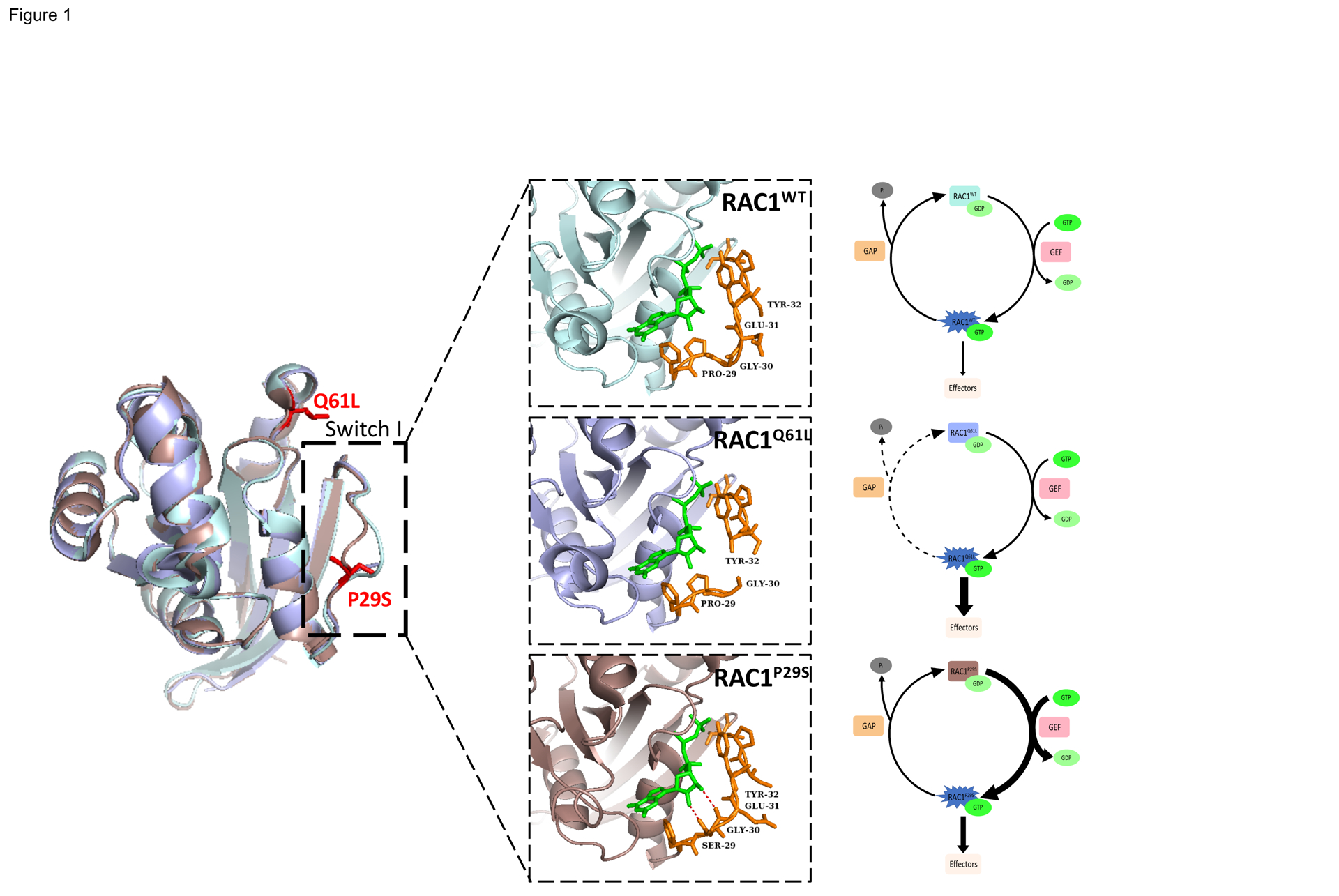
Comparison of crystal structures of RAC1WT (light blue, PDB 3TH5) with RAC1Q61L (light purple, PDB 4GZL), and RAC1P29S (dark pink, PDB 3SBE) in complex with the slow hydrolyzing GTP analog GMPPNP (green). RAC1P29S possesses a more flexible switch I domain than RACWT and RAC1Q61L. The proline to serine substitution enables hydrogen bonding between Ser-29 and Gly-30 of switch I and GTP and constitutes the difference between fast cycling mutations such as P29S and GTP-hydrolysis deficient mutations such as Q61L. Residue 29 and surrounding residues are displayed in orange stick format to illustrate hydrogen bonding (red).
RAC1P29S is the third most common driver mutation in sun-exposed melanoma behind BRAFV600E (~50%) and NRASQ61K/R/L (~30%), [23, 31, 43]. In melanoma, RAC1P29S is often found in combination with additional gain-of-function mutations in other oncogenes (e.g., BRAF or NRAS) and/or loss of function mutations in tumor suppressor genes (e.g., NF1, TP53, or PTEN), suggesting that RAC1P29S is not generally sufficient on its own to drive tumor formation. Mouse models are consistent with this view, as conditional expression of Rac1P29S in melanocytes did not result in the development of nevi or melanoma [46]. However, when combined with BrafV600E and loss of Tp53 or Nf1, the Rac1 mutation caused an acceleration in melanoma growth and reduced survival [46]. In humans, the co-occurrence of BRAF and RAC1 mutations confer a poor prognosis and resistance to BRAF inhibitor targeted therapy, underscoring the clinical significance of these combined mutations [31, 47]. In addition, an analysis of TCGA data suggests that RAC1 mutations are linked to elevated PD-L1 expression in melanoma cells, and thus might confer resistance to immunotherapy [48].
Additional mutations in RAC1 have been found that alter A159 (analogous to A146 in KRAS, which is associated with fast-cycling properties [49]), predominantly in head and neck cancers; N92, in melanoma, myeloma, and sarcoma [50, 51]; C157, and also at the canonical G12, mainly in prostate cancer; and Q61 sites, mainly in testicular germ cell cancer [52], respectively, as often found in HRAS and NRAS (Fig. 2). Other, less common mutations have been noted at additional sites, and these also appear to result in a gain of function. Some of these same gain-of-function mutations (e.g., N92T) have also been found in RAC2 (a gene expressed only in hematopoietic cells) and are associated with myeloid and lymphoid immunodeficiencies [53]. While extensive comparisons of the various RAC1 mutant proteins have not been reported, pull-down assays from HEK293 cells have shown that the Rac1A159V mutant GTPase is more active than RAC1P29S, but less active than RAC1Q61R [22]. However, whether the degree of RAC1 biochemical activation generally correlates with its in vivo effects is not known. As with the RAC1P29S/L mutation in melanoma, the RAC1A159V mutation found in head and neck squamous cell cancers (HNSCC) is associated with poor prognosis. While the relationship between this RAC1 mutation and clinical outcomes is not yet understood in detail, gene set enrichment analyses indicate that active RAC1 is associated with dysfunction of immune-related gene sets [54].
Figure 2. RAC1 Variations and Frequency in Cancer.
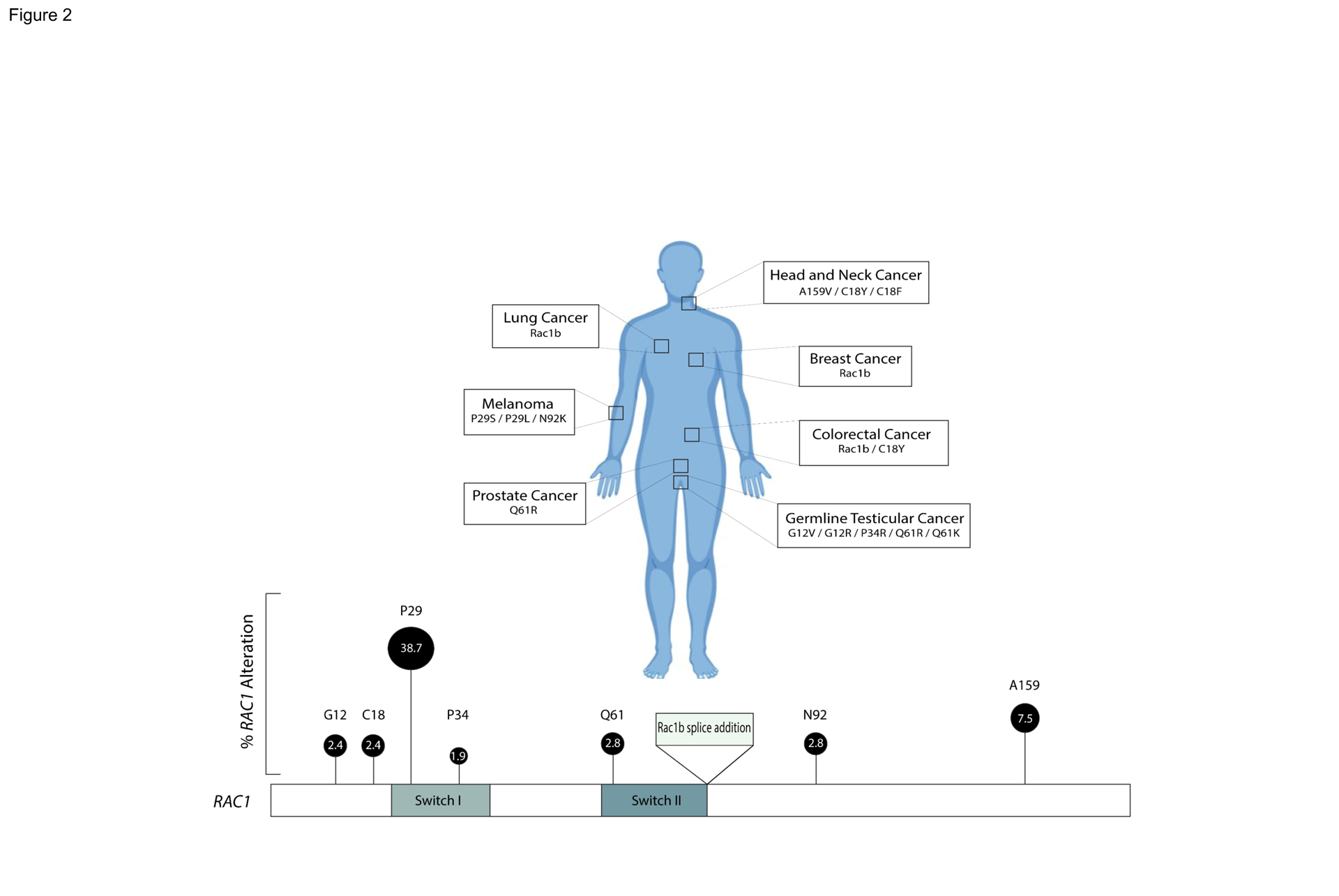
Representation of RAC1 hotspots and associated cancers. RAC1 point mutations associated with various types of cancer are depicted. Some of the most common cancer-associated mutations affect either the switch 1 (residues 26–45) or switch 2 (residues 59–74) region of RAC1. These regions represent key structural elements in the GTPase, mediated interactions with effectors and GEFs, respectively, and contain conserved residues important for nucleotide and Mg2+-ion coordination. The percentage of RAC1 hotspot mutations in RAC1-altered cancer is represented by height and size of locus marker. Frequencies are derived from cbioportal curated set of nonredundant studies.
The role of Rac1 effectors in oncogenesis
Like other small GTPases, RAC1 propagates its downstream biological effects by binding to and activating various effector proteins. Presumably, it is by activating one or more of these effectors that mutant RAC1 contributes to transformation. Among the most studied effectors are the Group A p21-activated protein kinases PAK1, −2, and −3, the lipid kinase phosphatidyl inositol-3 kinase (PI3K)-β, and actin assembly factors such as formins (e.g., mDIA2) and WASP-family verprolin-homologous protein 2 (WAVE2) [55] (Fig. 3). When activated by RAC1, Group A PAKs promote cell proliferation and survival primarily through phosphorylation of c-RAF at S338, MEK1 at S298, β-catenin at S675, and Merlin at S518 [56]. Other validated PAK substrates that are likely relevant to these cellular phenomena include Aurora kinase A and Polo-like kinase 1, which promote cell cycle progression, and BAD, which, when phosphorylated by PAK, dissociates from the pro-apoptotic protein BCL2-XL, thus promoting survival [56]. PAK1 also orchestrates cell invasion and metastasis by phosphorylating LIMK, which in turn phosphorylates cofilin and inhibits its ability to sever actin filaments. For these reasons, PAK likely represents an important effector for mutationally activated RAC1 in promoting transformation. In support of this view, genetic or pharmacological blockade of PAK function reduces the oncogenic effects of RAC1P29S in both cell-based models and animal models of melanoma [46, 57].
Figure 3. Signaling from RAC1 and Potential Vulnerabilities to Targeted Therapy.
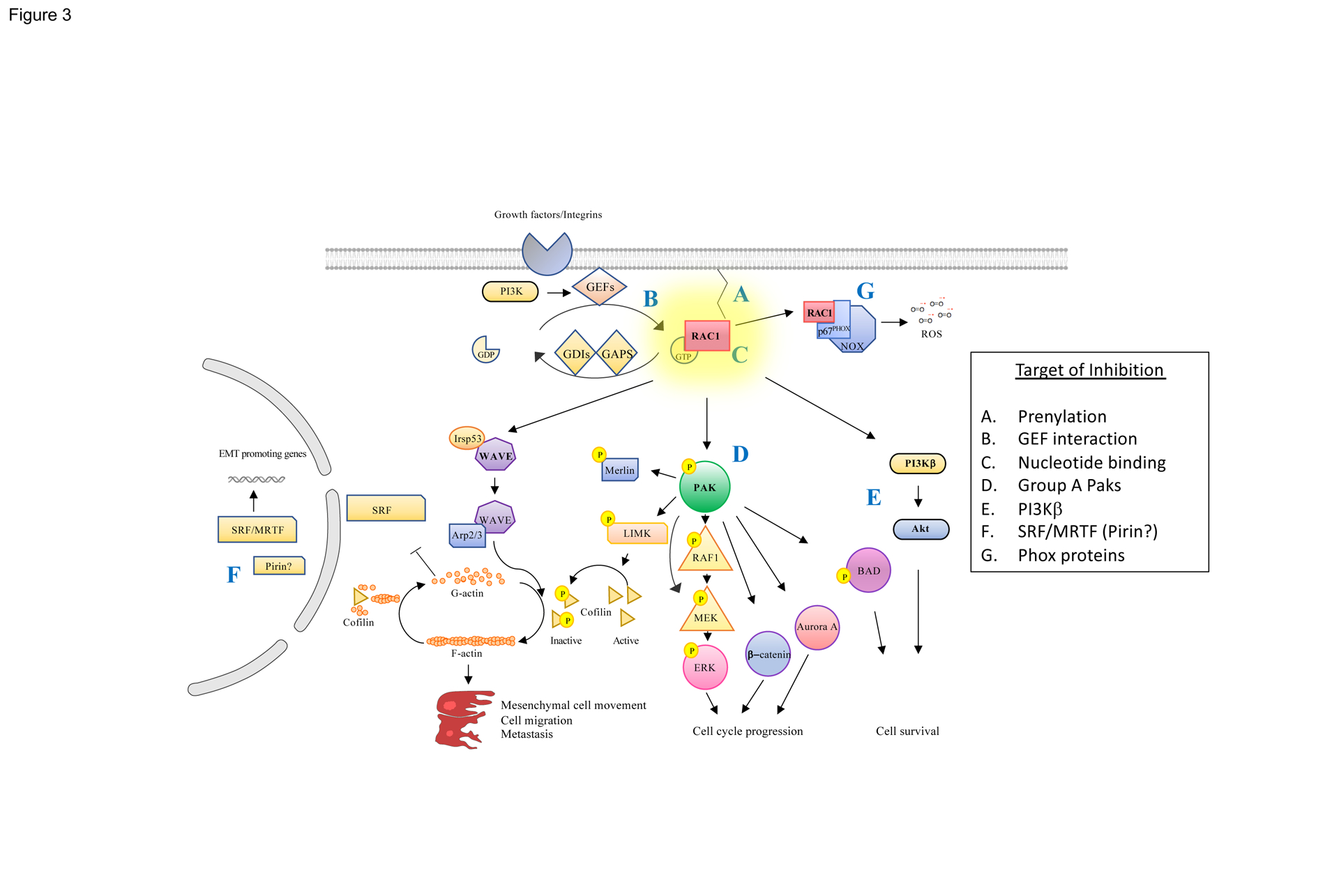
(A) RAC activation requires membrane localization mediated by prenyl modifications at its C-terminus, creating an opportunity for small inhibitors. (B) Fast-cycling mutants such as RAC1P29S, which rapidly exchange GDP for GTP, require stimulation by GEFs, and inhibitors of GEF/RAC interactions have been described, as have (C) molecules that displace bound GTP. (D) Effectors such as PAK (which regulates the activation of ERK, β-catenin, Aurora A, and BAD), (E) PI3Kβ, which regulates cell survival via AKT, and (F) WAVE2, which regulates actin polymerization and cytoskeletal structure, via IRSP53 and ARP2/3, as well as gene transcription via SRF/MRTF and, possibly, Pirin, are also potentially druggable. (G) ROS, generated by the RAC1 effectors p40Phox and p67Phox, may also play a role in oncogenic signaling from this small GTPase and are potentially targetable by small molecule inhibitors.
The WAVE complex and formin proteins represent additional effectors by which RAC1 controls actin polymerization. Active RAC1 activates WAVE2 by binding directly to the amino terminus of the small substrate for insulin receptor, IRSp53, which in turn binds to WAVE2 forming a trimolecular active complex [58, 59]. When activated, WAVE2 binds the ARP2/3 complex which leads to polymerization of monomeric (G-) actin to filamentous (F-) actin, forming lamellipodia. These changes in cytoskeletal structure alter the cellular distribution of the transcription factor Serum Response Factor (SRF) and its coactivator, Myocardin-Related Transcription Factor (MRTF) [60], because SRF/MRTF signaling is highly sensitive to the concentration of G-actin in cells. Under basal cellular conditions, G-actin binds SRF/MRTF and sequesters it in the cytoplasm, and when G-actin is depleted through polymerization SRF/MRTF translocates into the nucleus where it functions as a transcription factor, resulting in the expression of genes associated with motility. Interestingly, while SRF/MRTF had long ago been identified as a potential RAC1 effector [61, 62], it is far better known as an effector of a different small GTPase, RHOA [63]. It was therefore somewhat of a surprise when transcriptional analyses of RAC1P29S melanocytes revealed SRF and many of its targets as among the most upregulated genes compared to wild-type cells [46]. PAK and SRF likely represent independent effectors in RAC1 signaling, as previous mutational analysis of the RAC1 switch one region showed that mutants that selectively uncouple RAC1 from PAK nevertheless maintain lamellipodia formation and SRF activation [61]. The activation of SRF by RAC1 is likely to be independent of RHOA, as treatment of epithelial cells with a potent RHOA inhibitor did not RAC1-mediated activation of this transcription factor [62]. In melanoma, Lionarons et al. further showed that activated SRF/MRTF promotes transcription of epithelial-mesenchymal transition (EMT) genes such as CDH2, VIM, FN1, and COLl5A1 [46]. In melanomas bearing mutations in both BRAF and RAC1, the mesenchymal phenotype of transformed melanocytes may contribute to resistance to BRAF inhibitors. In this setting, combining a BRAF inhibitor and an SRF/MRTF inhibitor augmented drug sensitivity [46].
A similar signaling module may also underlie the recent findings of Mohan et al., who described RAC1P29S signaling within melanocyte lamellipodia [13]. In this schema, RAC1, likely acting via Group A PAKs, stimulates the phosphorylation and inactivation of Merlin, a tumor suppressor protein that, among other activities, regulates actin cytoskeletal remodeling. They found that the presence of these biochemically active, F-actin enriched lamellipodia drove proliferation in the absence of growth factors, independent of MAPK activity. This pathway also mediates resistance to BRAF inhibitors in BRAF/RAC1 double mutant melanoma cells, and such drug resistance can be reversed when actin polymerization is blocked by inhibitors of formins or ARP2/3. It is possible that these effects are mediated by a common mechanism, such as the release of SRF/MRTF to the nucleus, as suggested by Lionarons et al. In both cases, these results emphasize how the effects of Rac1 on the cytoskeleton are intimately linked to its effects on the cell cycle, and how RAC1-mediated changes in actin dynamics can influence tumor cell behavior.
Towards potential therapeutics
The discovery of RAC1 mutations in melanoma and other cancers may provide additional therapeutic targets in such tumors. Like other small GTPases, the mutant RAC1 protein is itself difficult to target directly, as it binds GTP with nM affinity and presents no obvious pockets for small molecule docking, and to date, no FDA-approved drugs are known to be effective in melanomas or other cancers bearing the RAC1P29S mutation. Nevertheless, tumors harboring this mutation might be sensitive to small molecules that impede membrane RAC1 localization, GEF/ RAC1 interactions, nucleotide binding, and/or the binding of effectors proteins.
Interfering with RAC1 localization
The signaling activities of Rac1 require membrane localization and this association is mediated by the addition of isoprenoid moieties to the C-terminus of the protein [64]. Small molecules targeting geranylgeranyl transferases type I (GGTI), the enzyme responsible for this prenylation, have been described [65], and some of these compounds have shown promising cell-based and preclinical results [66–68]. Unfortunately, the only GGTI that was evaluated in a clinical trial did not show efficacy [69, 70]. It should also be noted that, at least in some cell types, loss of GGTI may paradoxically activate RAC1 [71]. Intervening at earlier steps in isoprenoid synthesis, such as blocking HMG-CoA reductase with statins, also reduces RAC1 membrane association and activity [72]. However, this approach suffers from lack of specificity, as many proteins in addition to RAC1 depend on the synthesis of isoprenoid precursors [73, 74].
Inhibiting GEF/RAC1 interactions
Unlike most RAS mutants, RAC1 mutant proteins are usually fast-cycling rather than GTPase deficient. Given these enzymatic properties, it is attractive to target its upstream signaling elements, in particular RAC1 GEFs. In support of this idea, genetic or pharmacologic disruption of the RAC1 GEF DOCK1 has been shown to suppress Rac1P29S GDP/GTP exchange and to reduce matrix invasion and macropinocytosis in RAC1-mutant mouse embryonic fibroblasts, human melanoma IGR-1, and breast cancer MDA-MB-157 cells [45]. Thus, the general strategies for selecting treatment for tumors bearing fast-cycling RAC1 mutants might differ from those developed for RAS mutants, which most often result in near-total loss of GTPase activity and lack of response to GEFs.
A number of compounds have been described that interfere with GEF/ RAC1 interactions. Among these are NSC23766 and its derivatives such as EHop-016 and MBQ-167 [75]. NSC23766 inhibits TIAM1- and TRIO-mediated cell growth and transformation, reversing tumor cell phenotypes in prostate cancer cells without affecting CDC42 and RHOA activation [76]. However, NSC23766 has been shown to inhibit agonist signaling in Rac1−/− platelets [77] and to interfere with muscarinic acetylcholine receptors (mAChRs) in cardiac myocytes [78], raising serious questions as to its specificity and mechanism(s) of action. In addition, this compound has very low efficacy, making it poorly suited for clinical application [79]. A derivative compound, EHop-016, blocks RAC1/VAV2 rather than RAC1/TRIO binding, decreases RAC1-mediated activation of PAK1, and suppresses RAC1-driven directed cell migration of metastatic cancer cells. A drawback of this compound, however, is that, when used at high concentrations, it also targets CDC42 [80]. The latest molecule in this series, MBQ-167, is the first to display sub-micromolar IC50 values for RAC1 inhibition, although it too inhibits CDC42 activation [81]. MBQ-167 was shown to display profound effects on the growth of xenografted GFP-HER2-BM breast cancer cells in nude mice without causing undue toxicity, indicating that this compound has promise for further development. As this compound has only been tested in a limited number of cell types, it will be important to determine if its beneficial effects are related to RAC pathway activity in cancer cells. Other candidate molecules designed to interfere with GEF/RAC1 interactions include ZINC69391and its derivative 1A-116, which block the interaction of P-REX1, and perhaps other GEFs, with RAC1, and which have shown anti-metastatic effects in breast cancer models [82].
Impairing nucleotide binding
As small GTPases such as RAC1 have very high affinity for GTP, it seems unlikely that a small molecule could be designed to outcompete it. Nevertheless, a molecule that causes displacement of bound GTP from RAC, but not CDC42 or RHO, has been described [83]. This compound, EHT1864, binds to all RAC isoforms, displacing bound nucleotides and preventing GEF-mediated nucleotide exchange, as well as impeding RAC binding to downstream effectors. Unfortunately, like the GEF/GTPase blocker NSC23766, EHT1864 also has notable off-target effects in wild-type cells, including inhibition of mAChRs [77]. Despite these issues, EHT1864 remains in wide use in preclinical studies and, given its unique proposed mechanism of action that directly targets RAC1, might provide a basis for further development.
Targeting Rac effectors
Despite efforts to target Rac1 activators or RAC1 itself, as detailed above, to date the most effective small molecule approach for blocking RAC1 signaling has been to target its effectors and further downstream elements, in particular readily druggable enzymes such as protein or lipid kinases. For RAC1, this means considering protein kinases such as the aforementioned group A PAKs (PAK1, −2, and −3), MEK kinases (MEKKs), mixed lineage kinases (MLKs), and p70S6K; lipid modifying enzymes such as PI3Kβ, PI45K, DAG kinase, PLC, and PLD; proteins that promote ROS generation such as p47phox and p67phox, and actin-modifying proteins such as WAVE2 (via IRSp53) leading to activation of SRF/MRTF, formins, and IQGAP [84]. Of these effectors, PAK is the best characterized druggable target downstream of RAC1. PAKs contribute to the ERK, β-catenin, Aurora A, and Merlin activation, and melanoma cells and xenografts bearing the RAC1P29S mutation have been shown to be sensitive to PAK inhibitors [31, 57]. In addition, the Rasopathy-like developmental phenotypes conferred by RAC1P29S expression in zebrafish embryos can be reversed by PAK inhibitors [57]. Unfortunately, despite these promising data, a clinical path forward remains uncertain due to the essential role of PAK2 in cardiac function in adult organisms [85]. It remains possible that selective PAK1 inhibitors, such as that described by a group from Novartis [86], could be further developed for use in RAC1 mutant or -dependent tumors.
With respect to PI3K as a potential therapeutic target, RAC1 selectively engages the PI3Kβ isoform to activate AKT [87], and therefore isoform-specific inhibitors of this enzyme might be effective in RAC1-driven tumors. To date, the data are mixed on this point, with one study showing that selective PI3Kβ inhibitors impeded the growth and migration of melanoma cell lines driven by mutant RAC1 but not by mutant BRAF, while selective PI3Kα inhibitors had the opposite profile [88], and another study showing limited activity for any PI3K inhibitor in RAC1P29S melanocytes. In this latter study, however, an inhibitor of AKT3 was effective, implying some role for the PI3K/AKT signaling axis in such tumor cells [46].
RAC1P29S activates the SRF/MRTF transcriptional pathway and the WAVE2/ARP2/3 actin complex, which results in a melanocytic to mesenchymal phenotypic switch and the formation of filamentous actin that increases mesenchymal cell movement, migration and metastasis. Depleting MRTF using RNAi or using the specific SRF/MRTF inhibitors CCG-1423 and CCG-203971, respectively, suppressed the melanocytic to mesenchymal transition in melanoma cell lines. In mice, co-treatment with the SRF/MRTF inhibitor CCG-257081 and the BRAF inhibitor PLX4720 suppressed tumor growth [46], and in zebrafish embryos, an SRF/MRTF inhibitor (CCG-203971) reduced the abnormalities induced by the injection of RAC1P29S mRNA [88]. It should be noted that the exact molecular target of this series of SRF/MRTF pathway inhibitors is uncertain. Rather than affecting SRF or MRTF directly, a recent report showed that these compounds bind to Pirin, an iron-binding co-transcription factor not previously linked to SRF/MRTF [89]. Given these findings, whether Pirin also plays a role in oncogenic Rac1 signaling is a question well worth asking, especially as Pirin has previously been implicated in melanoma cell senescence, migration, and progression [90–92]. Of note, degrader versions of a Pirin inhibitor has already been described that effectively abolish Pirin expression in cells when used at nM concentrations [93]. Other small molecule inhibitors of actin nucleation and/or polymerization, such as those that target Arp2/3 or formins, might also be of use in RAC1 mutant tumors, as suggested by Mohan et al. [13].
Concluding remarks
In the short time since RAC1 activating mutations were first identified in malignant melanoma, the presence of such mutations has also been recognized in other human cancers and useful cell-based and animal models constructed. Importantly, the outline of the oncogenic signaling mechanisms employed by RAC1 has become clearer, emphasizing the role of some expected effectors such as the PAKs, while also revealing some surprising new candidates, in particular SRF/MRTF, which highlights an important but often ignored link between actin-induced cytoskeletal changes and oncogenic transcriptional events. These findings in turn suggest new therapeutic strategies that could be of use in cancers driven by this mutation. However, as neither PAK nor SRF/MRTF inhibitors are currently in clinical use or being tested in human trials, there is still much work to be done in translating these findings into practice. Future work will need to address several critical issues, such as the biology underlying the unusual spectrum of mutations encountered in RAC1-mutant cancers, their role in therapeutic resistance, and the possibility of blocking oncogenic RAC1 signaling via inhibiting specific GEFs, direct effectors such as PAKs and/or PI3K isoforms, and/or proteins that regulate actin polymerization. Finally, given the recent rapid progress in RAS-targeting inhibitors, it is also possible that direct inhibitors of mutant RAC1 might be developed. Such therapeutics might help address the drug-resistant phenotype associated with RAC1 mutations, thus providing a new weapon in our armamentarium against cancer.
Acknowledgments
This work is supported by NIH grants R01CA142928, R01CA148805, and R01CA227184, and a grant from the Melanoma Research Foundation. We apologize to our colleagues whose works are not cited because of space limitations.
References
- 1.Sorokina EM and Chernoff J (2005) Rho-GTPases: New members, new pathways. J Cell Biochem 94, 225–31. [DOI] [PubMed] [Google Scholar]
- 2.Mack NA et al. (2011) The diverse roles of Rac signaling in tumorigenesis. Cell Cycle 10, 1571–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Etienne-Manneville S and Hall A (2002) Rho GTPases in cell biology. Nature 420, 629–35. [DOI] [PubMed] [Google Scholar]
- 4.Qiu RG et al. (1995) An essential role for Rac in Ras transformation. Nature 374, 457–9. [DOI] [PubMed] [Google Scholar]
- 5.Marcar L et al. (2019) Acquired resistance of EGFR-mutated lung cancer to tyrosine kinase inhibitor treatment promotes PARP inhibitor sensitivity. Cell Rep 27, 3422–3432 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wertheimer E et al. (2012) Rac signaling in breast cancer: A tale of GEFs and GAPs. Cell Signal 24, 353–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berger MF et al. (2012) Melanoma genome sequencing reveals frequent PREX2 mutations. Nature 485, 502–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaneto N et al. (2014) Rac1 inhibition as a therapeutic target for gefitinib-resistant non-small-cell lung cancer. Cancer Sci 105, 788–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ryan MB et al. (2016) Erk/Mapk signaling drives overexpression of the Rac-Gef, Prex1, in BRAF- and NRAS-mutant melanoma. Mol Cancer Res 14, 1009–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clements ME and Johnson RW (2019) Prex1 drives spontaneous bone dissemination of ER+ breast cancer cells. Oncogene. [DOI] [PMC free article] [PubMed]
- 11.Zhong Y et al. (2019) Phosphatidylinositol-3,4,5-trisphosphate dependent Rac exchange factor 1 (Prex1) is a novel predictor of prognosis for breast cancer patients: A retrospective case series. Med Sci Monit 25, 6554–6562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lissanu Deribe Y et al. (2016) Truncating Prex2 mutations activate its GEF activity and alter gene expression regulation in Nras-mutant melanoma. Proc Natl Acad Sci U S A 113, E1296–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mohan AS et al. (2019) Enhanced dendritic actin network formation in extended lamellipodia drives proliferation in growth-challenged Rac1(P29S) melanoma cells. Dev Cell 49, 444–460 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ridley AJ et al. (1992) The small GTP-binding protein Rac regulates growth factor-induced membrane ruffling. Cell 70, 401–10. [DOI] [PubMed] [Google Scholar]
- 15.Joneson T et al. (1996) Stimulation of membrane ruffling and MAP kinase activation by distinct effectors of Ras. Science 271, 810–2. [DOI] [PubMed] [Google Scholar]
- 16.Kissil JL et al. (2007) Requirement for Rac1 in a K-ras induced lung cancer in the mouse. Cancer Res 67, 8089–94. [DOI] [PubMed] [Google Scholar]
- 17.Heid I et al. (2011) Early requirement of Rac1 in a mouse model of pancreatic cancer. Gastroenterology 141, 719–30, 730 e1–7. [DOI] [PubMed] [Google Scholar]
- 18.Wang Z et al. (2010) Rac1 is crucial for Ras-dependent skin tumor formation by controlling Pak1-Mek-Erk hyperactivation and hyperproliferation in vivo. Oncogene 29, 3362–73. [DOI] [PubMed] [Google Scholar]
- 19.Tian T et al. (2018) Rac1 is a novel therapeutic target in mantle cell lymphoma. Blood Cancer J 8, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang J et al. (2009) Overexpression of Rac1 in leukemia patients and its role in leukemia cell migration and growth. Biochem Biophys Res Commun 386, 769–74. [DOI] [PubMed] [Google Scholar]
- 21.Wang JY et al. (2013) Activation of Rac1 GTPase promotes leukemia cell chemotherapy resistance, quiescence and niche interaction. Mol Oncol 7, 907–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chang MT et al. (2016) Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat Biotechnol 34, 155–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krauthammer M et al. (2012) Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet 44, 1006–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davis MJ et al. (2013) Rac1P29S is a spontaneously activating cancer-associated gtpase. Proc Natl Acad Sci U S A 110, 912–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fritz G et al. (1999) Rho GTPases are over-expressed in human tumors. Int J Cancer 81, 682–7. [DOI] [PubMed] [Google Scholar]
- 26.Juric D et al. (2018) Phosphatidylinositol 3-kinase alpha-selective inhibition with alpelisib (BYL719) in PIK3ca-altered solid tumors: Results from the first-in-human study. J Clin Oncol 36, 1291–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ji J et al. (2015) Rac1 is correlated with aggressiveness and a potential therapeutic target for gastric cancer. Int J Oncol 46, 1343–53. [DOI] [PubMed] [Google Scholar]
- 28.Kamai T et al. (2004) Overexpression of Rhoa, Rac1, and Cdc42 gtpases is associated with progression in testicular cancer. Clin Cancer Res 10, 4799–805. [DOI] [PubMed] [Google Scholar]
- 29.Schnelzer A et al. (2000) Rac1 in human breast cancer: Overexpression, mutation analysis, and characterization of a new isoform, Rac1b. Oncogene 19, 3013–20. [DOI] [PubMed] [Google Scholar]
- 30.Lou S et al. (2018) Prognostic and clinicopathological value of Rac1 in cancer survival: Evidence from a meta-analysis. J Cancer 9, 2571–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Watson IR et al. (2014) The Rac1 P29S hotspot mutation in melanoma confers resistance to pharmacological inhibition of raf. Cancer Res 74, 4845–4852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu M et al. (2019) FLT3/ITD cooperates with Rac1 to modulate the sensitivity of leukemic cells to chemotherapeutic agents via regulation of DNA repair pathways. Haematologica. [DOI] [PMC free article] [PubMed]
- 33.Goka ET et al. (2019) Rac1b overexpression confers resistance to chemotherapy treatment in colorectal cancer. Mol Cancer Ther 18, 957–968. [DOI] [PubMed] [Google Scholar]
- 34.Skvortsov S et al. (2014) Rac1 as a potential therapeutic target for chemo-radioresistant head and neck squamous cell carcinomas (hnscc). Br J Cancer 110, 2677–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jordan P et al. (1999) Cloning of a novel human Rac1b splice variant with increased expression in colorectal tumors. Oncogene 18, 6835–9. [DOI] [PubMed] [Google Scholar]
- 36.Zhou C et al. (2013) The Rac1 splice form Rac1b promotes K-ras-induced lung tumorigenesis. Oncogene 32, 903–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matos P and Jordan P (2005) Expression of Rac1b stimulates NF-kappab-mediated cell survival and G1/S progression. Exp Cell Res 305, 292–9. [DOI] [PubMed] [Google Scholar]
- 38.Faria M et al. (2017) Rac1b overexpression stimulates proliferation and NF-kB-mediated anti-apoptotic signaling in thyroid cancer cells. PLoS One 12, e0172689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Radisky DC et al. (2005) Rac1b and reactive oxygen species mediate MMP-3-induced emt and genomic instability. Nature 436, 123–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Singh A et al. (2004) Rac1b, a tumor associated, constitutively active Rac1 splice variant, promotes cellular transformation. Oncogene 23, 9369–80. [DOI] [PubMed] [Google Scholar]
- 41.Ma Q et al. (2009) Antitumorigenesis of antioxidants in a transgenic Rac1 model of kaposi’s sarcoma. Proc Natl Acad Sci U S A 106, 8683–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dalton LE et al. (2013) Constitutive Rac activation is not sufficient to initiate melanocyte neoplasia but accelerates malignant progression. J Invest Dermatol 133, 1572–81. [DOI] [PubMed] [Google Scholar]
- 43.Hodis E et al. (2012) A landscape of driver mutations in melanoma. Cell 150, 251–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Killoran RC and Smith MJ (2019) Conformational resolution of nucleotide cycling and effector interactions for multiple small GTPases determined in parallel. J Biol Chem 294, 9937–9948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tomino T et al. (2018) Dock1 inhibition suppresses cancer cell invasion and macropinocytosis induced by self-activating Rac1(P29S) mutation. Biochem Biophys Res Commun 497, 298–304. [DOI] [PubMed] [Google Scholar]
- 46.Lionarons DA et al. (2019) Rac1(P29S) induces a mesenchymal phenotypic switch via serum response factor to promote melanoma development and therapy resistance. Cancer Cell 36, 68–83 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Van Allen EM et al. (2014) The genetic landscape of clinical resistance to Raf inhibition in metastatic melanoma. Cancer Discov 4, 94–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vu HL et al. (2015) Rac1 P29S regulates PD-L1 expression in melanoma. Pigment Cell Melanoma Res 28, 590–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Poulin EJ et al. (2019) Tissue-specific oncogenic activity of Kras(A146T). Cancer Discov 9, 738–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cerami E et al. (2012) The cbio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov 2, 401–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gao J et al. (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cbioportal. Sci Signal 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bagrodia A et al. (2016) Genetic determinants of cisplatin resistance in patients with advanced germ cell tumors. J Clin Oncol 34, 4000–4007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sharapova SO et al. (2019) Heterozygous activating mutation in RAC2 causes infantile-onset combined immunodeficiency with susceptibility to viral infections. Clin Immunol 205, 1–5. [DOI] [PubMed] [Google Scholar]
- 54.Ngan HL et al. (2019) RAC1 genomic aberrations as predictive biomarkers for head and neck squamous cell carcinoma (hnscc). Cancer Res 79, 4033. [Google Scholar]
- 55.Yang HW et al. (2012) Cooperative activation of PI3K by Ras and Rho family small gtpases. Mol Cell 47, 281–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Radu M et al. (2014) Pak signalling during the development and progression of cancer. Nat Rev Cancer 14, 13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Araiza-Olivera D et al. (2018) Suppression of Rac1-driven malignant melanoma by group a Pak inhibitors. Oncogene 37, 944–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Miki H and Takenawa T (2002) Wave2 serves a functional partner of IRSp53 by regulating its interaction with Rac. Biochem Biophys Res Commun 293, 93–9. [DOI] [PubMed] [Google Scholar]
- 59.Lane J et al. (2014) Structure and role of Wasp and Wave in Rho GTPase signalling in cancer. Cancer Genomics Proteomics 11, 155–65. [PubMed] [Google Scholar]
- 60.Posern G and Treisman R (2006) Actin’ together: Serum response factor, its cofactors and the link to signal transduction. Trends Cell Biol 16, 588–96. [DOI] [PubMed] [Google Scholar]
- 61.Westwick JK et al. (1997) Rac regulation of transformation, gene expression, and actin organization by multiple, pak-independent pathways. Mol Cell Biol 17, 1324–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Busche S et al. (2008) Epithelial cell-cell contacts regulate SRF-mediated transcription via Rac-actin-Mal signalling. J Cell Sci 121, 1025–35. [DOI] [PubMed] [Google Scholar]
- 63.Treisman R et al. (1998) Regulation of SRF activity by Rho family GTPases. Cold Spring Harb Symp Quant Biol 63, 643–51. [DOI] [PubMed] [Google Scholar]
- 64.Abdrabou A and Wang Z (2018) Post-translational modification and subcellular distribution of Rac1: An update. Cells 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ullah N et al. (2016) Protein geranylgeranyltransferase type 1 as a target in cancer. Curr Cancer Drug Targets 16, 563–71. [DOI] [PubMed] [Google Scholar]
- 66.Navarro-Lerida I et al. (2012) A palmitoylation switch mechanism regulates Rac1 function and membrane organization. EMBO J 31, 534–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cicha I et al. (2004) Monitoring the cellular effects of HMG-CoA reductase inhibitors in vitro and ex vivo. Arterioscler Thromb Vasc Biol 24, 2046–50. [DOI] [PubMed] [Google Scholar]
- 68.Ferri N et al. (2007) Simvastatin reduces MMP1 expression in human smooth muscle cells cultured on polymerized collagen by inhibiting rac1 activation. Arterioscler Thromb Vasc Biol 27, 1043–9. [DOI] [PubMed] [Google Scholar]
- 69.Baines AT et al. (2011) Inhibition of Ras for cancer treatment: The search continues. Future Med Chem 3, 1787–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ochocki JD and Distefano MD (2013) Prenyltransferase inhibitors: Treating human ailments from cancer to parasitic infections. Medchemcomm 4, 476–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Khan OM et al. (2011) Geranylgeranyltransferase type i (GGTase-I) deficiency hyperactivates macrophages and induces erosive arthritis in mice. J Clin Invest 121, 628–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Miller T et al. (2011) Simvastatin stimulates apoptosis in cholangiocarcinoma by inhibition of rac1 activity. Dig Liver Dis 43, 395–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhou Q and Liao JK (2009) Statins and cardiovascular diseases: From cholesterol lowering to pleiotropy. Curr Pharm Des 15, 467–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Draper JM and Smith CD (2009) Palmitoyl acyltransferase assays and inhibitors (review). Mol Membr Biol 26, 5–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Maldonado MDM and Dharmawardhane S (2018) Targeting Rac and Cdc42 gtpases in cancer. Cancer Res 78, 3101–3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gao Y et al. (2004) Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc Natl Acad Sci U S A 101, 7618–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dutting S et al. (2015) Critical off-target effects of the widely used rac1 inhibitors nsc23766 and eht1864 in mouse platelets. J Thromb Haemost 13, 827–38. [DOI] [PubMed] [Google Scholar]
- 78.Levay M et al. (2013) NSC23766, a widely used inhibitor of Rac1 activation, additionally acts as a competitive antagonist at muscarinic acetylcholine receptors. J Pharmacol Exp Ther 347, 69–79. [DOI] [PubMed] [Google Scholar]
- 79.Marei H and Malliri A (2017) Rac1 in human diseases: The therapeutic potential of targeting Rac1 signaling regulatory mechanisms. Small GTPases 8, 139–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Castillo-Pichardo L et al. (2014) The Rac inhibitor EHOP-016 inhibits mammary tumor growth and metastasis in a nude mouse model. Transl Oncol 7, 546–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Humphries-Bickley T et al. (2017) Characterization of a dual Rac/Cdc42 inhibitor MBQ-167 in metastatic cancer. Mol Cancer Ther 16, 805–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cardama GA et al. (2014) Preclinical development of novel Rac1-GEF signaling inhibitors using a rational design approach in highly aggressive breast cancer cell lines. Anticancer Agents Med Chem 14, 840–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shutes A et al. (2007) Specificity and mechanism of action of EHT 1864, a novel small molecule inhibitor of Rac family small gtpases. J Biol Chem 282, 35666–78. [DOI] [PubMed] [Google Scholar]
- 84.Cotteret S and Chernoff J (2002) The evolutionary history of effectors downstream of Cdc42 and Rac. Genome Biol 3, REVIEWS0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rudolph J et al. (2016) Chemically diverse group I p21-activated kinase (Pak) inhibitors impart acute cardiovascular toxicity with a narrow therapeutic window. J Med Chem 59, 5520–41. [DOI] [PubMed] [Google Scholar]
- 86.Karpov AS et al. (2015) Optimization of a dibenzodiazepine hit to a potent and selective allosteric Pak1 inhibitor. ACS Med Chem Lett 6, 776–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Fritsch R et al. (2013) Ras and Rho families of gtpases directly regulate distinct phosphoinositide 3-kinase isoforms. Cell 153, 1050–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Uribe-Alvarez C et al. Targeting effector pathways in Rac1P29S-driven malignant melanoma. Small GTPases (in press). [DOI] [PMC free article] [PubMed]
- 89.Lisabeth EM et al. (2019) Identification of pirin as a molecular target of the CCG-1423/CCG-100. [DOI] [PMC free article] [PubMed]
- 90.Licciulli S et al. (2011) Pirin inhibits cellular senescence in melanocytic cells. Am J Pathol 178, 2397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Miyazaki I et al. (2010) A small-molecule inhibitor shows that pirin regulates migration of melanoma cells. Nat Chem Biol 6, 667–73. [DOI] [PubMed] [Google Scholar]
- 92.Licciulli S et al. (2010) Pirin delocalization in melanoma progression identified by high content immuno-detection based approaches. BMC Cell Biol 11, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chessum NEA et al. (2018) Demonstrating in-cell target engagement using a pirin protein degradation probe (CCT367766). J Med Chem 61, 918–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Reinstein J et al. (1991) P21 with a phenylalanine 28----leucine mutation reacts normally with the GTPase activating protein GAP but nevertheless has transforming properties. J Biol Chem 266, 17700–6. [PubMed] [Google Scholar]
- 95.Zhang H et al. (2019) Gain-of-function rhoa mutations promote focal adhesion kinase activation and dependency in diffuse gastric cancer. Cancer Discov. [DOI] [PMC free article] [PubMed]